

Dyslipidemia is associated with atherosclerotic cardiovascular disease (ASCVD), the leading cause of death worldwide1. Plasma levels of low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and triglycerides are all highly correlated with ASCVD risk and are demonstrated heritable traits2. The study of individuals with extreme lipid phenotypes has led to the identification of rare variants in multiple genes that have a major influence on lipoprotein metabolism. In several cases, these discoveries have contributed to the validation of new therapeutic targets and ultimately to effective and approved therapeutics. For example, loss-of-function (LoF) mutations in MTTP and APOB that cause abetalipoproteinemia and hypobetalipoproteinemia, respectively, are associated with very low LDL-C and led to the development of lomitapide, a small molecule MTP inhibitor, and mipomersen, an antisense oligonucleotide (ASO) to APOB, both approved for homozygous familial hypercholesterolemia (HoFH)3. In a family with familial combined hypolipidemia (low LDL-C, HDL-C, and triglycerides), LoF mutations in ANGPTL3 provided validation of this secreted protein as a therapeutic target, and evinacumab is an inhibitory antibody approved for HoFH and being studied for treatment of hypertriglyceridemia4. An especially interesting example is that of PCSK9; initial studies in families with very high LDL-C identified rare missense variants in PCSK9 that are now known to be gain-of-function (GoF) variants, whereas some individuals with very low LDL-C levels were found to have classic LoF variants. PCSK9 is now a target of two approved antibodies and an approved siRNA therapeutic (inclisiran)5. The example of PCSK9 demonstrates the immense biological insights derived from allelic series that include GoF variants, and the lipid field has been interested in the identification of GoF variants in other genes known to affect lipoprotein metabolism. In this issue of Circulation, Dijk and colleagues report the first GoF variant in the LIPC gene which encodes the enzyme hepatic lipase (HL).
The investigators focused on DNA sequencing of individuals with extreme lipid phenotypes using a custom panel of known lipid-related genes. In one proband with very low levels of LDL-C and HDL-C they identified a private variant in LIPC: p.Glu97Gly (E97G). Within the extended family, carriers of the LIPC-E97G variant exhibited low LDL-C and HDL-C levels. The authors used homology-based modeling to hypothesize that the amino acid substitution modifies a conserved motif affecting substrate access to HL’s catalytic site. Experimental studies confirmed that the E97G mutation altered substrate specificity, leading to increased HL phospholipase activity with no effect on triglyceride lipase activity in vitro. Mice overexpressing LIPC-E97G recapitulated the combined hypocholesterolemic phenotype and provided key insight into the variant’s effect on lipoprotein kinetics: this novel GoF variant was shown to promote the clearance of cholesterol-containing lipoprotein remnants from blood. Surprisingly, much of this accelerated clearance in mice was by extra-hepatic tissues (namely, adipose tissue depots and oxidative tissues). The discovery of this private GoF variant LIPC-E97G demonstrates that HL phospholipase activity is specifically critical in apoB-lipoprotein and HDL-C metabolism.
LIPC-E97G represents the first GoF variant in LIPC/HL, and is shown to lead to hypocholesterolemia. Importantly, LoF variants in LIPC/HL were first identified by Hegele in 1991 as a rare cause of hypercholesterolemia6. Thus, LIPC joins PCSK9 as another lipid gene in which both LoF and GoF variants have been identified and result in plasma lipid levels at opposite ends of the spectrum, creating a full allelic series. Interestingly, the best-known GoF variant in the lipid field is a variant in lipoprotein lipase (LPL), a close relative of HL. Rare biallelic LoF mutations in LPL cause familial chylomicronemia syndrome (FCS) characterized by severe hypertriglyceridemia and risk of pancreatitis. In contrast, the relatively common LPL variant S447X is a surprising GoF variant which truncates the protein at the C-terminal by only two amino acids. This loss of terminal amino acids enhances LPL activity, thereby decreasing plasma triglycerides, increasing HDL-C, and modestly protecting against cardiovascular disease7. This fascinating experiment of nature proved that therapeutic intervention to increase LPL activity would not only reduce triglycerides but would also reduce risk of ASCVD and has fueled intense interest in developing other approaches to increasing LPL activity. It also led to the use of this GoF variant in a gene therapy approach for LPL deficiency in humans (alipogene tiparvovec) that was approved in Europe8.
Apolipoprotein C-III (gene APOC3) is an endogenous inhibitor of LPL that is transported on triglyceride-rich lipoproteins. LoF variants in APOC3 are proven to reduce triglyceride levels (through increased LPL activity) and provide protection against ASCVD. One missense variant in APOC3 (A43T) behaves like a LoF variant due to reduced lipid binding and rapid clearance of poorly lipid-bound protein, providing insight into ApoC-III structure-function9. APOC3-Q38K is an interesting potential GoF variant that may provide further insight. Like LIPC-E97G, this mutation was initially identified in a single family with increased plasma triglycerides10. APOC3-Q38K has been reported to increase very low-density lipoprotein (VLDL) assembly and secretion via de novo lipogenesis11. Recent work also demonstrates that the substitution increases the LPL inhibitory capacity of the protein and enhances ApoC-III’s affinity for ApoB-containing lipoproteins while reducing its affinity for HDL particles12. Other GoF variants in APOC3 are likely to be identified. In contrast to APOC3, Apolipoprotein A-V (gene APOA5) is an enhancer of LPL. LoF variants in APOA5 have been reported in association with hypertriglyceridemia and increased risk of pancreatitis and ASCVD13. To date, no GoF variants in APOA5 have been characterized, but their discovery will be of great interest in helping define the structure-function properties of ApoA-V and as a potential tool for APOA5-based therapeutics. Leverage of GoF variants in a therapeutic setting has influenced the development of gene therapy approaches as seen with Factor IX Padua in hemophilia14.
Continued identification and mechanistic study of other naturally-occurring GoF variants in lipid genes will provide expansion of the allelic series, insight into structure-function, and potentially alternative approaches to lipid-lowering therapeutic strategies (Figure). LIPC-E97G alters HL’s phospholipid substrate preference to resemble more closely that of its relative LIPG (endothelial lipase, EL). ANGPTL3 is an inhibitor of EL and the LDL-lowering effect of ANGPTL3 inhibition is apparently due to the increased activity of EL15. One can imagine how a GoF variant in EL (not yet described) might result in a hypocholesterolemic phenotype and further elucidate the role of phospholipase activity in lipoprotein catabolism. Detailed variant-driven structure-function assessment of other key players in the lipase pathways (e.g., apolipoproteins, chaperones, inhibitors) will add to our understanding of the biology of these proteins and enhance our therapeutic arsenal.
Figure. Observed or predicted effects on plasma lipids by gain-of-function (GoF) variants in selected lipid genes.
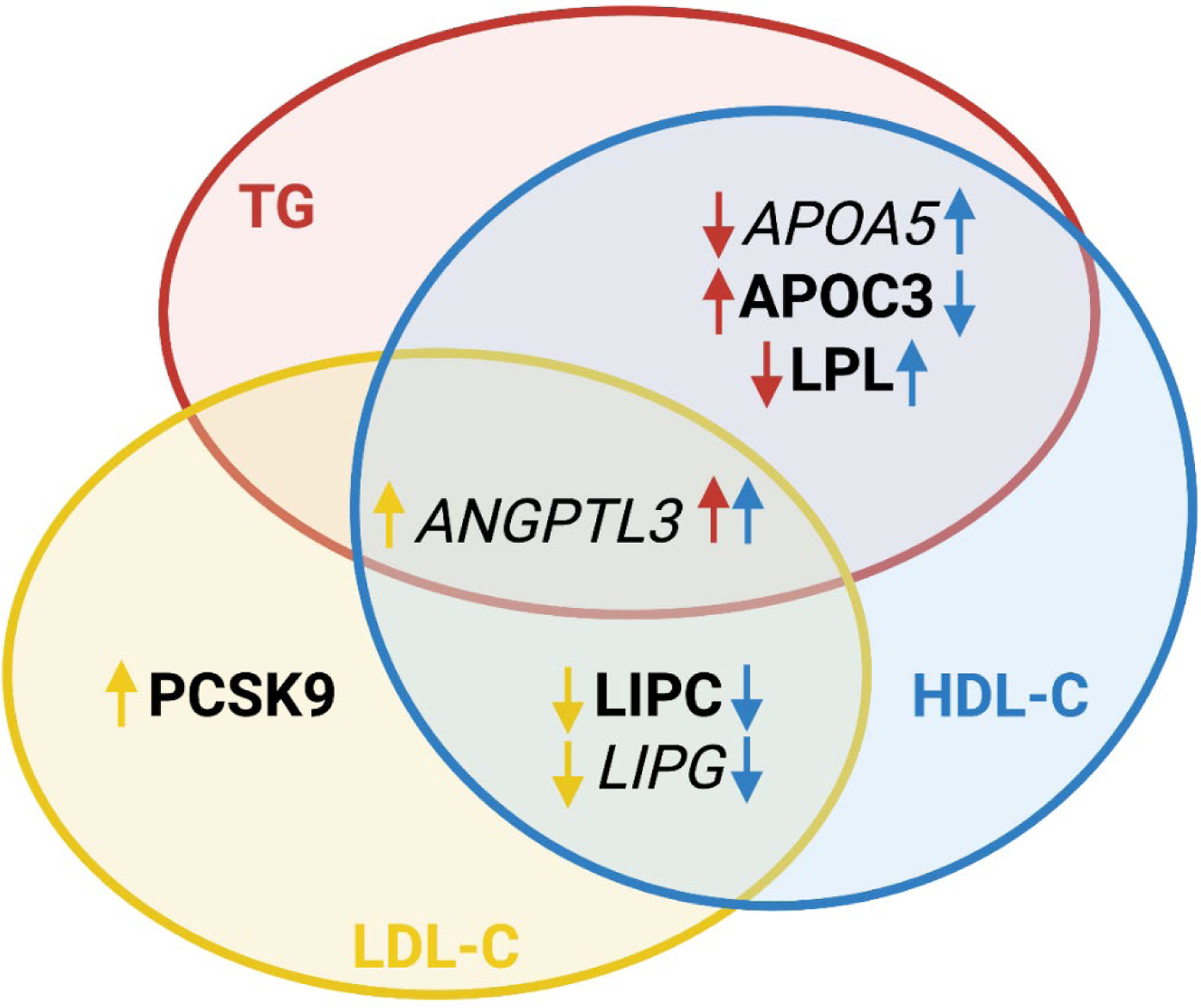
The diagram depicts the observed (bold) and predicted (italic) effects of GoF variants in selected genes on triglycerides (TG), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C). Color of the arrows refers to the lipid parameter affected by GoF variants in indicated genes: red - TG, blue - HDL-C, and yellow - LDL-C. Direction of the arrows indicates the direction of effect: ↑ increase or ↓ decrease of indicated lipid parameters. Gene symbols: APOA5, apolipoprotein A5; APOC3, apolipoprotein C3; LPL, lipoprotein lipase; ANGPTL3, angiopoietin like 3; PCSK9, proprotein convertase subtilisin/kexin type 9; LIPC, lipase C, hepatic type; LIPG, lipase G, endothelial type.
These interesting findings by Dijk, et al. arose from the discovery of a private variant identified in a single family with an extreme lipid phenotype. Careful mechanistic study of this variant revealed LIPC-E97G as the first GoF variant in HL and demonstrates the power of ‘N of 1’ discoveries in humans with extreme phenotypes. Structure-function analysis of HL and other proteins and their variants of interest continues to uncover novel and relevant biology with significant therapeutic implications. The insight gained from LIPC-E97G makes a strong case for detailed mechanistic investigations of other proteins and their naturally occurring mutations, leveraging human phenotypic and genotypic data to understand protein structure-function and provide insights into new therapeutic approaches.
Acknowledgements:
This work was supported in part by R01 HL133502 from the National Heart Lung and Blood Institute of the NIH.
Sources of Funding:
R01 HL133502 from the National Heart Lung and Blood Institute of the NIH
Footnotes
Disclosure Statement
None
References
- 1.Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022;145:e153–e639. doi: 10.1161/CIR.0000000000001052 [DOI] [PubMed] [Google Scholar]
- 2.Upadhyay RK. Emerging risk biomarkers in cardiovascular diseases and disorders. J Lipids 2015;2015:971453. doi: 10.1155/2015/971453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014;129:1022–1032. doi: 10.1161/CIRCULATIONAHA.113.001292 [DOI] [PubMed] [Google Scholar]
- 4.Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, Ali S, Banerjee P, Chan KC, Gipe DA, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med 2020;383:711–720. doi: 10.1056/NEJMoa2004215 [DOI] [PubMed] [Google Scholar]
- 5.Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol 2019;16:155–165. doi: 10.1038/s41569-018-0107-8 [DOI] [PubMed] [Google Scholar]
- 6.Hegele RA, Vezina C, Moorjani S, Lupien PJ, Gagne C, Brun LD, Little JA, Connelly PW. A hepatic lipase gene mutation associated with heritable lipolytic deficiency. J Clin Endocrinol Metab 1991;72:730–732. doi: 10.1210/jcem-72-3-730 [DOI] [PubMed] [Google Scholar]
- 7.Wittrup HH, Tybjaerg-Hansen A, Nordestgaard BG. Lipoprotein lipase mutations, plasma lipids and lipoproteins, and risk of ischemic heart disease. A meta-analysis. Circulation 1999;99:2901–2907. doi: 10.1161/01.cir.99.22.2901 [DOI] [PubMed] [Google Scholar]
- 8.Scott LJ. Alipogene tiparvovec: a review of its use in adults with familial lipoprotein lipase deficiency. Drugs 2015;75:175–182. doi: 10.1007/s40265-014-0339-9 [DOI] [PubMed] [Google Scholar]
- 9.Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, Landro JA, Barucci N, Zavadoski WJ, Sun Z, et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med 2017;23:1086–1094. doi: 10.1038/nm.4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pullinger CR, Malloy MJ, Shahidi AK, Ghassemzadeh M, Duchateau P, Villagomez J, Allaart J, Kane JP. A novel apolipoprotein C-III variant, apoC-III(Gln38-->Lys), associated with moderate hypertriglyceridemia in a large kindred of Mexican origin. J Lipid Res 1997;38:1833–1840. [PubMed] [Google Scholar]
- 11.Sundaram M, Curtis KR, Amir Alipour M, LeBlond ND, Margison KD, Yaworski RA, Parks RJ, McIntyre AD, Hegele RA, Fullerton MD, et al. The apolipoprotein C-III (Gln38Lys) variant associated with human hypertriglyceridemia is a gain-of-function mutation. J Lipid Res 2017;58:2188–2196. doi: 10.1194/jlr.M077313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vitali C, Stankov S, Khetarpal SA, Millar J, Mayne L, Englander SW, Hand NJ, Lund-Katz S, Phillips MC, Rader DJ. ApoC-III helical structure determines its ability to bind plasma lipoproteins and inhibit Lipoprotein Lipase-mediated triglyceride lipolysis. bioRxiv 2020. doi: 10.1101/2020.07.02.183178 [DOI] [Google Scholar]
- 13.Han Y, Dorajoo R, Chang X, Wang L, Khor CC, Sim X, Cheng CY, Shi Y, Tham YC, Zhao W, et al. Genome-wide association study identifies a missense variant at APOA5 for coronary artery disease in Multi-Ethnic Cohorts from Southeast Asia. Sci Rep 2017;7:17921. doi: 10.1038/s41598-017-18214-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, Cuker A, Sullivan LM, Majumdar S, Teitel J, et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N Engl J Med 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L, Soundarapandian MM, Castoreno AB, Millar JS, Rader DJ. LDL-Cholesterol Reduction by ANGPTL3 Inhibition in Mice Is Dependent on Endothelial Lipase. Circ Res 2020;127:1112–1114. doi: 10.1161/CIRCRESAHA.120.317128 [DOI] [PMC free article] [PubMed] [Google Scholar]