

Abstract
Nonalcoholic fatty liver disease (NAFLD) represents a spectrum of metabolic liver disease associated with obesity, ranging from relatively benign hepatic steatosis to nonalcoholic steatohepatitis (NASH). The latter is characterized by persistent liver injury, inflammation, and liver fibrosis that collectively increase the risk for end-stage liver disease such as cirrhosis and hepatocellular carcinoma. Recent work has shed new light on the pathophysiology of NAFLD/NASH, particularly the role of genetic, epigenetic, and dietary factors and metabolic dysfunctions in other tissues in driving excess hepatic fat accumulation and liver injury. In parallel, single-cell RNA sequencing studies have revealed unprecedented details on the molecular nature of liver cell heterogeneity, intrahepatic crosstalk, and disease-associated reprogramming of the liver immune and stromal vascular microenvironment. This review covers the recent advances in these areas, the emerging concepts of NASH pathogenesis, and potential new therapeutic opportunities.
Keywords: NASH, NAFLD, fatty liver, single-cell, steatosis, microenvironment, lipid metabolism
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a common liver manifestation of the metabolic syndrome that affects approximately 25% of the global population (131). A defining feature of NAFLD is the excess fat accumulation in the liver, which may exist in a clinically benign state known as simple steatosis. Approximately 20% of NAFLD patients will further progress to nonalcoholic steatohepatitis (NASH), a more severe metabolic liver disease characterized by persistent liver injury, inflammation, and fibrosis. NASH markedly increases the risk for end-stage liver diseases such as cirrhosis and hepatocellular carcinoma (HCC), which are emerging as leading indications for liver transplantation in the U.S. Despite this, there is currently no FDA-approved therapy specifically targeting NASH. NASH pathogenesis encompasses disruptions of hepatic lipid metabolism, hepatocyte injury and regeneration, and progressive remodeling of the liver immune and stromal microenvironment. Several recent reviews cover different aspects of NASH pathogenesis (7, 93, 129). Here we discuss the pathophysiological mechanisms underlying hepatic steatosis and the recent advances in single-cell transcriptomics that provide unprecedented details on liver cell heterogeneity, crosstalk, and reprogramming during disease progression.
De novo lipogenesis and hepatic steatosis
The liver acquires lipids via two routes: lipid uptake from the circulation and de novo lipogenesis (DNL). Neutral lipids such as triacylglycerol (TAG) and cholesteryl ester are stored in lipid droplets (LDs) in hepatocytes, which can be hydrolyzed to release free fatty acids (FFAs) via lipolysis or lipophagy (lysosome-based degradation). FFAs are metabolized through peroxisomal and mitochondrial fatty acid β-oxidation, which generates energy for hepatocytes and substrates for ketogenesis that provides metabolic fuel for extrahepatic tissues. LDs also provide a supply of lipids for very low-density lipoproteins (VLDL). An imbalance between lipid supply (DNL, dietary lipids, and adipose lipolysis) and utilization (fatty acid β-oxidation and VLDL secretion) drives pathogenic fat accumulation in the liver (Figure 1).
Figure 1. An overview of the pathophysiological mechanisms underlying NASH.
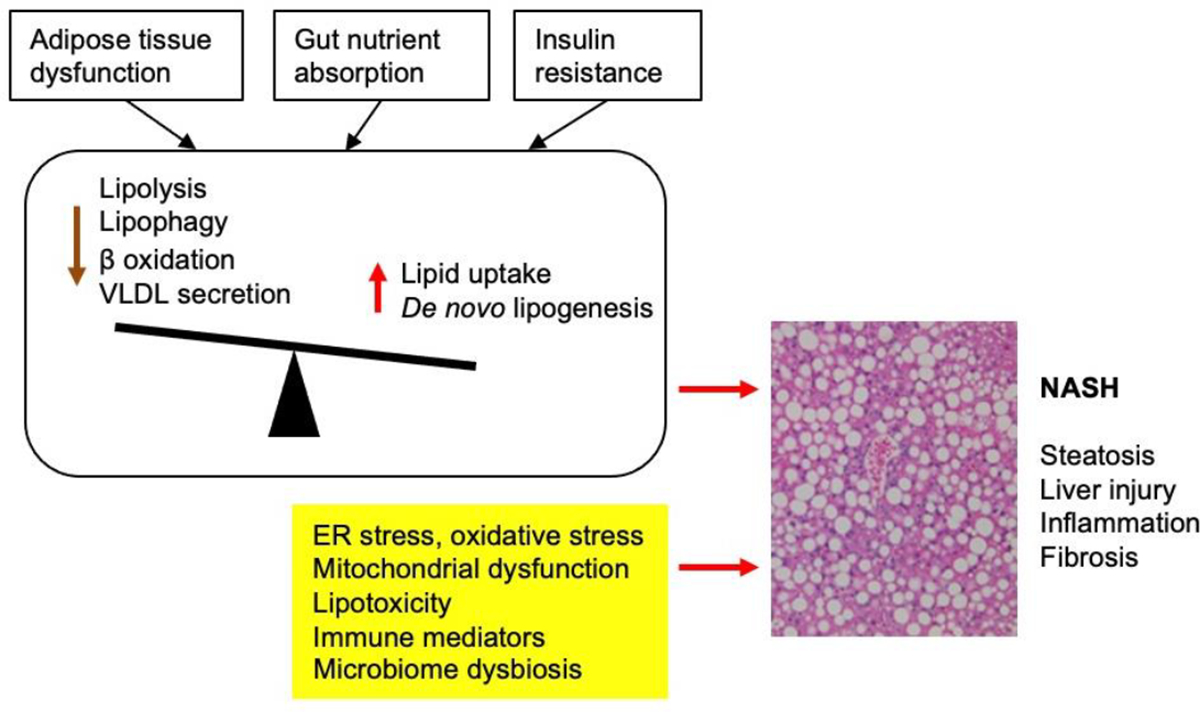
Hepatic steatosis results from imbalance between lipid supply (dietary lipids, adipose lipolysis and de novo lipogenesis) and utilization (fatty acid β oxidation and VLDL secretion) by hepatocytes. Insulin resistance, adipose tissue dysfunction, and excess dietary fructose drive hepatic steatosis, which predisposes hepatocytes to injury and cell death in the presence of other stress signals and pathogenic insults. Liver injury triggers remodeling of the immune and stromal vascular microenvironment that results in chronic tissue inflammation and liver fibrosis.
Hepatic DNL is elevated in humans with NAFLD, contributing to 11%, 19%, and 38% of intrahepatic TAG in lean, obese, and obese with NAFLD groups, respectively (75, 126, 137). Dietary sugars, particularly fructose, are potent inducers of lipid synthesis and contribute to hepatic steatosis when consumed in excess (52, 138). Glucose is converted to fatty acids through a chain of enzymatic reactions mediated by glucokinase, pyruvate carboxylase, ATP-citrate lyase, malic enzyme, acetyl-CoA carboxylase, and fatty acid synthase. Long-chain fatty acids (LCFAs) are activated by long chain acyl-CoA synthetases to generate LCFA-CoAs (124). The latter provides substrates for TAG synthesis by glycerol-3-phosphate acyltransferases, 1-acylglycerol-3-phosphate-O-acyltransferases, and diacylglycerol acyltransferases. Pharmacological inhibition of DNL using liver-directed ACC inhibitors (MK-4074 and PF-05221304) decreases liver steatosis in patients with NAFLD in a dose-dependent manner (15, 67). However, ACC inhibition leads to elevated plasma triglyceride levels, likely a consequence of increased VLDL secretion. This unexpected hypertriglyceridemic side effect remains an obstacle in therapeutic targeting of ACCs for NASH treatment.
Overconsumption of food containing high sugar content and high-fructose corn syrup has been linked to obesity, hepatic steatosis, and NASH. Fructose is absorbed by intestinal epithelial cells primarily via the glucose transporter GLUT5 (52, 138). In the liver, fructose is extracted from portal circulation and undergoes rapid phosphorylation by ketohexokinase to form fructose-1-phosphate. The latter generates substrates for DNL and serves as an allosteric activator of glucokinase that enhances glycolytic flux in hepatocytes. Genetic ablation of fructokinase protects mice from dietary fructose-induced metabolic disorders (3). Importantly, liver-specific inactivation of fructokinase nearly completely blocks hepatic steatosis in fructose-fed mice, underscoring the importance of hepatic fructose metabolism in driving liver fat accumulation under metabolic stress conditions. A recent study demonstrates that gut microbiota process fructose to generate acetate, providing a lipogenic substrate that exacerbates hepatic steatosis (172). The findings illustrate that dietary sugars contribute to elevated DNL through multiple physiological mechanisms.
Sterol response element-binding protein 1c (SREBP1c) and carbohydrate response element-binding protein (ChREBP) are two master transcriptional regulators of the lipogenic gene program (32, 158) (Figure 2). SREBP1c gene expression, posttranslational processing, and transcriptional activity are regulated by nutrient and hormonal signals. Liver X receptor activation strongly stimulates SREBP-1c and lipogenic gene expression (121, 128), whereas farnesoid X receptor elicits opposite effects (113). Aberrant activation of SREBP1c-mediated hepatic lipogenesis has been observed in mice and in humans with NAFLD (56, 71, 136). In the context of nutrient signaling, activation of AKT and mammalian target of rapamycin (mTOR) complex 1 (mTORC1) stimulates DNL, in part through SREBP1c and ChREBP (44, 76). ChREBP is highly responsive to glucose and fructose and hormonal signals; its function is regulated at the level of gene expression, subcellular localization, and transcriptional activity (1). ChREBP is regulated by the transcription factor ZBTB20, which is required for lipogenic induction and diet-induced hepatic steatosis (88). Both SREBP1c and ChREBP interact with an array of transcriptional coactivators and corepressors and serve as a nexus for crosstalk with the liver clock that drives diurnal rhythms of hepatic lipid metabolism (39, 168).
Figure 2. Regulation of hepatic de novo lipogenesis.
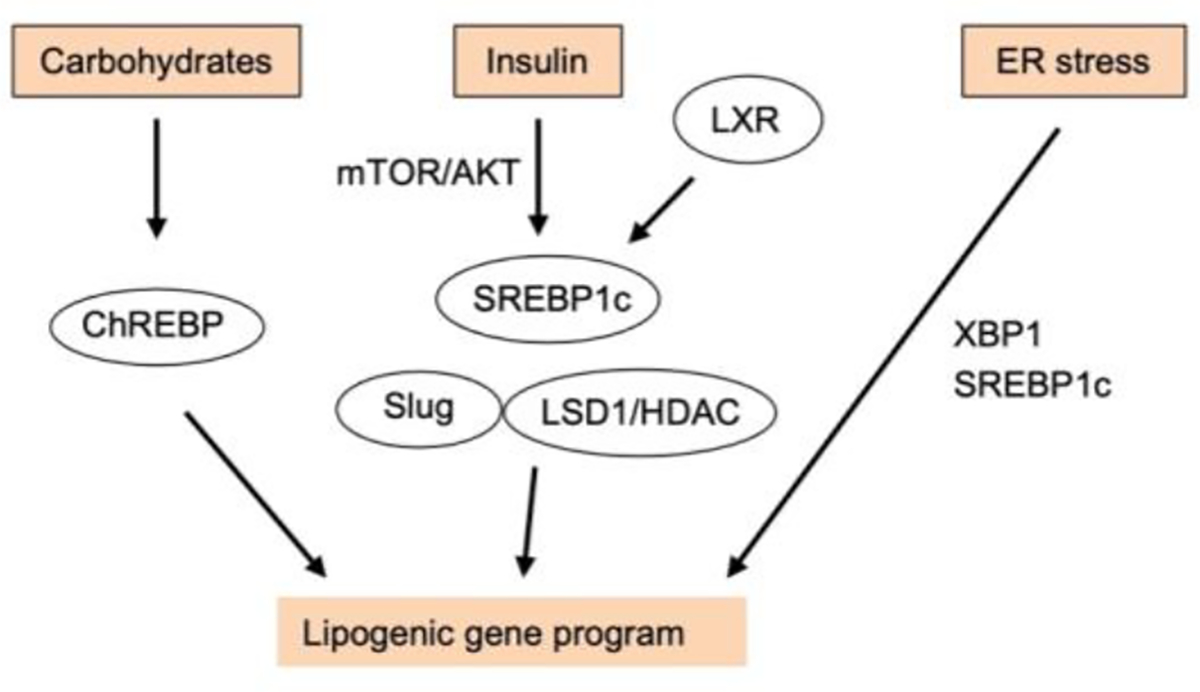
Hepatic DNL gene program is regulated by nutrients, hormonal cues, and pathophysiological signals, which act on several transcription factors and chromatin regulators that culminate in lipogenic gene induction. Carbohydrates such as fructose stimulate gene expression and transcriptional activity of ChREBP, whereas insulin and LXR activation leads to SREBP1c-mediated lipogenic induction. ER stress triggers activation of XBP1 and SREBP1c to regulate lipogenic gene expression.
Insulin potently stimulates de novo lipogenesis in the liver and adipose tissue, in part through induction of SREBP1c expression, processing, and activation downstream of the AKT/mTORC1 pathway (124). Insulin upregulates Slug in hepatocytes, which stimulates DNL and exacerbates hepatic steatosis through an epigenetic mechanism (91). Insulin stimulates glycolysis by inhibiting FOXO1, which recruits transcriptional corepressor proteins to suppress glucokinase expression and glycolysis (77). In insulin resistant state, insulin maintains its capability to stimulate hepatic lipogenesis even though its ability to suppress gluconeogenesis is diminished, lending to the concept of selective insulin resistance (85). The induction of pro-lipogenic signaling combined with an ample supply of lipogenic substrates are major drivers of hepatic steatosis in NAFLD.
Lipid delivery to the liver and hepatic steatosis
It is estimated that hepatic lipid uptake accounts for ~59% of liver fat in humans with NAFLD (28). Several plasma membrane FFA transporters have been implicated in liver lipid uptake, including CD36, fatty acid transport protein 2 (FATP2) and FATP5. Hepatocyte-specific deletion of CD36 mitigates high fat diet (HFD)-induced steatosis in mice (160), whereas knockdown of liver FATP2 or deletion of FATP5 ameliorates liver steatosis (26, 30). Several transcription factors, including peroxisome proliferator-activated receptor-ɣ (PPARɣ), pregnane X receptor, aryl hydrocarbon receptor, and testicular receptor 4, have been demonstrated to directly activate CD36 expression and promote liver steatosis (65, 80, 81, 173). PPARγ also increases expression of lipid droplet proteins to augment cellular lipid storage capacity, and hepatocyte-specific deletion of PPARγ decreases HFD-induced hepatic steatosis (100). Hepatic CD36 upregulation correlates with the release of FFAs from white adipose tissue (WAT), likely facilitating lipid trafficking from adipose tissue to the liver during NAFLD (124). In addition to circulating fatty acids, chylomicron and VLDL remnants also provide a source of lipids for hepatocytes (63).
Too little fat (lipodystrophy) and too much fat (obesity) both are associated with pathogenic liver fat accumulation. Blocking lipolysis (ablating adipocyte ATGL) protects against HFD-fed liver steatosis (127), whereas ATGL upregulation in fat (adipocyte-specific deletion of Snail1) aggravates liver steatosis (140). These findings illustrate the importance of WAT-liver trafficking of lipids in NAFLD. Brown and beige fat contain high mitochondrial content and contribute to systemic energy metabolism through uncoupling protein-1 (UCP1)-dependent and -independent thermogenesis (17, 48) and by secreting endocrine hormones and exosomes that act on other tissues (101, 154). Dysfunctions of brown and beige fat thermogenesis aggravate hepatic steatosis and liver disease progression (125). Ablation of UCP1 exacerbates alcohol-induced liver steatosis, injury, inflammation, and fibrosis in mice (133). A recent study demonstrates that leptin resistance in hypothalamic neurons suppresses sympathetic nerve outflow to BAT, leading to BAT dysfunctions, obesity, and liver steatosis (62). Beyond thermogenesis, the brown fat-derived endocrine factor Neuregulin 4 protects mice from diet-induced NASH pathogenesis through attenuating hepatic lipogenesis and preserving hepatocyte health under stress conditions (40, 155). Adiponectin signaling exerts multifaceted effects on hepatic metabolism and metabolic liver disease (54).
Hepatic fat oxidation and liver steatosis
Neutral lipids are stored within the core of lipid droplets, which are coated with a monolayer of phospholipids and proteins. LDs are born from the ER and heterogeneous in sizes and lipid composition (171). Lipase-mediated triglyceride hydrolysis and lipophagy, a specialized form of autophagy that directs lipids for lysosomal degradation, are two major pathways contributing to lipid catabolism in the liver (117, 145). Fatty acids released from LDs are targeted for peroxisomal and mitochondrial β-oxidation, which generates acetyl-CoA that is routed to several pathways. A major metabolic fate of acetyl-CoA is oxidation by the TCA cycle to produce ATP. Stable isotope tracer studies demonstrate that hepatic TCA cycle activity and acetyl-CoA oxidation are elevated in human NAFLD livers (33, 143), potentially an adaptive response to increased lipid and acetyl-CoA flux. Excess adipose lipolysis and hepatic acetyl-CoA production increase pyruvate carboxylase flux, leading to elevated hepatic gluconeogenesis (108). This mechanism contributes to impaired insulin suppression of hepatic glucose production in obesity with adipose tissue dysfunction where inhibition of lipolysis by insulin is inadequate. Interestingly, liver-specific activation of mitochondrial uncoupling reverses hepatic steatosis in NASH, presumably by increasing mitochondrial respiration and combusting fat (110). During starvation, acetyl-CoA is consumed for synthesis of ketone bodies that are secreted into the circulation as metabolic fuel for extrahepatic tissue. Blocking of ketogenesis reroutes acetyl-CoA for lipogenesis, inducing liver steatosis (19).
Glucagon plays an important role in liver steatosis, as demonstrated by findings that glucagon deficiency exacerbates liver steatosis whereas chronic glucagon infusion mitigates HFD-induced liver steatosis (46, 109). Interestingly, glucagon stimulates liver β oxidation in a PPARα-dependent mechanism (92, 124). The inositol triphosphate receptor 1 and its downstream Ca2+ signaling cascade are also required for glucagon action (109). Foxa2/HNF3β also acts downstream of glucagon to stimulate β oxidation and ketogenesis (153).
PPARα is the master transcriptional regulator of the β oxidation and ketogenesis gene programs. Hepatocyte-specific deletion of PPARα decreases liver β oxidation and ketogenesis, resulting in liver steatosis (99). Several transcriptional coactivators converge on PPARα to influence fatty acid oxidation and liver lipid content, including PGC-1α, BAF60a, SIRT1, and transducin beta-like 1 (TBL1) (74, 86). In addition, IRF9 binds to and increases PPARα activity, and hepatic overexpression of IRF9 decreases liver steatosis in obese mice (157). By contrast, G protein pathway suppressor 2 (GPS2) inhibits PPARα activity, and liver-specific deletion of Gps2 ameliorates diet-induced liver steatosis (87). Astrocyte elevated gene 1 suppresses PPARα by competing for RXR, and hepatocyte-specific deletion of this factor decreases HFD-induced NAFLD (139). Hepatic small ubiquitin-related modifier (SUMO)-Specific protease (SENP2) is upregulated in obesity and deSUMOylates PPARα to induce its ubiquitination and degradation, and hepatocyte-specific ablation of SENP2 mitigates HFD-induced liver steatosis (89).
Sirtuins (SIRTs) are a family of enzymes responsible for posttranslational protein modifications, particularly NAD+-dependent lysine deacetylation (21). SIRT1 deacetylates and activates PGC-1α, enhancing β oxidation. Hepatocyte-specific deletion of SIRT1 decreases expression of fatty acid oxidation genes and exacerbates diet-induced liver steatosis (114). Liver-specific deletion of SIRT2 or SIRT6 also augments HFD-induced hepatic steatosis (68, 120), but it remains to be determined if PPARα mediates SIRT2/6 actions. Mitochondrial SIRT3 deacetylates and activates multiple enzymes engaged in β oxidation and ketogenesis in an NAD+-dependent manner (24, 135). Liver SIRT3 and NAD+ levels are reduced in obesity, leading to suppression of β oxidation/ketogenesis and increased hepatic lipid accumulation. Supporting the notion that hepatic SIRT1/2/3/6 confer health benefits, dietary supplement of NAD+ precursor nicotinamide riboside reverses high fat/high sucrose-induced liver steatosis (36).
Epigenetic reprogramming and other pathogenic factors contributing to hepatic steatosis
Chromatin regulators exert potent effects on lipogenic gene expression via epigenetic mechanisms (Figure 2). Slug (encoded by Snai2) is a transcription factor that recruits several chromatin-remodeling complexes through its N-terminal SNAG domain, including histone deacetylase 1 (HDAC1), HDAC2, and lysine-specific demethylase 1 (LSD1) (174). Slug recruits LSD1 to the FASN promoter, leading to histone 3 lysine-9 demethylation and induction of lipogenic gene expression in hepatocytes (91). Hepatocyte-specific ablation of Slug prevents HFD-induced liver steatosis; conversely, liver-specific overexpression of wild-type Slug, but not an epigenetic-defective Slug mutant, aggravate diet-induced NAFLD. Unlike Slug, Snail1 represses lipogenic gene expression and attenuate hepatic steatosis by promoting histone deacetylation of H3K9 and H3K27 (90). HDAC3 exerts broad effects on transcriptional regulation of hepatic genes involved in lipogenesis and fat oxidation through its interaction with transcription factors including Rev-erb and HNF4α (31, 106, 142). Rhythmic chromatin association of HDAC3 plays an important role in orchestrating the circadian metabolic transcriptome in the liver. Given the pleiotropic role of these epigenetic regulators in gene expression across diverse tissues, whether they can be therapeutically targeted remains an open question.
Endoplasmic reticulum (ER) stress impacts NAFLD pathogenesis through multiple mechanisms (78). The ER membrane harbors diverse enzymes engaged in fatty acid elongation and desaturation, triglyceride, phospholipid and cholesterol biosynthesis, and lipid oxidation (the cytochrome P450 family). In addition, ER is the site of VLDL lipidation and secretion. Activation of unfolded protein response has been linked to lipogenic induction and hepatic steatosis through XBP1 activation (79), PERK/elF2α-dependent regulation of lipogenic transcription factors (105), and induction of noncanonical SREBP processing and activation by caspase 2/S1P (69). Recent studies highlight ER contacts with the plasma membrane and other intracellular organelles, such as mitochondrion-associated ER membrane (MAM); the latter mediates ER-to-mitochondria transfer of phosphatidylserine. Blocking phosphatidylserine transfer across the MAM induces liver steatosis and NASH in mice (53). Hepatocyte MAM levels are reduced in obesity (149), suggesting that impairment in MAM-based ER-mitochondria crosstalk contributes to liver steatosis. Beyond its role in disruption of hepatic lipid metabolism, ER stress is also linked to lipotoxicity and hepatocyte injury.
Transcriptomic signatures of NASH beyond steatosis
While hepatocyte fat accumulation is a defining feature of fatty liver disease, NASH pathogenesis is characterized by complex remodeling of liver microenvironment involving diverse immune and stromal vascular cell populations. Previous transcriptomic studies using bulk liver samples have uncovered several core gene signatures associated with human NASH, most prominently the pathways linked to hepatic metabolism, immune response, and fibrosis (4, 38, 82). Human NASH develops over the course of years to decades. It is remarkable that diet-induced NASH in mice, typically within a period of two to six months, recapitulates several major aspects of liver transcriptomic remodeling (18, 164), illustrate that studies using diet-induced NASH models may reveal insights highly relevant for human disease. Several gene sets have been identified that serve as molecular predictors of NAFLD/NASH severity. For example, a transcriptional signature of 25 differentially expressed genes was found to correlate with stages F2 to F4 of fibrosing steatohepatitis (38). In addition, unique gene signatures of lobular inflammation and fibrosis have been identified, some of which are upregulated during HSC activation. Significant sexual dimorphism of NASH liver gene signatures was observed in a large human dataset, underscoring the need for dissecting pathophysiological mechanisms that may operate in a sex-specific manner (152).
Transcriptional profiling studies have provided new insights into potential pathogenic mechanisms underlying NASH. Hepatic expression of Dermatopontin, a component of the extracellular matrix, is closely associated with liver fibrosis (82). Functional studies in mice demonstrate that Dermatopontin promotes collagen deposition and fibrosis in the setting of Ccl4-induced liver injury. The major pathways altered in human NASH appear to be conserved in diet-induced NASH in mice (18, 164). Similar to human NASH, mRNA and protein expression enriched for cytokine signaling, immune response, and extracellular matrix biology are highly increased in mouse livers upon diet-induced NASH.
In addition to transcriptomic analysis, mass spectrometry-based proteomic, metabolomic, and lipidomic tools have been used for NASH biomarker discovery and mechanistic investigation, as recently reviewed (104). Plasma proteomic profiling patients with and without NAFLD or cirrhosis identified Polymeric immunoglobulin receptor as a plasma protein highly associated with NAFLD and liver cirrhosis (104). SOMAscan analysis revealed that differentially regulated plasma proteins can be used in a 12-protein biomarker panel to distinguish early and advanced fibrosis in NAFLD patients (95). In a separate study, plasma AKR1B10 and GDF15 were found to strongly associate with disease activity and fibrosis stage (38). While the diagnostic value of these newly discovered biomarkers remains to be fully evaluated, it is conceivable that combinations of these plasma factors may offer a readily available and accurate assessment of NASH grades in patients.
The liver microenvironment at single-cell resolution
The mammalian liver comprises diverse cell types, including hepatocytes which constitute the liver parenchyma, and non-parenchymal cells (NPCs) such as liver sinusoidal endothelial cells (LSECs), the resident macrophage Kupffer cells (KCs), hepatic stellate cells (HSCs), cholangiocytes, and diverse immune cell types (Figure 3). Liver cell heterogeneity poses a major challenge for delineating cell type-specific transcriptomic landscape using bulk RNA sequencing (RNAseq) data. Recent advance in single-cell RNAseq, single-nucleus RNAseq, and spatial genomics has generated unprecedented insights into liver cell heterogeneity and disease-associated transcriptomic reprogramming at single-cell resolution (6, 116, 163).
Figure 3. Liver cell heterogeneity and reprogramming during NASH.
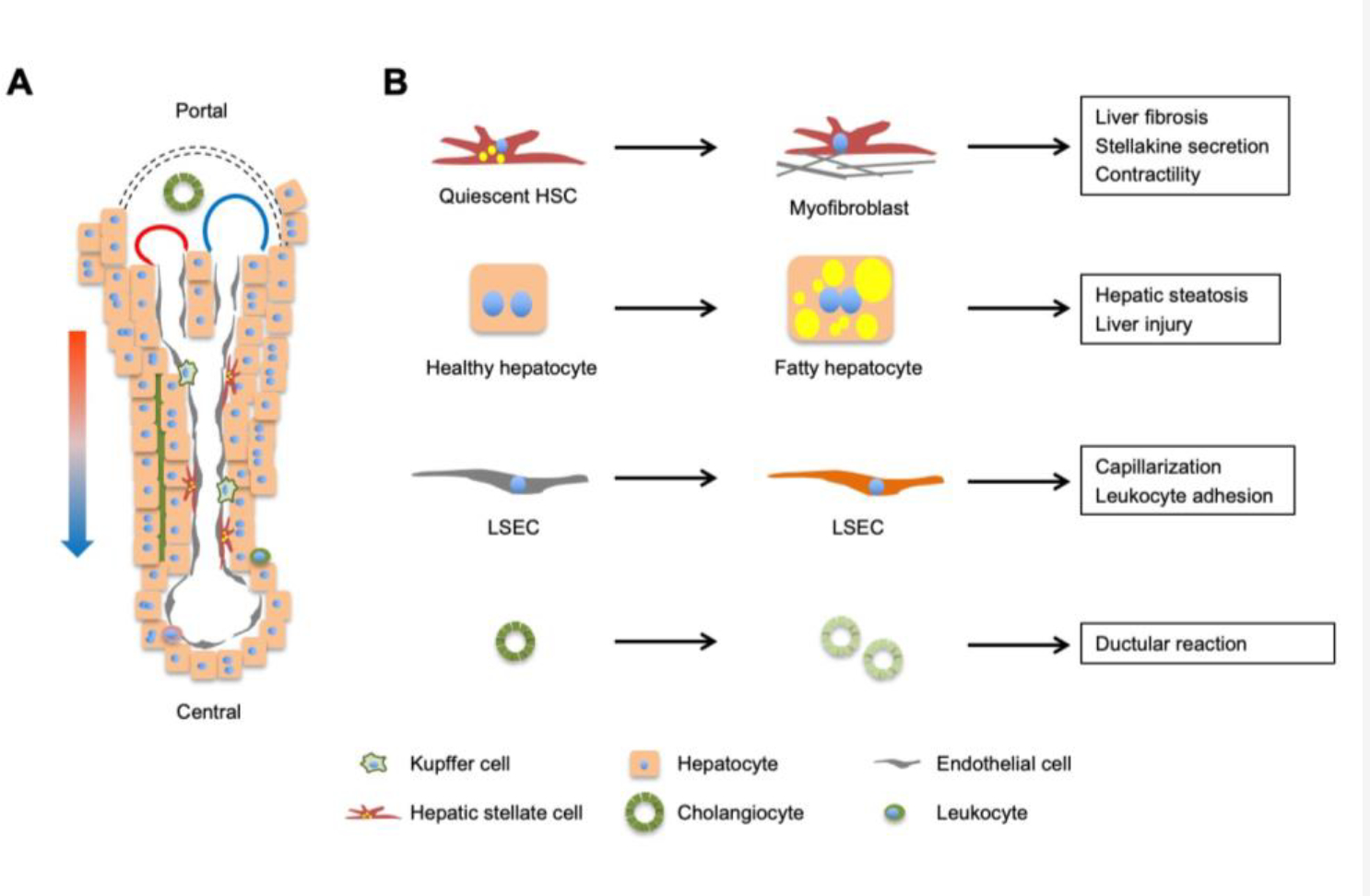
(A) A liver lobule contains parenchymal hepatocytes (60–70% of liver cells) and non-parenchymal cells (30–40%), including sinusoidal endothelial cells, hepatic stellate cells, cholangiocytes, and immune cells (Kupffer cells and leukocytes). Mixed blood from hepatic artery (red) and portal vein (blue) flows along the portal to central direction, creating gradients of oxygen, nutrients, hormones, and gut-derived factors that drive transcriptomic and functional zonation of cells in the liver. (B) Reprogramming of major cell types during NASH pathogenesis. Individual cell types in the liver undergo cell type-specific transcriptomic, metabolic, and functional reprogramming during NASH progression.
A prominent feature of liver cell transcriptome is the presence of zonation-associated gene expression patterns along the portal to central axis. Hepatocytes display gradient mRNA expression for an array of metabolic enzymes, plasma proteins, and signaling molecules in a spatially defined manner (43). This division of cellular metabolism and function among hepatocytes is highly conserved in mice and humans (2). A common feature of mammalian hepatocytes is their variations in ploidy, a phenomenon that is regulated during development and disease states (27). Single-nucleus RNA sequencing analysis has revealed unique molecular signatures and functional enrichment for hepatocytes containing 2n and 4n nuclei and crosstalk between gene dosage and spatial distribution of hepatocytes (122). Using Seq-Scope, a recently developed spatial barcoding technology, Cho et al. provided high-resolution transcriptomic visualization of portal-central hepatocyte zonation, non-parenchymal cell types and subtypes, and subcellular localization of transcripts (16). These studies illustrate intrinsic heterogeneity of the liver parenchyma that likely underlies tissue function and plasticity.
Liver endothelial cells and HSCs also exhibit remarkable transcriptomic and functional heterogeneity. Endothelial subtypes that correspond to portal, sinusoidal, and central endothelial cells have been identified, which express unique molecular markers (2, 42, 162). By combining spatial cell sorting with transcriptomics and quantitative proteomics/phosphoproteomics, a recent study provided in-depth molecular details into the spatially resolved proteome landscape of the liver endothelium and zonated mechanisms of vascular signaling (60). Remarkably, endothelial phosphoproteomes, as revealed by Serine, Threonine, and Tyrosine phosphorylation, exhibit clear zonation patterns along the portal to central axis. Similarly, single-cell transcriptomic analysis uncovered distinct stromal cell populations that represent portal vein-associated HSC, central vein-associated HSC, and fibroblasts in mouse liver (25, 123, 146). In human liver, two HSC populations with unique gene expression signature were identified and exhibit distinct localization in portal/central vein area and perisinusoidal space (107). The exact relationships between mouse and human HSC subtypes remain to be established.
Intercellular crosstalk is a fundamental feature of complex organs. Endocrine, paracrine, and adhesion signaling regulate diverse biological processes and serve a critical role in tissue homeostasis and function. The delineation of single-cell transcriptomes provides a valuable resource for constructing a blueprint for intrahepatic cell-cell signaling mediated by ligands and receptors. Single-cell secretome gene analysis has revealed cell type-restricted patterns of ligand and receptor gene expression that is highly conserved in mouse and human liver (162). HSCs are emerging as a signaling hub by secreting an array of biologically active factors (stellakines), including growth factors, cytokines, and chemokines, which are predicted to act on endothelial and immune compartments. Interestingly, HSCs express several classes of membrane receptors that mediate vasoactive signaling, suggesting that contractile activity of HSCs is tightly regulated under physiological conditions. In a separate study, the central vein-associated HSCs express Lysophosphatidic Acid Receptor 1 (LPAR1) and serve as dominant pathogenic collagen-producing cells in a mouse model of centrilobular fibrosis (25). Blockade of LPAR1 attenuates liver fibrosis in a mouse model of NASH. Together, these single-cell RNA sequencing studies provide a high-resolution transcriptomic map for all major liver cell types and illustrate cellular heterogeneity and crosstalk in mammalian liver.
Reprogramming of intrahepatic immune landscape during NASH pathogenesis
Chronic liver injury and inflammation are two defining features of the transition from simple steatosis to NASH (93). Both innate and adaptive immune cell types have been implicated in NASH progression by modulating intrahepatic immune microenvironment. The explosion of single-cell transcriptomic analysis of liver immune cell populations has painted a detailed picture of NASH-associated changes in immune cell type and cell state in the liver during disease progression. Studies performed using human liver samples and rodent liver with diet-induced NASH have delineated several common features of immune reprogramming during NASH progression. Most notably, macrophages and T cells adopt NASH-associated molecular signatures that influence their functions and disease pathogenesis (Figure 4).
Figure 4. Reprogramming of macrophages and intrahepatic T cells during NASH.
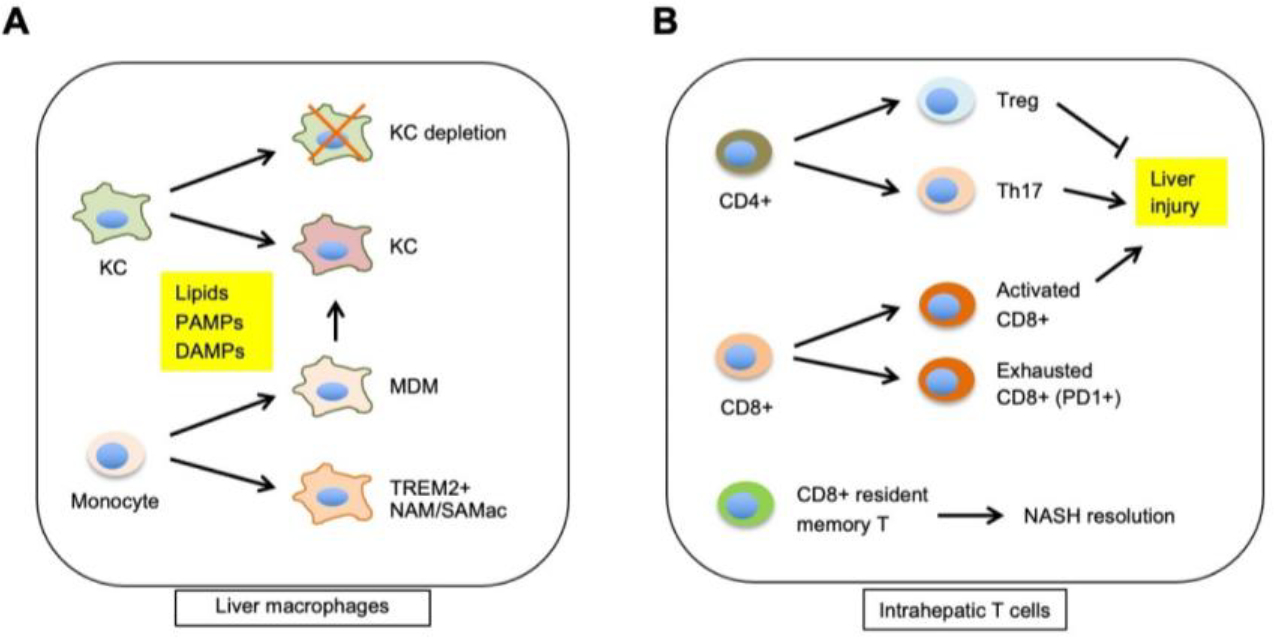
(A) NASH triggers infiltration of circulating monocytes into the liver, a subset of which polarize to a distinct macrophage subtype characterized by high levels of Trem2 expression (NASH-associated macrophage, NAM or scar-associated macrophage, SAMac). Monocyte-derived macrophages (MDM) contribute to the replenishment of depleted Kupffer cells during NASH. The transcriptomic and functional reprogramming of macrophages is regulated by exposure to lipids, PAMPs, and DAMPs. (B) Features of intrahepatic T cell reprogramming during NASH. Different T cell subtypes contribute to liver injury during NASH, fibrosis resolution, and response to immunotherapy agents.
Liver macrophage heterogeneity during NASH
Kupffer cells are resident liver macrophages that represent the major macrophage cell population in healthy liver. Chronic hepatocyte injury occurred during NASH triggers infiltration of circulating monocytes that subsequently acquire distinct molecular phenotypes and functional properties (66, 83). Several recent studies have uncovered a unique macrophage population that is marked by expression of Triggering Receptor Expressed on Myeloid Cells 2 (Trem2), CD9, and Gpnmb in NASH livers (20, 115, 130, 162). TREM2 is highly expressed in microglial cells in Alzheimer’s Disease (AD) brain and its polymorphisms have been associated with AD risk (150). TREM2 is a membrane protein required for clearance of amyloid plagues by microglia. Trem2-expressing macrophages are relatively rare in healthy liver and highly inducible in response to diet-induced NASH in mice (162). These NASH-associated macrophages (NAM) exhibit a transcriptional profile that is enriched in lysosomal function, phagocytosis, and antigen presentation, suggesting that NAM may be responsible for the clearance of injured/dead hepatocytes during NASH. TREM2 expression is strongly associated with NASH pathologies in humans, including liver injury/ballooning grade, inflammation grade, fibrosis stage, and NASH activity score. NAMs exhibit a transcriptional signature that closely resembles lipid-associated macrophages that form the crown-like structures (CLSs) in obese adipose tissue (55, 61). The NAM transcriptional signature is diminished upon NASH resolution induced by dietary and pharmacological treatments (162, 164). These Trem2-expressing macrophages exhibit high levels of Ccr2 and Cx3cr1 expression and are recruited to hepatic CLSs in the steatotic liver in a CCR2-dependent manner (20). The induction of this unique macrophage subpopulation may serve adaptive functions in the clearance of dead hepatocytes and tissue remodeling, at least at early stage of NASH.
A similar population of TREM2-expressing scar-associated macrophages (SAMac) was found to exist in collagen-positive scar regions and expand during fibrosis in human livers (115). Conditioned medium from sorted SAMac stimulates collagen gene expression in primary human HSC culture, indicating pro-fibrotic activity of SAMac in the context of liver injury. Paradoxically, Trem2 deficiency accelerates NAFLD progression in mice, in part through disruption of macrophage-hepatocyte crosstalk that results in mitochondrial dysfunction in hepatocytes (57). Trem2 knockout mice developed more severe liver injury and inflammation and worsened survival following treatment with CCL4 and acetaminophen (111). Trem2 expression has been detected in tumor-associated macrophages in several cancers, including hepatocellular carcinoma (102, 170), suggesting that induction of this unique macrophage population may contribute to the development of NASH-associated liver cancer. In support of this, Trem2 inactivation and anti-TREM2 monoclonal antibody treatment enhance anti-tumor immunity and anti-PD-1 immunotherapy efficacy (10, 98).
Lipid overloading results in depletion of embryonically derived resident Kupffer cells in the liver, which is gradually replaced by Ly-6C+ monocytes during NASH progression (147). These monocyte-derived KCs exhibit lower efficiency in promoting liver triglyceride storage and exacerbate liver damage during NASH. Among the embryonically derived KCs, two subpopulations distinguished by differential expression of CD206 and ESAM were identified (11). KCs with high levels of CD206 and ESAM exhibit enriched expression of genes involved in lipid metabolism and contribute to lipid overload-induced oxidative stress through the fatty acid transporter CD36. A recent study showed that iron-rich KCs exhibit proinflammatory and profibrotic phenotype during the development of NASH and form crown-like structures surrounding dead hepatocytes (64). Whether distinct KC subtypes differentially contribute to the formation of CLSs in NASH liver remains to be established.
Mechanistic role of liver macrophages in NASH progression
An important aspect of NASH-associated reprogramming of liver macrophages involves macrophage-derived secreted factors. Upon NASH induction, mRNA expression of S100 calcium-binding protein A8 (S100a8) and S100a9 was downregulated in monocyte-derived macrophages in the liver, compared to healthy liver (73). S100A8 and S100A9 proteins form a heterodimer called calprotectin that functions in part as a cytokine. The significance of reduced macrophage secretion of calprotectin in NASH pathogenesis remains to be explored. In contrast to calprotectin, expression of Osteopontin (Secreted phosphoprotein 1, Spp1) was found to increase in Trem2-expressing macrophages in NASH liver (119). Osteopontin has been previously shown to promote liver fibrotic response (5); as such, this factor may mediate crosstalk between macrophages and HSCs in the context of NASH liver fibrosis.
Certain macrophage plasma membrane proteins undergo regulated proteolysis to release soluble fragments, such as TREM2 (165) and macrophage c-mer tyrosine kinase (MERTK) (14). Soluble TREM2 (sTREM2) is capable of binding bioactive ligands and modulating immune response and serves as a cerebrospinal fluid biomarker for AD (165). Whether sTREM2 levels are elevated in plasma from NASH patients and its biological role in liver disease remain an interesting unresolved question. A recent study demonstrates that ADAM16-mediated MERTK cleavage on liver macrophages is decreased during steatosis to NASH transition and that mice with a cleavage-resistant MERTK mutant develop more severe NASH fibrosis (14). This study illustrates that shedding of macrophage membrane proteins may have important functional consequences in metabolic disease settings.
NASH-associated changes in liver macrophage phenotype and function can be partially attributed to altered lipid signaling. Transcriptomic analysis of sorted liver macrophages from mice fed diet containing different lipid compositions has revealed profound effects of dietary cholesterol on diet-induced macrophage infiltration and NASH pathologies (97). Treatment of murine and human macrophages with oxidized low-density lipoprotein recapitulates certain aspects of transcriptomic remodeling that occurs in NASH liver. In contrast, the proresolving mediator maresin 1, a metabolite of docosahexaenoic acid, attenuates liver inflammation and NASH progression (45). Injured hepatocytes release danger-associated molecular patterns, such as mitochondrial DNA, which are recognized by innate immune cells via a TLR9-dependent mechanism (35). Exposure of KCs to mitochondrial DNA triggers STING-mediated inflammatory response that results in exacerbation of NASH (167). Hepatic STING expression is elevated in human NASH and diet-induced NASH in mice; its activation augments HSC activation through macrophage secreted factors (94).
Gut mibrobiota exert multifaceted effects on nutrient metabolism, host immunity, mucosal barrier function, and signaling, and represent an integral aspect of metabolic physiology in mammals. NASH pathogenesis is linked to profound changes in the composition and phenotypic properties of gut microbiome in humans and mice (13, 72). In the context of NASH, gut microbiome promotes hepatic fat accumulation via several mechanisms, including increased production and absorption of nutrients such as short-chain fatty acids, remodeling of bile acids, signaling through the gut-liver axis, and modulating the liver immune and stromal microenvironment. Gut-derived endocrine factors such as FGF15/19 regulate hepatic lipogenesis and influence liver regeneration (8, 151). Gut microbiota-derived short-chain fatty acids from dietary fibers mitigate insulin resistance and hepatic steatosis by activating GPR41 and GPR43 receptors (23). Gut barrier disruption contributes to increased exposure of macrophages to bacterial products such as endotoxins, which triggers inflammasome activation and aggravates NASH pathologies (51, 58, 166).
T cell heterogeneity and role in NASH
T cell-mediated adaptive immunity impacts on the progression of NAFLD/NASH via multiple mechanisms, including hepatocyte injury, intrahepatic inflammation, HSC activation, and anti-tumor surveillance (144). The abundance of intrahepatic CD8+ T cells is increased in several models of diet-induced NASH mice, including high-fat high-carbohydrate diet and choline-deficient HFD (9, 161). CD8 staining in the liver from patients with NASH and cirrhosis positively correlates with α-smooth muscle actin, a marker of HSC activation (12). Ablation of CD8+ T cells using anti-CD8a monoclonal antibody reverses liver steatosis and improves liver NASH pathologies in mice (9, 12), illustrating a disease-promoting role of metabolically activated CD8+ T cells in NASH pathogenesis.
Recent single-cell transcriptomic studies have shed new light on CD8+ T cell heterogeneity, reprogramming, and function in the context of NASH and NASH-associated hepatocellular carcinoma. Single-cell RNA sequencing of cells expressing T cell receptor β chain from the livers of mice with NASH reveals a population of resident effector CD8+ T cells (CXCR6+) exhibiting molecular markers of T cell exhaustion, including Pdcd1 (encoding PD1) (29, 112). The induction of inhibitory receptor gene expression was found to be restricted to CD8+, but not CD4+ T cells, upon diet-induced NASH in mice (47). Importantly, this population of CXCR6+PD1+ CD8 T cells is increased in the livers from NASH patients, underscoring the conserved nature of this transcriptomic and functional reprogramming of CD8+ T cells in NASH liver. In the context of NASH, CXCR6+CD8+ T cells undergo metabolic activation, elicit auto-aggressive killing of cells in an MHC-I-independent manner, and exacerbate hepatocyte injury and liver immune pathologies (29). Despite increased PD1 expression on CD8+ T cells, anti-PD1 treatment failed to induce tumor regression; instead, it exacerbated tumorigenesis in a mouse model of NASH-HCC (112). Similarly, NASH impairs immunotherapy efficacy of anti-OX40 via a mechanism dependent on CD4+ T cells (50). Meta-analysis of three phase III clinical trials indicate that checkpoint inhibitors failed to improve survival in patients with advanced non-viral HCC, suggesting that NASH-associated HCC may be less responsive to immunotherapy owing to aberrant T cell activation and impaired immune surveillance in NASH liver microenvironment.
Several other T cell subtypes have been implicated in NASH pathogenesis. Single-cell transcriptomic analysis reveals enrichment of a population of liver resident memory CD8+ T cells during fibrosis resolution, whose maintenance requires IL-15 signaling (70). This subpopulation of T cells targets activated HSCs to FasL-Fas-mediated apoptosis, thereby restricting collagen deposition and promoting fibrosis resolution. Progression of hepatic steatosis to NASH is marked by increased hepatic IL-17 expression and induction of CD4+ T helper 17 (Th17) cells in both mice and humans (37, 118). Pharmacological suppression of Th17 differentiation, IL-17A blocking antibodies, and genetic ablation of IL-17A receptor in myeloid cells attenuate metabolic inflammation and ameliorate NASH severity and HCC development. NASH-associated accumulation of Th17 is regulated in part by OX40 signaling, which elicits effects on both innate and adaptive immunity in the liver (141). Regulatory CD4+ T cells (Tregs) have been implicated in the regulation of metabolic inflammation and cancer immunosurveillance (103). The abundance of intrahepatic Tregs is increased in mouse and human NASH livers and regulated by neutrophil extracellular traps, which contain web-like chromatin filaments during tissue injury, including NASH (156). Treg depletion improves NASH pathologies and suppresses HCC development in mice.
NASH-associated reprogramming of intrahepatic T cell landscape is linked to altered circulating and liver dendritic cells. Transcriptional network analysis and single-cell RNA sequencing studies reveal that conventional dendritic cells type 1 (cDC1s) are more abundant and assume a more activated molecular signature in human and mouse NASH (22, 41). These XCR1+ cDC1s physically interact with T cells, promote inflammatory T cell activation, and worsen NASH liver pathologies in mice. In a separate study, CD103+ cDC1s have been shown to attenuate liver damage and inflammation and play a protective role in murine NASH (49). Liver dendritic cell heterogeneity and different NASH models used may contribute to the discrepancies observed in these studies. Extrahepatic factors may influence infiltration of T cells into the liver. For instance, aberrant thymic activity induces severe T cell-based liver inflammation in mice, leading to fatal liver injury (132, 134).
Remodeling of the stromal microenvironment in NASH liver
Major cell types constituting the liver stroma include HSCs, endothelial cells, cholangiocytes, and portal fibroblasts (Figure 2). A defining feature of NASH pathogenesis is progressive liver fibrosis, resulting from collagen deposition by activated HSCs and myofibroblasts. Liver fibrosis alters the biophysical properties of extracellular matrix (ECM) and intrahepatic paracrine signaling that ultimately impair liver function and increase the risk for end-stage liver disease. The molecular mechanisms that govern HSC activation and myofibroblast differentiation have been discussed in recent reviews (129, 148). In addition to fibrosis, endothelial capillarization and ductular reaction are commonly associated with NASH pathogenesis. We focus this review on new insights gained from single-cell transcriptomics that illustrate stromal vascular cell heterogeneity and disease-associated reprogramming during NASH.
While it has been recognized that several cell types are capable of secreting matrix collagen in the liver, including HSCs, myofibroblasts, fibroblasts, smooth muscle cells, and mesothelial cells, their molecular and spatial heterogeneity and contribution to liver fibrosis in NASH remain poorly defined. Single-cell RNA sequencing analysis of healthy and cirrhotic human livers has identified four populations of liver mesenchymal cells, representing vascular smooth muscle cells (MYH11+), HSCs (RGS5+), mesothelial cells, and scar-associated mesenchymal cells (PDGFRA+) (115). The latter is characterized by high-level expression of PDGFRA, fibrillar collagens, and pro-fibrogenic genes. These scar-associated mesenchymal cells undergo expansion in cirrhotic livers and are mapped to the fibrotic niche. A separate study identified three ECM-producing cell populations that represent quiescent HSCs, activated HSCs, and mesothelial cells in six healthy human livers (159). Pseudotime trajectory analysis reveals diverse transcriptomic states of mesenchymal cells in both healthy livers and during liver fibrosis. Within the fibrotic niche, several ligand-receptor signaling pathways involving macrophages, endothelial cells, and mesenchymal cells were uncovered that likely serve to maintain and propagate the fibrogenic microenvironment.
Single-cell analysis of HSCs from a mouse model of NASH provides insights into the dynamic regulation of transcriptomic landscape of HSCs during disease progression and fibrosis resolution. A prominent HSC cluster representing activated myofibroblasts is only detected in NASH livers, but not healthy and NASH regressed livers (123). Interestingly, HSCs from the livers with resolving fibrosis have a gene expression signature that is similar to, but distinct, from quiescent HSCs in health livers. Whether activated HSCs acquire the quiescent molecular phenotype or new HSC populations emerge during NASH resolution remains to be established. An important aspect of HSC reprogramming during NASH is pathological changes in HSC secretome and receptor repertoire. Diet-induced NASH is associated with dysregulation of expression for a subset of stellakines and membrane receptors, which is in part mediated by increased IL11-STAT3 signaling in HSCs (162). Impaired signaling of vasoactive peptides may underlie myofibroblast contractility and vascular resistance in fibrotic liver. Along this line, HSC expression of plasmalemma vesicle-associated protein is suppressed in activated HSCs upon injury, potentially contributing to alterations of vascular permeability and immune cell recruitment to the injury sites (146). As such, both cell-intrinsic and -extrinsic mechanisms contribute to reprogramming of HSCs in NASH, which exert effects beyond HSC itself in diseases liver.
Vascular remodeling in NASH
Liver endothelial cells undergo morphological and functional transformation in disease states such as NASH (59). LSECs acquire the characteristics of capillary endothelial cells and exhibit altered metabolic and inflammatory properties; the latter is mediated by changes in gene expression for leukocyte adhesion molecules, cytokines, and chemokines. Single-cell secretome gene analysis of healthy and NASH liver endothelial cells reveals aberrant expression of genes involved in vascular signaling and intrahepatic crosstalk (162). This reprogramming of LSEC secretome is associated with induction of genes involved in lipid metabolism, likely a consequence of lipid overload in NASH liver. Integrated single-cell omics analysis of human cirrhotic liver and a minipig NASH model shed light on the landscape of vascular adaptation during disease progression (169). Notably, a molecular signature of sinusoidal to capillary maladaptation was observed in human cirrhotic liver that is linked to induction of the epigenetic regulators HDAC2 and DNMT1. In this context, NASH-associated vascular remodeling alters secretion of IGFBP7 and ADAMTS1 that collectively promote Th17 cells and fibrotic response.
Endothelial cells express diverse adhesion molecules that mediate immune cell adhesion and recruitment. Hepatic expression of vascular cell adhesion molecule 1 (VCAM-1) is induced upon diet-induced NASH in mice and in human NASH; its plasma levels are a predictor of liver fibrosis in NAFLD patients (34, 84). NASH-association increase in endothelial VCAM-1 expression is regulated by exposure of LSECs to lipotoxic stress. VCAM-1 inactivation and blockade by neutralizing antibody or pharmacological inhibition improve NASH, in part through attenuating the recruitment of proinflammatory monocytes to the liver. Endothelial dysfunction is frequently associated with platelet aggregation and activation in liver sinusoids in mouse and human NASH (96), a process dependent on Kupffer cells and CD44 binding. Antiplatelet treatment abrogates immune cell infiltration, improves NASH pathologies, and prevents NASH-associated HCC development. At the molecular level, platelet GPIbα is critically required for exacerbating NASH progression. These studies illustrate a pathogenic role of remodeling of the endothelial compartment in disease progression.
Concluding remarks
NASH pathogenesis is characterized by reprogramming of hepatic metabolism and liver microenvironment that together drive progressive development of liver pathologies. Metabolic stress-induced hepatocyte injury likely triggers homeostatic immune response and tissue repair during the simple steatosis stage of NAFLD, which represents a compensated and stable disease condition. However, persistent insults to the liver may lead to a tipping point where inflammatory and injury response may become maladaptive, thereby initiating the transition from simple steatosis to NASH. While this model is conceptually simple and supported by a large body of work, several critical questions remain unanswered. First, NASH encompasses a spectrum of metabolic liver disease influenced by genetic and environmental factors that in turn impinge on body metabolism and gut microbiome. It is therefore important to delineate how these factors contribute to the risk profile in NAFLD patients, i.e., transition from steatosis into progressive NASH. Second, recent single-cell transcriptomic and spatial transcriptomic studies have revealed a great deal of details on the molecular and cellular nature of NASH-associated reprogramming of liver microenvironment. The significance of the changes in intrahepatic crosstalk among parenchymal, stromal, and immune compartments that may influence different aspects of NASH pathologies remains to be established. Finally, what are the drivers of NASH pathogenesis that may serve as targets for therapeutic intervention? While the initiation and progression of NASH are driven by a complex array of pathogenic factors, it is likely that critical disease-associated regulatory nodes may exist and remains to be elucidated. Therapeutic agents targeting these regulatory nodes may effectively break the vicious cycles of hepatocyte destruction and tissue repair response and lead to restoration of normal liver function.
Acknowledgements
This work was supported by grants from the NIH (DK102456, DK118731, and DK112800 to J.D.L. and DK115646, DK114220, and DK127568 to L.R.).
Footnotes
The authors declare no conflict of financial interest.
References
- 1.Abdul-Wahed A, Guilmeau S, Postic C. 2017. Sweet Sixteenth for ChREBP: Established Roles and Future Goals. Cell Metab 26: 324–41 [DOI] [PubMed] [Google Scholar]
- 2.Aizarani N, Saviano A, Sagar Mailly L, Durand S, et al. 2019. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572: 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, et al. 2020. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab 32: 117–27 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arendt BM, Comelli EM, Ma DW, Lou W, Teterina A, et al. 2015. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 61: 1565–78 [DOI] [PubMed] [Google Scholar]
- 5.Arriazu E, Ge X, Leung TM, Magdaleno F, Lopategi A, et al. 2017. Signalling via the osteopontin and high mobility group box-1 axis drives the fibrogenic response to liver injury. Gut 66: 1123–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Moshe S, Itzkovitz S. 2019. Spatial heterogeneity in the mammalian liver. Nat Rev Gastroenterol Hepatol 16: 395–410 [DOI] [PubMed] [Google Scholar]
- 7.Bence KK, Birnbaum MJ. 2021. Metabolic drivers of non-alcoholic fatty liver disease. Mol Metab 50: 101143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatnagar S, Damron HA, Hillgartner FB. 2009. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J Biol Chem 284: 10023–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, et al. 2017. Hepatic Natural Killer T-cell and CD8+ T-cell Signatures in Mice with Nonalcoholic Steatohepatitis. Hepatol Commun 1: 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, et al. 2021. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep 37: 109844. [DOI] [PubMed] [Google Scholar]
- 11.Bleriot C, Barreby E, Dunsmore G, Ballaire R, Chakarov S, et al. 2021. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity 54: 2101–16 e6 [DOI] [PubMed] [Google Scholar]
- 12.Breuer DA, Pacheco MC, Washington MK, Montgomery SA, Hasty AH, Kennedy AJ. 2020. CD8(+) T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol 318: G211–G24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruneau A, Hundertmark J, Guillot A, Tacke F. 2021. Molecular and Cellular Mediators of the Gut-Liver Axis in the Progression of Liver Diseases. Front Med (Lausanne) 8: 725390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, et al. 2020. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab 31: 406–21 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, et al. 2021. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med [DOI] [PubMed] [Google Scholar]
- 16.Cho CS, Xi J, Si Y, Park SR, Hsu JE, et al. 2021. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184: 3559–72 e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chouchani ET, Kazak L, Spiegelman BM. 2019. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab 29: 27–37 [DOI] [PubMed] [Google Scholar]
- 18.Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, et al. 2013. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 305: G483–95 [DOI] [PubMed] [Google Scholar]
- 19.Cotter DG, Ercal B, Huang X, Leid JM, d’Avignon DA, et al. 2014. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J Clin Invest 124: 5175–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, et al. 2021. Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep 34: 108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dall M, Hassing AS, Treebak JT. 2021. NAD(+) and NAFLD - caution, causality and careful optimism. J Physiol [DOI] [PubMed] [Google Scholar]
- 22.Deczkowska A, David E, Ramadori P, Pfister D, Safran M, et al. 2021. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med 27: 1043–54 [DOI] [PubMed] [Google Scholar]
- 23.den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, et al. 2015. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 64: 2398–408 [DOI] [PubMed] [Google Scholar]
- 24.Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, et al. 2015. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab 21: 637–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, et al. 2019. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep 29: 1832–47 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doege H, Baillie RA, Ortegon AM, Tsang B, Wu Q, et al. 2006. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis. Gastroenterology 130: 1245–58 [DOI] [PubMed] [Google Scholar]
- 27.Donne R, Saroul-Ainama M, Cordier P, Celton-Morizur S, Desdouets C. 2020. Polyploidy in liver development, homeostasis and disease. Nat Rev Gastroenterol Hepatol 17: 391–405 [DOI] [PubMed] [Google Scholar]
- 28.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. 2005. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115: 1343–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, et al. 2021. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 592: 444–49 [DOI] [PubMed] [Google Scholar]
- 30.Falcon A, Doege H, Fluitt A, Tsang B, Watson N, et al. 2010. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab 299: E384–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng D, Liu T, Sun Z, Bugge A, Mullican SE, et al. 2011. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science 331: 1315–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferre P, Phan F, Foufelle F. 2021. SREBP-1c and lipogenesis in the liver: an update1. Biochem J 478: 3723–39 [DOI] [PubMed] [Google Scholar]
- 33.Fletcher JA, Deja S, Satapati S, Fu X, Burgess SC, Browning JD. 2019. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furuta K, Guo Q, Pavelko KD, Lee JH, Robertson KD, et al. 2021. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, et al. 2016. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 126: 859–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, et al. 2016. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 63: 1190–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, et al. 2016. Metabolic Inflammation-Associated IL-17A Causes Non-alcoholic Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 30: 161–75 [DOI] [PubMed] [Google Scholar]
- 38.Govaere O, Cockell S, Tiniakos D, Queen R, Younes R, et al. 2020. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci Transl Med 12 [DOI] [PubMed] [Google Scholar]
- 39.Guan D, Xiong Y, Borck PC, Jang C, Doulias PT, et al. 2018. Diet-Induced Circadian Enhancer Remodeling Synchronizes Opposing Hepatic Lipid Metabolic Processes. Cell 174: 831–42 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo L, Zhang P, Chen Z, Xia H, Li S, et al. 2017. Hepatic neuregulin 4 signaling defines an endocrine checkpoint for steatosis-to-NASH progression. J Clin Invest [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Molendi-Coste O, et al. 2019. Transcriptional Network Analysis Implicates Altered Hepatic Immune Function in NASH development and resolution. Nat Metab 1: 604–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halpern KB, Shenhav R, Massalha H, Toth B, Egozi A, et al. 2018. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat Biotechnol 36: 962–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halpern KB, Shenhav R, Matcovitch-Natan O, Toth B, Lemze D, et al. 2017. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542: 352–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han J, Wang Y. 2018. mTORC1 signaling in hepatic lipid metabolism. Protein Cell 9: 145–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han YH, Shin KO, Kim JY, Khadka DB, Kim HJ, et al. 2019. A maresin 1/RORalpha/12-lipoxygenase autoregulatory circuit prevents inflammation and progression of nonalcoholic steatohepatitis. J Clin Invest 129: 1684–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hancock AS, Du A, Liu J, Miller M, May CL. 2010. Glucagon deficiency reduces hepatic glucose production and improves glucose tolerance in adult mice. Mol Endocrinol 24: 1605–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansel C, Erschfeld S, Baues M, Lammers T, Weiskirchen R, et al. 2019. The Inhibitory T Cell Receptors PD1 and 2B4 Are Differentially Regulated on CD4 and CD8 T Cells in a Mouse Model of Non-alcoholic Steatohepatitis. Front Pharmacol 10: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harms M, Seale P. 2013. Brown and beige fat: development, function and therapeutic potential. Nat Med 19: 1252–63 [DOI] [PubMed] [Google Scholar]
- 49.Heier EC, Meier A, Julich-Haertel H, Djudjaj S, Rau M, et al. 2017. Murine CD103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol 66: 1241–50 [DOI] [PubMed] [Google Scholar]
- 50.Heinrich B, Brown ZJ, Diggs LP, Vormehr M, Ma C, et al. 2021. Steatohepatitis Impairs T-cell-Directed Immunotherapies Against Liver Tumors in Mice. Gastroenterology 160: 331–45 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, et al. 2012. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herman MA, Birnbaum MJ. 2021. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hernandez-Alvarez MI, Sebastian D, Vives S, Ivanova S, Bartoccioni P, et al. 2019. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 177: 881–95 e17 [DOI] [PubMed] [Google Scholar]
- 54.Heydari M, Cornide-Petronio ME, Jimenez-Castro MB, Peralta C. 2020. Data on Adiponectin from 2010 to 2020: Therapeutic Target and Prognostic Factor for Liver Diseases? Int J Mol Sci 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, et al. 2018. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A 115: E5096–E105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horton JD, Goldstein JL, Brown MS. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of clinical investigation 109: 1125–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hou J, Zhang J, Cui P, Zhou Y, Liu C, et al. 2021. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Invest 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoyles L, Fernandez-Real JM, Federici M, Serino M, Abbott J, et al. 2018. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat Med 24: 1070–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ibrahim SH. 2021. Sinusoidal endotheliopathy in nonalcoholic steatohepatitis: therapeutic implications. Am J Physiol Gastrointest Liver Physiol 321: G67–G74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inverso D, Shi J, Lee KH, Jakab M, Ben-Moshe S, et al. 2021. A spatial vascular transcriptomic, proteomic, and phosphoproteomic atlas unveils an angiocrine Tie-Wnt signaling axis in the liver. Dev Cell 56: 1677–93 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, et al. 2019. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178: 686–98 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang L, Su H, Wu X, Shen H, Kim MH, et al. 2020. Leptin receptor-expressing neuron Sh2b1 supports sympathetic nervous system and protects against obesity and metabolic disease. Nat Commun 11: 1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jo H, Choe SS, Shin KC, Jang H, Lee JH, et al. 2013. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 57: 1366–77 [DOI] [PubMed] [Google Scholar]
- 64.Kanamori Y, Tanaka M, Itoh M, Ochi K, Ito A, et al. 2021. Iron-rich Kupffer cells exhibit phenotypic changes during the development of liver fibrosis in NASH. iScience 24: 102032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang HS, Okamoto K, Kim YS, Takeda Y, Bortner CD, et al. 2011. Nuclear orphan receptor TAK1/TR4-deficient mice are protected against obesity-linked inflammation, hepatic steatosis, and insulin resistance. Diabetes 60: 177–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, et al. 2019. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 16: 145–59 [DOI] [PubMed] [Google Scholar]
- 67.Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, et al. 2017. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab 26: 394–406 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, et al. 2010. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12: 224–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, et al. 2018. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 175: 133–45 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, et al. 2021. CD8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun 12: 4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, et al. 2008. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. International journal of molecular medicine 21: 507–11 [PubMed] [Google Scholar]
- 72.Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E. 2019. The role of the microbiome in NAFLD and NASH. EMBO Mol Med 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krenkel O, Hundertmark J, Abdallah AT, Kohlhepp M, Puengel T, et al. 2019. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut [DOI] [PubMed] [Google Scholar]
- 74.Kulozik P, Jones A, Mattijssen F, Rose AJ, Reimann A, et al. 2011. Hepatic deficiency in transcriptional cofactor TBL1 promotes liver steatosis and hypertriglyceridemia. Cell Metab 13: 389–400 [DOI] [PubMed] [Google Scholar]
- 75.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. 2014. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146: 726–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lamming DW, Sabatini DM. 2013. A Central role for mTOR in lipid homeostasis. Cell Metab 18: 465–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langlet F, Haeusler RA, Linden D, Ericson E, Norris T, et al. 2017. Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell 171: 824–35 e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. 2018. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol 69: 927–47 [DOI] [PubMed] [Google Scholar]
- 79.Lee AH, Scapa EF, Cohen DE, Glimcher LH. 2008. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320: 1492–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee JH, Wada T, Febbraio M, He J, Matsubara T, et al. 2010. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139: 653–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee YJ, Ko EH, Kim JE, Kim E, Lee H, et al. 2012. Nuclear receptor PPARgamma-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc Natl Acad Sci U S A 109: 13656–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lefebvre P, Lalloyer F, Bauge E, Pawlak M, Gheeraert C, et al. 2017. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARalpha-regulated dermatopontin. JCI Insight 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lefere S, Tacke F. 2019. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep 1: 30–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lefere S, Van de Velde F, Devisscher L, Bekaert M, Raevens S, et al. 2017. Serum vascular cell adhesion molecule-1 predicts significant liver fibrosis in non-alcoholic fatty liver disease. Int J Obes (Lond) 41: 1207–13 [DOI] [PubMed] [Google Scholar]
- 85.Li S, Brown MS, Goldstein JL. 2010. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 107: 3441–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li S, Liu C, Li N, Hao T, Han T, et al. 2008. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab 8: 105–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liang N, Damdimopoulos A, Goni S, Huang Z, Vedin LL, et al. 2019. Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARalpha. Nat Commun 10: 1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu G, Zhou L, Zhang H, Chen R, Zhang Y, et al. 2017. Regulation of hepatic lipogenesis by the zinc finger protein Zbtb20. Nat Commun 8: 14824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Y, Dou X, Zhou WY, Ding M, Liu L, et al. 2021. Hepatic Small Ubiquitin-Related Modifier (SUMO)-Specific Protease 2 Controls Systemic Metabolism Through SUMOylation-Dependent Regulation of Liver-Adipose Tissue Crosstalk. Hepatology 74: 1864–83 [DOI] [PubMed] [Google Scholar]
- 90.Liu Y, Jiang L, Sun C, Ireland N, Shah YM, et al. 2018. Insulin/Snail1 axis ameliorates fatty liver disease by epigenetically suppressing lipogenesis. Nat Commun 9: 2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Y, Lin H, Jiang L, Shang Q, Yin L, et al. 2020. Hepatic Slug epigenetically promotes liver lipogenesis, fatty liver disease, and type 2 diabetes. J Clin Invest 130: 2992–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Longuet C, Sinclair EM, Maida A, Baggio LL, Maziarz M, et al. 2008. The Glucagon Receptor Is Required for the Adaptive Metabolic Response to Fasting. Cell Metabolism 8: 359–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Loomba R, Friedman SL, Shulman GI. 2021. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184: 2537–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo X, Li H, Ma L, Zhou J, Guo X, et al. 2018. Expression of STING Is Increased in Liver Tissues From Patients With NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 155: 1971–84 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luo Y, Wadhawan S, Greenfield A, Decato BE, Oseini AM, et al. 2021. SOMAscan Proteomics Identifies Serum Biomarkers Associated With Liver Fibrosis in Patients With NASH. Hepatol Commun 5: 760–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, et al. 2019. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 25: 641–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McGettigan B, McMahan R, Orlicky D, Burchill M, Danhorn T, et al. 2019. Dietary Lipids Differentially Shape Nonalcoholic Steatohepatitis Progression and the Transcriptome of Kupffer Cells and Infiltrating Macrophages. Hepatology 70: 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, et al. 2020. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 182: 886–900 e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Montagner A, Polizzi A, Fouche E, Ducheix S, Lippi Y, et al. 2016. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 65: 1202–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, et al. 2011. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J 25: 2538–50 [DOI] [PubMed] [Google Scholar]
- 101.Mori MA, Ludwig RG, Garcia-Martin R, Brandao BB, Kahn CR. 2019. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab 30: 656–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mulder K, Patel AA, Kong WT, Piot C, Halitzki E, et al. 2021. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 54: 1883–900 e5 [DOI] [PubMed] [Google Scholar]
- 103.Munoz-Rojas AR, Mathis D. 2021. Tissue regulatory T cells: regulatory chameleons. Nat Rev Immunol 21: 597–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niu L, Geyer PE, Wewer Albrechtsen NJ, Gluud LL, Santos A, et al. 2019. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol Syst Biol 15: e8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. 2008. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab 7: 520–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Papazyan R, Sun Z, Kim YH, Titchenell PM, Hill DA, et al. 2016. Physiological Suppression of Lipotoxic Liver Damage by Complementary Actions of HDAC3 and SCAP/SREBP. Cell Metab 24: 863–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Payen VL, Lavergne A, Alevra Sarika N, Colonval M, Karim L, et al. 2021. Single-cell RNA sequencing of human liver reveals hepatic stellate cell heterogeneity. JHEP Rep 3: 100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, et al. 2015. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell 160: 745–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Perry RJ, Zhang D, Guerra MT, Brill AL, Goedeke L, et al. 2020. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 579: 279–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. 2015. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347: 1253–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, et al. 2019. Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut 68: 533–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pfister D, Nunez NG, Pinyol R, Govaere O, Pinter M, et al. 2021. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 592: 450–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Preidis GA, Kim KH, Moore DD. 2017. Nutrient-sensing nuclear receptors PPARalpha and FXR control liver energy balance. J Clin Invest 127: 1193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. 2009. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 9: 327–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, et al. 2019. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575: 512–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR, Henderson NC. 2020. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat Rev Gastroenterol Hepatol 17: 457–72 [DOI] [PubMed] [Google Scholar]
- 117.Ramos VM, Kowaltowski AJ, Kakimoto PA. 2021. Autophagy in Hepatic Steatosis: A Structured Review. Front Cell Dev Biol 9: 657389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rau M, Schilling AK, Meertens J, Hering I, Weiss J, et al. 2016. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J Immunol 196: 97–105 [DOI] [PubMed] [Google Scholar]
- 119.Remmerie A, Martens L, Thone T, Castoldi A, Seurinck R, et al. 2020. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 53: 641–57 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ren H, Hu F, Wang D, Kang X, Feng X, et al. 2021. Sirtuin 2 Prevents Liver Steatosis and Metabolic Disorders by Deacetylation of Hepatocyte Nuclear Factor 4alpha. Hepatology 74: 723–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, et al. 2000. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes & development 14: 2819–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Richter ML, Deligiannis IK, Yin K, Danese A, Lleshi E, et al. 2021. Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy. Nat Commun 12: 4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rosenthal SB, Liu X, Ganguly S, Dhar D, Pasillas MP, et al. 2021. Heterogeneity of HSCs in a Mouse Model of NASH. Hepatology 74: 667–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rui L 2014. Energy metabolism in the liver. Compr Physiol 4: 177–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rui L 2017. Brown and Beige Adipose Tissues in Health and Disease. Compr Physiol 7: 1281–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Santos-Baez LS, Ginsberg HN. 2021. Nonalcohol fatty liver disease: balancing supply and utilization of triglycerides. Curr Opin Lipidol 32: 200–06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schoiswohl G, Stefanovic-Racic M, Menke MN, Wills RC, Surlow BA, et al. 2015. Impact of Reduced ATGL-Mediated Adipocyte Lipolysis on Obesity-Associated Insulin Resistance and Inflammation in Male Mice. Endocrinology 156: 3610–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, et al. 2000. Role of LXRs in control of lipogenesis. Genes & development 14: 2831–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schwabe RF, Tabas I, Pajvani UB. 2020. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 158: 1913–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, et al. 2020. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 52: 1057–74 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S. 2020. Nonalcoholic Steatohepatitis: A Review. JAMA 323: 1175–83 [DOI] [PubMed] [Google Scholar]
- 132.Shen H, Ji Y, Xiong Y, Kim H, Zhong X, et al. 2019. Medullary thymic epithelial NF-kB-inducing kinase (NIK)/IKKalpha pathway shapes autoimmunity and liver and lung homeostasis in mice. Proc Natl Acad Sci U S A 116: 19090–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shen H, Jiang L, Lin JD, Omary MB, Rui L. 2019. Brown fat activation mitigates alcohol-induced liver steatosis and injury in mice. J Clin Invest 130: 2305–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shen H, Sheng L, Xiong Y, Kim YH, Jiang L, et al. 2017. Thymic NF-kappaB-inducing Kinase (NIK) Regulates CD4+ T Cell-elicited Liver Injury and Fibrosis in Mice. J Hepatol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, et al. 2010. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab 12: 654–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shimomura I, Bashmakov Y, Horton JD. 1999. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. The Journal of biological chemistry 274: 30028–32 [DOI] [PubMed] [Google Scholar]
- 137.Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, et al. 2020. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest 130: 1453–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Softic S, Cohen DE, Kahn CR. 2016. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig Dis Sci 61: 1282–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Srivastava J, Robertson CL, Ebeid K, Dozmorov M, Rajasekaran D, et al. 2017. A novel role of astrocyte elevated gene-1 (AEG-1) in regulating nonalcoholic steatohepatitis (NASH). Hepatology 66: 466–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sun C, Jiang L, Liu Y, Shen H, Weiss SJ, et al. 2016. Adipose Snail1 Regulates Lipolysis and Lipid Partitioning by Suppressing Adipose Triacylglycerol Lipase Expression. Cell Rep 17: 2015–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sun G, Jin H, Zhang C, Meng H, Zhao X, et al. 2018. OX40 Regulates Both Innate and Adaptive Immunity and Promotes Nonalcoholic Steatohepatitis. Cell Rep 25: 3786–99 e4 [DOI] [PubMed] [Google Scholar]
- 142.Sun Z, Miller RA, Patel RT, Chen J, Dhir R, et al. 2012. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med 18: 934–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sunny NE, Parks EJ, Browning JD, Burgess SC. 2011. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 14: 804–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sutti S, Albano E. 2020. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 17: 81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tardelli M, Bruschi FV, Trauner M. 2020. The Role of Metabolic Lipases in the Pathogenesis and Management of Liver Disease. Hepatology 72: 1117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Terkelsen MK, Bendixen SM, Hansen D, Scott EAH, Moeller AF, et al. 2020. Transcriptional Dynamics of Hepatic Sinusoid-Associated Cells After Liver Injury. Hepatology 72: 2119–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tran S, Baba I, Poupel L, Dussaud S, Moreau M, et al. 2020. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity 53: 627–40 e5 [DOI] [PubMed] [Google Scholar]
- 148.Tsuchida T, Friedman SL. 2017. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14: 397–411 [DOI] [PubMed] [Google Scholar]
- 149.Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, et al. 2014. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63: 3279–94 [DOI] [PubMed] [Google Scholar]
- 150.Ulrich JD, Ulland TK, Colonna M, Holtzman DM. 2017. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 94: 237–48 [DOI] [PubMed] [Google Scholar]
- 151.Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, et al. 2013. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 62: 899–910 [DOI] [PubMed] [Google Scholar]
- 152.Vandel J, Dubois-Chevalier J, Gheeraert C, Derudas B, Raverdy V, et al. 2021. Hepatic Molecular Signatures Highlight the Sexual Dimorphism of Nonalcoholic Steatohepatitis (NASH). Hepatology 73: 920–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.von Meyenn F, Porstmann T, Gasser E, Selevsek N, Schmidt A, et al. 2013. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab 17: 436–47 [DOI] [PubMed] [Google Scholar]
- 154.Wang GX, Zhao XY, Lin JD. 2015. The brown fat secretome: metabolic functions beyond thermogenesis. Trends Endocrinol Metab 26: 231–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang GX, Zhao XY, Meng ZX, Kern M, Dietrich A, et al. 2014. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nat Med 20: 1436–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, et al. 2021. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol [DOI] [PubMed] [Google Scholar]
- 157.Wang XA, Zhang R, Jiang D, Deng W, Zhang S, et al. 2013. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology 58: 603–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang Y, Viscarra J, Kim SJ, Sul HS. 2015. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol 16: 678–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang ZY, Keogh A, Waldt A, Cuttat R, Neri M, et al. 2021. Single-cell and bulk transcriptomics of the liver reveals potential targets of NASH with fibrosis. Sci Rep 11: 19396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. 2016. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 157: 570–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, et al. 2014. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26: 549–64 [DOI] [PubMed] [Google Scholar]
- 162.Xiong X, Kuang H, Ansari S, Liu T, Gong J, et al. 2019. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell 75: 644–60 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xiong X, Kuang H, Liu T, Lin JD. 2020. A Single-Cell Perspective of the Mammalian Liver in Health and Disease. Hepatology 71: 1467–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xiong X, Wang Q, Wang S, Zhang J, Liu T, et al. 2019. Mapping the molecular signatures of diet-induced NASH and its regulation by the hepatokine Tsukushi. Mol Metab 20: 128–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yang J, Fu Z, Zhang X, Xiong M, Meng L, Zhang Z. 2020. TREM2 ectodomain and its soluble form in Alzheimer’s disease. J Neuroinflammation 17: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, et al. 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101 [DOI] [PubMed] [Google Scholar]
- 167.Yu Y, Liu Y, An W, Song J, Zhang Y, Zhao X. 2019. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest 129: 546–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang D, Tong X, Nelson BB, Jin E, Sit J, et al. 2018. The hepatic BMAL1/AKT/lipogenesis axis protects against alcoholic liver disease in mice via promoting PPARalpha pathway. Hepatology 68: 883–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang H, Ma Y, Cheng X, Wu D, Huang X, et al. 2021. Targeting epigenetically maladapted vascular niche alleviates liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med 13: eabd1206. [DOI] [PubMed] [Google Scholar]
- 170.Zhang Q, He Y, Luo N, Patel SJ, Han Y, et al. 2019. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179: 829–45 e20 [DOI] [PubMed] [Google Scholar]
- 171.Zhang X, Wang Y, Liu P. 2017. Omic studies reveal the pathogenic lipid droplet proteins in non-alcoholic fatty liver disease. Protein Cell 8: 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, et al. 2020. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579: 586–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, et al. 2006. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem 281: 15013–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou W, Gross KM, Kuperwasser C. 2019. Molecular regulation of Snai2 in development and disease. J Cell Sci 132 [DOI] [PMC free article] [PubMed] [Google Scholar]