


Abstract
Here, we present DeepBIO, the first-of-its-kind automated and interpretable deep-learning platform for high-throughput biological sequence functional analysis. DeepBIO is a one-stop-shop web service that enables researchers to develop new deep-learning architectures to answer any biological question. Specifically, given any biological sequence data, DeepBIO supports a total of 42 state-of-the-art deep-learning algorithms for model training, comparison, optimization and evaluation in a fully automated pipeline. DeepBIO provides a comprehensive result visualization analysis for predictive models covering several aspects, such as model interpretability, feature analysis and functional sequential region discovery. Additionally, DeepBIO supports nine base-level functional annotation tasks using deep-learning architectures, with comprehensive interpretations and graphical visualizations to validate the reliability of annotated sites. Empowered by high-performance computers, DeepBIO allows ultra-fast prediction with up to million-scale sequence data in a few hours, demonstrating its usability in real application scenarios. Case study results show that DeepBIO provides an accurate, robust and interpretable prediction, demonstrating the power of deep learning in biological sequence functional analysis. Overall, we expect DeepBIO to ensure the reproducibility of deep-learning biological sequence analysis, lessen the programming and hardware burden for biologists and provide meaningful functional insights at both the sequence level and base level from biological sequences alone. DeepBIO is publicly available at https://inner.wei-group.net/DeepBIO.
Graphical Abstract
Graphical Abstract.
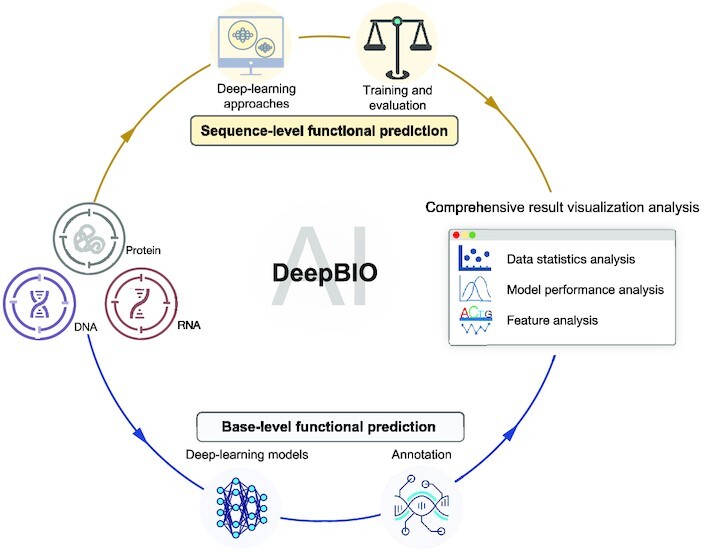
DeepBIO is an automated and interpretable deep-learning platform for high-throughput biological sequence level prediction, functional site annotation, and comprehensive visualization analysis.
INTRODUCTION
The development of next-generation sequencing techniques has led to an exponential increase in the amount of biological sequence data accessible, including genomic, transcriptomic and proteomic sequences. It naturally poses an important challenge—how to build the relationships from sequences to structures and functions (1). Given such large-scale data, traditional wet lab experimental methods are laborious, time-consuming and high cost for functional analysis. Alternatively, the booming development of machine-learning approaches has paved a new way to understand the complex mapping from biological sequences to their structures and functional mechanisms. Over the past few decades, data-driven machine-learning approaches have emerged as powerful methods to enable the automated and fast function prediction ab initio from sequences alone, providing a new perspective on studying the functional aspects of biological sequences (2–5).
The increasing use of machine-learning workflows in biological sequence analysis and the willingness to disseminate trained machine-learning models have pushed computer scientists to design more user-friendly solutions. In recent years, there have been an increasing number of web servers and software packages developed for this purpose. For instance, BioSeq-Analysis was the first platform to analyze various biological sequences via machine-learning approaches (6). Later, Liu et al. established BioSeq-Analysis2.0 (7) to automatically generate various predictors for biological sequence analysis at both the residue level and sequence level. Additionally, iLearnplus is a popular platform that provides a pipeline for biological sequence analysis, which comprises feature extraction and selection, model construction and analysis of prediction results (8). More recently, Liu et al. developed BioSeq-BLM, a web platform that introduces different biological language models for DNA, RNA and protein sequence analysis (9). In addition, other representative tools include iFeatureOmega (10), Rcpi (11) and protr (12). The platforms and tools have boosted the use of machine-learning solutions for biological sequence analysis tasks in biology. However, these traditional machine-learning workflows have some drawbacks. For instance, to train a good model, strong professional knowledge is usually required in terms of designing feature descriptors, selecting machine-learning algorithms as well as setting up the model parameters, which limit their usability in real applications to some extent; on the other hand, they cannot support large-scale prediction and analysis. Recently, deep learning has played a complementary role to traditional machine learning due to its excellent scalability and adaptivity. Therefore, some deep-learning analysis tools have been developed, such as Kipoi (13), Pysster (14) and Selene (15). Kipoi (13) is a repository of ready-to-use trained models for genomic data analysis. Pysster (14) is a Python package that supports training deep-learning models only with convolutional neural networks on biological sequence data. More recently, Chen et al. developed Selene, a PyTorch-based deep-learning library for quick and simple development, training and application of deep-learning model architectures for biological sequence data (15).
These tools make the use of deep learning more convenient for data-driven biological sequence analysis to some extent. However, there remain some key challenges that need to be addressed. First, despite the availability of deep-learning tools, they are not completely automated. Running the tools still faces some technical challenges for non-expert researchers. On the one hand, they require strong professional knowledge and programming skills to set up sophisticated software, which directly limits the generic use of deep learning in non-expert communities (i.e. researchers without any computer science training). On the other hand, it is extremely difficult for researchers to train deep-learning models with the enormous scale of data space, since the major downside of deep learning is its computational intensity, requiring high-performance computational resources and a long training time. Accordingly, a web platform that enables an automated deep-learning pipeline is greatly needed. Second, most of the existing tools cannot meet the high demand of the research community, since they provide few deep-learning architectures (seen in Supplementary Table S1), such as convolutional neural networks, for model construction. In fact, there are some state-of-the-art deep-learning models with successful applications in bioinformatics problems. For example, Graph Neural Networks (GNNs) demonstrated an ability to address complex biological problems, for example, the prediction of microRNA–disease interaction (16). Large-scale language pre-trained models such as DNABERT and ProtBERT showed strong capacities in discriminative feature learning in biological sequence analysis (5,17). Thus, it is necessary to enable the use of state-of-the-art deep-learning approaches in biology. Ultimately, existing tools lack comprehensive result analysis, which might limit users’ understanding of what the deep-learning models learn; how reliable the deep-learning predictions are; and why the deep learning performs well.
In this work, we present DeepBIO, an automated deep-learning platform for biological sequence prediction, functional annotation and result visualization analysis. To be specific, our DeepBIO distinguishes itself from other platforms with the following unique advantages. (i) One-stop-shop service. DeepBIO is the first-of-its-kind platform as a code-free web portal to ensure the reproducibility of deep-learning biological sequence analysis and to lessen the programming burden for biologists. (ii) Pure deep-learning platform. DeepBIO is a purely deep-learning platform that integrates >40 state-of-the-art mainstream deep-learning algorithms, including convolutional neural networks, advanced natural language processing models and GNNs (detailed in Supplementary Table S2), which enables researchers who are interested to train, compare and evaluate different architectures on any biological sequence data. (iii) Two predictive modules. DeepBIO is the first platform that supports not only sequence-level function prediction for any biological sequence data, but also allows nine base-level functional annotation tasks using pre-trained deep-learning architectures, covering DNA methylation, RNA methylation and protein binding specificity. Besides, we further offer an in-depth comparative analysis between the predictions by our models and by experimental data to validate the reliability of the predictions. Notably, empowered by high-performance computers, we demonstrate that DeepBIO supports fast prediction with up to million-scale sequence data, demonstrating its usability in real application scenarios. (iv) Comprehensive result visualization analysis. Aiming to help researchers to delve into more insights from the model predictions, DeepBIO offers a comprehensive visualization result analysis with a variety of interactive figures and tables, covering the following five aspects: statistical analysis of input data; evaluation and comparison of the performance of the prediction models; feature importance and interpretable analysis; model parameter analysis; and sequence conservation analysis. In particular, integrating interpretable mechanisms (e.g. attention heatmap and motif discovery) into deep-learning frameworks enables researchers to analyze which sequential regions are important for the predictions, addressing the issue of the ‘black box’ in deep learning and effectively building the relationship between biological sequences and functions.
MATERIALS AND METHODS
The overall framework of DeepBIO
The DeepBIO platform fully automates the model training process and applies >40 deep-learning approaches for sequence classification prediction and functional site analysis for genomic, transcriptomic, and proteomic sequence data. Figure 1A illustrates the overall framework of the proposed DeepBIO, which consists of four modules: (i) data input module; (ii) sequence-level functional prediction module; (iii) base-level functional annotation module; and (iv) result report module. The workflow of DeepBIO is described as follows. Firstly, DeepBIO takes biological sequence data from the data input module, where it can handle three biological sequence types: DNA, RNA and protein. Next, there are two functional modules for the two types of tasks: one is the sequence-level functional prediction module for binary classification tasks and the other is the base-level functional annotation module for functional annotation tasks. The detailed web techniques and the overview of the interaction between the front end and back end are shown in Figure 1B. In the sequence-level functional prediction module, we support users in automatically training, evaluating and comparing the deep-learning models with their input data. Specifically, there are four main steps (seen in Figure 1C) in this module: (i) data pre-processing; (ii) model construction; (iii) model evaluation; and (iv) visualization analysis. In the base-level functional annotation module, we provide the base-level functional annotation using deep-learning approaches, such as DNA methylation, RNA methylation and protein binding specificity prediction. Similarly, we also curated four steps for this module (seen in Figure 1C): (i) data selection; (ii) task selection; (iii) model loading; and (iv) result visualization. Ultimately, in the result report module, we provide a series of visualization analysis results with various kinds of data formats. Below, we emphasize the details of the four modules.
Figure 1.

The overview of the DeepBIO platform. (A) The architecture of the DeepBIO platform. DeepBIO contains four modules, comprising the data input module, the sequence-level functional prediction module, the base-level functional annotation module and the result report module. DeepBIO accepts DNA, RNA and protein sequence data from the data input module, with some additional user-selectable settings (e.g. sequence similarity setting, imbalanced data processing and data augmentation). The sequence-level functional prediction module provides the deep-learning biological sequence classification and analysis, while the base-level functional annotation module supports the prediction of functional sites for user-provided sequences. In the result report module, DeepBIO provides user-friendly, interactive and interpretable visualization representations to deliver results of the above two functional modules as a download-free report. (B) The web technique of DeepBIO. (C) The workflow of the two main functional modules. The sequence-level functional prediction module consists of four steps: (i) data pre-processing; (ii) model construction; (iii) model evaluation; and (iv) visualization analysis. DeepBIO outputs classification predictions of input sequences based on the four steps and displays the result reports with interactive graphs and tables. The base-level functional annotation module includes the following steps: (i) data selection; (ii) task selection; (iii) model loading; and (iv) result visualization. DeepBIO offers multiple methylation annotations for DNA and RNA sequences while supporting ligand-binding site recognition for protein sequences.
Data input module
DeepBIO serves as an automatically integrated analysis platform based on biological sequence data, including for DNA, RNA and protein, which can be entered in the online input box or uploaded with the standard file format. In addition, users can select deep-learning models and different settings (e.g. whether to turn on the data enhancement option). After obtaining the user's input data, DeepBIO further performs data cleaning to ensure that the user input data are ready and legal to pass into the next module and run properly.
Sequence-level functional prediction module
Step 1. Data pre-processing
This step is an optimization-based data-processing phase that uses a variety of approaches to handle and obtain the input sequence data that are suitable for model training, ensuring the robustness of the final results and improving the success rate of our service. We design four sections to pre-process the input data: (i) sequence legitimacy testing; (ii) sequence similarity setting; (iii) imbalanced data processing; and (iv) data augmentation. In Section (i), for the input sequence data, we first examine whether the input data have the legal data format (e.g. FASTA), and then check if the input sequences contain illegal characters (e.g. characters other than A, T, C and G in DNA sequences). In Section (ii), considering that the high sequence similarity in the input data might lead to a performance evaluation bias, we provide a commonly used tool named CD-HIT (18) to reduce the sequence similarity in the input data. The similarity threshold ranges from 0.7 to 1. In Section (iii), considering that the data imbalance issue might exist in a majority of datasets in real scenarios (i.e. the number of the positives and negatives are extremely imbalanced), we further provide several imbalanced data processing strategies that are commonly used in the machine-learning field, including Focal Loss (19), Synthetic Minority Oversampling Technique (SMOTE) (20) and Adaptive Synthetic (ADASYN) (21). By doing so, we can prevent the biased prediction of models due to the imbalanced data distribution and perhaps achieve an improvement in the overall predictive performance. Finally, in Section (iv), to deal with the few sample learning problem, we further provide some data augmentation methods, such as sequence replacement, sequence flipping and sequence cropping, which can improve the robustness and generalization ability of the deep-learning models.
Step 2. Model construction
The pre-processed sequence data are then fed into user-selected models to train and make the binary prediction. For a scenario in which users do not provide sequence data with training and testing labels, we randomly split the dataset into the training and testing set at a ratio of 8:2 for model training and evaluation. Table 1 summarizes all the state-of-the-art deep-learning models in our platform. There are a total of 42 deep-learning models, covering classic deep-learning methods [e.g. Deep Neural Networks (DNN) and Gate Recurrent Unit (GRU)], natural language processing (NLP) methods [e.g. Transformer and Bidirectional Encoder Representations from Transformers (BERT)] and GNN methods [e.g. Graph Convolutional Network (GCN)]. Notably, each of the above models can be exploited to deal with all three types (DNA, RNA and protein) of sequence analysis tasks. In addition, DeepBIO permits users to select multiple models simultaneously, and train and compare their performance on the same datasets. It is worth noting that we have also deployed several deep-learning models (e.g. DNABERT, RNABERT and ProtBERT) that are well pre-trained on large-scale biological background sequences. They can be utilized for fine-tuning the models on small datasets, if necessary, to alleviate the overfitting problem and increase the models' generalization capability.
Table 1.
Deep-learning models in DeepBIO
| Category | Methods | References |
|---|---|---|
| Classic deep-learning methods | DNN | (22) |
| RNN | (23) | |
| LSTM | (24) | |
| BiLSTM | (25) | |
| LSTM-Attention | (26) | |
| GRU | (27) | |
| TextCNN | (28) | |
| TextRCNN | (29) | |
| VDCNN | (30) | |
| RNN-CNN | (31) | |
| Natural language processing methods | Transformer | (32) |
| Reformer | (33) | |
| Performer | (34) | |
| Linformer | (35) | |
| RoutingTransformer | (36) | |
| DNABERT, RNABERT, ProtBERT | (5,17) | |
| BERT-Base | (37) | |
| BERT-CNN | (38) | |
| BERT-DPCNN | (39) | |
| BERT-RCNN | (40) | |
| BERT-RNN | (41) | |
| ERNIE | (42) | |
| Graph neural network methods | GCN | (43) |
| TextGCN | (44) | |
| GIN | (45) | |
| GAT | (46) | |
| GraphSage | (47) | |
| ChebGCN | (48) | |
| RECT-L | (49) | |
| LightGCN | (50) | |
| GNN-FILM | (51) | |
| HYPER-Conv | (52) | |
| HYPER-Attention | (53) | |
| APPNP | (54) | |
| TextRGNN | (55) | |
| TextSGC | (56) | |
| BertGCN | (57) | |
| BertGAT | (58) | |
| RoBERTaGCN | (59) | |
| RoBERTaGAT | (60) |
Step 3. Model evaluation
For the performance comparison of deep-learning models, we chose several commonly used evaluation metrics, including sensitivity (SN), specificity (SP), accuracy (ACC) and Matthews’ correlation coefficient (MCC). The formulas of these metrics are as follows:
 |
(1) |
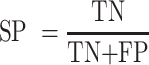 |
(2) |
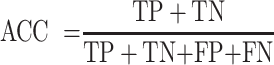 |
(3) |
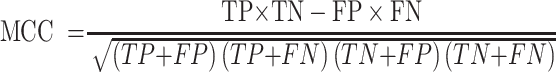 |
(4) |
where TP, FP, TN and FN represent the numbers of true positives, false positives, true negatives and false negatives, respectively. For the comprehensive performance comparison of different models, the area under the receiver operating characteristic (ROC) curve (AUC) and the area under the precision recall (PR) curve (AP), which range from 0 to 1, are calculated based on the ROC curve and the PR curve, respectively. The ROC curve shows the proportion of true positives versus false positives, in which the AUC equals the probability of ranking a randomly chosen true target higher than a randomly chosen decoy target. The PR curve is used more frequently to evaluate the performance of a model on imbalanced datasets (61). Similarly, we can calculate the AP, which equals the average of the interpolated precisions. The higher the AUC and AP values, the better the predictive performance of the underlying model.
Step 4. Visualization analysis
DeepBIO combines the prediction results and intermediate data analysis generated from above, and helps users to better understand the input data through statistical analysis and the model results through various types of visualization analyses. To provide users with intuitive and comprehensive result analysis, we have designed and illustrated multiple visual presentation formats, including the pie plot, histogram, sequence statistics graph, ROC and PR curves, kernel density plot and scatter plot. In Table 2, we present the four sections of visualization analysis, which are dataset statistical analysis, result analysis, feature analysis and parameter optimization analysis. The details of each section are described as follows.
Table 2.
Result visualization analysis in the prediction module
| Category | Type | Purpose |
|---|---|---|
| Statistical analysis | Pie plot | Sequence composition analysis |
| Sequence statistics graph | Sequence statistical analysis from the datasets | |
| Histogram | Sequence length distribution analysis | |
| Result analysis | ROC and PR curve | Performance of model prediction |
| Line chart | Performance of different epochs of models | |
| Kernel density plot | Prediction confidence by different models | |
| Venn diagram and Upset plot | Overlap of prediction results of different models | |
| Feature analysis | ROC and PR curve | Feature performance comparison between hand-crafted features and the features learned by selected models |
| UMAP plot | Feature analysis result by UMAP | |
| SHAP plot | Feature importance analysis by SHAP | |
| Attention heatmap | Feature representation attention analysis | |
| Motif discovery | The discovery of conservative sequence motifs by models | |
| Parameter optimization analysis | ROC and PR curve, histogram | The effect of different parameters (sequence similarity setting, data augmentation and imbalanced data processing) on predictive performance |
For dataset statistical analysis, we summarize the overall status of the input datasets to assist users in better comprehending the datasets themselves. There are the following three statistical analyses, namely sequence composition analysis, sequence motif analysis and the sequence length distribution analysis. In sequence composition analysis, DeepBIO plots the sequence composition histogram for the samples in the positive and negative dataset. In sequence motif analysis, users can obtain the conservation of each position along the sequences. Ultimately, the sequence length distribution analysis provides users with the length preference of the sequences in the dataset.
For result analysis, the DeepBIO visualization analysis includes plots considering all the evaluation metrics presented in step 3, to conduct detailed performance comparisons among different models, such as ROC and PR curves, the performance of different epochs and density distribution of model prediction confidence. In addition, we offer a Venn diagram and an Upset plot to illustrate the relationship among different models from the overlapping prediction results. It can be concluded that in this part users can obtain an intuitive comparison of prediction performance for different deep-learning models.
For feature analysis, we use a dimensionality reduction tool called Uniform Manifold Approximation and Projection (UMAP) (62) for feature space analysis, and a commonly used tool Shapley additive explanation (SHAP) (63) for feature importance analysis. UMAP can map and visualize the high-dimensional features learned from the deep-learning models to the low-dimensional space. It intuitively illustrates how good the learned features are. On the other hand, SHAP gives a good explanation of the feature importance, which quantitively measures the impact of each of the learned features on the predictive performance. In addition, for some specific models such as DNABERT and RNABERT, to further explore what the model learns, we employ the attention heatmap to visualize the information the model captures from either global or local perspectives. Furthermore, the motif discovery enables users to know the conservative sequential determinants which the models learn from the datasets. The details of the motif discovery process are described as follows. Firstly, we apply the attention mechanism of BERT-based models to identify key regions from the sequences. Then, for those identified regions, we further extract and visualize the corresponding motifs using the attention scores obtained from the model. In this way, we highlight the concerns of different models for different features and enhance the interpretability of the deep-learning models.
For parameter optimization analysis, we provide users with some parameter options to make their own choice to optimize the model training, including the effect of different sequence similarities in datasets, the effect of different data augmentation strategies and the effect of different imbalanced data processing methods on predictive performance. We also give some tabular information, including a summary of the input datasets and the performance of deep-learning models. This helps the users to compare the results of the same model with different parameters.
Base-level functional annotation module
The base-level functional annotation module is to predict the functions of the biological sequences at base level using deep learning. In this module, we provide nine annotation tasks, including the methylation annotation for DNA and RNA sequences, and the ligand-binding site recognition for protein sequences. The module comprises four steps: data selection, task selection, model loading and result visualization. The details of this module are as follows.
Step 1. Data selection
In this step, DeepBIO enables users to upload the sequences they want to annotate. In particular, for the DNA methylation site prediction task, we support two ways to input sequence data, i.e. (i) uploading sequence data by themselves and (ii) selecting a segment of our pre-set human genome by choosing a specific cell line and chromosome.
Step 2. Task selection
There are three types of functional annotation tasks: DNA methylation site prediction, RNA methylation site prediction and protein–ligand binding site prediction. For DNA methylation site prediction, there are DNA N4-methylcytosine (4mC) site prediction, DNA 5-hydroxymethylcytosine (5hmC) site prediction and DNA N6-methyladenine (6mA) site prediction. For RNA methylation site prediction, there are RNA 5-hydroxymethylcytosine (5hmC) site prediction, RNA N6-methyladenosine (m6A) site prediction and RNA N4-methylcytidine (m4C) site prediction. For protein–ligand binding site prediction, there are DNA-binding site prediction, RNA-binding site prediction and peptide-binding site prediction.
Step 3. Model loading
In this step, there are two modes for deep-learning model selection. One is the default and the other is customized. For the default mode, we provide several deep-learning models that are well pre-trained with large-scale biological data, such as DNABERT. The well-pre-trained models generally have good generalization ability and achieve good performance for different downstream tasks. For the customized mode, we allow users to upload the models they trained for the same tasks in the sequence-level functional prediction module.
Step 4. Result visualization
In this part, we provide users with the in-depth annotation result analysis based on the model predictions and biological experiment data (e.g. histone modification signals and protein 3D structural information). Specifically, for DNA, we designed two visualization sections, namely functional site annotation and integrative analysis. For base-level functional annotation, we predict the functional sites that match the selected task type (e.g. DNA 4mC site prediction) and perform the model-predicted confidence of the corresponding position. In the integrative analysis, we perform statistical analysis between DNA methylation predictions and different histone modification signals. For RNA, due to the constraint of data, we only perform the functional site annotation section the same as in DNA. In addition, for protein, to intuitively show the binding site annotation results, we visualize and annotate the binding residues on the 3D structure of a given protein sequence with PDB ID.
Result report module
We list the analysis results in the form of reports, which involve various kinds of data formats from the results of the two main functional modules. Users can view or download freely all of the result data and charts. Notably, each submitted task is immediately notified to the corresponding user once it is completed.
RESULTS AND DISCUSSION
Case study
We showcase real-world applications of DeepBIO with two bioinformatic scenarios: (i) the prediction of DNA 6mA methylation and (ii) the prediction of protein toxicity. We emphasize that the underlying objective is to illustrate how to use our platform for two such diverse applications, rather than securing the top predictive performance compared with the state-of-the-art methods.
DNA methylation is closely associated with a variety of key processes, such as genomic imprinting and carcinogenesis (64). Therefore, for the task of the DNA 6mA methylation prediction, we use the Drosophila melanogaster dataset with 11 191 methylation sequences (as positives) and 11 191 non-methylation sequences (as negatives) as an example dataset (65) for DNA sequence functional analysis. In addition, protein toxicity is closely related to most neurodegenerative diseases, and accurate toxicity prediction leads to significant promotion in new drug design. We choose a protein toxicity benchmark dataset (66) with 3379 toxic animal protein sequences (as positives) and 5464 non-toxic animal protein sequences (as negatives) for protein functional analysis. With the two example datasets, we first randomly split them into the training set and testing set and then submit them to our online platform to demonstrate the data analysis and various functions of DeepBIO. More details of the visualization results of the two cases are described below.
Dataset statistics
Figure 2 illustrates the dataset statistics of the protein toxicity dataset. The proportion and number of bases or amino acids in the dataset are counted and illustrated as a pie chart and composition histogram, respectively (Figure 2A), from which we can clearly see the difference between the positives and the negatives from the compositional perspective. In addition, we calculate and show the sequence length distribution (Figure 2B), which intuitively gives users the sequence length preference in the datasets. For example, it can be seen from Figure 2B that positive samples are mainly distributed in the range of 50–150 amino acids, while the negative samples have a relatively even distribution. Furthermore, we use a motif analysis tool called Weblogo to generate the sequence motifs of the datasets, which enables users to analyze the composition and conservation at the sequence level (Figure 2C).
Figure 2.

The statistical analysis on the protein toxicity dataset. (A) Sequence compositions of positive and negative samples in the training and testing sets. (B) Sequence length distribution of positive and negative samples in the training and testing sets. (C) Motifs of sequence statistics from the training and testing sets.
Model prediction analysis
DeepBIO provides detailed analysis for users to interpret and show comprehensive performance comparisons among different deep-learning models. Here, we select four models (DNABERT, DNN, LSTM and RNN) to train and compare their prediction performance on the DNA methylation dataset. Figure 3A shows the performances of the compared models in terms of ACC, sensitivity, specificity, AUC and MCC. It can be seen that DNABERT achieves the best performance with the ACC, sensitivity, specificity, AUC and MCC of 0.921, 0.896, 0.946, 0.967 and 0.844, respectively. For a better evaluation of models, Figure 3B illustrates the ROC and PR curves of the compared models. As seen in Figure 3B, DNABERT achieved the highest AUC and AP values of 0.968 and 0.973, respectively, which further confirms that DNABERT is better than the other compared models. To further study the generalization ability of the compared models, we draw the epoch plot in Figure 3C to show the trends of accuracy and calculation loss for each model during the training process. From Figure 3C, it is easy to see that the accuracy of three models (DNABERT, LSTM and RNN), but not DNN, increases rapidly at first and then the accuracy curves become smooth gradually. Meanwhile, their calculation losses converge to lower values, indicating that these models are well trained and their predictions are reliable. It is worth noting that during the training process, DNN always achieves relatively low accuracy, however with high calculation loss on the example dataset (seen in Figure 3C). This indicates that it is not a good and robust model after training. In addition, to intuitively show the relationship among different models from their prediction results, we further provide an Upset plot and a Venn diagram in Figure 3D and E. From Figure 3D and E, we can see that most positive samples are successfully predicted from the overlapping predictions of the compared models. Moreover, to clearly understand the prediction preference for each model, we visualize the density distribution of the prediction confidence of different deep-learning models in Figure 3F. As can be seen, DNN mainly focuses its prediction confidence on ∼0.5, which is the center part of the x-axis, illustrating its poor discrimination ability between positive and negative samples on the example dataset. In contrast, the other three models (DNABERT, LSTM and RNN) achieve higher prediction confidence on both positive and negative samples, indicating that they possess better classification ability on the example dataset. The comparison results for this part on the dataset of protein toxicity can be seen in Supplementary Figures S1–S6.
Figure 3.

The prediction result analysis from different models using DeepBIO. (A) Performance comparison among different models in terms of ACC, sensitivity, specificity, AUC and MCC. (B) ROC and PR curves of different deep-learning models. (C) The trends of performance and calculation loss with each epoch, showing ACC and the loss change process on different models. (D and E) Upset plot and Venn diagram to express the relationships of prediction results for different models. (F) Density distribution of the prediction confidence for different deep-learning models.
Feature analysis and visualization
To compare the features automatically learned by deep-learning models with the traditional hand-crafted features, we select some commonly used feature encodings, including accumulated nucleotide frequency (ANF) (67), binary (68), composition of K-spaced nucleic acid pairs (CKSNAP) (69) and dinucleotide composition (DNC) (70,71) for nucleotide sequences, and show the prediction performance in terms of different metrics in Figure 4A and B. It is worth noting that in Figure 4A and B, the compared deep-learning models, with the exception of DNN, outperform most of the traditional feature encodings, allowing us to easily conclude that the features learnt from the deep-learning methods generally are superior to hand-crafted features.
Figure 4.

The visualization of feature analysis and model interpretation using DeepBIO. (A) Prediction performance comparison between deep-learning models and traditional hand-crafted feature-based methods. (B) ROC and PR curves of deep-learning models and traditional hand-crafted feature-based methods. (C) UMAP feature visualization results of different deep-learning models. (D) SHAP feature importance analysis of different deep-learning models. (E) Attention heatmap of the user-chosen biological sequence. (F) Motif discovery by deep-learning models.
In addition, to intuitively evaluate the feature representations learnt from different deep-learning models, we employ UMAP to conduct the dimension reduction and feature visualization (Figure 4C). In Figure 4C, each point represents a sample in the dataset; the positive and negative samples are assigned with different colors. In this case, we can get a better sense of the classification ability of each model from the UMAP plot. Specifically, Figure 4C shows that compared with the other three models (DNABERT, LSTM and RNN), the samples belonging to different classes in the feature space of DNN are almost connected, making it difficult to distinguish the positives from the negatives. Moreover, the positives and negatives are distributed more clearly in two clusters the feature space of DNABERT than in that of RNN and LSTM. This demonstrates that DNABERT has stronger prediction ability than the other models. In addition, we also provide the SHAP plot (Figure 4D), assigning each feature a value to represent its importance for the model prediction. Each row represents the SHAP value distributions of a feature, and the x-axis refers to the SHAP value, where the value of SHAP >0 shows that the prediction favors the positive class, and a value <0 indicates that the prediction tends to be the negative class. The color of sample points in Figure 4D indicates the corresponding feature value: redder points mean higher feature importance values, while bluer points indicate lower feature values. The features are sorted according to the sum of SHAP values incorporating all the samples in the dataset. Therefore, the SHAP plot shows the relationship between the value of features and their impact on the model prediction. From Figure 4D, we can see that the high-dimension features play a predominant role for DNABERT; the top-ranked feature values in DNABERT are more easily distinguished in the color as compared with other models, indicating that DNABERT performs better in feature representation learning.
To improve the interpretability of the deep-learning models, in Figure 4E we calculate the importance of the base at each position on the user-given sequence from the attention matrix in the model and visualize it with normalized attention scores, enabling us to analyze which sequential region the model considers is more important for the prediction. To further study what the models learn from the training process, we extract the important regions with high attention scores through the attention mechanism of the models to find conservative sequence motifs in Figure 4F from the datasets, which might be related to biological functions. The detailed feature analysis results on the dataset of protein toxicity can be seen in Supplementary Figures S7–S9.
Functional annotation analysis
To intuitively illustrate in-depth annotation result analysis of biological sequences, we provide users with functional site prediction and result visualization analysis as illustrated in Figure 5. As seen in Figure 5A, we can predict DNA methylation sites with corresponding positions and prediction confidence based on user-provided DNA sequences or a segment of our pre-set human genome. In addition, if users select a segment from our pre-set human genome by choosing a specific cell line and chromosome, we will display the statistical analysis between our DNA methylation site predictions and relevant histone modification signals queried from the database (Figure 5B), assisting in mining the potential relationships between them. Moreover, for base-level annotation on protein sequences, we predict and visualize the annotation results on the 3D structure of the given protein sequence with its PDB ID. Specifically, Figure 5C shows the protein-peptide binding site prediction results of an example protein (PDB ID 1a81A), and the residues in red color on the protein 3D structure indicate the protein-peptide binding sites.
Figure 5.

The biological sequence functional annotation analysis using DeepBIO. (A) Prediction results of DNA methylation annotation. (B) Comparison and analysis between the DNA methylation site prediction results and relevant histone modification signals. (C) Prediction results of protein-peptide binding site annotation on an example protein (1a81A).
Webserver implementation
DeepBIO is empowered by the high-performance graphic processing architecture with million-scale calculation capability. Specifically, DeepBIO is deployed with the Ubuntu 18.04 Linux system, multiple Intel Xeon Silver 4210R CPUs, 256 GB of RAM, hundreds of TB Solid State Drives and NVIDIA RTX 3090 GPU clusters. Unlike other online platforms that use CPU for training models and making predictions, the models we provide are all trained and evaluated based on GPU, which significantly reduces the computational time. It can be seen from Supplementary Table S3 that we spent less time in optimizing deep-learning models and enable fast and accurate predictions. In addition, Supplementary Figures S10–S15 show that the front-end pages are carefully designed to give users an interactive visualization of the results while they can download each result graph freely. To be specific, we use React framework to set up the user interface, Spring Boot framework for server implementation in the back end, Python and R programming languages for the model's construction and visualization, and the MySQL database to manage the data storage. In addition, to provide a better Web experience for users, we also construct interactive graphs to visualize the analysis results on the front page. Our platform can be run stably on many browsers including Internet Explorer (≥v.7.0), Mozilla Firefox, Microsoft Edge, Safari and Google Chrome. In conclusion, with our DeepBIO platform, data scientists and researchers limited by equipment resources can now employ high-performance computers to tackle their challenging work.
DATA AVAILABILITY
As an online platform for biological sequence analysis, DeepBIO can be freely accessed without registration via https://inner.wei-group.net/DeepBIO. All code used in data analysis and preparation of the manuscript, alongside a description of necessary steps for reproducing results, can be found in a GitHub repository accompanying this manuscript: https://github.com/WLYLab/DeepBIO. The source code for DeepBIO is also freely available at Zenodo website (DOI: 10.5281/zenodo.7547847).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr. Martin de Jesus LOZA-LOPEZ from University of Tokyo for his valuable suggestions regarding the improvement of our platform.
Author contributions: L.W. conceived the basic idea and designed the DeepBIO framework. R.W., Y.J, J.J., C.Y., H.Y., F.W. and J.F. set up the server and visualized the result analysis. Y.J., R.W., L.W. and C.Y. designed the interface and tested the server. R.W., C.Y., J.J., Q.Z. and L.W. wrote the manuscript. K.N., R.S., Q.Z. and L.W. revised the manuscript.
Contributor Information
Ruheng Wang, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
Yi Jiang, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
Junru Jin, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
Chenglin Yin, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
Haoqing Yu, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
Fengsheng Wang, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
Jiuxin Feng, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
Ran Su, College of Intelligence and Computing, Tianjin University, Tianjin, China.
Kenta Nakai, Human Genome Center, Institute of Medical Science, University of Tokyo, Tokyo, Japan.
Quan Zou, Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China.
Leyi Wei, School of Software, Shandong University, Jinan, China; Joint SDU-NTU Centre for Artificial Intelligence Research (C-FAIR), Shandong University, Jinan, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The work was supported by the Natural Science Foundation of China [62250028 and 62071278].
Conflict of interest statement. None declared.
REFERENCES
- 1. Larranaga P., Calvo B., Santana R., Bielza C., Galdiano J., Inza I., Lozano J.A., Armananzas R., Santafe G., Perez A.et al.. Machine learning in bioinformatics. Brief .Bioinform. 2006; 7:86–112. [DOI] [PubMed] [Google Scholar]
- 2. Wang R., Jin J., Zou Q., Nakai K., Wei L.. Predicting protein–peptide binding residues via interpretable deep learning. Bioinformatics. 2022; 38:3351–3360. [DOI] [PubMed] [Google Scholar]
- 3. Jiang Y., Wang R., Feng J., Jin J., Liang S., Li Z., Yu Y., Ma A., Su R., Zou Q.et al.. Explainable deep graph learning accurately modeling the peptide secondary structure prediction. 2022; bioRxiv doi:10 August 2022, preprint: not peer reviewed 10.1101/2022.06.09.495580. [DOI] [PMC free article] [PubMed]
- 4. Jin J., Yu Y., Wang R., Zeng X., Pang C., Jiang Y., Li Z., Dai Y., Su R., Zou Q.. iDNA-ABF: multi-scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol. 2022; 23:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elnaggar A., Heinzinger M., Dallago C., Rehawi G., Wang Y., Jones L., Gibbs T., Feher T., Angerer C., Steinegger M.et al.. ProtTrans: towards cracking the language of lifes code through self-supervised deep learning and high performance computing. IEEE Trans. Pattern Anal. Mach. Intell. 2021; 44:7112–7127. [DOI] [PubMed] [Google Scholar]
- 6. Liu B. BioSeq-Analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2019; 20:1280–1294. [DOI] [PubMed] [Google Scholar]
- 7. Liu B., Gao X., Zhang H.. BioSeq-Analysis2. 0: an updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019; 47:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Z., Zhao P., Li C., Li F., Xiang D., Chen Y.Z., Akutsu T., Daly R.J., Webb G.I., Zhao Q.et al.. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021; 49:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li H.-L., Pang Y.-H., Liu B.. BioSeq-BLM: a platform for analyzing DNA, RNA and protein sequences based on biological language models. Nucleic Acids Res. 2021; 49:e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Z., Liu X., Zhao P., Li C., Wang Y., Li F., Akutsu T., Bain C., Gasser R.B., Li J.et al.. iFeatureOmega: an integrative platform for engineering, visualization and analysis of features from molecular sequences, structural and ligand data sets. Nucleic Acids Res. 2022; 50:W434–W447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cao D.-S., Xiao N., Xu Q.-S., Chen A.F.. Rcpi: R/Bioconductor package to generate various descriptors of proteins, compounds and their interactions. Bioinformatics. 2015; 31:279–281. [DOI] [PubMed] [Google Scholar]
- 12. Xiao N., Cao D.-S., Zhu M.-F., Xu Q.-S.. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics. 2015; 31:1857–1859. [DOI] [PubMed] [Google Scholar]
- 13. Avsec Ž., Kreuzhuber R., Israeli J., Xu N., Cheng J., Shrikumar A., Banerjee A., Kim D.S., Beier T., Urban L.. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat. Biotechnol. 2019; 37:592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Budach S., Marsico A.. Pysster: classification of biological sequences by learning sequence and structure motifs with convolutional neural networks. Bioinformatics. 2018; 34:3035–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen K.M., Cofer E.M., Zhou J., Troyanskaya O.G.. Selene: a PyTorch-based deep learning library for sequence data. Nat. Methods. 2019; 16:315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li C., Liu H., Hu Q., Que J., Yao J.. A novel computational model for predicting microRNA–disease associations based on heterogeneous graph convolutional networks. Cells. 2019; 8:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ji Y., Zhou Z., Liu H., Davuluri R.V.. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics. 2021; 37:2112––2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li W., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 19. Lin T.-Y., Goyal P., Girshick R., He K., Dollár P.. Focal loss for dense object detection. Proceedings of the IEEE international conference on computer vision. 2017; 2980–2988. [Google Scholar]
- 20. Chawla N.V., Bowyer K.W., Hall L.O., Kegelmeyer W.P.. SMOTE: synthetic minority over-sampling technique. J. Artificial Intelligence Res. 2002; 16:321–357. [Google Scholar]
- 21. He H., Bai Y., Garcia E.A., Li S.. ADASYN: Adaptive synthetic sampling approach for imbalanced learning. IEEE international joint conference on neural networks (IEEE world congress on computational intelligence). 2008; 1322–1328. [Google Scholar]
- 22. LeCun Y., Bengio Y., Hinton G.. Deep learning. Nature. 2015; 521:436–444. [DOI] [PubMed] [Google Scholar]
- 23. Rumelhart D.E., Hinton G.E., Williams R.J.. Learning representations by back-propagating errors. Nature. 1986; 323:533–536. [Google Scholar]
- 24. Hochreiter S., Schmidhuber J.. Long short-term memory. Neural Comput. 1997; 9:1735–1780. [DOI] [PubMed] [Google Scholar]
- 25. Graves A., Schmidhuber J.. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005; 18:602–610. [DOI] [PubMed] [Google Scholar]
- 26. Wang Q., Hao Y.. ALSTM: an attention-based long short-term memory framework for knowledge base reasoning. Neurocomputing. 2020; 399:342–351. [Google Scholar]
- 27. Dey R., Salem F.M.. Gate-variants of gated recurrent unit (GRU) neural networks. 2017 IEEE 60th international midwest symposium on circuits and systems (MWSCAS). 2017; 1597–1600. [Google Scholar]
- 28. dos Santos C., Gatti M. Deep convolutional neural networks for sentiment analysis of short texts. Proceedings of COLING 2014, the 25th International Conference on Computational Linguistics: Technical Papers. 2014; 69–78. [Google Scholar]
- 29. Lai S., Xu L., Liu K., Zhao J.. Recurrent convolutional neural networks for text classification. Twenty-ninth AAAI conference on artificial intelligence. 2015. [Google Scholar]
- 30. Simonyan K., Zisserman A.. Very deep convolutional networks for large-scale image recognition. 2015; arXiv doi:10 April 2015, preprint: not peer reviewedhttps://arxiv.org/abs/1409.1556.
- 31. Wang J., Yang Y., Mao J., Huang Z., Huang C., Xu W.. Cnn-rnn: A unified framework for multi-label image classification. Proceedings of the IEEE conference on computer vision and pattern recognition. 2016; 2285–2294. [Google Scholar]
- 32. Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I.. Attention is all you need. Advances in neural information processing systems. 2017; 5998–6008. [Google Scholar]
- 33. Kitaev N., Kaiser Ł., Levskaya A.. Reformer: The efficient transformer. Proceedings of ICLR. 2020. [Google Scholar]
- 34. Choromanski K., Likhosherstov V., Dohan D., Song X., Gane A., Sarlos T., Hawkins P., Davis J., Mohiuddin A., Kaiser L.. Rethinking attention with performers. 2022; arXiv doi:19 November 2022, preprint: not peer reviewedhttps://arxiv.org/abs/2009.14794.
- 35. Wang S., Li B.Z., Khabsa M., Fang H., Ma H.. Linformer: self-attention with linear complexity. 2020; arXiv doi:14 June 2020, preprint: not peer reviewedhttps://arxiv.org/abs/2006.04768.
- 36. Roy A., Saffar M., Vaswani A., Grangier D. Efficient content-based sparse attention with routing transformers. Transactions of the Association for Computational Linguistics. 2021; 9:53–68. [Google Scholar]
- 37. Devlin J., Chang M.-W., Lee K., Toutanova K.. BERT: Pre-training of deep bidirectional transformers for language understanding. Proceedings of NAACL. 2019; 4171–4186. [Google Scholar]
- 38. Safaya A., Abdullatif M., Yuret D. Kuisail at semeval-2020 task 12: Bert-cnn for offensive speech identification in social media. Proceedings of the Fourteenth Workshop on Semantic Evaluation. 2020; 2054–2059. [Google Scholar]
- 39. Li Y.-J., Zhang H.-J., Pan W.-M., Feng R.-J., Zhou Z.-Y.. Artificial intelligence in China. 2021; Springer; 524–530. [Google Scholar]
- 40. Nguyen Q.T., Nguyen T.L., Luong N.H., Ngo Q.H.. Fine-tuning bert for sentiment analysis of vietnamese reviews. 2020 7th NAFOSTED Conference on Information and Computer Science (NICS). 2020; 302–307. [Google Scholar]
- 41. Huang P., Zhu H., Zheng L., Wang Y.. Text Sentiment Analysis based on BERT and Convolutional Neural Networks. 2021 5th International Conference on Natural Language Processing and Information Retrieval (NLPIR). 2021; 1–7. [Google Scholar]
- 42. Zhang Z., Han X., Liu Z., Jiang X., Sun M., Liu Q.. ERNIE: Enhanced language representation with informative entities. Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics. 2019; 1441–1451. [Google Scholar]
- 43. Kipf T.N., Welling M.. Semi-supervised classification with graph convolutional networks. International conference on learning representations (ICLR ’17). 2017. [Google Scholar]
- 44. Zhu J., Cui Y., Liu Y., Sun H., Li X., Pelger M., Yang T., Zhang L., Zhang R., Zhao H.. Textgnn: Improving text encoder via graph neural network in sponsored search. Proceedings of the Web Conference 2021. 2021; 2848–2857. [Google Scholar]
- 45. Chen J., Xie Y., Wang K., Wang Z.H., Lahoti G., Zhang C., Vannan M.A., Wang B., Qian Z.. Generative invertible networks (GIN): Pathophysiology-interpretable feature mapping and virtual patient generation. International Conference on Medical Image Computing and Computer-Assisted Intervention. 2018; Springer; 537–545. [Google Scholar]
- 46. Wang K., Shen W., Yang Y., Quan X., Wang R.. Relational graph attention network for aspect-based sentiment analysis. Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics. 2020; 3229–3238. [Google Scholar]
- 47. Hamilton W., Ying Z., Leskovec J.. Inductive representation learning on large graphs. NIPS'17: Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017; 1025–1035. [Google Scholar]
- 48. Defferrard M., Bresson X., Vandergheynst P.. Convolutional neural networks on graphs with fast localized spectral filtering. NIPS'16: Proceedings of the 30th International Conference on Neural Information Processing Systems. 2016; 3844–3852. [Google Scholar]
- 49. Wang Z., Ye X., Wang C., Cui J., Philip S.Y.. Network embedding with completely-imbalanced labels. IEEE Trans. Knowl. Data Eng. 2020; 33:3634–3647. [Google Scholar]
- 50. He X., Deng K., Wang X., Li Y., Zhang Y., Wang M.. Lightgcn: Simplifying and powering graph convolution network for recommendation. Proceedings of the 43rd International ACM SIGIR conference on research and development in Information Retrieval. 2020; 639–648. [Google Scholar]
- 51. Brockschmidt M. Gnn-film: Graph neural networks with feature-wise linear modulation. International Conference on Machine Learning. 2020; 1144–1152. [Google Scholar]
- 52. Ma T., Dalca A.V., Sabuncu M.R.. Hyper-convolution networks for biomedical image segmentation. Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision. 2022; 1933–1942. [Google Scholar]
- 53. Duan W., He X., Zhou Z., Rao H., Thiele L.. Injecting descriptive meta-information into pre-trained language models with hypernetworks. Interspeech 2021. 2021; 3216–3220. [Google Scholar]
- 54. Klicpera J., Bojchevski A., Günnemann S.. Predict then propagate: Graph neural networks meet personalized pagerank. 7th International Conference on Learning Representations. 2019. [Google Scholar]
- 55. Chen J., Zhang B., Xu Y., Wang M.. TextRGNN: residual Graph Neural Networks for Text Classification. 2021; arXiv doi:30 December 2021, preprint: not peer reviewedhttps://arxiv.org/abs/2112.15060.
- 56. Wu F., Souza A., Zhang T., Fifty C., Yu T., Weinberger K.. Simplifying graph convolutional networks. International conference on machine learning. 2019; PMLR; 6861–6871. [Google Scholar]
- 57. Lin Y., Meng Y., Sun X., Han Q., Kuang K., Li J., Wu F.. BertGCN: Transductive Text Classification by Combining GCN and BERT. Findings of the Association for Computational Linguistics: ACL-IJCNLP 2021. 2021; 1456–1462. [Google Scholar]
- 58. Veličković P., Cucurull G., Casanova A., Romero A., Lio P., Bengio Y.. Graph attention networks. International conference on learning representations. 2018. [Google Scholar]
- 59. Wei M., He Y., Zhang Q.. Robust layout-aware IE for visually rich documents with pre-trained language models. Proceedings of the 43rd International ACM SIGIR Conference on Research and Development in Information Retrieval. 2020; 2367–2376. [Google Scholar]
- 60. Chandra S., Mishra P., Yannakoudakis H., Nimishakavi M., Saeidi M., Shutova E.. Graph-based modeling of online communities for fake news detection. 2020; arXiv doi:23 November 2020, preprint: not peer reviewedhttps://arxiv.org/abs/2008.06274.
- 61. Saito T., Rehmsmeier M.. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One. 2015; 10:e0118432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Becht E., McInnes L., Healy J., Dutertre C.-A., Kwok I.W., Ng L.G., Ginhoux F., Newell E.W.. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019; 37:38–44. [DOI] [PubMed] [Google Scholar]
- 63. Lundberg S.M., Lee S.-I.. A unified approach to interpreting model predictions. Proceedings of the 31st international conference on neural information processing systems. 2017; 4768–4777. [Google Scholar]
- 64. Richardson B.C. Role of DNA methylation in the regulation of cell function: autoimmunity, aging and cancer. J. Nutr. 2002; 132:2401S–2405S. [DOI] [PubMed] [Google Scholar]
- 65. Lv H., Dao F.-Y., Zhang D., Guan Z.-X., Yang H., Su W., Liu M.-L., Ding H., Chen W., Lin H.. iDNA-MS: an integrated computational tool for detecting DNA modification sites in multiple genomes. Iscience. 2020; 23:100991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pan X., Zuallaert J., Wang X., Shen H.-B., Campos E.P., Marushchak D.O., De Neve W.. ToxDL: deep learning using primary structure and domain embeddings for assessing protein toxicity. Bioinformatics. 2020; 36:5159–5168. [DOI] [PubMed] [Google Scholar]
- 67. Xu H., Hu R., Jia P., Zhao Z.. 6mA-Finder: a novel online tool for predicting DNA N6-methyladenine sites in genomes. Bioinformatics. 2020; 36:3257–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen Z., Chen Y.-Z., Wang X.-F., Wang C., Yan R.-X., Zhang Z.. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLoS One. 2011; 6:e22930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen Z., Zhao P., Li F., Marquez-Lago T.T., Leier A., Revote J., Zhu Y., Powell D.R., Akutsu T., Webb G.I.. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief. Bioinform. 2020; 21:1047–1057. [DOI] [PubMed] [Google Scholar]
- 70. Liu B., Liu F., Wang X., Chen J., Fang L., Chou K.-C.. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015; 43:W65–W71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu B., Liu F., Fang L., Wang X., Chou K.-C.. repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics. 2015; 31:1307–1309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
As an online platform for biological sequence analysis, DeepBIO can be freely accessed without registration via https://inner.wei-group.net/DeepBIO. All code used in data analysis and preparation of the manuscript, alongside a description of necessary steps for reproducing results, can be found in a GitHub repository accompanying this manuscript: https://github.com/WLYLab/DeepBIO. The source code for DeepBIO is also freely available at Zenodo website (DOI: 10.5281/zenodo.7547847).