

Abstract
Objective
Orexin-A (OX-A) is a neuropeptide produced selectively by neurons of the lateral hypothalamus. It exerts powerful control over brain function and physiology by regulating energy homeostasis and complex behaviors linked to arousal. Under conditions of chronic or acute brain leptin signaling deficiency, such as in obesity or short-term food deprivation, respectively, OX-A neurons become hyperactive and promote hyperarousal and food seeking. However, this leptin-dependent mechanism is still mostly unexplored. The endocannabinoid 2-arachidonoyl-glycerol (2-AG) is known to be implicated in food consumption by promoting hyperphagia and obesity, and we and others demonstrated that OX-A is a strong inducer of 2-AG biosynthesis. Here, we investigated the hypothesis that, under acute (6 h fasting in wt mice) or chronic (in ob/ob mice) hypothalamic leptin signaling reduction, OX-A-induced enhancement of 2-AG levels leads to the production of the 2-AG-derived 2-arachidonoyl-sn-glycerol-3-phosphate (2-AGP), a bioactive lipid belonging to the class of lysophosphatidic acids (LPAs), which then regulates hypothalamic synaptic plasticity by disassembling α-MSH anorexigenic inputs via GSK-3β-mediated Tau phosphorylation, ultimately affecting food intake.
Methods
We combined cell-type-specific morphological (CLEM and confocal microscopy), biochemical, pharmacological, and electrophysiological techniques to dissect the leptin- and OX-A/2-AGP-mediated molecular pathways regulating GSK-3β-controlled pT231-Tau production at POMC neurons of obese ob/ob and wild-type (wt) lean littermate mice and in an in vitro model of POMC neurons such as mHypoN41 neurons (N41).
Results
2-AGP is overproduced in the hypothalamus of obese leptin-deficient, or lean 6 h food-deprived mice, and promotes food intake by reducing α-MSH-expressing synaptic inputs to OX-A neurons via lysophosphatidic acid type-1 receptor (LPA1-R) activation, and pT231-Tau accumulation in α-MSH projections. This effect is due to the activation of the Pyk2-mediated pTyr216-GSK3β pathway and contributes to further elevating OX-A release in obesity. Accordingly, we found a strong correlation between OX-A and 2-AGP levels in the serum of obese mice and of human subjects.
Conclusions
Hypothalamic feeding pathways are endowed with 2-AGP-mediated synaptic plasticity according to their inherent functional activities and the necessity to adapt to changes in the nutritional status. These findings reveal a new molecular pathway involved in energy homeostasis regulation, which could be targeted to treat obesity and related disturbances.
Keywords: Hypothalamus, Obesity, α-MSH, Orexin/hypocretin, 2-Arachidonoyl-glycerol, 2-AG
Graphical abstract
Leptin deficiency caused by obesity provokes an increment OX-A levels in arcuate nucleus with an enhancement of 2-AG and 2-AGP levels in POMC neurons. The endocannabinoid-derived lysophospatidic acid (2-AGP) promotes Tau phosphorylation by binding the receptor LPA1-R. This effect is prevented by leptin or by selective antagonism of LPA1-R and OX-1R receptors.
Highlights
-
•
Under leptin signaling deficiency OX-A neurons promote food seeking by upregulating endocannabinoid 2-AG biosynthesis.
-
•
The enhancement of 2-AG levels leads to the biosynthesis of the 2-arachidonoyl-sn-glycerol-3-phosphate (2-AGP).
-
•
2-AGP promotes food intake by reducing α-MSH inputs to OX-A neurons via LPA1-R activation and pT231-Tau accumulation.
-
•
A strong correlation is found between OX-A and 2-AGP levels in the serum of human subjects and obese mice.
1. Introduction
Obesity has reached epidemic proportions globally and is a major contributor to the world burden of chronic diseases and disabilities related to this condition and affecting all ages and socioeconomic groups. From an evolutionary perspective, the functioning of the mammalian brain circuits regulating energy homeostasis is committed to seeking, securing, consuming, and storing nutrients to ensure the natural drive for the survival of the individuals and the species. A paradoxical consequence of this evolutionary drive is over-nutrition and obesity in humans. The hypothalamus is the brain hub integrating inputs from different neuronal networks encoding metabolic, behavioural, and environmental cues affecting energy homeostasis, food intake and arousal. The lateral hypothalamus (LH) contains orexin-A (OX-A)-expressing neurons, which are endowed with a short-lived form of synaptic plasticity according to their inherent functional adjustment to changes in the nutritional status [2,8,23,55,68]. Experimental and clinical studies report that chronic overnutrition leads to changes in brain connectivity and function. However, the mechanisms underlying these changes are not completely understood. Unraveling the adaptive and maladaptive pathways governing or deregulating the synaptic refinement of OX-A neurons in response to balanced or unbalanced energy intake may help understand some of the deranged behavioural responses that compromise metabolic health during obesity.
Lysophosphatidic acids (LPAs) are strong candidates to act as local messengers that rapidly affect synaptic homeostasis by regulating neurite Tau phosphorylation, neurotransmitter release, plasticity [29], and excitation/inhibition (E/I) balance in cortical networks [78]. The role of Tau phosphorylation as a regulatory mechanism of synaptic plasticity has not been fully addressed in in vivo physiological conditions, and previous data only refer to synaptic contacts between mossy fibers and hippocampal pyramidal neurons, which undergo changes mediated by fully and rapidly reversible phosphorylation of Tau at the Threonine 231 epitope (pT231-Tau) [3,56,57]. Although Tau phosphorylation at different residues (Ser199, Ser202/Ser 205 or AT8, Ser422, Ser396/Ser404 or PHF-1) produces irreversible Tau aggregation in the neurofibrillary tangles of Alzheimer's disease (AD) [30,40], pT231-Tau production is not neurotoxic per se as it closely matches the pattern of physiological neuronal plasticity in the adult brain by controlling Tau binding affinity to microtubules [[3], [4], [5]] and the synaptic architecture of axons [6,54,66,72]. Many subtypes of LPAs and selective LPA G-protein-coupled receptors (LPA1–6) are present in the brain [16]. The LPA species 2-arachidonoyl-sn-glycerol-3-phosphate (2-AGP) and its preferential receptor LPA1 (or EDG2), hereafter referred to as LPA1R, are prevalent in embryonic and adult mammalian brains, where 2-AGP can be biosynthesized also from the phosphorylation of the endocannabinoid 2-AG [51].
Among its several functions, OX-A was originally identified also as a positive regulator of feeding behavior [21,62] and energy consumption [32] in a circadian-dependent manner [48]. Furthermore, OX-A has been demonstrated to act as a strong inducer of 2-AG biosynthesis via the OX-1R-Gq-PLCβ-DAGLα cascade in in vitro [1,36,76] and in in vivo experiments [19,34]. In previous studies, we reported that hypothalamic 2-AG biosynthesis is enhanced in ob/ob and HFD-induced obese mice because of the increased OX-A release due to lack or inefficiency of leptin signaling [18,27,50].
Here, we investigated the hypothesis that, under acute (6 h fasting in wt mice) or chronic (in ob/ob mice) leptin signaling reduction/impairment in mice, OX-A-induced enhancement of 2-AG levels in the ARC leads to 2-AGP production, LPA1R activation and pT231-Tau accumulation in POMC neurons, with subsequent transient or irreversible disassembly of α-MSH synaptic inputs to OX-A neurons.
2. Materials and methods
2.1. Experimental model and subject details and sample collection
The study has been performed according to the ARRIVE Guidelines to improve the reporting of bioscience research using laboratory animals. Experiments were performed following the European Union animal welfare guidelines [European Communities Council Directive of September 22, 2010 (2010/63/EU)] and the Italian Decree n.26/2014 authorization n. 152/2020-PR and 589/2018. Maximal efforts were made to avoid or reduce any suffering as well as to reduce the number of animals used.
2.2. Subject population and sample collection
The study was approved by the Regional Health Authority of the University of Molise. Written informed consent was obtained from subjects. The ethical principles of the Declaration of Helsinki and the national and international guidelines for human research were followed. Sixty male participants (n = 30 normal weight and n = 30 obese subjects) were recruited from the Centre for Research and Training in Medicine for Aging (CeRMA) of the University of Molise (Italy). The division was carried out based on the Body Mass Index (BMI); subjects with BMI > 30 were classified as obese, according to the cut-off proposed by the WHO (World Health Organization). The patients on treatment with brain-active drugs underwent a washout period of at least 14 days before the assessment. Venous blood was collected with standard clinical procedures between 7:30 and 8:00 am after overnight fasting. For the collection we used vacutainer serum tubes (Becton & Dickinson, Milan, Italy); samples were coagulated at room temperature for 10 min, and then a 10-min centrifugation at 3000 g was applied. Supernatants were frozen in liquid nitrogen and stored at −80 °C until measurements. Serum was used to analyze OX-A and 2-AGP levels.
2.3. Animals
Male C57BL/6jmice from Charles River Laboratories were used starting at 16-weeks-old. Male mice with spontaneous nonsense mutation of the ob gene for leptin (ob/ob, JAX mouse strain) B6. Cg-Lepob/J and WT ob gene expressing homozygous siblings of different ages were obtained from breeding ob gene heterozygotes and genotyped with PCR. OREXIN-eGFP (Tg(Hcrt-eGFP/Rpl10a) JD218Jdd, Jackson Lab) and POMC-eGFP (C57BL/6 J-Tg(Pomc-eGFP)1Low/J, Jackson Lab) mice were used for a subset of experiments. Animals were maintained under a 12:12 h reverse light/dark cycle (lights on at 8:00) for at least 4 weeks before euthanizing at ZT20-22. Since orexin levels exhibit a diurnal fluctuation with an increment during the active phase (i.e., ZT23-24) and decrease during the rest phase [81]. Mice were housed in controlled temperature (22 ± 2 °C) and humidity conditions and received a standard chow diet and water ad libitum. All the animals were used in scientific experiments for the first time and were not previously exposed to any pharmacological treatments. The number of mice used in each experiment is indicated in each figure legend and statistical analysis. Wt and ob/ob mice were injected i.p. with several treatments as follows: vehicle-DPBS, OX-A (Tocris, 40 μg/kg, 1 h), leptin (Sigma Aldrich, 5 mg/kg, 1 h), SB334867 (Tocris, 30 mg/kg for wt and 60 mg/kg for ob/ob mice, 3 h alone or 1 h before OX-A injection), PF431396 (Tocris, 5μM/animal, 3 h alone or 1 h before OX-A injection), O-7460 (Cayman, 12 mg/kg, 3 h alone or 1 h before OX-A injection), 2-AG-(d8) (Tocris, 10 mg/kg, 1 h), 2-AGP (Avanti Polar Lipids, 10 mg/kg, 1 h), AM095 (MedChemExpress, 10 mg/kg, 3 h alone or 1 h before 2-AGP injection). To evaluate food intake the mice were housed, one per cage and the same known amount of pellet (5 g) was positioned per each cage and the remaining was weight registered after 24 h.
2.4. Tissue collection
At the end of the in vivo experiments, mice were killed to obtain either fresh or perfused tissues, as mentioned per procedure. For obtaining perfused brain tissue, mice were deeply anesthetized with an i.p. injection of sodium pentobarbital (300 μl/30 g Exagon mixed with 30 mg/kg lidocaine) and perfused transcardially with ice-cold phosphate buffer saline (PBS, pH 7.4), followed by 4% paraformaldehyde (PFA, Sigma–Aldrich, France). Whole brains were extracted and post-fixed in 4% PFA overnight at 4 °C, and frozen after cryo-protection with a 30% sucrose solution in PBS at 4 °C. Coronal sections (10 μm) were cut with a cryostat (CM1950, Leica, Germany), collected, and stored in anti-freeze solution (30% ethylene glycol, 30% glycerol in KPBS) at −20 °C until analysis. For fresh tissue sampling, mice were killed by cervical dislocation (for plasma collection) or dislocation, and tissues were collected, frozen on dry ice, and stored at −80 °C until needed for molecular and biochemical analysis. Lean and obese mice, treated as described previously, were killed by cervical dislocation, the brains removed and the ARC and LH were rapidly dissected.
2.5. Cell cultures
Hypothalamic mouse mHypoE-N41 cells were cultured in Dulbecco's modified Eagle’ s medium (Life Technologies) supplemented with 10% horse fetal bovine serum (Life Technologies), penicillin (50 units/ml), and streptomycin (50 μg/ml), at 37 °C in 100-mm culture dishes (Life Technologies) gassed with an atmosphere of 95% air, 5% CO2.
Primary cultures of hypothalamic ARC neurons, derived from neonatal 0 day-old POMC-eGFP transgenic mice (Charles River) were obtained as described elsewhere [53]. Briefly, the hypothalamic area of the ARC was quickly dissected and mechanically dispersed in Ca2+- and Mg2+-free buffered Hanks'balanced salt solution. Tissue was dissociated enzymatically (0.125% trypsin solution, 37 °C for 20 min) and mechanically. Cells were plated at a density of 2 × 104 cells/cm2 on polylysine-coated coverslips and grown in Neurobasal medium supplemented with 2% B27, 0.5 mM l-glutamine, penicillin (50 U/mL) and streptomycin (50 μg/mL). Cells were used between days 6th–8th in vitro.
2.6. Immunohistochemistry and quantification
Animals were euthanized under isoflurane anesthesia and perfused transcardially with 4% (wt/vol) paraformaldehyde/0.1 M phosphate buffer (PB) pH 7.4. Brains were removed, fixed and then transferred into a 30% sucrose (#S8501, Sigma Aldrich) and were cut with a Leica CM3050S cryostat into 10 μm-thick serial sections in the coronal plane, collected in three alternate series and then frozen until being processed. Alternate brain slices (10 μm of thickness) containing hypothalamic areas of mice were incubated overnight with the following primary antibodies: 1) goat anti-cFos (1:400, #sc-52-G, Santa Cruz Biotechnology); 2) rabbit anti-POMC (1:500, # EPR22534-165, ab254257, Abcam); 3) mouse anti LPA1R or EDG2 (1:400, #TA503737, Origene); 4) goat anti-OX-A (1:500, #sc-8070, Santa Cruz Biotechnology); 5) mouse anti-pThr231 Tau (1:500; # MN1040AT180, Thermo Fisher Scientific) rabbit anti-pThr231 Tau (1:500, #44746G, Invitrogen); 6) mouse anti-PSD95 (1:1000, #124 011, Synaptic Systems); 7) rabbit anti-α-MSH (1:200; Abcam). After incubation with primary antibodies, the sections were washed and then incubated with appropriate Alexa-488, -546, or -350 donkey anti IgGs (Invitrogen LifeTechnologies) secondary antibodies before being counterstained with nuclear dye DAPI (4′,6diamidino-2-phenylindole). Immunofluorescence was analyzed by the confocal microscopy Nikon Eclipse Ti2 and images were acquired with the digital camera DS-Qi2 (Nikon) and processed by Image analysis software NIS-Elements C (Nikon, Florence, Italy). N = 6–10 z stacks were collected through each analyzed section every 0.5 μm throughout the area of interest to be processed by the imaging deconvolution software. For the densitometric analysis of pT231-Tau immunoreactivity, 10-μm coronal pT231-Tau–ir section of the ARC were scanned by using a Leica Metamorph imaging software (Leica Meta-Morph ©, Germany) according to the method reported in [27]. The density of immunoreactivity was rendered as area of distribution by selecting 20 × 20 μm2 as the region of interest of the ARC nucleus, from the 3rd ventricle throughout the approximate posterior levels from Bregma −1.70 mm to Bregma −2.30 mm. The densitometric analysis of pT231-Tau immunoreactivity was performed by considering every three consecutive sections (10 μm thickness each) per mouse (n = 3 mice/gruoup) and n = 20 z-stacks/section (z stack = 0.5 μm thickness each). The quantitative analysis of the percentage of POMC-immunoreactive neurons colocalizing with c-fos or with pT231-Tau imunolabeling was performed per μm2 of coronal sections of each ARC nucleus by using Leica Metamorph imaging software (Leica Meta-Morph ©, Germany), according to the method reported in [27]. The percentage was obtained from the ratio between the number of double immunoreactive neurons and the number of POMC-immunoreactive neurons. With this purpose, z-stack plans of each selected section were reconstructed in 3D rendering by using the software image processing LAS-X Measurement (Leica©) for counting double immunoreactive cell number from a respective immunolabeled section. An area of 20 μm2/ARC nucleus selected from the 3rd ventricle throughout the approximate posterior levels, from Bregma −1.70 mm to Bregma −2.30 mm, was considered per each ARC nucleus. The quantification was performed by considering every three consecutive sections (10 μm thickness each) per mouse (n = 3 mice/group) and n = 20 z-stacks/section (z stack = 0.5 μm thickness each). If not verified in otherwise published works using the same antibodies, the control of the specificity of immunolabeling for the antibodies used in multiple or single immunofluorescence has been proven by omission of primary and secondary antibodies or by pre-absorption of primary antibodies with the respective blocking peptide as such in the case of anti-LPA1R antibody produced on our request by the company Origine (Source: Uniprot.Org, NCBI; Uniprot ID: Q92633 Gene ID). No labeling was detected in all the control samples. In detail, the specificity of mouse anti-LPA1R was also verified using the immunoperoxidase reaction. For this purpose, the sections were pretreated with 0.75% H2O2 in an aqueous solution for 5 min at room temperature (RT) to block the endogenous peroxidase activity and then incubated with TBS-T (0.4% Triton in tris buffered saline solution) blocking buffer, containing the primary antiserum (NGS), for 30 min at RT, to block nonspecific binding sites and obtain an optimal tissue permeabilization. The sections were incubated overnight in a humid box at RT with the primary mouse anti-LAP1R antibody diluted 1:400 in TBS-T, while adjacent brain sections containing the ARC nuclei were incubated in a mixture of anti-LAP1R saturated with selective blocking peptide from Origine©. The following day, the sections were rinsed with TBS and then incubated for 3 h at RT with biotin-conjugate secondary goat anti-mouse immunoglobulin (H + L) (Vector Laboratories, Burlingame, CA) diluted 1:100 in TBS-T. For the peroxidase reaction, the sections were rinsed again with TBS and incubated with avidin-biotin complex (ABC) for 1 h, followed by an incubation with the chromogen substrate 3,3′-diaminobenzidine-4 (DAB) (Sigma Fast, Sigma–Aldrich, Louis, MO U.S.A.) for 1 or 2 min when the intensity of the signal was appropriate for imaging. To finish the reaction, the slices were rinsed in TBS to stop the DAB reaction, dehydrated by subsequent slides immersion in alcohol 50% (2 min), 75% (2 min), 95% (2 min), 100% I (2 min), 100% II (2 min), clarified by immersion 2 times in xylene for 10 min each and then mounted with DPX (dibutyl phthalate xylene). The immunodensity of pThr231-Tau immunoreactivity was evaluated by quantifying the optical density (O.D.) of the immunoperoxidase signal by using the LAS AF MetaMorph Imaging Software (Leica, Wetzlar, Germany) according to the formula: O.D. = log10 (255/I), where the “I” was the pixel intensity value given by the imaging software. All the analyzed sections were exposed to the same parameters of brightness, contrast and magnification. No immunoreactive signals were detected in the sections immunoreacted with the mixture of anti-LPA1R pre-adsorbed with the blocking peptide (data shown in Supplementary Fig. 3)
2.7. Western blot analysis
Tissue samples were homogenized in 1xTNE buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA) containing 10% Triton X-100, protease and phosphatase inhibitor mixtures (Sigma Aldrich) for obtaining protein extracts. Protein concentrations were analyzed using Lowry protein assay (Bio-Rad Laboratories) to allow the loading of the same amount of proteins (20 μg).Proteins were separated in an SDS-polyacrylamide gel (4%–20%) by electrophoresis and transferred to PDVF membranes, which were then blocked for 1 h with 5% skim milk powder dissolved in 1xTBST (20 mM Tris, 137 mM NaCl, 0.1% Tween-20). After blocking, the membranes were incubated overnight at 4 °C with the following primary antibodies: rabbit anti-AKT (1:900, #9272 S, Cell Signalling); rabbit anti-pSer473 AKT (1:1000, #4060 S, Cell Signalling); rabbit anti-GSK3β (1:1000, #9315 S, Cell Signalling); rabbit anti-pTyr216 GSK3β (1:1000, #GWB-516365 Gene Way); rabbit anti-pSer9 GSK3β (1:1000, #9323 S, Cell Signalling); rabbit anti-PYK2 (1:1000, #3280 S, Cell Signalling); rabbit anti-pTyr402 PYK2 (#3291 S, Cell Signalling); mouse anti-Tau (1:900, #4019 S, Cell Signalling); rabbit anti-pThr231 Tau (1:1000, #44746G, Invitrogen); mouse anti-PSD95 (1:2000, #124011, Synaptic system), and mouse monoclonal anti-β-actine (1:4000, #A1978, Sigma Aldrich), which was used to check for equal protein loading. After washing in TBST, the membranes were incubated for 1 h at room temperature with HRP-conjugated goat anti-rabbit secondary antibody (1:4000, #1706515 Biorad) or goat anti-mouse (1:4000, #1706516, Biorad). Subsequently rinsed 3 times in TBST, immunoreactive bands were visualized using enhanced chemiluminescence (Clarity ECL, #1705061, Biorad) then exposed on a ChemiDoc MP Imaging System (# 17001402, Biorad). Bands were quantified using ImageJ software (NIH, USA). When necessary, membranes were stripped after protein detection for 10 min at 55 °C with a solution containing 62.5 mmol/l Tris–HCl, 100 mmol/l 2-mercaptoethanol, and 2% SDS, blocked, and reblotted with another primary antibody.
2.8. CLEM
pThr231-Tau, OX-A and α-MSH immunoreactivities in preembedding electron microscopy. Under isoflurane anaesthesia, mice were perfused transcardially with 3% PFA/0.5–1% glutaraldehyde (vol/vol) in PB. Multiple preembedding immunogold labeling was performed in the ARC or LH sections (50-μm-thick) to reveal LPA1R immunogold labeling in the ARC of POMC-eGFP mice or pT231-Tau/α-MSH double immunolabelling in the LH of ob/ob or OX-eGFP food-deprived mice. The sections were incubated free-floating overnight at 4 °C with primary antibodies containing LPA1R (mouse anti-LPA1R or EDG2-1:100, #TA503737, Origene) or pThr231Tau/α-MSH. For pThr231-Tau immunolabeling the free floating sections were firstly incubated overnight at +4 °C with mouse primary antibody (Thermo Fisher Scientific) diluted 1:100 in donkey serum blocking solution containing saponin 0.02% v/v and, after washing, the same sections were incubated with a specific biotinylated donkey anti-mouse secondary antibody which was revealed by DAB-Ni-enhancing method. After appropriate washing, the same sections were incubated in a mixture of rabbit anti-αMSH primary antibody (Abcam), diluted 1:100 in donkey serum blocking solution containing 0.02% saponin. Immunoreactivities were revealed by incubation with 10 nm donkey anti-rabbit (to α-MSH) or donkey anti mouse (to LPA1R) gold-conjugated secondary antibodies (Aurion), diluted 1:30 in donkey serum blocking solution with 0.02% saponin. Finally, the sections were treated with 0.5% OsO4 in PB for 10 min at 4 °C, dehydrated in an ascending series of ethanol and propylene oxide and embedded in TAAB 812 resin (TAAB). During dehydration, sections were treated with 1% uranyl acetate in 70% ethanol (vol/vol) for 15 min at 4 °C. Ultrathin (60 nm thickness) sections were cut by vibratome (Leica), collected on Formovar-coated single- or multiple-slot (50-mesh) grids and stained with 0.65% lead citrate. Appropriate ultrathin sections were analyzed by TEM after accurate selection of POMC-eGFP- or OX-eGFP-expressing neurons identified by fluorescence microscopy and specific anatomical reference points. Electron micrographs were taken with the TEM microscope (FEI Tecnai G2 Spirit TWIN). The observation was limited to a series sectioned up to 0.6–0.8 μm depth from the external surface of preembedded immunolabeled tissue. Additional sections were processed in parallel as controls of reaction by omitting both or one of the primary antibodies from the mixture. No labeling was detected in the control samples.
2.9. Lipid extraction and LC-MS quantification of 2-AG and 2-AGP levels
For the lipid extraction and LC-MS quantification of 2-AG and 2-AGP levels, the hypothalamic area containing the ARC nucleus was dissected from the brain of lean and obese mice killed by cervical dislocation. From each brain, the ARC tissue from one side and from the controlateral one were used for 2-AG and 2-AGP quantification, respectively. The tissues were homogenized in 5 vol chloroform/methanol/Tris HCl 50 mM (2:1:1 by volume) containing 50 pmol of d5-2-arachidonoylglycerol (d5-2-AG) as internal standards. Homogenates were centrifuged at 13,000×g for 16 min (4 °C), and the aqueous phase plus debris were collected and four times extracted with 1vol chloroform. The lipid-containing organic phases were dried and pre-purified by open-bed chromatography on silica columns eluted with increasing concentrations of methanol in chloroform. Fractions for 2-AG measurement were obtained by eluting the columns with 9:1 (by volume) chloroform/methanol and then analyzed by liquid chromatography-atmospheric pressure chemical ionization-mass spectrometry (LC-APCI-MS). LC-APCI-MS analyses were carried out in the selected ion monitoring mode, using m/z values of 384.35 and 379.35 (molecular ions +1 for deuterated 2-AG-(d8), and undeuterated 2-AG). Values are expressed as pmol/mL of lipid extracted. (2-AG pmol/mg lipids; 2-AGP pmol/mg lipids). LC-MS quantification of deuterated 2-AGP-(d8) in the ARC hypothalamic nuclei or in the POMC-eGFP neurons sorted by FACS from the hypothalami of adult mice i.p.-injected with vehicle or with deuterated 2-AG-(d8), was also performed as described above. The POMC-eGFP neurons were obtained using FACS assay as described by [42]. Briefly, adult POMC-eGFP mice were sacrificed by cervical dislocation and the hypothalami digested with papain (20 U/ml, for 30 min at 37 °C), and the cell suspension filtered through a 40 μm cell strainer. Influx Cell Sorter (BD Biosciences, San Jose, CA, USA) was set according to cell size and the fluorescence at 488 nm/532 nm. GFP-positive cells were collected in cold PBS/5% fetal bovine serum and then processed for LC-APCI-MS as described above.
2.10. ELISA for OX-A levels
OX-A levels were measured with commercial ELISA KIT (Phoenix Pharmaceuticals Inc, Burlingame, CA, USA) according to the manufacturer's instructions. Triplicate samples were assayed, and levels were determined against a known standard. OX-A levels were measured using an ultra-sensitive Fluorescent EIA Kit (Phoenix Pharmaceuticals Inc, Burlingame, CA, USA) following the manufacturer's instructions. Before measurement, serum OX-A was extracted using Sep-Pak C18 columns (Waters, Milford, MA). The samples, applied to the column, were eluted slowly with 80% acetonitrile. The samples were evaporated, and the dry residue was dissolved in water and used for Fluorescent EIA Kit.
2.11. Electrophysiology
Preparation of hypothalamic slices: OX/Hcrt-Tg(HcrteGFP/Rpl10a)JD218Jdd 4–6 weeks old of both sexes were used. After anaesthesia with an overdose of isoflurane, the mice were decapitated, and the brains were quickly removed. Coronal hypothalamic slices (200 μm) containing the LH were cut using a Leica VT1000 S Vibrating blade microtome at 3–5 °C in a solution containing (in mM): 87 NaCl, 25 NaHCO3, 2.5 KCl, 0.5 CaCl2, 7 MgCL2, 10 glucose, 75 sucrose, and saturated with 95% O2 and 5% CO2. After cutting, we let the slices recover for at least 30 min at 35 °C and for 10 min at room temperature in artificial cerebrospinal fluid (ACSF) containing in mM: 125 NaCl, 25 NaHCO3, 10 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCL2 (bubbled with 95% O2–5% CO2). Coronal slices were transferred to a recording chamber and continually perfused with gassed ACSF. Whole-cell recordings were performed with a Multiclamp 700B/Digidata1440 A system on visualized OX-eGFP neurons using a Leica DM6000 FS microscope equipped with a WAT-902H Ultimate camera. Recording pipettes pulled using a Sutter P- 1000 puller had a resistance of 4–7 MΩ when filled with k-gluconate solution. Data were acquired at 10 kHz sample frequency and filtered at 2 kHz with a low-pass Bessel filter. In all experiments, we used a high k-gluconate intracellular solution containing (in mM): 126 k-gluconate, 4 NaCl, 1 MgSO4, 0.02 CaCl2, 0.1 1,2-bis(o-aminophenoxy) ethane-N,N,N′, N′-tetraacetic acid (BAPTA), 15 glucose, 5 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 3 ATP, and 0.1 GTP. The pH was adjusted to 7.3 with KOH and osmolarity was adjusted to 290 mOsmol/L using sucrose. All whole-cell patch-clamp recording experiments were performed in the presence of 30 μM bicuculline methiodide, 5 μm CGP55845 (all from Tocris Bioscience, Ellisville, MO, U.S.A.) to inhibit GABAA and GABAB. Recordings of miniature excitatory postsynaptic currents (mEPSC) were done in presence of TTX 0.3 μM (Tocris). OX-eGFP were voltage clamped at −60 mV. Recordings with a series resistance >20 MΩ were discarded. One cell for slices was recorded and 2-AGP (250 nM) was incubated for 20 min after the baseline recording. To inhibit the LPA1R, we used AM095 (10 μM,MedChemExpress). Evoked excitatory postsynaptic currents (eEPSCs) were recorded in OX-eGFP neurons in response to extracellular stimulation (A-M Systems 2100, Sequim WA) with a monopolar glass electrode filled with ACSF placed in the proximity of the recorded cells. GABA receptors were inhibited using 30 μM Bicuculline and 5 μM CGP58845. The minimal response with no failures and a stable latency and shape was found at a stimulation intensity fixed at 100 μA, lasting between 50 and 500 μs. All data were acquired with pClamp 10.4 software (Molecular Devices) and analyzed offline with Clampfit 11.1 (Molecular Device), Excel, and GraphPad Prism (GraphPad Software, USA). mEPSCs amplitude and frequency were calculated by averaging the events in 3 min of recording. We considered the peak amplitude as the difference between the baseline and the peak of the eEPSCs. To calculate pulse ratio (PPR), two brief stimulation pulses were applied at inter-stimulus intervals ranging between 10 ms and 1 s (10 sweeps for each trial), and the ratio between the amplitude of the second (I2) and the first response (I1) was calculated.
2.12. Quantification and statistical analysis
Statistical analysis was carried out using GraphPad Prism Software version 8.0.2. In all cases, the test was significant when p < 0.05. Data are expressed as mean ± SEM. Sample sizes for experiments were determined based on sample sizes used in similar experiments reported previously in the literature. The statistical test used for each comparison is described in the figure legends corresponding to the specific figure. The normality distribution of the data was confirmed using D'Agostino-Pearson, Shapiro–Wilk, and Kolmogorov–Smirnov test. When comparing two unpaired groups, data were analyzed by a two-tailed Student's t-test (parametric) or a two-tailed Mann–Whitney U test (non-parametric). When two paired groups, data were analyzed by Wilcoxon matched-pairs signed rank test, two-tailed. When comparing three or more groups, data were analyzed by One-way ANOVA/Bonferroni, Kruskal–Wallis/Dunn's tests or 2-way ANOVA with Tukey post hoc were used to analyze data appropriately. Fisher's LS post hoc tests were run, when applicable, for identifying differences among groups. Pearson's correlation coefficient was used to analyze the correlation.
3. Results
3.1. Lack of leptin in mice leads to increased OX-A signalling in the ARC, 2-AG overproduction and consequent enhancement of 2-AGP levels
OX-A induces 2-AG biosynthesis via an OX-1R-Gq-PLC-DAGLα-dependent pathway, in both in vitro [19,36,41,76] and in vivo experiments [34]. 2-AGP is an LPA species that is both a precursor and a metabolite of 2-AG [51]. Here we found that 2-AG and 2-AGP are present in the ARC nucleus of the mouse brain, and their levels, measured by LC-MS, are positively correlated with each other in wt mice (Figure 1A; wt: R = 0.76, P < 0.005), whereas the levels of 2-AG and 2-AGP, and of OX-A and 2-AGP, were positively correlated in obese mice (Figure 1B; ob/ob: R = 0.79, P < 0.001 and Figure 1C; R = 0.75, p < 0.001). In support of the hypothesis that the OX-1R-Gq-PLC-DAGLα pathway, under the negative control of leptin [14,17], underlies the positive correlation between OX-A, 2-AG and 2-AGP, we found that: i) as previously reported in a different area of the brain [18,50], OX-A levels in the ARC of 6 h food-deprived wt mice and of ob/ob mice were higher than those of wt ad libitum fed mice, and in both cases, they were reduced after intraperitoneal (i.p.) leptin injection in ob/ob mice in comparison to their respective vehicle-injected control, while there was no effect of leptin injection in ad libitum fed wt mice (Supplementary Fig. 1); ii) 2-AG and 2-AGP levels in the ARC, were increased by i.p. OX-A injection (Figure 1D, group “OX-A”) in a manner prevented by the OX-1R antagonist SB334867 (Figure 1D, group “SB + OX-A”) or by treatment with O7460, an inhibitor of the major 2-AG biosynthesizing enzyme, DAGLα (Figure 1D, group “O7460+OX-A”); iii) the concentrations of 2-AG and 2-AGP were respectively ∼ 4-fold higher in the ARC of ob/ob compared to wt mice (wt: 2-AGP = 4.4 ± 0.5 vs 2-AG = 48.54 ± 3.8 the values are mean of pmol/mg lipids±SEM; and ob/ob: 2-AGP = 20.12 ± 0.9 vs 2-AG = 206.7 ± 2.7 the values are mean of pmol/mg lipids±SEM of the mean) (Figure 1D, group “veh”); iv) 2-AG and 2-AGP elevation in the ARC of ob/ob mice was counteracted by leptin treatment (Figure 1D, group “lept”). Importantly, the levels of 2-AGP were one tenth of those of 2-AG in both wt and ob/ob mice.
Figure 1.

OX-A and 2-AG derived 2-AGP positively correlate and are increased in obese patients: (A) Correlation between the levels of 2-AG and 2-AGP in the ARC of wt mice. n = 16 mice/group; correlation index R = 0.58. (B) Correlation between the levels of 2-AG and 2-AGP in the ARC of ob/ob mice. n = 20 mice/group; correlation index R = 0.61. (C) Correlation between the levels of OX-A and 2-AGP in the ARC of ob/ob mice. n = 20 mice/group; correlation index R = 0.75. Correlations were analyzed using Pearson's correlation coefficient. (D) 2-AG and 2-AGP levels in the ARC of wt or ob/ob mice treated with vehicle, leptin, OX-A, SB334867+OX-A or O7460+OX-A. Data are means ± SEM from n = 6 mice/group, two-way ANOVA with Tukey's multiple comparisons test. Each comparison is represented as follows: ∗ = wt vs ob/ob, # = wt vs wt treated, $ = ob/ob vs ob/ob treated. Overall significance is represented as follow: ∗∗, ##, $$p < 0.01, ∗∗∗, ###, $$$p < 0.001, ∗∗∗∗, ####, $$$$p < 0.0001. (E) Bar graph of OX-A levels in the serum of normal weight (n.w.) and obese subjects. two-tailed Student's t-test, n = 30 per group, 23.27 ± 0.3 pg/ml in n. w and 50.84 ± 1.5 pg/ml in obese. (F) Bar graph of 2-AGP levels in the serum of normal weight (n.w.) and obese human subjects. Two-tailed Student's t-test, n = 30 per group, 259.7 ± 25.41 pmol/mg lipids in n.w. and 593.1 ± 46.07 pmol/mg lipids in obese. (G) Correlation between the levels of OX-A and 2-AGP in the serum of n.w. men (n = 30), R = 0.06, p = 0.17. (H) Correlation between the levels of OX-A and 2-AGP in the serum of obese men (n = 30), R = 0.44.
To further investigate the relationship between 2-AG and 2-AGP, we performed an LC-MS-based determination of 2-AGP-(d8) production starting from 2-AG-(d8), both in in vivo, by i.c.v. 2-AG-(d8) injection in POMC-eGFP mice, and in in vitro by adding 2-AG-(d8) to the cell growth medium of mHypoE N41 cells, in the presence or absence of OMDM169, a potent inhibitor of 2-AG inactivation by monoacylglycerol lipase (MAGL). In both cases, we found a strong and statistically significant increase in 2-AGP-(d8) levels (Supplementary Fig. 2).
3.2. 2-AGP levels are elevated in the serum of obese humans
Despite the fact that changes in endocannabinoid and LPA plasma levels were positively associated with body mass index (BMI) [47,49], 2-AGP levels have not been analyzed before in the serum of obese humans. Here, we found that both OX-A and 2-AGP levels are enhanced in the serum of obese vs normal-weight male human subjects (Figure 1G,H) (n = 30 per group; BMI∼35 in obese and BMI ∼ 23 in normal-weight subjects; age = 65 ± 5 years) and positively correlated with each other (Figure 1E and Fig1F) and with BMI (Supplementary Figure 3).
Figure 3.

Changes of pT231-Tau immunoreactivity in the ARC of wt and ob/ob mice. (A) Confocal images of T231-Tau and POMC immunolabeling in the fibers and soma of neurons in the ARC of wt and ob/ob mice. Data are from n = 6 mice/group (Scale bar: 50 μm). (B) Percentage of pT231-Tau/POMC colocalizing cells in wt and ob/ob 6 h food deprived and wt and ob/ob ad libitum; Data are reported as means ± SEM (n = 6 mice/group). The p values were obtained via two-way ANOVA with Tukey's multiple comparisons test. Each comparison is represented as follows: ∗ = wt vs ob/ob, # = wt vs wt treated, $ = ob/ob vs ob/ob treated. ∗∗∗∗, ####, $$$$p < 0.0001. (C) Representative pT231-Tau immunofluorescence in the fibers and soma of the ARC in ad libitum fed wt and ob/ob mice, 6 h food-deprived wt mice or AM095 injected ob/ob mice or OX-A wt and ob/ob injected mice. Scale bar: 100 μm. (D) Optical density quantification of the pT231-Tau immunoperoxidase labeling in wt ad libitum fed mice, 6 h food-deprived mice, or ob/ob mice. Data represent means ± SEM (n = 60 sections/group, i.e. n = 20 sections/mouse; n = 3 mice/group), Kruskal–Wallis test with Dunn's multiple comparisons test, ∗∗∗∗p < 0.0001. (E) Representative immunoblots of pT231-Tau/Tau ratio show the effect of fasting or ad libitum re-feeding in the ARC of wt mice in comparison with ad libitum no fasted and re-fed wt mice. ANOVA followed by Tukey's test, ∗p < 0.05.
3.3. LPA drives feeding by acting at post-synaptic LPA1-R in mouse POMC neurons
LPA1-R is, among the six types of G-protein-coupled LPA receptors, the most selective for 2-AGP and the most abundant in the brain [16,82]. We detected immunolabeling of LPA1-R in POMC neurons of both wt (Figure 2A1–A2) and ob/ob (Figure 2A3–A4) mice. Diffuse LPA1-R immunoreactivity was also detected by immunoperoxidase labeling in the somata of the neuronal profile in the ARC of wt mice (Supplementary Fig. 4A), whereas no LPA1-R-immunoreactivity was detected after incubation with a mixture of LPA1R primary antibody and the LPA1-R blocking peptide (Supplementary Fig. 4B).
Figure 2.

2-AGP stimulates food intake by modulating POMC neurons. (A) Confocal images of LPA1-R/POMC/OX-A immunolabeling of the ARC nucleus of ad libitum fed) wt (A1–A2) and ob/ob (A3–A4) mice showing LPA1-R immunoexpression at POMC neurons. LPA1-R is expressed diffusely in POMC neurons and in the neuropil of the ARC. (A2, A4). High-power fluorescent micrographs of the resepctive cells inside the boxed area and with orthogonal stacks for wt (A2) and ob/ob (A4) mice; dotted lines and crosshair show 3D coordinates pointing out the LPA1-R/POMC/OXA colocalization. Scale bar: 20 μm (A1, A3) and 10 μm (A1, A3). (B) Effect of 2-AGP on food intake in wt and ob/ob mice. Data are means ± SEM from n = 10 mice/group, two-way ANOVA with Tukey's multiple comparisons test. Each comparison is represented as follows: ∗ = wt vs ob/ob, # = wt vs wt treated, $ = ob/ob vs ob/ob treated mice. (C) Representative images of c-Fos and POMC immunoreactivities in the ARC nucleus of wt and ob/ob mice treated with vehicle, 2-AGP, AM095+2AGP, AM095, and leptin. Scale bar: 100 μm. (D) Bar graph of the quantification of the percentage c-Fos/POMC colocalizing cells. Data are means ± SEM from n = 6 mice/group. The p values were obtained by two-way ANOVA with Tukey's multiple comparisons test. Each comparison is represented as follows: ∗ = wt vs ob/ob, # = wt vs wt treated, $ = ob/ob vs ob/ob treated. Overall significance is represented as follow: ∗, #, $p < 0.05, ∗∗∗∗, ####, $$$$p < 0.0001.
Since the endocannabinoid 2-AG and OX-A are both involved in the regulation of energy homeostasis and their levels are altered in obesity, we next investigated whether 2-AGP, by acting at the LPA1-R, plays a role in food intake. Administration of 2-AGP to wt mice increased food-intake by ∼40% by reaching similar values of food intake to those of untreated obese mice (Figure 2B). This effect was prevented by intra-peritoneal injection of AM095, an LPA1-R antagonist, 1 h before 2-AGP administration (Figure 2B). 2-AGP-injected obese mice did not become more hyperphagic than the vehicle-injected ones, possibly because of the constitutively high levels of 2-AGP in the ARC (Figure 2B). In fact, hyperphagia in ob/ob mice was partially blunted by AM095 when injected before 2-AGP, or completely abolished when the antagonist was administered per se (Figure 2B). These behavioural data were supported by immunohistochemical evidence of induction of pT231-Tau-immunoreactivity in POMC neurons of ad libitum fed wt and ob/ob mice i.p.-injected with 2-AGP and prevented by AM095 treatment before 2-AGP injection (Supplementary Fig. 5).
In line with its hyperphagic effect, 2-AGP injection in wt ad libitum fed mice reduced c-Fos expression in POMC neurons in comparison to the pattern of c-Fos in POMC neurons of vehicle-injected wt mice, whereas 2-AGP did not affect the pattern of c-Fos expression in ob/ob mice in comparison to vehicle-injected ones (Fig. 2C). The inhibitory activity of 2-AGP to POMC neurons of wt ad libitum fed mice was prevented by AM095 injection before 2-AGP treatment (Figure 2C), while this antagonist per se did not affect c-Fos expression in wt ad libitum fed mice (Figure 2C). Of interest, a significant increase of c-Fos immunoreactivity was found in POMC neurons when ob/ob mice were injected with AM095 alone, supporting a partial recovery of activity of POMC neurons following the counteraction of endogenous 2-AGP action in the ARC of these mice. Finally, leptin injection in either wt or ob/ob ad libitum fed mice was used as a positive control of POMC responsiveness to c-Fos expression since the hormone enhanced this latter signal in both cases (Figure 2C). These morphological data were supported by a quantitative evaluation of the percentage of POMC/c-Fos cells in the ARC at different experimental conditions reported before (Figure 2D).
3.4. Leptin deficiency- and OX-A enhancement-induced increase of pT231-Tau levels in POMC neurons occurs via 2-AGP
Since microtubule disassembly and neurite retraction are the most common effects exerted by LPAs via Tau-mediated signalling pathways in differentiated neuroblastoma cells [64,65,69], we hypothesized that 2-AGP, under the negative control of leptin and the positive control of OX-A (Figure 1D), could decrease the activity of POMC-derived projections to OX-A neurons by reducing their length through site-specific phosphorylation of Tau, which underlies microtubule disassembly. In line with this hypothesis, we found changes in pT231-Tau/POMC colocalization in the ARC by comparing wt food-deprived vs ad libitum fed mice or ob/ob vs ob/ob leptin-injected mice (Figure 3A). Quantification of the pT231-Tau/POMC co-localization ratio in food-deprived wt and ob/ob mice injected with leptin 1 h before sacrifice, revealed that this adipokine lowered pT231-Tau in POMC neurons of both groups to levels similar to those measured in vehicle-injected wt ad libitum fed mice (Figure 3B), in agreement with its anorexigenic effect [38]. By contrast, we observed the opposite effect following 2-AGP or OX-A injection in ad libitum fed wt and ob/ob mice, which led to the elevation of pT231-Tau immunoreactivity in POMC neurons (Figure 3B). The OX-A-mediated elevation of the number of pT231-Tau/POMC co-expressing neurons was prevented by OX-1R antagonism by SB334867 or by LPA-1R antagonism by AM095 (Figure 3B). Collectively, these data suggest a role of pT231-Tau in the negative regulation of the anorexigenic function of POMC neurons, during acute (6 h food-deprived mice) or chronic lack of leptin (ob/ob mice) (Figure 3B). Accordingly, by evaluating the changes of pT231-Tau optical density of immunoreactivity in ad libitum fed wt and ob/ob or in wt food-deprived mice, we found that pT231-Tau expression was constitutively elevated in POMC neurons in the ARC of ob/ob mice, similar to those of wt food deprived mice or of ad libitum fed wt mice after OX-A injection. The basal expression of pT231-Tau immunoreactivity in the ARC of ob/ob mice was significantly reduced by injection of AM095 then confirming the phosphorylation of Tau as effect of enhanced 2-AGP levels, associated with the lack of leptin signaling in the ARC of these mice (Fig. 3C). These findings were confirmed by optical density measurement of pT231-Tau immune-expression in ad libitum or food-deprived wt mice, and in ob/ob mice (Figure 3D), and were further supported by western blot analyses of pT231-Tau (Figure 3E). Additionally, pT213-Tau immuno-expression was also elevated in mHypoE N41 cells after incubation with 100 nM and 250 nM 2-AGP or with OX-A, in a manner counteracted, respectively, by AM095 and SB334867 (Supplementary Fig. 6).
By a correlative light and ultrastructural morphological study of hypothalamic sections, we confirmed the existence of the anatomical substrate underlying the putative effect of 2-AGP on the plasticity of POMC neurons since LPA1-R immunogold labelling was found in POMC-eGFP neurons of ad libitum fed mice (black arrows, Figure 4A). To support the regulatory role of leptin in hypothalamic pT231-Tau production, we also detected pT231-Tau immunolabeling (arrowheads, Figure 4B) in α-MSH asymmetrical (putative excitatory) inputs to orexin targets in the LH of both ob/ob mice (arrows, Figure 4B) and food-deprived wt mice (Figure 4C).
Figure 4.

Morphological evidence for LPA1-R immunoreactivity in POMC neurons and pT231-Tau accumulation in α-MSH projections to the LH (A). Representative CLEM images in the ARC and LH of POMC e-GFP mice, OX-eGFP fasted mice, and ob/ob mice showing expression of diverse immunogold signaling: (A) LPA1-R at POMC-eGFP plasma membrane. (B) pT231-Tau DAB-Nichel immunolabeling (arrowheads) inside a-MSH immunogold (10 nm, arrows) labeled fiber projecting to a putative excitatory synaptic target in the LH of obese mice. (C) pT231-Tau DAB-Nichel immunolabeling (arrowheads) and sparse or vesicle-dense a-MSH immunogold labeling (10 nm, arrows) in fiber projecting to OX-eGFP synaptic target in the LH of wt fasted mice (Scale bar: 0.5 μm).
Hyperphosphorylation of several serine and threonine amino acid residues of Tau additional to T231 has been described, including Ser199, Ser202/Thr205 or AT8, Ser396/404 or PHF-1 and Ser422, which are responsible for aggregation processes causing pathological Tau mislocalization in neurodegenerative disorders such as AD [66]. Therefore, we analyzed the phosphorylation of these sites through independent automated quantification based on digital image analysis that provides the respective phosphorylation profiles. We found that the elevated pT231-Tau phosphorylation in the ARC of ob/ob mice (Supplementary Fig. 7) was not accompanied by Tau phosphorylation on other potential amino acid acceptors of phosphate groups.
3.5. 2-AGP and leptin affect pT231-Tau phosphorylation in the ARC by regulating GSK-3β activity in opposite ways
As mentioned above, acute leptin treatment has been shown to decrease OX-A release into the ARC and subsequent 2-AG biosynthesis in POMC neurons [18,50], but this treatment is also known to reduce Tau phosphorylation in neuronal cells in vitro [31]. However, understanding whether or not 2-AGP is involved in hypothalamic pT231-Tau accumulation, and through what molecular mechanism, required further experiments, as described below.
GSK3β, a serine/threonine glycogen synthase kinase, has been implicated in pT231-Tau accumulation and microtubule disassembly [44,45,60,66]. Ser9 phosphorylation of GSK3β inhibits its catalytic activity, whereas Tyr216 phosphorylation has the opposite effect [25,79]. Here, by using western blot analyses, we found that enhanced 2-AGP levels in the ARC of ob/ob mice, or exogenous administration of this lipid in wt mice, are accompanied by phosphorylation/activation of the protein tyrosine kinase 2 (Pyk2), at Tyr402. Pyk2 activation, in turn, catalyzed phosphorylation/activation of GSK3β at Tyr216, thereby activating this latter kinase for T231-Tau phosphorylation, in line with in vitro studies [64]. These effects were prevented by AM095 (Figure 5A), thus implicating the LPA1-R. Likewise, enhancement of OX-A signaling in the ARC, as in ob/ob mice or ad libitum fed wt mice treated with OX-A, was accompanied by Pyk2 phosphorylation at Tyr202, subsequent Tyr216 phosphorylation/activation of GSK3β, and finally, pT231-Tau accumulation. These effects were prevented by leptin, SB-334867, and the DAGL-α inhibitor O-7460 (Figure 5B, Supplementary Fig. 8), suggesting the involvement of leptin deficiency, OX-1R and OX-A-induced 2-AG biosynthesis in this effect. Conversely, and in agreement with in vitro studies [31,77], we found that leptin caused GSK3β inactivation through PI3K/Akt-mediated phosphorylation at Ser9 (Figure 5B), an effect which was blunted by OX-A injection, but not by SB-334867 in combination with the neuropeptide (Figure 5B). Collectively, these data provide evidence that the pT231Tau/Tau ratio is transiently controlled by leptin and 2-AG-derived 2-AGP in opposite manners, through opposing effects on GSK3β activity, as summarized in the scheme of Figure 5C.
Figure 5.

Leptin and OX-A regulate Tau phosphorylation thereby modulating GSK3β, activity. (A) Representative immunoblots of pY402-Pyk2/Pyk2, pS47- Akt/Akt, pY216-GSK3β/GSK3β, and pT231-Tau/Tau ratios showing the effect of in vivo 2-AGP and AM095 treatment in the ARC of wt and ob/ob mice. Lanes from blots spliced together in a composite image are separated by a dotted black vertical line. Please note that these OD values are expressed as the ratio between phosphorylated and total protein signals. Data are means ± SEM (n = 3 mice/group). The p values were obtained via two-way ANOVA with Tukey's multiple comparisons test. (B) Representative immunoblots of pY402Pyk2/Pyk2, pS473Akt/Akt, pS9GSK3β/GSK3β, pY216GSK3β/GSK3β and pT231-Tau/Tau ratios showing the effect of in vivo leptin, OX-A and SB treatment in the ARC of wt and ob/ob mice. OD values are expressed as the ratio between phosphorylated and total protein signals. Data represent means ± SEM (n = 3/group). The p values were obtained via two-way ANOVA with Tukey's multiple comparisons test. Each comparison is represented as follows: ∗ = wt vs ob/ob, # = wt vs wt treated, $ = ob/ob vs ob/ob treated. Overall significance is represented as follow: ∗, #, $p < 0.05, ∗∗, ##, $$p < 0.01, ∗∗∗, ###, $$$p < 0.001, ∗∗∗∗, ####, $$$$ p < 0.0001. (C) Representative scheme showing the opposite effect of leptin and 2-AG-derived 2-AGP on GSK3β activity and pT231-Tau production.
3.6. 2-AGP reduces the excitatory drive onto OX-A neurons in the lateral hypothalamus
Endogenous 2-AGP signalling is known to mediate short-term tuning of excitatory synaptic transmission by decreasing the size of the ready-to-use synaptic vesicle pool at excitatory terminals [29,59], a plastic event essential for information processing at glutamatergic networks in the brain. Therefore, and following the present morphological observation of presynaptic α-MSH-expressing asymmetrical, i.e. putative excitatory, afferences to orexin neurons (Figure 4B), we next measured if 2-AGP could affect the overall excitatory drive to these neurons by influencing their functional activity. We found that 2-AGP, by acting at the presynaptic level, inhibits the excitatory inputs to OX-eGFP neurons, as shown by the reduction of the frequency of mEPSCs recorded in presence of 0.3 μM TTX (Figure 6A–C). The LPA1-R antagonist AM095 prevented this effect (Figure 6D–F). Subsequently, we performed whole-cell voltage-clamp recordings of OX-eGFP neurons in wt mice by placing an extracellular electrode in the proximity of the cell body to produce an evoked excitatory post synaptic current (eEPSC). We found a significant reduction of the eEPSC amplitude after 15 min of 2-AGP (250 nM) treatment (Figure 6G–H) and an increment of the paired pulse ratio (PPR) at a short inter-pulse interval (ISI: 10 ms) (Figure 6I), in agreement with previous work [29]. The incubation with AM095 before 2-AGP treatment in the ACSF prevented the reduction of the evoked excitatory transmission onto OX neurons (Figure 6J–L). These results confirm the role of LPA1-R in the dampening of the excitatory synapses to orexin neurons.
Figure 6.

2-AGP impairs excitatory transmission to orexin neurons. (A) Representative traces of mEPSCs recorded in OX-eGFP neurons before (black) and after 2-AGP (250 nM) (blue). (B) Effect of 2-AGP on the mEPSCs amplitude in n = 6 neurons from 3 mice; p = 0.1, Two-tailed paired Wilcoxon signed-rank test. (C) Effect of 2-AGP on the frequency of mEPSCs in n = 6 neurons from 3 mice; ∗p < 0.05, Two-tailed paired Wilcoxon signed-rank test. (D) Representative traces of mEPSCs recorded in OX-eGFP neurons preincubated with AM095 (10 μM, green trace) and after 2-AGP treatment (250 nM, blue trace). (E) Plot of the mEPSCs amplitude in AM095 and AM095+2-AGP; n = 6 neurons from 3 mice; p = 0.75, Two-tailed paired Wilcoxon signed-rank test. (F) Plot of the mEPSC frequency in AM095 and AM095+2-AGP; n = 6 neurons from 3 mice: p = 0.62, Two-tailed paired Wilcoxon signed-rank test. (G) Representative traces of eEPSCs evoked in OX-eGFP neurons using extracellular stimulation before (black circles) and after 2-AGP (250 nM) (blue squares), P1 and P2 represent the first and the second eEPSCs evoked in an interspike interval of 10 ms. (H) Amplitude of the first eIPSC (P1) before and after 2-AGP (250 nM); n = 6 neurons from 3 mice; ∗p < 0.05, Two-tailed paired Wilcoxon signed-rank test. (I) Ratio between two consecutive eIPSCs evoked between 10 ms and 1sec in control (black circle) and after 2-AGP (blue square) (n = 6 neurons from 3 mice; ∗p < 0.05, Two1005 tailed paired Wilcoxon signed-rank test). The ratio is plotted as a function of the inter-spike interval on a logarithmic scale. (J) Representative traces of paired-pulse at 10 ms in AM095 (10 mM) (green trace) and AM095 + 2-AGP (250 nM) (blue trace) in OX-eGFP neurons using extracellular stimulation. P1 and P2 represent the first and the second eEPSCs evoked in an interspike interval of 10 ms. (K) Amplitude of the first eIPSC (P1) in AM095 and after 2-AGP (250 nM); n = 6 neurons from 3 mice. (L) Ratio between two consecutive eIPSCs evoked between 10 ms and 1sec in AM095 (green circles) and after the treatment with 2-AGP (blue squares). The ratio is plotted as a function of the inter-spike interval on a logarithmic scale. n = 6 neuron from 3 mice; p = 0.8, Wilcoxon signed-rank test. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.7. 2-AGP- and lack of leptin-induced T231-Tau phosphorylation leads to retraction of POMC-derived projections to LH
Finally, we analyzed whether the stimulatory effect of 2-AGP on pT231-Tau and the likely subsequent microtubule disassembly, could result in changes in the length of POMC-eGFP-derived projections and of postsynaptic density of α-MSH anchoring sites at OX-eGFP target neurons. Treatment with 2-AGP alone diminished the mean length of in vitro POMC-eGFP projections by 2.1–2.8 μm in a time-dependent fashion (Figure 7A–B) and in a manner prevented by apreincubation with AM095 (Supplementary Fig. 9). In agreement with OX-A-induced pT231-Tau accumulation (Figures 3A–B, 4B–C and 5), and the consequent microtubule disassembly, OX-A induced a 2.1–1.6 μm reduction of the mean length of POMC-eGFP neuronal processes (Figure 7B), in a manner prevented by cell pre-incubation with either OX-1R antagonism (SB), DAGLα inhibition (O-7460), or the PYK2 inhibition (PF431396) (Supplementary Fig. 9). Conversely, leptin treatment promoted a 1.2–1.5 μm elongation of neuronal processes (Figure 7B), an effect blocked by a leptin receptor antagonist (Supplementary Fig. 9). Additional evidence from in vitro experiments were provided to support the effect of 2-AGP on the retraction of αMSH inputs to OX-eGFP neurons since LPA1-R immunoreactivity was detected in the same neuronal processes where pT231-Tau immunolabeling was induced by 1 h of 2-AGP administration in the bath (Figure 7, C- D). Moreover, anatomical evidence supported the 2-AGP-mediated effect of pT231-Tau on the disassembly of POMC-derived projections to the OX-eGFP target since we found a reduction of immunolabelling of PSD-95, a key excitatory scaffolding protein required for synaptic stabilization [9], in OX-eGFP neurons receiving α-MSH inputs, this effect being antagonized by AM095, as observed by western blot in the LH of 2-AGP-injected ad-lib fed wt mice and in ob/ob mice (Figure 7E). This effect is likely responsible for the reduction of OX-A-positive anchoring sites, as suggested by the decrease of α-MSH/PSD-95/OX-eGFP co-expression in the LH of 2-AGP-injected wt mice (Figure 7F).
Figure 7.

2-AGP promotes a time-dependent neurite retraction of POMC neurons. (A) Time-lapse fluorescence microscopy of in vitro cultured POMC-eGFP primary neurons showing the effect of 2-AGP, added in the cell medium, on the retraction of neurites (arrows). Scale bar: 20 μm. (B) Representative images showing a 30 min and 60 min time course effect of leptin, OX-A, and 2-AGP on the morphometric changes of the neuronal processes of POMC cultured neurons (range ± 2 mm) respect to the start time (t = 0) represented by the vertical dotted line. (C, D) High magnification of the cell indicated by the white dotted box area at 60 min of 2-AGP treatment showing pT231-Tau/LPA1-R co immunolabeling at the neurite indicated in the red dotted boxed (left image), (Scale bar: 10 μm). (E) Representative immunoblots of PSD95 and β-actin showing the effect of 2-AGP and AM095 on the postsynaptic density after 2-AGP treatment in wt or ob/ob mice. OD values are expressed as the ratio between total protein signal and β-actin. Data are mean ± SEM from n = 3 mice/group; p values are obtained via two-way ANOVA with Tukey's multiple comparisons test. (F) Representative confocal images showing α-MSH/PSD-95 colocalizing immunoreactivities at Hcrt-eGFP or OX-eGFP neurons of mice treated with 2-AGP. Data are from n = 6 mice/group. Scale bar = 30 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
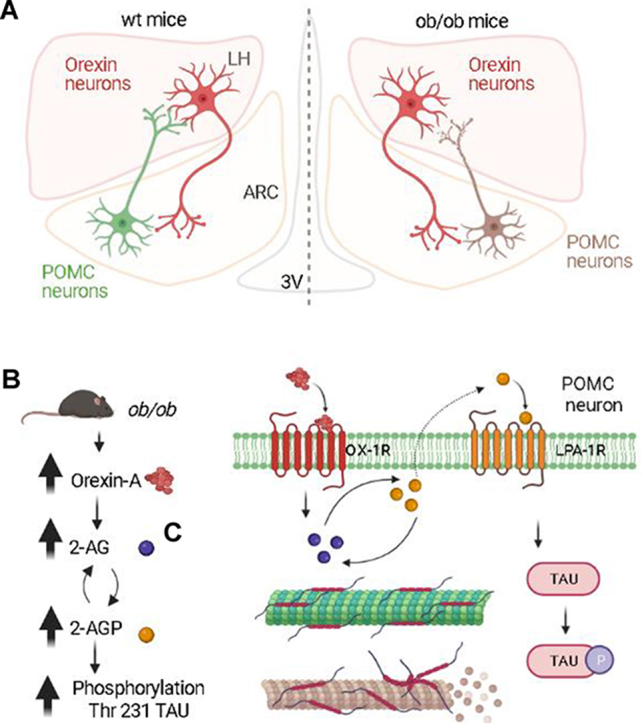
In the hypothalamus, POMC neurons represent an heterogeneous cell population including glutamatergic and GABAergic cells [13,22,42] probably playing a role in all aspects of feeding behaviour [10,15]. POMC glutamatergic α-MSH projections under the positive control of mTORC1 promote satiety [63], whereas the endocannabinoid 2-AG, under the negative control of leptin, instead induces mostly orexigenic effects by activating cannabinoid CB1 receptors [58,67]. Yet, in lean mice, activation of CB1, depending on its localization, can also produce effects that may lead to inhibition of food intake [7,63]. For example, the retrograde action of post-synaptically released endocannabinoids at CB1 receptors located on excitatory/glutamatergic, and mostly α-MSH-releasing, terminals innervating orexinergic neurons in the LH leads to their inhibition and potential hypophagic effects [18].
Obesity, and the central leptin signaling deficiency status that it normally carries, however, may alter this “yin and yang” action of CB1 signaling in food intake, and shift the balance towards hyperphagia. Accordingly, in both ob/ob and HFD obese mice, the alteration of the mTOR pathway and the elevation of 2-AG biosynthesis occurs in the ARC nucleus, concomitantly with a switch from CB1-controlled excitatory inputs (most of which are α-MSH expressing fibers) to inhibitory inputs (the ∼70% of which are NPY-expressing fibers) to OX-A neurons [18]. The latter neurons, while being normally inhibited by retrograde endocannabinoid signaling in lean mice, consequently become disinhibited in obesity due to an increment of depolarizing suppression of inhibition, thus contributing to hyperphagia and its consequences.
Here we present a mechanism that explains the reduction of glutamatergic α-MSH inputs to OX-A neurons in the LH during conditions of leptin signaling reduction. We show that, in POMC neurons, 2-AG produced by activation of the OX-1R-Gq-DAGLα-cascade is partly converted into 2-AGP, which, through activation of LPA1-R, triggers pT231-Tau phosphorylation, microtubule dysfunction and reversible or chronic loss of α-MSH-expressing excitatory inputs to OX-A neurons in the LH. This new form of 2-AG metabolism-dependent synaptic plasticity likely underlies the OX-A- and 2-AGP-mediated food intake observed following food deprivation in lean mice or in obesity, respectively, i.e. when leptin signaling in the hypothalamus is temporarily reduced or chronically defective. In line with the enhancement of 2-AGP levels in the serum of obese subjects, also different lysophosphatidic acids, such as 16:0 LPA [26], as well as the mRNA levels of distinct LPA receptors (LPA4, LPA5, and/or LPA6), have been found increased in the myocardial tissue and in peripheral circulating cells from overweight or obese humans [11].
More specifically, by combining biochemical, pharmacological, and cell-type-specific morphological and electrophysiological techniques, we dissected the 2-AGP-driven molecular pathways that underlie the loss of POMC-derived α-MSH inputs observed under conditions of reduced or impaired leptin signalling. First, we found that 2-AGP acts as an orexigenic autocrine messenger by inducing the retraction of α-MSH inputs to OX-A neurons via elevation of Tau phosphorylation. Next, we demonstrated that these effects: 1) result from activation of post-synaptic LPA1-R and tyrosine kinase Pyk2-mediated GSK3β phosphorylation of Tyr216, and subsequent pT231Tau elevation, and 2) are prevented by either antagonism of OX-1R, inhibition of 2-AG biosynthesis or leptin-controlled phosphorylation of Ser9-GSK3β via the PI3K/Akt pathway, the latter being a mechanism common to several microtubule associated proteins involved in microtubule aggregation [20,31,33,43,70]. These results, together with those obtained using correlative light and electron microscopy of OX-eGFP neurons, coupled to targeted patch-clamp electrophysiology, reveal for the first time the existence of 2-AG and 2-AGP-mediated remodelling of α-MSH projections to OX-A perikarya, which controls negatively the synaptic efficiency of melanocortin anorexigenic inputs.
This study was focused on the 2-AGP-mediated regulation of synaptic projections originating from POMC excitatory α-MSH neurons. In this regard, the production of 2-AGP from 2-AG in POMC neurons was demonstrated through in vitro and in vivo experiments based on the LC-MS quantification of 2-AGP-(d8) production in POMC neurons isolated from the brain of mice injected with deuterated (d8) 2-AG as precursor of deuterated (d8) 2-AGP. OX and POMC neurons represent both anatomical and neurochemical different specialized subsets of neurons strictly localized, respectively, in the hypothalamic lateral (LH) and the arcuate (ARC) nuclei (Acuna-Goycolea and van den Pol, 2009; Schwartz et al., 2000) and OX-A neurons send prominent projections to POMC somas (Horvath et al., 1999; Guan et al., 2001) where the released OX-A can activate OX-1R's therein located (Bäckberg et al., 2002; Muroya et al., 2003). Thus, neuroendocrine POMC neurons appear to be good candidates to mediate orexin actions including the here demonstrated effect on the pT231-Tau-controlled regulation of POMC-derived excitatory axonal projections, back to OX-A neurons. Indeed, the new form of endocannabinoid-mediated, but CB1 receptor-independent, regulation of synaptic plasticity described here may lead specifically to the loss of CB1-inhibited excitatory synapses within α-MSH inputs to OX-A neurons, with consequent impairment of synaptic anchoring and glutamate trafficking, thus strongly contributing to the rewiring of OX-A neurons [35,71], and the observed increase of OX-A release in obesity [18,27].
The Pyk2-GSK3β cascade has already been shown to be downstream of LPA and LPA1-R signaling [24,61], and to be implicated in the neurite retraction activity of this receptor [64]. However, what we describe here is a new perspective on LPA/LPA1-R signaling arising as the consequence of the metabolism (to a non-conventional type of LPA) of the endocannabinoid 2-AG, which is otherwise deeply involved in both food intake and orexinergic signaling via CB1 receptors. Indeed, endocannabinoid/CB1 signaling via 2-AG is usually terminated by the enzyme MAGL, which is expressed pre-synaptically to OX-A neurons in the LH [18] and converts the endocannabinoid into products that are per se inactive at CB1 [75]. It is, therefore, interesting to find that, during both acute (and physiological) and chronic (hence pathological) leptin signaling impairment, part of the 2-AG overproduced in ARC POMC neurons, rather than being directly inactivated by MAGL, is converted instead into a metabolite, i.e. 2-AGP. This latter mediator thus carries a pro-orexigenic signal via a different, non-CB1 receptor-mediated mechanism, by reducing the number of excitatory α-MSH-expressing inputs to OX-A neurons. The overexpression of DAGLα in the LH and ARC of obese mice [18], together with elevated OX-A signaling at OX-1R expressed in POMC neurons, which leads to 2-AG biosynthesis by DAGLα [50], can clearly sustain the production in these neurons of 2-AG levels sufficient to both activate CB1 receptors and produce 2-AGP to activate LPA1-R, thereby contributing to food intake via two different mechanisms, both of which can be enhanced by inhibition of 2-AG degradation by MAGL. Interestingly, in POMC neurons, others have reported that a population of CB1 receptors, expressed on the external membrane of mitochondria, promote, at least when activated by Δ9-tetrahydrocannabinol, POMC expression with the exclusive role of enhancing the release of food intake-promoting β-endorphin (but not α-MSH) in the hypothalamus of lean ad libitum fed mice [39]. The data described here may suggest that this latter mechanism, as well as other CB1-mediated effects in POMC neurons, could be partly blunted in ob/ob mice or food-deprived lean mice, by 2-AGP-induced α-MSH neurite retraction, regardless of the subcellular localization of CB1.
It is also interesting to note how, following the results of the present study, it is now possible to suggest that CB1 receptors and/or 2-AG intervene in all phases, i.e. release, action and (dys)regulation thereof, of OX-A/OX1-R signalling in obesity, i.e.: 1) as shown here, 2-AG, but not CB1, overproduced in POMC neurons, is partly responsible for the leptin signalling impairment-induced reduction of excitatory innervation of OX-A neurons (the increase of inhibitory NPY-afferents being instead due to the lack of leptin-inhibition of mTOR signaling [18]; 2) 2-AG overproduced from the somata of OX-A neurons enacts retrograde disinhibition of such neurons, by activating CB1 on their NPY-inhibitory afferents, thereby leading to enhanced OX-A release in LH output areas, including the ARC [18,50]; 3) 2-AG produced from OX1-R-Gq activation by OX-A, and its activation of CB1, underlie also all the actions described so far for this neuropeptide in LH neuron output areas [19,27,52]; and 4) CB1, by making heteromers with OX1-R, both in vitro and in vivo, sensitizes them to activation by OX-A [36,37,80].
In conclusion, we have described here a new form of regulation of hypothalamic synaptic plasticity that: 1) is 2-AG metabolism-dependent and CB1 receptor-independent, and 2) may account for part of the transient or chronic hyperphagia observed, respectively, following food deprivation or during obesity. Indeed, since we have shown here that it originates from reduced or impaired leptin signaling, this mechanism is very likely to occur also in diet-induced obesity, which is instead characterized by leptin insensitivity in the hypothalamus and, in mouse models, by the same subsequent overactivity of the OX-1R-Gq-PLC-DAGLα-2-AG cascade [18,27,50]. The present findings strengthen the general idea that 2-AG is a fundamental as well as multi-faceted downstream mediator of orexinergic signaling via several, not necessarily CB1 receptor-mediated, mechanisms, as shown also by a recent study in which OX-A-induced 2-AG biosynthesis in the amygdala mediates fear extinction deficits via activation of the cannabinoid receptor type-2 (CB2) in astrocytes [73]. Therefore, future studies are now required to investigate the possibility that the 2-AG/2-AGP/LPA1-R pathway, via effects on Tau phosphorylation and microtubule assembly, mediates also other forms of synaptic plasticity in other brain areas, possibly under pathological and irreversible conditions of 2-AG overproduction, such as in the hippocampus of mice with diet-induced obesity [27,46], thereby underlying some forms of obesity-related cognitive and affective dysfunctions. Our study, by establishing a causal relationship between morphological changes in αMSH excitatory inputs to OX-A cells from obese animals and network-level changes in the activity of the orexin system, may open new avenues of research on the mechanisms underlying not only hyperphagia and eating disorders, but also arousal and sleep/wake cycle dysregulation, and the consequent daytime sleepiness, reward seeking, altered stress response, and impaired cognitive tasks, which are emerging as characterizing dysfunctions accompanying obesity [12,27,28,71,74].
Author contributions
A.C. designed and performed western blot and biochemical analysis; N.F. designed and performed electrophysiological experiments and statistical analysis; L.T., R.I. performed immunohistochemical, immunocytochemical and TEM experiments; A.D.C. categorized and followed the enrolled subjects; L.P. performed biochemical and cellular analysis; F.P. performed biochemical analysis by LC-MS mass spectrometry; V.D. and L.C. conceptualized the study and wrote the final version of the manuscript; L.C. conceived and supervised the study. All authors analyzed and discussed the data, and edited and approved the final version of the manuscript.
Funding
EU financial support, grant PON IDF SHARID ARS01_01270 CUP B66G18000640005, to L.C., V.D. and N.F. Financial support by the Joint International Research Unit (JIRU) for Chemical and Biomolecular Research on the Microbiome and its impact on Metabolic Health and Nutrition (MicroMeNu) and by the Sentinelle Nord Program of Université Laval (to N.F, L.C., and V.D.), which in turn is funded by the Canada First/Apogée program of the Tri-Agency of the Canadian Federal Government. Financial support by the Italian MUR, grant PRIN #2017M42834_002 to L.C.
Acknowledgments
The authors are grateful to Dr. Giovanna Morello who performed pilot experiments at the initial phase of the study by producing biochemical analysis; Dr. Roberta Verde (ICB-CNR Pozzuoli, Italy) for the technical support to LC-MS mass spectrometry; Dr. Brenda Marfella and Dr. Alessandro Nicois (ICB-CNR Pozzuoli, Italy) the technical support in the cell culture; Dr. Elena Polishchuk (Telethon sition.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2023.101713.
Contributor Information
Vincenzo Di Marzo, Email: vincenzo.di-marzo.1@ulaval.ca.
Luigia Cristino, Email: luigia.cristino@icb.cnr.it.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Ammoun S., Lindholm D., Wootz H., Akerman K.E.O., Kukkonen J.P. G-protein-coupled OX1 orexin/hcrtr-1 hypocretin receptors induce caspase-dependent and -independent cell death through p38 mitogen-/stress-activated protein kinase. J Biol Chem. 2006;281:834–842. doi: 10.1074/jbc.M508603200. [DOI] [PubMed] [Google Scholar]
- 2.Andrews Z.B., Liu Z.-W., Walllingford N., Erion D.M., Borok E., Friedman J.M., et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arendt T. Synaptic plasticity and cell cycle activation in neurons are alternative effector pathways: the “Dr. Jekyll and Mr. Hyde concept” of Alzheimer's disease or the yin and yang of neuroplasticity. Prog Neurobiol. 2003;71:83–248. doi: 10.1016/j.pneurobio.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Arendt T. Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:167–179. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- 5.Arendt T., Holzer M., Gärtner U. Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer's disease. J Neural Transm. 1998;105:949–960. doi: 10.1007/s007020050104. [DOI] [PubMed] [Google Scholar]
- 6.Avila J., Pérez M., Lim F., Gómez-Ramos A., Hernández F., Lucas J.J. Tau in neurodegenerative diseases: tau phosphorylation and assembly. Neurotox Res. 2004;6:477–482. doi: 10.1007/BF03033284. [DOI] [PubMed] [Google Scholar]
- 7.Bellocchio L., Lafenetre P., Cannich A., Cota D., Puente N., Grandes P., et al. Bimodal control of stimulated food intake by the endocannabinoid system. Nat Neurosci. 2010;13:281–283. doi: 10.1038/nn.2494. [DOI] [PubMed] [Google Scholar]
- 8.Benani A., Hryhorczuk C., Gouaze A., Fioramonti X., Brenachot X., Guissard C., et al. Food intake adaptation to dietary fat involves PSA-dependent rewiring of the arcuate melanocortin system in mice. J Neurosci: Off J Soc Neurosci. 2012;32:11970–11979. doi: 10.1523/JNEUROSCI.0624-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berry K.P., Nedivi E. Spine dynamics: are they all the same? Neuron. 2017;96:43–55. doi: 10.1016/j.neuron.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandt C., Nolte H., Henschke S., Engström Ruud L., Awazawa M., Morgan D.A., et al. Food perception primes hepatic ER homeostasis via melanocortin-dependent control of mTOR activation. Cell. 2018;175:1321–1335.e20. doi: 10.1016/j.cell.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown A., Hossain I., Perez L.J., Nzirorera C., Tozer K., D'Souza K., et al. Lysophosphatidic acid receptor mRNA levels in heart and white adipose tissue are associated with obesity in mice and humans. PLoS One. 2017;12 doi: 10.1371/journal.pone.0189402. e0189402–e0189402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burdakov D., Karnani M.M. Ultra-sparse connectivity within the lateral hypothalamus. Curr Biol: CB. 2020;30:4063–4070.e2. doi: 10.1016/j.cub.2020.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell J.N., Macosko E.Z., Fenselau H., Pers T.H., Lyubetskaya A., Tenen D., et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat Neurosci. 2017;20:484–496. doi: 10.1038/nn.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casabiell X., Piñeiro V., Tomé M.A., Peinó R., Dieguez C., Casanueva F.F. Presence of leptin in colostrum and/or breast milk from lactating mothers: a potential role in the regulation of neonatal food intake. J Clin Endocrinol Metabol. 1997;82:4270–4273. doi: 10.1210/jcem.82.12.4590. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y., Lin Y.-C., Kuo T.-W., Knight Z.A. Sensory detection of food rapidly modulates arcuate feeding circuits. Cell. 2015;160:829–841. doi: 10.1016/j.cell.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi J.W., Chun J. Lysophospholipids and their receptors in the central nervous system. Biochim Biophys Acta. 2013;1831:20–32. doi: 10.1016/j.bbalip.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coppari R., Bjørbæk C. Leptin revisited: its mechanism of action and potential for treating diabetes. Nat Rev Drug Discovery. 2012;11:692–708. doi: 10.1038/nrd3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cristino L., Busetto G., Imperatore R., Ferrandino I., Palomba L., Silvestri C., et al. Obesity-driven synaptic remodeling affects endocannabinoid control of orexinergic neurons. Proc Natl Acad Sci USA. 2013;110:E2229–E2238. doi: 10.1073/pnas.1219485110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cristino L., Luongo L., Imperatore R., Boccella S., Becker T., Morello G., et al. Orexin-A and endocannabinoid activation of the descending antinociceptive pathway underlies altered pain perception in leptin signaling deficiency. Neuropsychopharmacology: Off Publ Am Coll Neuropsychopharmacol. 2016;41:508–520. doi: 10.1038/npp.2015.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 21.de Lecea L., Kilduff T.S., Peyron C., Gao X., Foye P.E., Danielson P.E., et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dicken M.S., Tooker R.E., Hentges S.T. Regulation of GABA and glutamate release from proopiomelanocortin neuron terminals in intact hypothalamic networks. J Neurosci. 2012;32:4042–4048. doi: 10.1523/JNEUROSCI.6032-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietrich M.O., Horvath T.L. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends Neurosci. 2013;36:65–73. doi: 10.1016/j.tins.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Dikic I., Tokiwa G., Lev S., Courtneidge S.A., Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 25.Doble B.W., Woodgett J.R. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fayyaz S., Japtok L., Schumacher F., Wigger D., Schulz T.J., Haubold K., et al. Lysophosphatidic acid inhibits insulin signaling in primary rat hepatocytes via the LPA3 receptor subtype and is increased in obesity. Cell Physiol Biochem: Int J Exp Cell Physiol, Biochem, Pharmacol. 2017;43:445–456. doi: 10.1159/000480470. [DOI] [PubMed] [Google Scholar]
- 27.Forte N., Boccella S., Tunisi L., Fernández-Rilo A.C., Imperatore R., Iannotti F.A., et al. Orexin-A and endocannabinoids are involved in obesity-associated alteration of hippocampal neurogenesis, plasticity, and episodic memory in mice. Nat Commun. 2021;12 doi: 10.1038/s41467-021-26388-4. 6137–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garau C., Blomeley C., Burdakov D. Orexin neurons and inhibitory Agrp→orexin circuits guide spatial exploration in mice. J Physiol. 2020;598:4371–4383. doi: 10.1113/JP280158. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Morales V., Montero F., Gonzalez-Forero D., Rodriguez-Bey G., Gomez-Perez L., Medialdea-Wandossell M.J., et al. Membrane-derived phospholipids control synaptic neurotransmission and plasticity. PLoS Biol. 2015;13 doi: 10.1371/journal.pbio.1002153. e1002153–e1002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goedert M., Spillantini M.G., Crowther R.A. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci USA. 1992;89:1983–1987. doi: 10.1073/pnas.89.5.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greco S.J., Sarkar S., Casadesus G., Zhu X., Smith M.A., Ashford J.W., et al. Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neurosci Lett. 2009;455:191–194. doi: 10.1016/j.neulet.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hara J., Beuckmann C.T., Nambu T., Willie J.T., Chemelli R.M., Sinton C.M., et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 33.Hartigan J.A., Johnson G.V. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase 3beta-dependent pathway. J Biol Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- 34.Ho Y.-C., Lee H.-J., Tung L.-W., Liao Y.-Y., Fu S.-Y., Teng S.-F., et al. Activation of orexin 1 receptors in the periaqueductal gray of male rats leads to antinociception via retrograde endocannabinoid (2-arachidonoylglycerol)-induced disinhibition. J Neurosci: Off J Soc Neurosci. 2011;31:14600–14610. doi: 10.1523/JNEUROSCI.2671-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horvath T.L., Gao X.-B. Input organization and plasticity of hypocretin neurons: possible clues to obesity's association with insomnia. Cell Metabol. 2005;1:279–286. doi: 10.1016/j.cmet.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Imperatore R., Palomba L., Morello G., Spiezio A.D., Piscitelli F., Marzo V.D., et al. Formation of OX-1R/CB1R heteromeric complexes in embryonic mouse hypothalamic cells: effect on intracellular calcium, 2-arachidonoyl-glycerol biosynthesis and ERK phosphorylation. Pharmacol Res. 2016;111:600–609. doi: 10.1016/j.phrs.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 37.Jantti M.H., Putula J., Turunen P.M., Nasman J., Reijonen S., Lindqvist C., et al. Autocrine endocannabinoid signaling through CB1 receptors potentiates OX1 orexin receptor signaling. Mol Pharmacol. 2013;83:621–632. doi: 10.1124/mol.112.080523. [DOI] [PubMed] [Google Scholar]
- 38.Klok M.D., Jakobsdottir S., Drent M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: a review. Obes Rev. 2007;8:21–34. doi: 10.1111/j.1467-789X.2006.00270.x. [DOI] [PubMed] [Google Scholar]
- 39.Koch M., Varela L., Kim J.G., Kim J.D., Hernandez-Nuno F., Simonds S.E., et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519:45–50. doi: 10.1038/nature14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kowall N.W., Kosik K.S. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer's disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 41.Kukkonen J.P. G-protein-dependency of orexin/hypocretin receptor signalling in recombinant Chinese hamster ovary cells. Biochem Biophys Res Commun. 2016;476:379–385. doi: 10.1016/j.bbrc.2016.05.130. [DOI] [PubMed] [Google Scholar]
- 42.Lam B.Y.H., Cimino I., Polex-Wolf J., Nicole Kohnke S., Rimmington D., Iyemere V., et al. Heterogeneity of hypothalamic pro-opiomelanocortin-expressing neurons revealed by single-cell RNA sequencing. Mol Metabol. 2017;6:383–392. doi: 10.1016/j.molmet.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lesort M., Greendorfer A., Stockmeier C., Johnson G.V., Jope R.S. Glycogen synthase kinase-3beta, beta-catenin, and tau in postmortem bipolar brain. J Neural Transm. 1999;106:1217–1222. doi: 10.1007/s007020050235. [DOI] [PubMed] [Google Scholar]
- 44.Li T., Paudel H.K. Glycogen synthase kinase 3beta phosphorylates Alzheimer's disease-specific Ser396 of microtubule-associated protein tau by a sequential mechanism. Biochemistry. 2006;45:3125–3133. doi: 10.1021/bi051634r. [DOI] [PubMed] [Google Scholar]
- 45.Lin Y.-T., Cheng J.-T., Liang L.-C., Ko C.-Y., Lo Y.-K., Lu P.-J. The binding and phosphorylation of Thr231 is critical for Tau's hyperphosphorylation and functional regulation by glycogen synthase kinase 3beta. J Neurochem. 2007;103:802–813. doi: 10.1111/j.1471-4159.2007.04792.x. [DOI] [PubMed] [Google Scholar]
- 46.Massa F., Mancini G., Schmidt H., Steindel F., Mackie K., Angioni C., et al. Alterations in the hippocampal endocannabinoid system in diet-induced obese mice. J Neurosci. 2010;30:6273–6281. doi: 10.1523/JNEUROSCI.2648-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michalczyk A., Budkowska M., Dołęgowska B., Chlubek D., Safranow K. Lysophosphatidic acid plasma concentrations in healthy subjects: circadian rhythm and associations with demographic, anthropometric and biochemical parameters. Lipids Health Dis. 2017;16 doi: 10.1186/s12944-017-0536-0. 140–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mignot E. A commentary on the neurobiology of the hypocretin/orexin system. Neuropsychopharmacology: Off Publ Am Coll Neuropsychopharmacol. 2001;25:S5–S13. doi: 10.1016/S0893-133X(01)00316-5. [DOI] [PubMed] [Google Scholar]
- 49.Monteleone A.M., Di Marzo V., Monteleone P., Dalle Grave R., Aveta T., Ghoch M.E., et al. Responses of peripheral endocannabinoids and endocannabinoid-related compounds to hedonic eating in obesity. Eur J Nutr. 2016;55:1799–1805. doi: 10.1007/s00394-016-1153-9. [DOI] [PubMed] [Google Scholar]
- 50.Morello G., Imperatore R., Palomba L., Finelli C., Labruna G., Pasanisi F., et al. Orexin-A represses satiety-inducing POMC neurons and contributes to obesity via stimulation of endocannabinoid signaling. Proc Natl Acad Sci USA. 2016;113:4759–4764. doi: 10.1073/pnas.1521304113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakane S., Oka S., Arai S., Waku K., Ishima Y., Tokumura A., et al. 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch Biochem Biophys. 2002;402:51–58. doi: 10.1016/S0003-9861(02)00038-3. [DOI] [PubMed] [Google Scholar]
- 52.Palomba L., Motta A., Imperatore R., Piscitelli F., Capasso R., Mastroiacovo F., et al. Role of 2-arachidonoyl-glycerol and CB1 receptors in orexin-A-mediated prevention of oxygen-glucose deprivation-induced neuronal injury. Cells. 2020;9 doi: 10.3390/cells9061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palomba L., Silvestri C., Imperatore R., Morello G., Piscitelli F., Martella A., et al. Negative regulation of leptin-induced reactive oxygen species (ROS) formation by cannabinoid CB1 receptor activation in hypothalamic neurons. J Biol Chem. 2015;290:13669–13677. doi: 10.1074/jbc.M115.646885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Papasozomenos S.C., Binder L.I. Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil Cytoskeleton. 1987;8:210–226. doi: 10.1002/cm.970080303. [DOI] [PubMed] [Google Scholar]
- 55.Pinto S., Roseberry A.G., Liu H., Diano S., Shanabrough M., Cai X., et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 56.Popov V.I., Bocharova L.S. Hibernation-induced structural changes in synaptic contacts between mossy fibres and hippocampal pyramidal neurons. Neuroscience. 1992;48:53–62. doi: 10.1016/0306-4522(92)90337-2. [DOI] [PubMed] [Google Scholar]
- 57.Popov V.I., Bocharova L.S., Bragin A.G. Repeated changes of dendritic morphology in the hippocampus of ground squirrels in the course of hibernation. Neuroscience. 1992;48:45–51. doi: 10.1016/0306-4522(92)90336-z. [DOI] [PubMed] [Google Scholar]
- 58.Quarta C., Claret M., Zeltser L.M., Williams K.W., Yeo G.S.H., Tschöp M.H., et al. POMC neuronal heterogeneity in energy balance and beyond: an integrated view. Nature Metabol. 2021;3:299–308. doi: 10.1038/s42255-021-00345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roza C., Campos-Sandoval J.A., Gómez-García M.C., Peñalver A., Márquez J. Lysophosphatidic acid and glutamatergic transmission. Front Mol Neurosci. 2019;12:138. doi: 10.3389/fnmol.2019.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ryder J., Su Y., Ni B. Akt/GSK3beta serine/threonine kinases: evidence for a signalling pathway mediated by familial Alzheimer's disease mutations. Cell Signal. 2004;16:187–200. doi: 10.1016/j.cellsig.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 61.Ryu J.M., Han H.J. Autotaxin-LPA axis regulates hMSC migration by adherent junction disruption and cytoskeletal rearrangement via LPAR1/3-dependent PKC/GSK3β/β-catenin and PKC/Rho GTPase pathways. Stem Cells. 2015;33:819–832. doi: 10.1002/stem.1882. [DOI] [PubMed] [Google Scholar]
- 62.Sakurai T., Amemiya A., Ishii M., Matsuzaki I., Chemelli R.M., Tanaka H., et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 63.Saucisse N., Mazier W., Simon V., Binder E., Catania C., Bellocchio L., et al. Functional heterogeneity of POMC neurons relies on mTORC1 signaling. Cell Rep. 2021;37:109800. doi: 10.1016/j.celrep.2021.109800. [DOI] [PubMed] [Google Scholar]
- 64.Sayas C.L., Ariaens A., Ponsioen B., Moolenaar W.H. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell. 2006;17:1834–1844. doi: 10.1091/mbc.e05-07-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sayas C.L., Moreno-Flores M.T., Avila J., Wandosell F. The neurite retraction induced by lysophosphatidic acid increases Alzheimer's disease-like Tau phosphorylation. J Biol Chem. 1999;274:37046–37052. doi: 10.1074/jbc.274.52.37046. [DOI] [PubMed] [Google Scholar]
- 66.Schwalbe M., Kadavath H., Biernat J., Ozenne V., Blackledge M., Mandelkow E., et al. Structural impact of tau phosphorylation at threonine 231. Structure. 2015;23:1448–1458. doi: 10.1016/j.str.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 67.Silvestri C., Di Marzo V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metabol. 2013;17:475–490. doi: 10.1016/j.cmet.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 68.Sternson S.M., Shepherd G.M.G., Friedman J.M. Topographic mapping of VMH--> arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci. 2005;8:1356–1363. doi: 10.1038/nn1550. [DOI] [PubMed] [Google Scholar]
- 69.Sun Y., Kim N.-H., Yang H., Kim S.-H., Huh S.-O. Lysophosphatidic acid induces neurite retraction in differentiated neuroblastoma cells via GSK-3beta activation. Mol Cell. 2011;31:483–489. doi: 10.1007/s10059-011-1036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi M., Tomizawa K., Kato R., Sato K., Uchida T., Fujita S.C., et al. Localization and developmental changes of tau protein kinase I/glycogen synthase kinase-3 beta in rat brain. J Neurochem. 1994;63:245–255. doi: 10.1046/j.1471-4159.1994.63010245.x. [DOI] [PubMed] [Google Scholar]
- 71.Tan Y., Hang F., Liu Z.-W., Stoiljkovic M., Wu M., Tu Y., et al. Impaired hypocretin/orexin system alters responses to salient stimuli in obese male mice. J Clin Invest. 2020;130:4985–4998. doi: 10.1172/JCI130889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tancredi V., D'Antuono M., Cafè C., Giovedì S., Buè M.C., D'Arcangelo G., et al. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem. 2000;75:634–643. doi: 10.1046/j.1471-4159.2000.0750634.x. [DOI] [PubMed] [Google Scholar]
- 73.Ten-Blanco M., Flores Á., Pereda-Pérez I., Piscitelli F., Izquierdo-Luengo C., Cristino L., et al. Amygdalar CB2 cannabinoid receptor mediates fear extinction deficits promoted by orexin-A/hypocretin-1. Biomed Pharmacother. 2022;149:112925. doi: 10.1016/j.biopha.2022.112925. [DOI] [PubMed] [Google Scholar]
- 74.Tunisi L., D'Angelo L., Fernández-Rilo A.C., Forte N., Piscitelli F., Imperatore R., et al. Orexin-A/Hypocretin-1 controls the VTA-NAc mesolimbic pathway via endocannabinoid-mediated disinhibition of dopaminergic neurons in obese mice. Front Synaptic Neurosci. 2021;13 doi: 10.3389/fnsyn.2021.622405. 622405–622405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Turcotte C., Chouinard F., Lefebvre J.S., Flamand N. Regulation of inflammation by cannabinoids, the endocannabinoids 2-arachidonoyl-glycerol and arachidonoyl-ethanolamide, and their metabolites. J Leukoc Biol. 2015;97:1049–1070. doi: 10.1189/jlb.3RU0115-021R. [DOI] [PubMed] [Google Scholar]
- 76.Turunen P.M., Jantti M.H., Kukkonen J.P. OX1 orexin/hypocretin receptor signaling through arachidonic acid and endocannabinoid release. Mol Pharmacol. 2012;82:156–167. doi: 10.1124/mol.112.078063. [DOI] [PubMed] [Google Scholar]
- 77.Valerio A., Ghisi V., Dossena M., Tonello C., Giordano A., Frontini A., et al. Leptin increases axonal growth cone size in developing mouse cortical neurons by convergent signals inactivating glycogen synthase kinase-3beta. J Biol Chem. 2006;281:12950–12958. doi: 10.1074/jbc.M508691200. [DOI] [PubMed] [Google Scholar]
- 78.Vogt J., Yang J.-W., Mobascher A., Cheng J., Li Y., Liu X., et al. Molecular cause and functional impact of altered synaptic lipid signaling due to a prg-1 gene SNP. EMBO Mol Med. 2016;8:25–38. doi: 10.15252/emmm.201505677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Q.M., Fiol C.J., DePaoli-Roach A.A., Roach P.J. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994;269:14566–14574. [PubMed] [Google Scholar]
- 80.Ward R.J., Pediani J.D., Milligan G. Heteromultimerization of cannabinoid CB(1) receptor and orexin OX(1) receptor generates a unique complex in which both protomers are regulated by orexin A. J Biol Chem. 2011;286:37414–37428. doi: 10.1074/jbc.M111.287649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshida Y., Fujiki N., Nakajima T., Ripley B., Matsumura H., Yoneda H., et al. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14:1075–1081. doi: 10.1046/j.0953-816x.2001.01725.x. [DOI] [PubMed] [Google Scholar]
- 82.Yung Y.C., Stoddard N.C., Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res. 2014;55:1192–1214. doi: 10.1194/jlr.R046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.