

Abstract
Ehm2/1, an Ehm2 transcript variant, regulates the cytoskeleton by binding to plasma membrane proteins. However, the role of Ehm2/1 in breast cancer development remains poorly understood. This study shows that, the expression of Ehm2/1 was decreased in breast cancer and that patients with low Ehm2/1 expression had a significantly poorer prognosis than those with high expression of Ehm2/1. Overexpression of Ehm2/1 in MCF-7 breast cancer cells inhibited cell migration and invasion. Ehm2/1 markedly increased the stability and half-life of E-cadherin. Moreover, Ehm2/1 was collocated with E-cadherin in the plasma membrane of MCF-7 cells. Furthermore, downregulation of Ehm2/1 promoted ubiquitination of E-cadherin, whereas overexpression of Ehm2/1 inhibited ubiquitination of E-cadherin. These results suggest that Ehm2/1 could suppress the migration and invasion of breast cancer cells by increasing E-cadherin stability.
Ehm2/1 was significantly lower in patients with breast cancer who had a poor prognosis. Ehm2/1 increased the stability of E-cadherin by inhibiting its polyubiquitination and proteasomal degradation and could suppress the migration and invasion of breast cancer cells.
Graphical abstract
Graphical Abstract.

Introduction
Breast cancer is the most common malignancy in women worldwide, with the highest incidence and mortality rates. It is estimated that approximately 2.3 million new cases of breast cancer occurred in 2020, causing 685 000 deaths in women (1). With the advent of molecular classification in breast cancer, the 5-year survival rate for patients diagnosed with localized tumors has reached 90%; however, for patients diagnosed with advanced tumors, the 5-year survival rate was less than 30% (2–4). Breast cancer mortality is primarily due to chemoresistance and metastasis to other organs. The increased motility and metastasis of breast cancer cells are associated with epithelial–mesenchymal transition (EMT), wherein the cells lose their epithelial phenotype and obtain migratory and mesenchymal characteristics (5). EMT is a key step in promoting the migration and invasion of stationary tumor cells. The hallmarks of the EMT program are loss of adherence junctions, apical–basal polarity and acquisition of the mesenchymal phenotype (5,6). Multiple factors have been reported to regulate the development of breast cancer. For example, SET8 promotes EMT and enhances the invasive capacity of breast cancer cells by interacting with TWIST (7). Moreover, OSR1 promotes EMT and metastasis in breast cancer by phosphorylating the Smad2/3 linker region and inducing TGF-β1 autocrine (8).
E‐cadherin, an epithelial marker, is a calcium‐dependent cell‐to‐cell adhesion molecule and tumor suppressor protein (9). E‐cadherin plays a critical role in establishing and maintaining cell polarity and in preventing the EMT process through intercellular adhesion complexes (10,11). Tumor progression and the capacity to invade and metastasize to distant sites in breast cancer are strongly associated with the loss of E-cadherin (11,12). E3 ligase-NRBE3 inhibits the transcription of E-cadherin in breast cancer cells and promotes metastasis in them (13). ZEB1 and ELK3 can collaborate to repress E-cadherin expression in triple-negative breast cancer cells (14). Owing to the complexity of EMT in the tumor process, it is necessary to identify new markers to reveal the mechanism of tumor metastasis.
Ehm2, which is expressed in highly metastatic cells, is a metastasis-associated gene that belongs to the four-point-one protein, ezrin, radixin, moesin (FERM) superfamily and has two protein isoforms, Ehm2/1 and Ehm2/2 (15). As a FERM protein, Ehm2 contains a highly conserved FERM domain that mediates protein–protein interactions by binding to the cytoplasmic tails of transmembrane proteins (16,17). A previous study demonstrated that Ehm2 binds to the FERM-binding motif of Crb3 in the plasma membrane and activates ARHGEF18/p114RhoGEF to control the morphology and cohesion of cancer cells (18). Others have shown that Ehm2 is also involved in steroid-regulated cytoskeletal reorganization in human fibrosarcoma cells (19). In addition, Ehm2 is often dysregulated in cancers and plays a role in cancer metastasis by interacting with transmembrane and cytoskeletal proteins (20–22). In our previous study, we found that the splice variant Ehm2/1 interacts with β-catenin and increases its localization to the plasma membrane, and that overexpression of Ehm2/1 inhibits the migration ability of MCF-7 cells (23). Thus, Ehm2/1 might play a critical role in the EMT process in breast cancer; however, there are only a few studies on the role of Ehm2/1 in the EMT process in breast cancer.
In this study, we investigated the expression levels of Ehm2 transcript variant 1 in breast cancer tissues and found that Ehm2/1 was expressed at low levels in breast cancer. In vitro cell function assays showed that Ehm2/1 inhibited the migration and invasion of MCF-7 cells. Further, mechanistic research showed that Ehm2/1 increased the stability of E-cadherin by inhibiting its polyubiquitination and proteasomal degradation.
Materials and methods
Dataset
Breast tumor datasets were downloaded from www.synapse.org (unc.edu_BRCA_IlluminaHiSeq_RNASeqV2.isoformExp). All patients were divided into lymph node (LN)-positive and LN-negative groups based on lymph node(s) examined number or tumor size, metastasis to distant organs and clinical stage. Data for the Kaplan–Meier survival analysis were obtained from http://kmplot.com/analysis, and log-rank tests were used for statistical analysis.
Human breast cancer specimens
A total of five breast cancer tissues and paired normal tissues were collected immediately after surgery at Beijing Friendship Hospital, Capital Medical University and snap frozen in liquid nitrogen until further use. The size of cancer and normal tissues was confirmed by a pathologist, and the background tissues were free of tumor deposits. All the protocols were approved by the local ethics committee.
Cell culture and transfection
MCF-7 human breast cancer cell lines and HEK 293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) in 2017. Breast cancer cells were authenticated by 20-STR analysis in 2020. Cells were routinely maintained in Dulbecco’s modified Eagle medium (DMEM)-F12 supplemented with 10% fetal calf serum and 1 × penicillin/streptomycin (Gibco BRC, Paisley, Scotland) and were cultured at 37°C in a 5% CO2 and 95% humidified atmosphere. The expression plasmid Ehm2/1 (NM_018424.2) tagged with FLAG was purchased from OriGene Technologies (Rockville, MD). Cells were transfected with pCMV-Entry and pCMV-FLAG-Ehm2/1 using TurboFect Transfection Reagent (Thermo Fisher Scientific, Waltham, MA). The siRNA of Ehm2 was synthesized by GenePharma Company (Shanghai, China) and transfected with Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific, Waltham, MA). The siEhm2 sequence was CCUGCUUAUGCUUUACACUtt.
Cohort and immunohistochemistry analysis
Human breast cancer tissue microarrays (HBre-Duc060CS-02) were purchased from Shanghai Outdo Biotechnology Company Ltd. Included in the tissue microarrays were 30 cases of paraffin-embedded breast cancer tissue samples and matching adjacent normal tissues. The pathological diagnosis of breast cancer patients was performed according to the manufacturer’s instructions. Patients’ detailed information is provided in Supplementary Table 1, available at Carcinogenesis Online. After dewaxing and rehydration, the paraffin-embedded microarrays were antigen-retrieved with citrate buffer. The tissue microarrays were blocked and then incubated with an antibody against human Ehm2/1 from rabbit (1:200) (AP338037, OriGene, MD) and with the relevant secondary antibody (ZSGB Biotechnology, Beijing, China). Microarrays were visualized using diaminobenzidine (Cell Signal Technology, Danvers, MA), and the nuclei were counterstained with hematoxylin. All these processes were automatically performed using a fully automated immunohistochemistry and in situ hybridization system, Leica BOND-MAX (Leica Biosystems, Richmond, IL). Staining was assessed by two investigators blinded to the clinical data using a quantitative imaging method, and the percentage of positive cells and staining intensity were recorded. H-score was calculated using the following formula: H-score = Σ (PP × SI). PP represents the percentage of positive cells versus all cells, scored as 0, 1, 2, 3 and 4, representing less than 1%, 1%–25%, 26%–50%, 51%–75% and 76%–100% of positive cells. SI represents the staining intensity, which was graded as 0 = negative (no cells stained); 1 = weak staining; 2 = moderate staining; and 3 = strong staining. And the symbol of “Σ” represents the sum of PP × SI.
Immunoprecipitation and western blotting
For immunoprecipitation assays, cells were washed with cold phosphate‑buffered saline (PBS) and lysed for 30 min at 4°C with cold lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.25% sodium deoxycholate) supplemented with protease inhibitor cocktail (1183617000, Roche, Basel, Switzerland). Thereafter, whole cellular extracts were incubated with appropriate specific antibodies or normal rabbit/mouse immunoglobin G at 4°C overnight with constant rotation, followed by the addition of protein A/G Sepharose beads and incubation for 2 h at 4°C. The beads were washed with cell lysis buffer. The immune complexes and input were subjected to SDS-PAGE, followed by immunoblotting with primary antibodies to identify target proteins. Immunodetection was performed using enhanced chemiluminescence (ECL System, Thermo Fisher Scientific, Waltham, MA), according to the manufacturer’s instructions.
Protein samples were prepared as follows for western blotting of protein extracts from frozen tissues. First, the tissues were disrupted in protein extraction buffer (10 mM Tris–HCl [pH 7.4], 160 mM NaCl, 1% Triton X-100, 1 mM EGTA, 1 mM EDTA and Roche Complete protease inhibitors) and incubated for 30 min on ice. After centrifugation at 16 000 g for 30 min, the supernatants were collected, aliquoted and stored at −80°C. The protein concentration was measured using a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA), and equal amounts of proteins were separated by SDS-PAGE, followed by immunoblotting with primary antibodies to identify the target proteins.
Immunofluorescence Analysis
The MCF-7 cells were transfected with His-E-cadherin and FLAG-Ehm2/1 plasmids. Coverslips were placed in a 6-well culture plate. MCF-7 cells were seeded in 6-well culture plates. Coverslips were harvested when the confluence reached 60–80%. The cells were fixed in 4% paraformaldehyde in a petri dish for 15 min. After washing 3 times with ice-cold PBS, the cells were permeabilized and blocked with a blocking buffer containing 1% BSA and 0.05% Triton X-100 for 30 min, and then incubated with anti-FLAG (1:100; F3165, Sigma, St. Louis, MO) or anti-E-cadherin (1:100; ab1416, Abcam, Cambridge, UK) antibodies at room temperature for 1 h, followed by further incubation at room temperature for 1 h with Alexa Fluor® 488‑conjugated goat anti‑mouse or 594-conjugated goat anti-rabbit secondary antibodies (1:200; Thermo Fisher Scientific, Waltham, MA). The slides were mounted using ProLong® Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, Waltham, MA). A Leica SP8 confocal microscope (Leica Microsystems) was used to photograph the slides.
Quantitative reverse transcription-PCR
Total cellular RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA). The first strand of cDNA was synthesized using Transcriptor First Strand cDNA Synthesis Kit (Promega, Madison, WI). Next, cDNA was mixed with 1 μl forward and 1 μl reverse primers (5 μM each), 4.5 μl RNase-free water and 7.5 μl 2 × PCR SYBR Green Mix buffer in a 20-μl reaction. Relative gene expression levels were determined using real-time SYBR Green fluorescence (Roche), which was run in the ABI PRISM 7500 Fast Sequence Detection System (Applied Biosystems, Foster City, CA). The process was set to 40 cycles of PCR, which were conducted at 95°C for 15 s and 60°C for 1 min within each cycle. Relative expression was quantified by the comparative Ct method (2−ΔΔCt) with GAPDH used as an internal control. The primer pairs used are listed in Supplementary Table 2, available at Carcinogenesis Online.
Cell invasion and migration assays
For the cell invasion assay, Transwell chamber filters were precoated with Matrigel (BD Biosciences, Franklin Lakes, NJ). and MCF-7 (1 × 105) cells were resuspended in 200 μl serum-free medium and pipetted into the upper chamber of the Transwell apparatus. The chambers were then transferred to wells containing 500 µL of medium with 10% FBS. MCF-7 cells were incubated for 18 h and 48 h, respectively. The cells in the top wells were removed by wiping the top of the membrane with cotton swabs. The membranes were then stained with crystal violet, and the remaining cells were counted. Five high-power fields were counted for each membrane. The protocol for the cell migration assay was the same as that for the cell invasion assay, except that the Transwell chamber filters were not coated with Matrigel.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 8. Cell results are presented as mean ± SD, and clinical results are presented as mean ± SEM. All results were obtained from at least three independent experiments unless otherwise noted. Comparisons were performed using a two-tailed unpaired Student’s t-test for statistical analysis. P values < 0.05 were considered statistically significant, with *P < 0.05, **P < 0.01, ***P < 0.001.
Results
Ehm2/1 expression was low in breast cancer and was associated with poor prognosis of breast cancer patients
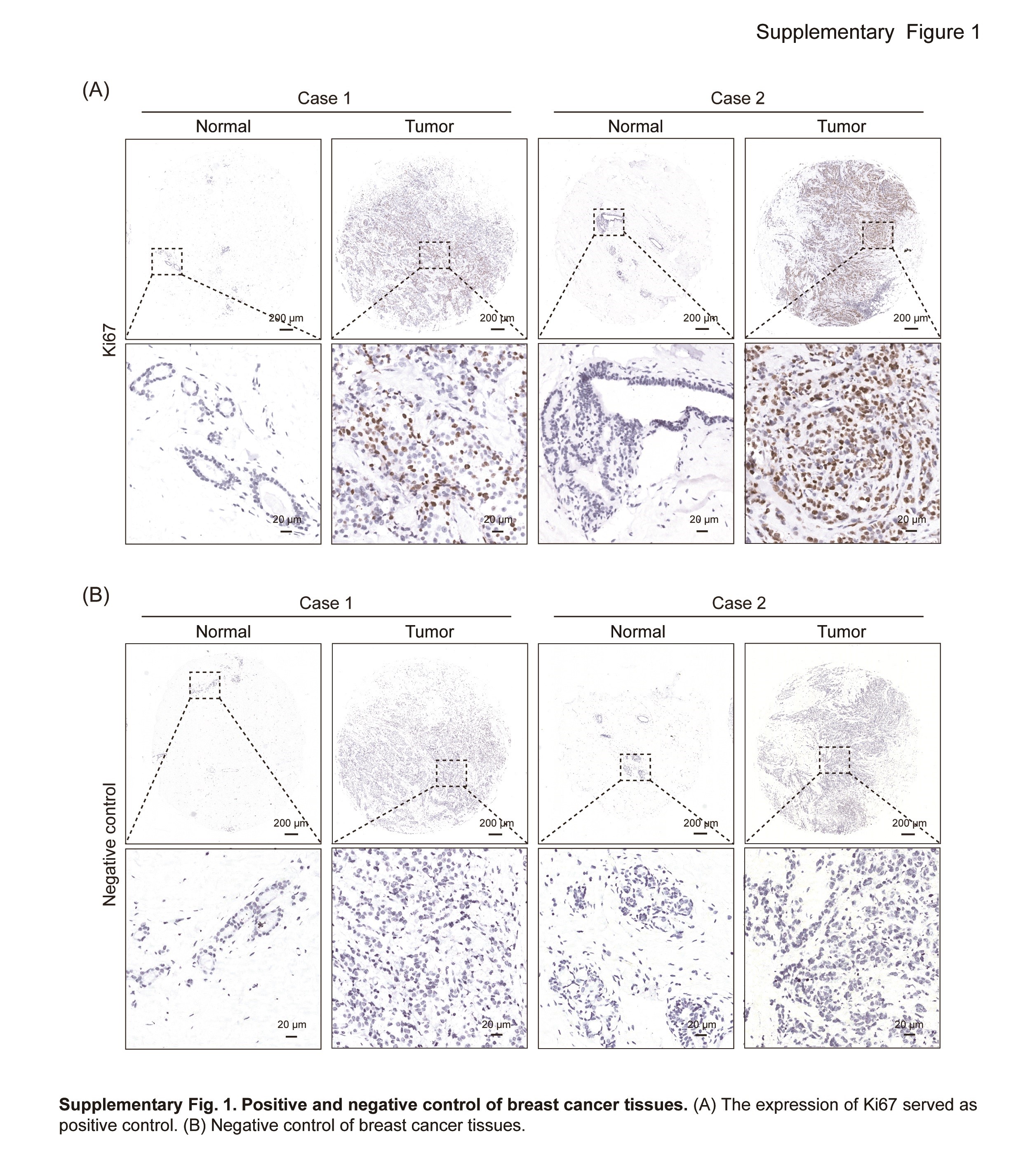
To investigate the role of Ehm2/1 in breast cancer progression, we first analyzed its expression levels using GEO database (GSE36295 and GSE42568) and TCGA dataset downloaded from SYNAPS (www.synapse.org, unc.edu_BRCA_IlluminaHiSeq_RNA SeqV2.isoformExp). We found that Ehm2/1 was downregulated in breast cancer tissues compared with normal breast tissues (Figure 1A and B). The relationship between Ehm2/1 expression and metastasis to distant organs was also analyzed using GEO database (GSE46563). Ehm2/1 expression level was significantly decreased in breast cancer with distant metastasis (Figure 1C). Furthermore, we analyzed the association of Ehm2/1 (Affy ID 220524_at) with breast cancer patient survival using the Kaplan–Meier survival analysis (http://kmplot.com/analysis) and found that breast cancer patients with higher Ehm2/1 expression exhibited better overall survival (Figure 1D) and disease-free survival (Figure 1E). Then, immunohistochemical staining of Ehm2/1 was performed using a tissue microarray, which included 30 breast cancer specimens paired with normal tissues. The results revealed that Ehm2/1 protein expression significantly decreased in breast cancer tissues (Figure 2A and B). The positive and negative control were shown in Supplementary Figure 1, available at Carcinogenesis Online. The relevance of Ehm2/1 protein levels in the stage of cancer development was further verified. In general, the levels of Ehm2/1 were higher in adjacent normal tissues and lower in breast cancer tissues, and there were no significant differences among the different grades (Figure 2C and D). Besides, the protein levels of Ehm2/1 in five pairs of frozen breast cancer tissues and adjacent normal tissues were measured. The protein levels of Ehm2/1 markedly decreased in the breast cancer tissues (Figure 2E and F). Collectively, these results suggest that Ehm2/1 was expressed at low levels in breast cancer and correlated with poor prognosis of breast cancer patients.
Figure 1.

Decreased Ehm2/1 expression is associated with poor prognosis of breast cancer patients. (A) Relative mRNA levels of Ehm2/1 in breast cancer and normal tissues in GEO datasets (GSE36295 and GSE42568). (B) Relative mRNA levels of Ehm2/1 in breast cancer and adjacent breast tissue in TCGA datasets. (C) Relative RNA levels of Ehm2/1 in breast cancer patients with metastasis and without metastasis in GEO datasets (GSE46563). (D) Kaplan–Meier analysis of the association of Ehm2/1 expression with breast cancer patients’ overall survival (http://kmplot.com/analysis). (E) Kaplan–Meier analysis of the association of Ehm2/1 expression with breast cancer patients’ disease-free survival. Data are shown as means ± SEM. P values were calculated by Student’s t-test. **P < 0.05, ***P < 0.001.
Figure 2.

Ehm2/1 is lowly expressed in breast cancer patients. (A) Representative images from immunohistochemical staining of Ehm2/1 in breast cancer tissue microarray containing 30 breast cancer specimens and paired normal tissues. Black arrows represent positive epithelial cells. (B) Ehm2/1 expression levels were shown as box plots. (C) Representative images from immunohistochemical staining of Ehm2/1 in breast cancer specimens in different grades were shown. Black arrows represent positive epithelial cells. (D) Semiquantitative analysis of Ehm2/1 expression score in different grades. Data were shown as means ± SEM. (E) The protein expression of Ehm2/1 in breast cancer patients was measured by western blotting (n = 5). (F) Protein expression of (E) was quantified by gray scanning. Data were shown as means ± SD. P values were calculated by Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Ehm2/1 suppressed breast cancer cell migration and invasion in vitro
To explore the function of Ehm2/1 in breast cancer, we sought to characterize the altered cellular phenotypes in breast cancer cells following overexpression or knockdown of Ehm2/1. Overexpression and knockdown of Ehm2/1 in MCF-7 cells were verified by western blotting and Quantitative reverse transcription PCR (qRT-PCR) (Figure 3A and B). Overexpression of Ehm2/1 significantly impaired cells migration and invasion, as determined by Transwell assays (Figure 3C). Knockdown of Ehm2/1 significantly impaired cells migration and invasion as determined in Figure 3D. These results indicated that Ehm2/1 played an important role in the migration and invasion of breast cancer cells.
Figure 3.

Ehm2/1 suppresses cell migration and invasion in vitro. (A) Ehm2/1 protein levels were analyzed in Ehm2/1 overexpressed and knocked down MCF-7 cells using western blotting. (B) The mRNA expression levels of Ehm2/1 were analyzed in Ehm2/1 overexpressed and knocked down MCF-7 cells using Quantitative reverse transcription PCR (qRT-PCR). Cell migration assay and cell invasion assay were performed in Ehm2/1 overexpressed (C) and knocked down MCF-7 cells (D). Results were from a representative experiment performed in triplicate. Image J was used to perform quantitative analysis. Data were shown as means ± SD. P values were calculated by Student’s t-test. *P <0.05, **P < 0.01.
Ehm2/1 increased the stability of E-cadherin protein
To elucidate the mechanism underlying the suppressive role of Ehm2/1 in breast cancer, we attempted to identify downstream targets that could directly regulate the migration and invasion ability of breast cancer cells. In our previous study, we found that overexpression of Ehm2/1 increased E-cadherin protein levels and inhibited cell migration (23). Here, we determined the effect of Ehm2/1 on E-cadherin expression by western blotting and qRT–PCR. The results indicated that forced expression of Ehm2/1 was enhanced, whereas knockdown of Ehm2/1 suppressed the protein expression of E-cadherin (Figure 4A and B). However, E-cadherin mRNA levels were not significantly changed neither Ehm2/1 overexpressed nor knocked down (Figure 4A and B). This result indicated that Ehm2/1 might regulate E-cadherin degradation. Thus, the half-life of E-cadherin was measured using a cycloheximide assay. Overexpression of Ehm2/1 dramatically extended the half-life of E-cadherin (Figure 4C and D), and depletion of Ehm2/1 markedly reduced the half-life of E-cadherin (Figure 4E and F). Together, these results suggest that Ehm2/1 could stabilize E-cadherin protein level.
Figure 4.

Ehm2/1 enhances the protein stability of E-cadherin. (A) Protein and mRNA levels of E-cadherin derived from MCF-7 cells with the overexpression of Ehm2/1 were measured by western blotting or Quantitative reverse transcription PCR (qRT-PCR). (B) Protein and mRNA expressions of E-cadherin derived from MCF-7 cells with knockdown of Ehm2/1 were measured by western blotting or qRT-PCR. (C) Expression of E-cadherin in MCF-7 cells transfected with Ehm2/1 express plasmid and treated with cycloheximide (CHX) for the indicated time points was measured by western blotting. (D) The graph shows the quantitative results of (C) by Image J. (E) Expression of E-cadherin in MCF-7 cells transfected with Ehm2/1 siRNA and treated with CHX for the indicated time points was measured by western blotting. (F) The graph shows the quantitative results of (E) by Image J. Data are shown as means ± SD. P values were calculated by Student’s t-test, **P < 0.01.
Ehm2/1 inhibited the ubiquitination of E-cadherin
Ubiquitination is a posttranslational modification of proteins with ubiquitin in an ATP-dependent enzyme-catalyzed cascade process, and the ubiquitin-proteasome system is involved in the degradation of more than 80% of proteins in cells (24). To explore whether Ehm2/1 extended the half-life of E-cadherin protein through the ubiquitin-proteasome system, the proteasome inhibitor MG-132 was used to treat MCF-7 cells with overexpression and knockdown of Ehm2/1. In the presence of the proteasome inhibitor MG-132, Ehm2/1 overexpression did not further increase E-cadherin protein levels (Figure 5A), however, inhibition of the proteasome by MG-132 prevents degradation of E-cadherin by knockdown of Ehm2/1 (Figure 5B), suggesting that Ehm2/1 stabilized E-cadherin protein levels by inhibiting its proteasomal degradation. To test whether Ehm2/1 decreases endogenous E-cadherin polyubiquitination, Ehm2/1 was silenced using siRNA in MCF-7 cells. As shown in Figure 5C, knockdown of Ehm2/1 remarkably improved endogenous E-cadherin protein polyubiquitination levels compared with the control cells. At the same time, we cotransfected Flag-Ehm2/1, HA-Ub and His-E-cadherin in HEK-293T cells. As shown in Figure 5D, overexpression of Ehm2/1 decreased E-cadherin polyubiquitination. These data collectively supported the notion that Ehm2/1 stabilized E-cadherin mainly by inhibiting its polyubiquitination.
Figure 5.

Ehm2/1 inhibites the ubiquitination of E-cadherin. (A) The improvement of E-cadherin protein level induced by Ehm2/1 overexpression was blocked by proteasome inhibitor MG-132. Western blotting analysis of E-cadherin and Ehm2/1 derived from MCF-7 cells treated with 10 mM MG-132 for 6 h after Ehm2/1 overexpression. (B) MG-132 inhibited the decrease in E-cadherin protein level caused by Ehm2/1 knockdown. Western blotting analysis of E-cadherin and Ehm2/1 derived from MCF-7 cells treated with 10 mM MG-132 for 6 h after knocking down Ehm2/1. (C) Ehm2/1 knockdown in MCF-7 cells increased the endogenous E-cadherin ubiquitination. The cells were transfected with control or Ehm2/1 siRNAs for 48 h. siRNA was used as the negative control. Western blotting analysis of anti-E-cadherin immunoprecipitate and WCL was used to detect the ubiquitinated E-cadherin protein levels using anti-ubiquitin antibody after the cells were treated with MG-132 for 6 h. (D) Ehm2/1 decreased E-cadherin ubiquitination. HEK-293T cells were cotransfected with Flag-Ehm2/1, HA-Ub and His-E-cadherin and were treated with MG-132 for 6 h. Western blotting of anti-His immunoprecipitate and WCL was used to detect the ubiquitinated E-cadherin protein.
Ehm2/1 interacted with E-cadherin
To test whether Ehm2/1 interacted with E-cadherin, His-E-cadherin and FLAG-Ehm2/1 were cotransfected into HEK-293T cells and co-immunoprecipitated using an anti-His antibody. The results indicated that FLAG-Ehm2/1 co-immunoprecipitated with His-E-cadherin (Figure 6A). Endogenous co-immunoprecipitation assays demonstrated that Ehm2/1 interacted with E-cadherin (Figure 6B). Furthermore, Immunofluorescence staining results showed that Ehm2/1 was colocalized with E-cadherin in the plasma membrane (Figure 6C). Taken together, these data suggested that Ehm2/1 could interact with E-cadherin.
Figure 6.

Ehm2/1 interacted with E-cadherin. (A) Exogenous Ehm2/1 and E-cadherin proteins interact with each other in HEK-293T cells. FLAG-Ehm2/1 and His-E-cadherin were cotransfected in HEK-293T cells. His-E-cadherin was immunoprecipitated by an anti-His antibody. (B) Endogenous Ehm2/1 and E-cadherin proteins interacted with each other in MCF-7 cells. E-cadherin was immunoprecipitated by an anti-E-cadherin antibody. (C) Ehm2/1 was colocalized with E-cadherin at the cell membrane. Immunofluorescence staining was used to test the subcellular localization of Ehm2/1 and E-cadherin in MCF-7 cells transfected with FLAG-Ehm2/1.
Discussion
In this study, we identified that Ehm2/1 mediated the degradation of E-cadherin by inhibiting its ubiquitination. First, we found that Ehm2/1 decreased and was associated with a high survival rate in breast cancer patients. Next, the inhibited migration and invasion abilities of Ehm2/1 were demonstrated. Our results indicate that Ehm2/1 could stabilize the protein level of E-cadherin by inhibiting the ubiquitination of E-cadherin. Taken together, our results suggest that Ehm2/1 is a tumor suppressor that stabilizes E-cadherin and maintains epithelial homeostasis to inhibit tumor progression.
Ehm2 is upregulated in prostate cancers and promotes prostate cancer progression and metastasis (25). Ehm2 was also increased in breast cancer and was correlated with poor prognosis and metastasis (26). These results were controversial in our study. This must have been caused by the alternative splicing of Ehm2. Ehm2 can produce two transcript variants, encoding two protein isoforms, Ehm2/1 and Ehm2/2. In comparison with Ehm2/2, 382 amino acids are absent from the carboxyl-terminal region of Ehm2/1 (20). The two Ehm2 isoforms have distinct expression patterns in various cancers, including lung adenocarcinoma (LUAD) and breast cancers (22). Our previous study demonstrated that the two isoforms of Ehm2 played different roles in LUAD. Overexpression of Ehm2/1 exerted inhibitory effects, while knockdown of Ehm2/1 promoted the growth, invasion and migration of A549 cells in vitro (22). In this study, we focused on the role of Ehm2/1 in breast cancer. In accordance with our previous results, Ehm2/1 was expressed at low levels in breast cancers and exerted inhibitory roles in breast cancer cells, inhibiting cell migration and invasion.
Cadherins are transmembrane glycoproteins that are responsible for cell–cell adhesion and maintenance of normal tissue architecture. Classical cadherins include epithelial-, placental- and neural-cadherin (E-, P- and N-cadherin, respectively) (27). In our previous study, we found that Ehm2/1 could colocalize with β-catenin and E-cadherin at the cell membrane, and overexpression of Ehm2/1 upregulated the protein levels of E-cadherin (23). E-cadherin and β-catenin form a cadherin-catenin complex, which maintains cell shape and polarity. Based on this, we analyzed the effects of Ehm2/1 on E-cadherin expression. Similar to the effect of Ehm2/1 on β-catenin expression, Ehm2/1 increased E-cadherin protein levels without affecting its mRNA levels. Later, we found that E-cadherin was stabilized by Ehm2/1 to inhibit the migration and invasion of breast cancer cells. In carcinomas, EMT is a pivotal process of morphogenesis, with increased invasion and metastasis abilities (28,29). A typical phenomenon of EMT is the loss of E-cadherin expression during cancer progression (30). In non-small cell lung cancer, E-cadherin is a target gene of TBC1D2b, which is stimulated by the ZEB1/NuRD complex and promotes cancer cell metastasis (31). E-cadherin also plays an important role in the regulation of HDACs in human pancreatic cancers. ZEB1 recruits HDACs to the E-cadherin promoter and inhibits their expression (32). Thus, our study supports that Ehm2/1 might impede the EMT process in breast cancer by inhibiting E-cadherin degradation in the proteasome.
Ubiquitination is the process of ligating an ubiquitin molecule, a 76-amino acid protein that is ubiquitously expressed in all tissues, to a substrate protein, which in turn regulates protein stability and translocation (33). Importantly, direct and indirect evidence indicates that ubiquitination actively participates in the regulation of core proteins required for epithelial morphogenesis (34,35). Many proteins can be involved in cell motility and invasiveness through regulating ubiquitination of EMT-associated proteins. For example, GSK3β controls EMT and tumor metastasis by mediating the phosphorylation of Slug and facilitating Slug protein turnover via chromatin immunoprecipitation (36). One study showed that CD147 promotes hepatocyte depolarization by inducing E-cadherin degradation and mediates hepatocyte polarity loss (37). Similar to our results, we found that overexpression of Ehm2/1 decreased, while knocking down Ehm2/1 increased the ubiquitination of E-cadherin.
In the process of protein ubiquitination, E3 ligases have pivotal roles in conferring specificity to ubiquitination, since they select substrates and modification sites and govern the types of modifications that determine substrate specificity. Many E3 ligases play critical roles in EMT. For example, the Ub E3 ligase Pellino-1 can mediate EMT by upregulating Vimentin, Slug and Snail expression and downregulating E-cadherin and β-catenin expression (38). At present, knowledge of the E3 ligases responsible for E-cadherin ubiquitination is limited (39). The first suggested E3 ligase is Hakai, which targeted E-cadherin for endocytosis, and was identified in a yeast two-hybrid screening as an interactor of phosphorylated E-cadherin, but not of N- or OB-cadherin (40). In contrast to Hakai, which targets E-cadherin for endocytosis and degradation in the lysosome, MDM2, the E3 ligase responsible for p53 ubiquitination and degradation, targets E-cadherin for proteasomal degradation (41). A recent study demonstrated that lncRNA UCA1 can inhibit the degradation of E-cadherin induced by MDM2 by influencing the interaction between E-cadherin and MDM2 in primary prostate cancer (42). Another E3 ligase, RNF43, has also been reported to ubiquitinate and degrade E-cadherin to facilitate EMT in LUAD (43). In our ongoing study, we screened for a new E3 ligase that can target E-cadherin for proteasomal degradation. We also found that Ehm2/1 regulated E3 ligase-mediated E-cadherin degradation.
In conclusion, our study demonstrated that Ehm2/1 could inhibit the migration and invasion of breast cancer cells. This process was achieved through the interaction between E-cadherin and Ehm2/1, and inhibited the degradation of E-cadherin by Ehm2/1. Unfortunately, the Ub E3 ligase has not yet been clarified in this study. However, a detailed and complex mechanism is being studied further.
Supplementary material
Supplementary data are available at Carcinogenesis online.
Supplementary Fig. 1. Positive and negative control of breast cancer tissues. (A) The expression of Ki67 served as positive control. (B) Negative control of breast cancer tissues.
{kind=link}
Acknowledgements
We would like to thank Dongwei Fan and Zhicheng Ge for providing clinical breast cancer samples.
Abbreviations
- ATCC
American Type Culture Collection
- DMEM
Dulbecco’s modified Eagle medium
- ECL
enhanced chemiluminescence
- EMT
epithelial-mesenchymal transition
- FERM
four-point-one protein, ezrin, radixin, moesin
- LN
lymph node
- qRT–PCR
Quantitative reverse transcription PCR
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
Contributor Information
Xin Yin, Beijing Key Laboratory of Cancer Invasion and Metastasis Research, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Gen Li, Beijing Key Laboratory of Cancer Invasion and Metastasis Research, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Dongwei Fan, Department of Orthopaedics, Peking University Third Hospital, Beijing 100191, China.
Zhicheng Ge, Department of General Surgery, Beijing Friendship Hospital, Capital Medical University, Beijing 100050, China.
Tianshu Yang, Beijing Key Laboratory of Cancer Invasion and Metastasis Research, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Yaxin Shang, Beijing Key Laboratory of Cancer Invasion and Metastasis Research, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Tianyu Ma, Key Laboratory of Cancer and Microbiome, State Key Laboratory of Molecular Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China.
Baowen Yuan, Key Laboratory of Cancer and Microbiome, State Key Laboratory of Molecular Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China.
Wei Huang, Beijing Key Laboratory of Cancer Invasion and Metastasis Research, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Xu Teng, Beijing Key Laboratory of Cancer Invasion and Metastasis Research, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Hefen Yu, Beijing Key Laboratory of Cancer Invasion and Metastasis Research, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China.
Funding
This work was supported by the National Natural Science Foundation of China (81672726, 81902960, 41931291); the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT310027); the State Key Laboratory of Molecular Oncology (SKLMO-KF2021-21); and the Nature Science Foundation of Beijing (7204241).
Conflict of Interest Statement
None declared.
Data availability
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
References
- 1. Sung, H., et al. (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin., 71, 209–249. [DOI] [PubMed] [Google Scholar]
- 2. Xu, B., et al. (2020) Chinese expert consensus on the clinical diagnosis and treatment of advanced breast cancer (2018). Cancer, 126, 3867–3882. [DOI] [PubMed] [Google Scholar]
- 3. Bao, J., et al. (2019) Outcomes in patients with small node-negative invasive breast cancer. Breast J., 25, 638–643. [DOI] [PubMed] [Google Scholar]
- 4. Inoue, H., et al. (2019) Breast-conserving surgery without radiation in elderly women with early breast cancer. Surg. Oncol., 31, 22–25. [DOI] [PubMed] [Google Scholar]
- 5. Mittal, V. (2018) Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol., 13, 395–412. [DOI] [PubMed] [Google Scholar]
- 6. Pastushenko, I., et al. (2019) EMT transition states during tumor progression and metastasis. Trends Cell Biol., 29, 212–226. [DOI] [PubMed] [Google Scholar]
- 7. Yang, F., et al. (2012) SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J., 31, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li, Y., et al. (2021) OSR1 phosphorylates the Smad2/3 linker region and induces TGF-β1 autocrine to promote EMT and metastasis in breast cancer. Oncogene, 40, 68–84. [DOI] [PubMed] [Google Scholar]
- 9. Corso, G., et al. (2018) Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. J. Med. Genet., 55, 431–441. [DOI] [PubMed] [Google Scholar]
- 10. Yeung, K.T., et al. (2017) Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol., 11, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Corso, G., et al. (2020) E-cadherin deregulation in breast cancer. J. Cell. Mol. Med., 24, 5930–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Padmanaban, V., et al. (2019) E-cadherin is required for metastasis in multiple models of breast cancer. Nature, 573, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zheng, T., et al. (2018) NRBE3 promotes metastasis of breast cancer by down-regulating E-cadherin expression. Biochim. Biophys. Acta, Mol. Cell. Res., 1865, 1869–1877. [DOI] [PubMed] [Google Scholar]
- 14. Cho, H.-J., et al. (2019) ZEB1 Collaborates with ELK3 to repress E-Cadherin expression in triple-negative breast cancer cells. Mol Cancer Res., 17, 2257–2266. [DOI] [PubMed] [Google Scholar]
- 15. Chauhan, S., et al. (2003) Androgen regulation of the human FERM domain encoding gene EHM2 in a cell model of steroid-induced differentiation. Biochem. Biophys. Res. Commun., 310, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Badouel, C., et al. (2009) The FERM-domain protein expanded regulates Hippo pathway activity via direct interactions with the transcriptional activator Yorkie. Dev. Cell, 16, 411–420. [DOI] [PubMed] [Google Scholar]
- 17. Pearson, M.A., et al. (2000) Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell, 101, 259–270. [DOI] [PubMed] [Google Scholar]
- 18. Loie, E., et al. (2015) CRB3A controls the morphology and cohesion of cancer cells through Ehm2/p114RhoGEF-dependent signaling. Mol. Cell. Biol., 35, 3423–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chauhan, S., et al. (2003) Androgen regulation of the human FERM domain encoding gene EHM2 in a cell model of steroid-induced differentiation. Biochem. Biophys. Res. Commun., 310, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shimizu, K., et al. (2000) Molecular cloning of a novel NF2/ERM/4.1 superfamily gene, ehm2, that is expressed in high-metastatic K1735 murine melanoma cells. Genomics, 65, 113–120. [DOI] [PubMed] [Google Scholar]
- 21. Hashimoto, Y., et al. (1996) Identification of genes differentially expressed in association with metastatic potential of K-1735 murine melanoma by messenger RNA differential display. Cancer Res., 56, 5266–5271. [PubMed] [Google Scholar]
- 22. Li, S., et al. (2019) Differential expression and functions of Ehm2 transcript variants in lung adenocarcinoma. Int. J. Oncol., 54, 1747–1758. [DOI] [PubMed] [Google Scholar]
- 23. Yu, H., et al. (2016) The splice variant Ehm2/1 in breast cancer MCF-7 cells interacted with b-catenin and increased its localization to plasma membrane. RSC Adv., 6, 78436–78444. [Google Scholar]
- 24. Sun, T., et al. (2020) The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer, 19, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang, J., et al. (2006) Increased expression of the metastasis-associated gene Ehm2 in prostate cancer. Prostate, 66, 1641–1652. [DOI] [PubMed] [Google Scholar]
- 26. Yu, H., et al. (2010) Clinical implications of the influence of Ehm2 on the aggressiveness of breast cancer cells through regulation of matrix metalloproteinase-9 expression. Mol. Cancer Res., 8, 1501–1512. [DOI] [PubMed] [Google Scholar]
- 27. Kaszak, I., et al. (2020) Role of cadherins in cancer-a review. Int. J. Mol. Sci., 21, 7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nieto, M.A., et al. (2016) EMT: 2016. Cell, 166, 21–45. [DOI] [PubMed] [Google Scholar]
- 29. Das, V., et al. (2019) The basics of epithelial-mesenchymal transition (EMT): a study from a structure, dynamics, and functional perspective. J. Cell. Physiol., 234, 14535–14555. [DOI] [PubMed] [Google Scholar]
- 30. Gloushankova, N., et al. (2018) Role of epithelial-mesenchymal transition in tumor progression. Biochemistry (Mosc)., 83, 1469–1476. [DOI] [PubMed] [Google Scholar]
- 31. Manshouri, R., et al. (2019) ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat. Commun., 10, 5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aghdassi, A., et al. (2012) Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut, 61, 439–448. [DOI] [PubMed] [Google Scholar]
- 33. Deng, L., et al. (2020) The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther., 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheng, X., et al. (2018) Degradation for better survival? Role of ubiquitination in epithelial morphogenesis. Biol. Rev. Camb. Philos. Soc., 93, 1438–1460. [DOI] [PubMed] [Google Scholar]
- 35. Cai, J., et al. (2018) The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell., 9, 754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kao, S., et al. (2014) GSK3β controls epithelial-mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene, 33, 3172–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu, M., et al. (2018) Basolateral CD147 induces hepatocyte polarity loss by E-cadherin ubiquitination and degradation in hepatocellular carcinoma progress. Hepatology., 68, 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jeon, Y., et al. (2017) Pellino-1 promotes lung carcinogenesis via the stabilization of Slug and Snail through K63-mediated polyubiquitination. Cell Death Differ., 24, 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Niño, C., et al. (2019) When ubiquitin meets E-cadherin: plasticity of the epithelial cellular barrier. Semin. Cell Dev. Biol., 93, 136–144. [DOI] [PubMed] [Google Scholar]
- 40. Fujita, Y., et al. (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol., 4, 222–231. [DOI] [PubMed] [Google Scholar]
- 41. Yang, J., et al. (2006) MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol. Cell. Biol., 26, 7269–7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao, X., et al. (2020) LncRNA UCA1 maintains the low-tumorigenic and nonmetastatic status by stabilizing E-cadherin in primary prostate cancer cells. Mol. Carcinog., 59, 1174–1187. [DOI] [PubMed] [Google Scholar]
- 43. Zhang, Y., et al. (2019) RNF43 ubiquitinates and degrades phosphorylated E-cadherin by c-Src to facilitate epithelial-mesenchymal transition in lung adenocarcinoma. BMC Cancer, 19, 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.