

CONSPECTUS:
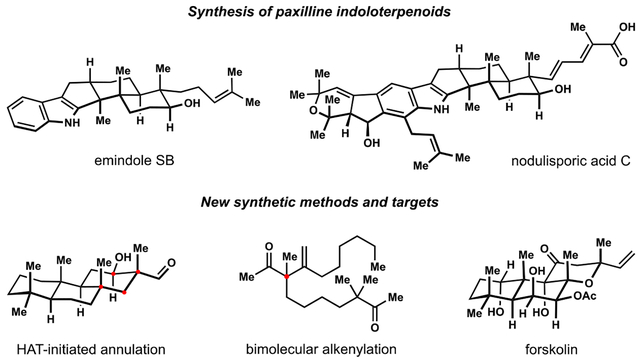
Indoloterpenoids of the paxilline type belong to a large family of secondary metabolites that exhibit unique molecular architectures and a diverse set of biological activities. More than 100 congeners identified to date share a common structural motif that contains an indole moiety fused to a rearranged diterpenoid fragment. The representative physiological and cellular effects attributed to this family of natural products include neurological and insecticidal activities, modulation of lipid balance, and inhibition of mitosis. The uniting polycyclic motif combined with the diversity of individual structural features of paxilline indoloterpenoids and the broad scope of their biological activities have fascinated organic chemists for the past four decades and have led to the development of numerous syntheses. In this Account, we describe our contributions to this field and how they in turn shape new directions that are developing in our laboratory.
We begin with the discussion of our strategy for the synthesis of the shared indoloterpenoid core. To address stereochemical challenges encountered in earlier reports, we planned to leverage a suitably substituted cyclopentanone in a polycyclization to form the desired trans-decalin motif. This polycyclization relied on a radical-polar crossover cascade initiated by hydrogen atom transfer. The original process exhibited poor diastereoselectivity, but we discovered an efficient solution to this problem that took advantage of intramolecular tethering effects, culminating in short synthesis of emindole SB. During these studies, we also identified indium-mediated alkenylation of silyl enol ethers with alkynes as a suitable method for the synthesis of highly substituted β,γ-unsaturated ketones that was critical to achieving brevity of our route. We subsequently developed a catalytic version of this transformation that allowed for a formal bimolecular ene reaction that exhibited unusual and potentially useful selectivity in construction of quaternary centers.
To test the scope and limitations of our approach to paxilline indoloterpenoids and identify potential improvements, we developed a synthesis of the more complex congener nodulisporic acid C. The convergent assembly of this natural product was enabled by identification of new elements of stereocontrol in the radical-polar crossover polycyclization en route to the polycyclic terpenoid motif and development of a highly diastereoselective enyne cycloisomerization to access the indenopyran motif and a ketone arylation protocol to unite the two complex fragments.
In subsequent studies, we expanded the radical-polar crossover cascade underlying our approach to paxilline indoloterpenoids to a bimolecular setting, which allowed for annulation of two unsaturated carbonyl components to produce functionalized cyclohexanes. This transformation is particularly well suited for installation of fully substituted carbons and can be complementary to the venerable Diels–Alder reaction. The utility of the new annulation was tested in the synthesis of forskolin, allowing for rapid construction of the complex polycyclic motif in this densely functionalized labdane diterpenoid.
Over the past five years, our initial forays into the synthesis of paxilline indoloterpenoids have grown into a program that incorporates development of new synthetic methods and pursues artificial assembly of terpenoid natural products from several different families. We are encouraged by the increasing diversity of structural motifs made accessible by application of this chemistry and continue to discover new aspects of the underlying reactivity.
Graphical Abstract
INTRODUCTION
Chemical synthesis of natural products and other complex molecules provides a continuous supply of scientific innovations. These advances include development of new biochemical tools and chemotherapeutic agents and design of new synthetic strategies and chemical methods.5 The interdisciplinary nature of the former can lead to broad recognition by the scientific community, but the impact of the latter is no less profound and becomes apparent after careful analysis of subsequent applications. Natural products exhibit an astonishing diversity of structural motifs and offer a seemingly endless supply of synthetic challenges that cannot be altered in the academic exercise of total synthesis, but only surmounted through ingenuity, driving the development of new solutions.6 Examples are numerous and range from invention of protecting groups to discovery of new reactivity patterns, inspiring generations of chemists to embark upon studies in the synthesis of natural products.5b,7,8
Over the past five years, our laboratory has reported efforts in the synthesis of several secondary metabolites (Figure 1).1,3,4,9 The pursuit of these largely unrelated compounds was driven by their distinct structural motifs and the associated synthetic challenges, both the readily apparent ones and those identified in previous studies. Our investigations have prompted discovery of new methods to construct some of the key bonds in the target compounds that can in turn enable new synthetic approaches to other natural products.2,4,10 In the following sections, we discuss studies related to paxilline indoloterpenoids and highlight causal relationships between separate projects.
Figure 1.
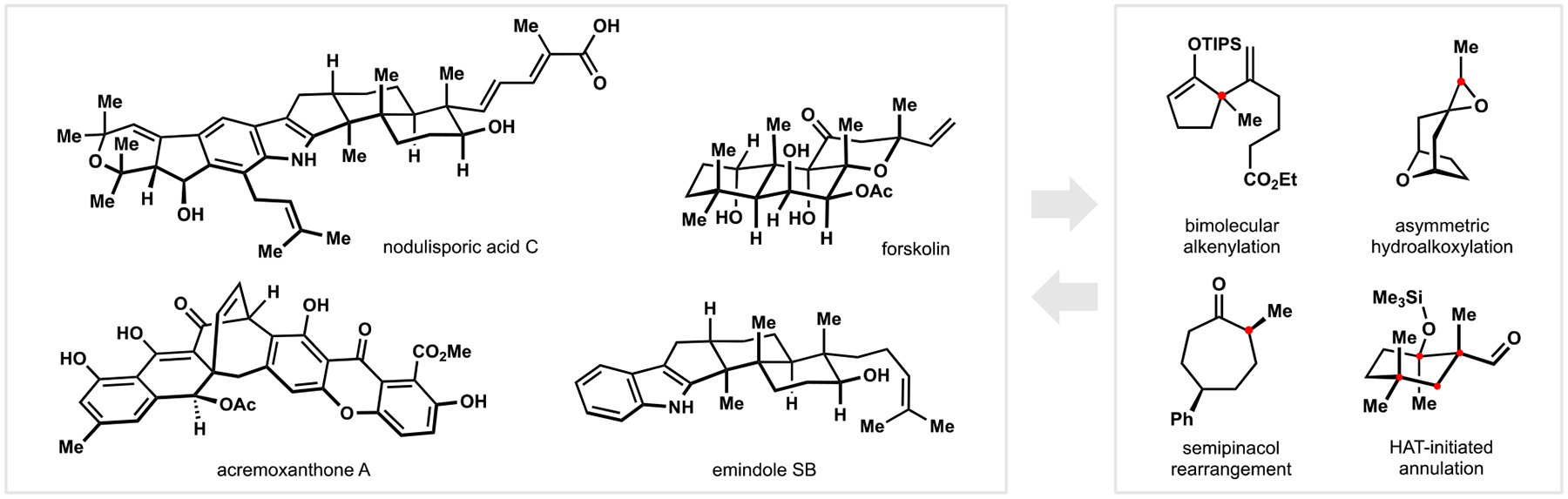
Examples of natural products (left) whose syntheses led to the development or were enabled by new chemical methods (right) in the Pronin laboratory (2015–2019; red dots highlight carbon atoms involved in key bond-forming events).
Indoloterpenoids of paxilline type constitute a large group of fungal metabolites of terrestrial origin.11 For a single family of natural products, these compounds exhibit impressive diversity of documented biological effects, although in many cases the exact molecular mechanism of action remains unknown and it is not clear whether the underlying cellular interactions have uniting traits.12 The individual congeners are readily identified by the presence of unique connectivity pattern in the shared tricarbocyclic motif, which, together with the indole fragment, can contain a variety of substituents and functional group arrays (Scheme 1; highlighted in red). Studies in the biosynthesis of paxilline indoloterpenoids indicate that the common core originates from an unusual cationic polycyclization of a prenylated indole derivative.13,14 This transformation involves an apparent anti-Markovnikov cyclization of the prenyl subunit proximal to the indole fragment followed by an alkyl shift and subsequent nucleophilic capture of the resulting tertiary carbocation, which produces a strained hexahydroindene motif containing vicinal quaternary stereocenters. Attempts to mimic the polycyclization event in a chemical reaction have yet to achieve the desired connectivity pattern.15 Recent study of a relevant intramolecular alkylation of an indole by the Newhouse group demonstrated success in forging the corresponding quaternary center in their remarkably short synthesis of paspaline but also echoed previous observations that alternative, undesired connectivity patterns were prevalent among the products of these chemical reactions.16
Scheme 1.
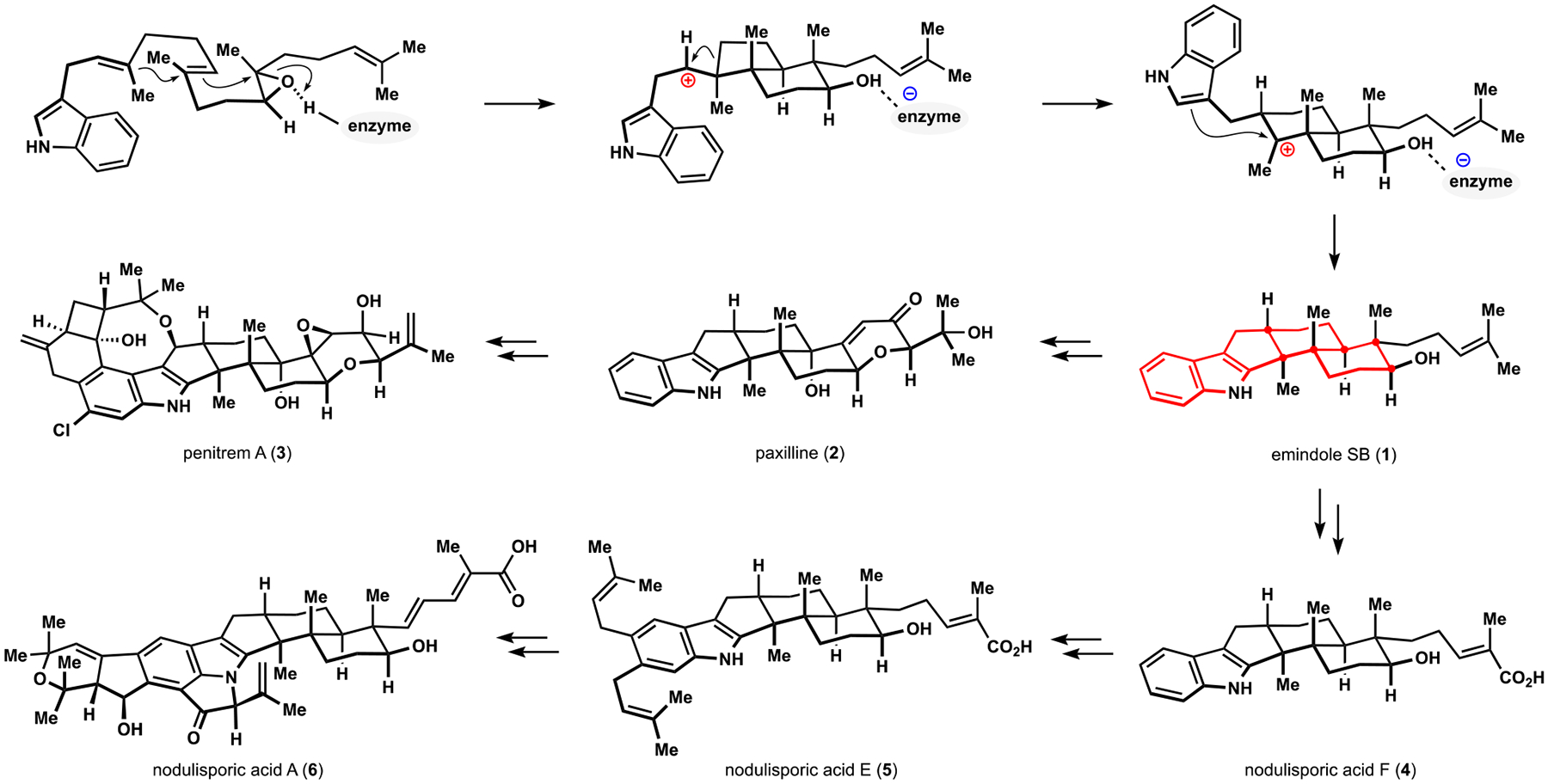
Representative Biosynthetic Pathways to Paxilline Indoloterpenoids That Highlight Structural Diversity of Congeners
Nearly four decades of fascination with the structural features and biological activities of the paxilline indoloterpenoids among organic chemists have led to the development of numerous impressive syntheses and approaches to different congeners. These advances were championed by extensive and remarkable efforts from the Smith group that yielded several strategies for the assembly of the indoloterpenoid motif and culminated in the landmark synthesis of penitrem D and, more recently, nodulisporic acids B, C, and D.17 Early studies from the Saxton group established an efficient and highly diastereoselective protocol for construction of the vicinal quaternary centers in the shared terpenoid fragment.18 Variants of Saxton’s approach were subsequently employed in several efforts by other laboratories, including the syntheses reported by the Kuwahara group.19 An alternative approach to the highly stereocontrolled assembly of the hexahydroindene motif was disclosed by the Johnson group and employed clever desymmetrization tactics to install several stereocenters and structural fragments in their synthesis of paspaline.20 Notably, comparison of these synthetic routes using various metrics such as step count, number of functional group interconversions, and redox and protecting group manipulations suggests that the artificial assembly of the paxilline indoloterpenoids remains a significant challenge. While this observation motivated initiation of our own inquiries, the tremendous knowledge of reactivity accumulated during the above-mentioned investigations also had a significant impact on our synthetic planning and is certain to inform future efforts in this area.
SYNTHESIS OF EMINDOLE SB
Our initial investigations focused on the development of a concise and stereoselective assembly of the shared terpenoid fragment represented by ketoaldehyde 7 (Scheme 2). This intermediate was expected to provide rapid access to several congeners of interest. Recognizing that formation of the trans-hexahydroindene motif had often been a stumbling block in the previous syntheses,17b,19,20 we sought to establish the desired stereochemical relationship at the fusion bond prior to constructing the remainder of the polycyclic core. To this end, we planned to build the trans-decalin motif starting with a properly substituted cyclopentanone. We were particularly intrigued by the possibility of performing intramolecular addition of a tertiary alkyl radical 9 derived from 1,1-disubstituted alkene 8 to the pendant enal motif followed by an aldol reaction, or its equivalent, to complete the desired double cyclization. This proposal was inspired by the timely report from the Baran group who demonstrated Giese addition of alkyl radicals, which were generated by chemoselective hydrogen atom transfer (HAT) to alkenes from putative iron hydrides.21 The Baran group proposed that the conjugate addition was followed by reduction of the resulting enol radical to generate the corresponding enolate, which we in turn intended to intercept with an intramolecular aldol reaction of intermediate 10. Notable challenges associated with implementation of the proposed radical-polar crossover cascade included formation of the quaternary stereocenter during the conjugate addition and rapid access to the polycyclization precursor. The requisite β,γ-unsaturated ketone was expected to arise from alkenylation of an enolate, or its equivalent, derived from a substituted cyclopentanone. While only limited precedent existed for selective formation of quaternary centers from unsymmetrical dialkyl ketones,22 we were encouraged by the emergence of new and highly relevant cross-coupling protocols.23
Scheme 2.
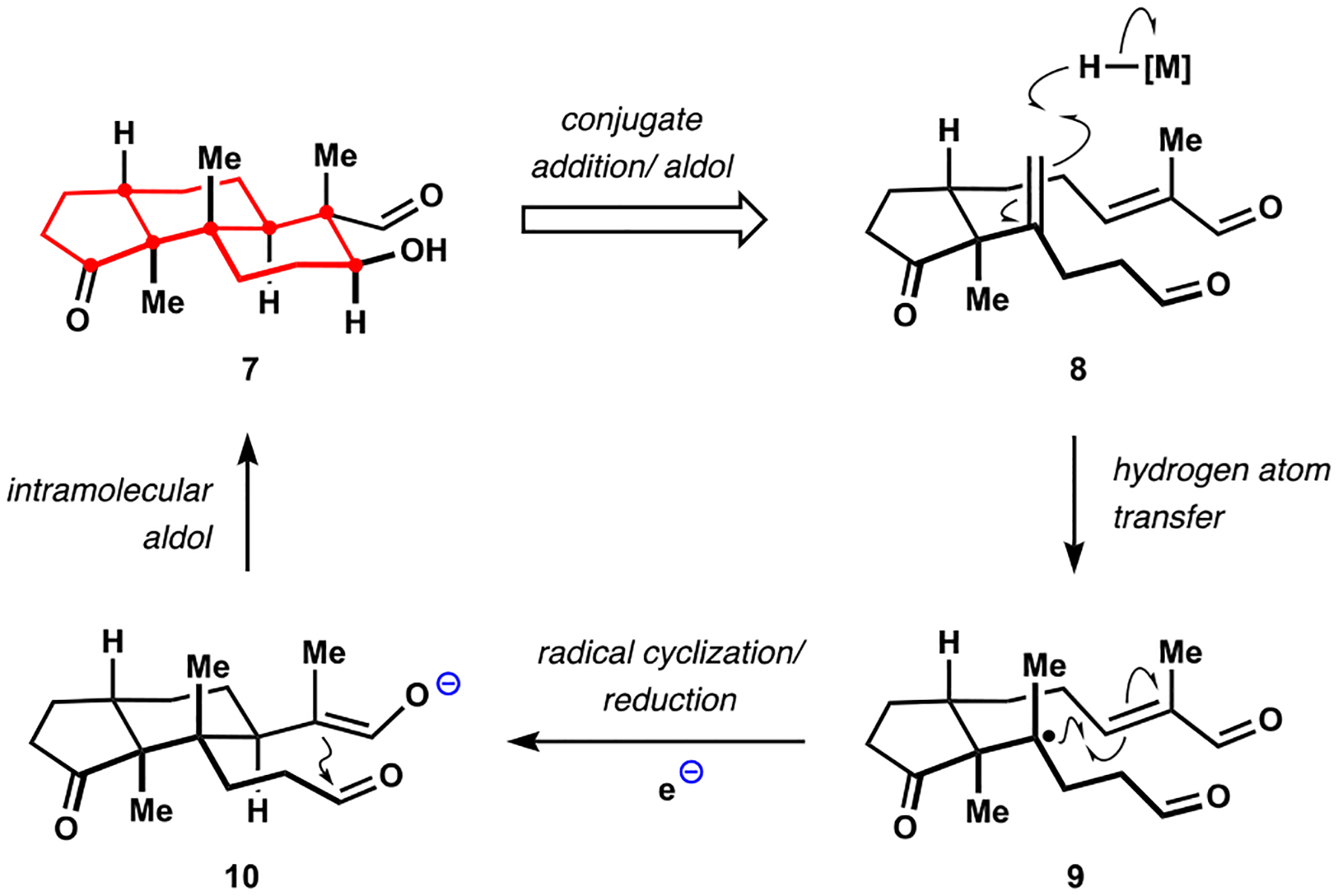
Our Approach to the Common Terpenoid Motif
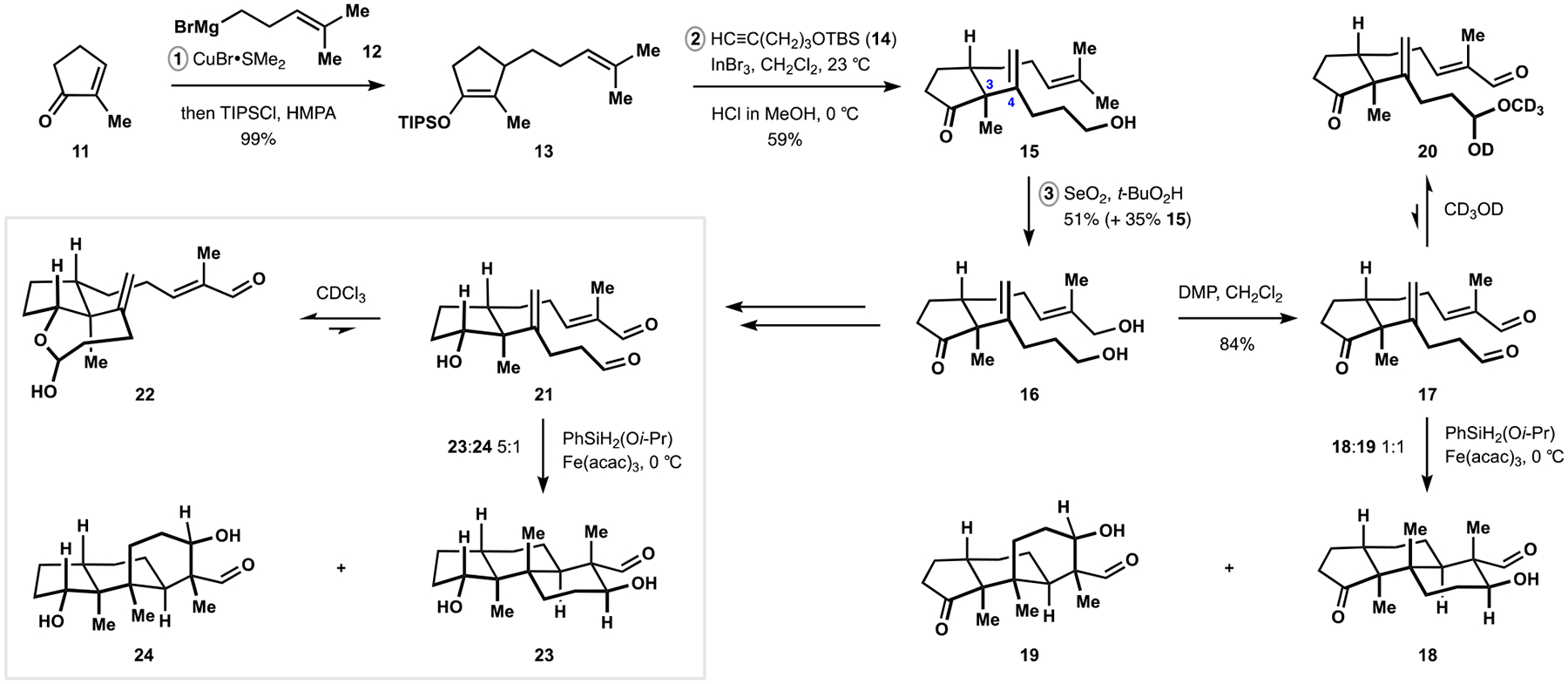
We began with a copper-catalyzed conjugate addition of homoprenyl Grignard reagent 12 to commercially available methylcyclopentenone 11 and trapping the resulting magnesium enolate as silyl enol ether 13 (Scheme 3).1 After extensive experimentation, we identified carboindation of protected pentynol 14 with triisopropylsilyl enol ether 13 and indium tribromide as suitable means for construction of the C3–C4 bond, providing access to ketone 15 on multigram scale. This transformation was based on work from the Baba group who reported related alkenylations of ketene silyl acetals and demonstrated intermediacy of alkenylindium derivatives.24 Sharpless allylic hydroxylation of intermediate 15 followed by double oxidation of diol 16 completed construction of the polycyclization precursor.25
Scheme 3.
Synthesis of the Polycyclization Precursor and Discovery of the Tethering Effect
Subjection of dialdehyde 17 to the conditions of iron-catalyzed HAT in the presence of (isopropoxy)phenylsilane induced the desired bond-forming events but also resulted in equimolar mixture of trans- and cis-decalins 18 and 19. While we were unable to improve the ratio of products to any significant extent, we also discovered that the aliphatic aldehyde fragment in intermediate 17 underwent complete hemiacetalization in alcohols. We thought that introduction of a hydroxy group in the cyclopentane fragment could allow for transient intramolecular tethering of aliphatic aldehyde through the reversible formation of the corresponding lactol. This tethering could restrict the available conformations of the disubstituted alkene as well as the corresponding tertiary alkyl radical and allow for enhanced diastereocontrol during the initial cyclization event.
Our studies with hydroxydialdehyde 21 revealed a significant extent of lactolization in solutions and its superior performance in the polycyclization, delivering good diastereoselectivity and preference for the desired trans-decalin 23. Blocking lactol formation by methylation of the secondary alcohol or conducting the reaction in ethanol resulted in deterioration of diastereomeric ratios, suggesting that the transient tethering was contributing to the improved outcomes. Application of (isopropoxy)phenylsilane, identified by the Shenvi group as an exceptional hydride source in HAT-mediated reactions, was critical to the success of our investigations as it allowed us to perform the polycyclization in a broad range of solvents and temperature regimes.26
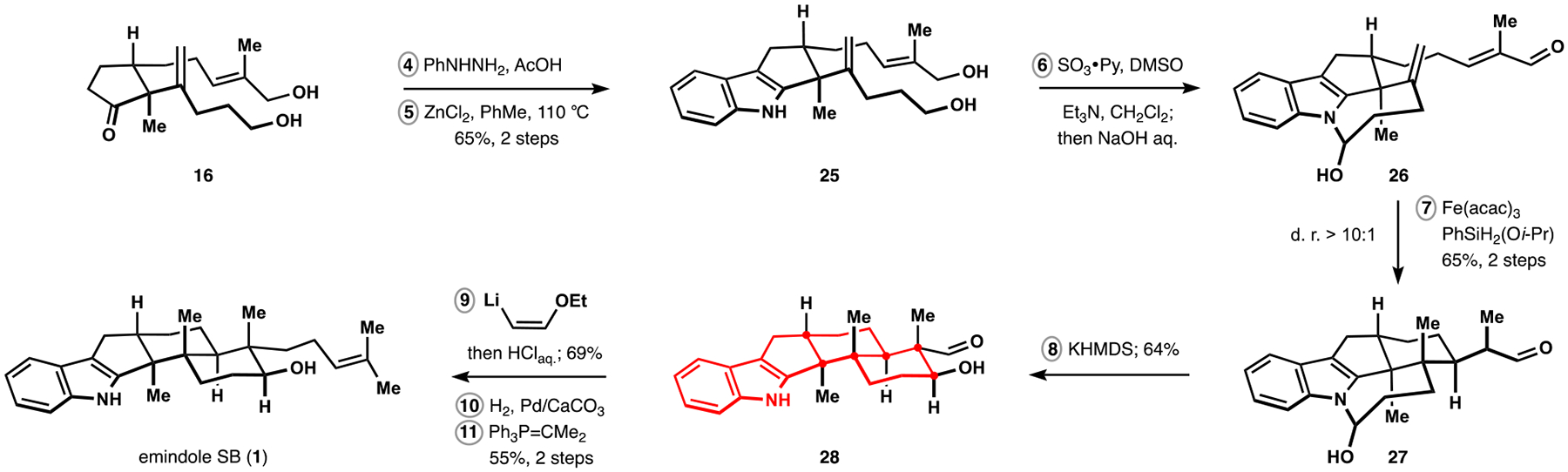
Encouraged by these developments, we sought to leverage the tethering effect while avoiding the need for multiple functional group interchanges imposed by cyclopentanol 21 during subsequent elaborations. With this in mind, Parikh–Doering oxidation of diol 25, prepared by Fischer indole synthesis from ketone 16 according to the Wood protocol, followed by treatment of the reaction mixture with mild aqueous base resulted in direct formation of hemiaminal 26 (Scheme 4).27,28 In accord with our findings in the polycyclization of hydroxydialdehyde 21, radical cyclization of substrate 26 provided efficient and highly diastereoselective entry into the desired configuration of the vicinal quaternary centers. At the same time, stability of the hemiaminal motif precluded complete propagation of the desired cascade and required treatment of aldehyde 27 with base to induce the final cyclization. The choice of metal counterion was critical to the success of the intramolecular aldol reaction with potassium showing optimal performance. Ultimately, indole 28 was converted to the simplest member of paxilline indoloterpenoids, emindole SB (1), in three straightforward operations, providing a proof of concept for application of our synthetic approach.29 In this final sequence, application of the Schlosser homologation was critical to the success of our efforts because it avoided epimerization of the aldol motif that was observed with other olefination methods.30
Scheme 4.
Synthesis of Emindole SB
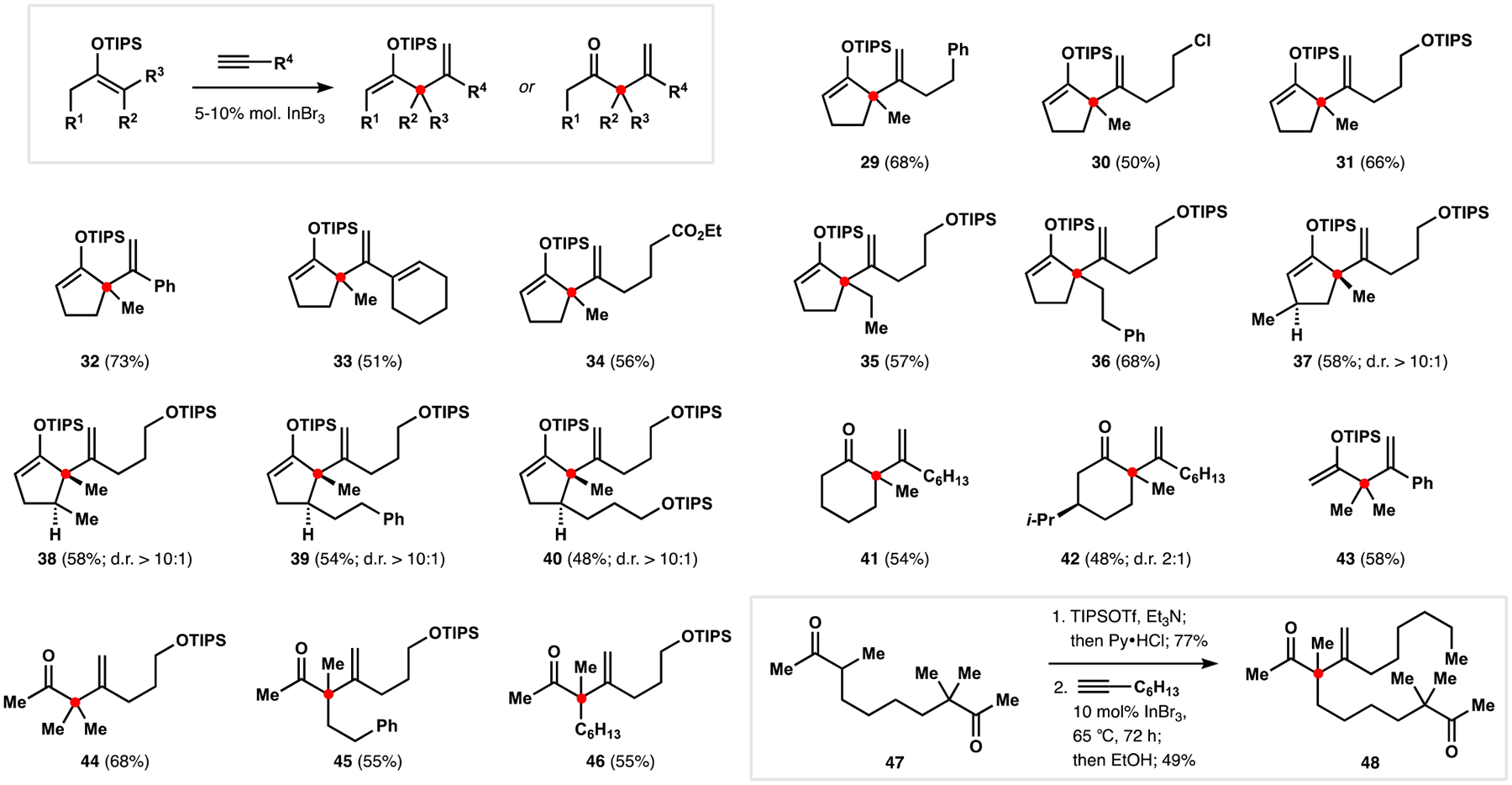
To improve upon the alkenylation protocol en route to unsaturated ketone 15 and its potential for application outside our synthetic efforts, we sought to develop a catalytic version of this transformation. Our initial attempts to effect turnover with protic polar cosolvents, akin to gold-catalyzed intramolecular alkenylations developed by the Toste group, resulted in nearly complete loss of the desired reactivity, which could not be rescued by changing the Lewis acid to those commonly employed in electrophilic activation of alkynes.31 During these studies, we also observed small amounts of 2-siloxy-1,4-diene in addition to the expected β,γ-unsaturated ketone in reactions with stoichiometric indium tribromide that suggested a possibility of turnover in the original system. After extensive optimization, we found that catalytic amounts of the same Lewis acid provided optimal performance in promoting formal bimolecular ene reaction upon heating of ketone-derived silyl enol ethers and terminal alkynes (compounds 29–46; Scheme 5).2 This transformation exhibited unusual selectivity for the construction of quaternary centers from unsymmetrical dialkyl ketones that prevented participation of products in a second alkenylation event and was most evident in derivatization of substrate 47 to produce enedione 48. The resulting 2-siloxy-1,4-dienes were readily hydrolyzed upon treatment with aqueous acid, providing a new approach to the catalytic synthesis of β,γ-unsaturated ketones that was complementary to the previously established protocols.32 While it did not offer improvements to our construction of unsaturated ketone 15, the reaction tolerated a variety of functionalities and exhibited appreciable substrate scope in preparation of challenging structural motifs. Notable, though not unexpected, limitations were diminished reactivity with substrates containing Lewis basic functionalities, including nitriles, amides, and sulfonamides, and competing side reactions of aldehydes, presumably involving Mukaiyama aldol pathways.
Scheme 5.
Catalytic Bimolecular Alkenylation of Ketone-Derived Silyl Enol Ethers with Terminal Alkynes
SYNTHESIS OF NODULISPORIC ACID C
Our reliance on the hemiaminal fragment to achieve diastereoselective construction of the polycyclic core posed a significant obstacle in the development of a general approach to the paxilline indoloterpenoids because it required early installation of the indole moiety, which precluded convergent assembly of complex congeners. In contrast, the originally planned application of tricyclic ketone 7 as the common precursor to selected targets was devoid of these limitations (see Scheme 2). Notably, a report from the Smith group that appeared during our studies featured the same intermediate and underscored its versatility in the synthesis of nodulisporic acid D.17b While polycyclization of hydroxydialdehyde 21 showed promise in accessing intermediates related to tricyclic ketone 7 (see Scheme 3), the expected need for protecting and functional group interchanges during subsequent manipulations prompted us to develop alternative solutions that came to fruition in our synthesis of nodulisporic acid C.
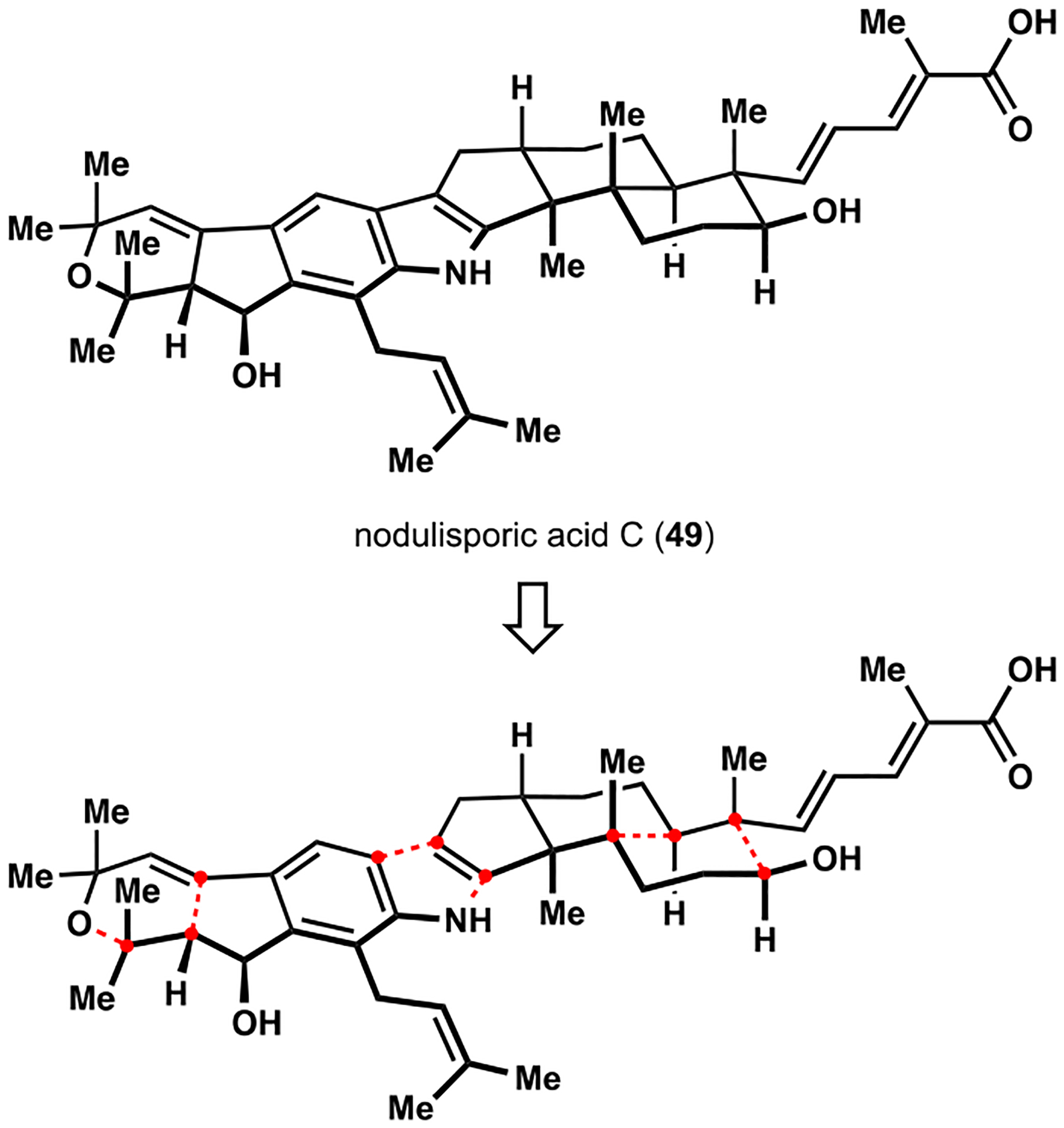
Nodulisporic acids were isolated from an endophytic fungus by Merck Laboratories during their search for new insecticidal agents.33 These indoloterpenoids exhibit potent activity against several ectoparasites, which has been attributed to specific activation of a subset of ligand-gated chloride ion channels found in arthropods.34 The magnitude of biological effects appears to correlate positively with structural complexity of the congeners, and the flagship member of the family, nodulisporic acid A (6; see Scheme 1), has demonstrated high efficacy against fleas while lacking overt toxicity in dogs.35 Ultimately, medicinal chemistry efforts have resulted in promising lead compounds for the development of new antiflea medications for companion animals.36
We chose nodulisporic acid C (49) as our primary target, partly because we were interested in elucidating its potential synthetic relationship to nodulisporic acid A and planned to access the indole motif in the final steps of the synthesis (Scheme 6). Notably, existing reports from the Smith group emphasized the challenge associated with similar late-stage manipulations but also identified an efficient solution to the problem that relied on the Barluenga indole synthesis.17b,c,37 Assembly of the tricarbocyclic terpenoid fragment was expected to take advantage of our previously developed strategy with appropriate modifications to address the stereochemical issues while preserving the requisite arrangement of functional groups. We also envisioned a cycloisomerization of a properly disposed enyne moiety to construct the indenopyrane motif, which is unique to several indoloterpenoids of the paxilline type. An earlier report by the Fensterbank group documented two instances of formation of dihydropyrans in gold-catalyzed reactions of functionalized enynes.38 However, the Sanz group subsequently demonstrated a strong preference for an alternative cyclization pattern in phenylacetylenes related to our study, precluding definitive prediction of the regiochemical outcome in the proposed cascade.39
Scheme 6.
Our Approach to Nodulisporic Acid C
Building on the polycyclization of hydroxydialdehyde 21 (see Scheme 3), we prepared and evaluated other derivatives containing properly disposed hydroxyl groups in the cyclopentane fragment.3 Notably, the radical-polar crossover cascade with participation of cyanohydrine 50 produced encouraging levels of diastereocontrol in the preparation of aldol 51 (Scheme 7).
Scheme 7.
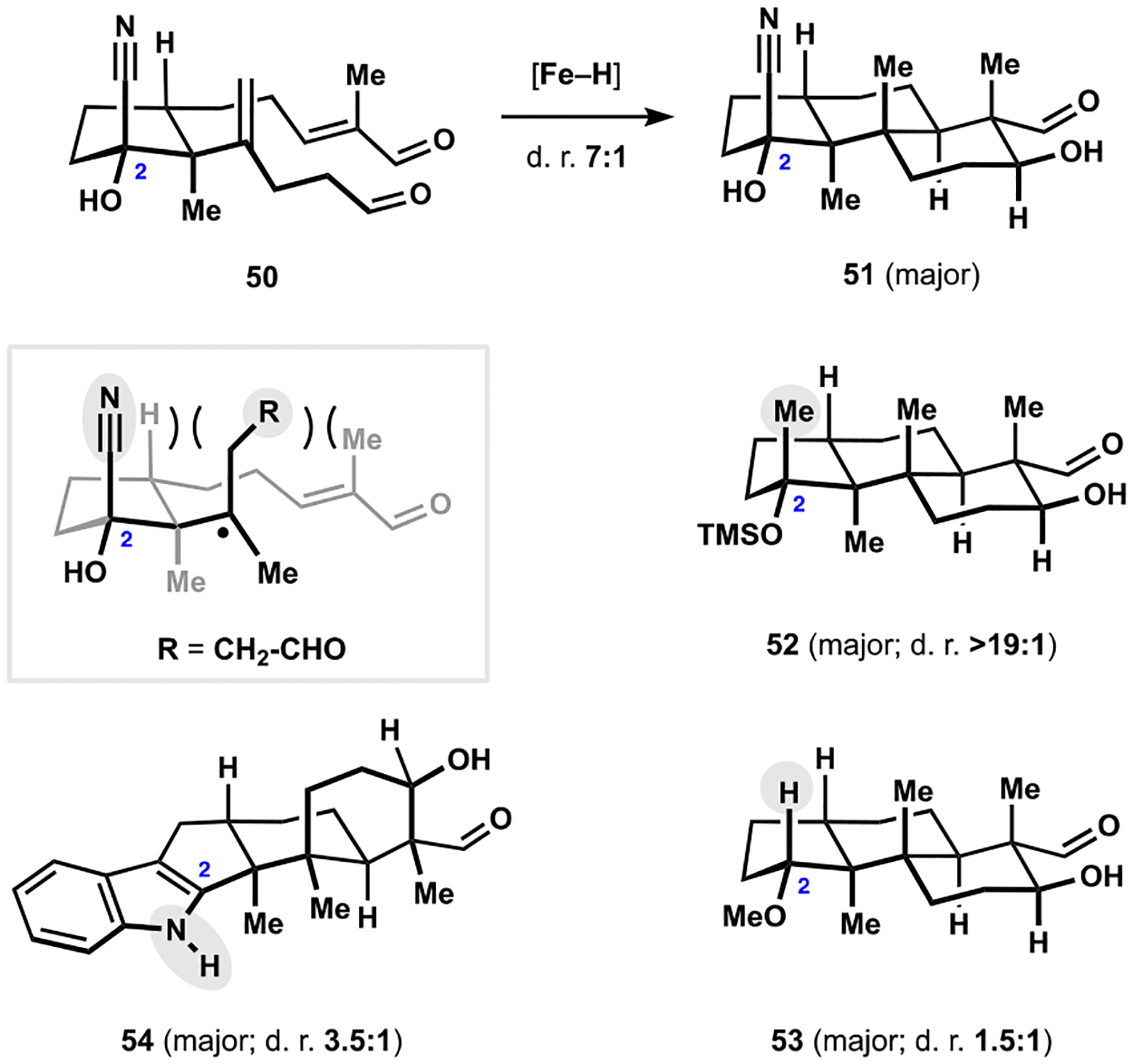
Diastereocontrol in Radical-Polar Crossover Polycyclizations
Spectroscopic analysis of the starting material did not detect any appreciable lactolization in various solvents, suggesting that different factors imparted the desired selectivity profile. Further studies revealed that presence of additional substituents at C2 had a significant effect on the outcome of the reaction. For example, desired trans-decalin 52 was formed as the only detectable product in polycyclization of the corresponding dialdehyde, which was in stark contrast to the low diastereoselectivity observed in the synthesis of derivative 53 lacking a fully substituted C2 position. We attributed the improved selectivity in the initial cyclization event to the pseudodiaxial interactions between the substituents at C2 and the radical center that, in the undesired transition state, would force rotation of the formylethyl fragment toward the enal incurring a steric penalty not associated with the methyl substituent being oriented in this pseudoaxial position. In a similar vein, the predominant formation of cis-decalin 54 in the polycyclization of the corresponding indole can be viewed as manifestation of similar steric interactions resulting from the NH-group en route to the desired pentacyclic motif.
Our synthesis of nodulisporic acid C required enantioselective access to silyl enol ether 13 (Scheme 8). Guided by studies from the Minnaard group, we identified Josiphos derivative 55 as a suitable ligand for asymmetric conjugate addition of alkylmagnesium bromide 12 to cyclopentenone 11, which produced the desired product in excellent yield and in a scalable manner.40,41 Although only moderate stereoinduction was achieved, we expected to be able to improve the levels of enantiopurity at later stages via recrystallization of suitable intermediates (see below). Following the alkenylation of silyl enol 13, the resulting cyclopentanone was derivatized to protected cyanohydrin 56, which was subsequently converted to dialdehyde 57 according to the previously established sequence of oxidations. In agreement with our findings in polycyclizations of related substrates, subjection of siloxynitrile 57 to the conditions of iron-catalyzed HAT followed by sequential treatment of the reaction mixture with aqueous acid and base provided highly diastereoselective access to cyclopentanone 7. Notably, attempts at direct olefination of this intermediate were unsuccessful and led to partial epimerization of the secondary alcohol. As a corollary, this observation also highlighted the advantages of the polycyclization cascade over stepwise construction of the sensitive aldol motifs. Tricyclic intermediate 7 was eventually protected as the corresponding pivalate, which allowed for recrystallization to provide material with significantly enhanced enantioenrichment, and successful Horner–Wadsworth–Emmons olefination with allylic phosphonate 58 completed construction of denoted fragment 59 in eight steps from commercially available cyclopentenone 11.
Scheme 8.
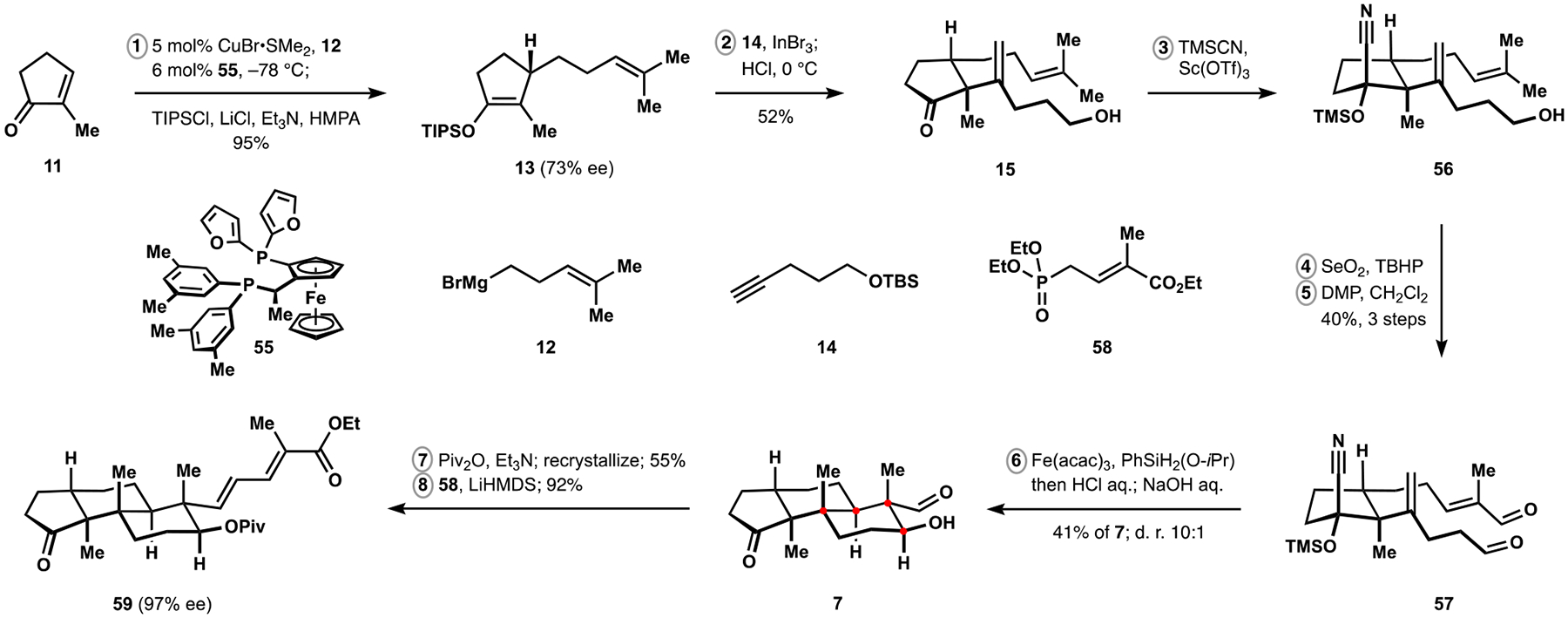
Synthesis of the Dienoate Fragment of the Nodulisporic Acids
Our synthesis of the indenopyran fragment required careful choreography during the assembly of the desired polycyclization precursor to achieve efficient and selective installation of substituents. After several iterations, we identified benzaldehyde 61 as a suitable starting point for our elaborations (Scheme 9). This intermediate was accessible in three steps from aniline 60 and underwent an efficient enantioselective addition of alkenylzinc 62 in the presence of Nugent’s ligand 63 according to the procedure developed by the Walsh group.42,43 Sonogashira cross-coupling of dibromide 64 proceeded regioselectively, and protection of the allylic alcohol as a silyl ether set the stage for construction of the indenopyran motif. Any modifications to the order of events and identity of the halogen substituents resulted in a precipitous loss of regioselectivity and efficiency in the synthesis of enyne 66. The vast body of literature on relevant transformations allowed us to quickly identify an appropriate catalyst for the desired cycloisomerization.44 In practice, treatment of substrate 66 with a cationic gold complex allowed for efficient construction of tricyclic product 67 without any detectable formation of the alternative cyclization patterns (see discussion above). In a simplified representation, this reaction involves nucleophilic attack of the isobutenyl fragment at the alkyne–gold complex in intermediate 70 followed by trapping of the resulting tertiary carbocation 71 with the pendant hydroxyl group and subsequent proton transfer to regenerate the catalyst. We also observed that performing the reaction with rigorous exclusion of moisture led to accumulation of hydroxyallene 72, which was rectified by adding small amounts of water to the reaction mixture. The robust protecting group at the allylic alcohol proved necessary for achieving high efficiency of the polycyclization. The striking preference for the desired configuration at the newly formed stereocenter prompted our investigation into the origins of selectivity. We eventually found that the presence of the substituent at C26 was crucial to obtaining high levels of diastereocontrol in a broad range of substrates evaluated in this transformation. We attributed the observed selectivity to the allylic strain between the benzyl ether and the ortho substituent, which can be expected to force rotation of the silyl group toward the alkene, increasing steric interactions with the propylidene group in the undesired transition state of the initial cyclization and reducing contribution of this pathway (see products 73 and 74). Chemoselective Stille cross-coupling of aryl bromide 67 completed construction of the indenopyran fragment found in nodulisporic acid C and set the stage for the final assembly–a challenging task that ultimately identified and leveraged unusual reactivity profiles in these complex systems.45
Scheme 9.
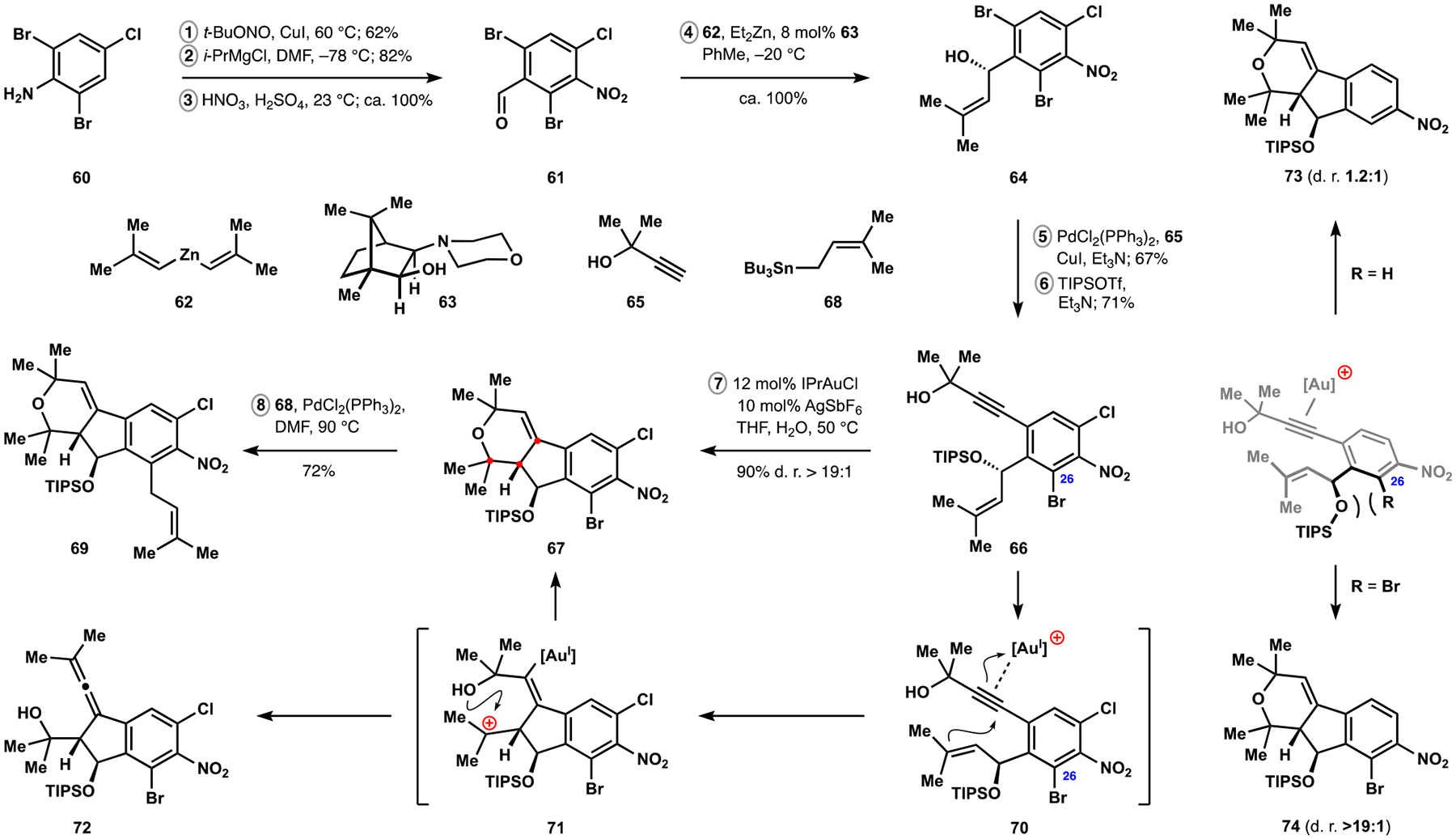
Synthesis of the Indenopyran Fragment of Nodulisporic Acid C
Our attempts at fragment union initially led to utter frustration with the inability to form either of the structural bonds upon application of relevant, previously developed protocols and appropriate precursors, including the variant of the Barluenga indole assembly developed by the Smith group in their synthesis of nodulisporic acid D.17b Small amounts of desired products were eventually observed during palladium-catalyzed cross-coupling of an enolate derived from ketone 59 with aryl chloride 69 (Scheme 10). Optimization efforts identified several conditions that led to increased production of the arylation products, which were formed with broadly varying levels of diastereocontrol. Having originally discounted the selectivity as inconsequential for the subsequent transformations, we were surprised to discover that the epimeric nitroaryl ketones displayed dramatically different reactivity patterns during the reductive cyclization to the desired indole. Thus, reduction of diastereomer 76 produced only aminoketone 77, and subsequent dehydration required forcing conditions that invariably triggered exhaustive elimination in the indenopyrane motif. At the same time, diastereomer 75 underwent rapid reductive cyclization to the indole under mild conditions. Perplexed by these developments, we revisited the arylation protocol and identified conditions that favored formation of epimer 75. Ultimately, tributyltin enolate generated in situ from tricyclic ketone 59 underwent cross-coupling with nearly stoichiometric amounts of aryl chloride 69 in the presence of Buchwald’s RuPhos ligand and palladacycle RuPhosG4 as a precatalyst to produce the desired product with moderate diastereoselectivity and efficiency.46 Treatment of nitroarene 75 with zinc in the presence of acetic acid followed by desilylation and double saponification completed our synthesis of nodulisporic acid C (49), which proceeded in 12 steps from commercially available starting materials in either linear branch of this highly convergent sequence. Notably, the unexpected reactivity and selectivity patterns identified during our campaign can inform subsequent efforts in the synthesis of paxilline indoloterpenoids, paving the way to development of a general approach to structurally diverse members of this fascinating family and a potential entry into their otherwise unavailable analogs for relevant interdisciplinary studies.
Scheme 10.
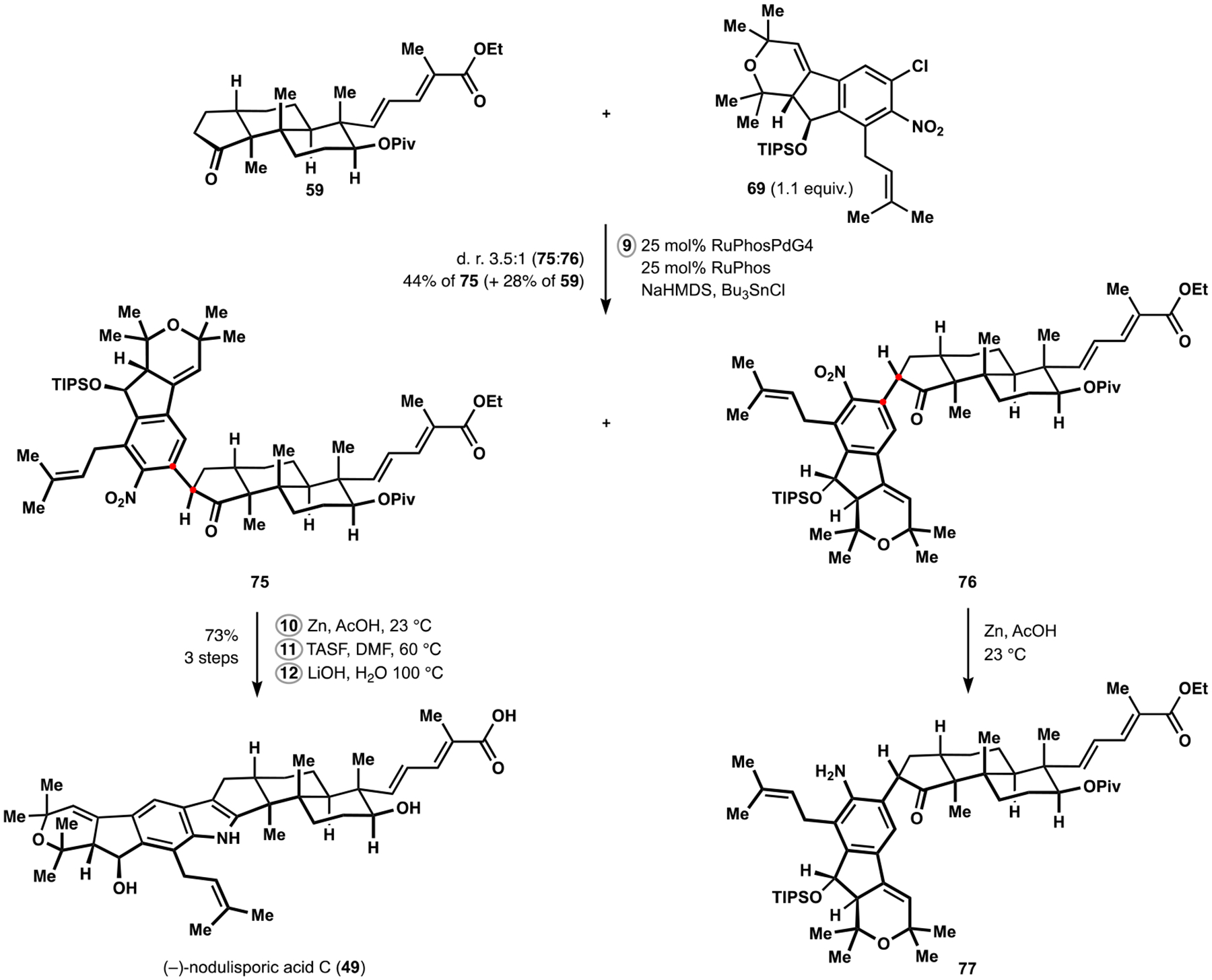
Synthesis of (−)-Nodulisporic Acid C
SYNTHESIS OF FORSKOLIN
Encouraged by the developments in our synthesis of paxilline indoloterpenoids, we considered expansion of the underlying radical-polar crossover cascade (see Scheme 2) to a bimolecular setting. The corresponding annulation of two unsaturated carbonyl components (Scheme 11) could offer a new approach to construction of six-membered carbocyclic motifs found in a broad range of terpenoids, including the large families of labdane and scalarane natural products. Should the facile assembly of quaternary centers in our polycyclization events translate to the bimolecular variant, it could prove complementary to the venerable Diels–Alder reaction, where the requirement for the diene component to adopt the s-cis conformation often imposes significant limitations on substitution patterns in bimolecular settings.47,48
Scheme 11.
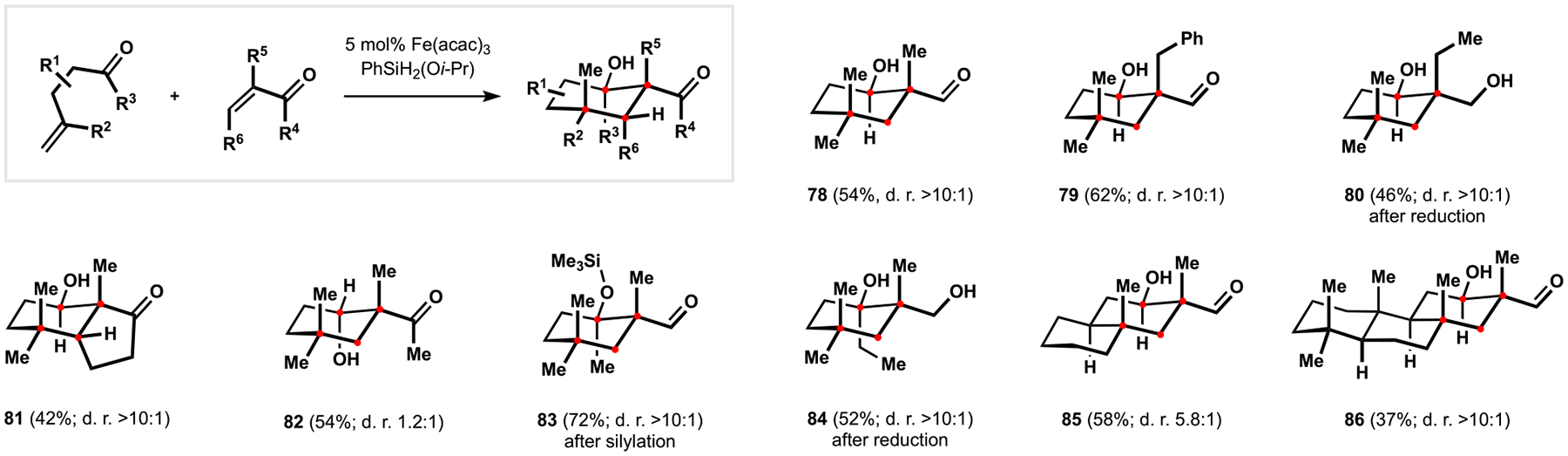
Development of Radical-Polar Crossover Annulation
Our studies in the development of a suitable annulation protocol identified the proper stoichiometry of reactants and mixing parameters to be critical to achieving optimal efficiency and attenuation of competing reductive aldol pathways.4,49 Observation of the latter was curious because it suggested that the selectivity of HAT from putative iron hydrides toward electron-rich alkenes was more nuanced than we had anticipated. Indeed, our ongoing studies determined these alternative reactivity patterns as one major factor contributing to inferior performance of conjugated cyclohexenones as annulation partners. Cyclopentenone derivatives and α-substituted acroleins were less prone to reductive aldol reactions and underwent conversion to the desired products in a highly diastereoselective manner (products 78–81, 85, 86). Preferential formation of syn aldol motifs was attributed to the intermediacy of iron enolates with E configuration.21b In contrast, the presumed unselective formation of the corresponding E and Z enolate isomers in reactions of acyclic α,β-unsaturated ketones was reflected in production of nearly equimolar mixtures of diastereomeric products (product 82). Presence of α-substituents in the α,β-unsaturated carbonyl component was required for the intramolecular aldolization, and only products of the corresponding Giese reaction were observed in all other cases. We also found that γ,δ-unsaturated aldehydes and ketones both served as suitable partners in the annulation events, where efficient and highly diastereoselective formation of tertiary alcohols was particularly striking (products 83 and 84). The fully substituted aldol motifs proved exceptionally labile and required derivatization to facilitate isolation and characterization, once again highlighting the advantages of the radical-polar crossover cascade over the stepwise construction of these sensitive intermediates.
Eager to probe the utility of the new annulation, we turned our attention to the synthesis of forskolin (104, Scheme 12).4,50 This densely functionalized labdane diterpenoid exhibits allosteric activation of adenylate cyclases and has become an important biochemical tool for interrogation of cellular processes.51 A water-soluble derivative of forskolin NHK477 is currently approved in Japan for the treatment of acute heart failure.52 The parent natural product has been the subject of multiple synthetic studies that led to accumulation of a vast body of knowledge of reactivity in this complex system.53 Once again, we were fortunate to have support from prior discoveries and observations in our own strategic planning. We were particularly intrigued by the studies from the Kienzle group in their assembly of another labdane diterpenoid, erigerol, and subsequent efforts from the Svenda group that leveraged Kienzle’s findings in their recent synthesis of forskolin.53e,54 These investigations demonstrated the remarkable utility of a common enedione motif as a starting point for elaboration of polycyclic systems found in the target natural products but also highlighted the difficulty of constructing the shared intermediate in the realm of existing methods.55 We believed that the new annulation was uniquely positioned to address those challenges and would pave the way to a rapid and efficient access to forskolin and related compounds.
Scheme 12.
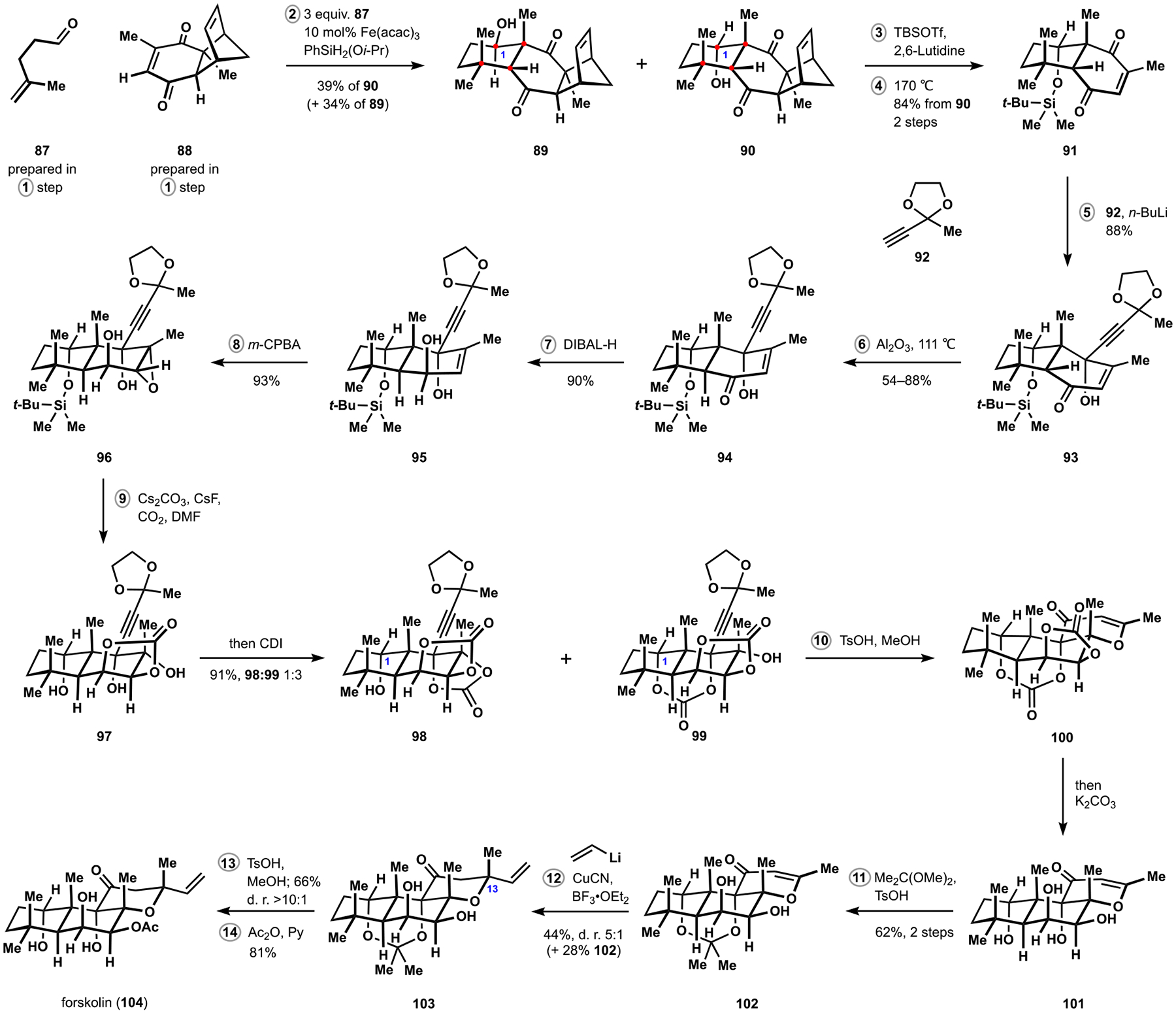
Synthesis of Forskolin
We identified enedione 88 to be an excellent partner in our radical-polar crossover cascades, which efficiently produced the desired connectivity pattern with unsaturated aldehyde 87. The bicycloheptene motif of substrate 88 could be established in a highly enantioselective manner upon application of the catalytic Diels–Alder reaction developed by the Corey group and provided a suitable handle for stereochemical relay in our synthesis.56 At the same time, formation of the aldol motif exhibited low selectivity and delivered a nearly equimolar mixture of secondary alcohols 89 and 90. However, two straightforward redox manipulations corrected the stereochemical configuration at C1 of the undesired intermediate 89. Initial attempts at retro-Diels–Alder extrusion of cyclopentadiene from the desired annulation product led to significant epimerization at the secondary alcohol, which was readily addressed by a protecting group. Ultimately, highly enantioenriched enedione 91 could be prepared on multigram scale in four steps and 30% overall yield from 2,6-dimethylbenzoquinone, a dramatic improvement over the previous protocols and a strong indication of potential advantages of the new annulation in construction of densely substituted cyclohexanes.
Subsequent steps in our synthesis of forskolin were notably influenced by the previous reports in this area. Addition of an alkynyllithium to enedione 91 followed by epimerization of the ring juncture in intermediate 93 was conducted according to protocols from the Liotta and Kienzle groups and provided regio- and diastereoselective access to unsaturated ketone 94.54,57 Inspired by the work from the Lett group, we planned to leverage intramolecular displacement of epoxide 96 for installation of the functionalization pattern found in the target natural product.53d Preparation of substrate 96 included reduction of ketone 94 and epoxidation of resulting allylic alcohol 95. Initial attempts at nucleophilic capture of the epoxide with the pendant monoalkylcarbonate generated in situ according to the Myers protocol indicated that deprotection of the alcohol at C1 was a prerequisite for achieving appreciable rates of conversion and was best performed in the presence of a fluoride source.58 At the same time, protection of the 1,3-diol motif was necessary for subsequent assembly of the dihydropyranone fragment and was accomplished by in situ derivatization of intermediate triol 97, delivering dioxanone 99 along with its isomer 98. The observed interconversion of carbonates 98 and 99 under various conditions allowed us to converge their mixtures to desired dihydropyrone 100 upon treatment with strong acid. At this stage, attempted conjugate addition of vinylcopper revealed overwhelming preference for the undesired configuration at C13, not entirely unexpected as prior syntheses of forskolin had demonstrated drastically different stereochemical outcomes depending on the nature of dihydropyrone and the organocopper reagent.59,53c,d After evaluating a variety of precursors in the conjugate addition, we identified acetonide 102 as suitable substrate for achieving the desired stereochemical arrangement in tetrahydropyrone 103. Preparation of intermediate 102 was accomplished upon selective protection of tetraol 101 obtained after methanolysis of the reaction mixture containing carbonate 100. Presence of the unprotected 1,2-diol in intermediate 102 did not interfere with the conjugate addition, allowing us to avoid challenges associated with deprotection of this motif encountered in prior syntheses, and delivered the target natural product after two additional manipulations.53,60 Overall, these studies secured rapid enantioselective access to forskolin and bolstered application of the radical-polar crossover annulation strategy in the synthesis of other terpenoid motifs, a subject of ongoing efforts in our laboratory. Notably, our approach may allow for structural modifications that have been inaccessible in the previous routes and by semisynthesis, suggesting potential value of this development to the relevant biological fields.53,61
CONCLUSIONS
We have been and continue to be fascinated by the paxilline family of indoloterpenoids with its awe-inspiring levels of molecular complexity and an incredible selection of compelling biological effects. These important considerations notwithstanding, it was the analysis of the status quo in the corresponding area of natural product synthesis and its evolution over the past decades that strongly motivated us to initiate our own inquiry. It seemed as if every effort in the field achieved better understanding of chemical reactivity in these complex systems and yet also uncovered a plethora of new challenges. We began with an idea that establishing the necessary arrangement of what we deemed to be key stereocenters in the shared terpenoid motif early in the synthesis would allow for efficient and rapid construction of the target compounds. Our modest contributions demonstrate that this approach is feasible, but its generality and advantages have yet to be determined, and a unifying strategy that allows access to structurally diverse congeners with only minimal modifications to the core reaction sequence continues to be the subject of future investigations. Perhaps even more rewarding for us is the realization that the underlying reactivity patterns may have utility outside their originally intended purposes as exemplified by our radical-polar crossover annulations that were inspired by the synthesis of emindole SB and nodulisporic acid C, a notion that resonates strongly with the premise of our research.
ACKNOWLEDGMENTS
We gratefully acknowledge contributions of past and present members of the Pronin group. This work was supported by the National Institutes of Health (R01GM121678), the Hellman Foundation, and the University of California, Irvine.
Biographies
William P. Thomas received his B.S. degree in biochemistry from Santa Clara University. He is currently a graduate student in the Pronin group where he investigates synthesis of terpenoid natural products.
Sergey V. Pronin is an Associate Professor in the Department of Chemistry at The University of California, Irvine.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.0c00809
The authors declare no competing financial interest.
Contributor Information
William P. Thomas, Department of Chemistry, University of California, Irvine, California 92697-2025, United States
Sergey V. Pronin, Department of Chemistry, University of California, Irvine, California 92697-2025, United States
REFERENCES
- (1).George DT; Kuenstner EJ; Pronin SV A concise approach to paxilline indole diterpenes. J. Am. Chem. Soc 2015, 137, 15410. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report described our initial foray into the synthesis of paxilline indoloterpenoid, established the validity of our approach to the common polycyclic motif, and demonstrated synthesis of emindole SB as a proof-of-concept for application of our strategy.
- (2).Holmbo SD; Godfrey NA; Hirner JJ; Pronin SV A catalytic intermolecular formal ene reaction between ketone-derived silyl enol ethers and alkynes. J. Am. Chem. Soc 2016, 138, 12316. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report described the development of a catalytic method for bimolecular alkenylation of ketone-derived silyl enol ethers with alkyne, a transformation that exhibits unusual selectivity in construction of quaternary centers and was key to success in our synthesis of emindole SB.
- (3).Godfrey NA; Schatz DJ; Pronin SV Twelve-step asymmetric synthesis of (−)-nodulisporic acid. J. Am. Chem. Soc 2018, 140, 12770. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report detailed implementation of our strategy in a highly convergent and enantioselective synthesis of a more complex congener, nodulisporic acid C.
- (4).Thomas WP; Schatz DJ; George DT; Pronin SV A Radical-Polar Crossover Annulation to Access Terpenoid Motifs. J. Am. Chem. Soc 2019, 141, 12246. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report detailed expansion of the radical-polar crossover cascade employed in our synthesis of paxilline indoloterpenoids to a bimolecular setting, allowing for annulation of two unsaturated carbonyl components that enabled a brief synthesis of forskolin.
- (5).For examples, see:; (a) Sheehan JC; Henery Logan KR A General Synthesis of the Penicillins. J. Am. Chem. Soc 1959, 81, 5838. [Google Scholar]; (b) Corey EJ; Weinshenker NM; Schaaf TK; Huber W Stereo-Controlled Synthesis of Prostaglandins F2α and E2 (dl). J. Am. Chem. Soc 1969, 91, 5675. [DOI] [PubMed] [Google Scholar]; (c) Aicher TD; Buszek KR; Fang FG; Forsyth CJ; Jung SH; Kishi Y; Matelich MC; Scola PM; Spero DM; Yoon SK Total Synthesis of Halichondrin B and Norhalichondrin B. J. Am. Chem. Soc 1992, 114, 3162. [Google Scholar]; (d) Corey EJ; Gin DY; Kania RS Enantioselective Total Synthesis of Ecteinascidin 743. J. Am. Chem. Soc 1996, 118, 9202. [Google Scholar]
- (6).For an excellent discussion, see:; Nicolaou KC; Vourloumis D; Winssinger N; Baran PS The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century. Angew. Chem., Int. Ed 2000, 39, 44. [PubMed] [Google Scholar]
- (7).For examples from the Corey synthesis of prostaglandins, see:; (a) Corey EJ; Bakshi RK; Shibata S; Chen C-P; Singh VK A Stable and Easily Prepared Catalyst for the Enantioselective Reduction of Ketones. Application to Multistep Syntheses. J. Am. Chem. Soc 1987, 109, 7925. [Google Scholar]; (b) Corey EJ; Imwinkelried R; Pikul S; Xiang YB Practical Enantioselective Diels-Alder and Aldol Reactions Using a New Chiral Controller System. J. Am. Chem. Soc 1989, 111, 5493. [Google Scholar]
- (8).For examples from the Overman synthesis of rearranged spongian diterpenes, see:; (a) Schnermann MJ; Overman LE A Concise Synthesis of (–)-Aplyviolene Facilitated by a Strategic Radical Conjugate Addition. Angew. Chem., Int. Ed 2012, 51, 9576. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lackner GL; Quasdorf KW; Overman LE Direct Construction of Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of tert-Alkyl N-Phthalimidoyl Oxalates. J. Am. Chem. Soc 2013, 135, 15342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jamison CR; Overman LE Fragment Coupling with Tertiary Radicals Generated by Visible-Light Photo-catalysis. Acc. Chem. Res 2016, 49, 1578. [DOI] [PubMed] [Google Scholar]
- (9).Holmbo SD; Pronin SV A Concise Approach to Anthraquinone–Xanthone Heterodimers. J. Am. Chem. Soc 2018, 140, 5065. [DOI] [PubMed] [Google Scholar]
- (10).(a) Touney EE; Foy NJ; Pronin SV Catalytic Radical-Polar Crossover Reactions of Allylic Alcohols. J. Am. Chem. Soc 2018, 140, 16982. [DOI] [PubMed] [Google Scholar]; (b) Discolo CA; Touney EE; Pronin SV Catalytic Asymmetric Radical-Polar Crossover Hydroalkoxylation. J. Am. Chem. Soc 2019, 141, 17527. [DOI] [PubMed] [Google Scholar]
- (11).Minami A; Liu C; Oikawa H Total biosynthesis of fungal indole diterpenes using cell factory. Heterocycles 2016, 92, 397. [Google Scholar]
- (12).For selected examples, see:; (a) Knaus H-G; McManus OB; Lee SH; Schmalhofer WA; Garcia-Calvo M; Helms LMH; Sanchez M; Giangiacomo K; Reuben JP; Smith AB III; Kaczorowski GJ; Garcia ML Tremorgenic Indole Alkaloids Potently Inhibit Smooth Muscle High-Conductance Calcium-Activated Potassium Channels. Biochemistry 1994, 33, 5819. [DOI] [PubMed] [Google Scholar]; (b) Meinke PT; Smith MM; Shoop WL Nodulisporic Acid: Its Chemistry and Biology. Curr. Top. Med. Chem 2002, 2, 655. [DOI] [PubMed] [Google Scholar]
- (13).For initial reports, see:; de Jesus AE; Gorst-Allman CP; Steyn PS; van Heerden FR; Vleggaar R; Wessels PL; Hull WE Tremorgenic mycotoxins from Penicillium crustosum. Biosynthesis of penitrem A. J. Chem. Soc., Perkin Trans. 1 1983, 1863. [Google Scholar]
- (14).Byrne KM; Smith KS; Ondeyka JG Biosynthesis of Nodulisporic Acid A: Precursor Studies. J. Am. Chem. Soc 2002, 124, 7055. [DOI] [PubMed] [Google Scholar]
- (15).For example, see:; Rainier JD; Smith AB III Polyene cyclizations to indole diterpenes. The first synthesis of (+)-emindole SA using a biomimetic approach. Tetrahedron Lett. 2000, 41, 9419. [Google Scholar]
- (16).Kim DE; Zweig JE; Newhouse TR Total Synthesis of Paspaline A and Emindole PB Enabled by Computational Augmentation of a Transform-Guided Retrosynthetic Strategy. J. Am. Chem. Soc 2019, 141, 1479. [DOI] [PubMed] [Google Scholar]
- (17).For selected examples, see:; (a) Smith AB III; Kanoh N; Ishiyama H; Hartz RA Total synthesis of (−)-penitrem D. J. Am. Chem. Soc 2000, 122, 11254. [DOI] [PubMed] [Google Scholar]; (b) Zou Y; Melvin JE; Gonzales SS; Spafford MJ; Smith AB III. Total synthesis of (−)-nodulisporic acid D. J. Am. Chem. Soc 2015, 137, 7095. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zou Y; Li X; Yang Y; Berritt S; Melvin J; Gonzales S; Spafford M; Smith AB III. Total synthesis of (−)-nodulisporic acids D, C, and B: evolution of a unified synthetic strategy. J. Am. Chem. Soc 2018, 140, 9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Guile SD; Saxton JE; Thornton-Pett M Synthetic studies towards paspalicine. Part 2. An alternative approach to the synthesis of the C/D ring system. J. Chem. Soc., Perkin Trans. 1 1992, 1992, 1763. [Google Scholar]
- (19).For selected example, see:; Enomoto M; Morita A; Kuwahara S Total synthesis of the tremorgenic indole diterpene paspalinine. Angew. Chem., Int. Ed 2012, 51, 12833. [DOI] [PubMed] [Google Scholar]
- (20).Sharpe RJ; Johnson JS A global and local desymmetrization approach to the synthesis of steroidal alkaloids: stereocontrolled total synthesis of paspaline. J. Am. Chem. Soc 2015, 137, 4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).For initial discovery, see:; (a) Lo JC; Yabe Y; Baran PS A practical and catalytic reductive olefin coupling. J. Am. Chem. Soc 2014, 136, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; For mechanistic considerations, see; (b) Kim D; Rahaman SMW; Mercado BQ; Poli R; Holland P Roles of iron complexes in catalytic radical alkene cross-coupling: a computational and mechanistic study. J. Am. Chem. Soc 2019, 141, 7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For pioneering report see:; Kosugi M; Hagiwara I; Migita T 1-Alkenylation on α-Position of Ketone: Palladium-Catalyzed Reaction of Tin Enolates and 1-Bromo-1-Alkenes. Chem. Lett 1983, 12, 839. [Google Scholar]
- (23).For selected examples, see:; Grigalunas M; Ankner T; Norrby P-O; Wiest O; Helquist P Ni-Catalyzed Alkenylation of Ketone Enolates under Mild Conditions: Catalyst Identification and Optimization. J. Am. Chem. Soc 2015, 137, 7019. [DOI] [PubMed] [Google Scholar]
- (24).Nishimoto Y; Moritoh R; Yasuda M; Baba A Regio- and Stereoselective Generation of Alkenylindium Compounds from Indium Tribromide, Alkynes, and Ketene Silyl Acetals. Angew. Chem., Int. Ed 2009, 48, 4577. [DOI] [PubMed] [Google Scholar]
- (25).Umbreit MA; Sharpless KB Allylic oxidation of olefins by catalytic and stoichiometric selenium dioxide with tert-butyl hydro-peroxide. J. Am. Chem. Soc 1977, 99, 5526. [Google Scholar]
- (26).Obradors C; Martinez RM; Shenvi RA Ph(i-PrO)SiH2: an exceptional reductant for metal-catalyzed hydrogen atom transfers. J. Am. Chem. Soc 2016, 138, 4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Parikh JR; Doering W. v. E. Sulfur trioxide in the oxidation of alcohols by dimethyl sulfoxide. J. Am. Chem. Soc 1967, 89, 5505. [Google Scholar]
- (28).Vaswani RG; Day JJ; Wood JL Progress Toward the Total Synthesis of (±)-Actinophyllic Acid. Org. Lett 2009, 11, 4532. [DOI] [PubMed] [Google Scholar]
- (29).Nozawa K; Nakajima S; Kawai K -i.; Udagawa, S.-i. Isolation and structures of indoloditerpenes, possible biosynthetic intermediates to the tremorgenic mycotoxin, paxilline, from Emericella striata. J. Chem. Soc., Perkin Trans. 1 1988, 2607. [Google Scholar]
- (30).Lau KSY; Schlosser M Selective syntheses with organometallics. 7. (Z)-2-Ethoxyvinyllithium: a remarkably stable and synthetically useful 1,2-counterpolarized species. J. Org. Chem 1978, 43, 1595. [Google Scholar]
- (31).For selected examples, see:; Staben ST; Kennedy-Smith JJ; Huang D; Corkey BK; LaLonde RL; Toste FD Gold(I)-Catalyzed Cyclizations of Silyl Enol Ethers: Application to the Synthesis of (+)-Lycopladine A. Angew. Chem., Int. Ed 2006, 45, 5991. [DOI] [PubMed] [Google Scholar]
- (32).For a contemporary review of alkenylation of enolates, see:; Ankner T; Cosner CC; Helquist P Palladium- and Nickel-Catalyzed Alkenylation of Enolates. Chem. - Eur. J 2013, 19, 1858. [DOI] [PubMed] [Google Scholar]
- (33).For initial discovery, see:; (a) Ondeyka JG; Helms GL; Hensens OD; Goetz MA; Zink DL; Tsipouras A; Shoop WL; Slayton L; Dombrowski AW; Polishook JD; Ostlind DA; Tsou NN; Ball RG; Singh SB Nodulisporic acid A, a novel and potent insecticide from a Nodulisporium sp. Isolation, structure determination, and chemical transformations. J. Am. Chem. Soc 1997, 119, 8809. [Google Scholar]; Nodulisporic acid C:; (b) Ondeyka JG; Byrne K; Vesey D; Zink DL; Shoop WL; Goetz MA; Singh SB Nodulisporic Acids C, C1, and C2: A series of D-ring-opened nodulisporic acids from the fungus Nodulisporium sp. J. Nat. Prod 2003, 66, 121. [DOI] [PubMed] [Google Scholar]
- (34).Ludmerer SW; Warren VA; Williams BS; Zheng Y; Hunt DC; Ayer MB; Wallace MA; Chaudhary AG; Egan MA; Meinke PT; Dean DC; Garcia ML; Cully DF; Smith MM Ivermectin and nodulisporic acid receptors in Drosophila melanogaster contain both γ-aminobutyric acid-gated Rdl and glutamate-gated GluClα chloride channel subunits. Biochemistry 2002, 41, 6548. [DOI] [PubMed] [Google Scholar]
- (35).Shoop WL; Gregory LM; Zakson-Aiken M; Michael BF; Haines HW; Ondeyka JG; Meinke PT; Schmatz DM Systemic efficacy of nodulisporic acid against fleas on dogs. J. Parasitol 2001, 87, 419. [DOI] [PubMed] [Google Scholar]
- (36).Meinke PT; Colletti SL; Fisher MH; Wyvratt MJ; Shih TL; Ayer MB; Li C; Lim J; Ok D; Salva S; Warmke LM; Zakson M; Michael BF; deMontigny P; Ostlind DA; Fink D; Drag M; Schmatz DM; Shoop WL Discovery of the development candidate N-tert-butyl nodulisporamide: a safe and efficacious once monthly oral agent for the control of fleas and ticks on companion animals. J. Med. Chem 2009, 52, 3505. [DOI] [PubMed] [Google Scholar]
- (37).Smith AB III; Kürti L; Davulcu AH; Cho YS; Ohmoto K Indole diterpene synthetic studies: development of a second-generation synthetic strategy for (+)-nodulisporic acids A and B. J. Org. Chem 2007, 72, 4611. [DOI] [PubMed] [Google Scholar]
- (38).Harrak Y; Simonneau A; Malacria M; Gandon V; Fensterbank L Gold(I)-catalyzed cycloisomerization of 1,6-enynes into functionalized allenes. Chem. Commun 2010, 46, 865. [DOI] [PubMed] [Google Scholar]
- (39).Sanjuán AM; Martínez A; García-García P; Fernández-Rodríguez MA; Sanz R Gold(I)-catalyzed 6-endo hydroxycyclization of 7-substituted-1,6-enynes. Beilstein J. Org. Chem 2013, 9, 2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Calvo BC; Madduri AVR; Harutyunyan SR; Minnaard AJ Copper-catalysed conjugate addition of Grignard reagents to 2-methylcyclopentenone and sequential enolate alkylation. Adv. Synth. Catal 2014, 356, 2061. [Google Scholar]
- (41).Schatz DJ; Li W; Pronin SV Catalytic enantioselective conjugate addition en route to paxilline indoloterpenoids. Tetrahedron 2019, 75, 3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nugent WA MIB: an advantageous alternative to DAIB for the addition of organozinc reagents to aldehydes. Chem. Commun 1999, 35, 1369. [Google Scholar]
- (43).Lurain AE; Maestri A; Kelly AR; Carroll PJ; Walsh PJ Highly enantio- and diastereoselective one-pot synthesis of acyclic epoxy alcohols with three contiguous stereocenters. J. Am. Chem. Soc 2004, 126, 13608. [DOI] [PubMed] [Google Scholar]
- (44).For an excellent review, see:; Dorel R; Echavarren AM Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev 2015, 115, 9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Stille JK Palladium catalyzed coupling of organotin reagents with organic electrophiles. Pure Appl. Chem 1985, 57, 1771. [Google Scholar]
- (46).Bruno NC; Niljianskul N; Buchwald SL N-substituted 2-aminobiphenylpalladium methanesulfonate precatalysts and their use in C–C and C–N cross-couplings. J. Org. Chem 2014, 79, 4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).For selected examples of bimolecular Diels–Alder reactions with highly substituted 1,3-dienes and relevant discussions, see:; Jung ME; Ho D; Chu HV Synthesis of highly substituted cyclohexenes via mixed Lewis acid-catalyzed Diels-Alder reactions of highly substituted dienes and dienophiles. Org. Lett 2005, 7, 1649. [DOI] [PubMed] [Google Scholar]
- (48).For example of an elegant solution to this problem, see:; Zutterman F; Krief A Synthesis and Diels-Alder reactions of allylidenecyclopropane. J. Org. Chem 1983, 48, 1135. [Google Scholar]
- (49).For relevant reductive aldol reactions, see:; (a) Isayama S; Mukaiyama T Cobalt(II) catalyzed coupling reaction of α,β-unsaturated compounds with aldehydes by the use of phenylsilane. New method for preparation of β-hydroxy nitriles, amides, and esters. Chem. Lett 1989, 18, 2005. [Google Scholar]; (b) Baik T-G; Luis AL; Wang L-C; Krische MJ Diastereoselective cobalt-catalyzed aldol and Michael cycloreductions. J. Am. Chem. Soc 2001, 123, 5112. [DOI] [PubMed] [Google Scholar]
- (50).Bhat SV; Bajqwa BS; Dornauer H; do Scusa NJ; Fehlhaber H-W Structures and stereochemistry of new labdane diterpenoids from Coleus forskohlii briq. Tetrahedron Lett. 1977, 18, 1669. [Google Scholar]
- (51).Insel PA; Ostrom RS Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell. Mol. Neurobiol 2003, 23, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hosoda S; Motomiya T; Katagiri T; Takano T; Sasayama S; Toshima H; Ogawa N Acute effect of NHK477, a novel forskolin derivative, in patients with acute heart failure. Rinsho Yakuri 1997, 28, 583. [Google Scholar]
- (53).For syntheses of forskolin preceding our work, see:; (a) Ziegler FE; Jaynes BH; Saindane MT A synthetic route to forskolin. J. Am. Chem. Soc 1987, 109, 8115. [Google Scholar]; (b) Hashimoto S; Sakata S; Sonegawa M; Ikegami S A total synthesis of (±)-forskolin. J. Am. Chem. Soc 1988, 110, 3670. [Google Scholar]; (c) Corey EJ; Jardine P. d. S.; Rohloff JC Total synthesis of (±)-forskolin. J. Am. Chem. Soc 1988, 110, 3672. [Google Scholar]; (d) Delpech B; Calvo D; Lett R Total synthesis of forskolin – part II. Tetrahedron Lett. 1996, 37, 1019. [Google Scholar]; (e) Hylse O; Maier L; Kučera R; Perečko T; Svobodová A; Kubala L; Paruch K;Švenda J A concise synthesis of forskolin. Angew. Chem., Int. Ed 2017, 56, 12586. [DOI] [PubMed] [Google Scholar]
- (54).Kienzle F; Stadlwieser J; Rank W; Schönholzer P Die synthese des labdanditerpenes erigerol und analoger verbindungen. Helv. Chim. Acta 1990, 73, 1108. [Google Scholar]
- (55).For alternative asymmetric preparation of the enedione motif, see:; Nagasawa S; Jones KE; Sarpong R Enantiospecific Entry to a Common Decalin Intermediate for the Syntheses of Highly Oxygenated Terpenoids. J. Org. Chem 2019, 84, 12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Liu D; Canales E; Corey EJ Chiral oxazaborolidine–aluminum bromide complexes are unusually powerful and effective catalysts for enantioselective Diels-Alder reactions. J. Am. Chem. Soc 2007, 129, 1498. [DOI] [PubMed] [Google Scholar]
- (57).Liotta D; Saindane M; Sunay U; Jamison WCL; Grossman J; Phillips P Acetylide additions to enediones. Regioselectivity based on stereoelectronic control. J. Org. Chem 1985, 50, 3241. [Google Scholar]
- (58).Myers AG; Widdowson KL Direct transformation of 2,3-epoxy alcohols into hydroxyl carbonates under mildly basic conditions. Tetrahedron Lett. 1988, 29, 6389. [Google Scholar]
- (59).(a) Delpech B; Lett R Retrosynthetic studies with forskolin. Tetrahedron Lett. 1987, 28, 4061. [Google Scholar]; (b) Ziegler FE; Jaynes BH Formation of (±)-forskolin via a cuprate addition to a synthetic dihydropyran-4-one. Tetrahedron Lett. 1988, 29, 2031. [Google Scholar]
- (60).Bhat SV; Bajwa BS; Dornauer H; de Souza NJ Reactions of forskolin, a biologically active diterpenoid from Coleus forskohlii. J. Chem. Soc., Perkin Trans. 1 1982, 1982, 767. [Google Scholar]
- (61).For relevant discussion of forskolin analogues and SAR studies, see:; (a) Seamon KB; Daly JW; Metzger H; de Souza NJ; Reden J Structure-Activity Relationships for Activation of Adenylate Cyclase by the Diterpene Forskolin and Its Derivatives. J. Med. Chem 1983, 26, 436. [DOI] [PubMed] [Google Scholar]; (b) Pinto C; Papa D; Hübner M; Mou TC; Lushington GH; Seifert R Activation and Inhibition of Adenylyl Cyclase Isoforms by Forskolin Analogs. J. Pharmacol. Exp. Ther 2008, 325, 27. [DOI] [PubMed] [Google Scholar]