

Abstract
Rearranged indole diterpenes of the paxilline type comprise a large group of fungal metabolites that possess diverse structural features and potentially useful biological effects. The unique indoloterpenoid motif, which is common to all congeners, was first confirmed by crystallographic studies of paxilline. This family of natural products has fascinated organic chemists for the past four decades and has inspired numerous syntheses and synthetic approaches. The present review highlights efforts that have laid the foundation and introduced new directions to this field of natural product synthesis. The introduction includes a summary of biosynthetic considerations and biological activities, the main body of the manuscript provides a detailed discussion of selected syntheses, and the review concludes with a brief outlook on the future of the field.
1. Introduction
In 1975, Clardy and co-workers disclosed their crystallographic studies of paxilline (1, Fig. 1), a tremorgenic metabolite previously isolated from the cultures of Penicillium paxilli by Wells and co-workers.1 This report marked the first unambiguous structural characterization of an unusual polycyclic natural product containing an indole moiety fused to a rearranged diterpene fragment. The same connectivity pattern was previously identified by Arigoni and co-workers in their spectroscopic and chemical degradation studies of several metabolites from Claviceps paspali.2–4 Among those compounds was paspaline (2), which contained the angular methyl group at C12 and provided early insight into the biosynthetic origins of these natural products.1,5 The shared indolotricarbocyclic core was later confirmed in a broad range of fungal metabolites, and over 150 related natural products have been characterized to date. Penitrem A (3), lolitrem B (4), and shearinine A (5) exemplify the remarkable levels of structural complexity that can be found among the congeners.6–8 These diterpenoids belong to the family of natural products that is sometimes referred to as either “indole diterpenes” or “indoloditerpenes”. Both names represent collective terms for secondary metabolites that arise biosynthetically from the oxidation and subsequent cyclization of the 3-geranylgeranylindole and include structurally distinct aflavazole, tubingensins, and other compounds beyond the scope of this review.9,10 Early efforts to recognize the diversity of the resulting connectivity patterns and to introduce subdivisions into this large family were made by Nozawa, who distinguished paspaline-type natural products from the rest of indole-containing diterpenoid motifs according to their structural and proposed biogenetic relationships to the namesake metabolite.9 This terminology, however, has since been used only sparingly, and the ever-increasing diversity of the newly isolated congeners often resulted in further fragmenting of this subfamily of indole diterpenes in the current literature.11,12 A representative example includes nodulisporic acid A (6) and related nodulisporanes, which arise from a new branch of the biosynthetic pathway established after formation of the characteristic indolotricarbocyclic motif, and are often classified into a standalone group of fungal metabolites.13 Isolation of lecanindole A (7), sespendole (8), and several other indole-containing sesquiterpenoids further expanded the scope of structurally related metabolites.14,15 Strictly for the purposes of this review and to signify the uniting features that expand beyond the paspaline derivatives, we have chosen to refer to all compounds relevant to our discussion as paxilline indole diterpenes or indole diterpenes of the paxilline type, as the title compound was the first to receive full and unambiguous structural characterization. The uniting polycyclic motif and the diversity of individual structural features within this group of natural products combined with a broad scope of biological effects have fascinated organic chemists for the past four decades and have inspired numerous syntheses and synthetic approaches.16–31 This review is intended to highlight the diversity of strategies and tactics that have been employed in this area of organic synthesis, from its inception to the present. An overview of the biosynthesis precedes the discussion of selected chemical syntheses to serve as an introduction to the challenges associated with these compounds. A brief survey of the reported biological activity of these indole diterpenes, which is not intended to be comprehensive but accentuates the significance of the existing chemical studies, is also included.
Fig. 1.
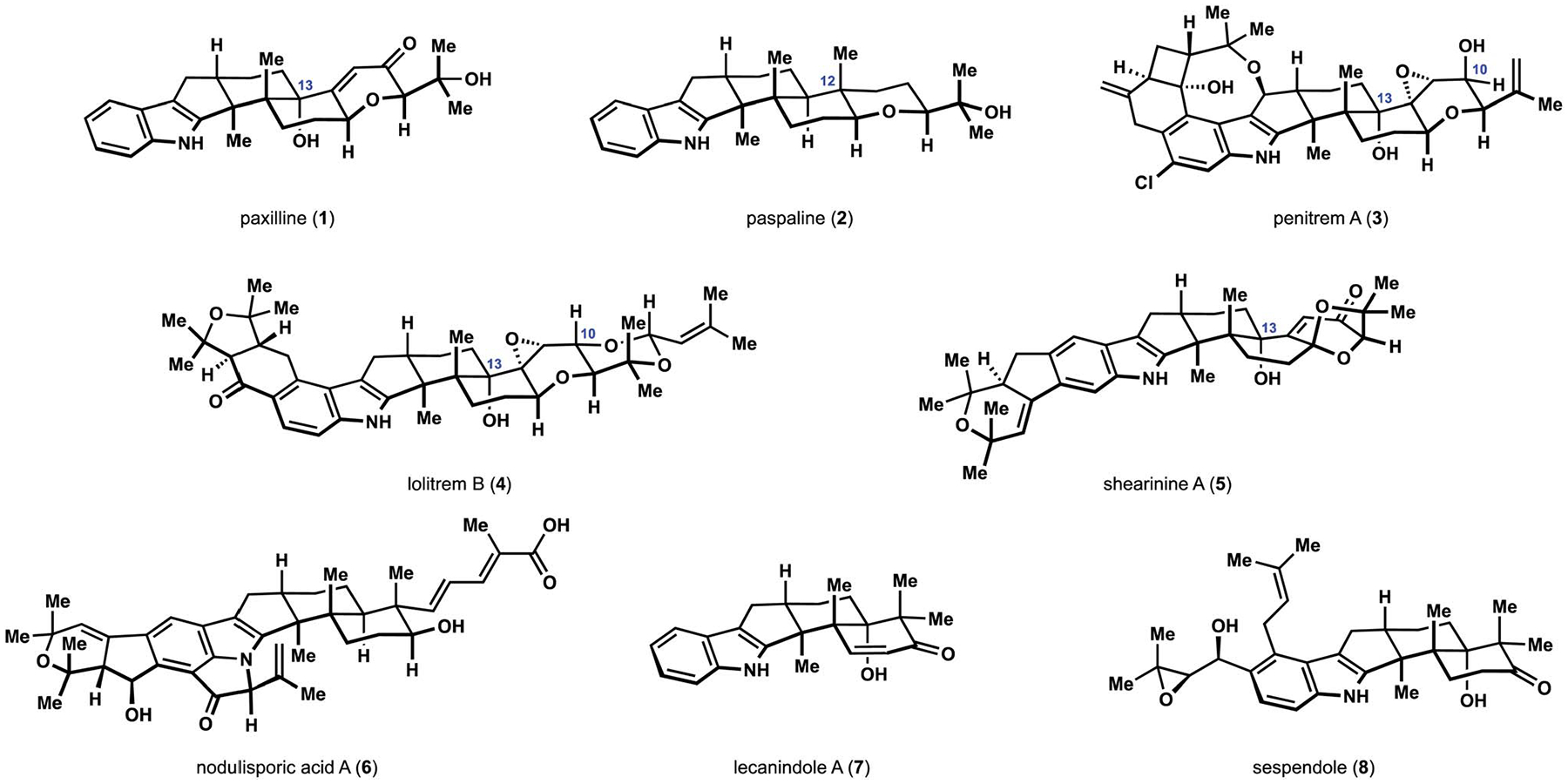
Representative natural products containing rearranged indoloterpenoid core.
2. Biosynthetic considerations
Numerous studies into the biosynthesis of the indole diterpenes appeared since the first reports of structural characterization of these unusual natural products. Several reviews provide excellent coverage of the field, including book chapters by Sings and Singh, and Parker and Scott, a comprehensive account and overviews by Oikawa and co-workers, and a detailed survey of biosynthetic production of relevant natural products reported by Molnár and co-workers.11,12,32,33 Early investigations by Arigoni and co-workers into the biosynthetic incorporation of isotopically labelled acetate and tryptophan suggested that the polycyclic motif found in paspaline arises from 3-geranylgeranylindole via an unusual polycyclization.34 Related studies of the biosynthesis of penitrem A provided additional support for the terpenoid origin of the indolotricarbocyclic motif that is shared among the congeners.35 Subsequent efforts in the identification and reconstitution of the biosynthetic gene cluster responsible for the production of paxilline have further refined the original proposal.36–38 Epoxide 9 is presently recognized as the precursor to the cationic polycyclization event that ultimately leads to production of emindole SB (12), which is a starting point for subsequent diversification of the common indoloteprenoid motif (Scheme 1). Construction of the pentacyclic scaffold appears to involve anti-Markovnikov cyclization of the prenyl unit proximal to the indole fragment and formation of secondary carbocation 10.13,34,35,39 A subsequent alkyl shift results in expansion of the hydrindane motif to generate decalinyl cation 11, which in turn undergoes Friedel–Crafts alkylation of the pendant indole to forge the strained indoloterpenoid core of the natural product. This unique cascade has become a subject of extensive investigation by synthetic chemists, but has proven difficult to execute in an artificial setting. Attempted polycyclizations of precursors related to epoxide 9 have invariably resulted in the selective formation of the alternative terpenoid motifs found in emindole SA and related metabolites. Mimicking the biosynthetic construction of the indolotricarbocyclic core thus remains a largely unsolved challenge in synthetic organic chemistry.29,40,41
Scheme 1.
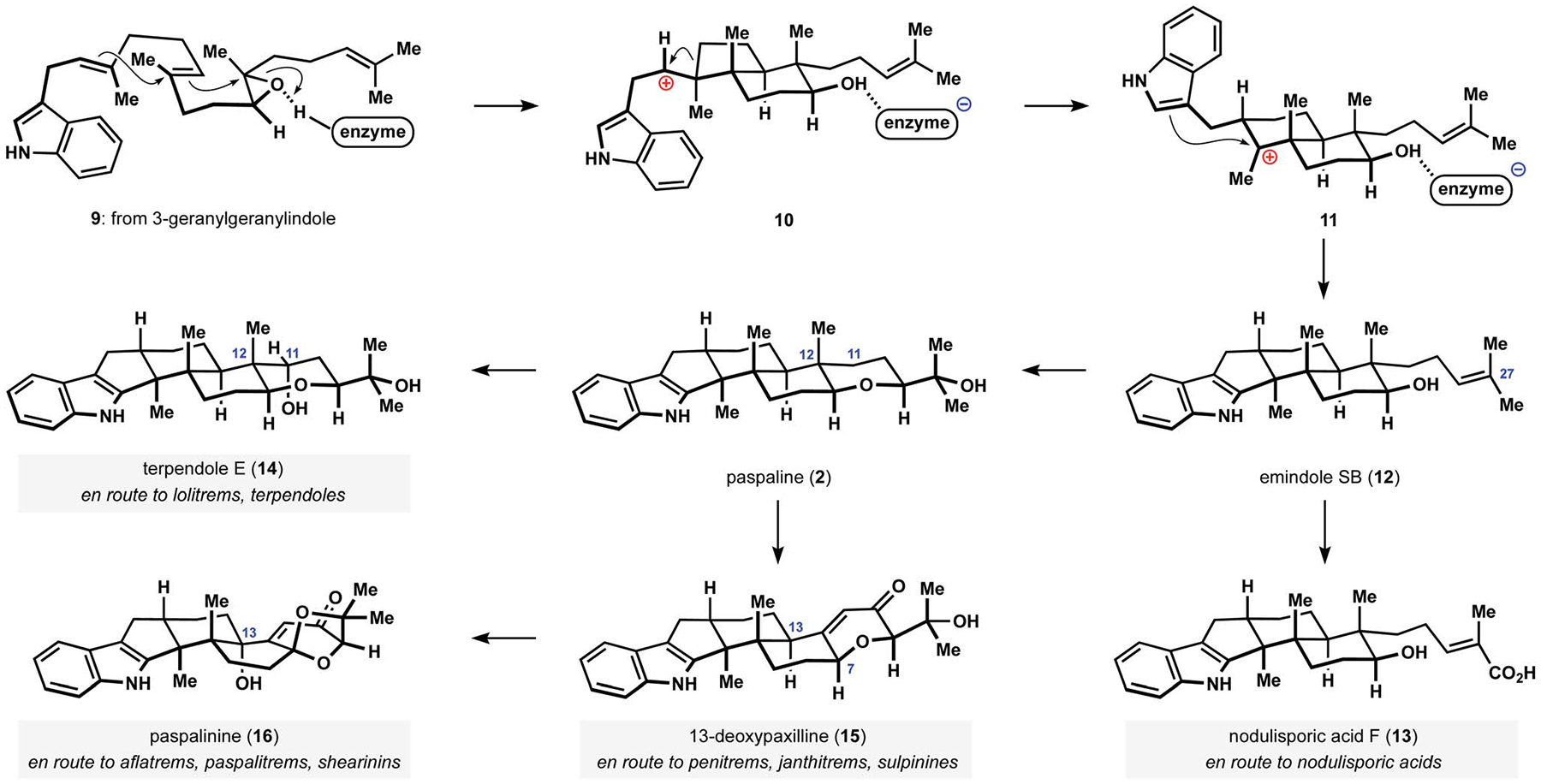
Biosynthetic construction and modification of the shared indoloterpenoid core.
The product of polycyclization, emindole SB (12), serves as a common intermediate en route to all indole diterpenes of relevance to this review.38,42 Subsequent functionalization of the remaining prenyl group depends on the producing organism and provides the first point of diversion in the biosynthesis of these fungal metabolites. Oxidation of the C27 methyl group in Hypoxylon pulicicidum results in the formation of nodulisporic acid F (13), which undergoes subsequent modifications of the indole and alkenoic acid fragments to produce increasingly complex congeners, of which nodulisporic acid A is the flagship species (6; see Fig. 1).13,42–44 Complex indole moieties that arise from multiple prenylations of the aromatic ring and subsequent oxidation events feature prominently in many other indole diterpenes, including penitrem A (3), lolitrem B (4), and shearinine A (5, see Fig. 1). Other species of endophytic fungi affect epoxidation of the alkene at C27, and subsequent intramolecular nucleophilic displacement affords paspaline (2).38 The resulting hexacyclic motif serves as a precursor to all congeners containing the corresponding tetrahydropyran fragment or derivatives thereof. Hydroxylation at C11 in Tolypocladium album produces terpendole E (14), and a homologous process is proposed to occur in Neotyphodium lolii.45,46 The hydroxylation is followed by an oxidative cleavage of the methyl group at C12, and additional oxidations and modifications of the indole motifs result in a rapid increase of complexity to produce lolitrems and terpendoles, amongst others.45,47 The characteristic configuration of the functionality at C10 is the uniting feature of highly oxidized indoloditerpenoids that arise from this branch of the biosynthetic pathway, which includes lolitrem B (4, see Fig. 1). Species from Claviceps, Penicillium, and Aspergillus genera affect oxidative cleavage of paspaline (2) at C12 and eventually produce 13-deoxypaxilline (15) as another branching point en route to congeners derived from the paxilline motif and paspalinine (16) after hydroxylation at C13 and a subsequent or preceding oxidative cyclization at C7.36,48,49 Shearinines, aflatrems, and other metabolites produced along the paspalinine pathway are readily identified by the presence of the dioxolane motif.48,50–52 The paxilline pathway delivers some of the most complex members of the family, including penitrems, janthitrems, and related compounds.53 Among the distinguishing structural features of this subfamily is the functionality at C10 with stereochemical configuration opposite to that of lolitrems and terpendoles, as seen in the example of lolitrem B (3, see Fig. 1).
3. Biological activity
Studies have long attributed instances of muscle tremors in grazing livestock to contamination of the diet with various fungi that produce indole diterpenes of the paxilline type. Reports of these neurotoxic effects implicate paspalinine (16, see Scheme 1) and paspalitrems A and B from C. paspali as causative agents of Dallis grass poisoning, commonly known as Paspalum staggers.54 Other notable examples include ryegrass stagger disorders in New Zealand due to the presence of lolitrem B (4, see Fig. 1) from N. lolii and Bermuda grass tremors induced by paspalitrem B from C. cynodontis.55,56 Similar neurotoxicity was observed upon poisoning by soil and food molds from Aspergillus and Penicillium genera that produce aflatrems and penitrems.57 Early structure–activity relationship (SAR) studies suggested that the presence of the hydroxy group at C13 confers tremorgenic effects.58 Studies with neuromuscular tissue preparations suggest that the tremorgenic effects of the indole diterpenes may be linked to an increased release of neuro-transmitters.59 Potent and selective inhibition of high-conductance calcium-activated potassium (BK) channels by several indole diterpenes have shown little dependence on the oxidation at C13 across various congeners, which suggests that the effect of the indole diterpenes on potassium conductivity may have diverse manifestations.60 In subsequent studies, paxilline (1) and lolitrem B (4) did not demonstrate any tremorgenic and ataxic activity in animal models lacking BK channels, thus providing strong evidence for a link between the inhibitory effects and the neurotoxic properties.61
The biological activity of the nodulisporic acids is also attributed to activation of ion channels. The flagship member of the family, nodulisporic acid A (6; see Fig. 1), was discovered during the search for new antiparasitic agents.62 This fungal metabolite exhibits insecticidal activity against fleas that has been linked to inhibition of a specific subset of ligand-gated chloride ion channels found in arthropods.63 The high isoform selectivity renders nodulisporic acids harmless to mammals and, combined with high potency in flea feeding assays, has prompted the development of these natural products as new anti-flea medications for companion animals.64 Nodulisporic acid A (6) is the most active member of the family, where the presence of both the indenopyran and the dihydropyrroloindole motifs confers the strongest insecticidal effects.65 Extensive SAR studies identified the dienoic acid fragment as a suitable modification point, and subsequent medicinal chemistry efforts produced a series of unsaturated amides with improved potencies and pharmacokinetic properties.66 Notably, modifications of the indole-containing fragment were not tolerated, in agreement with the SAR trends observed among the natural congeners.
Several fungal metabolites arising from the terpendole E branch of the biosynthetic pathway (see Scheme 1) were found to inhibit acyl-CoA:cholesterol acyltransferase, which is an enzyme that facilitates cholesterol homeostasis in tissues.67 Congeners containing prenyl ether functionality at C27 exhibited the highest levels of activity and also demonstrated low general cytotoxicity. In the following whole cell and enzymatic investigations, terpendole C did not demonstrate significant isoform selectivity, which is desirable for the modulation of pathways contributing to atherosclerosis and Alzheimer’s disease.68,69 A more recent study found that terpendole E (14) and its oxidation product 11-ketopaspaline disrupt separation of mitotic spindles in phenotypic assays.70,71 Mechanistic analysis indicated that the effects on cell division are linked to inhibition of mitotic kinesin Eg5, which has been a target of interest in the development of new anti-cancer therapeutics.72 No cross-resistance with other known inhibitors of Eg5 was observed in enzymatic assays, suggesting a new mode of interaction with the target protein, although the molecular basis for the inhibitory action is not well understood.71
Several reports describe antiproliferative activities of penitrem A (3, see Fig. 1) in malignant cells.73–75 These effects may be linked to the potent inhibition of BK channels, which can ultimately result in impaired nutrient transport and starvation in glioma cell lines as well as upregulation of TNF-a and the ensuing apoptosis in breast cancer cell lines.73,74 Notably, normal mammalian cells were found to be significantly less sensitive to penitrem A (3), but the tremorgenic and ataxic activity also associated with inhibition of BK channels remains a point of concern in the context of potential medicinal applications. In contrast, the reported attenuation of Wnt/β-catenin pathway by penitrem A (3) that may also contribute to its antiproliferative activity appears to be separate from the effects on potassium channels.75 Thus, other natural congeners lacking inhibitory activity towards BK channels retained the suppressing effect on the intracellular levels of β-catenin.
4. Chemical synthesis
The unique and diverse structural features, along with the multitude of biological activities associated with indole diterpenes of the paxilline type have made these fungal metabolites a subject of frequent scientific inquiry by organic chemists. To date, there have been sixteen reports describing syntheses of different congeners.16–29 Other publications detailed synthetic approaches and other chemistry studies relevant to these indoloterpenoids.30,31 A recent account by Smith and co-workers and a minireview by Garg and co-workers offer excellent discussion of selected relevant contributions.76,77 In the following sections, we will highlight efforts that have laid the foundation and introduced new directions to this field of natural product synthesis, and we conclude with a brief analysis and outlook on the challenges that lie ahead for the synthetic practitioner.
4.1. The Smith synthesis of paspaline
In 1985, the laboratory of Amos Smith III communicated their pioneering efforts in the landmark synthesis of paspaline (2, see Scheme 1).16 This report provided early insight into the reactivity of the unique polycyclic motif and also identified some of the key challenges associated with artificial assembly of this and related indole diterpenes. Smith and Mewshaw envisioned that establishment of the trans-hydrindane motif and the associated vicinal quaternary centers would take advantage of reductive alkylation of the corresponding cyclopentenone precursor, akin to the diastereoselective conversion of cyclopentenone 17 to hydrindanone 18 reported earlier by the Trost group in their synthesis of aphidicolin (Scheme 2).78 Ultimately, translation of this precedent to the synthesis of paspaline was met with modest success, and stereocontrolled construction of the desired stereotriad found in the indole diterpenes would become the subject of multiple investigations by the Smith and other laboratories in the next several decades.30,79–83 In contrast, clever solutions to the installation of the trans-decalin, indole, and tetrahydropyran fragments identified during the first synthesis of paspaline would enable many subsequent efforts in this field (see discussion below).
Scheme 2.
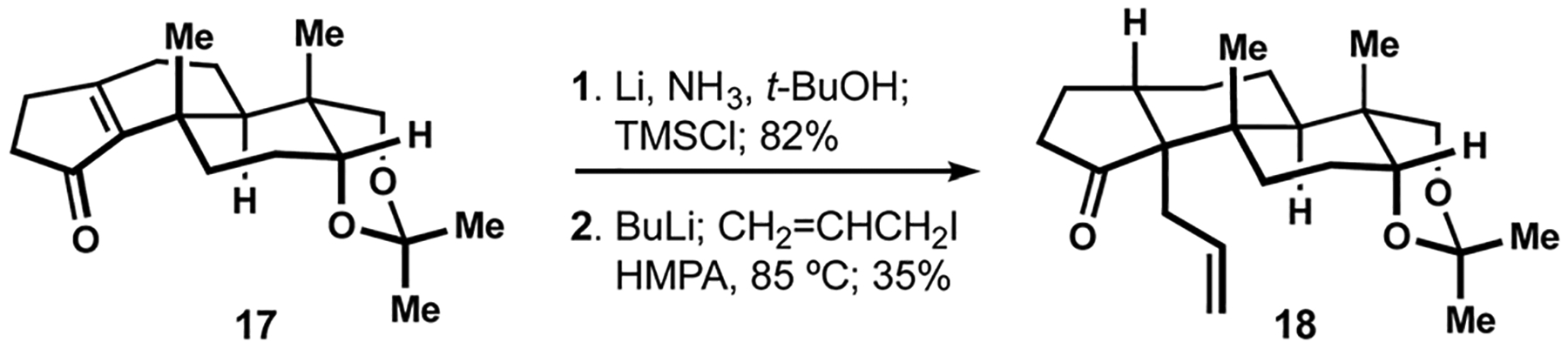
Reductive allylation en route to aphidicolin.
The synthesis commenced with selective protection of Wieland–Mieschler ketone (19) at C3 in the acid-catalyzed transketalization with dioxolane 20 (Scheme 3).84 Phenylthiomethylation of the unsaturated ketone 21 set the stage for a reductive allylation of the resulting sulfide 22 that proceeded with high diastereoselectivity and a preference for the desired trans-decalin motif containing quaternary stereocenter at C12. Reduction of ketone 23 resulted in good levels of stereocontrol, and subsequent hydrolysis of the dioxolane produced hydroxyketone 24. Assembly of the cyclopentenone fragment took advantage of the protocol developed by the Raphael group in their synthesis of strigol.85 Thus, addition of the alkynyllithium derived from a protected propargyl alcohol to ketone 24 and deprotection in the presence of dilute acid delivered triol 25 as an inconsequential mixture of diastereomers at C3. Brief exposure of the products to concentrated sulfuric acid presumably induced dehydration and rearrangement of the butynediol motif to the corresponding dienone intermediate, which was followed by a Nazarov cyclization to afford cyclopentenone 26 with modest efficiency. Protection of the secondary alcohol delivered silyl ether 27, thus setting the stage for installation of the trans-hydrindane motif. Initial experiments indicated a strong preference for the undesired stereochemical outcome during alkylation of the intermediate enolate. Desired ketone 28 was eventually obtained as a minor component of the product mixtures when methylation was performed at elevated temperatures. Although attempts to improve on the unexpected stereochemical outcome proved unsuccessful, this sequence provided access to the shared tricarbocyclic motif of the paxilline indole diterpenes, and allowed for methodological inquiry into the assembly of the indole and tetrahydropyran fragments. Elaboration of the latter fragment began with a formal anti-Markovnikov hydration of the allyl substituent to deliver alcohol 29, which was oxidized to the corresponding aldehyde 30. Wittig olefination proceeded chemoselectively and produced disubstituted alkene 31. Epoxidation of intermediate 31 followed by treatment of the reaction mixture with catalytic amounts of strong acid produced a mixture of diastereomeric alcohols 32 arising from intramolecular displacement of the intermediate epoxides. Oxidation to the mixture of corresponding ketones followed by treatment of base allowed for stereoselective access to diketone 33, amending the unselective installation of the stereocenter at C26 during the epoxidation event. Addition of methyl Grignard reagent to diketone 33 proceeded regioselectively and completed construction of the fully elaborated terpenoid fragment 34. Final assembly of the indole fragment proved to be a nontrivial undertaking, and attempts at formation of the corresponding hydrazone for Fischer indole synthesis were unsuccessful, pointing to the sterically demanding nature of the cyclopentatone motif. A suitable solution was identified in Gassman indole synthesis, which began with installation of the thioether functionality at C18.86 Treatment of the inconsequential mixture of diastereomeric methylthioethers 35 with N-chloroaniline was followed by reductive desulfurization to produce aminoketone 36 with an undetermined stereochemical configuration at C18. Contrary to expectations, dehydration of intermediate 36 required forcing conditions, and (−)-paspaline (2) was only obtained after prolonged exposure to strong acid at elevated temperatures. Similar challenges were noted in the subsequent synthetic studies, and became particularly exacerbated during the assembly of congeners containing elaborate indole motifs.28 The Gassman protocol originally executed for paspaline (2) was later employed in the Smith synthesis of paspalicine and paspalinine (16, see Scheme 1), and the Johnson synthesis of paspaline (2). The completion of the first synthesis of paspaline (2) prominently established stereoselective assembly of the hydrindane motif as one of the main challenges in the artificial assembly of the indole diterpenes.
Scheme 3.
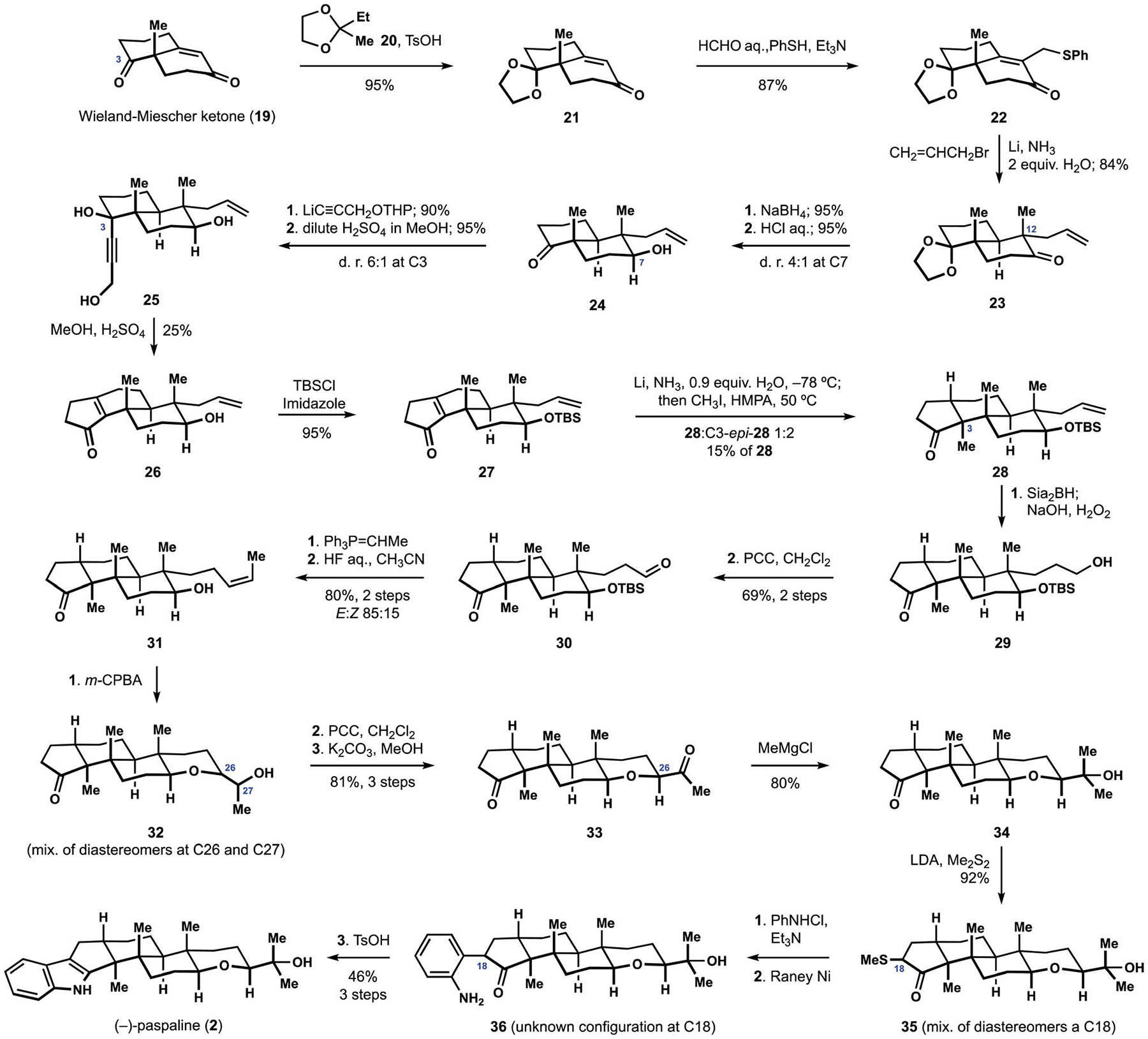
The Smith synthesis of (−)-paspaline (2).
Three years later, the Smith group reported the first of many efforts to develop a solution to the selective installation of the stereocenter at C3 (Scheme 4).80,87 The revised approach to the tricyclic fragment 28 relied on excellent stereocontrol in a conjugate addition to cyclohexenone 38, which was obtained from protected Wieland-Miescher ketone 37 by Robinson annulation with methyl vinyl ketone. Treatment of unsaturated ketone 38 with dimethylzinc in the presence of a nickel catalyst followed by trapping of the intermediate enolate with chlorosilane resulted in the efficient formation of enol ether 39 with the desired configuration at C16 and C3. Although highly diastereoselective, the new sequence also necessitated a ring contraction to establish the desired cyclopentanone motif. To this end, ozonolysis of enoxysilane 39 and esterification of the intermediate carboxylic acid produced ester 40, which underwent an intramolecular aldol reaction with the pendant aldehyde in the presence of strong base. Oxidation of alcohol 41 followed by hydrolysis and decarboxylation of the intermediate ketoester completed construction of cyclopentanone 42. Deprotection of the C7 ketone was accompanied by migration of the alkene to produce conjugated alkenone 43. Installation of the quaternary center at C12 took advantage of the previously developed sequence, to include formation of phenyl thioether 44 and subsequent reductive allylation to afford hydroxyketone 45 with undetermined stereochemical configuration at C2. Reduction of the protected hydroxyketone 46 and silylation of the newly formed secondary alcohol produced differentially protected diol 47 with good diastereoselectivity at C7. Ultimately, deprotection of the trimethylsilyl ether followed by oxidation of the intermediate alcohol afforded cyclopentanone 28, intercepting the previously established synthetic sequence to paspaline (2). Stereoselective access to intermediate 42 also provided robust platform for the subsequent synthesis of (+)-paspalicine and (+)-paspalinine (16, see Scheme 1).17 At the same time, the planned construction of elaborate indole motifs found in penitrems required a different set of functionalities in the terpenoid motif, leading to the development of alternative approaches.
Scheme 4.
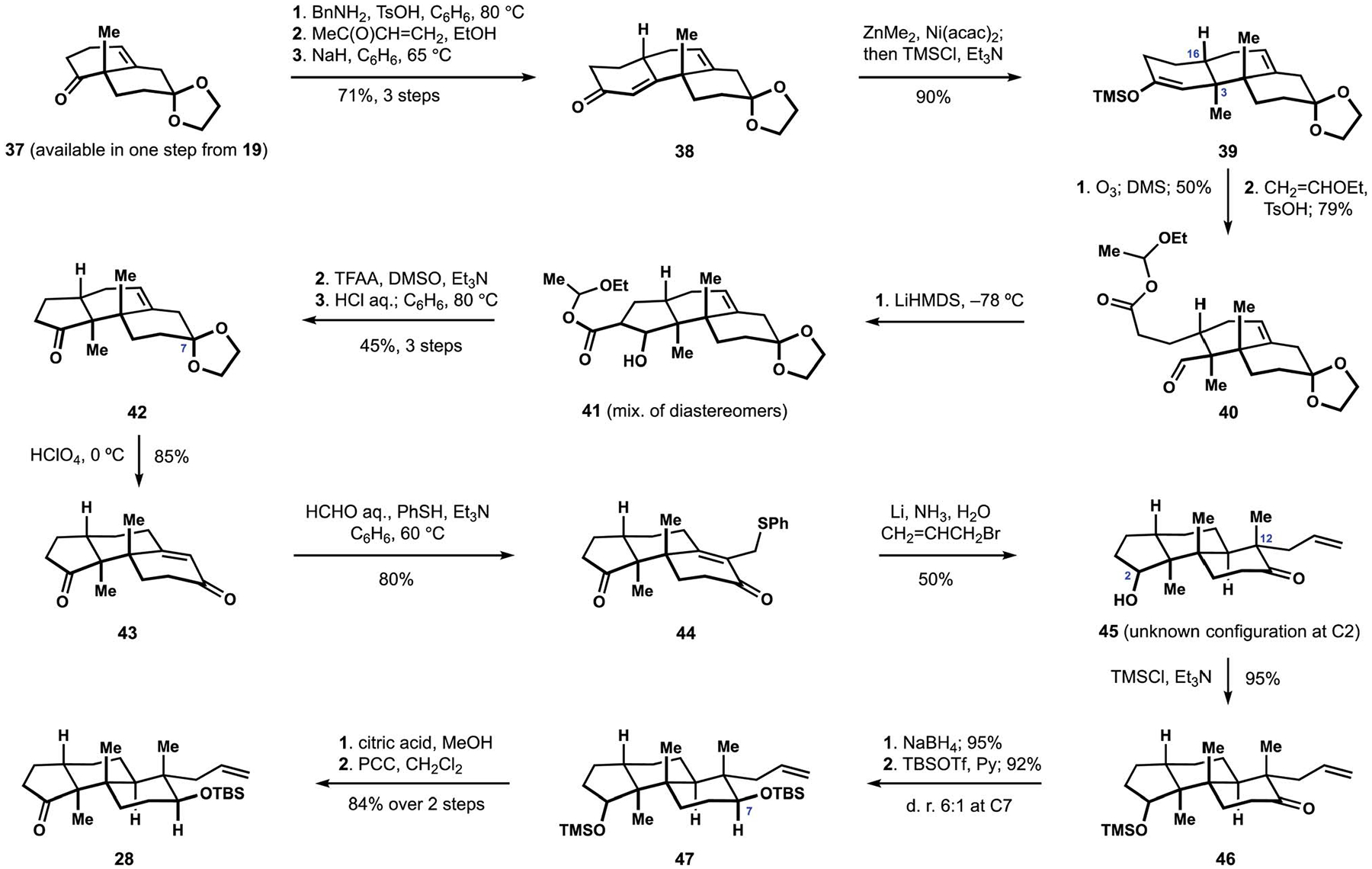
Revised synthesis of the tricyclic terpenoid motif by the Smith group.
4.2. The Smith synthesis of penitrem D
The Smith group’s successes in the synthesis of penitrems marked a tremendous milestone.18 Appropriately referred to as the most formidable accomplishment in the artificial assembly of indoloterpenoids, these monumental efforts provided new-insight into chemical reactivity of complex heterocyclic scaffolds.78 The cornerstone of synthetic strategy towards the penitrems was the convergent construction of the polycyclic indole fragment. To this end, the Smith group developed a new method that took inspiration from the Madelung synthesis.88 Initial studies demonstrated that a dilithio derivative of silylated toluidine 48 underwent efficient annulation with butyrolactone 49 to furnish indole 50 (Scheme 5), where the newly formed primary alcohol could serve as a handle for subsequent assembly of the fused hydrindane fragment.89 This strategy was subsequently applied in construction of other indole diterpenes, including (−)-21-isopentenylpaxilline and (+)-nodulisporic acid F (13, see Scheme 1).19,20
Scheme 5.
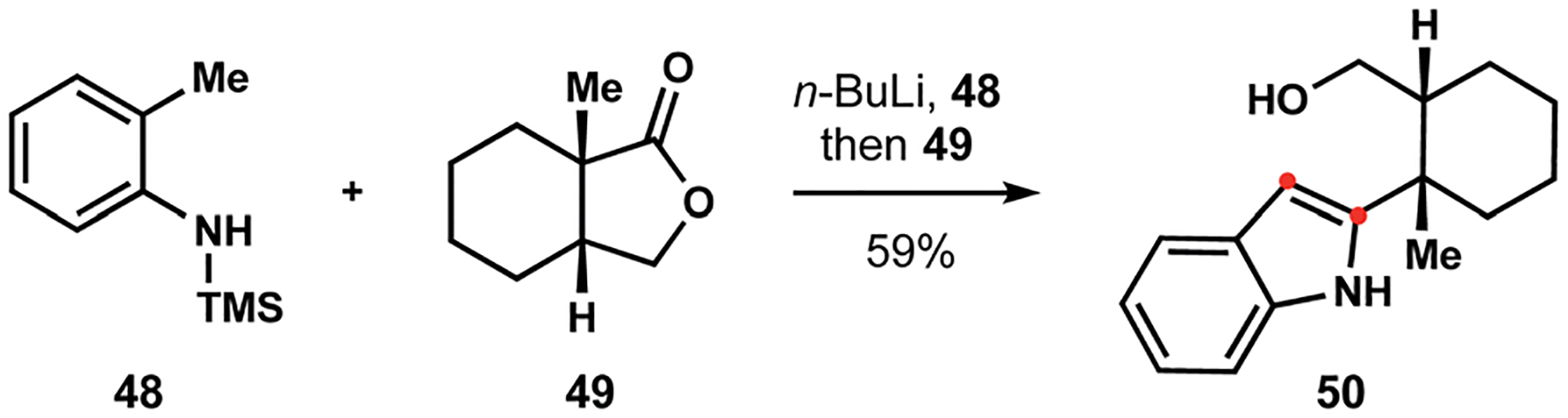
New indole synthesis developed by the Smith group.
In a continued effort to address the stereoselective installation of the vicinal quaternary centers at C3 and C4, the Smith group devised a new approach to the terpenoid fragment that was also suitable for the planned assembly of the indole motif.82 Construction of the requisite annulation precursor began with alkylation of the conjugated siloxydiene derived from enone 51 with dithienium salt 52 and proceeded with moderate diastereoselectivity, but nonetheless provided ready access to intermediate 53 in decagram quantities (Scheme 6).90 Reduction of the ketone followed by acetylation of the resulting alcohol furnished ester 54, which was subjected to deprotection of the aliphatic aldehyde and subsequent reduction to deliver alcohol 55. Two additional protecting group manipulations secured silyl ether 56, where the stereochemical configuration of the secondary alcohol was critical for installation of the methyl group at C3. Thus, cyclopropanation of alkene 56 proceeded in a highly diastereoselective manner by leveraging the directing effect of the adjacent hydroxy group. Subsequent oxidation delivered ketone 57, which underwent cleavage of the cyclopropane fragment to produce the desired methyl substituent upon a dissolving metal reduction. The resulting sodium enolate was trapped as the corresponding enol triflate, and displacement with Gilman reagent furnished alkene 58 with the requisite configuration of the ring fusion. A related strategy for the stereoselective installation of vicinal quaternary centers in the tricarbocyclic motif of the indole diterpenes was previously developed by the Saxton group, and later adopted by the Kuwahara group in their synthesis of paspalinine (16, see Scheme 1) and paspalicine as discussed below.21,30 Oxidative cleavage of alkene 58 was followed by an acid-catalyzed lactonization, which was accompanied by hydrolysis of the dioxo-lane. Intramolecular aldol condensation of the intermediate diketone established the carbocyclic motif found in the terpenoid fragment of the penitrems. Installation of the tetrahydropyran moiety followed the approach developed earlier by the Smith group for their synthesis of (+)-paspalicine and (+)-paspalinine (16, see Scheme 1).17 In three synthetic manipulations, lactone 59 was converted to acetal 60, which was produced as a mixture of diastereomers at C2.18 Preparation of the corresponding hydrazone 61 enabled Stork alkylation with epoxide 63, which was prepared in five steps from glyceraldehyde acetonide.91,92 Critical to the success this transformation was the addition of sacrificial hydrazone 62, which presumably promoted formation of the requisite metalloenamine. Benzoylation of intermediate secondary alcohol 64 followed by hydrolysis of the hydrazone allowed for isolation of acetal 65, which was converted to corresponding lactone 66 in two straightforward manipulations. After significant optimization, reductive cyclization of unsaturated ketone 66 could be accomplished with good efficiency and high diastereoselectivity, to deliver the desired tetrahydropyran 67. Attempts at direct installation of the tertiary allylic alcohol at C13 upon reaction with singlet oxygen were unsuccessful, and an alternative solution to this problem was devised. Hydrolysis of benzoate 67, accompanied by partial cleavage of the lactone that was remedied by intramolecular esterification, was followed by oxidation of the resulting alcohol 68 to ketone 69. The presence of the carbonyl functionality allowed for aerobic oxidation of extended enolate derived from the b,g-unsaturated ketone under mild conditions, and subsequent reduction of the intermediate hydroperoxide furnished the desired tertiary alcohol 70. The protected secondary alcohol at C10 was reestablished in two synthetic manipulations, to deliver the annulation partner 71.
Scheme 6.
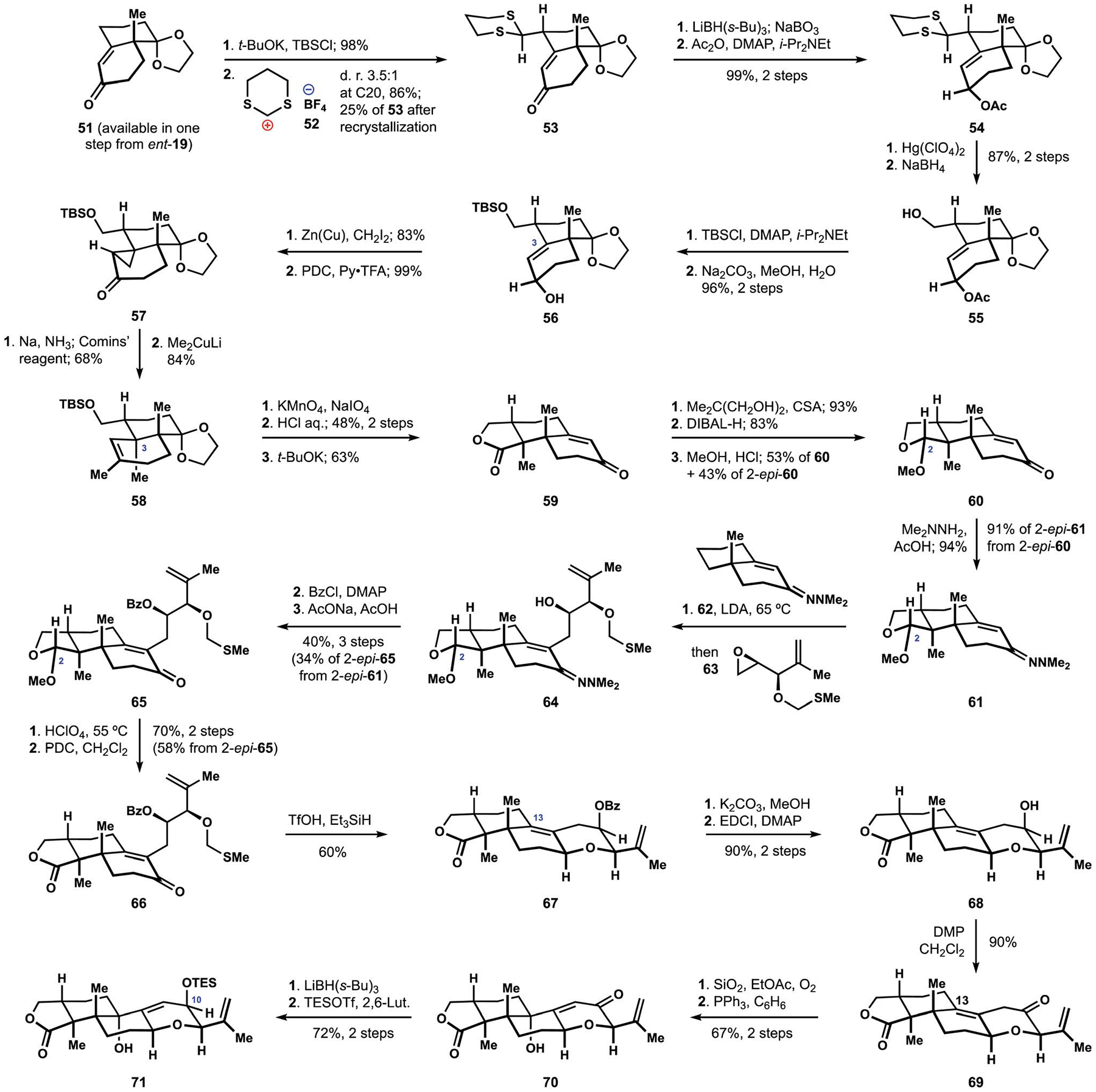
Synthesis of the polycyclic terpenoid motif of penitrems by the Smith group.
Assembly of the toluidine component in the synthesis of penitrem D relied on installation of the fused cyclobutane and aromatic motifs on cyclohexenone precursor 74 (Scheme 7).81 The enantioselective synthesis of unsaturated ketone 74 took advantage of desymmetrizing enolization of cyclohexanone 73, which was prepared in three straightforward manipulations from commercially available precursor 72.93 Photocycloaddition of alkene 74 with methyl acrylate delivered cyclobutane 75, where the ester functionality served as a starting point for construction of dimethylcarbinol 76.18 Claisen condensation of the protected derivative with ethyl formate set the stage for Robinson annulation of ketoaldehyde 77 to produce tricyclic motif 78. Semmler-Wolff aromatization of ketone 78 established toluidine fragment 79, and a series of protecting group exchanges ultimately produced annulation partner 80, which contained a combination of functionalities deemed suitable for subsequent elaborations.94
Scheme 7.
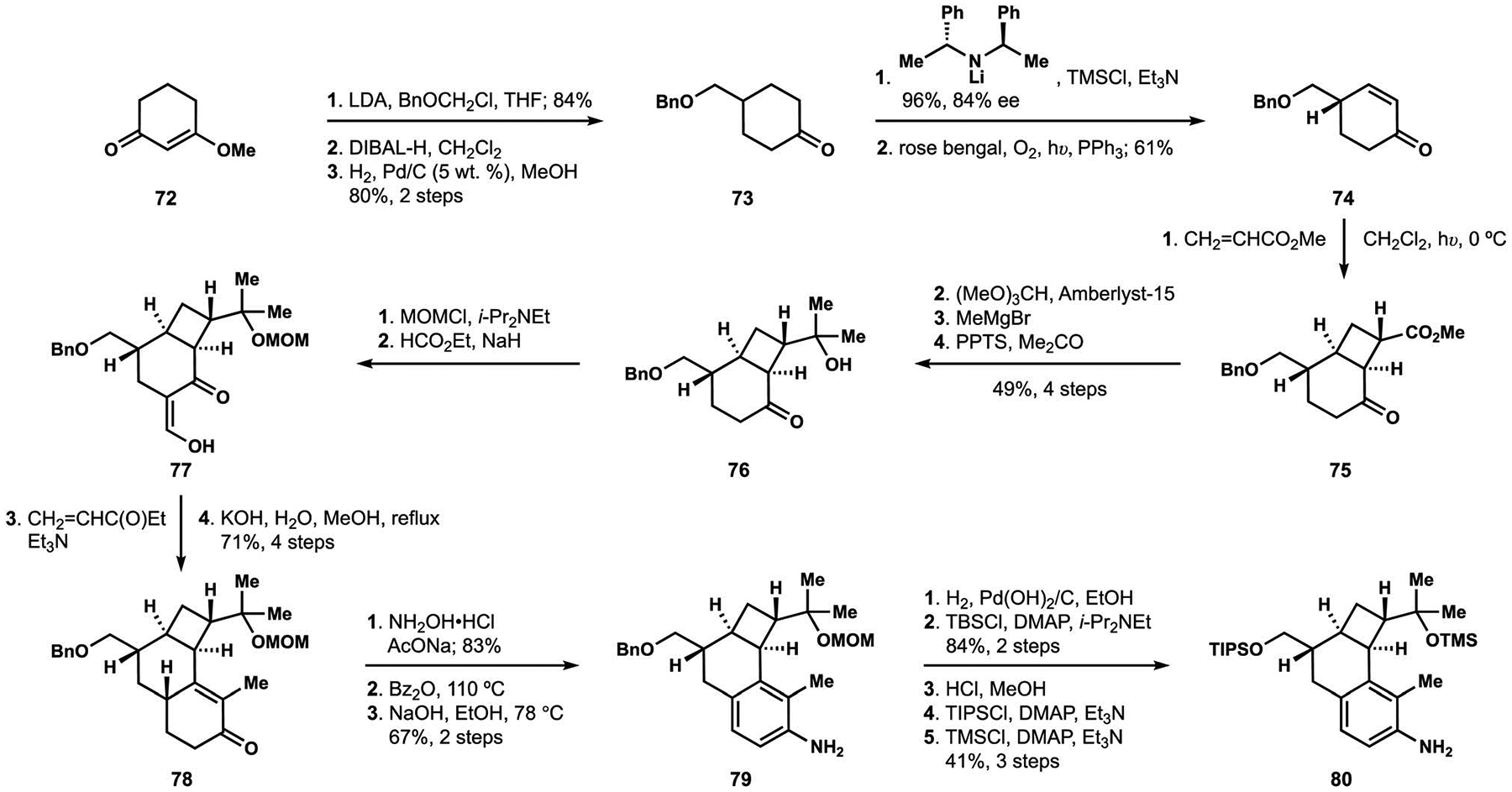
Construction of the toluidine fragment by the Smith group.
The union of the terpenoid and the aniline fragments necessitated treatment of lactone 71 with a large excess of a dilithio derivative obtained from toluidine 80 following in situ formation of the corresponding N-trimethylsilyl amine (Scheme 8). Brief exposure of the product mixture to silica gel ensured complete formation of desired indole 81 with the necessary handles for installation of the oxocane motif. Oxidation of the primary alcohol was followed by hydrolytic removal of the silyl ethers, and the resulting aldehyde underwent double cyclization upon exposure to a strong Lewis acid at elevated temperatures to produce oxocane 82. Protecting group manipulations allowed for dehydration of the primary alcohol according to the Grieco elimination protocol, and hydrolysis of the temporarily installed acetate completed the synthesis of (−)-penitrem D (83).18,95 To date, this accomplishment stands as the only synthesis of a penitrem and highlights the challenges associated with artificial assembly of these remarkable indoloterpenoid targets.
Scheme 8.
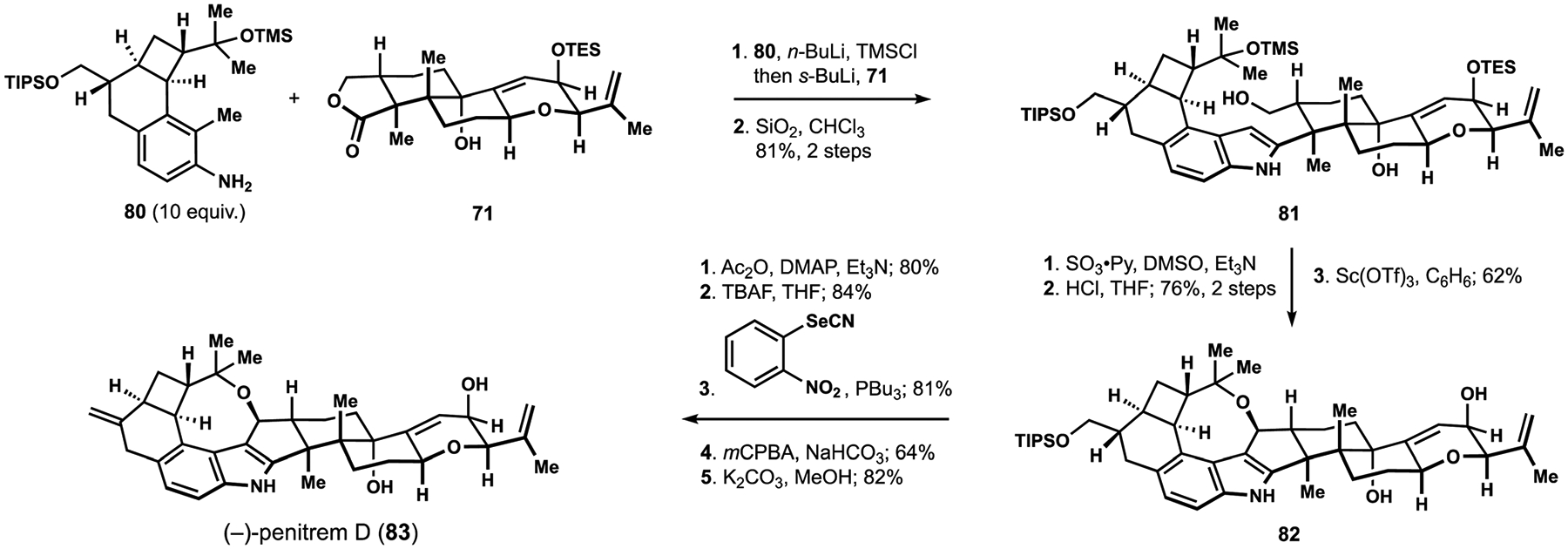
The Smith synthesis of (−)-penitrem D (83).
4.3. The Kuwahara synthesis of paspalinine and the saxton approach to the terpenoid core
Subsequent to the report from the Smith group detailing their synthesis of paspalicine and paspalinine (16, see Scheme 1), the Saxton group disclosed their own solution to the stereoselective assembly of the tricarbocyclic motif found in these natural products.30 Installation of the angular methyl group relied on cyclopropanation of a cyclopentene motif and took advantage of the directing effect in allylic alcohol 87 (Scheme 9). Preparation of this intermediate commenced with propargylation of ketone 37 with iodide 84 to deliver alkyne 85, which underwent hydration to the corresponding b-ketophosphonate, and intramolecular Horner–Wadsworth–Emmons olefination completed construction of cyclopentenone 86. Reduction of the ketone with a sterically-demanding hydride complex produced desired alcohol 87, providing the necessary handle for stereocontrol during the cyclopropanation. Treatment of the alkoxide derived from intermediate 87 with Simmons-Smith reagent accomplished the highly diastereoselective constriction of the new carbon–carbon bond at C3 of cyclopropane 88. Oxidation of the resulting secondary alcohol reestablished the carbonyl functionality at C18 and allowed for reductive cleavage of the cyclopropane motif to complete installation of the critical angular methyl at C3 in ketone 89. The Saxton group speculated that trapping of the intermediate enolate would allow for subsequent regioselective installation of the indole moiety, but did not report relevant experimental studies.
Scheme 9.
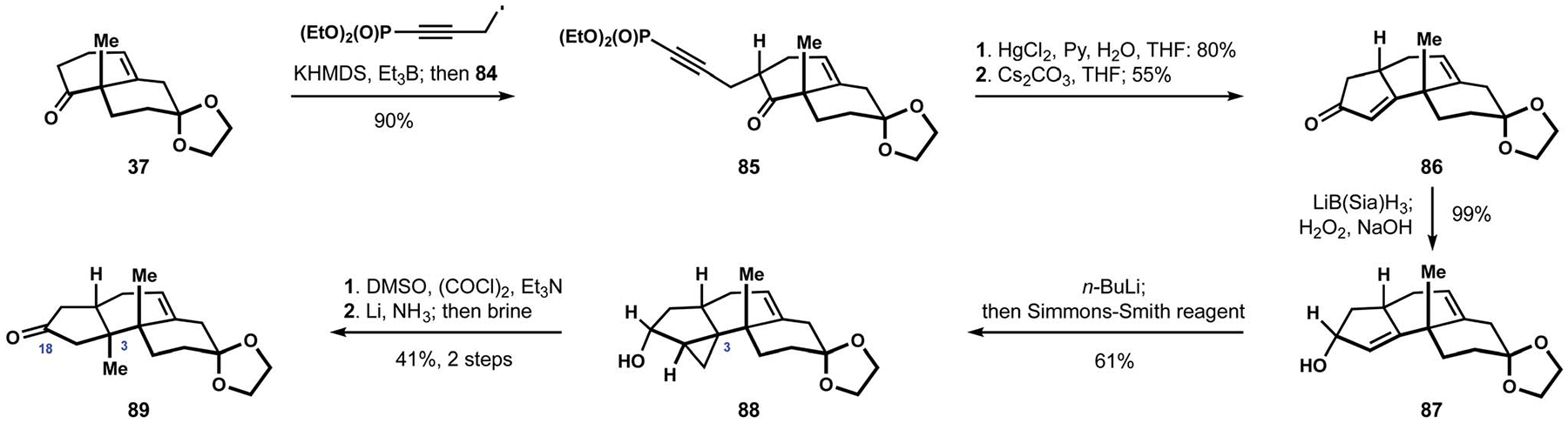
The Saxton approach to the terpenoid core.
This clever solution to the assembly of trans-hexahydrindane motif was subsequently employed by the Kuwahara group in their synthesis of (−)-paspalinine (16).21 In a closely related sequence, the Kuwahara group prepared cyclopentanone 92 from ketone 37 and allylic bromide 90 (Scheme 10). The enol ether in alkylating agent 90 was chosen as an alternative surrogate to the b-ketophosphonate fragment of intramolecular olefination precursor 91, which was obtained with similar efficiency. Reductive cleavage of cyclopropyl ketone 92 was followed by sulfonylation of the resulting sodium enolate to produce triflate 93. The regiospecific generation of the enol sulfonate provided a suitable starting point for construction of the indole fragment. To this end, the Stille cross-coupling with arylstannane 94 produced aniline derivative 95, and a subsequent oxidative cyclization at the styrene motif delivered the pentacyclic indoloterpenoid core 96.96 Installation of the bicyclic ketal found in the target natural product commenced with preparation of allyl enol carbonate 97, which then underwent a Tsuji–Trost reaction to deliver terminal alkene 98.97 Following cross metathesis with 2-methyl-3-buten-2-ol, a sharpless asymmetric dihydroxylation of the resulting allylic alcohol 99 proceeded with low stereoselectivity, but the desired intermediate 100 constituted the major component in the inseparable mixture of diastereomeric triols. Intramolecular ketalization and oxidation of the remaining secondary alcohol allowed for isolation of ketone 101 as a single diastereomer. Installation of the tertiary alcohol at C13 was accomplished by phenylselenylation of the extended enolate followed by oxidation and rearrangement of the intermediate selenoxide. Ultimately, deprotection of the indole fragment delivered (−)-paspalinine (16).21 These efforts informed subsequent studies by the Kuwahara group in their synthesis of lecanindole D and terpendole E (14, see Scheme 1).23
Scheme 10.
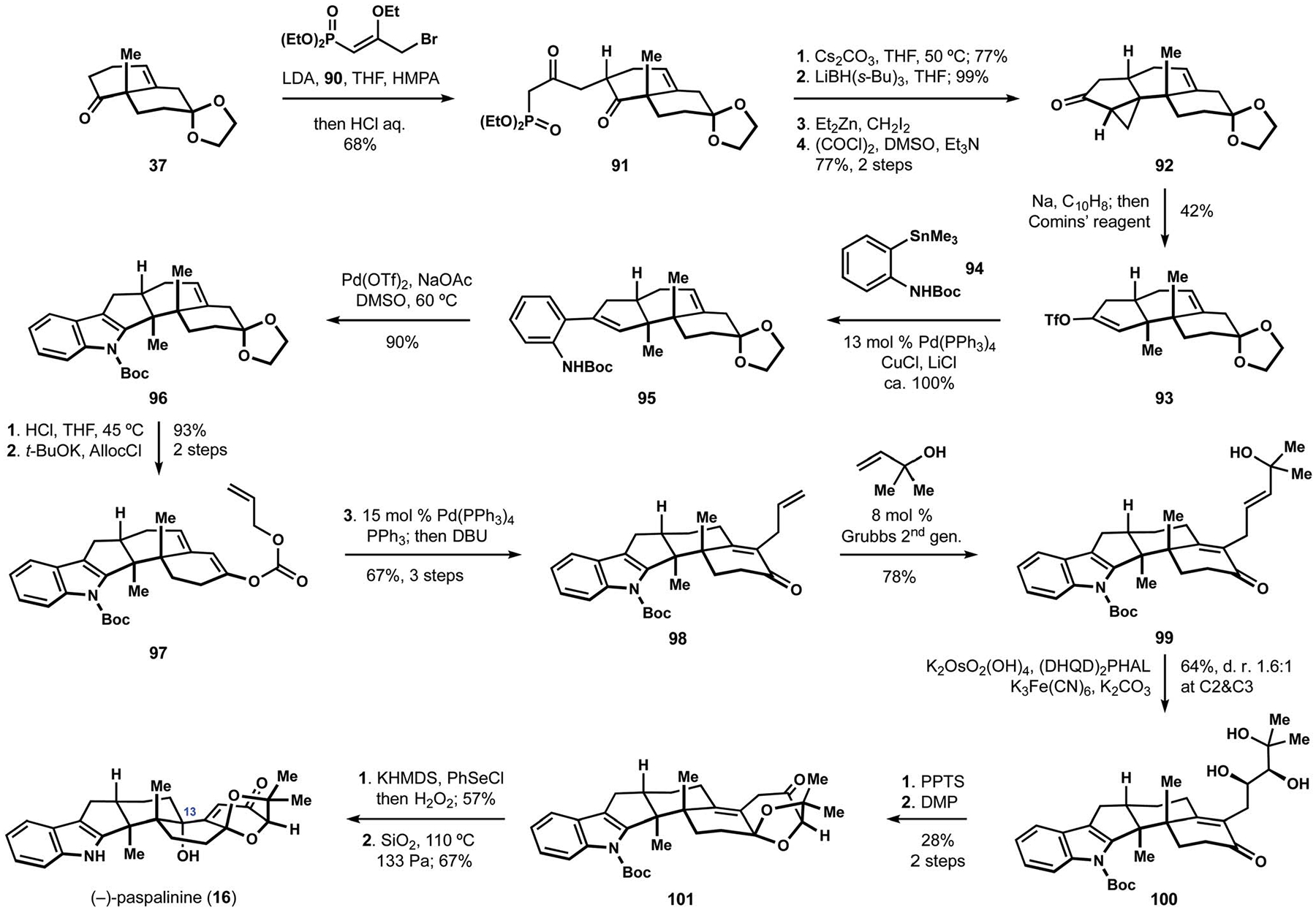
The Kuwahara synthesis of (−)-paspalinine (16).
4.4. The Johnson synthesis of paspaline
In contrast to the previous studies in the field, the synthesis of paspaline by the Johnson group was based on a markedly different approach and aimed to establish the tetrahydropyran motif early in the synthetic sequence.24 To this end, reductive desymmetrization of diketone 102 secured access to hydroxyketone 103, which provided the necessary functional groups for construction of the heterocyclic fragment (Scheme 11). Experiments with epoxidation of the alkene revealed that the conversion of the remaining ketone to the corresponding hydrazone was critical to obtaining high levels of diastereoselectivity and enhanced reactivity in the subsequent intramolecular displacement. Ultimately, tetrahydropyran 104 was produced with a strong preference for the desired configuration at C9 after treatment of the intermediate epoxide with catalytic amounts of a pyridinium salt. Protection of the tertiary alcohol was followed by methylation of a dilithio derivative of the corresponding hydrazone, and a subsequent Shapiro reaction allowed for trapping of the intermediate alkenyllithium with formaldehyde to deliver allylic alcohol 105. Assembly of the hydrindane motif commenced with Ireland–Claisen rearrangement of silyl ketene acetal derived from isobutyrate 106, where the desired stereochemical outcome was attributed to the presence of angular methyl substituent at C12. The resulting unsaturated acid 107 was converted to corresponding unsaturated ketone 108, which underwent hydroboration and subsequent oxidation of the intermediate alkylborane to deliver product 109 as an inconsequential mixture of diastereomeric secondary alcohols. Oxidation of diol 106 followed by an intramolecular aldol condensation of the corresponding ketoaldehyde produced cyclohexenone 110. Assembly of the cyclopentane ring required selective functionalization of the equatorial methyl substituent at C3, which was accomplished by application of the Sanford protocol for directed acetoxylation.98 The requisite oxime 111 was prepared by hydrogenation of unsaturated ketone 110 and subsequent condensation with benzyloxyamine. In accord with the prediction, functionalization of substrate 111 proceeded with high selectivity for the equatorial methyl substituent to generate the desired acetate 112. Hydrolytic cleavage of the oxime, the ester, and the silyl ether was followed by oxidation of the intermediate triol to produce ketoaldehyde 113, and double addition of a vinyl nucleophile afforded diene 114 as a mixture of diastereomers. Ring-closing metathesis of intermediate 114 delivered tetracyclic triol 115, which underwent acid-catalyzed dehydration to unsaturated ketone 116. While attempts at hydrogenation of the trisubstituted alkene resulted exclusively in the undesired configuration at C16, reduction of the carbonyl group provided an opportunity for leveraging the directing effects of alcohol 117. Thus, exposure of this intermediate to Crabtree’s catalyst under an atmosphere of hydrogen provided the desired trans-hydrindane motif with high diastereoselectivity, and subsequent oxidation of the secondary alcohol produced cyclopentanone 34, intercepting the Smith route to (−)-paspaline (2).16,99 Application of the Gassman indole synthesis according to the previously developed protocol delivered the target natural product. The Johnson synthesis of (−)-paspaline (2) reaffirmed that directing effects are capable of providing a high degree of diastereocontrol during the assembly of the challenging trans-hydrindane motif.24 These findings echoed earlier observations from the Saxton and the Smith groups and emphasized the utility of related stereochemical strategies in the synthesis of paxilline indole diterpenes.18,30,82
Scheme 11.
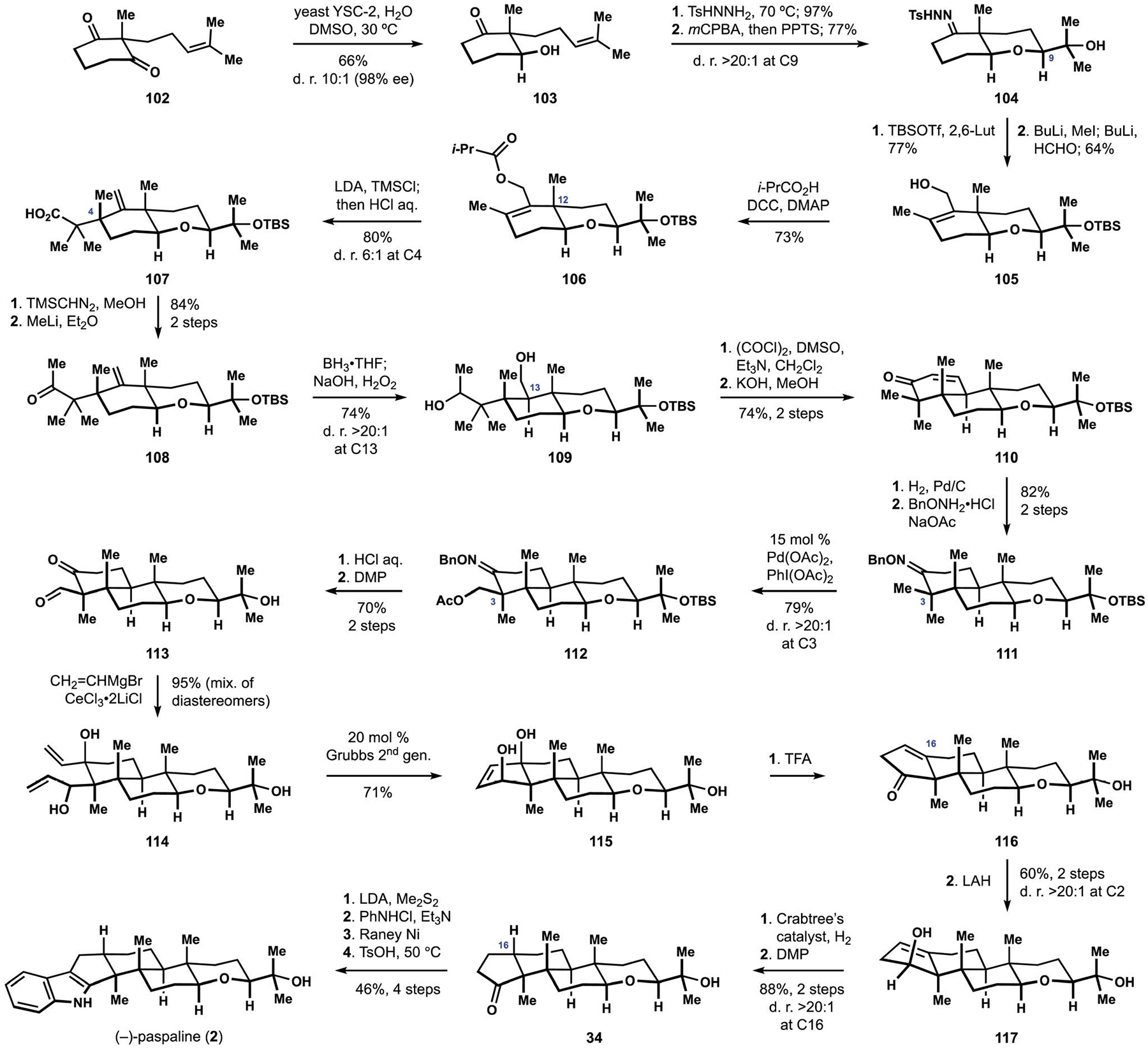
The Johnson synthesis of (−)-paspaline (2).
4.5. The Smith synthesis of nodulisporic acids B, C and D
Continued efforts by the Smith group in the synthesis of the indole diterpenes recently culminated in the development of convergent routes to nodulisporic acids B, C, and D.25,27 These congeners possess elaborate and labile indole moieties that hinder application of the previous methods, including the Gassman and the modified Madelung synthesis, that were successful en route to other compounds from this family of natural products.16,18 In their search for alternative approaches to indole assembly, the Smith group discovered that the Barluenga protocol provided efficient access to strained pyrroloindole motifs akin to those found in complex nodulisporic acids.100 Thus, Buchwald-Hartwig amination of enol triflate 118 with chloroindoline 119 resulted in efficient formation of polycyclic indole 120 (Scheme 12).25 This strategy proved generally applicable to the construction of relevant motifs to potentially address one of the long-standing challenges in the synthesis of the paxilline indole diterpenes.
Scheme 12.
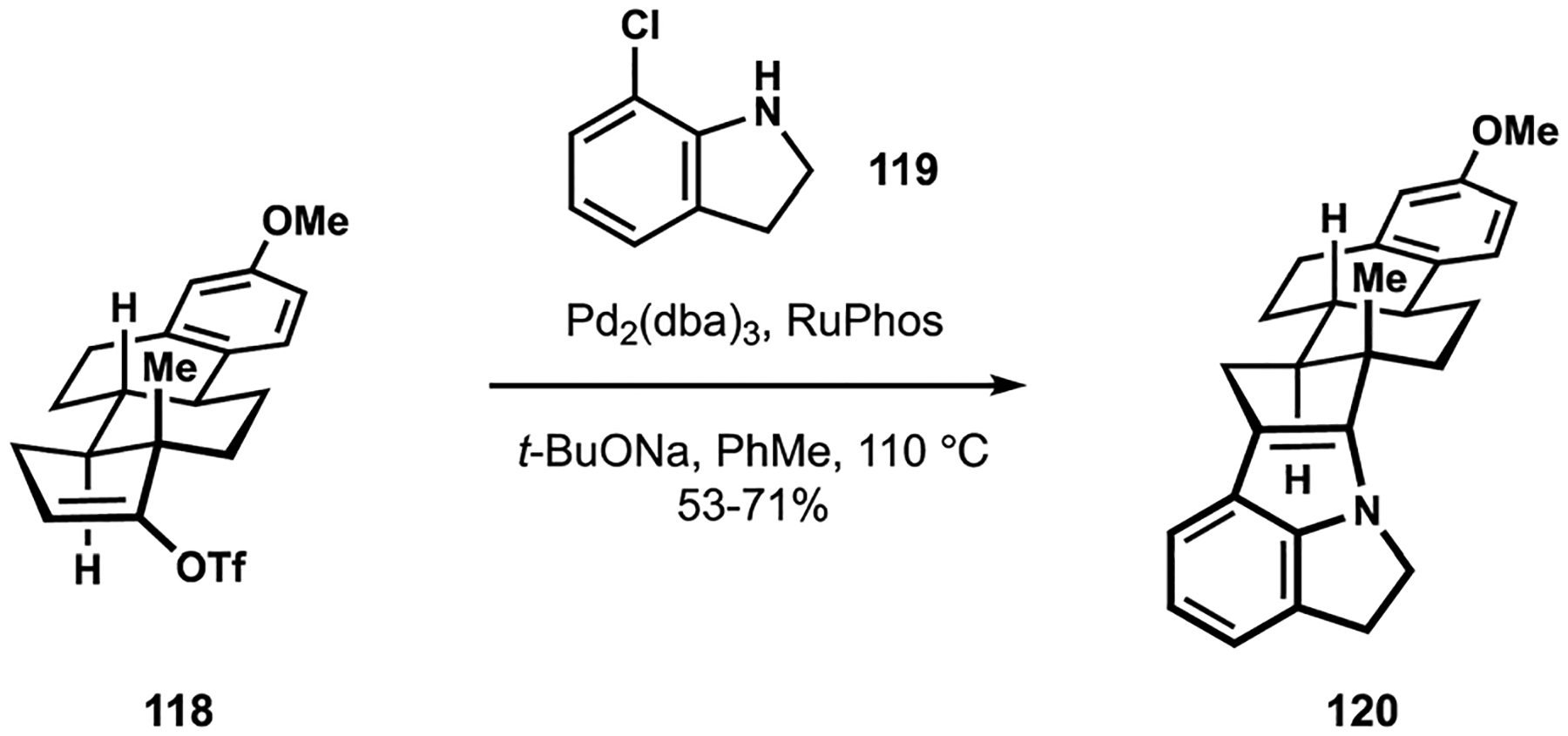
Fragment coupling en route to nodulisporic acids developed by the Smith group.
The assembly of the requisite terpenoid fragment built on the previous routes from the Smith group, beginning with dissolving metal reduction of unsaturated ketone 22 and hydroxymethylation of the intermediate silyl enol ether (Scheme 13). Directed reduction of aldol 121 followed by a sequence of protecting group manipulations delivered ketone 122 with the desired stereochemical configuration at C7. Carbonylation of the corresponding enol triflate produced unsaturated aldehyde 123, which was previously employed by the Smith group in their synthesis of (+)-nodulisporic acid F (13, see Scheme 1).20 In an improved protocol that avoided the need for a chiral auxiliary and epimerization at C12, substrate 123 underwent addition of a vinyl cuprate, delivering enoxysilane 124 in a highly diastereoselective manner.25 Methylation of the lithium enolate generated from enol derivative 124 proceeded efficiently under carefully optimized conditions and afforded unsaturated aldehyde 125 as the desired stereoisomer at C3. Installation of the five-membered carbocycle was accomplished by ring-closing metathesis of a diene obtained upon addition of a vinyl Grignard reagent to aldehyde 125, and subsequent oxidation of the intermediate allylic alcohol delivered cyclopentenone 126. Notably, formation of the cyclopentanone motif could also be accomplished by intramolecular hydroformylation of the vinyl substituent in the presence of a rhodium complex. Although attempts to render this transformation catalytic in metal were unsuccessful, it was capable of providing an appealing starting point for securing rapid access to the tricyclic terpenoid core. Hydrogenation of alkene 126 was followed by deprotection of the 1,3-diol motif and selective oxidation of the primary alcohol to produce hydroxyaldehyde 127. Silylation of the secondary alcohol and conversion of the ketone to the corresponding enol triflate produced intermediate 128, which, together with related derivatives of aldol 127, would become a valuable coupling partner in the synthesis of several nodulisporic acids.
Scheme 13.
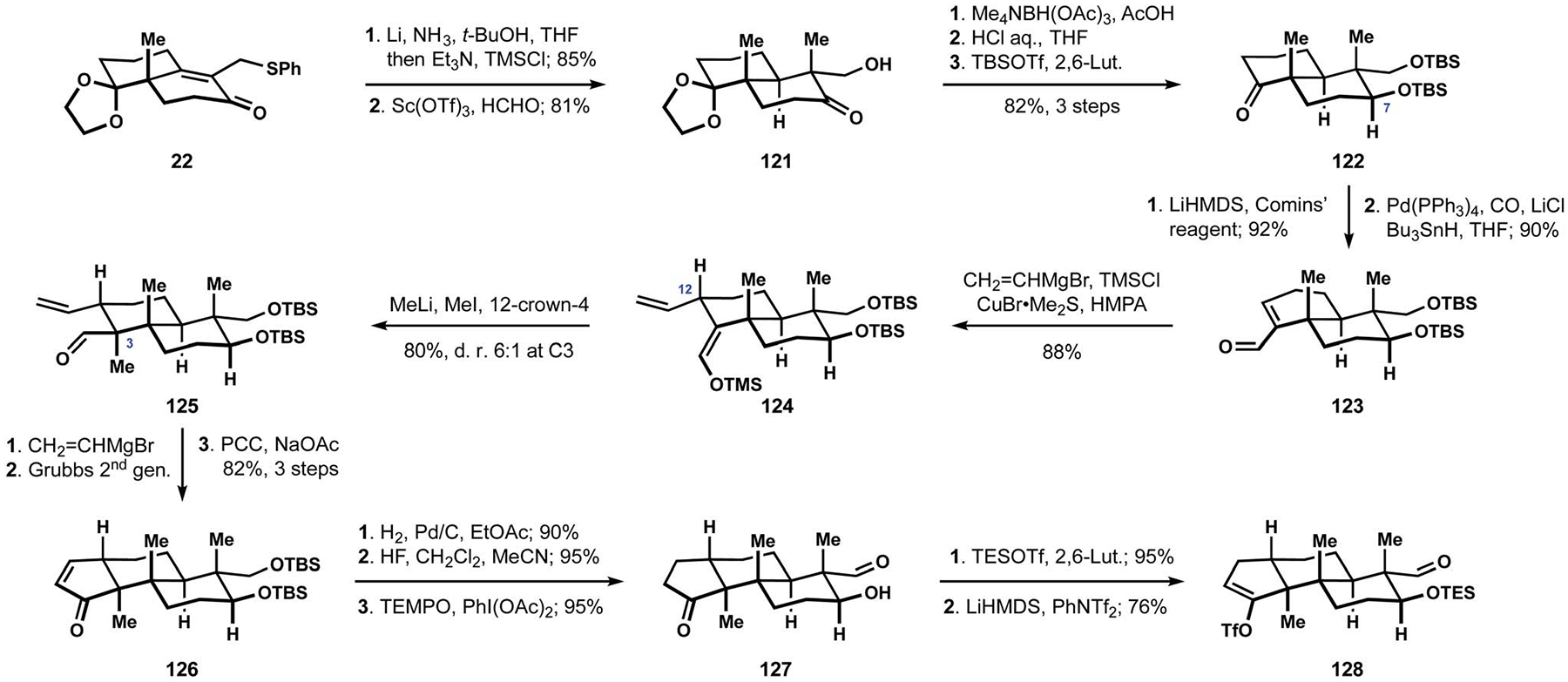
Refined and abbreviated construction of the tricyclic terpenoid motif by the Smith group.
Assembly of the aniline component of nodulisporic acid D (137) commenced with an Enders alkylation of hydrazone 129 with benzyl iodide 130, which was prepared in two steps from the corresponding benzoic acid (Scheme 14).101,102 Oxidative removal of the auxiliary delivered ketone 131. Installation of the enol triflate was followed by reduction of the nitroarene, enabling iodination of the resulting aniline to deliver iodide 132. Stille–Kelly reaction allowed for an intramolecular reductive coupling of the alkenyl triflate and the aryl iodide and produced desired annulation partner 133.103 The critical union of fragments 128 and 133 took advantage of conditions that had been identified earlier during model studies and afforded the complete indoloterpenoid motif 134, which is found in the structure of nodulisporic acid D (137).25 The silyl-protected aldol motif proved challenging to engage in an olefination event and required protecting group interchange to generate acetate 135. These observations informed subsequent studies and prompted revision of the optimal annulation partner for the Buchwald–Hartwig amination (see below). Horner–Wadsworth–Emmons olefination of the aldehyde with phosphonate 136 followed by saponification completed synthesis of nodulisporic acid D (137), marking the first entry into these complex metabolites.
Scheme 14.
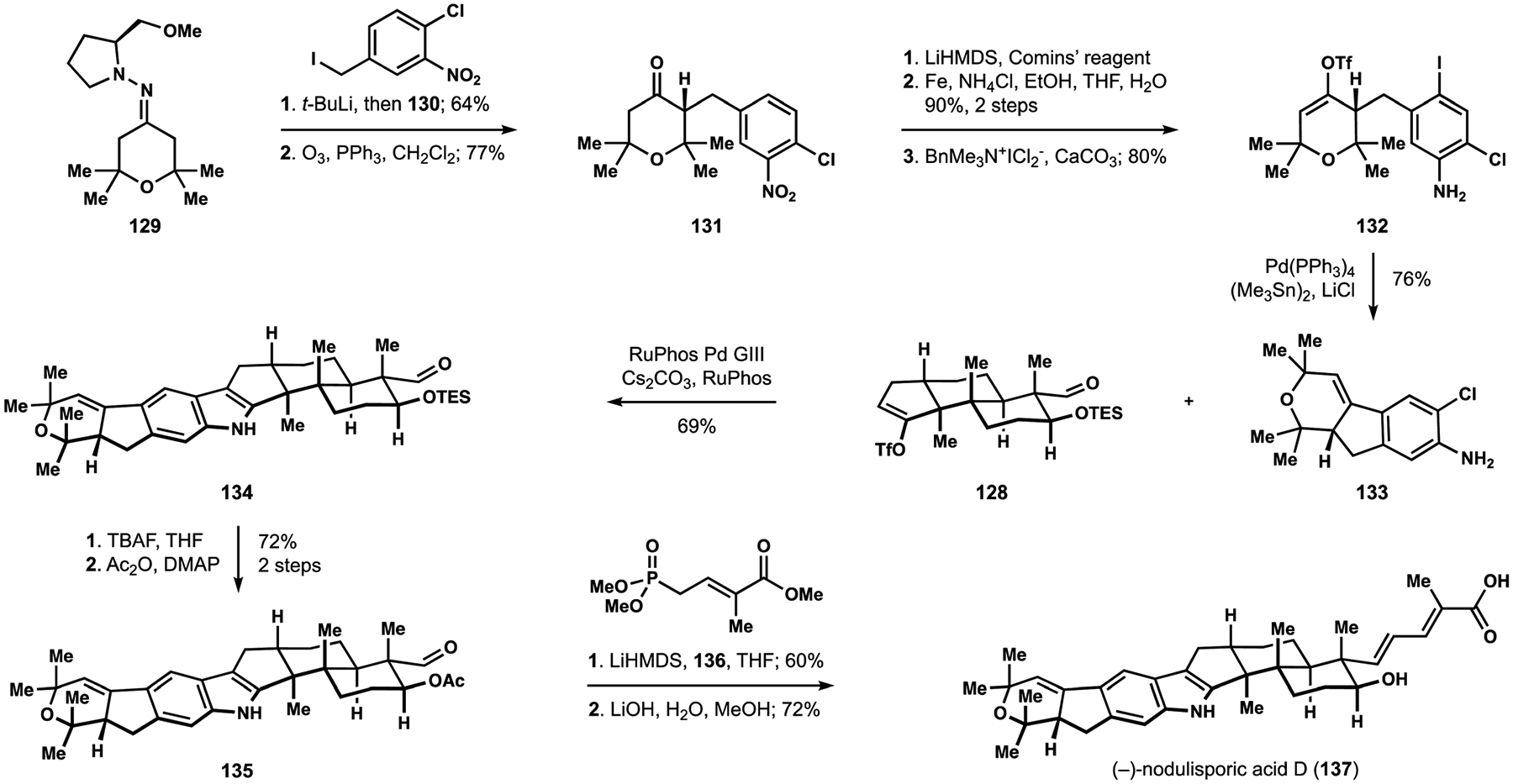
The Smith synthesis of (−)-nodulisporic acid D (137).
Incorporation of a secondary alcohol and an additional substituent in the indenopyran fragment of nodulisporic acids A, B, and C required modifications to the assembly of the aniline annulation partners.27 Enders aldol reaction between hydrazone 129 and benzaldehyde 138, accessible in three steps from a commercially available material, delivered secondary alcohol 139 with the desired stereochemical configuration (Scheme 15).104 Oxidative cleavage of the hydrazone provided starting material for an intramolecular Nozaki–Hiyama–Kishi reaction to generate diol 140, which was subjected to selective protection of the secondary alcohol and dehydration to styrene 141.105 Attempts at metalation of arene 141 suggested that free alcohol 142 was an optimal substrate for generation of nucleophilic species at C23. Thus, treatment of intermediate 142 with a large excess of tert-butyllithium followed by addition of a copper salt allowed for efficient prenylation to deliver alkene 143. Reestablishment of the silyl ether followed by deprotection of the aniline provided annulation partner 144 containing the substitution pattern found in nodulisporic acid C (148). Direct translation of the previously developed protocol to the Buchwald–Hartwig amination to aniline 144 and newly prepared alkenyl trifiate 145 was unsuccessful, but extensive evaluation of alternative phosphine ligands allowed for identification of APhos as a superior performer en route to annulation product 146.106 Completion of the synthesis followed the established Horner–Wadsworth–Emmons olefination with phosphonate 136, and saponification of intermediate ester 147 with subsequent cation exchange secure access to the sodium salt of nodulisporic acid C (148), which facilitated characterization of the product.27 Successful installation of the prenyl substituent at C23 also provided a starting point for assembly of the more complex congener, nodulisporic acid B (158, Scheme 16).
Scheme 15.
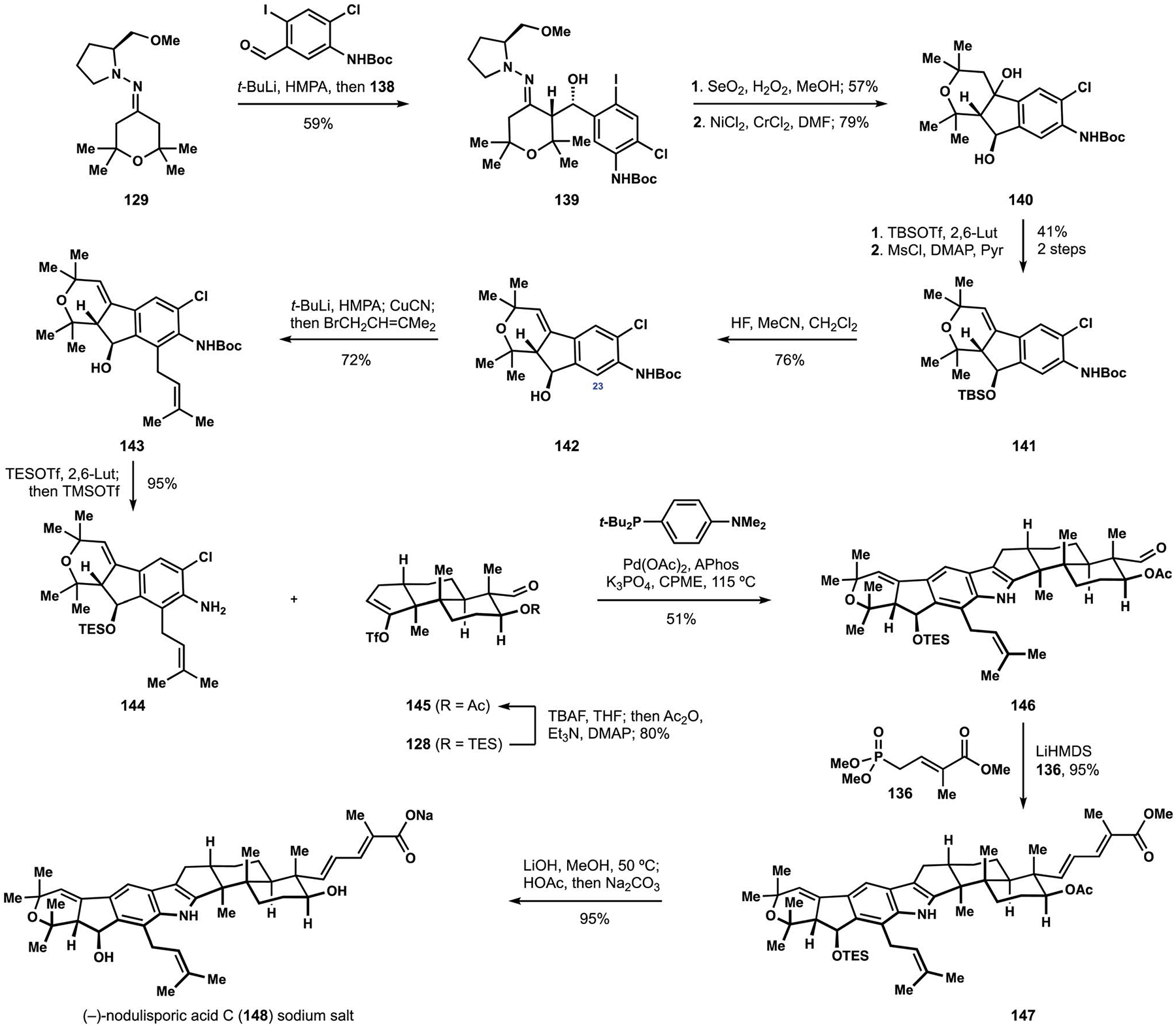
The Smith synthesis of (−)-nodulisporic acid C (148).
Scheme 16.
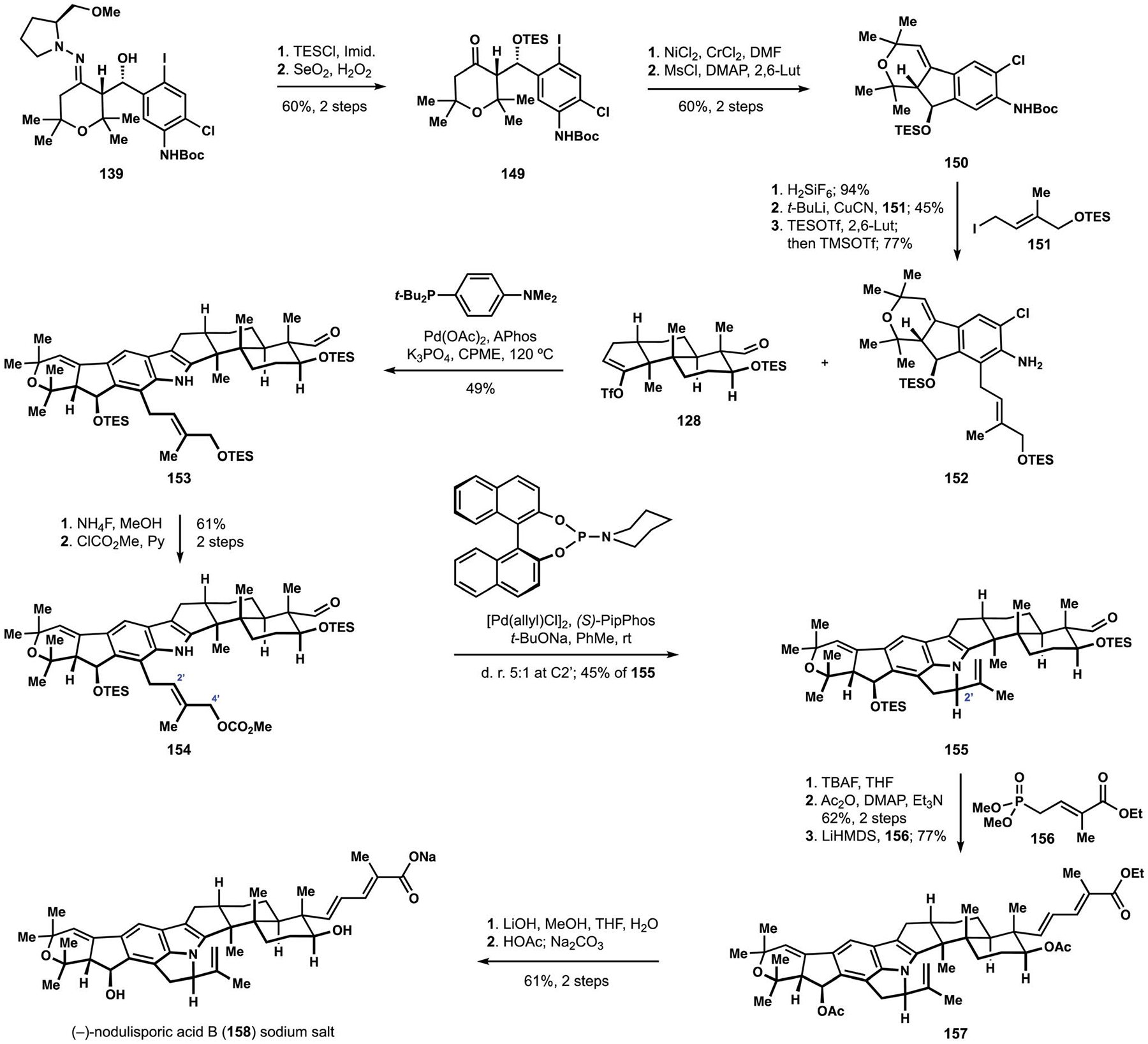
The Smith synthesis of (−)-nodulisporic acid B (158).
The envisioned construction of the pyrroloindole motif in the target natural product required access to intermediate 152, which contained the necessary functionality for an intramolecular conjugate displacement in the prenyl fragment.27 Synthesis of aniline 152 proceeded in a manner closely related to the assembly of intermediate 144 en route to nodulisporic acid C (148), but with several modifications that increased the overall efficiency of the sequence. Thus, protection of the secondary alcohol of aldol product 139 allowed for improved outcomes in subsequent auxiliary removal by attenuating elimination to the unsaturated ketone. Nozaki–Hiyama–Kishi cyclization of substrate 149 followed by elimination of the intermediate tertiary alcohol delivered styrene 150 as single regioisomer without competing formation of the corresponding indene that was observed in the previous studies. Cleavage of the silyl ether enabled alkylation with iodide 151 according to the established protocol, affording aniline 152 after re-protection of the secondary alcohol and removal of the carba-mate. In this setting, alkenyl triflate 128 was chosen as an annulation partner to address the apparent instability of the secondary acetate in the tricyclic terpenoid fragment 145 that was employed during the early investigations into the construction of the pyrroloindole motif. Selective deprotection of the allylic alcohol in indole 153 followed by installation of the corresponding carbonate delivered cyclization precursor 154. Initial attempts at the intramolecular Tsuji–Trost amination led exclusively to the corresponding diene resulting from b-hydride elimination in the presumed allylpalladium intermediate. Extensive optimization revealed that the outcome of the cyclization was dependent on the ligand, and varying ratios of octacyclic indole 155 and its epimer at C2′ could be obtained, together with the corresponding azepane motif arising from cyclization at C4′. The diastereomeric products were found to interconvert under the reaction conditions, eventually leading to formation of the corresponding azepine motif, driven presumably by relief of the angle strain associated with the desired pyrroloindole fragment. Ultimately, application of Pip-Phos developed by Feringa and co-workers delivered optimal performance and secured diastereoselective access to intermediate 155.107 Further elaboration to the target natural product followed the previously established sequence, including protecting group exchanges and olefination with phosphonate 156 en route to unsaturated ester 157. Saponification of the penultimate intermediate furnished the sodium salt of nodulisporic acid B (158).27
4.6. The Pronin synthesis of emindole SB and nodulisporic acid C
A new approach to the indole diterpenes developed by the Pronin group was based on a rapid assembly of the common tricarbocyclic fragment found in these natural products.26,28 Initial studies revealed that diene 159 could be engaged in a polycyclization cascade to produce the desired decalin motif (Scheme 17).26 These efforts were enabled by the earlier findings from the Baran group that iron complexes mediated chemo-selective hydrogen atom transfer (HAT) to electron-rich alkenes.108 In a postulated mechanism, HAT to the 1,1-disubstituted alkenes was followed by an intramolecular Giese addition of the resulting tert-alkyl radical 160 to the pendant unsaturated aldehyde. Subsequent reduction of the resulting oxallyl radical and an intramolecular aldol reaction would deliver intermediate 127, which was contemporaneously employed by the Smith group in their synthesis of nodulisporic acid D (137, see Scheme 13).25 Direct generation of trans-decalin 127 from diene 159 was accompanied by formation of cis-decalin 161 in equimolar quantities, but further experimentation revealed that polycyclization of the secondary alcohol 163 delivered the desired tricyclic motif 164 with good selectivity over cis-fused product 165.26 These observations were attributed to the transient formation of lactol 162, where the hydroxy group of the cyclopentanol moiety served as a tethering point for the aliphatic aldehyde fragment thereby restricting the available conformations of the reactive intermediates. The same effect was expected to be conferred by the hemiaminal motif 170, which was indeed employed with success to achieve a highly diastereoselective synthesis of emindole SB (12, Scheme 18). Synthesis of intermediate 170 commenced with conjugated addition of Grignard reagent 167 to cyclopentenone 166. Extensive experimentation with installation of the alkenyl substituent identified carboindation of alkyne 169 with silyl enol ether 168, when followed by treatment with acid as a suitable means for preparation of hydroxyketone 170.109 Allylic oxidation of the trisubstituted alkene was followed by a Fisher synthesis of indole 172 from the corresponding cyclopentanone 171. Oxidation of the diol under Parikh–Doering conditions produced hemiaminal 173, thus setting the stage for installation of the desired vicinal quaternary centers.110 This substrate provided further improvement on the diastereoselectivity observed with secondary alcohol 163 and underwent reductive cyclization with exclusive preference for the desired configuration at C4. While stability of the hemiaminal motif precluded direct formation of the decalin fragment, treatment of intermediate 174 with a strong base triggered release of the aliphatic aldehyde, along with an ensuing aldol reaction to complete construction of the pentacyclic indoloterpenoid core. Hydroxyaldehyde 175 was converted to emindole SB (12) in three straightforward manipulations.26 These studies laid the foundation for the synthesis of a more complex indole diterpene, (−)-nodulisporic acid C (148).
Scheme 17.
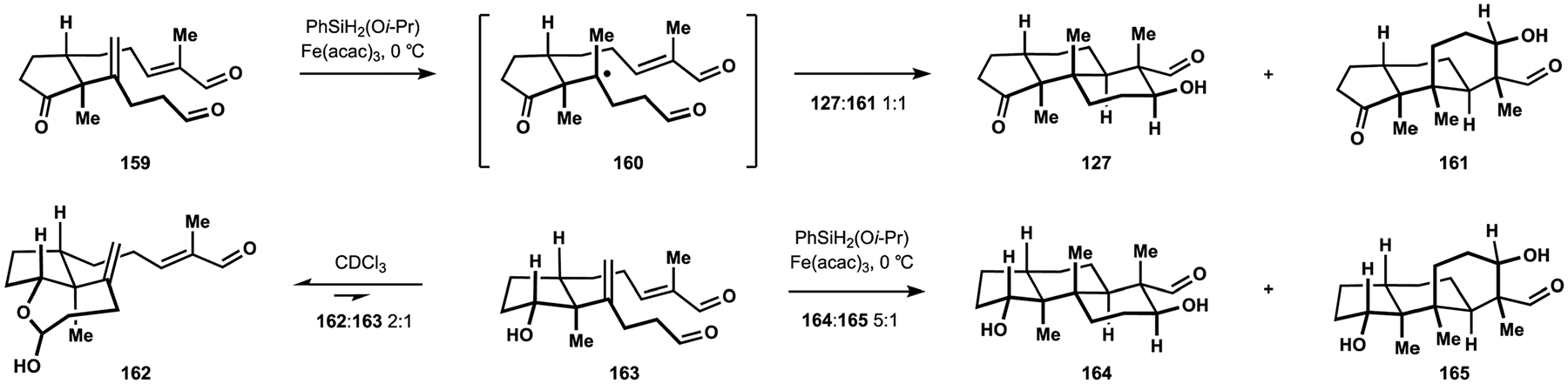
HAT-initiated polycyclization en route to the tricyclic terpenoid core developed by the Pronin group.
Scheme 18.
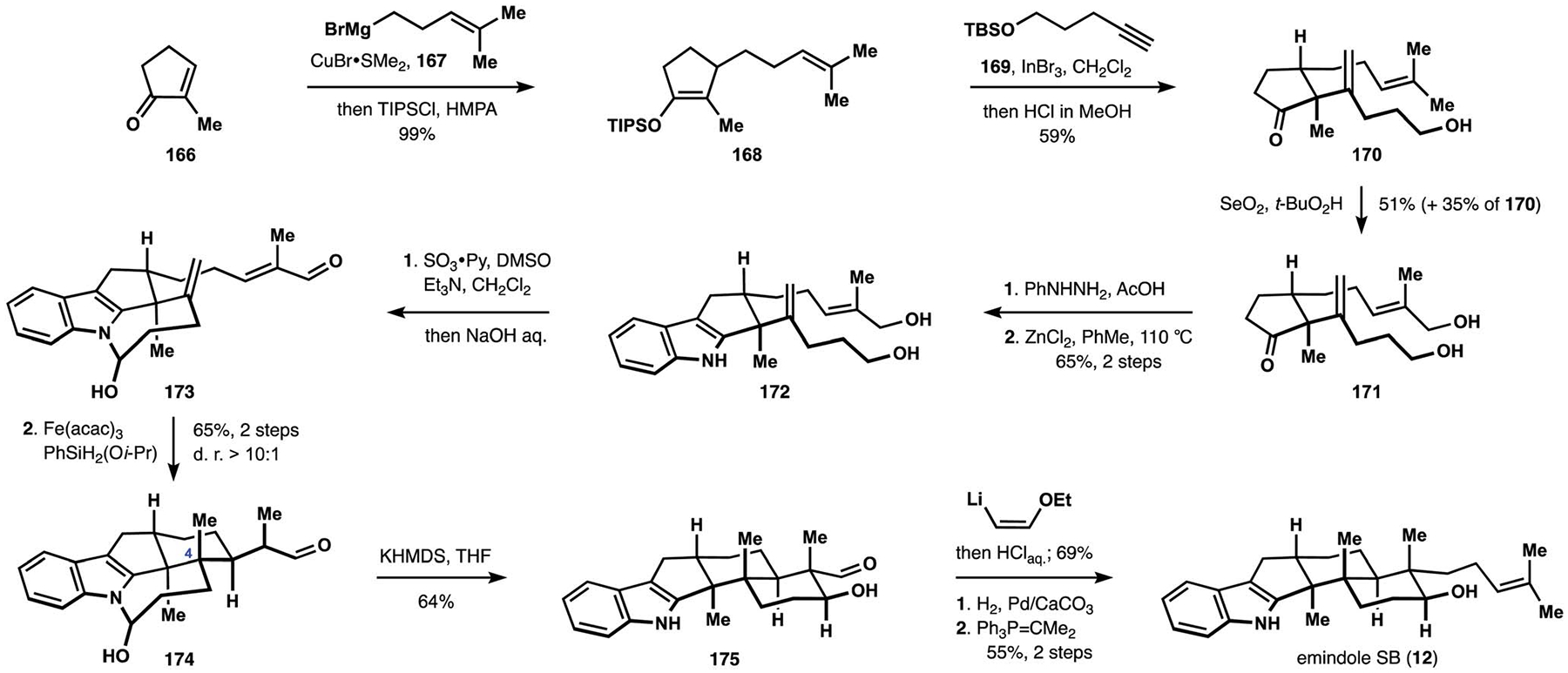
The Pronin synthesis of emindole SB (12).
To this end, the construction of the tricyclic terpenoid motif found in the nodulisporic acids required enantioselective access to silyl enol ether 168 (Scheme 19).28 Extensive optimization identified Josiphos derivative 176 as the optimal ligand and delivered the desired intermediate with useful levels of enantioenrichment.111 Installation of the 1,1-disubstituted alkene according to the previously developed protocol produced hydroxyketone 170. At this point, the cyclic ketone was converted to the corresponding protected cyanohydrin, and a subsequent allylic hydroxylation was followed by a double oxidation of the intermediate diol to synthesize polycyclization precursor 177. In contrast to parent ketone 159, cyanohydrin 177 underwent cyclization with a pronounced preference for the trans-decalin motif. Treatment of the reaction mixture with acid followed by base afforded ketoaldehyde 127 in a highly diastereoselective manner. The presence of a pseudoaxial substituent at C2 was found to be critical for the desired outcome in the polycyclization and provided an improvement to the original stereochemical strategy by securing ready access to more complex congeners. Protection of the secondary alcohol in intermediate 127 allowed for a recrystallization that significantly improved the level of enantioenrichment of the corresponding pivalate. Horner–Wadsworth–Emmons olefination with phosphonate 178 completed construction of dienoic acid fragment 179.
Scheme 19.
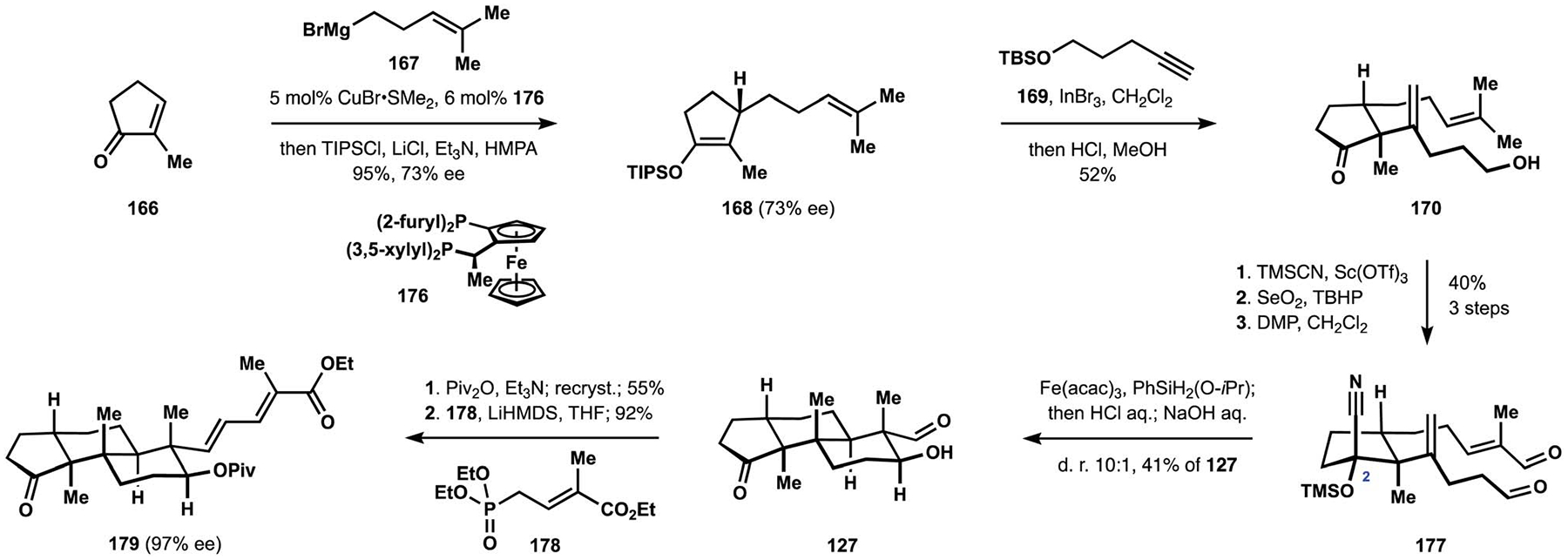
Synthesis of the dienoic acid fragment by the Pronin group.
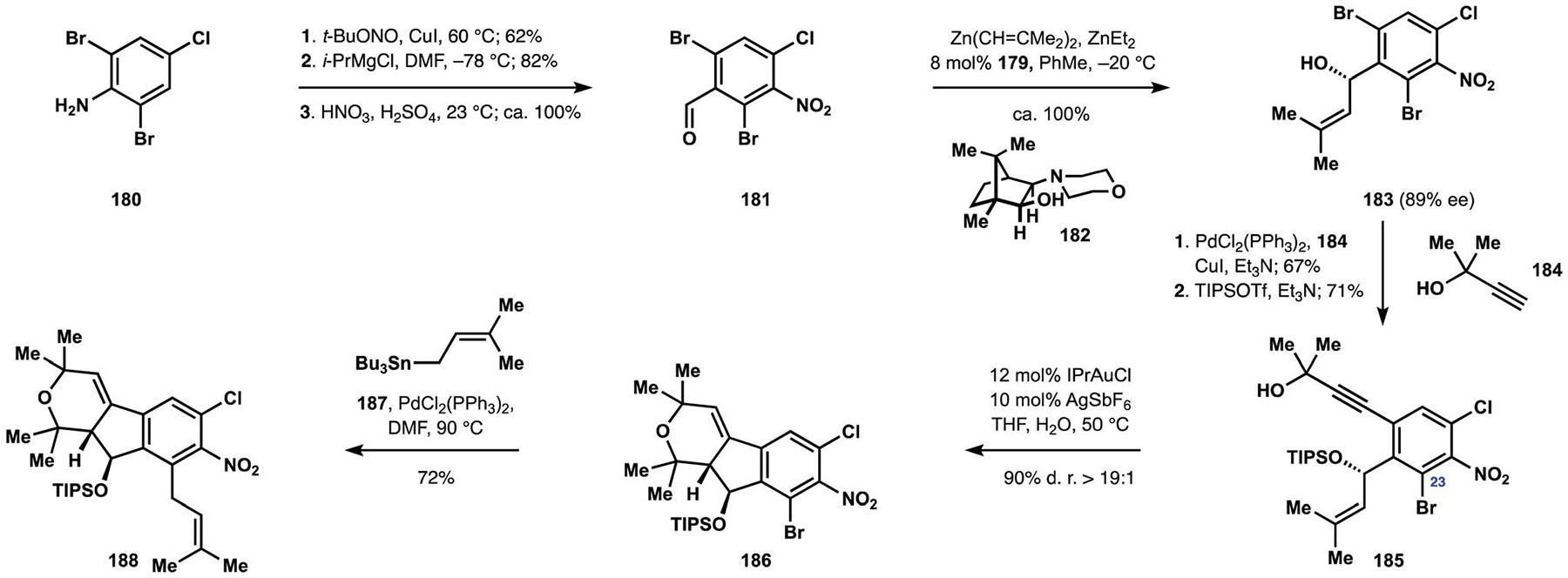
Assembly of the indenopyran motif was based on a cycloisomerization of propargylic alcohol 185 with the proximal trisubstituted alkene (Scheme 20).28 Synthesis of this intermediate commenced with addition of diisobutenylzinc to aromatic aldehyde 181, which was prepared in three steps from aniline 180. Catalysis by Nugent’s aminoalcohol 182 secured access to benzyl alcohol 183 in a highly enantioselective manner.112 Chemo- and regioselective Sonogashira coupling of the aryl bromide with alkyne 184 followed by protection of the benzylic hydroxy group provided the desired cycloisomerization precursor.113 Treatment of enyne 185 with a cationic gold complex resulted in highly diastereoselective formation of indenopyran motif 186. Substitution at C23 was found to be a critical stereocontrol element and attributed to the associated allylic strain in the benzyl ether fragment. Chemoselective Stille coupling of the aryl bromide with prenylstannane 187 completed installation of substituents found in the indole fragment of nodulisporic acid C (148), securing access to nitroarene 188.
Scheme 20.
Synthesis of the indenopyran fragment by the Pronin group.
Extensive optimization identified an optimal catalytic system for the coupling of a tin enolate derived from ketone 179 with aryl chloride 188 (Scheme 21).28,114 Notably, useful levels of efficiency could be obtained with nearly equimolar ratios of reactants and allowed for increased material throughput during these late-stage manipulations. The arylation proceeded in a diastereoselective manner favoring the configuration at C18 found in product 189, which was critical to the success of reductive cyclization en route to the desired indole under mild conditions. Thus, attempted conversion of the epimeric arylation product to the target heterocycle resulted solely in reduction of the nitro group, and the intramolecular condensation could only be accomplished under forcing conditions, which led to decomposition of the indenopyran motif. At the same time, conversion of ketone 189 to the corresponding indole proceeded readily under mild conditions, which also stood in contrast to the previous examples in the field.16,24 Ultimately, cleavage of the silyl ether produced diester 190, and saponification delivered the target natural product, (−)-nod ulisporic acid C (148).28
Scheme 21.
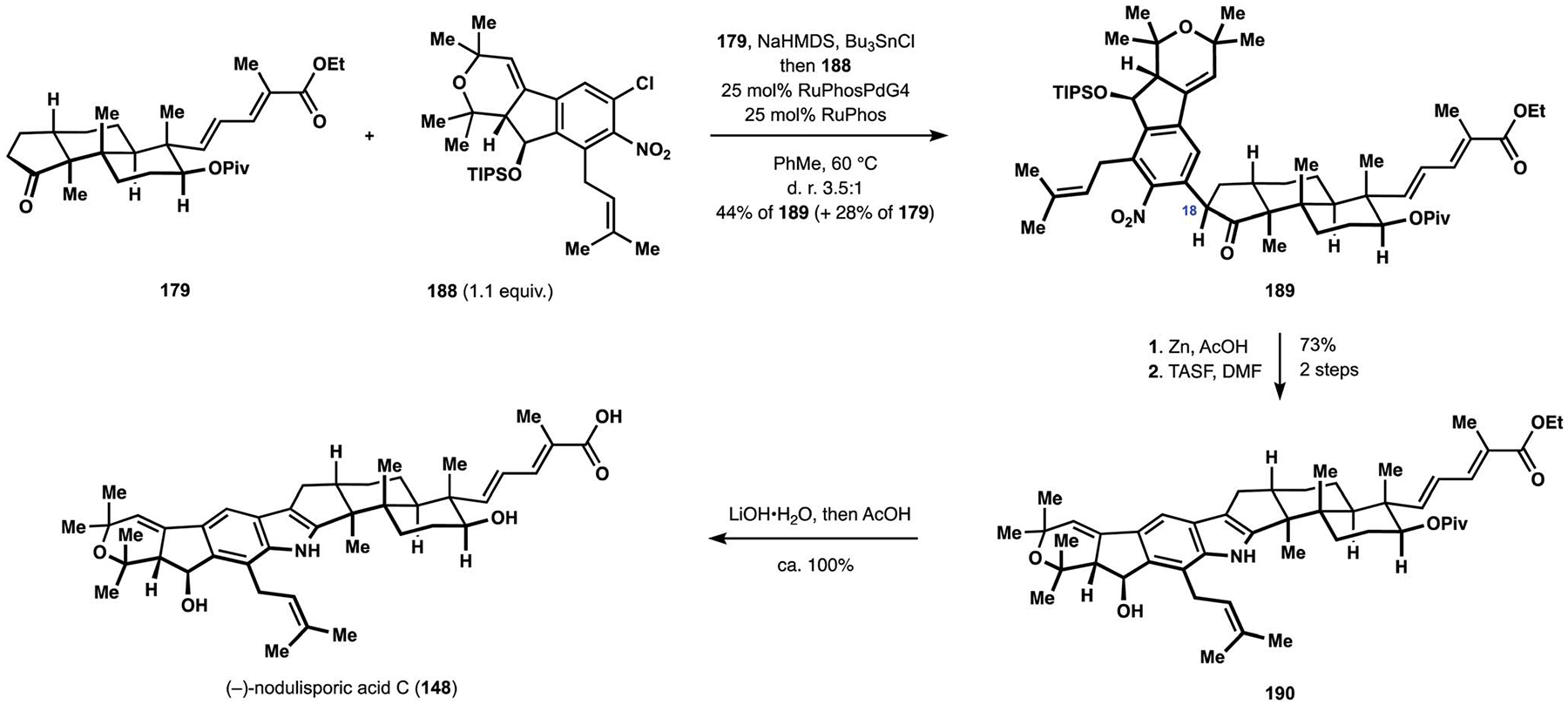
The Pronin synthesis of (−)-nodulisporic acid C (148).
4.7. The Newhouse synthesis of paspaline
Guided by the biosynthesis of the indole diterpenes, the Newhouse group aimed to elucidate the possibility of constructing the C2–C3 bond in a biomimetic fashion en route to the trans-hydrindane motif.29 The proposed cyclization would mimic one of the postulated elementary steps in the biosynthetic assembly of the indoloterpenoid core. Computational evaluation of the possible reaction pathways associated with the carbocation 191 and similar intermediates suggested that the desired Friedel–Crafts cyclization may be associated with a lower activation barrier when compared to the alternative migration of the neighbouring methyl substituent (Fig. 2). The latter pathway would lead to formation of the indoloterpenoid motif associated with a different group of secondary metabolites. It was anticipated that success of the intramolecular alkylation would ensure rapid entry into paspaline (2) since a synthetic equivalent of intermediate 191 could be prepared in a concise manner.
Fig. 2.
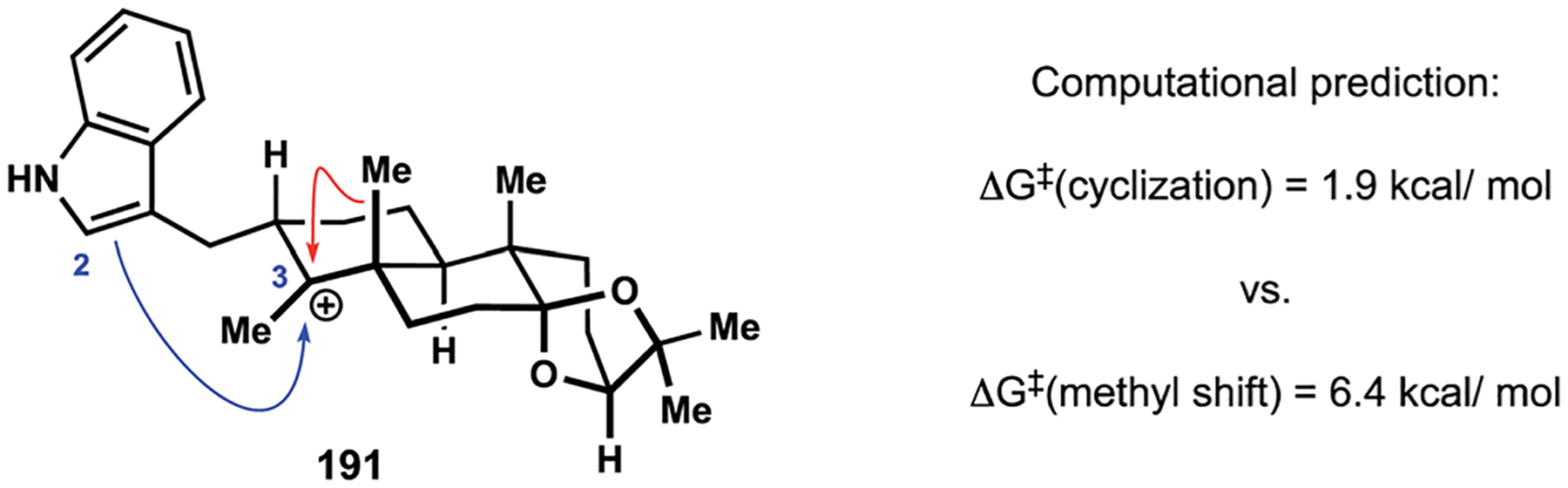
Computational evaluation of a biomimetic Friedel–Crafts cyclization by the Newhouse group.
Construction of the Friedel–Crafts cyclization precursor began with enantioselective synthesis of substituted Wieland–Miescher ketone 193, itself available in two steps from cyclohexanedione 192 (Scheme 22).29 Alkylation of the potassium enolate delivered diene 194, and dihydroxylation of the prenyl substituent proceeded with moderate diastereoselectivity, but nonetheless secured access to diol 195. Intramolecular ketalization was followed by HAT-mediated reduction of the endo-cyclic alkene, which established the desired trans-decalin motif.115 Alkylation of ketone 196 with iodide 197 and subsequent addition of methyllithium to the resulting cycloalkanone afforded precursor to the planned cyclization in the form of tertiary alcohol 198. Extensive experimentation revealed that the desired connectivity pattern could be obtained upon treatment of the substrate with a strong Lewis acid. Contrary to the computational prediction for the partitioning of competing reactivity patterns for carbocation 191, intermediate 200 was produced only as a minor component of product mixtures, where rearranged alkene 199 was the favoured constituent. These observations suggest the contribution from a concerted migratory displacement en route to product 199 that avoids the formation of carbocations, pointing to potential benefit of exploring alternative approaches to generation of intermediate 191. Nevertheless, reduction of the dioxolane moiety allowed for efficient conversion of ketal 200 to the target natural product, thus completing the synthesis of (−)-paspaline (2). Alkene 199 was in turn converted to emindole PB – a member of a different group of indoloterpenoids – in several synthetic manipulations. These studies provide a starting point for the development of new approaches to biomimetic assembly of the indoloterpenoid core.
Scheme 22.
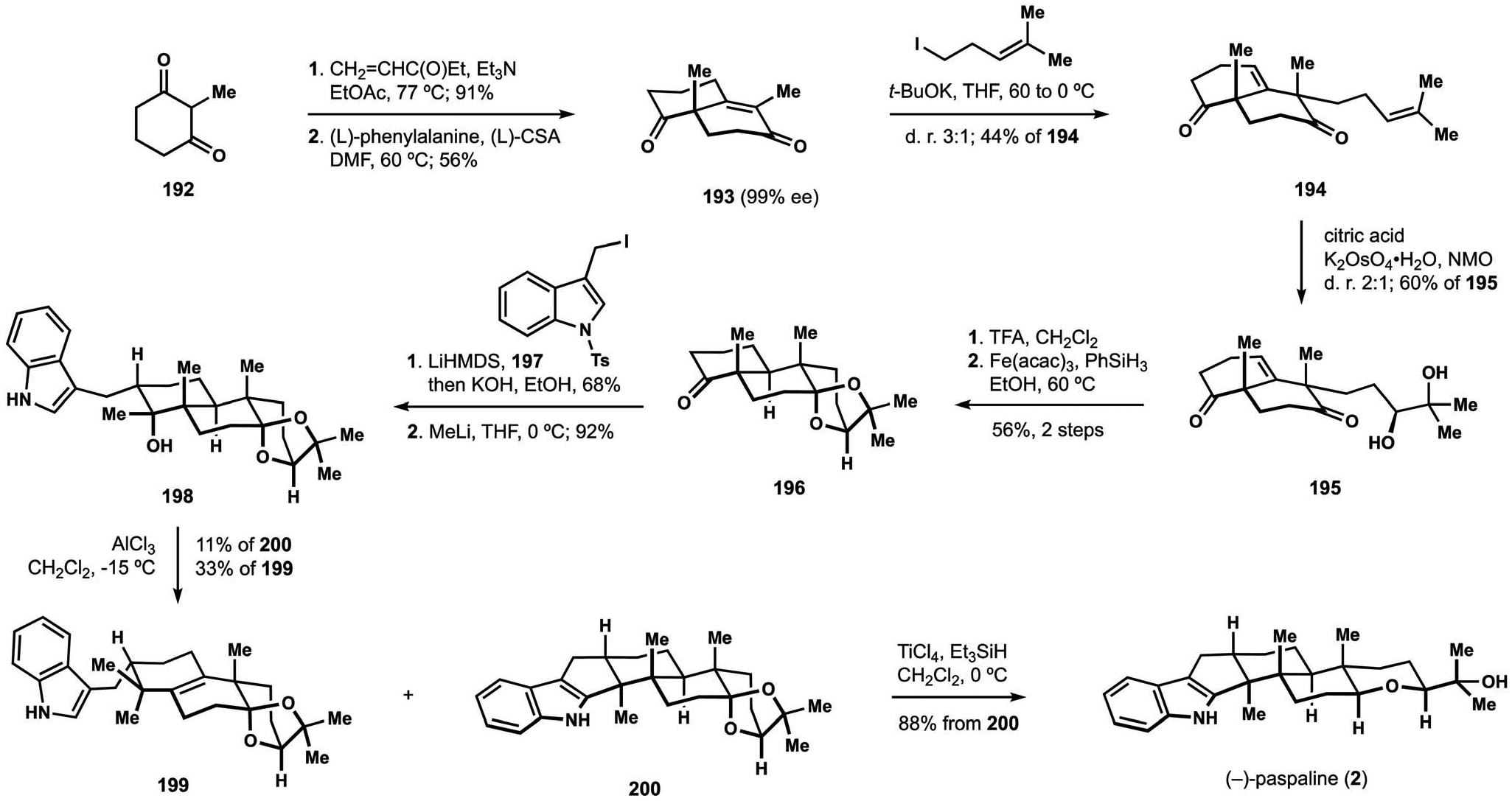
The Newhouse synthesis of (−)-paspaline (2).
5. Conclusions
Continued efforts in the synthesis of indole diterpenes have generated new knowledge in the reactivity of these complex chemical systems, introduced new tactics to the arsenal of synthetic organic chemists, and led to the successful assembly of several congeners with different structural patterns. The construction of the shared terpenoid core has proven to be one of the key challenges and has prompted the development of several tactics for the stereoselective installation of the trans-hydrindane motif. In this context, exploitation of directing effects have provided excellent performance, but new approaches have offered potentially attractive alternatives. Good to excellent levels of stereocontrol in the latter strategies have also resulted in significant improvements in the brevity of synthetic sequences. Recent work in biomimetic assembly of the rearranged indoloterpenoid motifs have also provided initial insight to inform future studies in this challenging yet fascinating direction. These developments notwithstanding, rapid construction of diverse congeners continues to present a significant challenge. While sheer complexity of these compounds imposes fundamental limitations on the levels of concision attainable in their synthesis, a general and flexible route could enable access to structural modifications without significant impacts on the longest linear sequence. This idealized approach would be particularly attractive in the context of chemical biology and biochemical studies of the target metabolites that have invariably been identified among the driving forces behind the synthetic efforts. Notably, application of the relevant findings in chemical reactivity to further the understanding of biological effects associated with the indole diterpenes has been only sporadic, despite the growing number of completed syntheses.60,66 Addressing these challenges will not only result in continued expansion of the chemistry knowledge but will also provide new opportunities for impact at the interface with other relevant fields of science.
Acknowledgements
The authors acknowledge support from The University of California, Irvine, NIH (R01GM121678), NSF (CHE-1848076) and Amgen.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Springer JP, Clardy J, Wells JM, Cole RJ and Kirksey JW, Tetrahedron Lett., 1975, 16, 2531. [Google Scholar]
- 2.(a) Stamm G, PhD dissertation No. 4418, Eidgenössische Technische Hochschule, Zürich, Switzerland, 1969; [Google Scholar]; (b) Gysi RP, PhD dissertation No. 4990, Eidgenössische Technische Hochschule, Zürich, Switzerland, 1973; [Google Scholar]; (c) Leutwiler A, PhD dissertation No. 5163, Eidgenössische Technische Hochschule, Zürich, Switzerland, 1973. [Google Scholar]
- 3.For crystallographic studies, see:; (a) Springer JP and Clardy J, Tetrahedron Lett, 1980, 21, 231; [Google Scholar]; (b) Gallagher RT, Finer J and Clardy J, Tetrahedron Lett, 1980, 21, 235. [Google Scholar]
- 4.For relevant isolation studies, see:; Fehr Th. and Acklin W, Helv. Chim. Acta, 1966, 49, 1907. [DOI] [PubMed] [Google Scholar]
- 5. To avoid confusion, paxilline numbering will be used whenever possible. See ref. 1.
- 6.de Jesus AE, Steyn PS, Van Heerden FR, Vleggaar R and Wessels PL, J. Chem. Soc., Chem. Commun, 1981, 289. [Google Scholar]
- 7.Gallagher RT, Hawkes AD, Steyn PS and Vleggaar R, J. Chem. Soc., Chem. Commun, 1984, 614. [Google Scholar]
- 8.Belofsky GN, Gloer JB, Wicklow DT and Dowd PF, Tetrahedron, 1995, 51, 3959. [Google Scholar]
- 9.Nozawa K, Mycotoxins, 1993, 37, 17. [Google Scholar]
- 10.Tang M-C, Lin H-C, Li D, Zhou Y, Li J, Xu W, Cacho RA, Hillenmeyer ME, Garg NK and Tang Y, J. Am. Chem. Soc, 2015, 137, 13724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sings H and Singh S, Alkaloids - Chem. Biol, 2003, 60, 51. [DOI] [PubMed] [Google Scholar]
- 12.Parker EJ and Scott BD, Handbook of industrial mycobiology, Marcel Dekker, New York, 2005, vol. 22, pp. 405–426. [Google Scholar]
- 13.Byrne KM, Smith SK and Ondeyka JG, J. Am. Chem. Soc, 2002, 124, 7055. [DOI] [PubMed] [Google Scholar]
- 14.Uchida R, Kim Y-P, Nagamitsu T, Tomoda H and Ōmura S, J. Antibiot, 2006, 59, 338. [DOI] [PubMed] [Google Scholar]
- 15.Roll DM, Barbieri LR, Bigelis R, McDonald LA, Arias DA, Chang L-P, Singh MP, Luckman SW, Berrodin TJ and Yudt MR, J. Nat. Prod, 2009, 72, 1944. [DOI] [PubMed] [Google Scholar]
- 16.(a) Mewshaw R and Smith III AB, J. Am. Chem. Soc, 1985, 107, 1769; [Google Scholar]; (b) Mewshaw RE, Taylor MD and Smith III AB, J. Org. Chem, 1989, 54, 3449. [Google Scholar]
- 17.(a) Smith III AB, Sunazuka T, Leenay TL and Kingery-Wood J, J. Am. Chem. Soc, 1990, 112, 8197; [Google Scholar]; (b) Smith III AB, Kingery-Wood J, Leenay TL, Nolen EG and Sunazuka T, J. Am. Chem. Soc, 1992, 114, 1438. [Google Scholar]
- 18.(a) Smith III AB, Kanoh N, Haruaki I and Hartz RA, J. Am. Chem. Soc, 2000, 122, 11254; [Google Scholar]; (b) Smith III AB, Kanoh N, Haruaki I, Minakawa N, Rainier JD, Hartz RA, Cho YS, Cui H and Moser WH, J. Am. Chem. Soc, 2003, 135, 8228. [DOI] [PubMed] [Google Scholar]
- 19.(a) Smith III AB and Cui H, Org. Lett, 2003, 5, 587; [DOI] [PubMed] [Google Scholar]; (b) Smith III AB and Cui H, Helv. Chim. Acta, 2003, 86, 3908. [Google Scholar]
- 20.(a) Smith III AB, Davulcu AH and Kürti L, Org. Lett, 2006, 8, 1665; [DOI] [PubMed] [Google Scholar]; (b) Smith III AB, Davulcu AH, Cho YS, Ohmoto K, Kürti L and Ishiyama H, J. Org. Chem, 2007, 72, 4596. [DOI] [PubMed] [Google Scholar]
- 21.Enomoto M, Morita A and Kuwahara S, Angew. Chem., Int. Ed, 2012, 51, 12833. [DOI] [PubMed] [Google Scholar]
- 22.Asanuma A, Enomoto M, Nagasawa T and Kuwahara S, Tetrahedron Lett, 2013, 54, 4561. [Google Scholar]
- 23.(a) Teranishi T and Kuwahara S, Tetrahedron Lett, 2014, 55, 1486; [Google Scholar]; (b) Teranishi T, Murokawa T, Enomoto M and Kuwahara S, Biosci. Biotechnol. Biochem, 2015, 79, 11. [DOI] [PubMed] [Google Scholar]
- 24.(a) Sharpe RJ and Johnson JS, J. Am. Chem. Soc, 2015, 137, 4968; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sharpe RJ and Johnson JS, J. Org. Chem, 2015, 80, 9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou Y, Melvin JE, Gonzales SS, Spafford MJ and Smith III AB, J. Am. Chem. Soc, 2015, 137, 7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.George DT, Kuenstner EJ and Pronin SV, J. Am. Chem. Soc, 2015, 137, 15410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou Y, Li X, Yang Y, Berritt S, Melvin J, Gonzales S, Spafford M and Smith III AB, J. Am. Chem. Soc, 2018, 140, 9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Godfrey NA, Schatz DJ and Pronin SV, J. Am. Chem. Soc, 2018, 140, 12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim DE, Zweig JE and Newhouse TR, J. Am. Chem. Soc, 2019, 141, 1479. [DOI] [PubMed] [Google Scholar]
- 30.Guile SD, Saxton JE and Thornton-Pett M, J. Chem. Soc., Perkin Trans 1, 1992, 1763. [Google Scholar]
- 31.For relevant synthetic approaches and studies also see:; (a) Ali A, Guile SD, Saxton JE and Thornton-Pett M, Tetrahedron, 1991, 47, 6407; [Google Scholar]; (b) Magnus P and Mansley TE, Tetrahedron Lett, 1999, 40, 6909; [Google Scholar]; (c) England DB, Magolan J and Kerr MA, Org. Lett, 2006, 8, 2209; [DOI] [PubMed] [Google Scholar]; (d) Curruca F, Fousteris M, Ishikawa Y, Rekowski MW, Hounsou C, Surrey T and Giannis A, Org. Lett, 2010, 12, 2096; [DOI] [PubMed] [Google Scholar]; (e) Oikawa M, Hashimoto R and Sasaki M, Eur. J. Org. Chem, 2011, 538; [Google Scholar]; (f) Sugino K, Nakazaki A, Isobe M and Nishikawa T, Synlett, 2011, 647; [Google Scholar]; (g) Adachi M, Higuchi K, Thasana N, Yamada Y and Nishikawa T, Org. Lett, 2012, 14, 114; [DOI] [PubMed] [Google Scholar]; (h) Feldman KS, Gonzalez IY and Glinkerman CM, J. Am. Chem. Soc, 2014, 136, 15138; [DOI] [PubMed] [Google Scholar]; (i) Feldman KS, Gonzalez IY and Glinkerman CM, J. Org. Chem, 2015, 80, 11849; [DOI] [PubMed] [Google Scholar]; (j) Yoshi Y, Otsu T, Hosokawa N, Takasu K, Okano K and Tokuyama H, Chem. Commun, 2015, 51, 1070; [DOI] [PubMed] [Google Scholar]; (k) Ono Y, Nakazaki A, Ueki K, Higuchi K, Sriphana U, Adachi M and Nishikawa T, J. Org. Chem, 2019, 84, 9750. [DOI] [PubMed] [Google Scholar]
- 32.(a) Minami A, Liu C and Oikawa H, Heterocycles, 2016, 92, 397; [Google Scholar]; (b) Liu C, Minami A, Ozaki T and Oikawa H, Chem. Biol, 2020, 2, 446; [Google Scholar]; (c) Ryo T, Atsushi M, Hiroki O and Oikawa H, Nat. Prod. Rep, 2020, 37, 1098. [DOI] [PubMed] [Google Scholar]
- 33.Kozák L, Szilágyi Z, Tóth L, Pócsi I and Molnár I, Appl. Microbiol. Biotechnol, 2019, 103, 1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acklin W, Weibel F and Arigoni D, Chimia, 1977, 31, 63. [Google Scholar]
- 35.de Jesus AE, Gorst-Allman CP, Steyn PS, van Heerden FR, Vieggaar R, Wessels PL and Hull WE, J. Chem. Soc., Perkin Trans 1, 1983, 1863. [Google Scholar]
- 36.(a) Young C, McMillan L, Telfer E and Scott B, Mol. Microbiol, 2001, 39, 754; [DOI] [PubMed] [Google Scholar]; (b) McMillan LK, Carr RL, Young CA, Astin JW, Lowe RGT, Parker EJ, Jameson GB, Finch SC, Miles CO, McManus OB, Schmalhofer W, Garcia ML, Kaczorowski GJ, Goetz M, Tkacz JS and Scott B, Mol. Genet. Genomics, 2003, 270, 9; [DOI] [PubMed] [Google Scholar]; (c) Saikia S, Parker EJ, Koulman A and Scott B, FEBS Lett, 2006, 580, 1625; [DOI] [PubMed] [Google Scholar]; (d) Saikia S, Parker EJ, Koulman A and Scott B, J. Biol. Chem, 2007, 282, 16829. [DOI] [PubMed] [Google Scholar]
- 37.Fueki S, Tokiwano T, Toshima H and Oikawa H, Org. Lett, 2004, 6, 2697. [DOI] [PubMed] [Google Scholar]
- 38.Tagami K, Liu C, Minami A, Noike M, Isaka T, Fueki S, Shichijo Y, Toshima H, Gomi K, Dairi T and Oikawa H, J. Am. Chem. Soc, 2013, 135, 1260. [DOI] [PubMed] [Google Scholar]
- 39.Uchida R, Tomoda H and Ōmura S, J. Antibiot, 2006, 59, 298. [DOI] [PubMed] [Google Scholar]
- 40.Rainier JD and Smith III AB, Tetrahedron Lett., 2000, 41, 9419. [Google Scholar]
- 41.(a) Clark JS, Myatt J, Wilson C, Roberts L and Walshe N, Chem. Commun, 2003, 1546; [PubMed] [Google Scholar]; (b) Clark JS, Myatt J, Wilson C, Roberts L and Walshe N, Synlett, 2005, 697. [Google Scholar]
- 42.Singh SB, Ondeyka JG, Jayasuriya H, Zink DL, Ha SN, Dahl-Roshak A, Greene J, Kim JA, Smith MM, Shoop WL and Tkacz JS, J. Nat. Prod, 2004, 67, 1496. [DOI] [PubMed] [Google Scholar]
- 43.Van de Bittner KC, Nicholson MJ, Bustamante LY, Kessans SA, Ram A, van Dolleweerd CJ, Scott B and Parker EJ, J. Am. Chem. Soc, 2018, 140, 582. [DOI] [PubMed] [Google Scholar]
- 44.Van de Bittner KC, Cameron RC, Bustamante LY, Bundela R, Kessans SA, Vorster J, Nicholson MJ and Parker EJ, RSC Med. Chem, 2019, 10, 1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Motoyama T, Hayashi T, Hirota H, Ueki M and Osada H, Chem. Biol, 2012, 19, 1611. [DOI] [PubMed] [Google Scholar]
- 46.Philippe G, Toxins, 2016, 8, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saikia S, Takemoto D, Tapper BA, Lane GA, Fraser K and Scott B, FEBS Lett, 2012, 586, 2563. [DOI] [PubMed] [Google Scholar]
- 48.Nicholson MJ, Koulman A, Monahan BJ, Pritchard BL, Payne GA and Scott B, Appl. Environ. Soil Sci, 2009, 75, 7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kozák L, Szilágyi Z, Vágó B, Kakuk A, Tóth L, Molnár I and Pócsi I, Appl. Microbiol. Biotechnol, 2018, 102, 3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tagami K, Minami A, Fujii R, Liu C, Tanaka M, Gomi K, Dairi T and Oikawa H, ChemBioChem, 2014, 15, 2076. [DOI] [PubMed] [Google Scholar]
- 51.Nicholson MJ, Eaton CA, Sätrkel C, Tapper BA, Cox MP and Scott B, Toxins, 2015, 7, 2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu C, Minami A, Dairi T, Gomi K, Scott B and Oikawa H, Org. Lett, 2016, 18, 5026. [DOI] [PubMed] [Google Scholar]
- 53.Liu C, Tagami K, Minami A, Matsumoto T, Frisvad JC, Suzuki H, Ishikawa J, Gomi K and Oikawa H, Angew. Chem., Int. Ed, 2015, 54, 5748. [DOI] [PubMed] [Google Scholar]
- 54.Cole RJ, Dorner JW, Lansden JA, Cox RH, Pape C, Cunfer B, Nicholson SS and Bedell DM, J. Agric. Food Chem, 1977, 25, 1197. [DOI] [PubMed] [Google Scholar]
- 55.Gallagher RT, Hawkes AD, Steyn PS and Vleggaar R, J. Chem. Soc., Chem. Commun, 1984, 614. [Google Scholar]
- 56.Uhlig S, Botha CJ, Vrålstad T, Rolén E and Miles CO, J. Agric. Food Chem, 2009, 57, 11112. [DOI] [PubMed] [Google Scholar]
- 57.(a) Wilson BJ and Wilson CH, Science, 1964, 144, 177; [DOI] [PubMed] [Google Scholar]; (b) Wilson BJ, Wilson CH and Hayes AW, Nature, 1968, 220, 77. [DOI] [PubMed] [Google Scholar]
- 58.Dorner JW, Cole RJ, Cox RH and Cunfer BM, J. Agric. Food Chem, 1984, 32, 1069. [Google Scholar]
- 59.(a) Wilson BJ, Hockman T and Dettbarn WD, Brain Res., 1972, 40, 540; [DOI] [PubMed] [Google Scholar]; (b) Norris PJ, Smith CCT, DeBelleroche J, Bradford HF, Mantle PG, Thomas AJ and Penny RHC, J. Neurochem, 1980, 34, 33. [DOI] [PubMed] [Google Scholar]
- 60.Knaus H-G, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LMH, Sanchez M, Giangiacomo K, Reuben JP, Smith AB III, Kaczorowski GJ and Garcia ML, Biochemistry, 1994, 33, 5819. [DOI] [PubMed] [Google Scholar]
- 61.Imlach WL, Finch SC, Dunlop J, Meredith AL, Aldrich RW and Dalziel JE, J. Pharmacol. Exp. Ther, 2008, 327, 657. [DOI] [PubMed] [Google Scholar]
- 62.Ondeyka JG, Helms GL, Hensens OD, Goetz MA, Zink DL, Tsipouras A, Shoop WL, Slayton L, Dombrowski AW, Polishook JD, Ostlind DA, Tsou NN, Ball RG and Singh SB, J. Am. Chem. Soc, 1997, 119, 8809. [Google Scholar]
- 63.Ludmerer SW, Warren VA, Williams BS, Zheng Y, Hunt DC, Ayer MB, Wallace MA, Chaudhary AG, Egan MA, Meinke PT, Dean DC, Garcia ML, Cully DF and Smith MM, Biochemistry, 2002, 41, 6548. [DOI] [PubMed] [Google Scholar]
- 64.Shoop WL, Gregory LM, Zakson-Aiken M, Michael BF, Haines HW, Ondeyka JG, Meinke PT and Schmatz DM, J. Parasitol, 2001, 87, 419. [DOI] [PubMed] [Google Scholar]
- 65.Singh SB, Ondeyka JG, Jayasuriya H, Zink DL, Ha SN, Dahl-Roshak A, Greene J, Kim JA, Smith MM, Shoop WL and Tkacz JS, J. Nat. Prod, 2004, 67, 1496. [DOI] [PubMed] [Google Scholar]
- 66.Meinke PT, Colletti SL, Fisher MH, Wyvratt MJ, Shih TL, Ayer MB, Li C, Lim J, Ok D, Salva S, Warmke LM, Zakson M, Michael BF, deMontigny P, Ostlind DA, Fink D, Drag M, Schmatz DM and Shoop WL, J. Med. Chem, 2009, 52, 3505. [DOI] [PubMed] [Google Scholar]
- 67.(a) Huang X-H, Tomoda H, Nishida H, Masuma R and Ōmura S, J. Antibiot, 1995, 48, 793; [DOI] [PubMed] [Google Scholar]; (b) Tomoda H, Tabata N, Yang D-J, Takayanagi H and Ōmura S, J. Antibiot, 1995, 48, 793. [DOI] [PubMed] [Google Scholar]
- 68.Ohshiro T, Rudel LL, Ōmura S and Tomoda H, J. Antibiot, 2007, 60, 43. [DOI] [PubMed] [Google Scholar]
- 69.Chang T-Y, Li B-L, Chang CCY and Urano Y, Am. J. Physiol.: Endocrinol. Metab, 2009, 297, E1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakazawa J, Yajima J, Usui T, Ueki M, Takatsuki A, Imoto M, Toyoshima YY and Osada H, Chem. Biol, 2003, 10, 131. [DOI] [PubMed] [Google Scholar]
- 71.Tarui Y, Chinen T, Nagumo Y, Motoyama T, Hayashi T, Hirota H, Muroi M, Ishii Y, Kondo H, Osada H and Usui T, ChemBioChem, 2014, 14, 934. [DOI] [PubMed] [Google Scholar]
- 72.Rath O and Kozielski F, Nat. Rev. Cancer, 2012, 12, 527. [DOI] [PubMed] [Google Scholar]
- 73.(a) Sallam AA, Houssen WE, Gissendanner CR, Orabi KY, Foudah AI and El Sayed KA, RSC Med. Chem, 2013, 4, 1360; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goda AA, Siddique AB, Mohyeldin M, Ayoub NM and El Sayed KA, Mar. Drugs, 2018, 16, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Niklasson M, Maddalo G, Sramkova Z, Mutlu E, Wee S, Sekyrova P, Schmidt L, Fritz N, Dehnisch I, Kyriatzis G, Krafcikova M, Carson BB, Feenstra JM, Marinescu VD, Segerman A, Haraldsson M, Gustavsson A-L, Hammarström LGJ, Jensen AJ, Uhrbom L, Altelaar AFM, Linnarsson S, Uhlén P, Trantirek L, Vincent CT, Nelander S, Enger PØ and Andäng M, Cancer Res, 2017, 77, 1741. [DOI] [PubMed] [Google Scholar]
- 75.Sallam AA, Ayoub NM, Foudah AI, Gissendanner CR, Meyer SA and El Sayed KA, Eur. J. Med. Chem, 2013, 70, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zou Y and Smith III AB, J. Antibiot, 2018, 71, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corsello MA, Kim J and Garg NK, Chem. Sci, 2017, 8, 5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Trost BM, Nishimura Y and Yamamoto K, J. Am. Chem. Soc, 1979, 101, 1328. [Google Scholar]
- 79.Smith III AB and Leenay TL, Tetrahedron Lett, 1988, 29, 2787. [Google Scholar]
- 80.Smith III AB, Nolen EG, Shirai R, Blase FR, Ohta M, Chida N, Hartz RA, Fitch DM, Clark WM and Sprengeler PA, J. Org. Chem, 1995, 60, 7837. [Google Scholar]
- 81.Smith III AB, Hartz RA, Spoors PG and Rainier JD, Isr. J. Chem, 1997, 37, 69. [Google Scholar]
- 82.Smith III AB, Cho YS and Ishiyama H, Org. Lett, 2001, 3, 3971. [DOI] [PubMed] [Google Scholar]
- 83.Smith III AB, Kürti L, Davulcu AH and Cho YS, Org. Process Res. Dev, 2007, 11, 19. [Google Scholar]
- 84.Gutzwiller J, Buchschacher P and Fürst A, Synthesis, 1977, 167. [Google Scholar]
- 85.MacAlpine GA, Raphael RA, Shaw A, Taylor AW and Wild H-J, J. Chem. Soc., Perkin Trans 1, 1976, 410. [Google Scholar]
- 86.Gassman PG, van Bergen TJ, Gilbert DP and Cue BW Jr, J. Am. Chem. Soc, 1974, 96, 5495. [Google Scholar]
- 87.(a) Smith III AB and Leenay TL, Tetrahedron Lett, 1988, 29, 2791; [Google Scholar]; (b) Smith III AB and Leenay TL, J. Am. Chem. Soc, 1989, 111, 5761. [Google Scholar]
- 88.Smith III AB, Visnick M, Haseltine JN and Spengeler PA, Tetrahedron, 1986, 42, 2957. [Google Scholar]
- 89.Smith III AB, Haseltine JN and Visnick M, Tetrahedron, 1989, 45, 2431. [Google Scholar]
- 90.Corey EJ and Walinski SW, J. Am. Chem. Soc, 1972, 99, 8932. [Google Scholar]
- 91.Smith III AB, Ohta M, Clark WM and Leahy JW, Tetrahedron Lett, 1993, 34, 3033. [Google Scholar]
- 92.Stork G and Benaim J, J. Am. Chem. Soc, 1971, 93, 5938. [DOI] [PubMed] [Google Scholar]
- 93.Whitesell JK and Felmen SW, J. Org. Chem, 1980, 45, 755. [Google Scholar]
- 94.(a) Semmler W, Chem. Ber, 1892, 25, 3352; [Google Scholar]; (b) Wolff L, Annalen, 1902, 322, 351. [Google Scholar]
- 95.(a) Grieco PA, Gilman S and Nishizawa M, J. Org. Chem, 1976, 41, 1485; [Google Scholar]; (b) Sharpless KB and Young MW, J. Org. Chem, 1975, 40, 947. [Google Scholar]
- 96.(a) Stille JK, Pure Appl. Chem, 1985, 57, 1771; [Google Scholar]; (b) Han X, Stoltz BM and Corey EJ, J. Am. Chem. Soc, 1999, 121, 7600. [Google Scholar]
- 97.Tsuji J, Minami I and Shimizu I, Tetrahedron Lett, 1983, 24, 1793. [Google Scholar]
- 98.Desai LV, Hull KL and Sanford MS, J. Am. Chem. Soc, 2004, 126, 9542. [DOI] [PubMed] [Google Scholar]
- 99.Crabtree RH and Davis MW, J. Org. Chem, 1986, 51, 2655. [Google Scholar]
- 100.Barluenga J, Fernández MA, Aznar F and Valdés C, Chem. - Eur. J, 2005, 11, 2276. [DOI] [PubMed] [Google Scholar]
- 101.Smith III AB, Ishiyama H, Cho YS and Ohmoto K, Org. Lett, 2001, 3, 3967. [DOI] [PubMed] [Google Scholar]
- 102.Enders D, Zamponi A, Raabe G and Runsink J, Synthesis, 1993, 725. [Google Scholar]
- 103.(a) Kelly TR, Li Q and Bhushan V, Tetrahedron Lett, 1990, 31, 161; [Google Scholar]; (b) Sheffy FK, Godschalx JP and Stille JK, J. Am. Chem. Soc, 1984, 106, 4833. [Google Scholar]
- 104.Enders D and Eichenauer H, Angew. Chem., Int. Ed, 1976, 15, 549. [Google Scholar]
- 105.(a) Takai K, Kimura K, Kuroda T, Hiyama T and Nozaki H, Tetrahedron Lett, 1983, 24, 5281; [Google Scholar]; (b) Jin H, Uenishi J, Christ WJ and Kishi Y, J. Am. Chem. Soc, 1986, 108, 5644. [Google Scholar]
- 106.Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ and Reider PJ, Org. Lett, 2006, 8, 1787. [DOI] [PubMed] [Google Scholar]
- 107.Teichert JF and Feringa BL, Angew. Chem., Int. Ed, 2010, 49, 2486. [DOI] [PubMed] [Google Scholar]
- 108.(a) Lo JC, Yabe Y and Baran PS, J. Am. Chem. Soc, 2014, 136, 1304; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo JC, Gui J, Yabe Y, Pan C-M and Baran PS, Nature, 2014, 516, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.For catalytic version see:; (a) Holmbo SD, Godfrey NA, Hirner JJ and Pronin SV, J. Am. Chem. Soc, 2016, 138, 12316. [DOI] [PMC free article] [PubMed] [Google Scholar]; For pioneering studies on alkenylation of ketene silyl acetals see:; (b) Nishimoto Y, Moritoh R, Yasuda M and Baba A, Angew. Chem., Int. Ed, 2009, 48, 4577. [DOI] [PubMed] [Google Scholar]
- 110.Parikh JR and Doering W. v. E., J. Am. Chem. Soc, 1967, 89, 5505. [Google Scholar]
- 111.Schatz DJ, Li W and Pronin SV, Tetrahedron, 2019, 75, 3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nugent WA, Chem. Commun, 1999, 35, 1369. [Google Scholar]
- 113.Sonogashira K, J. Organomet. Chem, 2002, 653, 46. [Google Scholar]
- 114.Bruno NC, Niljianskul N and Buchwald SL, J. Org. Chem, 2014, 79, 4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.(a) Iwasaki K, Wan KK, Oppedisano A, Crossley SW and Shenvi RA, J. Am. Chem. Soc, 2014, 136, 1300; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) King SM, Ma X and Herzon SB, J. Am. Chem. Soc, 2014, 136, 6884. [DOI] [PubMed] [Google Scholar]