

Summary
Cell type-specific expression of the developmental gene is conferred by distinct enhancer elements. Current knowledge about mechanisms in Nkx2-5 transcriptional regulation and its specific roles in multistage heart morphogenesis is limited. We comprehensively interrogate enhancers U1 and U2 in controlling Nkx2-5 transcription during heart development. Serial genomic deletions in mice reveal U1 and U2 function redundantly to confer Nkx2-5 expression at early stages, but U2 instead of U1 supports its expression at later stages. Combined deletions markedly reduce Nkx2-5 dosage as early as E7.5, despite being largely reinstated two days later, displaying heart malformations with precocious differentiation of cardiac progenitors. Cutting-edge low-input chromatin immunoprecipitation sequencing (ChIP-seq) confirmed that not only genomic NKX2-5 occupancy but also its regulated enhancer landscape is mostly disturbed in the double-deletion mouse hearts. Together, we propose a model that the temporal and partially compensatory regulatory function of two enhancers dictates a transcription factor (TF)’s dosage and specificity during development.
Subject areas: Biological sciences, Developmental genetics, Developmental biology
Graphical abstract
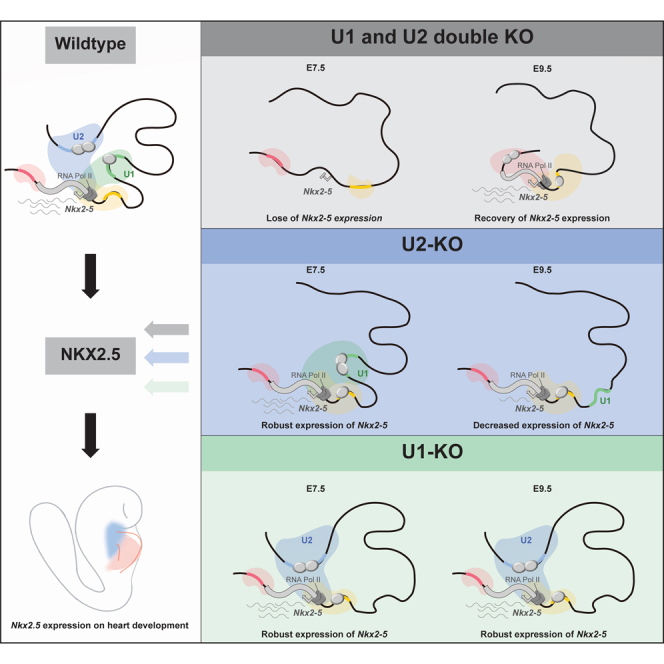
Highlights
-
•
U1 and U2 enhancers work hierarchically to promoter Nkx2-5 expression
-
•
DKO of U1 and U2 results in precocious differentiation of CPCs in the SHF
-
•
The spatiotemporal expression of TSs is key to establish the correct chromatin states
Biological sciences; Developmental genetics; Developmental biology
Introduction
Enhancers are cis-regulatory DNA elements conferring cell type-specific gene expression through recruiting specific transcription factor (TF) during normal development and disease.1,2,3,4,5 Enhancers ensure complementary, spatiotemporal gene expression in an additive manner, offering transcriptional robustness and functional redundancy for tissue development.6,7 Recent advances in next-generation sequencing technologies provide an unprecedented chance for unbiased profiling of potential enhancers genome wide, such as H3K27ac ChIP-seq (chromatin immunoprecipitation followed by sequencing),8 ATAC-seq (assay for transposase-accessible chromatin using sequencing),9 and DNase-seq (DNase I hypersensitive sites sequencing).10 Enhancers were identified to be located within intergenic regions, gene bodies, and even on distinct chromosomes.11,12,13 Recent genome-wide profiling studies have estimated that the transcription of one human gene is regulated by an average of 4.9 enhancers.14 Although the exact number may vary among different genes, it is obvious that regulation of most of the protein-coding genes is achieved by multiple enhancers.
Prior studies showed that enhancers regulate complementary spatiotemporal characteristics of gene expression in an additive manner, offering transcriptional robustness and functional redundancy for tissue development.6 However, many important questions remain open. For example, what types of enhancers mapped by genome-wide assays truly function in anticipated tissues in development; why among these enhancers validated with the in vivo activity by genetic reporter assays, only a limited number of them have been proven to be essential for animal development? In line with this, loss-of-function assays have uncovered the enhancer hierarchy with differential requirements for the regulation of a given gene.6,15,16,17
Heart development is finely controlled by the coordinated execution of spatiotemporal GRN (gene regulatory network).18 The cardiac GRNs are centered on several master cardiac TFs including NKX2-5, GATA4, SRF, and TBX5.19,20 Depletion of each of these TFs leads to heart malformations.21 However, how their transcriptional specificity and dosage are licensed during heart development is far from clear. NKX2-5 is a master cardiac TF. Studies have documented several enhancers in driving its spatiotemporal expression during early heart development.22,23,24,25 Despite having been well characterized with the spatiotemporal specificity,25,26 whether these enhancers are truly functional in vivo and how these distinct enhancers cooperate to orchestrate Nkx2-5 transcription and heart development remain elusive. Here, we combined genetic studies and molecular profiling to interrogate the functional hierarchy of two enhancers in transcriptional regulation of Nkx2-5 expression during heart development. Our work evidenced temporal, one-way additive regulatory mechanism of enhancers, U1 and U2, safeguarding the transcriptional level and specificity of NKX2-5 TF during development.
Results
Characterization of novel enhancers U1 and U2 at the Nkx2-5 locus
Super-enhancers have been identified as large clusters of enhancers that drive the expression of lineage-specific TFs.27 To identify potential super-enhancers responsible for the spatial-temporal orchestration of Nkx2-5 transcription during early heart development, we analyzed the genome-wide H3K27ac peaks (a mark for active enhancers)28 by ChIP-seq from E8.5 to E16.5 hearts and identified super-enhancers U1 and U2 next to the Nkx2-5 locus.29 Specifically, U1 enhancer fell within the previously identified upstream homology region 6 (UH6) enhancer,30 partially overlapping with the AR1 enhancer (Figure 1).31 The distal U2 enhancer was located ∼19 kb upstream of Nkx2-5. As the binding of TFs is required for the activation of enhancers,32 we examined the enrichment of TF motifs in the U1 and U2 enhancer regions relative to randomly selected genomic backgrounds and found a number of cardiac-specific TFs with detected expression, including NKX2-5, GATA4, TBX20, HAND1, and TBX5 (Figure S1 and Table S1). The above results indicate potential regulatory function of U1 and U2 in Nkx2-5 transcriptional regulation.
Figure 1.

Characterization of regulatory regions around Nkx2-5 during early heart development
RNA-seq tracks for E8.25 (EBI: ERR332388), E9.5 (GEO: GSE104670), E12.5 (GEO: GSE82471), and E16.5 (GEO: GSE78317) hearts were in red. H3K27ac ChIP-seq tracks for E9.5 OFT, E12.5 (GEO: GSE82449), and E16.5 (GEO: GSE82973) hearts were in purple. The AR1, AR2, and AR3 enhancers (Schwartz & Olson, 1999) were in magenta, and the G-S enhancer (III et al., 2004) was in orange, as well as the UH5 and UH6 enhancers (Chi et al., 2005) were in red and blue, respectively. The newly identified U1 and U2 enhancers were in light green and light blue, respectively. OFT, outflow tract. See also Figure S1.
Transiently missing but later fully reinstated Nkx2-5 expression in U1/U2 double-deletion mice with heart developmental defects
To investigate the requirement for enhancers U1 and U2 in fine-tuning Nkx2-5 expression during early heart development, we used CRISPR-Cas9 genome editing to generate a double knockout (DKO) of U1 (deletion of 4.3 kb) and U2 (deletion of 9.4 kb) mice (Figure 2A). All DKO mice died after E9.5 with underdeveloped and shortened outflow tracts (OFTs) and nearly missing right ventricles and open atrioventricular chambers (Figures 2B and 2C), reminiscent of Nkx2-5-null mice.33
Figure 2.

Temporal alteration of Nkx2-5 expression in U1/U2 double-deletion mice during heart developmental progression
(A) Schematic of the strategy for the double-knockout (DKO) mice generation.
(B) Gross morphological examination of E9.5 hearts from DKO mice. DKO mice showed severe SHF defects. The arrow indicated the underdeveloped outflow tract. The asterisk indicated the open atrioventricular chamber. The arrowhead indicated the single ventricle. Scale bar, 200 μm.
(C) Survival analysis of DKO strains determined by heterozygote intercrosses at three indicated developmental stages. p-value was calculated by chi-squared test.
(D) RT-qPCR for Nkx2-5 in WT, Het (Heterozygotes), and DKO embryonic hearts at E8.25, E8.75, and E9.5. RNA expression levels were normalized to 18s rRNA. Data were mean ± s.e.m. p-value was calculated by Student’s t-test.
(E, G, I, and K) Whole-mount in situ hybridization of Nkx2-5 in WT and DKO embryos at E7.5 (E), E8.25 (G), E8.75 (I), and E9.5 (K). Scale bar, 500 μm in (K) and 200 μm in (E, G, and I).
(F, H, J, and L) Immunofluorescence staining of NKX2-5 (red), TNNI3 (green), and DAPI (blue) in WT and DKO at E7.5 (F), E8.25 (H), E8.75 (J), and E9.5 (L). Insets of heart regions were shown in higher magnification on the right. Scale bar, 100 μm.
Given that DKO appeared to be a phenocopy of Nkx2-5-null mutants (Figure 2B),33 we measured NKX2-5 dosage at distinct developmental stages. We performed whole-mount in situ hybridization and immunofluorescence staining (Figures 2D–2L), finding that both Nkx2-5 mRNA and protein were undetected in E7.5 DKO compared to the wild-type (WT) littermates (Figures 2D–2F). RT-qPCR and whole-mount in situ hybridization confirmed a profound reduction in Nkx2-5 mRNA in E8.25 and E8.75 DKO mouse hearts compared to WT (Figures 2D, 2G, and 2I). Likewise, NKX2-5 protein also largely diminished in E8.25, but to a lesser degree in E8.75 DKO mice, as demonstrated by immunofluorescence staining (Figures 2H and 2J). Interestingly, both Nkx2-5 mRNA and protein in E9.5 DKO hearts recovered to a comparable level as in WT (Figures 2D, 2K, and 2L). Together, simultaneous deletion of enhancers U1 and U2 resulted in the missing expression of Nkx2-5 in hearts before E8.25. While the expression of Nkx2-5 recovered in E9.5 hearts, DKO embryos died of cardiac abnormalities as reported elsewhere in Nkx2-5-null mutants.33,34,35
Double deletion of U1 and U2 promotes precocious differentiation of cardiac progenitor cells in the second heart field (SHF)
We next leveraged single-cell RNA sequencing (RNA-seq) to explore the cellular mechanism underlying these morphological defects. The heart tube (HT) as the first heart field (FHF) and the OFT as the SHF from E8.25 DKO and littermate control hearts were collected and subjected to single-cell RNA-seq (Figure S2A). A total of 256 and 280 DKO and WT heart cells passing quality control were used for further analysis. To accurately discern the identity of DKO and WT heart cells, we took advantage of the published single-cell data across different heart developmental stages to construct a reference lineage differentiation trajectory. By reducing dimensionalities of single-cell RNA-seq data at E7.75, E8.25, E8.75, and E9.25 hearts with UMAP (Uniform Manifold Approximation and Projection),36,37 we revealed three major populations across four developmental stages (Figures 3A and 3B), corresponding to known cardiac cell types: cardiac progenitors (CPs), endothelial cells (ECs), and cardiomyocytes (CMs), as demonstrated by feature gene expression (Figure 3C) and gene ontology (GO) term analysis (Figures S2B and S2C).
Figure 3.

Double deletions result in precocious differentiation of cardiac progenitor cells from SHF
(A and B) UMAP visualization of all single cells from published data (GSE108963) colored by cell types in (A) and embryonic stages in (B). CP, cardiac progenitor cells; EC, endothelial cells; CM, cardiomyocytes.
(C) Shown were the expression patterns of representative genes highly enriched in each cell type.
(D and E) 3-Dimensional PCA showing single cells from SHF in (D) and HT in (E) of WT and DKO projected onto reference lineages in (A).
(F) Heatmaps showing differentially expressed genes in single cells from SHF between WT and DKO (left) and top enriched GO terms in each group (right). See also Figure S2.
When projecting spatially dissociated single cells onto the reference lineage trajectory described above, we observed SHF cells from WT and DKO were clustered separately. Specifically, DKO cells were all projected onto differentiating CMs, while WT cells represented a continuum of subpopulations spanning from CPs to CMs (Figures 3D, 3E, and S2D), suggesting potentially precocious differentiation of CPs in DKO. This finding was also supported by GO term analysis, showing DKO cells in SHF enriched for CM differentiation-related gene functions (Figure 3F). Accordingly, DKO SHF cells exhibited higher expression of CM-specific genes than WT SHF cells (Figure S2E), while the transcriptional level of these genes was mostly indistinguishable between WT and DKO cells in HT (Figures 3E, S2D, and S2E).
To interrogate the epigenetic mechanisms underlying the precocious differentiation of CPs in SHF but not in FHF of DKO hearts, we performed in situ ChIP-seq, a recently developed method38 for low-input samples with high signal-to-noise ratio, to examine H3K27ac-marked enhancer dynamics in both FHF and SHF of E8.25 WT and DKO hearts. The high quality of H3K27ac data were supported by the reproducibility between independent experiments and the fraction of reads in peaks (Figure S3 and Table S2). Specific inspection of the Nkx2-5 loci confirmed the loss of H3K27ac signals around U1 and U2 in FHF and SHF of E8.25 DKO hearts (Figure 4A). Consistent with the precocious differentiation of CPs toward CMs in SHF of DKO hearts by single-cell RNA sequencing (scRNA-seq) data analysis (Figure 3D), a marked increase in H3K27ac signals around the distal enhancer region of Tbx20 was observed (Figure 4B).
Figure 4.

Differential enhancer dynamics between FHF and SHF of E8.25 DKO and WT hearts
(A) Track view showing H3K27ac signals in FHF and SHF of E8.25 WT/DKO hearts around Nkx2-5 locus. U1 and U2 enhancers were highlighted by green and blue shading, respectively.
(B) Track view showing the H3K27ac in situ ChIP-seq signals in FHF and SHF of E8.25 WT/DKO hearts around the Tbx20 locus. Stronger H3K27ac signals around the Tbx20 enhancer in SHF of DKO hearts were highlighted by gray shading.
(C) Heatmaps showing H3K27ac signals at 60,836 peaks in E8.25 FHF of WT and DKO hearts. Groups consist of K-means clusters: cluster I (increased; n = 1,357), cluster II (unchanged; n = 58,413), and cluster III (decreased; n = 1,066). The heatmaps and aggregate plots were both generated with DeepTools.
(D) Aggregate plots of H3K27ac signals at ± 2 kb of peak centers of cluster I and cluster III in (C).
(E) Top 6 enriched GO terms of genes nearby upregulated and downregulated peaks in (C). p-value was calculated by Binomial test.
(F) Heatmaps showing H3K27ac signals at 44,375 peaks in E8.25 SHF of WT and DKO hearts. Groups consist of K-means clusters: cluster I (increased; n = 1,038), cluster II (unchanged; n = 42,694), and cluster III (decreased; n = 643). Heatmaps and aggregate plots were generated with DeepTools.
(G) Aggregate plots of H3K27ac signals at ± 2 kb of peak centers of cluster I and cluster III in (F).
(H) Top 6 enriched GO terms of genes nearby upregulated and downregulated peaks in (F). p-value was calculated by Binomial test. See also Figure S3.
U1 and U2 double deletions led to 1,357 and 1,038 regions with increased H3K27ac signals, as well as 1,066 and 643 peaks with downregulated signals, in FHF and SHF, respectively (Figures 4C–4H). However, the vast majority of peaks (58,413 in FHF and 42,694 in SHF) remained stable after deletions (Figures 4C and 4F), presumably consistent with the apparently normal heart morphology of E8.25 DKO hearts. Examining the biological function of the genes associated with the nearest differential peaks in DKO, we found that upregulated H3K27ac peaks in DKO FHF were enriched for GO terms related to non-cardiac development (Figures 4D and 4E). Interestingly, peaks with increased H3K27ac signals in DKO SHF were associated with genes participating in transcriptional and translational regulation (Figures 4G and 4H), in line with previous reports supporting that the differentiation process demands global elevation of the transcript and protein synthesis.39,40 Although only 643 peaks were downregulated in DKO SHF (Figure 4G), they exhibited more significant enrichments for GO terms related to heart development than these in DKO FHF, especially for heart conduction system development (Figures 4E and 4H), consistent with the atrioventricular conduction defects observed in humans with Nkx2-5 mutation and mouse Nkx2-5 knockout (KO) models.41 Thus, this evidence partially accounted for the developmental vulnerability in DKO SHF.
Genome-wide active heart enhancers were decommissioned in DKO
To identify the molecular basis underlying the dysregulated cardiac gene program in DKO with delayed Nkx2-5 re-expression, we performed H3K27ac in situ ChIP-seq38 of E9.5 WT and DKO hearts. The high quality of H3K27ac ChIP-seq data was supported by the reproducibility between independent experiments (r = 0.97 and 0.94 for both WT and DKO) (Figure S4A and Table S2).
We identified an average of 47,914 H3K27ac peaks in WT and 18,072 in DKO hearts, with more than half of H3K27ac peaks absent in DKO (Figures 5A and S4B). Of note, 15,213 significantly differentially bound peaks (DBPs) were identified (Figure S4C), by which WT and DKO hearts were largely distinguished in the first two principal components (Figure S4D). The nearby genes of 4,931 upregulated DBPs in DKO were enriched for biological processes associated with transcriptional and translational regulation, consistent with the increased RNA and protein synthesis in E8.25 DKO SHF (Figures 4H, S4E, and S4F). Further, 10,282 downregulated DBPs in DKO were related to heart muscle development, such as actin filament-based process, ventricular cardiac muscle tissue development, and regulation of heart contraction (Figures S4E, S4F, and Table S3).
Figure 5.

Genome-wide decommissioning of heart enhancers is associated with perturbation of NKX2-5 binding in DKO mice
(A) Venn diagram showing overlapping of H3K27ac peaks between WT and DKO.
(B) Track view showing H3K27ac in situ ChIP signals in DKO and WT hearts around Nkx2-5 locus. U1 and U2 enhancers were highlighted by light gray, and the highlighted green shading represented increased enhancer signals after U1 and U2 double knockout.
(C) Dot plots showing de novo motifs in downregulated DBPs discovered using Homer. Besides were matched known TFs. p-value was calculated by the Binomial test.
(D) Non-promoter H3K27ac signals around 52,251 non-promoter H3K27ac peaks in E9.5 WT and DKO hearts. Groups consisted of K-means clusters: cluster I (increased; n = 2,046) and cluster II (reduced; n = 50,205). The heatmaps and aggregate plots were both generated with DeepTools.
(E) Aggregate plots of H3K27ac ChIP-seq signals at ± 2 kb of peak centers in (D).
(F) Top 10 GO terms enriched in genes nearby increased and reduced peaks in (D). p-value was calculated by Binomial test.
(G) Heatmap showing NKX2-5 ChIP-seq signals around downregulated H3K27ac peaks in DKO hearts (n = 21,688 for downregulated NKX2-5 peaks and n = 28,517 for stable NKX2-5 peaks).
(H) Aggregation plot of NKX2-5 ChIP-seq signals at ± 1 kb of H3K27ac peak centers in (G).
(I) Track view showing decreased NKX2-5 binding signals at distal enhancers of Hand2 in DKO hearts.
(J) Heatmap displaying the strengths of most interactions between downregulated H3K27ac peaks in E9.5 DKO, and the promoters of heart developmental gene were weakened in DKO. ANOVA was performed for identifying significantly variable interactions.
(K) Violin plot showing the significantly decreased NKX2-5 binding signals on disrupted interactions between enhancers and promoters in (J).
(L) Visualization of chromatin interactions, H3K27ac, and NKX2-5 binding signals around Nkx2-5, Tbx5, and Hand1 loci. The highlighted red and green shading represented increased and decreeased interactions in DKO, respectively. See also Figure S4.
Specific inspection of the Nkx2-5 loci reinforced the loss of H3K27ac signals around U1 and U2 in E9.5 DKO hearts (Figure 5B). Interestingly, a marked increase of H3K27ac signals around the transcriptional termination sites of Nkx2-5 was observed (Figure 5B), likely associated with the recovery of Nkx2-5 expression in E9.25 DKO hearts. To identify NKX2-5-associated co-TFs, we performed de novo TF motif discovery in up- and downregulated DBPs. Several key heart-enriched TFs were highly over-represented in downregulated DBPs,36,42 such as MEF2C, GATA4, TEAD1,43 SMAD4,44 and NKX2-5 (Figure 5C), consistent with the physical interaction between NKX2-5 and each factor.45 The upregulated DBPs were enriched for TF motifs matching MAFK,46 NFATC3,47 PBX1,48 and HAND1 or TCF349 (Figure S4G). Loss of co-occurrence of identified co-TFs in DKO hearts indicated the inactivation of enhancers upon U1/U2 double deletion.
We next focused on the distal H3K27ac peaks as potential active enhancers. K-means clustering revealed two categories of enhancers including upregulated enhancers (cluster I) and downregulated enhancers (cluster II) (Figures 5D and 5E). As expected, the downregulated enhancers were associated with genes enriched for biological terms related to heart development, such as cardiac septum morphogenesis, consistent with the downregulated DBPs in E9.5 DKO (Figures 5E, 5F, S4E, S4F and Table S3). Collectively, the comparative analysis of H3K27ac-marked enhancers demonstrated that NKX2-5, a pivotal lineage-specific TF, activated heart developmental enhancers, decommissioning of which caused heart malformations.
We hypothesized that delayed expression of NKX2-5 under already dismantled chromatin states in DKO might not be able to fully engage target regions as in WT. To test this, we performed NKX2-5 in situ ChIP-seq in WT and DKO E9.5 hearts (Figure S4H)38 and observed that 27.9% (14,024/50,205) peaks identified in E9.5 WT hearts overlapped with E11.5 NKX2-5 peaks identified by Dupays et al. (Figure S4I).50 The high discordance of peaks by these two studies may be reflected by either the stage difference or the higher data quality of our NKX2-5 in situ ChIP-seq as we observed better signal-to-noise ratio values as shown by track view of the genomic loci (Figure S4J) and heatmap signals at the peak center regions (Figure S4K). A significant loss of NKX2-5 peaks in DKO was observed, in accordance with the loss of H3K27ac peaks (Figure S4L). Specifically, in DKO hearts, NKX2-5 signals reduced by nearly half at DKO downregulated distal H3K27ac peak regions (21,688/50,205) (Figures 5G and 5H). We also validated this change by the specific inspection of NKX2-5 signals at representative Hand2 loci (Figure 5I).
To confirm the hypothesis that the pre-disturbed chromatin architecture at earlier stages in DKO may induce barriers for lately expressed NKX2-5 binding, we performed genome-wide Hi-C experiments in E8.5 heart tubes of WT and DKO mice and obtained high-quality data with sufficient depth of ∼200 million non-duplicated valid pairs in each library (Table S2). Correlation analysis verified the reproducibility of biological replicates (Figure S4M). In order to explore the mechanisms underlying the developmental defects, though the expression of Nkx2-5 is recovered, we examined the interactions between downregulated H3K27ac ChIP-seq peaks in E9.5 DKO hearts (Figure 5D) and promoters of heart development-related genes (Table S4). The results showed that 324 interactions were decreased while 194 interactions were increased upon U1 and U2 deletion (Figure 5J). Further examining the NKX2-5 binding on these disrupted interactions, we observed decrease NKX2-5 binding signals on both upregulated and downregulated interactions (Figure 5K). For example, the NKX2.5 exhibited decreased binding capacity on the enhancers around Nkx2.5, Tbx5, and Hand1 loci (Figure 5L), which exhibited disrupted interactions with the promoters of their host genes. Together, our data supported that delayed expression of NKX2-5 in E9.5 DKO was not able to normally engage key heart developmental targets under the dysregulated chromatin states.
Loss of enhancer U1 manifested no overt heart phenotype
The misexpression of Nkx2-5 in DKO hearts confirmed the spatiotemporal role of enhancers U1 and U2 for transcriptional regulation. To understand the functional hierarchy, we delete U1 and U2 separately to evaluate the time course requirement of U1 and U2 for the Nkx2-5 transcriptional level. U1-KO (Figure S5A) mice obtained by heterozygote intercrosses displayed the expected Mendelian ratio from fetus to adult (Figure S5B). U1-KO mice exhibited no overt heart phenotype during embryonic stages and were viable through the age of 2–6 months with apparently normal heart function (Figures S5C–S5G). We posited that Nkx2-5 expression may also remain unaltered in U1-KO at various stages of heart development. Expectedly, Nkx2-5 appeared normal in E8.5 and E9.5 U1-KO (Figure S5H), and NKX2-5 protein remained normally distributed from E7.75 to E9.5 (Figure S5I). We further tested the hypothesis that the reliance on rewired neighboring enhancers or the increased activity of U2 might compensate for the loss of U1 enhancer, therefore resulting in a normal Nkx2-5 expression level. Indeed, chromatin conformation capture (3C) assay supported that the interactions between both U2 and proximal enhancers were significantly increased after U1 deletion (Figures S5J and S5K), while H3K27ac enrichment around U2 loci exhibited no significant difference between WT and U1-KO hearts as measured by ChIP-qPCR (Figure S5L). Thus, we concluded that enhancer U1 might sit in the lower place of the enhancer hierarchy.
U2-KO mice exhibited severe heart malformations with reduced NKX2-5 expression
Distinct from U1-KO mice, U2-KO embryos were observed during mid-gestation at the expected Mendelian ratio before E16.5 but died prenatally (Figures 6A, 6B, and S6A). Histologic examination of E16.5 U2-KO embryo hearts revealed severe ventricular septal defect (VSD) and significantly thinner ventricle walls than control (Figures 6C and 6D). To explore the cellular basis for the hypoplasia, we stained E16.5 U2-KO and control hearts with phosphorylated histone H3 (PH3), a cell cycle M-phase marker.51 U2-KO showed a marked decrease in mitotic cells by one-third (Figures S6B and S6C). Accordingly, we observed a gradual loss of Nkx2-5 mRNA in U2-KO hearts from E8.5 to E16.5 (Figure 6E). NKX2-5 proteins were also markedly diminished in E16.5 U2-KO ventricles, though detected in several sporadic regions (Figure S6D). Thus, the decreasing NKX2-5 expression accounted for the prenatal lethality and severe heart malformations of U2-KO mice.
Figure 6.

Characterization of U2-KO heart malformation and chromatin states
(A) Schematic of the strategy for the U2-knockout (U2-KO) mice generation.
(B) Survival analysis of U2-KO strains determined by heterozygote intercrosses at three indicated developmental stages. p-value was calculated by chi-squared test.
(C) Gross heart morphology and H&E staining of sections of E16.5 U2-KO and WT hearts. Arrows indicate ventricular septal defect (VSD) and thinner ventricle wall. Scale bar, 500 μm.
(D) Quantification analysis of the right ventricle thickness in (C). Data were mean ± s.e.m. p-value was calculated by Student’s t-test.
(E) RT-qPCR of Nkx2-5 in WT and U2-KO embryonic hearts at E8.5, E9.5, E12.5, and E16.5. Data were mean ± s.e.m. p-value was calculated by Student’s t-test.
(F) Track view showing ATAC-seq and H3K27ac ChIP-seq signals at Nkx2-5 or Myh7 locus in E16.5 heart apex.
(G) Heatmaps showing ATAC-seq signals at non-duplicated 36,282 peaks from all replicates. Each column was plotted at the 10 kb regions of the peak centers, and rows were sorted by log2 (WT+1)/log2(U2-KO+1) ATAC-seq signals.
(H) Heatmaps showing H3K27ac ChIP-seq signals at non-duplicated peaks from all replicates. Each column was plotted at the 10 kb regions of the peak centers, and rows were sorted by log2 (WT+1)/log2(U2 -KO+1) signals. log2 (ChIP signals) were shown in 15,501 peaks with WT/U2-KO > 1 and 5,095 peaks with WT/U2-KO ≤ 1.
(I) Bar plot showing top 15 GO terms enriched in genes nearby 5,709 significantly (FDR ≤ 0.05) downregulated H3K27ac peaks in U2-KO hearts. p-value was calculated by the Binomial test.
(J) H3K27ac ChIP-seq signals at TF regions whose motifs were enriched in down/upregulated H3K27ac peaks in E16.5 U2-KO hearts. TF motif p-value was calculated by Binomial test, and the H3K27ac signals were scaled by row Z score. Several cardiac feature genes were highlighted by blue, and the hematopoietic feature genes were highlighted by red.
(K) A working model showing the regulatory mechanisms of U1 and U2 regulating Nkx2-5 transcription during heart development. As early as E7.5, U1 and U2 work additively to sustain Nkx2-5 transcription, both of which could compensate for the loss of each other. From E12.5 and onward, U1 becomes insufficient to support Nkx2-5 expression. See also Figures S6 and S7.
The decreased enhancer activation but with unaltered chromatin accessibility in U2-KO hearts
Next, we asked how chromatin states changed upon NKX2-5 protein reduction in U2-KO. ATAC-seq and H3K27ac ChIP-seq were performed to characterize alterations of chromatin accessibility and active enhancers in E16.5 U2-KO and WT hearts (Table S2). Correlation analysis verified the reproducibility of biological replicates (Figures S7A–S7D). Complete loss of H3K27ac signals at the Myh7 locus was consistent with a severe defect in heart contraction in E16.5 U2-KO (Figure 6F).52 Indeed, H3K27ac signals but not chromatin accessibility (by ATAC-seq) signals largely decreased in U2-KO compared to WT hearts (Figures 6G and 6H), indicating the decreased activation of enhancers. The differential analysis further identified 5,709 significantly downregulated H3K27ac peaks, and these peaks were near genes enriched for terms involved in heart developmental processes such as cardiac ventricular muscle tissue development and coronary vasculature development (Figure 6I and Table S3). Nevertheless, 5,709 downregulated H3K27ac peaks exhibited no significant difference in chromatin accessibility (Figures S7E–S7G). We further performed de novo TF motif discovery and identified several cardiac TFs, such as TBX20, MEF2B, NKX2-5, and GATA4, in downregulated H3K27ac peaks, whose transcriptional activity also decreased upon U2 depletion (Figure 6J). These results suggested that temporally decreased NKX2-5 in U2 deletion disturbed the global enhancer landscape, therefore undermining chromatin occupancy of key cardiac TFs and subsequently resulting in the disarrayed transcriptional network in the hearts.
Together, our data demonstrated the temporal and partially compensatory regulatory function of enhancer U1 and U2 in fine-tuning host gene Nkx2-5 expression, thus sustaining normal heart development. Specifically, U1 and U2 function redundantly in Nkx2-5 transcriptional regulation at early stages, but U2 instead of U1 is necessarily required for its expression at later stages of heart chamber formation and maturation (Figure 6K).
Discussion
Extensive studies characterized several cis-regulatory elements that drive cardiac-specific Nkx2-5 expression in developing mouse hearts, including enhancer AR1-AR3,31 G-S,53 and UH5-UH6 (Figure 1).30 They mainly focused on regions of 5–10 kb upstream of the transcription start site (TSS), including the U1 enhancer. Examination of the transgenic reporter mouse lines confirmed expected expression domains of those enhancers in embryonic hearts. Nonetheless, whether those regulatory elements are necessarily required for heart development remains unexplored. Our data demonstrate that previously unknown distal U2 enhancer can compensate for loss of function of U1, likely through increasing interaction frequency with Nkx2-5 TSS. In addition, depletion of U1 caused a short linear distance between U2 enhancer and Nkx2-5 promoter, in part accounting for the unchanged Nkx2-5 transcription. This may also be consistent with two recent studies in preprint demonstrating that the shorter distance between enhancer and promoter can increase transcriptional activity and stability.54,55 However, we propose that U1 is able to compensate for transient U2 loss at the onset of Nkx2-5 transcriptional activation but later insufficient to maintain its expression as developmental progression. An alternative explanation is that the function of U2 at later stages may be independent of its enhancer activity. However, the specific regulatory mechanisms of U2 at later development stage demand further characterization. Although enhancer U1 sits in the lower, less important place of the enhancer hierarchy, it works together with U2 in an additive fashion to promote Nkx2-5 transcription before E9.5.
Analyzing differential H3K27ac ChIP-seq peaks between WT and DKO in both E8.25 and E9.5 hearts, we found that only a minority of peaks in E8.25 hearts were down- or up-regulated, but nearly half of the peaks in E9.5 were disrupted in DKO (Figures 4C, 4F, and 5D). This difference is pertinent to the developmental defect as the gross morphology of E8.25 hearts appears normal, but overt heart defects are apparent in E9.5 DKO.
Through the integration of H3K27ac and NKX2-5 ChIP-seq datasets in WT and DKO hearts, our study illustrates a model that temporal coupling of enhancers coordinates Nkx2-5 transcription. In this model, transiently missing NKX2-5 expression during early development dismantles the enhancer landscape, which cannot be rescued through later NKX2-5 re-expression. This is in part explained by the incomplete gain of NKX2-5 chromatin occupancy as in WT hearts.
In sum, our work presents a model that two hierarchical enhancers jointly control the dynamic transcription of nearby target genes during development. A generally accepted concept emerges that enhancers are often arranged in clusters, offering functional redundancy and transcriptional steadiness to fine-tune the target gene dosage.6,15,56 Our results demonstrated a functional hierarchy and partial compensation of two enhancers controlling Nkx2-5 transcriptional specificity and robustness during early heart development, with differential requirements for these regulatory elements at different stages of development to ensure development robustness (Figure 6K).
Limitations of the study
In this study, we comprehensively interrogate enhancers U1 and U2 in controlling Nkx2-5 transcription during heart development. Several limitations remain in this study. First, single-cell RNA-seq shows that double deletion of U1 and U2 promotes precocious differentiation of CP cells in the SHF. However, it has not been validated by experiments. Moreover, the mechanisms of why SHF is more susceptible to NKX2-5 deletion have not been systematically investigated. Further ChIP-seq for NKX2-5 in FHF and SHF of E8.25 hearts may help to understand the molecular basis. Second, in DKO hearts, Nkx2-5 expression recovers at E9.5, but the expression of Nkx2-5 gradually decreases after E9.5 in U2-KO hearts. How the other enhancers function to contribute to the divergent Nkx2-5 expression pattern in these two enhancer-KO models demands further exploration.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac | Abcam | Cat# ab4729; RRID: AB_2118291 |
| H3K27ac | Cell Signaling Technology | Cat# 8173S; RRID: AB_10949503 |
| NKX2-5 | R&D | Cat# AF2444; RRID: AB_ 355269 |
| Rabbit Control IgG antibody | ABclonal | Cat# AC005; RRID: AB_ 2771930 |
| Cardiac Troponin I | Abcam | Cat# ab56357; RRID: AB_ 880622 |
| WGA | Invitrogen | Cat# w32464; RRID: AB_ 2924839 |
| PH3 | Cell Signaling Technology | Cat# 9701S; RRID: AB_ 2924838 |
| Bacterial and virus strains | ||
| Bl21 (DE3) | TransGen Biotech | Cat# CD601 |
| Trans5α Chemically Competent Cell | TransGen Biotech | Cat# CD201 |
| Chemicals, peptides, and recombinant proteins | ||
| Formaldehyde Solution | SIGMA | Cat# F8775-500ML |
| Glycine | AMRESCO | Cat# 0167-1KG |
| Phosphate Buffered Daline (PBS) | Hyclone | Cat# SH30256.01 |
| HBSS, no Calcium, no Magnesium, no Phenol Red | Gibco | Cat# 14175103 |
| Sodium butyrate | SIGMA | Cat# 303410-100G |
| Ampicillin Na | AMRESCO | Cat# VT0236 |
| IPTG | INALCO | Cat# 1758-1400 |
| HEPES free acid | AMRESCO | Cat# 0511-1KG |
| Potassium hydroxide (KOH) | XiLong SCIENTIFIC | Cat# 1310-58-3 |
| Sodium chloride (NaCl) | SIGMA | Cat# S7653 |
| Glycerol | SIGMA | Cat# 49767-100ML |
| CA-630 | SIGMA | Cat# 56741 |
| Triton X-100 | SIGMA | Cat# T8787-50ML |
| EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 04693132001 |
| Phenyl Methane Sulfonyl Fluoride (PMSF) | AMRESCO | Cat# 0754-5G |
| Poly(ethyleneimine) solution (PEI) | SIGMA | Cat# P3143-100ML |
| Ni-NTA Agarose | QIAGEN | Cat# 30210 |
| Imidazole | SIGMA | Cat# 792527-100G |
| EDTA DISODIUM SALT DIHYDRATE | AMRESCO | Cat# 0105-1KG |
| Dithiothreitol (DTT) | INALCO | Cat# 1758-9030 |
| TRIS | AMRESCO | Cat# 0497-5KG |
| TAPS | SIGMA | Cat# T-5130 |
| Magnesium chloride solution (MgCl2) | SIGMA | Cat# 68475-100ML |
| N,N-Dimethylformamide (DMF) | SIGMA | Cat# D4551-250ML |
| SODIUM DODECYL SULFATE (SDS) | AMRESCO | Cat# 0227-1KG |
| Spermidine | SIGMA | Cat# S2501-1G |
| BioMag Plus Concanavalin A | Bangs Laboratories | Cat# BP531 |
| Potassium chloride (KCl) | XiLong SCIENTIFIC | Cat# 7447-40-7 |
| Calcium chloride dihydrate (CaCl2) | XiLong SCIENTIFIC | Cat# 10035-04-8 |
| Manganese (II) chloride tetrahydrate (MnCl2) | XiLong SCIENTIFIC | Cat# 13446-34-9 |
| Digitonin | SIGMA | Cat# D141-500MG |
| Bovine Serum Albumin | SIGMA | Cat# A1933-100G |
| Proteinase K | AMRESCO | Cat# 0706-100MG |
| KAPA HiFi HotStart ReadyMix | BIOSYSTEMS | Cat# KM2602 |
| AMPure XP | BECKMAN COULTER | Cat# A63881 |
| MboI | NEB | Cat# R0147M |
| T4 DNA ligase | NEB | Cat# M0202S |
| T7 RNA polymerase | Roche | Cat# 10881767001 |
| DIG RNA labeling mix | Roche | Cat# 11277073910 |
| TRIzol | Thermo Fisher | Cat# 15596018 |
| Critical commercial assays | ||
| cDNA synthesis kit | Vazyme | Cat# R223-01 |
| AceQ Universal SYBR qPCR Master Mix | Vazyme | Cat# R511-02 |
| TIANquick Mini Purification Kit | TIANGEN | Cat# DP203-02 |
| Deposited data | ||
| Raw and analyzed data | This paper | Database: GSE133760 |
| Single cell RNA-seq for E7.25-E9.25 heart cells | Xiong et al.57 | Database: GSE108963 |
| RNA-seq data for E8.25 hearts | ENA | Database: ERR332388 |
| RNA-seq data for E9.5 hearts | Papoutsi et al.58 | Database: GSE104670 |
| RNA-seq data for E12.5 hearts | Rahmanian et al.59 | Database: GSE82471 |
| RNA-seq data for E16.5 hearts | Rahmanian et al.59 | Database: GSE78317 |
| H3K27ac ChIP-seq data for E12.5 hearts | The ENCODE Project Consortium | Database: GSE82449 |
| H3K27ac RNA-seq data for E16.5 hearts | The ENCODE Project Consortium | Database: GSE82973 |
| NKX2-5 ChIP-seq data for E9.5 hearts | Wang et al.60 | Database: GSE129335 |
| NKX2-5 ChIP-seq data for E11.5 hearts | Dupays et al.50 | Database: GSE44576 |
| E8.25 Hi-C | figshare | Database: https://doi.org/10.6084/m9.figshare.22300177 |
| Oligonucleotides | ||
| See Table S1 for primer sequence used in this study | N/A | N/A |
| Software and algorithms | ||
| Bowtie2 | Langmead et al.61 |
http://bowtie-bio.sourceforge.net/bowtie2/index.shtml; RRID:SCR_016368 |
| Samtools | Li et al.62 | http://www.htslib.org/; RRID:SCR_002105 |
| BEDTools | Quinlan et al.63 | http://bedtools.readthedocs.io/en/latest/; RRID:SCR_006646 |
| deepTools | Ramírez et al.64 |
https://deeptools.readthedocs.io/en/develop/; RRID:SCR_016366 |
| MACS2 | Zhang et al.65 | https://github.com/taoliu/MACS; RRID:SCR_013291 |
| Picard | N/A | http://broadinstitute.github.io/picard/; RRID:SCR_006525 |
Resource availability
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the lead contact, Shanshan AI (aishanshan233@smu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Animal studies were performed according to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Mouse care and procedures were performed according to the protocol (no. IMM-HeAB-1) approved by the Institutional Animal Care and Use Committee of Peking University. The mice we used were on a C57BL/6J background. Mice of both sexes were used for all experiments and were analyzed at multiple ages from E7.5 to 6 months as indicated in the text and Fig. legends for each experiment. Embryos were staged by somite counts. Animals were randomly assigned to the control groups or experimental groups and all in vivo experiments were based on blinding procedures. All mutants were analyzed, relative to littermates. For heart collection, mice were euthanized by cervical dislocation. For echocardiography, mice were anaesthetized with isoflurane in oxygen (2% for induction and 1% for maintenance).
Experimental model and subject details
Generation of CRISPR/Cas9-mediated deletion mouse line
Cas9 mRNA and sgRNAs were co-injected into mouse zygotes. We used two sgRNAs for U1 knockout (sg1 and sg2), two sgRNAs for U2 knockout (sg3 and sg4), and all four sgRNAs for a compound double knockout. After genetic deletion was confirmed by genotyping, the germline transmission was performed for at least two generations by mating with C57BL/6J wild-type mice. F2 mice and later generations were used for heterozygote intercrosses to perform experiments. Knockout adults and embryos were genotyped by PCR using DNA extracted from tails and yolk sacs respectively. The primers for genotyping by PCR were provided (supplemental information online, Table S5).
Quantitative analysis of chromatin conformation capture (3C-PCR)
The 3C-PCR experiments were performed as described previously57,66 with minor changes. E8.5, E9.5, E12.5, E16.5 wildtype heart regions were used for 3C-PCR experiments. Samples were freshly dissected and cross-linked using 1% formaldehyde at room temperature (RT) for 10 min, followed by quenching by 0.125 M glycine. Fixed tissues were lysed in hypotonic buffer (20 mM HEPES pH = 7.5, 10 mM KCl, 1 mM EDTA, 10% glycerol, 0.2% CA-630) on ice for 30 min. After washing, the nucleus pellets were incubated in 0.5% SDS at 62°C for 10 min, then quenched by 3% Triton X--X 100 at 37°C for 15 min. Opened chromatin was digested by 20 U MboI (NEB, R0147M), rotated in NEB Buffer 2.1 at 37°C overnight, followed by heat inactivation at 62°C for 20 min, and ligated by T4 DNA ligase (NEB, M0202S) at RT for 6 h. Ligated chromatin was reverse-crosslinked with proteinase K at 55°C for 3 h, prior to incubation at 68°C overnight. Next, DNA was purified by phenol-chloroform. The proximal ligation events were detected by PCR. The primers are provided in Table S5. PCR products were detected by electrophoresis in 1.5% agarose gel and gel images were quantified using software ImageJ. The interaction frequencies in embryonic hearts were normalized to that of naked DNA control.
Histologic analyses
For hematoxylin & eosin (H&E) staining, E16.5 embryonic hearts were fixed in 4% PFA for 24 h–72 h at RT, dehydrated through a graded ethanol series (50%, 75%, 90%, 95%, and 100%), placed into xylene and paraffin-embedded. After sectioning of the four-chamber view (7 μm) by the Microtome (Leica, RM2245), tissues were deparaffinized in xylene and rehydrated through a graded ethanol series (100%, 95%, 90%, 75%, and 50%), then stained with Hematoxylin and Eosin. The thickness of the E16.5 ventricle wall was calculated as the ventricular compact myocardial area divided by its inner circumference. The relative thickness of E16.5 ventricle wall was calculated as thickness of E16.5 ventricle wall divided by outer circumference and normalized. After selection of myocardial areas, data were measured by Adobe Photoshop CC2014.
Immunostaining
Whole E7.5 to E9.5 specimens were fixed in 4% PFA at 4°C for 2 h. For E16.5 specimens, hearts were dissected out of the chest before fixation in 4% PFA overnight at 4°C. All specimens, including embryos and hearts, were rinsed in PBS, and dehydrated by 30% sucrose. After equilibrated in Tissue-Tek Optimal Cutting Temperature (OCT) (SAKURA) for at least 1 hour at 4°C, specimens were frozen using dry ice. Frozen blocks were equilibrated in the cryostat at −20°C for at least 30 min before sectioning. Transverse sections with a thickness of 7 μm were collected on a freezing microtome (MICROM, HM550VP) and stored at −80°C. Immunostaining was performed with indicated primary antibodies against TNNI3 (Abcam, ab56357, 1:200), NKX2-5 (R&D Systems, AF2444, 1:500) and ISL1 (DSHB, 39.4D5, 1:200). Primary antibodies were visualized by fluorescence-conjugated secondary antibodies as shown in Table S2. All the slides were imaged on a confocal microscope (Zeiss LSM710 NLO & DuoScan System).
RNA in situ hybridization (ISH)
Whole-mount RNA in situ hybridization with digoxigenin-labeled antisense RNA probes was performed as previously described with minor modifications.67,68 In brief, antisense probes against Nkx2-5 were transcribed from cDNA of mouse embryonic heart in vitro using T7 RNA polymerase (Roche, 10881767001) with DIG RNA labeling mix (Roche, 11277073910) according to the manufacturer’s guide. Embryos for ISH were fixed in 4% PFA at 4°C overnight, dehydrated through a graded methanol series (50%, 75%, and 100%) or can be stored in 100% methanol at −20°C for years. For the following experiments, the embryos were bleached in a solution containing 6% H2O2 for 2 h, rehydrated through a graded methanol series (100%, 75% and 50%) and washed in PBST (0.1% Tween-20). After proteinase K treatment for 10 min at RT, the embryos were post-fixed in 4% PFA for 20 min at RT. Embryos were pre-hybridized by the hybridization buffer (50% formamide, 5× SSC, 500 μg/ml yeast RNA, 50 μg/ml heparin and 0.1% Tween-20) for 4 h at 65°C, and 0.25 ng/μl to 1 ng/μl denatured specific DIG-labeled RNA probe was added and hybridized overnight at 65°C. After washing and 1 h RT blocking in 10% sheep serum, 2% blocking reagent (Roche, 11096176001) (diluted in maleic acid buffer containing Tween 20), embryos were incubated with anti-DIG-AP antibody (Roche, 11093274910, 1:3000) in blocking solution as above overnight at 4°C. Last, embryos were transferred to NTMT [100 mM NaCl, 100 mM Tris-HCl (pH 9.5), 50 mM MgCl2 and 0.1% Tween-20] for the post-antibody washing, and visualized in BM Purple AP Substrate (Roche, 11442074001). PCR primers for cloning RNA probes are provided in Table S5.
Reverse transcription and quantitative PCR (RT-qPCR)
Tissues and cells were washed by DEPC-PBS and harvested by TRIzol reagent (Life Technologies, #15596018) according to the procedure provided by the manufacturer. Total RNA (0.2 to 1 μg) was reverse transcribed using a cDNA synthesis kit (Vazyme, R223-01). qPCR was performed using AceQ Universal SYBR qPCR Master Mix (Vazyme, R511-02) on a Bio-Rad CFX96 TOUCH RealTime System. RT-qPCR analysis data yielded the expression relative to 18s rRNA expression. Primers for RT-qPCR are provided in Table S5. To estimate Nkx2-5 transcript RNA molecules per cell, we calculated the PCR efficiency of RT-qPCR primers and counted cell number used for PCR reaction as previously described.69
Echocardiography
Echocardiography was performed on U1KO and WT mice from 2-month-old through 6-month-old. Non-invasive, echocardiographic parameters were measured using a Vevo 2100 high-frequency ultrasound system (Visual Sonics). Two dimensional short-axis views were obtained for guided M-mode measurements of the left ventricular internal diameter at end diastole (LVIDd) and end systole (LVIDs). We measured LVIDd and LVIDs in at least three beats from each projection and averaged them. The fractional shortening (FS) represented the relative change of left ventricular diameters during the cardiac cycle, which was calculated by the following formula: FS (%) = [(LVIDd−LVIDs)/LVIDd] ×100%.
Low-input chromatin immunoprecipitation-sequencing (in situ ChIP-seq)
ChIP-seq was performed as previously described with minor modifications. E8.25 and E9.5 DKO/WT mouse hearts or E16.5 U2-KO/WT mice ventricles were harvested and washed twice with Wash Buffer (20 mM HEPES pH = 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1X Protease Inhibitor Cocktail and 10 mM sodium butyrate). Concanavalin A coated magnetic beads (Con-A beads) (Bangs Laboratories, BP531) were activated with Binding Buffer (20 mM HEPES pH = 7.9, 10 mM KCl, 1 mM CaCl2, 1 mM MnCl2) twice, and resuspended with adequate Binding Buffer and applied to the heart tissues. The tissue-bead mixture was incubated at RT for 20 min and collected by the magnet stand. Next, before the mixture was resuspended with Antibody Buffer (20 mM EDTA, 0.01% Digitonin, 0.2% TritonX-100 in Wash Buffer supplemented with 1X Protease Inhibitor Cocktail, 10 mM sodium butyrate) containing anti-Histone H3 (acetyl K27) antibody (Abcam, ab4729, 1:200). The cell-antibody mixture was incubated at 4°C for 2 h. The mixture was washed twice by DT-Wash Buffer (0.01% Digitonin, 0.2% TritonX-100 in Wash Buffer) and finally resuspended with DT-Wash Buffer containing 3 μg/ml pA-Tn5-emzyme. The mixture was incubated at 4°C for 1 h followed by wash three times. The reaction was activated by resuspending the mixture with cold Reaction Buffer (10 mM TAPS-NaOH pH = 8.3, 5 mM MgCl2, 10% DMF and supplemented with 1X Protease Inhibitor Cocktail, 10 mM sodium butyrate), followed by gently flicking and incubation at 25°C for 1 h. 20 mM EDTA was added to stop the reaction. The mixture was incubated at 65°C for 1 h with 0.2 % SDS and 0.5 mg/ml Proteinase K to release DNA fragments and then incubated at 85°C for 15 min to deactivate Proteinase K. DNA was purified by phenol-chloroform.
Assay for transposase-accessible chromatin followed by sequencing (ATAC-seq)
ATAC-seq was performed with E16.5 U2-KO and WT mice ventricles. Con-A beads were activated with Binding Buffer twice and were then resuspended with adequate Binding Buffer and applied to the heart tissues. The tissue-bead mixture was incubated at RT for 20 min and collected by the magnet stand. Next, the mixture was resuspended with DT-Wash Buffer containing 3 μg/ml pA-Tn5-emzyme, incubated at 4°C for 1 h, and washed. The reaction was activated by resuspending the mixture with cold Reaction Buffer, gently flicking, and incubating at 25°C for 1 h. 20 mM EDTA was added to stop the reaction. The mixture was incubated at 65°C for 1 h with 0.2 % SDS and 0.5 mg/ml Proteinase K to release DNA fragments and then incubated at 85°C for 15 min to deactivate Proteinase K DNA was purified by phenol-chloroform.
Hi-C
The Hi-C experiment was processed according to the previous description.70 Briefly, E8.5 hearts for Hi-C were fixed with 1% formaldehyde solution for 10 min at room temperature. MboI (NEB, R0147M) was used for digestion. Extaq (Takara) was used for library preparation with Nextera indexes. Libraries were sequenced on NovaSeq 6000 platform (Illumina PE-150).
Nextera library preparation and sequencing strategy of low-input ChIP-seq and ATAC-seq
PCR was performed using i5 index primers and i7 index primers. The reaction was set up at 72°C for 5 min as initial extension, 98°C for 45 s as initial denaturation, 13-18 cycles of amplification (98°C for 15 s, 63°C for 30 s, 72°C for 1 min), and the final elongation is 72°C for 5 min. After PCR, the resulting library was purified with 1.2X AMPure XP beads once. Size selection was carried out by first 0.6X AMPure beads to remove fragments more than 1 kb, and second 0.6X AMPure beads to the supernatant to obtain 200-800 bp fragments for sequencing. The libraries were sequenced with paired-end 150-bp reads on NovaSeq 6000 platform (Illumina).
Quantification and statistical analysis
ChIP-seq and ATAC-seq data processing
Firstly, we evaluated the quality of ChIP-seq raw sequencing data by FastQC (version 0.11.5). We used cutadapt (version 1.11) to remove sequencing adapters and trim low-quality bases at the end of sequence reads. We then used Bowtie 2 (version 2.2.9) to align the clean reads to mouse reference genome mm10 with parameters “--dovetail --very-sensitive-local --no-unal --no-mixed --no-discordant -X 2000” for sensitive and fast alignment as previously described.38 Uniquely mapped (MAPQ > 30), non-duplicated (PCR duplicates were removed by Picard) reads were used for subsequent analyses.
ChIP-seq and ATAC-seq data analyses and visualization
We calculated H3K27ac signals, ATAC signals, or NKX2-5 signals in consecutive 5 kb non-overlapping windows and normalized to total 10,000,000 reads per replicate using bamCoverage function in deepTools package.65 Integrative Genomics Viewer (version 2.3.59) was used to generate tracks. H3K27ac peaks, ATAC, and NKX2-5 peaks were identified using MACS2 (version 2.1.1).71 We performed hierarchical clustering, correlation analysis and PCA analysis using multiBigWigSummary function in deepTools. We utilized GREAT72 for GO enrichment of differential peak nearby genes. Differential analyses were performed using DiffBind package.73 De novo motifs were discovered by Homer.74 Plots were generated using R (https://www.r-project.org) and deepTools. Gene expression activity was calculated by summing the H3K27ac ChIP-seq signal around the targeted gene region (TSS±10 kb).
Single-cell RNA-seq data generation and processing
The single-cell library was prepared using a modified Smart-seq2 protocol, and the data processing followed by the method in Xiong et al.57 Briefly, for DKO and littermate WT embryos, single cells from heart tube or outflow tract were directly picked into lysis buffer (0.05 μl RNase Inhibitor(40U/ul), 0.095 μl 10% Triton X-100, 1.055 μl Nuclease-free water, 0.5 μl dNTP(10 mM), 0.20 μl Barcode primer(5uM), RNase-free water to total 2 μl) by mouth pipette after trypsin digestion. Paired-end sequencing reads were split based on the cell-specific barcode information in reads 2. Simultaneously, the unique molecular identifiers (UMI) sequences in reads 2 were attached to reads 1 and subsequently used for further analysis. The abundance of the transcripts was estimated by counting the uniquely mapped reads to each gene with HTSeq (13). Downstream single-cell analysis was based on the gene expression matrix. We used Seurat (version 2.3.4) (14) to cluster cells, perform dimension reduction of UMAP and identify differentially expressed genes with default parameters. Gene ontology analysis was performed to identify biological processes using DAVID (https://david.ncifcrf.gov/).
Hi-C data analysis
HiCExplorer (v.3.4.2)75 was used for Hi-C data processing. Non-duplicated reads were saved to generate the interaction matrix at 10 kb resolution with cooler (v.0.8.5).76 Knight-Ruiz (KR) was performed to balance interaction matrices. Observed/expected normalization was applied to eliminate background influences. The hicAggregateContacts was used to quantify interaction strengths between any two lists of genomic regions, with “--transform obs/exp” parameter. Visualized interactions and histone modification signals at different regions were displayed by PyGenomeTracks (v.1.0).
Statistical information
P value in RT-qPCR and quantification of ventricular wall thickness were calculated by two-tailed paired student’s t-test. P value in mouse survival analysis was calculated by chi-square test. All bioinformatic analysis was performed with R version 3.6.1. ANOVA and two-sample Wilcoxon Rank Sum test was used for the comparison of histone modification/TF ChIP-seq or ATAC-seq signal between WT and KO samples. P < 0.05 was referred to indicate a statistically significant value (if not specified).
Acknowledgments
We thank all members of the He lab for their critical comments on this manuscript. A.H was supported by grants from the National Key R&D Program of China (2021YFA1100101 and 2019YFA0801802), the National Natural Science Foundation of China (32025015 and 31771607), and the Peking-Tsinghua Center for Life Sciences. S.A. was supported by grants from the National Key R&D Program of China (2021YFA1102700 and 2022YFA1106200), the National Natural Science Foundation of China (82270307 and 32200660), the Natural Science Foundation of Guangdong Province (2022A1515011325), and the Natural Science Foundation of Guangzhou City (202201011012).
Author contributions
A.H. conceived and designed the study. J.Z., C.L., S.A., Q.W., and X.L. designed and performed experiments. C.L., S.A., and Y.L. performed the computational analyses. S.A., C.L., J.Z., Y.L., and A.H. wrote the paper with input from all other authors. All participated in data discussion and interpretation.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: March 29, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106509.
Contributor Information
Aibin He, Email: ahe@pku.edu.cn.
Shanshan Ai, Email: aishanshan233@smu.edu.cn.
Supplemental information
Data and code availability
-
•
All sequencing data generated in this study have been deposited in the GEO database. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Ong C.T., Corces V.G. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine M. Transcriptional enhancers in animal development and evolution. Curr. Biol. 2010;20:R754–R763. doi: 10.1016/j.cub.2010.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoenfelder S., Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019;20:437–455. doi: 10.1038/s41576-019-0128-0. [DOI] [PubMed] [Google Scholar]
- 4.Cashman T.J., Trivedi C.M. Super enhancers: enhancing human cardiogenesis. Circ. Res. 2020;127:1156–1158. doi: 10.1161/CIRCRESAHA.120.318000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wamstad J.A., Wang X., Demuren O.O., Boyer L.A. Distal enhancers: new insights into heart development and disease. Trends Cell Biol. 2014;24:294–302. doi: 10.1016/j.tcb.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Osterwalder M., Barozzi I., Tissières V., Fukuda-Yuzawa Y., Mannion B.J., Afzal S.Y., Lee E.A., Zhu Y., Plajzer-Frick I., Pickle C.S., et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018;554:239–243. doi: 10.1038/nature25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas H.F., Kotova E., Jayaram S., Pilz A., Romeike M., Lackner A., Penz T., Bock C., Leeb M., Halbritter F., et al. Temporal dissection of an enhancer cluster reveals distinct temporal and functional contributions of individual elements. Mol. Cell. 2021;81:969–982.e13. doi: 10.1016/j.molcel.2020.12.047. [DOI] [PubMed] [Google Scholar]
- 8.Li W., Notani D., Rosenfeld M.G. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 9.Bozek M., Cortini R., Storti A.E., Unnerstall U., Gaul U., Gompel N. ATAC-seq reveals regional differences in enhancer accessibility during the establishment of spatial coordinates in the Drosophila blastoderm. Genome Res. 2019;29:771–783. doi: 10.1101/gr.242362.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song L., Crawford G.E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spilianakis C.G., Lalioti M.D., Town T., Lee G.R., Flavell R.A. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 12.Tsai P.F., Dell'Orso S., Rodriguez J., Vivanco K.O., Ko K.D., Jiang K., Juan A.H., Sarshad A.A., Vian L., Tran M., et al. A muscle-specific enhancer RNA mediates cohesin recruitment and regulates transcription in trans. Mol. Cell. 2018;71:129–141.e8. doi: 10.1016/j.molcel.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birnbaum R.Y., Clowney E.J., Agamy O., Kim M.J., Zhao J., Yamanaka T., Pappalardo Z., Clarke S.L., Wenger A.M., Nguyen L., et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012;22:1059–1068. doi: 10.1101/gr.133546.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T., et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Will A.J., Cova G., Osterwalder M., Chan W.L., Wittler L., Brieske N., Heinrich V., de Villartay J.P., Vingron M., Klopocki E., et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog) Nat. Genet. 2017;49:1539–1545. doi: 10.1038/ng.3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin H.Y., Willi M., HyunYoo K., Zeng X., Wang C., Metser G., Hennighausen L. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 2016;48:904–911. doi: 10.1038/ng.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J., Li K., Cai W., Liu X., Zhang Y., Orkin S.H., Xu J., Yuan G.C. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 2018;9:943. doi: 10.1038/s41467-018-03279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satou Y., Satoh N. Gene regulatory networks for the development and evolution of the chordate heart. Genes Dev. 2006;20:2634–2638. doi: 10.1101/gad.1485706. [DOI] [PubMed] [Google Scholar]
- 19.Davidson E.H., Erwin D.H. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- 20.Yang X.H., Nadadur R.D., Hilvering C.R., Bianchi V., Werner M., Mazurek S.R., Gadek M., Shen K.M., Goldman J.A., Tyan L., et al. Transcription-factor-dependent enhancer transcription defines a gene regulatory network for cardiac rhythm. Elife. 2017;6:e31683. doi: 10.7554/eLife.31683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruneau B.G. Transcriptional regulation of vertebrate cardiac morphogenesis. Circ. Res. 2002;90:509–519. doi: 10.1161/01.res.0000013072.51957.b7. [DOI] [PubMed] [Google Scholar]
- 22.Lien C.L., Wu C., Mercer B., Webb R., Richardson J.A., Olson E.N. Control of early cardiac-specific transcription of Nkx2-5 by a GATA-dependent enhancer. Development. 1999;126:75–84. doi: 10.1242/dev.126.1.75. [DOI] [PubMed] [Google Scholar]
- 23.Harvey R.P., Lai D., Elliott D., Biben C., Solloway M., Prall O., Stennard F., Schindeler A., Groves N., Lavulo L., et al. Homeodomain factor Nkx2-5 in heart development and disease. Cold Spring Harb. Symp. Quant. Biol. 2002;67:107–114. doi: 10.1101/sqb.2002.67.107. [DOI] [PubMed] [Google Scholar]
- 24.Salamon I., Serio S., Bianco S., Pagiatakis C., Crasto S., Chiariello A.M., Conte M., Cattaneo P., Fiorillo L., Felicetta A., et al. Divergent transcription of the Nkx2-5 locus generates two enhancer RNAs with opposing functions. iScience. 2020;23:101539. doi: 10.1016/j.isci.2020.101539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz R.J., Olson E.N. Building the heart piece by piece: modularity of cis-elements regulating Nkx2-5 transcription. Development. 1999;126:4187–4192. doi: 10.1242/dev.126.19.4187. [DOI] [PubMed] [Google Scholar]
- 26.Wu S.M., Fujiwara Y., Cibulsky S.M., Clapham D.E., Lien C.L., Schultheiss T.M., Orkin S.H. Developmental origin of a bipotential myocardial and smooth muscle cell precursor in the mammalian heart. Cell. 2006;127:1137–1150. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 27.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D., et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chi X., Chatterjee P.K., Wilson W., 3rd, Zhang S.X., Demayo F.J., Schwartz R.J. Complex cardiac Nkx2-5 gene expression activated by noggin-sensitive enhancers followed by chamber-specific modules. Proc. Natl. Acad. Sci. USA. 2005;102:13490–13495. doi: 10.1073/pnas.0504295102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lien C.L., McAnally J., Richardson J.A., Olson E.N. Cardiac-specific activity of an Nkx2-5 enhancer requires an evolutionarily conserved Smad binding site. Dev. Biol. 2002;244:257–266. doi: 10.1006/dbio.2002.0603. [DOI] [PubMed] [Google Scholar]
- 32.Singh G., Mullany S., Moorthy S.D., Zhang R., Mehdi T., Tian R., Duncan A.G., Moses A.M., Mitchell J.A. A flexible repertoire of transcription factor binding sites and a diversity threshold determines enhancer activity in embryonic stem cells. Genome Res. 2021;31:564–575. doi: 10.1101/gr.272468.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyons I., Parsons L.M., Hartley L., Li R., Andrews J.E., Robb L., Harvey R.P. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L., Nomura-Kitabayashi A., Sultana N., Cai W., Cai X., Moon A.M., Cai C.L. Mesodermal Nkx2.5 is necessary and sufficient for early second heart field development. Dev. Biol. 2014;390:68–79. doi: 10.1016/j.ydbio.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prall O.W.J., Menon M.K., Solloway M.J., Watanabe Y., Zaffran S., Bajolle F., Biben C., McBride J.J., Robertson B.R., Chaulet H., et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ai S., Xiong H., Li C.C., Luo Y., Shi Q., Liu Y., Yu X., Li C., He A. Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol. 2019;21:1164–1172. doi: 10.1038/s41556-019-0383-5. [DOI] [PubMed] [Google Scholar]
- 37.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 38.Wang J., Chen X., Shen D., Ge D., Chen J., Pei J., Li Y., Yue Z., Feng J., Chu M., Nie Y. A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J. Mol. Cell. Cardiol. 2019;127:105–114. doi: 10.1016/j.yjmcc.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Sampath P., Pritchard D.K., Pabon L., Reinecke H., Schwartz S.M., Morris D.R., Murry C.E. A hierarchical network controls protein translation during murine embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2008;2:448–460. doi: 10.1016/j.stem.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jay P.Y., Harris B.S., Maguire C.T., Buerger A., Wakimoto H., Tanaka M., Kupershmidt S., Roden D.M., Schultheiss T.M., O'Brien T.X., et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J. Clin. Invest. 2004;113:1130–1137. doi: 10.1172/JCI19846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akerberg B.N., Gu F., VanDusen N.J., Zhang X., Dong R., Li K., Zhang B., Zhou B., Sethi I., Ma Q., et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 2019;10:4907. doi: 10.1038/s41467-019-12812-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Z., Friedrich G.A., Soriano P. Transcriptional enhancer factor 1 disruption by a retroviral gene trap leads to heart defects and embryonic lethality in mice. Genes Dev. 1994;8:2293–2301. doi: 10.1101/gad.8.19.2293. [DOI] [PubMed] [Google Scholar]
- 44.Song L., Yan W., Chen X., Deng C.X., Wang Q., Jiao K. Myocardial smad4 is essential for cardiogenesis in mouse embryos. Circ. Res. 2007;101:277–285. doi: 10.1161/CIRCRESAHA.107.155630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He A., Kong S.W., Ma Q., Pu W.T. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. USA. 2011;108:5632–5637. doi: 10.1073/pnas.1016959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katsuoka F., Motohashi H., Onodera K., Suwabe N., Engel J.D., Yamamoto M. One enhancer mediates mafK transcriptional activation in both hematopoietic and cardiac muscle cells. EMBO J. 2000;19:2980–2991. doi: 10.1093/emboj/19.12.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossow C.F., Minami E., Chase E.G., Murry C.E., Santana L.F. NFATc3-induced reductions in voltage-gated K+ currents after myocardial infarction. Circ. Res. 2004;94:1340–1350. doi: 10.1161/01.RES.0000128406.08418.34. [DOI] [PubMed] [Google Scholar]
- 48.Chang C.P., Stankunas K., Shang C., Kao S.C., Twu K.Y., Cleary M.L. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development. 2008;135:3577–3586. doi: 10.1242/dev.022350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McFadden D.G., Barbosa A.C., Richardson J.A., Schneider M.D., Srivastava D., Olson E.N. The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development. 2005;132:189–201. doi: 10.1242/dev.01562. [DOI] [PubMed] [Google Scholar]
- 50.Dupays L., Shang C., Wilson R., Kotecha S., Wood S., Towers N., Mohun T. Sequential binding of MEIS1 and NKX2-5 on the Popdc2 gene: a mechanism for spatiotemporal regulation of enhancers during cardiogenesis. Cell Rep. 2015;13:183–195. doi: 10.1016/j.celrep.2015.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eastman A.E., Guo S. The palette of techniques for cell cycle analysis. FEBS Lett. 2020 doi: 10.1002/1873-3468.13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han P., Li W., Lin C.-H., Yang J., Shang C., Nuernberg S.T., Jin K.K., Xu W., Lin C.-Y., Lin C.-J., et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown C.O., 3rd, Chi X., Garcia-Gras E., Shirai M., Feng X.H., Schwartz R.J. The cardiac determination factor, Nkx2-5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. J. Biol. Chem. 2004;279:10659–10669. doi: 10.1074/jbc.M301648200. [DOI] [PubMed] [Google Scholar]
- 54.Zuin J., Roth G., Zhan Y., Cramard J., Redolfi J., Piskadlo E., Mach P., Kryzhanovska M., Tihanyi G., Kohler H., et al. Nonlinear control of transcription through enhancer-promoter interactions. bioRxiv. 2021 doi: 10.1101/2021.04.22.440891. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rinzema N.J., Sofiadis K., Tjalsma S.J.D., Verstegen M., Oz Y., Valdes-Quezada C., Felder A.-K., Filipovska T., van der Elst S., de Andrade dos Ramos Z., et al. Building regulatory landscapes: enhancer recruits cohesin to create contact domains, engage CTCF sites and activate distant genes. bioRxiv. 2021 doi: 10.1101/2021.10.05.463209. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bahr C., von Paleske L., Uslu V.V., Remeseiro S., Takayama N., Ng S.W., Murison A., Langenfeld K., Petretich M., Scognamiglio R., et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018;553:515–520. doi: 10.1038/nature25193. [DOI] [PubMed] [Google Scholar]
- 57.Xiong H., Luo Y., Yue Y., Zhang J., Ai S., Li X., Wang X., Zhang Y.L., Wei Y., Li H.H., et al. Single-cell transcriptomics reveals chemotaxis-mediated intraorgan crosstalk during cardiogenesis. Circ. Res. 2019;125:398–410. doi: 10.1161/CIRCRESAHA.119.315243. [DOI] [PubMed] [Google Scholar]
- 58.Papoutsi T., Luna-Zurita L., Prados B., Zaffran S., de la Pompa J.L. Bmp2 and Notch cooperate to pattern the embryonic endocardium. Development. 2018;145:dev163378. doi: 10.1242/dev.163378. [DOI] [PubMed] [Google Scholar]
- 59.Rahmanian S., Murad R., Breschi A., Zeng W., Mackiewicz M., Williams B., Davis C.A., Roberts B., Meadows S., Moore D., et al. Dynamics of microRNA expression during mouse prenatal development. Genome Res. 2019;29:1900–1909. doi: 10.1101/gr.248997.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Q., Xiong H., Ai S., Yu X., Liu Y., Zhang J., He A. CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell. 2019;76:206–216.e7. doi: 10.1016/j.molcel.2019.07.015. [DOI] [PubMed] [Google Scholar]
- 61.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dekker J., Rippe K., Dekker M., Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 67.Wei Q., Manley N.R., Condie B.G. Whole mount in situ hybridization of E8.5 to E11.5 mouse embryos. J. Vis. Exp. 2011 doi: 10.3791/2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scotti M., Kmita M. Recruitment of 5' Hoxa genes in the allantois is essential for proper extra-embryonic function in placental mammals. Development. 2012;139:731–739. doi: 10.1242/dev.075408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han X., Zhang J., Liu Y., Fan X., Ai S., Luo Y., Li X., Jin H., Luo S., Zheng H., et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development. 2019;146:dev176198. doi: 10.1242/dev.176198. [DOI] [PubMed] [Google Scholar]
- 70.Li C.C., Zhang G., Du J., Liu D., Li Z., Ni Y., Zhou J., Li Y., Hou S., Zheng X., et al. Pre-configuring chromatin architecture with histone modifications guides hematopoietic stem cell formation in mouse embryos. Nat. Commun. 2022;13:346. doi: 10.1038/s41467-022-28018-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R., et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramírez F., Bhardwaj V., Arrigoni L., Lam K.C., Grüning B.A., Villaveces J., Habermann B., Akhtar A., Manke T. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 2018;9:189. doi: 10.1038/s41467-017-02525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abdennur N., Mirny L.A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36:311–316. doi: 10.1093/bioinformatics/btz540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All sequencing data generated in this study have been deposited in the GEO database. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.