

Summary
Adeno-associated virus (AAV) vectors are promising tools for gene therapy. The current AAV vector system produces an abundance of empty capsids that are eliminated before clinical use, leading to increased costs for gene therapy. In the present study, we established an AAV production system that regulates the timing of capsid expression using a tetracycline-dependent promoter. Tetracycline-regulating capsid expression increased viral yield and reduced empty capsids in various serotypes without altering AAV vector infectivity in vitro and in vivo. The replicase expression pattern change observed in the developed AAV vector system improved viral quantity and quality, whereas timing control of capsid expression reduced empty capsids. These findings provide a new perspective on the development of AAV vector production systems in gene therapy.
Subject areas: Virology, Biotechnology
Graphical abstract
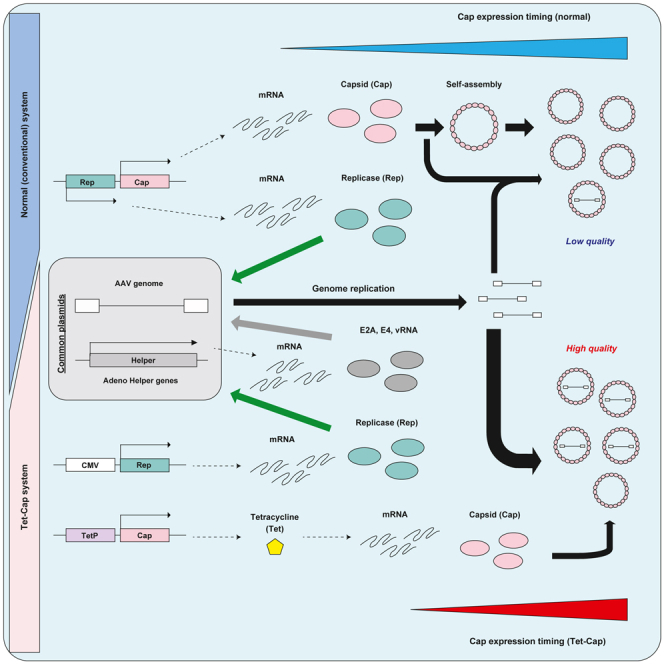
Highlights
-
•
Novel AAV vector system controlling Cap expression timing (Tet-Cap) was established
-
•
Tet-Cap system was applicable for various serotypes in AAV vector production
-
•
Rep expression pattern change affected the AAV vector yield and quality
-
•
Controlling Cap expression timing mainly improved the AAV vector quality
Virology; Biotechnology
Introduction
The adeno-associated virus (AAV) vector is thought to be a promising tool for gene therapy, and many clinical trials are ongoing globally.1,2,3,4,5,6 However, there are still several issues in current AAV vector system, such as generation of large numbers of empty capsids during vector production,7,8 toxicity because of broad tissue tropism of the AAV vector,9,10,11 and AAV elimination by pre-existing neutralizing antibodies.12,13 In particular, the low degree of tissue-specific delivery necessitates an increased dose for optimal treatment, resulting in higher costs to prepare large amounts of AAV vectors for gene therapy.14,15,16,17 In addition, the current AAV vector system produces an abundance of empty capsids during vector propagation, leading to a large-scale production system that requires multistep purifications for the elimination of empty capsids for clinical use. Hence, because cost is a major factor in gene therapy, an AAV production system that can reduce costs by improving productive efficiency would be favorable.
A reduction in the number of generated empty capsids can reduce the scale of AAV production and purification steps required to eliminate them, thereby reducing the process cost. Many studies have attempted to improve the AAV vector production system, particularly to increase the total yield of vectors.7,18,19,20 To increase AAV vector yield, the optimizing helper genes and replicase/capsid (Rep/Cap) expression (particularly the Rep gene), change of producer cells and helper viruses from adenovirus to herpes virus, and production system in baculovirus have been well analyzed.7,18,20 However, for improvement in AAV vector quantity and quality, modifications to the AAV vector system focusing on the nature of the Cap protein is limited.
The Cap protein is a fundamental component of viruses. It encapsulates and protects the genome of enveloped and non-enveloped viruses and plays a vital role in target cell entry in non-enveloped viruses.21,22 Cap proteins can naturally self-assemble because of their ability to form particles.23,24,25,26 This is considered a reason for the generation of empty capsids (empty particles). In fact, there are many reports that Cap expression creates a substantial number of empty capsids or virus-like particles (VLPs) in various viruses including AAV, human immunodeficiency virus type 1 (HIV-1), human papillomavirus (HPV), and human hepatitis B virus.27,28,29 VLPs, including empty capsids, are useful for analyzing viral life cycle in laboratory research; however, these are unnecessary or harmful in clinical applications because viral factors can act as immunogens that activate immunity or trigger inflammation.28,29,30 Therefore, empty capsids should be eliminated before using AAV vectors in gene therapy.
The mechanism of self-assembly by single Cap expression in cells is thought to be the same as that of particle formation in the viral life cycle. Therefore, many studies have used VLP formation to analyze the molecular mechanism of Cap proteins in vitro.31,32,33,34,35,36,37,38 Meanwhile, manufacturing an efficient AAV vector production system requires improvements using Cap biology along the viral life cycle for the reduction of empty capsids. Mature viruses (full particles), which contain viral genomes, are generated under proper conditions and timing in cells. It is assumed that fewer viral genomes in cells results in the generation of empty capsids rather than mature viruses. Because Rep, which regulates viral gene expression and transcribes viral genomes, and Cap genes are placed in one plasmid in the current AAV vector production system, it is predicted that a considerable amount of Cap protein is expressed before sufficient preparation of the viral genome, resulting in an increase of empty capsids. This raises the possibility that controlling Cap expression timing until sufficient genome is prepared can improve AAV vector productivity, particularly by reducing the number of empty capsids.
Therefore, in this study, we attempted to establish a novel AAV vector production system that regulates the timing of Cap expression to improve vector quantity and quality.
Results
Split expression of replicase and capsid does not improve AAV vector production
To control Rep and Cap expression, the Rep2 and Cap2 genes, which are expressed from one AAV genome cassette in a universal, commercialized AAV vector construct (pAAV-RC2), were independently cloned into a CMV promoter-regulating plasmid. The Rep52 protein was mainly expressed in pAAV-RC2, whereas Rep78 primarily in the CMV-regulating Rep construct (CMV-Rep) (Figure 1A). Because AAV protein expression is regulated by the original AAV promoters, a helper plasmid is required for Rep expression in pAAV-RC2 but not CMV-Rep. In contrast, no difference in Cap expression pattern was observed between pAAV-RC2 (RC2) and the CMV-regulating Cap2 plasmid (CMV-Cap2) transfection (Figure 1B). Increasing the transfection volume increased the expression levels of Rep Cap proteins in a dose-dependent manner (Figures 1C and 1D). It is thought that efficient viral production should be related to efficient particle formation by Cap; thus, its dose was changed for AAV vector production. The AAV vector yield was increased by the Cap transfection volume in a dose-dependent manner, but its total amount in the Cap systems did not exceed that of the conventional (normal) system even with the highest transfection dose (Figures 1E and 1F), indicating that split expression of CMV-Rep and Cap is not sufficiently strong to increase the AAV vector production.
Figure 1.

Split expression of replicase and capsid does not improve AAV vector production
(A) Western blot of replicase (Rep) proteins in HEK293 cells. Rep and β-actin proteins were detected using anti-Rep and anti-β-actin antibodies, respectively. Helper = pHelper plasmid containing adenovirus genes; RC2 = pAAV-RC2; CMV-Rep2 = pCMV-Rep2.
(B) Western blot of capsid (Cap) proteins in HEK293 cells. Cap and β-actin proteins were detected using anti-Cap and anti-β-actin antibodies, respectively. CMV-Cap2 = pCMV-Cap2.
(C) Dose-dependency of Rep expression in HEK293 cells. Rep and β-actin proteins were detected using anti-Rep and anti-β-actin antibodies, respectively. White and black triangles indicate non-specific and specific bands, respectively.
(D) Dose-dependency of Cap expression in HEK293 cells. Cap and β-actin proteins were detected using anti-Cap and anti-β-actin antibodies, respectively.
(E and F) Adeno-associated virus (AAV) vector yield using the split Rep and Cap expression system. (E) Amount of AAV vector in solution (vg/μL). vg = vector genome. (F) Fold differences in AAV vector yield among various Cap doses. Data were calculated from three independent experiments and normalized with the value of the normal control (RC2). The normal vector is the conventional AAV vector system using pAAV-RC2. Letters and asterisks in the panel indicate the following: n.s. = not significant, ∗ = p< 0.05, ∗∗ = p< 0.01. Error bars indicate the standard error of the mean.
Control of Cap expression timing increases total AAV vector yield
Cap expression itself can produce viral particles even without other viral components,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38 indicating the possibility that controlling Cap expression timing could be effective for improving the AAV vector production. To assess this possibility, the Cap gene was cloned into a plasmid that controls its expression with a tetracycline (Tet)-dependent promoter (Figure 2A). As shown in Figure 2B, Cap expression was regulated by the addition of doxycycline (Dox), a derivative of Tet, and its expression pattern similar to that of pAAV-RC2. Next, AAV vector production was performed as indicated by the time schedule in Figure 2C, and differences in viral production efficiency were compared. Transfection efficiency because of the change in constructs was not observed (Figure S1). The Tet-regulating Cap expression (Tet-Cap) system showed a 4–9-fold increase in AAV vector yield for the Rep 0.1 and 0.5 μg-transfected groups (means: 1.25 × 107 vg/μL and 4.09-fold in the Rep 0.1 group, 1.15 × 107 vg/μL and 8.70-fold in the Rep 0.5 group) (Figures 2D–2G). However, incomplete regulation of Cap expression by the Tet-promoter was observed without Dox stimulation in the Rep 0.1 μg transfection group (Figure 2H).
Figure 2.

Control of Cap expression timing increases total AAV vector yield
(A) Diagram of adeno-associated virus (AAV) vector constructs. CMV = CMV promoter; Tet = tetracycline-dependent promoter; Rep2 = AAV2 replicase (Rep) gene; Cap2 = AAV2 capsid (Cap) gene.
(B) Western blot for Cap expression of the Tet-Cap2 construct in HEK293 cells. Cap and β-actin proteins were detected using anti-Cap and anti-β-actin antibodies, respectively. Dox = doxycycline (derivative of Tet).
(C) Time schedule for AAV vector production using the normal control (RC2) and Tet-Cap2 systems. GOI = gene of interest; emp = empty. (D-G) AAV vector yield of the Tet-Cap2 system.
(D and E) AAV vector yield in solution (vg/μL) during Rep 0.1 or 0.5 μg-transfection with a change in medium and Dox stimulation at 12 h after transfection.
(F and G) Fold differences in AAV vector yield during Rep 0.1 μg or 0.5 μg-transfection with a change in medium and Dox stimulation for the Tet-Cap2 system. Data were normalized with the measurements of RC2.
(H) Western blot of Cap expression during AAV vector production in HEK293 cells. Cap and β-actin proteins were detected using anti-Cap and anti-β-actin antibodies, respectively. Experiments were independently performed at least three times for statistical analysis. Asterisks in the panel indicate the following: ∗ = p< 0.05, ∗∗ = p< 0.01. Error bars indicate the standard error of the mean.
These data indicate that the control of Cap expression timing can significantly increase total AAV vector yield, transfection with 0.5 μg Rep and a Dox stimulation time of 12 h was optimal.
Control of Cap expression timing improves the full/empty particle ratio
We have shown an improvement in AAV vector quantity using the Tet-Cap system. It is known that current commercialized AAV vector production systems generate empty AAV particles, in which an AAV genome is not incorporated, at a ratio of approximately 70–90%, thereby requiring multistep purifications.39,40 To investigate whether our novel viral production system could improve the quality of the AAV vector, immunoprecipitation was performed using the same number of AAV vectors calculated from the qPCR data in Figures 1D and 1E. The Tet-Cap system substantially reduced Cap content in viral solutions compared to the normal system (Figures 3A–3C), indicating that our viral production system efficiently produced full particles, which are mature AAV vectors containing a genome. In addition, the averaged data of VP1, 2 and 3 band intensities compared with the conventional system (RC2) revealed that reduction of Cap was significantly higher in Rep 0.5 μg than 0.1 μg transfection groups (Figure 3D). Although little reduction was observed in Cap amount for the group of Dox stimulation at early time points, such as at 6 h (Figure 3E), the total in viral solution as calculated using ELISA was strikingly low for the Tet-Cap system, especially for the Rep 0.5 μg-transfected group with Dox stimulation at 12 h (Figure 3F). Furthermore, the Tet-Cap system substantially improved the qPCR/ELISA ratio corresponding to the full/empty particle (F/E) ratio in all groups (Figures 3G and 3H). In particular, improvement in the F/E ratio was consistent when Dox stimulation was performed for 12 h during transfection with 0.5 μg Rep.
Figure 3.

Control of Cap expression timing improves the full/empty particle ratio
(A) Western blot of capsid (Cap) proteins after immunoprecipitation. The same titer of adeno-associated virus (AAV) vector (2 × 108 vg/sample) calculated using qPCR was subjected to immunoprecipitation using A20 antibodies and protein A and G (A/G) magnetic beads before western blotting. Cap proteins were detected using anti-Cap antibodies.
(B–D) Differences in Cap protein band intensities after immunoprecipitation. (B and C) Differences in individual band intensities for VP1, VP2, and VP3 after transfection with 0.1 μg (B) and 0.5 μg (C) replicase (Rep). (D) Differences in band intensity after averaging that of VP1, VP2, and VP3.
(E and F) Fold differences of Cap proteins measured with ELISA between the normal control (RC2) and Tet-Cap2 systems during Rep transfection, a change in medium, and doxycycline (Dox) stimulation at 6 h (E) and 12 h (F) after transfection. Data were normalized with the value measured for RC2.
(G and H) Values of qPCR/ELISA corresponding with the full/empty particle (F/E) ratio between RC2 and Tet-Cap2 after Rep transfection, a change in medium, and Dox stimulation at 6 h (G) and 12 h (H) after transfection. Experiments were independently performed at least three times for statistical analysis. Asterisks in the panel indicate the following: ∗ = p< 0.05, ∗∗ = p< 0.01. Error bars indicate the standard error of the mean.
These data suggest that the Tet-Cap AAV vector production system developed in this study significantly improved the total vector yield and F/E ratio, at least in AAV2.
Novel AAV vector production system is applicable for other serotypes
After establishing an efficient AAV production system for AAV2, we investigated whether the Tet-Cap system can improve AAV vector production in general by applying it to various serotypes. From the data in Figures 1, 2, and 3, the transfection condition containing a 1:1:1:0.25 ratio of pAAV-GOI:pHelper:pAAV-RC or pTet-Cap:pCMV-emp or pCMV-Rep2, as well as Dox stimulation at 12 h after transfection, were used for all experiments. To exclude the possibility of plasmid backbone interference, the Cap2 gene in the pAAV2-RC construct was changed to various others (named pAAV-RCX; X = serotype). Thereafter, Cap genes from various serotypes were cloned downstream to the Tet-regulating promoters (named Tet-CapX; X = serotype). There was no obvious difference in Cap expression pattern between conventional RC and Tet-Cap transfection groups; further, the change of Rep78/52 expression patterns in various Tet-Cap transfection groups during AAV production was the same as that observed for AAV2 production, as described in Figure 1A (Figure 4A). Although the augmented level of viral yield was different among serotypes (Figures 4B–4E and S2A–S2D), the Tet-Cap system increased yield by 1.8–6-fold in all serotypes, indicating that the Tet-Cap system has a universal effect on the improvement of AAV vector yield. Although the degree of improvement was different, immunoprecipitation using the same number of vectors for various serotypes showed that improved F/E ratios were achieved using the Tet-Cap system for most serotypes, except AAV8 (Figures 4F–4I and S3A–S3D). In addition, we observed an increase in the total AAV genome in cells during AAV vector production in the Tet-Cap system (Figures S2E–S2J), suggesting the possibility that an increase in AAV genome affects, to some extent, the augment of AAV vector yield and improvement of F/E ratio in various serotypes.
Figure 4.

Novel AAV vector production system is applicable for other serotypes
(A) Western blot of replicase (Rep) and capsid (Cap) proteins during adeno-associated virus (AAV) vector production for various serotypes in HEK293 cells. Rep, Cap, and β-actin proteins were detected using anti-Rep, anti-Cap, and anti-β-actin antibodies, respectively.
(B–E) AAV vector yield in various serotypes for the Tet-Cap system. Fold difference of AAV vector yield in AAV7 (B), AAV8 (C), AAV9 (D), and AAVrh10 (E) after a change in medium and doxycycline stimulation at 12 h after transfection. Data were normalized with the value of the normal control (RC) samples.
(F–I) Western blot of Cap proteins after immunoprecipitation for various serotypes. The same titer of AAV vector (1 × 109–4 × 109 vg/sample) calculated with qPCR was subjected to immunoprecipitation using ADK8 (AAV7, AAV8, and rh10) and ADK8/9 (AAV9) antibodies, as well as protein A/G magnetic beads, before western blotting. The left-hand figures show representative western blotting images, and the right-hand figures the fold difference of band intensities for VP1, VP2, and VP3 proteins between RC and Tet-Cap samples in each panel. These panels indicate the data for AAV7 (F), AAV8 (G), AAV9 (H), and AAVrh10 (I), respectively. Experiments were independently performed at least three times for statistical analysis. Asterisks in each panel indicate the following: ∗ = p< 0.05, ∗∗ = p< 0.01. Error bars indicate the standard error of the mean.
These data denote that the Tet-Cap AAV vector production system generally increases AAV vector yield in all serotypes and improves its quality in many serotypes.
Novel AAV vector production system does not affect viral infectivity in vitro and in vivo
Although the Tet-Cap system improved the quantity and quality of AAV vectors, the change in viral production system could also affect viral infectivity. Therefore, the difference in infectivity was first analyzed in vitro, and results showed no change in viral infectivity between the conventional system and our novel viral production system in various serotypes (Figures 5A–5E and S4A–S4D). Next, to check viral infectivity in vivo, we upscaled the Tet-Cap system and found that it did not affect the yield, quality, and infectivity of the AAV2 vector (Figures S5A–S5C). In addition, the VP1:VP2:VP3 particle ratio was comparable at least between vectors derived from the Tet-Cap system and commercial purified vectors (Figures S5D and S5E). Moreover, augmentation of the AAV vector yield was confirmed even with further scale-up of the Tet-Cap system (Cell Factory flask) for in vivo experiments using AAV9 (Figure S5F). By using AAV9 vectors produced in a large-scale system, those injected into the retro-orbital vein of C57BL/6J mice showed similar distributions at several time points (4, 6, and 8 weeks) after injection in the normal and novel AAV vector production systems (Figure 5F). Further analysis also showed no obvious difference in AAV genome distribution at various tissues between conventional and Tet-Cap systems (Figure 5G).
Figure 5.

Novel AAV vector production system does not affect viral infectivity in vitro and in vivo
(A–E) (A–D) Infectivity data of adeno-associated virus (AAV) vectors for various serotypes in vitro. The 2v6.11 cells were infected with AAV2 (500 vg/cell), AAV7 (1000 vg/cell), AAV8 (1000 vg/cell), AAV9 (1000 vg/cell), and AAVrh10 (1000 vg/cell). Cells were observed at 96 h after infection. Data indicate the infectivity of the AAV vector (EGFP; green) in AAV2 (A), AAV7 (B), AAV8 (C), AAV9 (D), and AAVrh10 (E). The blue signal shows nuclei (Hoechst 33342). White bar shows 100 μm.
(F) Data for AAV vector distribution in vivo. C57BL/6 mice were injected with AAV9 vectors carrying an EGFP-P2A-Nluc gene (5 × 1010 vg/mouse) derived from normal control (RC9) and Tet-Cap9 systems through the retro-orbital vein. Luciferase signals were monitored using IVIS-CT after injection of luciferase substrate through the retro-orbital vein at 4, 6, and 8 weeks. Data shows representative mice of RC9 (n = 4) and Tet-Cap9 (n = 4). Bar colors correspond to luciferase signals with a range of 0–5000 photons/s.
(G) AAV vector genome detection in indicated organs at 3 weeks after injection. Data were normalized with the AAV vector (EGFP) and mouse genomic GAPDH values. White triangles and black rhombuses indicate data of each mouse for RC9 (n = 4) and Tet-Cap9 (n = 4), respectively. Letters in the panel indicate the following: n.s. = not significant. Error bars indicate the standard error of the mean.
Thus, our data reveal that the control of Cap expression timing improves the quality and quantity of AAV vectors at all scales without reducing infectivity.
Difference in the Rep78/Rep52 expression ratio improves AAV vector quantity and quality
We have shown that the Tet-Cap system improves the quantity and quality of AAV vector production. However, the detailed mechanism responsible for this is unclear. In the present study, we have shown that the Rep78/52 ratio changed because Rep/Cap genes were separated into two plasmids to regulate Cap expression timing (Figure 1A). This suggests the possibility that differences in Rep78/52 ratio and Cap expression timing control could improve AAV vector yield and the F/E ratio during vector production. Therefore, we analyzed whether the Rep78/52 ratio is involved in improvement of AAV vector yield and quality. To control the Rep78/52 expression pattern, we constructed several plasmids (Figure 6A). Rep78-IRES-Rep52 (78-I-52) showed high expression of Rep78 similar to CMV-Rep, Rep52-IRES-Rep78 (52-I-78) was comparable to pAAV-RC2, and Rep52-P2A-Rep78 (52-2A-78) equally expressed Rep78 and Rep52 proteins (Figure 6B). During AAV vector production, different Cap expression patterns were not observed in each group (Figure 6C). Although all Rep constructs increased AAV vector yield compared with the original commercialized system, the increase was similar between CMV-Rep and 78-I-52 groups (Figure 6D). Meanwhile, the 52-I-78 and 52-2A-78 groups showed a reduction in viral yield compared with that of CMV-Rep and 78-I-52. The improvement in F/E ratio was similar for CMV-Rep and 78-I-52, whereas that of 52-I-78 was significantly reduced (Figure 6E). Intriguingly, 52-2A-78 also improved the F/E ratio similar to CMV-Rep and 78-I-52, although producing a lower yield. In addition, we observed a significant increase in the total AAV genome in cells during AAV vector production in CMV-Rep and 78-I-52 compared with conventional RC2 and 52-I-78 (Figures S6A and S6B). There was no obvious difference in infectivity of the AAV vectors among each group (Figure 6F).
Figure 6.

Difference in the Rep78/Rep52 expression ratio improves AAV vector quantity and quality
(A) Diagram of various replicase (Rep) constructs. CMV = CMV promoter; Rep2 = full length AAV2 Rep gene; 78 = Rep78 gene; 52 = Rep52 gene; IRES = internal ribosome entry site sequence; P2A = 2A sequence of porcine teschovirus-1.
(B) Western blot of Rep proteins using various Rep constructs in HEK293 cells. Rep and β-actin proteins were detected using anti-Rep and anti-β-actin antibodies, respectively. The white triangle shows Rep52-P2A-Rep78 fusion proteins.
(C) Western blot of capsid (Cap) proteins during adeno-associated virus (AAV) vector production using various Rep constructs in HEK293 cells. Cap and β-actin proteins were detected using anti-Cap and anti-β-actin antibodies, respectively.
(D) AAV vector yields for various Rep expressions. Fold difference in AAV vector yields between the normal control (RC2) and various Rep-transfected groups after a change in medium and doxycycline stimulation at 12 h after transfection.
(E) Fold differences in qPCR/ELISA corresponding with the full/empty particle ratio during various Rep expressions. Data were normalized with the value of the RC2 sample to calculate fold difference.
(F) Infectivity data for AAV vectors produced using various Rep constructs. The 2v6.11 cells were infected with AAV2 (500 vg/cell) and observed at 96 h after infection. These data indicate infectivity of the AAV vector (EGFP; green) in AAV2 produced using various Rep transfections. The blue signal shows nuclei (Hoechst 33342). White bar shows 100 μm. Experiments were independently performed at least three times for statistical analysis. Asterisks in each panel indicate the following: ∗ = p< 0.05, ∗∗ = p< 0.01. Error bars indicate the standard error of the mean.
These data indicate that, to some extent, the change in Rep78/Rep52 expression pattern plays an important role in improvement of AAV vector quantity and quality in the Tet-Cap AAV production system.
Control of Cap expression timing mainly improves AAV vector quality
Finally, we investigated the detailed effect of controlling Cap expression timing during AAV vector production by establishing HEK293 cells that stably carry the Tet-regulating Cap gene cassette. Although Cap expression level in each clone was quite low, we obtained stable clones (clones 1, 4 and 10) capable of expressing Cap proteins in response to Dox stimulation (Figure 7A). To evaluate differences in AAV vector yield and quality, transfection and Dox stimulation were performed in established clones with test conditions as indicated in Figure 7B. Yield of the AAV vector was slightly increased after Dox stimulation at early time points (3–12 h) after transfection, but decreased at 24 h, indicating that the optimal time for Dox stimulation was likely at approximately 12 h (Figures 7C and S7A). The amount of Cap proteins was 15–18% lower in the Dox stimulation group at 12 h after transfection compared with that of immediate stimulation after transfection, as calculated using the average of three clones (Figure 7D). Timing control of Cap expression increased the F/E ratio by 1.5-fold in stable clones (Figure 7E), indicating that it could improve AAV vector quality. Although the amount of Cap was reduced after Dox stimulation at 24 h, there was no significant difference in F/E ratio compared with that at 12 h, conceivably because of lower AAV vector productivity (Figures S7B and S7C).
Figure 7.

Control of Cap expression timing mainly improves AAV vector quality
(A) Establishment of HEK293 cells carrying the Tet-Cap2 gene cassette. Clones were lysed and subjected to western blotting. Capsid (Cap) and β-actin proteins were detected using anti-Cap and anti-β-actin antibodies, respectively. Letters above data indicate the clone number and doxycycline (Dox).
(B) Scheme of transfection and Dox stimulation. TF = transfection.
(C) Fold differences in AAV vector yield at indicated Dox stimulation timing after transfection in the HEK293 cells carrying the Tet-Cap2 gene cassette. Data were normalized with measurements from the 0 h Dox stimulation sample.
(D) Fold differences in Cap proteins using ELISA analysis after stimulation of Cap expression at 12 h in clones. Data were normalized with the measurement at 0 h (same = at the same time of transfection). Dox stimulation sample shows the average of three different clones (C1, C4, and C10).
(E) Fold difference in qPCR/ELISA corresponding with the full/empty particle ratio in clones. Data were normalized with the value measured at 0 h. Dox stimulation sample shows the average of three different clones. Experiments were independently performed at least three times for statistical analysis. Asterisks in each panel indicate the following: ∗ = p< 0.05, ∗∗ = p< 0.01. Error bars indicate the standard error of the mean.
These data denote that the timing control of Cap expression mainly affected improvement of AAV vector quality. Thus, our newly established AAV vector production system (Tet-Cap system) considerably improved AAV vector yield and quality by an additive effect through changing Rep expression pattern and controlling Cap expression timing.
Discussion
In the present study, we have established a novel AAV vector production system using Tet-Cap expression after separating the Rep and Cap genes onto two plasmids. This system could significantly increase total AAV vector yield for all serotypes (Figures 2, 4B–4E, S2A–S2D, S5A, and S5F) and substantially decrease the empty capsid ratio for many of them (Figures 3, 4F–4I, S3, and S5B) without reducing AAV vector infectivity in vitro and in vivo (Figures 5, S4, and S5C). In addition, the VP1:VP2:VP3 particle ratio was almost identical between vectors derived from the Tet-Cap and conventional system (Figures S5D and S5E). Analyses of the molecular mechanism revealed that the increased Rep78 expression ratio compared with the commercialized AAV vector system related to augmentation of total AAV vector yield and 3.5- to 4-fold improvement of the F/E ratio (Figure 6). Timing control of Cap expression showed a 1.5-fold improvement in the F/E ratio (Figure 7). These results indicate that our novel AAV vector production system improves total AAV vector yield and F/E ratio by an additive effect through changing Rep expression pattern and controlling Cap expression timing.
Our Tet-Cap system successfully increased total AAV vector yield and reduced the number of empty capsids. These effects seemed superior during transfection with 0.5 μg Rep compared with that of 0.1 μg Rep (Figures 2D–2G and 3). One explanation for this is that regulation of Cap expression is defective in the presence of low amounts of Rep proteins (Figure 2H). The Rep protein has a bifunctional ability that upregulates and downregulates transcription of the AAV genome,41 and the Rep78 protein represses AAV and other promoters, such as HIV-LTR and HPV-URR, suggesting that AAV Rep may also repress various promoters.42 Thus, higher Rep78 expression in the Tet-Cap system likely regulates Cap expression through repressive binding of the Tet promoter concomitantly with Tet-dependent expression.
The Tet-Cap AAV vector production system showed a difference in Rep78/52 expression ratio compared with the universal commercialized Rep/Cap construct (Figures 1A and 4A). The universal Rep/Cap constructs, which are globally used for AAV production, had an original p5 promoter moved from upstream to downstream of the Rep/Cap genes, which differs from the wildtype AAV genome structure. The universal Rep/Cap construct shows high Rep52 expression compared with the Rep78 protein in cells,43,44 whereas the AAV wild-type has an equal Rep78/52 ratio or stronger expression of Rep78, similar to the present study.45,46 This indicates that higher expression of Rep78 is likely not problematic because of the nature of AAV. In addition, some studies showed that large Rep proteins, such as Rep78 and Rep68, can efficiently encapsulate an AAV genome.47,48 These results strongly support our data that higher Rep78 expression improves the total vector yield and F/E ratio (Figure 6).
In the present study, the Tet-Cap system decreased empty capsids during AAV vector production. The ability of viral capsids to self-assemble is useful for analyzing the molecular mechanism and developing vesicles to deliver drugs and genes to targets,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38 although it is an obstacle for the safe development of gene therapy tools. Cap expression at late time points, such as 24 h after transfection, significantly improved the quantity of empty capsids (Figure S7B), but not the quality of AAV vectors (Figure S7C). This is likely because of the reduction in total AAV vector yield (Figure S6A). Although detailed reasons are unknown, decreased cell viability may have occurred during Rep expression; cell division in long-term cultures may have reduced the number of retained plasmids in cells; or AAV vectors may have collapsed during long-term culture. Additional analyses and optimization of AAV vector production could further improve the productivity of novel AAV production systems in the future.
The Tet-Cap system increased total AAV vector yield for all serotypes, although the magnitude differed among them. The amounts of empty capsids could not be reduced for some serotypes by Tet-Cap system in the present study (Figures 2, 3, 4, S2, and S3). Immunoprecipitation followed by western blotting may not have been sufficient to measure small differences in the F/E ratio. The small improvement in AAV vector yield and slight reduction of empty capsids seems correlative, suggesting that the improvement of AAV vector production by Tet-Cap system is serotype-dependent to some extent. There are differences in characteristics between serotypes, such as tissue tropism, classification on the evolutionary tree, and ability of vector secretion from cells during AAV vector production, which all seem to be mostly dependent on Cap sequences.49,50,51,52 In particular, the difference in vector secretion from cells between serotypes may affect reduction of empty capsids in the Tet-Cap system because secretion or accumulation of AAV vectors is likely related to the molecular feature of Cap proteins, including protein trafficking mediated by cellular machinery. Further analyses are required to address this point.
Stable cells carrying the Tet-regulating Cap gene cassette directly revealed the effect of Cap expression timing on reduction of empty capsids (Figure 7). This indicates that an efficient AAV vector production system using stable cells carrying Tet-Cap can be developed. However, expression level of the Cap protein in stable cells was weak compared with that in plasmid-transfected cells. This is likely dependent on copy number of the Cap gene in the cell. Copy number of the gene cassette in a transfected cell must be markedly higher than that of the stable clone established in this study. Therefore, there is the potential to establish a novel AAV vector production system by improving copy number of the Tet-Cap gene cassette in stable cells.
Overall, we established a novel AAV vector production system that increased quantity and improved quality of the AAV vector by regulating expression timing of the Cap gene. Our findings shed light on the new aspect of developing AAV vector production systems and may help improve the accessibility of gene therapy by reducing costs.
Limitations of the study
We have shown that the Tet-Cap AAV vector production system, which separated Rep and Cap genes to control Cap expression timing, significantly increased total AAV vector yield and improved its quality without altering vector features. Because our study used a commercialized AAV vector production system as the experimental control, it is still unclear whether the Tet-Cap system has a similar advantage over AAV vector production using Rep/Cap constructs modified by researchers in a laboratory that have different vector backbones. In addition, the present study has not yet fully shown detailed features of the Tet-Cap AAV vector systems and requires further studies to unravel them.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AAV1 intact particle antibody, ADK1a | PROGEN | Cat# 610150 |
| AAV2 intact particle antibody, A20 | PROGEN | Cat# 65155 |
| AAV5 intact particle antibody, ADK5a | PROGEN | Cat# 610148 |
| AAV8 intact particle antibody, ADK8 | PROGEN | Cat# 610160 |
| AAV8/9 intact particle antibody, ADK8/9 | PROGEN | Cat# 651161 |
| AAV VP1/VP2/VP3 rabbit polyclonal, VP51 | PROGEN | Cat# 61084 |
| AAV2 Replicase mouse monoclonal, 303.9 | PROGEN | Cat# 61069 |
| β-Actin (13E5) Rabbit | Cell Signaling Technology | Cat# 4970; RRID:AB_2223172 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Bacterial and virus strains | ||
| AAV1-EGFP | This paper | N/A |
| AAV2-EGFP | This paper | N/A |
| AAV2-EGFP-P2A-Nluc | This paper | N/A |
| AAV3B-EGFP | This paper | N/A |
| AAV5-EGFP | This paper | N/A |
| AAV6-EGFP | This paper | N/A |
| AAV7-EGFP | This paper | N/A |
| AAV8-EGFP | This paper | N/A |
| AAV9-EGFP | This paper | N/A |
| AAV9-EGFP-P2A-Nluc | This paper | N/A |
| AAVrh10-EGFP | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| E-MEM | FUJIFILM Wako | 051-07615 |
| FBS (HyClone) | Cytiva | SH30910.03 |
| MEM non-essential amino acid (×100) | FUJIFILM Wako | 139-15651 |
| Penicillin-Streptomycin Solution (×100) | FUJIFILM Wako | 168-23191 |
| Nano Glo substrate (Nano-Glo Luciferase Assay System) | Promega | N1110 |
| HilyMax transfection reagent | DOJINDO | H357-10 (342-91103) |
| Doxycycline | Takara Bio | 631311 (Z1311N) |
| PEI MAX | Polysciences | 24765-1 |
| Polyethylene glycol 8000 | Santa Cruz Biotechnology | SC-281693 |
| Pluronic F68 | ThermoFisher (GIBCO) | 24040-32 |
| Opti-Prep | Serumwerk | 1893 |
| TURBO DNase | ThermoFisher | AM2238 |
| 4–20% Mini-PROTEAN® TGX™ Precast Protein Gels | Bio-Rad Laboratories | 4561096 |
| Clarity Western ECL substrate | Bio-Rad Laboratories | 1705061 |
| Dynabeads™ Protein A for Immunoprecipitation | ThermoFisher | 10001D |
| Dynabeads™ Protein G for Immunoprecipitation | ThermoFisher | 10003D |
| Hoechst 33342 | ThermoFisher | H3570 |
| Blasticidin | InvivoGen | ant-bl-05 |
| Zeocin | InvivoGen | ant-zn-05 |
| Benzonase | Merck | 70746-3CN |
| Critical commercial assays | ||
| AllPrep DNA/RNA/Protein Mini Kit | QIAGEN | 80004 |
| TB Green® Premix Ex Taq™ II | Takara Bio | RR820A |
| AAV2 titration ELISA 2.0R kit | PROGEN | PRAAV2R |
| Experimental models: Cell lines | ||
| HEK293 cells | RIKEN cell bank | Cat# RCB1637; RRID:CVCL_0045 |
| 2v6.11 cells | Mohammadi et al.53 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 (JAX Mice Strain, B6) | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| Inverted terminal repeat (ITR)-specific 5′ primer: 5′-GGAACCCCTAGTGATGGAGTT-3′ | Aurnhammer et al.54 | N/A |
| Inverted terminal repeat (ITR)-specific 3′primer: 5′-CGGCCTCAGTGAGCGA-3′ | Aurnhammer et al.54 | N/A |
| EGFP-92 5′ primer: 5′-ggccacaagttcagcgtgtc-3′ | This paper | N/A |
| EGFP -200 3′ primer:5′-taggtcagggtggtcacgag-3′ | This paper | N/A |
| Mouse GAPDH-gDNA-3308F: 5′-caccaccatggagaaggccg-3′ | This paper | N/A |
| Mouse GAPDH- gDNA-3458R: 5′-tgacccttttggctccaccc-3′ | This paper | N/A |
| Recombinant DNA | ||
| pAAV-EGFP | This paper | N/A |
| pAAV-EGFP-P2A-Nluc | This paper | N/A |
| pAAV-RC2 (pAAV-RC) | Agilent | 240071 |
| pAAV-RC1 | This paper | N/A |
| pAAV-RC3B | This paper | N/A |
| pAAV-RC5 (pRC5) | Takara Bio | 6650 |
| pAAV-RC6 | This paper | N/A |
| pAAV-RC7 | This paper | N/A |
| pAAV-RC8 | This paper | N/A |
| pAAV-RC9 | This paper | N/A |
| pAAV-RCrh10 | This paper | N/A |
| pTet-Cap1 (pCW-AAV Cap1-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Cap2 (pCW-AAV Cap2-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Cap3B (pCW-AAV Cap3B-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Cap5 (pCW-AAV Cap5-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Cap6 (pCW-AAV Cap6-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Cap7 (pCW-AAV Cap6-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Cap8 (pCW-AAV Cap6-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Cap9 (pCW-AAV Cap6-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pTet-Caprh10 (pCW-AAV Cap6-WPRE-hPGK-Hyg-2A-rTetR) | This paper | N/A |
| pHelper | Takara Bio | 6650 |
| pcDNA3.1(+)-Rep2 | This paper | N/A |
| pcDNA3.1(+)-Cap2 | This paper | N/A |
| pRep78-I-52 | This paper | N/A |
| pRep52-I-78 | This paper | N/A |
| pRep52-P2A-78 | This paper | N/A |
| pcDNA4/TO-Cap2 | This paper | N/A |
| pcDNA6/TR | ThermoFisher | V102520 |
| Software and algorithms | ||
| IVIS Spectrum CT | PerkinElmer | https://www.perkinelmer.com/product/ivis-instrument-spectrum-ct-120v-128201 |
| Living Image Software | PerkinElmer | https://www.perkinelmer.com/product/spectrum-200-living-image-v4series-1-128113 |
| Luminescent image analyzer LAS-4000 mini | Cytiva | N/A |
| ImageQuant™ TL | Cytiva | N/A |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Operetta CLS | PerkinElmer | https://www.perkinelmer.com/category/operetta-cls-high-content-analysis-system |
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Resource availability
Lead contact
Further information and requests of resources and reagents should be directed to the lead contact, Kenji Ohba (ohbak@jichi.ac.jp).
Materials availability
Antibodies were obtained from commercial sources described in the STAR Methods key resources table. All unique materials generated in this study will be available from the lead contact after completing a Material Transfer Agreement.
Experimental model and subject details
Cell culture
HEK293 cells were obtained from the Cell Engineering Division (RIKEN cell bank, Tsukuba, Japan). HEK293 and 2v6.11 cells53 were cultured in Eagle’s minimum essential medium (FUJIFILM Wako, Osaka, Japan) supplemented with 10% fetal bovine serum (ThermoFisher, Waltham, MA, USA), 1× MEM non-essential amino acids (FUJIFILM Wako), and 1× penicillin-streptomycin (FUJIFILM Wako).
Animal studies
Animal experiments were conducted in a humane manner after receiving approval from the Institutional Animal Experiment Committee of the Jichi Medical University (Japan), and in accordance with the Institutional Regulation for Animal Experiments and Fundamental Guideline for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology (Japan).
Male C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). AAV9 vectors carrying EGFP-P2A-NLuc produced using RC9 or Tet9 constructs were injected into 4-6-week-old mice at a concentration of 5 × 1010 vg/mouse through the retro-orbital vein while under anesthesia. Distribution of the AAV9 vector in mice was monitored using the IVIS Spectrum CT In Vivo Imaging System (PerkinElmer, Waltham, MA, USA) at 1 min after injection of Nano Glo substrate (2.5 μL substrate in 150 μL phosphate-buffered saline per mouse) (Promega, Madion, WI, USA) through the retro-orbital vein every week while under anesthesia. Bioluminescent images were analyzed using Living Image Software (PerkinElmer). Genomic DNA and total RNA of tissues were extracted from mice at three weeks post-injection using an AllPrep DNA/RNA/Protein Mini Kit (QIAGEN, Venlo, Netherlands) according to the manufacturer’s instruction.
Method details
Plasmid construction
AAV serotypes 1, 7, 8, 9, and rh10 capsid (Cap) plasmids (pAAV2/1, pAAV2/7, pAAV2/8, pAAV2/9n, and pAAV2/rh10) were gifted by James M. Wilson (Addgene plasmids: 112862, 112863, 112864, 112865, and 112866). AAV serotype 3B and 6 Cap plasmids (pDGM3B and pDGM6) were gifted by David Russell (Addgene plasmids: 110809 and 110660).55,56 To adjust the plasmid backbone, the Cap gene of serotype 2 in pAAV-RC2 (Agilent, Santa Clara, CA, USA) was changed to a Cap gene from each serotype mentioned above and named pAAV-RC1, 3B, 6, 7, 8, 9, and rh10, respectively. The AAV serotype 5 Cap plasmid (pAAV-RC5) was obtained from TAKARA Bio (Kusatsu, Japan).
The AAV serotype 2 replicase (Rep) gene with 15 nucleotides (nt) before the start codon and 92 nt after the stop codon was cloned into pcDNA3.1(+) (ThermoFisher) and named pCMV-Rep. The AAV serotype 2 Cap gene with 350 nt before the start codon and 55 nt after the stop codon was cloned into pcDNA3.1(+) and named pCMV-Cap.
For tetracycline (Tet)-inducible Cap constructs, the multi-cloning site (MCS) and woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) were cloned upstream of the phosphoglycerate kinase promoter in pCW57.1 that was gifted by David Root (Addgene plasmid: 41393), and the puromycin resistance gene changed to a hygromycin resistance gene (pCW-MCS-WPRE-Hyg). Cap genes of various serotypes with 350 nt before the start codon and 55–166 nt after the stop codon were cloned into the MCS of pCW-MCS-WPRE-Hyg and named pTet-Cap1, 2, 3B, 5, 6, 7, 8, 9, and rh10, respectively.
For Rep78 and Rep52 expression constructs, these genes were cloned in this order or with swapped orientation (Rep52 then Rep78) upstream and downstream of the internal ribosome entry site (IRES) in pIRES (Takara Bio USA, Inc., Mountain View, CA, USA), respectively. Rep78 and Rep52 linked by the P2A sequence derived from porcine enterovirus polioencephalomyelitis were cloned into pcDNA3.1(+). These plasmids were called pRep78-I-52, pRep52-I-78, and pRep52-P2A-Rep78, respectively.
For the reporter constructs, EGFP or EGFP-P2A-NanoLuc (NLuc) genes (Promega) were cloned into pAAV-MCS (Agilent) and named pAAV-EGFP or pAAV-EGFP-P2A-Nluc, respectively.
For Cap-inducible stable cells, AAV serotype 2 Cap with 350 nt before the start codon and 55 nt after the stop codon was cloned into pcDNA4/TO (ThermoFisher) and named pcDNA4/TO-Cap2.
Plasmid transfection and AAV vector production
For small-scale studies using culture plates, HEK293 cells were seeded at concentrations of 1 × 106 or 2 × 106 cells/well into 12- or 6-well plates, respectively, one day before transfection. Under normal conditions, 2 μg pAAV-EGFP or pAAV-EGFP-P2A-Nluc, 2 μg pHelper (TAKARA), 2 μg pAAV-RC, and 0.5 μg pcDNA3.1(+) were co-transfected into cells by using HilyMax transfection reagent (DOJINDO, Kumamoto, Japan) in 6-well plates according to the manufacturer’s instructions. For the Tet-inducible Cap condition, 2 μg pAAV-EGFP or pAAV-EGFP-P2A-Nluc, 2 μg pHelper, 2 μg Tet-Cap, and 0.5 μg pCMV-Rep were co-transfected into cells using HilyMax transfection reagent in 6-well plates. All conditions were the same for co-transfection in 12-well plates except 0.25 μg pcDNA3.1(+), 0.25 μg pCMV-Rep, and 1 μg all other components were used. For both conditions, media were replaced with fresh medium containing 2 μg/mL doxycycline (Dox; TAKARA Bio) 6, 12, 24, and 36 h after transfection. At 72 h after medium change, cells and supernatants were collected in tubes. To harvest the AAV vector, collected cells and supernatants were frozen and thawed four times, and the supernatants collected as AAV vector solutions after centrifugation and filtration using a 0.45 μm filter.
For large-scale studies using a T-225 flask or 4-layer EasyFill Cell Factory system (ThermoFisher) under normal conditions, 70 or 600 μg pAAV-EGFP or pAAV-EGFP-P2A-Nluc, 70 or 600 μg pHelper, 70 or 600 μg pAAV-RC, and 17.5 or 150 μg pcDNA3.1(+) were mixed with 796.25 or 6825 μg PEI MAX (Polysciences, Warrington, PA, USA) in 20 or 50 mL 150 mM sodium chloride (NaCl) solution. After 15 min of incubation at room temperature (RT = 25°C), transfection mix was added to the cell culture. For Tet-inducible Cap conditions, 70 or 600 μg pAAV-EGFP or pAAV-EGFP-P2A-Nluc, 70 or 600 μg pHelper, 70 or 600 μg pTet-Cap, and 17.5 or 150 μg CMV-Rep were mixed with 796.25 or 6825 μg PEI MAX in 20 or 50 mL 150 mM NaCl solution. After 15 min of incubation at RT, transfection mix was added to the cell culture. At 12 h post-transfection, culture medium was replaced with fresh medium containing 2 μg/mL Dox for both samples. Cells and supernatants were separately collected 96 h after medium change. The AAV vector was purified as described previously.57 Briefly, four volumes of supernatant were mixed with one volume 40% polyethylene glycol 8000 (Santa Cruz Biotechnology, Dallas, TX, USA) containing 0.5 M NaCl, followed by centrifugation. Pellets of supernatants were resuspended in AAV Lysis buffer (phosphate-buffered saline containing 0.001% Pluronic F68 and 200 mM NaCl) and mixed with cell lysates. These were then frozen and thawed four times to extract AAV vectors, followed by centrifugation. Supernatants were collected and treated with 50 units/mL Benzonase (Merck, Darmstadt, Germany) as the final concentration at 37°C for 2 h. Solutions containing AAV vectors after centrifugation were ultracentrifuged using a 15%, 25%, 40%, and 54% Opti-Prep (Serumwerk, Bernburg, Germany) gradient. Samples were collected at the 40% layer after ultracentrifugation and purified using an Amicon Ultra 100 kDa MWCO column (Merck). The purified samples were filtered through a 0.45 μm filter, and virus solutions kept at −20°C before use. The titer of purified AAV vectors was quantified using qPCR analysis.
AAV vector genome detection and titration using qPCR
For AAV vector genome detection in cell lysates, collected samples were directly applied for qPCR analysis to monitor the total replicated AAV genomic DNA in cells during AAV vector production. For AAV vector titration, collected samples were treated with TURBO DNase (ThermoFisher) according to the manufacturer’s instructions before qPCR analysis. The total AAV vector genome and titer were quantified using qPCR with an inverted terminal repeat-specific primer set (5′-GGAACCCCTAGTGATGGAGTT-3′, 5′-CGGCCTCAGTGAGCGA-3′)54 and TB Green®Premix Ex Taq II (TAKARA Bio) according to the manufacturer’s instructions.
Western blotting
Cells were collected and lysed using cell lysis buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, and 1% NP-40). One volume 2× sodium dodecyl sulfate (SDS) sample buffer (125 mM Tris-HCl at pH 6.8, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, and 0.01% bromophenol blue) was added to the lysed samples and boiled for 10 min. Samples were subjected to a 4–20% gradient TGX precast gel (Bio-Rad, Hercules, CA), followed by the transfer of proteins to Immobilon-P polyvinylidene fluoride membranes (Millipore, Burlington, MA, USA) using the Trans-Blot Turbo Transfer System (Bio-Rad). After blocking membranes with 5% skim milk/TBS-T (50 mM Tris-HCl at pH 7.6, 150 mM NaCl, and 0.05% Tween 20), primary antibodies were allowed to react at RT for 1 h, followed by secondary antibody reactions at RT for 1 h. Proteins were detected using a Clarity Western ECL substrate (Bio-Rad) and the luminescent image analyzer, LAS-4000 mini (Cytiva, Marlborough, MA, USA). The band intensity was analyzed using ImageJ software.
Immunoprecipitation of AAV particles
Intact AAV particle antibodies (ADK1a, A20, ADK5a, ADK8, and ADK8/9; PROGEN, Heidelberg, Germany) were mixed with protein A and G (A/G) magnetic beads (Dynabeads; ThermoFisher) in immunoprecipitation buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, and 0.05% Tween 20) at RT for 1 h. Equal amounts of AAV vectors from qPCR data (2 × 108–4 × 109 vg/sample) were reacted with an antibody-protein A/G magnetic beads mixture at RT for 1 h with rotation. After the reaction, samples were placed in a magnetic stand to discard the buffer, and each sample washed with fresh immunoprecipitation buffer. After three washes, pellets were boiled in 1× SDS sample buffer diluted with cell lysis buffer for 10 min, followed by western blotting. AAV capsids were detected using anti-AAV VP1/VP2/VP3 rabbit polyclonal VP51 (PROGEN) and anti-rabbit IgG-HRP (Cell Signaling technologies Inc, Danvers, MA, USA) antibodies.
Calculation of the full/empty particle ratio
The amount of AAV2 Cap in samples was measured using the AAV2 titration ELISA 2.0R kit (PROGEN) according to the manufacturer’s instructions. The full/empty particle (F/E) ratio was calculated using the qPCR value (vg/μL)/ELISA value (Cap/μL) ratio. The calculated values were normalized with those of the pAAV-RC2 group.
AAV vector infection and immunofluorescent microscopy analysis
AAV vectors were added to 2v6.11 cells with the same titer of viruses calculated from the qPCR data in 96-well plates. At 3–5 days post-infection, the infectivity of AAV vectors carrying the EGFP reporter was observed with fluorescent or confocal microscopes (Operetta CLS; PerkinElmer) after nuclear staining with Hoechst 33342 (ThermoFisher).
Quantification of AAV vectors in various tissues
To quantify the AAV genome and mRNA in extracted genomic DNA and total RNA from tissues, 50 or 5 ng genomic DNA was subjected to qPCR analysis using primer sets for EGFP (5′-GGCCACAAGTTCAGCGTGTC-3′ and 5′-TAGGTCAGGGTGGTCACGAG-3′) or mouse GAPDH (5′-CACCACCATGGAGAAGGCCG-3′ and 5′-TGACCCTTTTGGCTCCACCC-3′) genes, respectively. Data were normalized by dividing EGFP values by the GAPDH values.
Establishment of inducible Cap2-expressing HEK293 cells
To establish inducible Cap2-expressing HEK293 cells, stable Tet repressor (TetR)-expressing HEK293 cells were obtained by transfection with the pcDNA6/TR plasmid and selection with blasticidin (InvivoGen, San Diego, CA, USA). Inducible Cap2-expressing HEK293 cells were then established by transfection with pcDNA4/TO-Cap2 and selection with blasticidin and Zeocin (InvivoGen). The established cells were used to analyze AAV2 production.
Quantification and statistical analysis
All in vitro experiments were conducted at least three times under similar conditions. Animal experiments were performed with four mice in each group. Graphs and statistical analyses were performed using GraphPad Prism v8 software (GraphPad, San Diego, CA, USA). Means of two groups were compared using the t-test with Welch’s correction. Error bars represent standard error of the mean. Statistical significance is indicated in figures as follows: ∗p< 0.05, ∗∗p< 0.01, n.s. = not significant.
Acknowledgments
We thank all our laboratory members, especially Akemi Takada who supported the handling of our budgets. This work was funded by the Japan Society for the Promotion of Science KAKENHI, Grant-in-Aid for Scientific Research [Grant numbers: 20K07681, 21H02768], and a Joint Research Fund sponsored by Takara Bio Inc.
Author contributions
K.O.1 conceived and designed the research and performed all experiments and data analysis; Y.S. supported the animal experiments. K.O.,1 T.E., and J.M. interpreted the results of all experiments. K.O.1 prepared the manuscript. K.O.,1 K.O.,2 and H.M edited the manuscript. All authors reviewed and approved the manuscript.
1Kenji Ohba, 2Keiya Ozawa.
Declaration of interests
T.E. and J.M. are the operating officer and director, respectively, and have treasury stocks in Takara Bio Inc. This work was partly funded by a Joint Research Fund sponsored by Takara Bio Inc. K.O. 1 and TAKARA bio Inc. have applied for the international patent based on present study.
1Kenji Ohba.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: March 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106487.
Supplemental information
Data and code availability
The data reported in this paper will be shared by the lead contact (ohbak@jichi.ac.jp) upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Kuzmin D.A., Shutova M.V., Johnston N.R., Smith O.P., Fedorin V.V., Kukushkin Y.S., van der Loo J.C.M., Johnstone E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021;20:173–174. doi: 10.1038/d41573-021-00017-7. [DOI] [PubMed] [Google Scholar]
- 2.Mendell J.R., Al-Zaidy S.A., Rodino-Klapac L.R., Goodspeed K., Gray S.J., Kay C.N., Boye S.L., Boye S.E., George L.A., Salabarria S., et al. Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 2021;29:464–488. doi: 10.1016/j.ymthe.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batty P., Lillicrap D. Advances and challenges for hemophilia gene therapy. Hum. Mol. Genet. 2019;28:R95–R101. doi: 10.1093/hmg/ddz157. [DOI] [PubMed] [Google Scholar]
- 4.Crudele J.M., Chamberlain J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019;28:R102–R107. doi: 10.1093/hmg/ddz128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lugin M.L., Lee R.T., Kwon Y.J. Synthetically engineered adeno-associated virus for efficient, safe, and versatile gene therapy applications. ACS Nano. 2020;14:14262–14283. doi: 10.1021/acsnano.0c03850. [DOI] [PubMed] [Google Scholar]
- 6.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penaud-Budloo M., François A., Clément N., Ayuso E. Pharmacology of recombinant adeno-associated virus production. Mol. Ther. Methods Clin. Dev. 2018;8:166–180. doi: 10.1016/j.omtm.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnödt M., Büning H. Improving the quality of adeno-associated viral vector preparations: the challenge of product-related impurities. Hum. Gene Ther. Methods. 2017;28:101–108. doi: 10.1089/hgtb.2016.188. [DOI] [PubMed] [Google Scholar]
- 9.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berns K.I., Muzyczka N. AAV: an overview of unanswered questions. Hum. Gene Ther. 2017;28:308–313. doi: 10.1089/hum.2017.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapti K., Grimm D. Adeno-associated viruses (AAV) and host immunity - a race between the hare and the hedgehog. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.753467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronzitti G., Gross D.A., Mingozzi F. Human immune responses to adeno-associated virus (AAV) vectors. Front. Immunol. 2020;11:670. doi: 10.3389/fimmu.2020.00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verdera H.C., Kuranda K., Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brennan T.A., Wilson J.M. The special case of gene therapy pricing. Nat. Biotechnol. 2014;32:874–876. doi: 10.1038/nbt.3003. [DOI] [PubMed] [Google Scholar]
- 15.Young C.M., Quinn C., Trusheim M.R. Durable cell and gene therapy potential patient and financial impact: US projections of product approvals, patients treated, and product revenues. Drug Discov. Today. 2022;27:17–30. doi: 10.1016/j.drudis.2021.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Ragni M. Royal gene therapy at a royal cost. Blood. 2021;138:1645–1646. doi: 10.1182/blood.2021012129. [DOI] [PubMed] [Google Scholar]
- 17.Cornetta K., Bonamino M., Mahlangu J., Mingozzi F., Rangarajan S., Rao J. Gene therapy access: global challenges, opportunities, and views from Brazil, South Africa, and India. Mol. Ther. 2022;30:2122–2129. doi: 10.1016/j.ymthe.2022.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aponte-Ubillus J.J., Barajas D., Peltier J., Bardliving C., Shamlou P., Gold D. Molecular design for recombinant adeno-associated virus (rAAV) vector production. Appl. Microbiol. Biotechnol. 2018;102:1045–1054. doi: 10.1007/s00253-017-8670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balakrishnan B., Jayandharan G.R. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr. Gene Ther. 2014;14:86–100. doi: 10.2174/1566523214666140302193709. [DOI] [PubMed] [Google Scholar]
- 20.Samulski R.J., Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu. Rev. Virol. 2014;1:427–451. doi: 10.1146/annurev-virology-031413-085355. [DOI] [PubMed] [Google Scholar]
- 21.Dimitrov D.S. Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004;2:109–122. doi: 10.1038/nrmicro817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garoff H., Hewson R., Opstelten D.J. Virus maturation by budding. Microbiol. Mol. Biol. Rev. 1998;62:1171–1190. doi: 10.1128/MMBR.62.4.1171-1190.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aljabali A.A., Hassan S.S., Pabari R.M., Shahcheraghi S.H., Mishra V., Charbe N.B., Chellappan D.K., Dureja H., Gupta G., Almutary A.G., et al. The viral capsid as novel nanomaterials for drug delivery. Future Sci. OA. 2021;7:FSO744. doi: 10.2144/fsoa-2021-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeevanandam J., Pal K., Danquah M.K. Virus-like nanoparticles as a novel delivery tool in gene therapy. Biochimie. 2019;157:38–47. doi: 10.1016/j.biochi.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Garmann R.F., Goldfain A.M., Manoharan V.N. Measurements of the self-assembly kinetics of individual viral capsids around their RNA genome. Proc. Natl. Acad. Sci. USA. 2019;116:22485–22490. doi: 10.1073/pnas.1909223116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buzón P., Maity S., Roos W.H. Physical virology: from virus self-assembly to particle mechanics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020;12:e1613. doi: 10.1002/wnan.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pushko P., Pumpens P., Grens E. Development of virus-like particle technology from small highly symmetric to large complex virus-like particle structures. Intervirology. 2013;56:141–165. doi: 10.1159/000346773. [DOI] [PubMed] [Google Scholar]
- 28.Grgacic E.V.L., Anderson D.A. Virus-like particles: passport to immune recognition. Methods. 2006;40:60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kushnir N., Streatfield S.J., Yusibov V. Virus-like particles as a highly efficient vaccine platform: diversity of targets and production systems and advances in clinical development. Vaccine. 2012;31:58–83. doi: 10.1016/j.vaccine.2012.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nooraei S., Bahrulolum H., Hoseini Z.S., Katalani C., Hajizade A., Easton A.J., Ahmadian G. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021;19:59. doi: 10.1186/s12951-021-00806-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryo A., Tsurutani N., Ohba K., Kimura R., Komano J., Nishi M., Soeda H., Hattori S., Perrem K., Yamamoto M., et al. SOCS1 is an inducible host factor during HIV-1 infection and regulates the intracellular trafficking and stability of HIV-1 Gag. Proc. Natl. Acad. Sci. USA. 2008;105:294–299. doi: 10.1073/pnas.0704831105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buzón P., Maity S., Christodoulis P., Wiertsema M.J., Dunkelbarger S., Kim C., Wuite G.J.L., Zlotnick A., Roos W.H. Virus self-assembly proceeds through contact-rich energy minima. Sci. Adv. 2021;7:eabg0811. doi: 10.1126/sciadv.abg0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Payne E., Bowles M.R., Don A., Hancock J.F., McMillan N.A. Human papillomavirus type 6b virus-like particles are able to activate the Ras-MAP kinase pathway and induce cell proliferation. J. Virol. 2001;75:4150–4157. doi: 10.1128/JVI.75.9.4150-4157.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo K., Liu B., Xiao Z., Yu Y., Yu X., Gorelick R., Yu X.F. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 2004;78:11841–11852. doi: 10.1128/JVI.78.21.11841-11852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnston G.P., Contreras E.M., Dabundo J., Henderson B.A., Matz K.M., Ortega V., Ramirez A., Park A., Aguilar H.C. Cytoplasmic motifs in the Nipah virus fusion protein modulate virus particle assembly and egress. J. Virol. 2017;91 doi: 10.1128/JVI.02150-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chlanda P., Mekhedov E., Waters H., Sodt A., Schwartz C., Nair V., Blank P.S., Zimmerberg J. Palmitoylation contributes to membrane curvature in influenza A virus assembly and hemagglutinin-mediated membrane fusion. J. Virol. 2017;91 doi: 10.1128/JVI.00947-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plescia C.B., David E.A., Patra D., Sengupta R., Amiar S., Su Y., Stahelin R.V. SARS-CoV-2 viral budding and entry can be modeled using BSL-2 level virus-like particles. J. Biol. Chem. 2021;296 doi: 10.1074/jbc.RA120.016148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng T., Qiu X., Tan L., Yu S., Yang B., Dai J., Liu X., Sun Y., Song C., Liu W., et al. Ubiquitination on lysine 247 of newcastle disease virus matrix protein enhances viral replication and virulence by driving nuclear-cytoplasmic trafficking. J. Virol. 2022;96 doi: 10.1128/JVI.01629-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crosson S.M., Dib P., Smith J.K., Zolotukhin S. Helper-free production of laboratory grade AAV and purification by iodixanol density gradient centrifugation. Mol. Ther. Methods Clin. Dev. 2018;10:1–7. doi: 10.1016/j.omtm.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tustian A.D., Bak H. Assessment of quality attributes for adeno-associated viral vectors. Biotechnol. Bioeng. 2021;118:4186–4203. doi: 10.1002/bit.27905. [DOI] [PubMed] [Google Scholar]
- 41.Pereira D.J., McCarty D.M., Muzyczka N. The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J. Virol. 1997;71:1079–1088. doi: 10.1128/JVI.71.2.1079-1088.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hörer M., Weger S., Butz K., Hoppe-Seyler F., Geisen C., Kleinschmidt J.A. Mutational analysis of adeno-associated virus Rep protein-mediated inhibition of heterologous and homologous promoters. J. Virol. 1995;69:5485–5496. doi: 10.1128/JVI.69.9.5485-5496.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Satkunanathan S., Wheeler J., Thorpe R., Zhao Y. Establishment of a novel cell line for the enhanced production of recombinant adeno-associated virus vectors for gene therapy. Hum. Gene Ther. 2014;25:929–941. doi: 10.1089/hum.2014.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Satkunanathan S., Thorpe R., Zhao Y. The function of DNA binding protein nucleophosmin in AAV replication. Virology. 2017;510:46–54. doi: 10.1016/j.virol.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owens R.A., Weitzman M.D., Kyöstiö S.R., Carter B.J. Identification of a DNA-binding domain in the amino terminus of adeno-associated virus Rep proteins. J. Virol. 1993;67:997–1005. doi: 10.1128/JVI.67.2.997-1005.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J., Samulski R.J., Xiao X. Role for highly regulated rep gene expression in adeno-associated virus vector production. J. Virol. 1997;71:5236–5243. doi: 10.1128/JVI.71.7.5236-5243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hölscher C., Kleinschmidt J.A., Bürkle A. High-level expression of adeno-associated virus (AAV) Rep78 or Rep68 protein is sufficient for infectious-particle formation by a rep-negative AAV mutant. J. Virol. 1995;69:6880–6885. doi: 10.1128/JVI.69.11.6880-6885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q., Wu Z., Zhang J., Firrman J., Wei H., Zhuang Z., Liu L., Miao L., Hu Y., Li D., et al. A robust system for production of superabundant VP1 recombinant AAV vectors. Mol. Ther. Methods Clin. Dev. 2017;7:146–156. doi: 10.1016/j.omtm.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rayaprolu V., Kruse S., Kant R., Venkatakrishnan B., Movahed N., Brooke D., Lins B., Bennett A., Potter T., McKenna R., et al. Comparative analysis of adeno-associated virus capsid stability and dynamics. J. Virol. 2013;87:13150–13160. doi: 10.1128/JVI.01415-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wörner T.P., Bennett A., Habka S., Snijder J., Friese O., Powers T., Agbandje-McKenna M., Heck A.J.R. Adeno-associated virus capsid assembly is divergent and stochastic. Nat. Commun. 2021;12:1642. doi: 10.1038/s41467-021-21935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonntag F., Köther K., Schmidt K., Weghofer M., Raupp C., Nieto K., Kuck A., Gerlach B., Böttcher B., Müller O.J., et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011;85:12686–12697. doi: 10.1128/JVI.05359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett A., Patel S., Mietzsch M., Jose A., Lins-Austin B., Yu J.C., Bothner B., McKenna R., Agbandje-McKenna M. Thermal stability as a determinant of AAV serotype identity. Mol. Ther. Methods Clin. Dev. 2017;6:171–182. doi: 10.1016/j.omtm.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohammadi E.S., Ketner E.A., Johns D.C., Ketner G. Expression of the adenovirus E4 34k oncoprotein inhibits repair of double strand breaks in the cellular genome of a 293-based inducible cell line. Nucleic Acids Res. 2004;32:2652–2659. doi: 10.1093/nar/gkh593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aurnhammer C., Haase M., Muether N., Hausl M., Rauschhuber C., Huber I., Nitschko H., Busch U., Sing A., Ehrhardt A., Baiker A. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods. 2012;23:18–28. doi: 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- 55.Khan I.F., Hirata R.K., Russell D.W. AAV-mediated gene targeting methods for human cells. Nat. Protoc. 2011;6:482–501. doi: 10.1038/nprot.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gregorevic P., Blankinship M.J., Allen J.M., Crawford R.W., Meuse L., Miller D.G., Russell D.W., Chamberlain J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ayukawa S., Kamoshita N., Nakayama J., Teramoto R., Pishesha N., Ohba K., Sato N., Kozawa K., Abe H., Semba K., et al. Epithelial cells remove precancerous cells by cell competition via MHC class I-LILRB3 interaction. Nat. Immunol. 2021;22:1391–1402. doi: 10.1038/s41590-021-01045-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this paper will be shared by the lead contact (ohbak@jichi.ac.jp) upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.