

SUMMARY
WNT/β-catenin signaling controls gene expression across biological contexts from development and stem cell homeostasis to diseases including cancer. How β-catenin is recruited to distinct enhancers to activate context-specific transcription is unclear, given that most WNT/ß-catenin-responsive transcription is thought to be mediated by TCF/LEF transcription factors (TFs). With time-resolved multi-omic analyses, we show that SOX TFs can direct lineage-specific WNT-responsive transcription during the differentiation of human pluripotent stem cells (hPSCs) into definitive endoderm and neuromesodermal progenitors. We demonstrate that SOX17 and SOX2 are required to recruit β-catenin to lineage-specific WNT-responsive enhancers, many of which are not occupied by TCFs. At TCF-independent enhancers, SOX TFs establish a permissive chromatin landscape and recruit a WNT-enhanceosome complex to activate SOX/ß-catenin-dependent transcription. Given that SOX TFs and the WNT pathway are critical for specification of most cell types, these results have broad mechanistic implications for the specificity of WNT responses across developmental and disease contexts.
In brief
Mukherjee et al. show that during WNT-induced differentiation of human pluripotent stem cells, SOX transcription factors can recruit β-catenin to lineage-specific enhancers independent of TCFs, the commonly accepted β-catenin binding partner. This helps explain how WNT signaling can regulate different transcriptional programs in different biological contexts.
Graphical Abstract
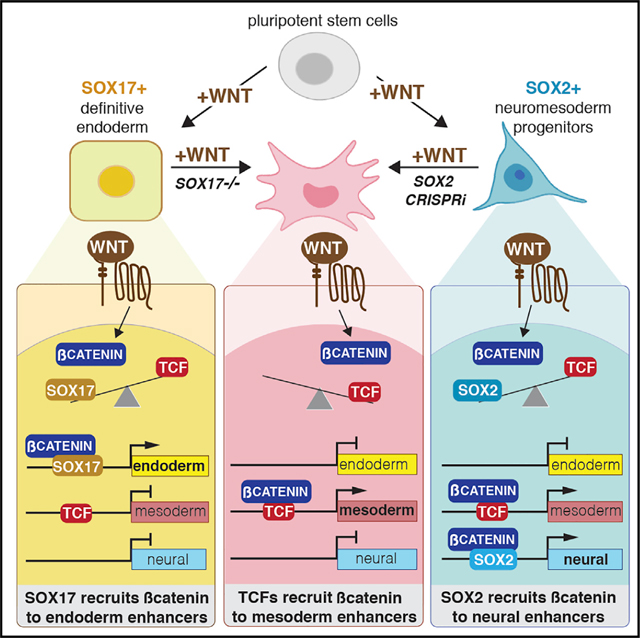
INTRODUCTION
WNT/β-catenin signaling is used reiteratively in all metazoans and is critical during developmental processes from cell fate determination in embryogenesis, organogenesis, and tissue regeneration to adult stem cell homeostasis (Nusse and Clevers, 2017). Dysregulation of the WNT pathway is associated with human diseases including cancer and neurodegenerative disorders (Ng et al., 2019). Despite being the subject of intense study for decades, how the WNT pathway executes its context-dependent roles through the selective transcription of distinct target genes remains poorly understood. (Masuda and Ishitani, 2017; Söderholm and Cantù, 2021)
In the canonical WNT pathway, recruitment of β-catenin (CTNNB1) to enhancers is the key event initiating transcription (Nakamura et al., 2016). In the absence of a WNT signal, cytosolic β-catenin is phosphorylated by glycogen synthase kinase-3β (GSK3β) and targeted for proteosomal degradation. The binding of WNT ligands to FZD/LRP receptors leads to the inactivation of the β-catenin destruction complex, allowing non-phosphorylated β-catenin to accumulate and localize to the nucleus. There, β-catenin associates with TCF/LEF (hereafter TCF) transcription factors (TFs), where it inactivates TLE/ Groucho co-repressors leading to the activation of WNT target genes (Söderholm and Cantù, 2021). Recent studies have provided a more integrated view of this transcription complex, termed the “WNT-enhanceosome,” which is assembled on WNT-responsive enhancers (WREs), where β-catenin interacts on chromatin with multiple cofactors, including BCL9, PYGOPUS, CHIP/LDB/SSDP, and the BAF complex (Gammons and Bienz, 2018). Current models propose that upon WNT activation, recruitment of β-catenin results in a conformational change in the WNT-enhanceosome, an association of WREs with their cognate promoters to recruit RNA polymerase II, and transcription initiation (Gammons and Bienz, 2018). Yet, how β-catenin, with no intrinsic DNA-binding activity, is recruited to distinct WREs in different cellular contexts remains a mystery.
The TCF family of HMG-box TFs are core mediators of WNT-responsive transcription by physically interacting with β-catenin and recruiting it to WREs. The four mammalian TCFs—TCF7, TCF7L1, TCF7L2, and LEF1—all bind in vitro as monomers to nearly identical 5′-C/GATCAAGC/G-3′ DNA sequences (Cadigan and Waterman, 2012), have redundant functions (Gerner-Mauro et al., 2020; Moreira et al., 2017), and co-occupy similar genomic loci (Blassberg et al., 2022; Guo et al., 2021). A few mechanisms have been proposed to account for a degree of variability in DNA binding, such as alternative RNA-splicing variants of TCF (Weise et al., 2010), but this is insufficient to account for the diversity of WNT-responsive transcription. Emerging evidence suggests that β-catenin can interact with TFs other than TCFs including OCT4, TBX3, HIF1α, SMAD, and SOX (Funa et al., 2015; Kaidi et al., 2007; Kelly et al., 2011; Zimmerli et al., 2020). One of the strongest candidates for an alternative TF family that could mediate WNT-responsive transcription is the SOX family. Related to TCFs, the twenty SOX TFs are conserved HMG-box TFs that recognize distinct variations of a core A/TA/ TCAAA/T motif (Badis et al., 2009). Many SOX TFs can bind to β-catenin in vitro and antagonize or potentiate WNT-responsive transcription of an artificial TOP:flash reporter in overexpression conditions (Corada et al., 2019; Kormish et al., 2010; Zorn et al., 1999). In mouse neural progenitor cells, Sox2 and b-cat/Lef1 repress the pro-proliferative WNT target CyclinD1 (Hagey and Muhr, 2014), and we recently showed in Xenopus gastrulae that Sox17 and β-catenin synergistically regulate the spatiotemporal expression of WNT-responsive endodermal genes (Mukherjee et al., 2020). However, whether SOX TFs can recruit β-catenin to distinct WREs in different cell types and regulate the specificity of WNT-responsive transcription is unclear.
In this study, we used genomic analyses of differentiating human pluripotent stem cells (hPSCs) to demonstrate that SOX TFs are required to recruit β-catenin to lineage-specific WREs, many of which are not occupied by TCFs. Ectopic expression of SOX17 is sufficient to recruit β-catenin to novel loci in a human kidney cell line that lack all four TCFs. Finally, at a subset of TCF-independent WREs, SOX TFs establish a permissive chromatin landscape and recruit a WNT-enhanceosome complex to direct SOX/ß-catenin-dependent transcription. These results establish SOX TFs as lineage-specific regulators of the WNT pathway, and they provide important insights into how the combination of TCFs and lineage-specific SOX TFs regulate distinct transcriptional programs downstream of WNT signaling across many biological contexts.
RESULTS
β-catenin binds dynamically to lineage-specific enhancers during endoderm differentiation
To characterize lineage-specific genomic recruitment of β-catenin during development, we performed a time course analysis of hPSCs differentiated to definitive endoderm (DE). Using a well-established protocol, we treated hPSCs for 3 days with recombinant ACTIVIN A and the GSK3b inhibitor CHIR99021 to stimulate the WNT/β-catenin pathway (Figure 1A). This protocol mimics the activity of NODAL and WNT signaling in gastrulating embryos and generated mesendoderm cells on day 1, endoderm progenitors on day 2, and a highly homogeneous (>90%) population of SOX17-expressing DE cells on day 3 (Figures 1A and 1B). Immunostaining and western blots confirmed that transcriptionally active, K49-acetylated “active” β-catenin (Hoffmeyer et al., 2017) was present in the nucleus of all cells from days 1–3 (Figures 1B and S1C)). We then performed chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) every 24 h to profile β-catenin genomic binding during the progressive differentiation from pluripotency to DE.
Figure 1. Dynamic genomic binding of β-catenin during DE differentiation.

(A) Schematic of DE differentiation.
(B) Immunostaining of active β-catenin and SOX17 during differentiation (scale bar represents 100 mm).
(C) Heatmap of β-catenin ChIP-seq showing five categories of temporally distinct peaks, n = 2 biological replicates.
(D) β-catenin binding dynamics during DE differentiation.
(E) Overlap of WNT-regulated genes with genes associated with β-catenin bound peaks. *Significant based on hypergeometric test, day 1: p = 4.75 × 10−105; day 2: p = 3.63 × 10−138; day 3: p = 2.52 × 10−48.
(F–J) Genome browser view of β-catenin binding for each of the five categories of peaks. Numbers indicate ChIP-seq signal scaling.
(K–O) Line graph of average normalized β-catenin (purple) and ATAC-seq (green) read density. Error bars represent standard error of mean. Dotted line represents the tag count threshold for significant ATAC-seq peaks. See also Figure S1 and Table S2.
In the day 0 “WNT-OFF” pluripotency state, we detected negligible β-catenin binding with 749 peaks, which increased upon CHIR treatment to 4,517 peaks at day 1, 18,608 peaks on day 2, and 11,684 peaks on day 3 (Figure 1D). β-catenin binding was observed at distinct genomic regions on different days, suggesting that it regulated distinct transcriptional programs as lineage specification proceeded. To identify these dynamic binding patterns, we merged all significantly called peaks across all days and performed k-means clustering (Figures 1C and 1F–1J). This identified five distinct patterns: common peaks shared across all time points (n = 1972), day 1 enriched peaks (n = 1,304), day 2 enriched peaks (n = 8,364), peaks specific to days 2 and 3 (n = 7,065), and day 3 enriched peaks (n = 2,611). To ensure that CHIR-mediated DE differentiation indeed mimicked WNT activation, we repeated the differentiation replacing CHIR99021 with recombinant WNT3A and validated that the expression of known DE WNT target genes and b-catenin genomic binding patterns were almost indistinguishable between CHIR99021 and WNT3A (Figures S1A, S1B, and S1D, see key resources table for additional supplementary data).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Beta-catenin | Invitrogen | Cat#71–2700; RRID:AB_2533982 |
| Beta-catenin K49ac | Cell Signaling Technology | Cat#9030; RRID:AB_2797689 |
| Beta-catenin | BD Biosciences | Cat#610154; RRID:AB_397555 |
| BCL9 | Abcam | Cat#ab37305; RRID:AB_2227890 |
| BRG1 | Bethyl Laboratories | Cat#A303–877A; RRID:AB_2615369 |
| BRG1 | Cell Signaling Technology | Cat#49360; RRID:AB_2728743 |
| FOXA2 | Abnova | Cat#H00003170-M01; RRID:AB_538933 |
| H3K27ac | Active Motif | Cat#39133; RRID:AB_2561016 |
| LEF1 | Cell Signaling Technology | Cat#76010; RRID:AB_2799877 |
| MED12 | Bethyl Laboratories | Cat#A300–774A; RRID:AB_669756 |
| NIPBL | Bethyl Laboratories | Cat#A301–779A; RRID:AB_1211232 |
| p300 | Abcam | Cat#ab14984; RRID:AB_301550 |
| PYGO2 | Novus Biologicals | Cat#NBP1–46171; RRID:AB_10009403 |
| SMC1 | Bethyl Laboratories | Cat#A300–055A; RRID:AB_2192467 |
| SOX17 | R&D Systems | Cat#AF1924; RRID:AB_355060 |
| SOX2 | R&D Systems | Cat#AF2018; RRID:AB_355110 |
| TBXT | Cell Signaling Technology | Cat#81694; RRID:AB_2799983 |
| TCF7 | Cell Signaling Technology | Cat#2203; RRID:AB_2199302 |
| TCF7L1 | Active Motif | Cat#61125; RRID:AB_2793517 |
| TCF7L1 | R&D Systems | Cat#AF6116; RRID:AB_10573292 |
| TCF7L2 | Cell Signaling Technology | Cat#2569; RRID:AB_2199816 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| NEB® 5-alpha Competent E. coli | New England Biolabs | Cat#C2987H |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| mTESR | STEMCELL Technologies | Cat#85851 |
| Essential 8 | Gibco | Cat#A1517001 |
| Essential 6 | Gibco | Cat#A1516401 |
| Opti-MEM | Gibco | Cat#31985062 |
| Matrigel | Corning | Cat#354277 |
| Versene | Gibco | Cat#15040066 |
| ReleSR | STEMCELL Technologies | Cat#05872 |
| VTN-N | Gibco | Cat#A14700 |
| Activin A | Shenandoah Biotechnology | Cat#800–01 |
| CHIR99021 | R&D Systems | Cat#4423 |
| FGF8b | Peprotech | Cat#100–25 |
| Y-27632 | STEMCELL Technologies | Cat#72304 |
| C59 | Tocris | Cat#5148 |
| PowerUP SYBR Green MasterMix | Applied Biosystems | Cat#A25777 |
| DAPI | Sigma Aldrich | Cat# D1388 |
| Doxycycline hyclate | Sigma Aldrich | Cat# D9891 |
| Protein G Dynabeads | Invitrogen | Cat# 10004D |
| Ampure XP | Beckman Coulter | Cat# A63880 |
| Alt-R® S.p. HiFi Cas9 Nuclease V3 | Integrated DNA Technologies | Cat# 1081060 |
| ChIP-qPCR primers | This paper | Table S1 |
| Alt-R® S.p. HiFi Cas9 Nuclease V3 | Integrated DNA Technologies | Cat# 1081060 |
| Lipofectamine 3000 | Invitrogen | Cat# L3000015 |
| Lipofectamine STEM Reagent | Invitrogen | Cat# STEM00001 |
| CloneR | STEMCELL Technologies | Cat# 05888 |
| DSP (dithiobis(succinimidyl propionate)), Lomanťs Reagent | Thermo Scientific | Cat# 22585 |
| Benzonase nuclease | Millipore | Cat# 70664 |
| EGS (ethylene glycol bis(succinimidyl succinate)) | Thermo Scientific | Cat# 21565 |
| DMP (dimethyl pimelimidate) | Thermo Scientific | Cat# 21666 |
| NEBNext High-Fidelity 2x PCR Mastermix | New England Biolabs | Cat# M0541L |
| Ampure RNAClean XP | Beckman Coulter | Cat# A63987 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Nucleospin RNA Extraction Kit | Machery Nagel | Cat# 740955 |
| Neon Transfection System | Invitrogen | Cat# MPK10096 |
| TruSeq® Stranded mRNA Library Prep | Illumina | Cat# 20020595 |
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat# E1910 |
| QIAquick PCR Purification Kit | Qiagen | Cat# 28104 |
| Qubit High-Sensitivity DS DNA Assay kit | lnvitrogen | Cat# Q32851 |
| SMARTer ThruPLEX DNA-Seq kit | Takara Bio | Cat# R400674 |
| Illumina Tagment DNATDE1 Enzyme and Buffer Kit | lllumina | Cat# 20034197 |
|
| ||
| Deposited data | ||
|
| ||
| Additional Supplementary Data | This paper | Mendeley:https://doi.org/10.17632/7jxtwztwxg.1 |
| RNA-Seq | This paper | GEO: GSE182842 |
| ATAC-Seq | This paper | GEO: GSE182842 |
| ChlP-Seq: Beta-catenin | This paper | GEO: GSE182842 |
| ChlP-Seq: SOX17 | This paper | GEO: GSE182842 |
| ChlP-Seq: SOX2 | This paper | GEO: GSE182842 |
| ChlP-Seq: TCF7 | This paper | GEO: GSE182842 |
| ChlP-Seq: TCF7L1 | This paper | GEO: GSE182842 |
| ChlP-Seq: TCF7L2 | This paper | GEO: GSE182842 |
| ChlP-Seq: LEF1 | This paper | GEO: GSE182842 |
| ChlP-Seq: BRG1 | This paper | GEO: GSE182842 |
| ChlP-Seq: H3K27ac | This paper | GEO: GSE182842 |
| ChlP-Seq: H3K4me1 | (Gifford etal., 2013) | GEO: GSM772971 |
| RNA-Seq: ectoderm | (Gifford et al., 2013) | GEO: GSM1112846, GSM1112844 |
| RNA-Seq: mesoderm | (Gifford et al., 2013) | GEO: GSM1112835, GSM1112833 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| hESC: H1 | WiCell | NIH:hESC-10–0043 |
| WT iPSC: iPSC72.3 | CCHMC Pluripotent Stem Cell Facility | N/A |
| SOX17KO iPSC | This paper | N/A |
| SOX17 Rescue iPSC | This paper | N/A |
| SOX2 CRlSPRi | (Mandegar et al., 2016) | N/A |
| HEK293T Parental | (Doumpas et al., 2019) | N/A |
| 4TCFKO HEK293T | (Doumpas et al., 2019) | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| RT-qPCR primers | This paper | Table S1 |
| ChIP-qPCR primers | This paper | Table S1 |
| hSoxI 7 HMG L1 (to amplify CRISPR/Cas9 targeted region): AGT GAC GAC CAG AGC CAG AC | This paper | N/A |
| hSoxI 7 HMG R1 (to amplify CRISPR/Cas9 targeted region): CAG AGC CTT AAG AAA GGACGTG | This paper | N/A |
| hSoxI 7 gRNAI L: CAC CTG AAA GCG TTC ATC GGC CGC | This paper | N/A |
| hSoxI 7 gRNAI R: AAA CGC GGC CGA TGA ACG CTT TCA | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSpCas9(BB)-2A-GFP (pX458) | Addgene | Cat#48138; RRID:Addgene_48138 |
| pGL4.23[luc2/miniP] | Promega | Cat# E841A |
| pcDNA/Myc DeltaN TCF4 | Addgene | Cat#16513; RRID:Addgene_16513 |
| pTET-Tight | (Fisher et al., 2017) | N/A |
| pTET-Tight-SOX17–3xFLAG | This paper | N/A |
| SOX17-V5 | (Sinner et al., 2007) | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| R | R Core Team (2020) | http://www.R-project.org/ |
| GraphPad Prism | www.graphpad.com | Version 9 |
| HOMER | (Heinz et al., 2010) | Version 4.4;http://homer.ucsd.edu/homer/ |
| FASTQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | Version 0.1.1.8 |
| Cutadapt | (Martin, 2011) | Version 3.2;https://cutadapt.readthedocs.io/en/stable/ |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | (Li et al., 2009) | http://www.htslib.org |
| Salmon | (Patro et al., 2017) | Version 1.1.0;https://combine-lab.github.io/salmon/ |
| DESeq2 | (Love et al., 2014) | Version 1.36.0;https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ggplot2 | Wickham (2016) | https://ggplot2.tidyverse.org/ |
| DiffBind | (Ross-Innes et al., 2012) | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| Pheatmap | https://github.com/raivokolde/pheatmap | Version 1.0.12 |
| Enhancedvolcano | https://github.com/kevinblighe/EnhancedVolcano | N/A |
| Bedtools | (Quinlan and Hall, 2010) | Version 2.27.1;https://bedtools.readthedocs.io/en/latest/ |
| Genrich | https://github.com/jsh58/Genrich | N/A |
| Nucleoatac | (Schep et al., 2015) | Version 0.3.4;https://nucleoatac.readthedocs.io/en/latest/ |
| Deeptools | (Ramirez et al., 2016) | Version 3.0.2;https://deeptools.readthedocs.io/en/develop/ |
| IGV | (Thorvaldsdottir et al., 2013) | Version 2.8.0;https://software.broadinstitute.org/software/igv/ |
| MEME | (Bailey et al., 2009) | https://meme-suite.org/meme/ |
| Gene Ontology | (Gene Ontology Consortium, 2021) | http://geneontology.org |
We determined which β-catenin binding events were associated with WNT-responsive transcription by performing RNA-seq analysis every 24 h on cultures treated with ACTIVIN and either a WNT agonist (CHIR99021) or WNT antagonist (C59) at different times (Figure S1F). Differential expression analysis of the WNT-ON and WNT-OFF states identified WNT-responsive transcripts at each day of differentiation (Figures S1G–S1I). Principal component (PCA) analysis showed that when WNT signaling was inhibited from the onset of differentiation, cells retained a pluripotency-like transcriptional signature, whereas inhibiting WNT from days 1 to 2 or days 2 to 3 resulted in an intermediate transcriptional program between mesendoderm and DE, indicating that continued WNT signaling is required for progressive DE specification (Figure S1J).
Integrating the ChIP-seq and RNA-seq data identified likely direct WNT targets and confirmed that the dynamic genomic binding of β-catenin was indeed associated with different transcriptional programs on different days (Figure 1E). Unsupervised hierarchical clustering of all WNT activated genes associated with β-catenin binding (Figures S1K–S1N) identified several temporally distinct groups of direct WNT target genes. These included “common” WNT-responsive genes activated by CHIR irrespective of the day of differentiation (n = 130) and genes specifically activated by CHIR on day 1 (n = 121), day 2 (n = 857), and day 3 (n = 338) (Figures S1K–S1N). Gene ontology (GO) enrichment analysis of “common” genes associated with β-catenin binding and CHIR-dependent expression at all days included well-known WNT targets such as SP5 (Huggins et al., 2017) and AXIN2 and were expectedly associated with biological processes like “WNT pathway” and “cell fate specification” (Figure 1K). In contrast, WNT/β-catenin targets on days 1 and 2 were enriched for terms related to processes like gastrulation, mesoderm, and endoderm formation. These included primitive streak genes such as FGF4, ISL1, and EPHA4 (Nakajima et al., 2006; Narkis et al., 2012) (Figures S1L and S1M). Day 3-specific WNT targets were associated with genes required to maintain epithelial integrity such as CTNND1 and ITGB1 (Brafman et al., 2013) (Figure S1N). Thus, progressive DE specification is associated with rapid relocalization of β-catenin to distinct lineage-specific genomic loci and dynamic target gene expression.
Chromatin accessibility is not sufficient to account for dynamic β-catenin binding
One possible explanation for the dynamic β-catenin binding was changes in the underlying chromatin landscape during differentiation (Chen and Dent, 2014). For example, day 1-specific β-catenin bound loci may only be accessible on day 1 but not on days 2–3. To test this hypothesis, we performed time course assay for transposase-accessible chromatin with sequencing (ATAC-seq) experiments and compared this to β-catenin binding (Figures 1K–1O). This revealed that although newly gained β-catenin binding sites were associated with increased chromatin accessibility, in general, most ß-catenin-bound loci were accessible with significant ATAC-seq signal at all days (Figures 1K–1O). This was exemplified in peaks enriched specifically at days 1 and 2, where β-catenin binding was lost on day 3 even though the chromatin remained accessible (Figure 1L and 1M). Thus, changes in chromatin accessibility are not sufficient to account for the dynamic shifts in β-catenin occupancy at line-age-specific WNT-responsive loci.
SOX17 and β-catenin co-occupy an increasing number of sites during DE differentiation
Next, we investigated the extent to which dynamic localization of β-catenin could be explained by underlying occupancy of TCFs or SOX17: the SOX TF regulating DE development (Zorn and Wells, 2009). RNA-seq showed that all four TCF/LEFs were expressed during DE differentiation (Figure 2A), so we performed ChIP-seq experiments every 24 h to profile the binding dynamics of each TCF and SOX17 and compared this to β-catenin. Peak overlap analysis revealed that 96% of β-catenin binding events (4,328/4,501) in day 1 mesendoderm cells could be accounted for by occupancy of at least one TCF, consistent with TCFs being predominant mediators of β-catenin binding as cells exit pluripotency (Moreira et al., 2017) (Figure 2B).
Figure 2. Dynamic co-localization of β-catenin with SOX17 and TCFs.

(A) Normalized RNA expression levels of SOX17, TCF7, TCF7L1, TCF7L2, and LEF1 during DE differentiation, n = 3 biological replicates.
(B–D) Venn diagrams showing ChIP-seq peak overlap of β-catenin, TCFs, and SOX17 during differentiation. TCF peaks are bound by at least one of the four TCFs. *Significant based on hypergeometric test, p < 0.0001..
(E) Heatmaps showing four categories of β-catenin, TCF, and SOX17 co-occupancy at day 3..
(D) Percentage of β-catenin peaks that overlap with TCFs and/or SOX17 at each day..
(G–I) Genome browser view of representative loci for each peak category. Numbers indicate ChIP-seq signal scaling..
(J–M) Enrichment of TCF and SOX DNA-binding motifs for each category of β-catenin peaks during differentiation. The circle size represents the proportion of peaks with TCF or SOX motifs. See also Figure S2, Tables S2 and S3.
Surprisingly, as differentiation progressed, we observed a progressive shift in co-localization of β-catenin from TCFs to SOX17. In day 2 cells, TCFs colocalized with β-catenin at 87% (16,272/18,608) of loci, but by day 3, TCFs accounted for only 57% of β-catenin binding (6,731/11,864) (Figures 2B–2F and S2A–S2F). In contrast, β-catenin/SOX17 co-occupied loci increased from 16% at day 1, to 63% at day 2, and 84% in day 3 DE cells (Figures 2B–2F and S2A–S2F). Focusing on day 3 DE, we identified four categories of peaks (Figure 2E): those co-occupied only by β-catenin and SOX17 but not TCFs such as BMP4 (4,224/11,684, 36%), loci bound by β-catenin/ SOX17/TCF such as TBX3 (5,544/11,684, 47%), peaks cobound by β-catenin and at least one TCF, but not SOX17 including LHX8 (1,187/11,684, 10%), and β-catenin peaks where we could not detect co-binding of either TCFs or SOX17 (n = 762/ 11,717, 7%) (Figures 2E–2I). De novo motif analysis revealed, other than TCF and SOX, an enrichment of endodermal TFs such as GATA, FOX, and SMAD motifs as expected for most AC-TIVIN/WNT-induced DE enhancers (Zorn and Wells, 2009) (Figure 2G). Time-resolved analyses of TCF and SOX motif enrichment showed a relative increase of SOX DNA-binding motifs correlating with increased SOX17/β-catenin co-occupancy (Figures 2J–2M). These data indicate that TCFs alone do not account for the diversity of β-catenin recruitment during progressive lineage specification and indicate that DE-specific β-catenin binding events are associated with increased SOX17co-occupancy.
SOX17 is required to recruit β-catenin to DE-specific WNT-responsive enhancers
We next sought to assessed whether SOX17 was required to recruit β-catenin to chromatin. Therefore, we generated a CRISPR-mediated SOX17−/− homozygous null mutant iPSC line (Figure S3A). qRT-PCR, immunostaining, and western blots confirmed a loss of SOX17 mRNA and protein in SOX17 knockout (KO) cells, and that loss of SOX17 did not affect levels of β-catenin, TCF7, TCF7L1, or TCF7L2, but LEF1 expression was modestly increased (Figures 3C–3G). Immunostaining and RNA-seq analysis confirmed that DE specification was compromised in SOX17KO cells treated with ACTIVIN and CHIR for day 3 differentiation (Figures S3D, S4A, and S4B), and this was independently validated using a SOX17-CRISPRi cell line (see key resources table for additional supplementary data). PCA analysis showed that day 3 SOX17KO cells were similar to day 1 mesendoderm cells (Figure S4C). Accordingly, mesoderm markers such as TBXT, TBX6, and FOXF1 were upregulated upon loss of SOX17 (Figure S4A). Indeed, GO analysis showed that transcripts downregulated in SOX17KO (n = 1,613) were enriched for biological processes relevant to endoderm specification, whereas transcripts upregulated by the loss of SOX17 (n = 1261) were enriched for mesoderm patterning processes (Figures S4A and S4B).
Figure 3. SOX17 is required to recruit β-catenin to chromatin.

(A–C) Heatmap and metaplots of β-catenin ChIP-seq signal in WT, SOX17KO or SOX17KO + SOX17 rescue cells at day 3. Differential binding analysis identified three categories of β-catenin binding: (A) β-catenin peaks lost in SOX17KO cells, (B) peaks unchanged between WT and SOX17KO cells, and (C) new β-catenin peaks gained in SOX17KO cells (log2FC > 1, p-adj < 0.05). Tables show the percentage of each peak set bound by β-catenin, SOX17, or any TCF (ND = not determined).
(D–F) Genome browser view of β-catenin and TCF7L2 binding at representative loci in WT, SOX17KO, and SOX17KO + SOX17 rescue cells. Numbers indicate ChIP-seq signal scaling.
(G–H) Integration of day 3 RNA-seq (n = 3) and ChIP-seq (n = 2) datasets to identify genes co-bound and coregulated by β-catenin and SOX17. *Significant overlap based on hypergeometric test, ChIP-seq overlap: p = 1.03 3 10–553; RNA-seq overlap: p = 2.07 × 10−61.
(I) Diagram showing proportion of coregulated peaks that are solely bound by SOX17/β-catenin or bound by SOX17/β-catenin/TCFs.
(J) Stacked bar graphs showing the percentage of day 3 genes co-bound and coregulated by SOX17/β-catenin (activated, Act; repressed, Rep) that are enriched in endoderm, mesoderm, or ectoderm based on Gifford et al. (2013).
(K) Model summarizing SOX17/β-catenin interactions during the DE-mesoderm cell fate decision. See also Figures S3 and S4, Tables S3 and S4.
We next performed β-catenin ChIP-seq on day 3 wild-type (WT) and SOX17KO cells, identifying 13,131 peaks bound by β-catenin in WT and 41,058 β-catenin peaks in SOX17KO cells. Differential binding analysis revealed three distinct categories of β-catenin binding: (1) β-catenin peaks lost in SOX17KO such as that associated with BMP7 (n = 4,337), (2) β-catenin peaks that remained unchanged in WT and SOX17KO like DKK1 (n = 3,894), and (3) a surprisingly large number of β-catenin peaks gained in SOX17KO cells such as near the mesoderm-specific gene MEOX1 (n = 24,096) (Figures 3A–3F and S4D). To test the specificity of the CRISPR-mutagenesis, and to demonstrate the requirement of SOX17 in orchestrating genome-wide β-catenin binding patterns, we introduced a doxycycline (dox)-inducible SOX17 transgene into the SOX17KO cell line. Addition of dox from days 1 to 3 induced SOX17 expression comparable to WT levels (Figures S3B and S3C), which rescued DE specification based on immunostaining and RNA-seq (Figures S3D and S4C). ChIP-seq analysis confirmed that adding back SOX17 to the SOX17KO cells largely restored the WT genomic β-catenin binding pattern; β-catenin peaks lost in the SOX17KO were restored, while de novo β-catenin binding events gained in the SOX17KO were lost upon SOX17 re-expression (Figures 3A–3F and S4F–S4H).
We next assessed the extent to which SOX17-dependent changes in β-catenin binding correlated with TCF co-occupancy by performing ChIP-seq for all TCFs in both WT and SOX17KO cells. We then quantified SOX17 and TCF enrichment at the three groups of β-catenin peaks by differential binding analysis (Figures 3A–3C). This revealed that while 96% (4,136/4,337) of β-catenin binding events lost in SOX17KO cells were co-occupied by SOX17, only 30% (n = 1,318/4,337) were co-bound by TCFs. Thus, the majority of β-catenin peaks lost in the SOX17KO cells were not significantly bound by TCFs. In contrast, 54% (n = 2,088/3,894) of β-catenin peaks that were unchanged between WT and SOX17KO were co-occupied by TCFs (and SOX17) in WT cells, and this increased to 90% TCF binding in SOX17KO. The striking increase in de novo β-catenin binding events in SOX17KO cells was also associated with a dramatic increase in TCF-co-occupancy from 18% (n = 4,373/24,096) in WT DE to 83% (n = 20,008/24,096) in SOX17KO cells as exemplified by the mesodermal MEOX1 enhancer (Figures 3C–3F). Consistent with this, GO analysis indicated that genes associated with lost β-catenin peaks were enriched for endoderm organogenesis, whereas genes associated with gained β-catenin peaks were enriched for processes such as cardiac mesoderm and epithelial to mesenchymal transition (Figure S4E). Moreover, de novo motif analysis of lost β-catenin peaks showed an enrichment of SOX motifs, while gained β-catenin peaks were enriched for TCF, TBX, and SMAD DNA-binding motifs. This is in line with the known role of TBXT/SMAD interactions in delineating endoderm versus mesoderm gene regulatory networks (Faial et al., 2015). Collectively, these data demonstrate that SOX17 is required to recruit β-catenin to specific genomic loci and that loss of SOX17 leads to a widespread relocalization of β-catenin binding and the activation of an alternative mesoderm-like transcriptome, likely due to the recruitment of β-catenin to different enhancers by TCFs.
Integrating the ChIP-seq and RNA-seq datasets, we identified 664 genes (corresponding to 1,329 peaks) that were co-occupied and coregulated by SOX17 and β-catenin (Figures 3G, 3H, and S4K) in day 3 DE. These SOX17/β-catenin co-bound peaks were enriched for H3K4me1 and H3K27ac, histone marks indicative of poised and/or active enhancers (Figure S4I) (Gifford et al., 2013). Of these SOX17/β-catenin coregulated enhancers, 41% had little to no evidence of TCF binding, while the remaining 59% were co-occupied by SOX17, β-catenin, and TCF (Figures 3I and S4J). Further analysis revealed that the vast majority of the direct SOX17/β-catenin target genes that were activated by SOX17 had endoderm-enriched expression, whereas a substantial number of the direct SOX17/β-catenin target genes repressed by SOX17 were normally expressed in mesectodermal lineages (Figure 3J; see STAR Methods). This was consistent with our recent finding in Xenopus gastrulae that Sox17 promotes endoderm fate while repressing mesectoderm identity (Mukherjee et al., 2020).
Together these data demonstrate that SOX17 is required to recruit β-catenin to a subset of endoderm-specific WREs, many of which have no evidence of TCF binding. On the other hand, β-catenin/TCF interactions tend to promote an alternate mesodermal identity, suggesting that competition between SOX17 and TCFs for recruitment of β-catenin regulates this cell fate decision (Figure 3K).
SOX17 is required to establish a permissive chromatin landscape at a subset of TCF-independent DE enhancers
There is evidence that SOX TFs can act as pioneering factors by directly engaging nucleosomes to regulate chromatin accessibility (Meers et al., 2019; Soufi et al., 2015), which might explain the loss of β-catenin binding in SOX17KO cells. To address this possibility, we performed ATAC-seq in day 3 WT and SOX17KO cells. Differential peak analysis revealed 13,737 genomic regions with significantly increased accessibility based on ATAC-seq, 29,580 regions that were significantly less accessible in SOX17KO cells, and 41,124 loci where accessibility was not significantly altered (Figure S5A). We focused our analysis on those enhancers that lost β-catenin binding in SOX17KO cells and were enriched for SOX17 but not TCF occupancy (n = 4,337); we termed these “TCF-independent” enhancers (Figure 3A). Of these, 36% enhancers (1,577/4,337) displayed decreased chromatin accessibility in SOX17KO cells (termed Class I enhancers), exemplified by the SALL1 enhancer, while the others did not display any significant SOX17-dependent changes in accessibility, such as the PRIMA1 enhancer (termed Class II enhancers) (Figures 4A and 4F). Nucleoatac software (Schep et al., 2015) showed the expected dip in nucleosome occupancy at ATAC peak centers for both Class I and Class II enhancers in WT cells. However, in SOX17KO cells, there was a significant increase in nucleosomes occupancy at Class I but not Class II enhancers, indicating that SOX17 is required for chromatin accessibility at some, but not all of the TCF-independent enhancers (Figures 4B and 4C).
Figure 4. Chromatin accessibility only partially accounts for loss of β-catenin binding in SOX17KO cells.

(A) Heatmaps showing ATAC-seq signal (n = 2) in WT and SOX17KO cells for two classes of SOX17-dependent β-catenin bound peaks; Class I enhancers with reduced accessibility in SOX17KO and Class II enhancers where accessibility is unchanged.
(B) Metaplots showing nucleosome occupancy signals and (C) quantification of ATAC-seq read densities in WT (purple) and SOX17KO (pink) cells for Class I and II enhancers. p values based on Wilcoxon rank-sum test.
(D) Metaplots showing average H3K27ac ChIP-seq signal for Class I and II enhancers and (E) violin plots quantifying tag density. p values based on Wilcoxon rank- sum test.
(F) Genome browser view showing representative Class I and Class II enhancers.
(G) Model of SOX17-dependent β-catenin recruitment. Dotted lines in (C) and (E) represent the tag count threshold for significant ATAC-seq peaks. See also Figure S5.
Despite this difference in SOX17-regulated chromatin accessibility, there was a significant decrease of the histone mark H3K27ac (Creyghton et al., 2010) in both Class I and Class II enhancers, indicating reduced transcriptional activity (Figures 4D and 4E). This indicates that loss of chromatin accessibility in SOX17KO cells cannot alone account for the loss of β-catenin chromatin recruitment and reduced activity of these WREs. A similar ATAC-seq analysis of peaks with unchanged β-catenin binding in WT and SOX17KO showed few, if any, changes in chromatin accessibility or H3K27ac deposition (Figures S5B–S5F). In contrast, 33% (7,908/24,096) of the loci that gained β-catenin and TCF binding in SOX17KO cells exhibited significantly increased chromatin accessibility and elevated H3K27ac (Figures S5G–S5K), consistent with activation of alternative transcriptional programs.
Collectively, these results indicate that SOX17 regulates lineage-specific WNT-responsive transcription both by regulating chromatin accessibility and by recruiting β-catenin to a subset of TCF-independent endodermal enhancers (Figure 4G).
Lineage-specific recruitment of β-catenin is a general feature of SOX TFs
To evaluate whether other SOX TFs also have the ability to regulate β-catenin genomic recruitment and lineage-specific WNT-responsive transcription, we examined neuromesodermal progenitors (NMPs) where antagonistic interactions between SOX2, WNT/β-catenin, and TBXT control neural versus mesoderm cell fate decision (Gouti et al., 2014; Koch et al., 2017). We directed the differentiation of hPSCs toward the NMP lineage by addition of FGF8b and the WNT agonist CHIR99021 as previously reported (Lippmann et al., 2015), generating relatively pure (>70%) populations of NMPs co-expressing SOX2 and TBXT after 3 days of culture (Figures 5A and 5B). To knock down (KD) SOX2, we used a dox-inducible CRISPRi iPSC line where deactivated Cas9 fused to the KRAB repressor domain is targeted to the SOX2 promoter to repress its expression (Mandegar et al., 2016). We treated cultures +/—dox after exit from pluripotency but before NMP specification (Figure 5A). Immunostaining and qRT-PCR confirmed loss of SOX2 in +dox SOX2KD cells and showed that there was no change in β-catenin, TCF7, TCF7L1, or TCF7L2 while LEF1 protein levels were moderately increased (Figures 5B and S6B–S6F).
Figure 5. SOX2 is required for β-catenin recruitment in neuromesodermal progenitors.

(A) Schematic of NMP differentiation in SOX2KD CRISPRi cells.
(B) Immunostaining for SOX2, TBXT, and active β-catenin on day 3 of NMP differentiation (scale bar represents 100 mm, n = 3).
(C and D) Heatmap and metaplots of β-catenin ChIP-seq signal in WT and SOX2KD NMP cells at β-catenin peaks (n = 2) (C) that are lost upon SOX2 knockdown and (D) peaks that are unchanged. Table shows the percentage of peaks bound by β-catenin, SOX2, or TCF (either TCF7L1, LEF1, or both).
(E) Integration of RNA-seq (n = 3) and ChIP-seq datasets. *Significant overlap based on hypergeometric test, ChIP-seq peak overlap: p = 1.07 × 10−459; RNA-seq overlap: p = 5.64 × 10−352.
(F and H) Metaplots showing nucleosome occupancy from ATAC-seq data at (F) β-catenin peaks lost in SOX2KD or (H) β-catenin peaks that are unchanged.
(G and I) Violin plots quantifying ATAC-seq read densities. p values based on Wilcoxon rank-sum test. Dotted lines in (G) and (I) represent tag count threshold for significant ATAC-seq peaks.
(J) Genome browser view of representative β-catenin peaks of each category.
(K) Model of SOX2 and WNT/β-catenin interactions during NMP specification. See also Figure S6 and Table S5.
ChIP-seq experiments in SOX2KD NMP cells +/−dox revealed that in WT conditions (–Dox), 92% (5,664/6,137) of β-catenin bound genomic loci were also occupied by SOX2 (Figure 5E). Differential peak analysis of WT versus SOX2KD NMP cells revealed a significant SOX2-dependent loss of β-catenin binding at 4,946 loci, while β-catenin binding was unchanged at 2,711 loci, with only a gain of 214 new β-catenin peaks in SOX2KD (Figures 5C, 5D, and S6H). To identify SOX2 and WNT target genes in NMPs, we performed RNA-seq on WT and SOX2KD cells as well as on cultures where CHIR was replaced with C59 to inhibit WNT signaling (Figures S6A–S6C). Differential expression analysis identified 835 SOX2-regulated and 2,862 WNT-regulated transcripts. Integrating the ChIP-seq and RNA-seq data identified 209 putative enhancers, corresponding to 119 genes that were coordinately co-occupied and coregulated by SOX2 and β-catenin (Figure 5E). Consistent with an antagonistic relationship between SOX2 and WNT/β-catenin in NMP development, 42% of co-occupied and coregulated genes (50/119) were repressed by SOX2 but WNT activated, whereas 29% (34/119) were SOX2 activated and WNT repressed (Figure S6G). GO analyses of the SOX2 and WNT-regulated genes as well as the peaks associated with SOX2-dependent β-catenin binding were consistent with the idea that SOX2 promotes neural fate in NMPs, while WNT favors mesoderm differentiation (Figures S6D, S6E, and S6I).
ChIP-seq for TCF7L1 and LEF1, the two TCFs most highly expressed in NMPs, revealed that 50% (2,483/4,946) of the loci that lost β-catenin peaks in SOX2KD cells were also co-occupied by TCFs such as the mesoderm marker MESP1, while the other half had no evidence of TCF7L1 or LEF1 binding such as the ectoderm gene TSHZ3 (Figures 5E–5J). We then assessed chromatin accessibility at SOX2-dependent β-catenin loci but did not observe any appreciable differences in WT and SOX2KD cells (Figures 5F–5I).
Collectively, these genomic analysis of SOX2/β-catenin in NMPs and SOX17/β-catenin in DE demonstrate that SOX TFs are required to regulate β-catenin recruitment and lineage-specific WREs, and that antagonistic activities between SOX and TCF TFs drive WNT-mediated cell fate choice (Figure 5K).
SOX17 is sufficient to recruit β-catenin to chromatin in TCF-null cells
Our data indicate that SOX17 and SOX2 are required to recruit β-catenin to thousands of lineage-specific enhancers that have little if any detectable TCF binding. However, it was formally possible that a low level of TCF at these enhancers was still required to recruit β-catenin. To test this, we ectopically expressed a SOX17-V5 construct into human epithelial kidney (HEK) 293T cells where all four TCF/LEF genes have been deleted (4TCFKO) (Doumpas et al., 2019) (Figure 6A). Optimizing transfections, we obtained a level of SOX17-V5 expression that was at levels comparable to endogenous SOX17 in hPSC-DE cells (Figure 6B). We then performed β-catenin and SOX17 ChIP-seq on WT, 4TCFKO, and 4TCFKO + SOX17 cells, +/− the WNT agonist CHIR99021. WT cells had 4,115 WNT-dependent β-catenin binding peaks, 82% (3,389/4,115) of which were lost in TCF-null cells, as previously reported (Doumpas et al., 2019) (Figure 6C). However, we detected 5,233 β-catenin peaks in WNT-stimulated 4TCFKO cells expressing SOX17-V5, 53% (2,787/5,233) of which were not present in WT cells (Figures 6C–6E). Of these novel β-catenin bound loci, 87% (2,436/2,787) were co-occupied by SOX17 in 4TCFKO cells, as exemplified by novel SOX17/β-catenin binding at the SOX5 and ATXN1 loci in 4TCFKO (Figures 6E and 6F). Comparing this to our human DE data, we found that 191 genes associated with SOX17/WNT-dependent β-catenin binding in 4TCFKO cells were coregulated by WNT and SOX17 in DE (Figure 6D). Despite being bound by SOX17 and β-catenin, most of these DE genes were not transcribed in 293T cells, likely due to the lack of ACTIVIN/SMAD stimulation, which is essential for DE gene expression. These data demonstrate that SOX17 is sufficient to recruit β-catenin to lineage-specific enhancers independent of TCFs.
Figure 6. SOX17 is sufficient recruit β-catenin to chromatin in the absence of TCFs.

(A) Experimental design.
(B) Western blots comparing SOX17 transfection amounts in HEK cells to hPSC-derived DE.
(C) Venn diagrams showing overlap of β-catenin ChIP-seq peaks in WT + WNT, 4KO + WNT and 4KO + SOX17 + WNT cells (n = 2).
(D) Venn diagram showing overlap of the number of genes associated with SOX17/β-catenin binding in 4KO + SOX17 + WNT cells and genes regulated by SOX17/ WNT in DE.
(E) Heatmap showing β-catenin (purple) and SOX17 (green) ChIP-seq signal in control, WT-WNT, WT + WNT, 4KO + WNT, WT + SOX17 + WNT, and 4KO + SOX17 + WNT cells at three categories of β-catenin peaks: those bound only in WT + WNT 293T cells, those bound both in WT and 4KO + WNT + SOX17 cells, and those that are newly bound in 4KO + SOX17 + WNT cells.
(F) Genome browser view of representative β-catenin peaks.
(G) Schematic of enhancer luciferase constructs with β-catenin/SOX17-dependent enhancers (CXCR4 and SALL1) and β-catenin/TCF-dependent enhancers (SP5 and AXIN2).
(H–I) Relative luciferase activity of constructs containing wild-type sequences (gray), sequences with SOX-binding sites mutated (pink), or with TCF-binding sites mutated (blue) in (H) WT or 4KO 293T cells +/— SOX17-V5 and (I) day 3 WT DE and SOX17KO cells, +/— WNT3A and +/— dnTCF7L2. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 and ns = not significant based on two-tailed Student’s t test. See also Table S6 and File S1.
SOX17 and β-catenin coordinately activate TCF-independent WNT-responsive enhancers
To further understand the TCF-independent activity of SOX17/ β-catenin regulated enhancers, we focused on a −60-kb CXCR4 and a —2.2-kb SALL1 putative WRE. CXCR4 is a well-established DE marker co-expressed with SOX17 (Loh et al., 2014; Nair and Schilling, 2008), while SALL1 is a direct SOX17 target in the uterine epithelium (Wang et al., 2018). In our analysis, CXCR4 and SALL1 were coregulated by SOX17/WNT, and the putative WREs were co-occupied by SOX17 and β-catenin, with little evidence of TCF binding. DNA sequence analysis confirmed that both WREs had several SOX sites but no TCF-binding sites (Table S1). We also assessed WREs of the well-defined TCF/ β-catenin target genes: SP5 (Fujimura et al., 2007) and AXIN2 (Leung et al., 2002). We cloned each enhancer into luciferase reporter constructs as well as versions where the putative SOX17 or TCF DNA-binding sites were mutated (ΔSOX or ΔTCF). We transfected the reporters into WT and 4TCFKO HEK293T cells +/−CHIR and +/−SOX17. As expected, the SP5 and AXIN2 reporters exhibited robust activity only in WT cells upon WNT stimulation, which was abolished in DTCF constructs. Consistent with previous reports (Sinner et al., 2007), SOX17 overexpression antagonized the WNT-dependent activity of SP5 and AXIN2 WREs in WT cells, an effect that was abolished by deletion of SOX17 sites in the case of SP5. On the other hand, the CXCR4 and SALL1 reporters were activated in both WT and 4TCFKO only when the cells were both stimulated with CHIR and cotransfected with SOX17, and this activation was abolished with the ΔSOX reporters. This demonstrates that the CXCR4 and SALL1 enhancers are bona fide SOX-dependent WREs (Figures 6G and 6H).
We repeated these luciferase assays in endogenous contexts in hPSC-derived WT and SOX17KO DE, with or without a dominant-negative isoform of TCF7L2 (dnTCF7L2). The well-characterized dnTCF7L2 lacks the N-terminal β-catenin binding domain and acts as a constitutive WNT repressor preventing recruitment of β-catenin to classic WREs (Korinek et al., 1997). As TCF7L2 is the most abundant TCF in DE cells, we reasoned that dnTCF7L2 overexpression would inhibit the activity of TCF-dependent but not TCF-independent enhancers. All four reporter constructs required WNT treatment to be expressed in WT DE, confirming that they are indeed WREs. As expected, the dnTCF7L2 suppressed the WNT stimulated activity of the SP5 and AXIN2 reporters in DE, as did mutation of the TCF DNA-binding sites. In contrast the dnTCF7L2 did not impact the WNT-dependent activity of the CXCR4 and SALL1 enhancers, but their expression was largely abolished in SOX17KO cells or by mutation of the SOX sites in WT cells. (Figure 6I).
Together, these data demonstrate that these SOX17/ß-catenin-bound enhancers are bona fide SOX-dependent, TCF-independent WREs and that competitive TCF and SOX interactions likely contribute to the specificity of WNT-responsive transcription.
SOX17 assembles a WNT-responsive transcription complex at TCF-independent enhancers
To better understand how SOX17 and β-catenin interact at WREs, we performed reciprocal coimmunoprecipitation (coIP) experiments from DE cultures treated with benzonase; this precludes the possibility that any observed interactions are simply due to proximity on DNA. We detected a physical interaction between SOX17 and β-catenin at endogenous levels in DE cells (Figures 7A and 7B). As expected, TCF7L2 and β-catenin also co-precipitated in both WT and SOX17KO cells (Figure S7A). Next, we performed reciprocal ChIP-reChIP-qPCR experiments, further confirming that SOX17 and β-catenin directly interact at SOX-dependent but not TCF-dependent enhancers (Figures 7C and S7B).
Figure 7. SOX17 assembles a TCF-independent WNT-enhanceosome complex.

(A and B) Western blots of co-immunoprecipitations from DE (n = 2) showing that BRG1 and SMC1 physically interact with (A) SOX17 and (B) β-catenin.
(C) Representative ChIP-reChIP-qPCR, showing the relative enrichment at SOX-dependent (CXCR4 and SALL1) or TCF-dependent (SP5 and NKD1) enhancers after a first β-catenin ChIP followed by reChIP with either SOX17, TCF7L2, or IgG, across three biological replicates.
(D and E) BRG1 ChIP-seq in day 3 WT DE or SOX17KO cells. (D) Metaplots showing average peak intensity and (E) violin plots quantifying BRG1 tag intensity at “TCF-independent” enhancers that exhibit SOX17-dependent β-catenin binding. p values based on Wilcoxon rank-sum test, p < 2.2 × 10–16. Dotted line in (E) represents the tag count threshold for significant peak calling.
(F–I) ChIP-qPCR (n = 3) of (F) p300, (G) SMC1, (H) BCL9, and (I) PYGO2 showing relative fold enrichment at SOX-dependent or TCF-dependent enhancers in day 3 WT DE or SOX17KO cells. Two-tailed Student’s t test. ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
(J). Model depicting SOX17-dependent assembly of a WNT-responsive transcription complex at TCF-independent DE enhancers. See also Figure S7.
We next tested the hypothesis that SOX-dependent WREs can serve as a scaffold for recruitment of a WNT-enhanceosome complex. CoIP assays demonstrated that in DE cells both β-catenin and SOX17 physically interact with the chromatin remodeler BRG1 and the cohesin subunit SMC1 (Figures 7A and 7B), known to be critical for transactivation of TCF/β-catenin target genes (Kagey et al., 2010; Kim et al., 2006). Moreover, BRG1 ChIP-seq revealed a substantial loss of BRG1 binding at TCF-independent enhancers in SOX17KO cells relative to WT (Figures 7D and 7E). We then performed ChIP-qPCR in WT and SOX17KO cells for known components of the WNT-enhanceosome including BCL9 and PYGO (Carrera et al., 2008; Kramps et al., 2002), the histone acyteltransferase p300 (Hecht, 2000), the cohesin subunit SMC1, cohesin loading protein NIPBL (Estará s et al., 2015), and the mediator subunit MED12 (Kim et al., 2006) (Figures 7F–7I, S7D, and S7E). In each case, SOX17 was required for efficient recruitment to the TCF-independent, SOX17/β-catenin-regulated DE enhancers CXCR4 and SALL1. In contrast, SOX17 was not required for the recruitment of these coactivators to the TCF-dependent WREs SP5, NKD1, and CDX2 (Figures 7F–7I, S7D, and S7E). qRT-PCR confirmed that expression levels of these coactivators did not change in SOX17KO cells, suggesting that SOX17 recruits them to enhancers (Figure S7C).
Collectively, our data show that SOX17 is required to recruit β-catenin to a subset of lineage-specific enhancers and assemble a TCF-independent transcription complex to activate endoderm-specific WNT-responsive transcription (Figure 7J).
DISCUSSION
How β-catenin is recruited to distinct enhancers to regulate lineage-specific WNT target genes was previously unclear. We demonstrate that SOX TFs are lineage-specific regulators of WNT-responsive transcription. During early germ layer specification, the recruitment of β-catenin to lineage-associated enhancers is dynamic and regulated by interactions of TCF and SOX TFs. Our data indicate that SOX17 in DE and SOX2 in NMPs are required to recruit β-catenin to specific enhancers, many of which have no evidence of TCF binding. While SOX TFs modulate chromatin accessibility at some enhancers, this alone cannot account for SOX-dependent β-catenin chromatin binding. We confirmed that SOX17 was sufficient to recruit β-catenin to specific chromatin loci in cells that lack all four TCF proteins. Analysis of histone marks and functional reporter assays confirmed that these SOX17/ß-catenin-bound enhancers are bona fide SOX-dependent, TCF-independent WREs. Finally, we show that SOX17 and WNT/β-catenin activate these WREs by recruiting a WNT-enhanceosome complex with BCL9, PYGO, and transcriptional coactivators p300, BRG1, MED12, and SMC1.
While the central role of TCFs in mediating WNT-responsive transcription is not in doubt, the notion that alternative TFs can, in some contexts, regulate WNT-responsive transcription has been controversial (Schuijers et al., 2014). We show that β-catenin binding and transcription of early mesendoderm or “universal” WNT target genes like AXIN2 are mediated primarily by TCFs. In contrast, in DE or NMP cells, SOX17 and SOX2 account for a major proportion of β-catenin binding and WNT-regulated transcription. One possibility is that while β-catenin/TCF interactions are predominant in well-known processes like cell proliferation, β-catenin/SOX or β-catenin/SOX/TCF interactions might be more prevalent during cell fate decisions where reiterative WNT signaling must dynamically re-establish transcriptional programs. Indeed, since several SOX genes are direct WNT targets (Kormish et al., 2010), it is possible that they act as feedback modulators tuning transcriptional response as cells progressively differentiate from prolonged WNT exposure.
Although we have focused on the most novel subset of enhancers where SOX TFs recruit β-catenin independently of TCFs, it is likely that the interplay between SOX and TCF TFs is complex and involves both cooperative and competitive interactions. The observation that many WNT-responsive genomic loci gain de novo TCF/β-catenin binding upon loss of SOX TFs suggests that SOX and TCF TFs can competing for a limited pool of nuclear β-catenin. It is also possible that the amount of β-catenin recruited to distinct WREs, and hence the level of transcription, is influenced by mass action and the number of SOX versus TCF DNA-binding sites at a specific enhancer. The fact that many WREs are co-occupied by SOX, TCF, and β-catenin also suggests that all three proteins might interact cooperatively. In vitro protein binding assays using recombinant Sox17, Tcf7l2, and β-catenin show that they can form a trimeric complex (Sinner et al., 2007). Although we did not explicitly test this possibility, we did not detect a physical interaction between endogenous SOX17 and TCF7L2 in DE. Sequence analysis of enhancers co-occupied by SOX17 and TCFs showed that their binding sites are often >50 bp away, so they are unlikely to form heterodimers. However, since they are both HMG-box TFs that bend DNA (Billin et al., 2000; Hou et al., 2017), it is conceivable that despite being distant to each other on linear DNA, SOX and TCF could still physically interact with the same β-catenin molecule.
Another possibility is that SOX TFs might recruit coactivators or corepressors to potentiate or antagonize β-catenin/TCF interactions or affect their stability, nuclear localization, or ability to interact with one another or p300 (Gao et al., 2014). For example, SOX9 enhances the phosphorylation and turnover of nuclear β-catenin in chondrocytes (Akiyama et al., 2004; Yano et al., 2005). While we did not observe differences in nuclear β-catenin levels between WT and SOX17KO or SOX2KD cells, it is possible that SOX TFs modulate WNT responses by recruiting enzymes that regulate β-catenin methylation or acetylation (Hoffmeyer et al., 2017), such as Kdm2a/b that can demethylate β-catenin to regulate its nuclear stability (Lu et al., 2015).
In general, SOX TFs acquire DNA-binding specificity by forming heterodimers with other TFs (Kondoh and Kamachi, 2010). Motif analyses of the SOX17/β-catenin peaks also showed an enrichment of GATA, FOXA, and SMAD motifs. This is not surprising since SOX17, GATA4–6, and FOXA TFs interact in a positive feedback loop downstream of NODAL/SMAD2 to promote each other’s expression, define the chromatin landscape, and activate the DE transcriptome (Zorn and Wells, 2009). It is possible that SOX-GATA or SOX-FOXA interactions provide additional DNA sequence specificity to SOX-dependent β-catenin recruitment at select WREs. Consistent with this idea, β-catenin/TCF7L2/GATA4 and TCF7L2/GATA1 interactions have been reported in cardiac and hematopoietic lineages (Iyer et al., 2018; Trompouki et al., 2011).
In summary, we show that SOX TFs are context-dependent regulators of WNT-responsive transcription during early cell fate decisions. SOX/WNT interactions are also widely implicated in cancer; for example, SOX17 expression levels are often anti-correlated with WNT activity in the breast, skin, and brain tumors (Moradi et al., 2017; Zhao et al., 2016; Zhou et al., 2014). Our data point to opportunities for selectively targeting WNT responses in cancer. It should be possible, in principle, to identify compounds that selectively inhibit or promote SOX/β-catenin but not TCF/β-catenin interactions. Indeed, the SOX17/β-catenin targets CXCR4 and SALL1 have oncogenic activity, are overexpressed in some cancer contexts, and are currently targets of druggable anticancer therapeutics (Chi et al., 2019; Song et al., 2021). Given that most, if not all cell types, express at least one of the 20 SOX TFs in the human genome, we anticipate that our results may have previously unappreciated widespread implications for WNT-responsive gene regulation across developmental and disease contexts.
Limitations of the study
Consistent with our previous findings in Xenopus gastrulae, we show that SOX17 activates the endoderm gene regulatory network while suppressing mesectoderm fates. However, future studies will be important to determine how SOX TFs recruit β-catenin during embryonic development in vivo. Our data indicate that SOX17 and TCFs are likely to compete for finite amounts of nuclear β-catenin. Although we did not explicitly test this possibility, future genomic and biochemical experiments with careful titration of SOX and TCF levels will be important to determine how different SOX TF families interact with β-catenin/TCF. Further investigations into the mechanisms through which SOX and TCFs interact to control the genomic specificity of β-catenin in different cellular contexts might open the possibility of targeting ß-catenin-SOX interactions for therapeutic purposes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aaron Zorn (aaron. zorn@cchmc.org).
Materials availability
All cell lines and reagents generated in this study will be made available upon request to the lead contact.
Data and code availability
Source data related to Figures 1 and 3 are available on Mendeley at https://doi.org/10.17632/7jxtwztwxg.1 The datasets generated during this study are available at the Gene Expression Omnibus (GEO) under the accession number GSE182842. The following public datasets were downloaded from GEO and reanalyzed: GSM772971, GSM1112846, GSM1112844, GSM1112835 and GSM1112833. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contactupon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Pluripotent stem cell culture
Human embryonic stem cell line WA01 (H1) was purchased from WiCell and induced pluripotent stem cell line iPS72.3 was obtained from CCHMC Pluripotent Stem Cell Facility. The CRISPRi-SOX2 line was a kind gift from Dr. Bruce Conklin (Gladstones Institutes, UCSF). hESCs and iPSCs were maintained in feeder-free cultures. Cells were plated on hESC-qualified Matrigel (Corning; 354277) and maintained on mTESR1 (StemCell Technologies; 85851) media at 37°C with 5% CO2. Media was changed daily, and cells were routinely passaged every 4 days using ReleSR (StemCell Technologies; 05872). CRISPRi-SOX2 cells were plated on vitronectin-coated (ThermoFisher; A14700) plates and maintained in Essential 8 Medium (Gibco; A1517001). These lines were routinely passaged every 3–4 days using Versene (ThermoFisher; 15040066). All lines were routinely screened for differentiation and tested for mycoplasma contamination.
HEK293T cell culture
Cells were cultured in high-glucose DMEM (Gibco; 41966029) with 10% FBS (Gibco). For SOX17 overexpression experiments, Lipofectamine 3000 was used to transfect 20ug of V5 epitope-tagged wild type SOX17 in WT or 4KO 293T cells per manufacturer’s instructions. Cells were harvested 48hours post transfection for downstream analysis. The V5-tagged SOX17 has previously been described (Sinner, 2004). For WNT activation experiments, 10um CHIR99021 (R&D Systems; 4423) was added to fresh media and cells were harvested 24 h later.
METHOD DETAILS
Generation of SOX17KO cell line
gRNA Validation: Two CRISPR/Cas9 guide RNAs targeting the first exon of the SOX17 gene were cloned into pX458 (Addgene 48138) and validated in HEK293T cells (ATCC CRL-3216) by the Transgenic Animal and Gene Editing core at CCHMC. The CRISPR targeted region was amplified with Phusion Polymerase (ThermoFisher; F531) and each amplicon was digested with T7 Endonuclease I (NEB; M0302S). Digested amplicons were run on agarose gels to quantify relative gRNA activity. Nucleofection: RNP complex assembly of the validated gRNA was performed by combining 20ug Alt-R® S.p. HiFi Cas9 Nuclease V3 (IDT; 1081060) with 16ug sgRNA (Synthego) in vitro for 45 min at room temperature. The RNP complex was electroporated into parental iPS 72.3 cells using a Lonza 4D Nucleofector. Isolated clones were lysed and amplified using Phusion polymerase and clones of interest were submitted for Sanger sequencing to the CCHMC DNA Sequencing Core.
Generation of pTET-SOX17FLAG rescue cell line
The pTET-Tight construct to generate the SOX17 Rescue line has previously been described (Fisher et al., 2017) and was a kind gift from Dr. Stephen Duncan. The rescue line was generated as described in (Heslop et al., 2021). Briefly, the SOX17–3xFLAG sequence was synthesized by GeneArt (Thermofisher, CA). 50 ug of the plasmid was then linearized with 5ul of Pvu1-HF (NEB R3150L) and purified using AMPure RNAClean XP (Beckman Coulter, A63987) beads. 20ug of linearized DNA was used to electroporate 20 × 106 cells using the Neon Transfection System (ThermoFisher, CA). After, electroporated cells were replated on Matrigel-coated plates in mTeSR supplemented with CloneR (StemCell Technologies, 05888) and Y-27632 (StemCell Technologies, 72304) and media was refreshed daily. After 72 h, 1ug/mL puromycin was added to the media and used thereafter. Puromycin resistant clones were then expanded and the function and successful integration of the SOX17–3xFLAG plasmid was verified through immunostaining and Western blot experiments. For all experiments shown, SOX17 was reexpressed by adding 50ng/mL doxycycline from days 1–3 of endoderm differentiation.
Definitive endoderm differentiation
Confluent cells were passaged to single cells using Accutase (Sigma Aldrich; A6964) and plated on Matrigel-coated plates using mTESR1 and Y-27632 (StemCell Technologies, 72304). The following data, basal media was replaced, and cells were washed with PBS. DE differentiation was then carried out in RPMI-1640 media (Thermo Fisher; 11875–093) supplemented with non-essential amino acids (ThermoFisher; 11140050). Cells were treated with 100ng/mL Activin A (Shenandoah, 800–01) and 2um CHIR99021 for 24hrs. In the next two days, cells were treated with 100ng/mL Activin A and 2 mm CHIR99021 in RPMI-1640 with increasing concentrations (0.2% on day 2, 2% on day 3) of ES-grade FBS (GE; SH30070.02). To identify Wnt-responsive genes at each day, cells were treated with 1um of the Wnt inhibitor C59 (Tocris; 5148) during either day 0, 1 2 of differentiation to identify day 1, 2 or 3 specific differential regulation.
Neuromesodermal progenitor differentiation
Confluent cells were passaged using Accutase and plated on vitronectin coated plates using E8 and Y-27632. NMP differentiation was carried out largely as described previously (Lippmann et al., 2015). Briefly, media was changed to Essential 6 Medium (Gibco; A1516401), 24 h later cells were treated with 200ng/mL FGF8b (PeproTech; 100–25) in E6 media. After a further 24 h, cells were treated with 200ng/mL FGF8b and 3mm CHIR99021 in E6 media. To knock down SOX2 levels, the CRISPRi-SOX2 cells were treated with 1mg/mL doxycycline (dox) on days 2 and 3 of differentiation. To identify Wnt regulated genes, NMP cultures were treated with either 3mm CHIR99021 or 1um C59 on day 3 of differentiation.
mRNA extraction. RT-qPCR and RNA-Seq
Total RNA was extracted using the Nucleospin RNA Extraction kit (Machery-Nagel; 740955) and reverse-transcribed to cDNA using SuperScript VILO (ThermoFisher; 1177250) according to manufacturer’s instructions. qPCR was performed using PowerUp SYBR Green MasterMix (ThermoFisher; A25777) and QuantStudio 3 Flex Real-Time PCR system. Relative mRNA expression was normalized to that of housekeeping gene PPIA (peptidylprolyl isomerase A) and calculated using the ΔΔCt method. For RNA-Seq experiments, three biological replicates were sequenced per condition. 300ng of total RNA, as determined by Qubit High-Sensitivity spectrofluorometric measurement, was poly-A selected and reverse transcribed using Illumina’s TruSeq stranded mRNA library preparation kit (Illumina; 20020595). Samples were incubated with unique Illumina-compatible adapters for multiplexing. After 15 cycles of amplification, libraries were paired end sequenced on a NovaSeq 6000 with a 2 × 100 read length.
Immunofluorescence
Cells were plated at a density of 10,000 cells/mL on Matrigel or vitronectin coated Ibidi 8-well chamber slides (Ibidi; 80826). Cells were washed once with PBS and fixed in 4% paraformaldehyde for 30 min at room temperature. If necessary, antigen retrieval was performed by adding 1x Citrate Buffer warmed to 55°C and incubating slides at 65°C for 45 min. Slides were then blocked with 5% normal donkey serum (NDS) for an hour. Primary antibodies were added in 5% NDS in PBS and incubated overnight at 4°C. The following day, cells were washed thrice in PBS and incubated with secondary antibodies and DAPI for an hour at room temperature. Slides were again washed in PBS before imaging. Images were taken using a Nikon A1R inverted confocal microscope and analyzed using NIS Elements (Nikon). Antibodies and dilutions used are listed in Table S2.
Cell fractionation
Nuclear isolation was performed as previously described (Sierra et al., 2018). Briefly, cells were dissociated using Accutase and counted using a Bio Rad TC20 Automated Cell Counter. 10 million cells were then lysed in 1mL of cytoplasmic buffer (50mM Tris-HCl pH 7.5, 10% glycerol, 0.5% Triton X-100, 137.5mM NaCl) supplemented with protease (ThermoFisher; A32953) and phosphatase (ThermoFisher; A32957) inhibitors and incubated on ice for 15min. Cells were then pelleted by centrifugation for 5 min at 16,000 rpm at 4°C. Nuclei were then resuspended in 10mM HEPES pH 7.8, 0.5 M NaCl, 0.1% NP-40 supplemented with 1mM DTT and fresh protease/phosphatase inhibitors. Samples were sonicated for two 10s pulses on ice. Nuclei were cleared by centrifugation for 10mins at 16,000 rpm at 4°C.
Co-immunoprecipitation
CoIP assays were performed as previously described (Cattoglio et al., 2020) with minor modifications. After differentiation to the desired stage, 20 million cells were washed on the plates with PBS and crosslinked with 1.5mM DSP (ThermoFisher; 22585) for 30 min at room temperature. The crosslinking reaction was quenched by 30mM Tris pH 7.4 in PBS and incubated for 20 min. Cells were then scraped in ice-cold PBS supplemented with protease inhibitors and resuspended in 1mL cytoplasmic buffer (10mM HEPES pH 7.9, 10mM KCl, 340mM sucrose, 3mM MgCl2, 10% glycerol, 0.1% Triton X-100) supplemented with 1mM DTT and fresh protease inhibitors. Cells were incubated on ice for 10 min and centrifuged for 10 min at 4000 rpm. The nuclear pellet was then resuspended in 500mL CoIP wash buffer (100mM NaCl, 25mM HEPES pH 7.9, 1mM MgCl2, 0.2mM EDTA, 0.5% NP-40) supplemented with protease inhibitors. Samples were sonicated for two 10s pulses, treated with 600U/mL benzonase (Millipore; 70,664) and incubated at 4°C with end over end rotation for 4 h. Afterward, the concentration of NaCl in the samples was adjusted to 200mM and samples were incubated for an additional 30 min. Nuclear extracts were then cleared by centrifuging for 30 min at max speed at 4°C. Nuclear lysates were then quantified by BCA assays and protein concentrations of the lysates were adjusted to either 500ug/mL or 1mg/mL by diluting with CoIP wash buffer. Lysates were then precleared with Protein G Dynabeads (ThermoFisher; 10004D) at 4°C for an hour. 10% input samples were collected from the precleared lysates and stored at −20°C. Then samples were transferred to fresh tubes and incubated with relevant antibodies overnight at 4°C with end-over-end rotation. The following day, lysates and antibody complexes were added to precleared Protein-G Dynabeads and allowed to incubate at 4°C for 2 h with end-over-end rotation. The antibody/beads complexes were then washed with ice-cold CoIP wash buffer 8 times at 4°C. Lysates were then briefly centrifuged to remove any residual wash buffer and the beads were resuspended in 60mL 2x LDS loading buffer (ThermoFisher; NP0007). Proteins were eluted from beads on a thermomixer at 65°C for 15 min at 1000 rpm. Immunoprecipitations with antibody and IgG were performed parallelly. A list of antibodies and associated dilutions can be found in Table S2.
Western blots
Nuclear lysates were quantified by BCA and equal concentrations of protein samples were loaded for all experiments. Samples were resuspended in 4x LDS loading buffer supplemented with fresh 100mM DTT and boiled for 10mins. Proteins were separated on 4–12% Bis-Tris or 7% Tris-Acetate gels and transferred to PVDF membranes. Membranes were blocked in LI-COR TBS Intercept Blocking Buffer (LiCor; 927–60001) for an hour and then incubated with primary antibodies overnight at 4°C. The next day, membranes were probed with relevant secondary antibodies and imaged on an LI-COR Odyssey Clx scanner and processed using LI-COR Image Studio Lite. A list of antibodies and associated dilutions can be found in Table S2.
Transfections and reporter assays
To design enhancers constructs for reporter assays, SOX and TCF motif scanning was performed at putative SOX17/β-catenin or TCF/β-catenin enhancers (the sequences +/—100bp across peak summits of relevant SOX17/β-catenin or TCF/β-catenin were used) using FIMO with default parameters and the CIS-BP (Weirauch et al., 2014) ‘Homo sapiens’ database as reference. The identified SOX or TCF binding sites were then scrambled to generate the ‘SOX17 mutated’ or ‘TCF mutated’ enhancers. Details of sequences used for reporter assays are available in Supplementary data.
Putative SOX17-dependnent or TCF-dependent enhancers were synthesized (Genscript) and cloned into the pGL4.23 (luc2/miniP; Promega) vector. For transfections, hESCs were dissociated into 2–3 cell clumps using Versene and plated at a density of 60,000 cells/mL using mTESR and RevitaCell supplement (Gibco; A2644501). DE differentiations were carried out as described above. On the completion of day 2 of differentiation, cells were washed with PBS and supplemented with fresh day 3 differentiation media and incubated at 37°C for 30 min 50mL Opti-MEM (Gibco; 31985062), 1mL Lipofectamine STEM Transfection Reagent (Invitrogen; STEM00001) and 500 ng DNA (495ng enhancer/luc, 5ng Renilla) were then added to the cells and they were incubated for 24hrs at 37°C. The next day, cells were washed with PBS, lysed and assayed using the Dual-Luciferase Assay System (Promega; E1910) according to manufacturer’s instructions.
For experiments with dnTCF7L2 overexpression, the pcDNA/Myc DeltaN TCF4 plasmid was obtained from Addgene (#16513, gift from Bert Vogelstein) and cotransfected with the enhancer luciferase constructs described above in equimolar amounts.
ChIP-qPCR, ChIP-reChIP and ChIP-Seq
Most ChIP experiments were performed in biological duplicates as in (Schuijers et al., 2014) with several modifications. After differentiation to the desired stage, approximately 20 million cells were dual crosslinked in plate, first with 1.5mM EGS (ThermoFisher; 21565) for 20 min, followed by supplementation with 1% formaldehyde for an additional 20 min at room temperature. The crosslinking reaction was quenched with 125mM glycine for 15 min at room temperature. Cells were then washed twice and scraped in ice-cold PBS and if needed, flash frozen in dry ice until future use. ChIP samples were resuspended in 1mL sonication buffer (20mM HEPES pH 7.4, 150mM NaCl, 0.1% SDS, 1% Triton X-100, 1mM EDTA, 0.5mM EGTA) supplemented with fresh protease inhibitors. Chromatin was sonicated using a Diagenode Bioruptor Pico instrument for 45 cycles of 30 s ON, 60 s OFF, to generate 200–400 bp sheared fragments.
Chromatin was then precleared with Protein G Dynabeads for an hour with end-over-end rotation at 4°C. A volume of the pre-cleared chromatin corresponding to 1% of the total volume was set aside as input. The rest of the samples were transferred to fresh tubes containing preblocked Protein G Dynabeads; relevant antibodies (see Table S2) were added and samples were incubated overnight at 4°C with end-over-end rotation. The next day, the beads were washed serially with 150mM salt wash buffer (20mM HEPES pH 7.4, 150mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 1mM EDTA, 0.5 mM EGTA), 500mM salt wash buffer (20mM HEPES pH 7.4, 500mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 1mM EDTA, 0.5 mM EGTA), 1M salt wash buffer (20mM HEPES pH 7.4, 1M NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 1mM EDTA, 0.5 mM EGTA), 2M salt wash buffer (20mM HEPES pH 7.4, 2M NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 1mM EDTA, 0.5 mM EGTA) and LiCl wash buffer (20mM HEPES pH 7.4, 0.5M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1mM EDTA, 0.5 mM EGTA). Each wash buffer was supplemented with fresh 1mM DTT and protease inhibitors, and each wash was performed for 20 min at 4°C. The beads were then washed twice in 1×TE buffer supplemented with fresh protease inhibitors. The beads were then resuspended in DNA elution buffer (1% SDS, 0.1M NaHCO3). Elution was performed twice on a thermomixer at 65°C at 1,200 rpm. Eluates from two rounds of elution were combined and supplemented with 1/10 volume of 5M NaCl. Simultaneously, an equal volume of DNA elution buffer and 5M NaCl were added to the input samples. All samples were reverse crosslinked overnight at 65°C. The following day, samples were treated with RNase A for an hour at 37°C and digested with Proteinase K for 2 h at 55°C. DNA was then purified using the Qiagen QIAquick Purification Kit (Qiagen; 28,104) using manufacturer’s instructions and eluted in 20mL Elution Buffer; 1mL was used to quantify DNA concentrations using a Qubit High-Sensitivity DS DNA Assay kit (Invitrogen; Q32851).
For ChIP-Seq experiments, DNA libraries were prepared using 1–5 ng of starting material using the SMARTer ThruPLEX DNA-Seq kit (Takara; R400674) according to manufacturer’s instructions. After library amplification, DNA was purified using AMPure XP beads (Beckman-Coulter; A63880) and size-selected to retain 200–600 bp fragments. DNA fragment traces were analyzed on a Bioanalyzer. Suitable libraries were then paired-end sequenced on a Illumina NovaSeq 6000 with a 2 × 75 read length.
For ChIP-reChIP experiments, before the first ChIP, antibodies were crosslinked to Protein G Dynabeads. Briefly, beads were washed with 0.2M sodium borate pH9, and antibodies were crosslinked to beads by using 20mM DMP (Pierce; 21,666) dissolved in 0.2M sodium borate. The crosslinking reaction was carried out at room temperature for 40 min. The reaction was then quenched using 0.2M ethanolamine pH 8.0 for an hour. Residual IgGs were removed by washing the antibody/beads complex with 0.58%v/v acetic acid +150mM NaCl. The beads were then added to processed chromatin samples and ChIP experiments were performed as described above. After the first ChIP, samples were eluted in DNA elution buffer supplemented with 10mM DTT. The samples were then diluted in 10 volumes of sonication buffer and the second ChIP was carried out as described above.
ChIP-qPCR was performed using PowerUp SYBR Green MasterMix and the QuantStudio 3 Flex Real-Time PCR system using default protocols. Primers were designed to span relevant SOX17 or β-catenin peak centers and relative expression was normalized to that of a ‘negative’ control gene desert genomic region. Relative fold change was calculated using the ΔΔCt method. Primer sequences are listed in Table S2.
ATAC-seq
ATAC-Seq experiments were largely performed as previously described (Buenrostro et al., 2015). Briefly, 50,000 cells were collected following differentiation to the desired stage, and lysed in 50mL of ATAC-lysis buffer (10mM Tris-HCl, pH 7.4. 10mM NaCl, 3mM MgCl2, 0.1% NP-40) to obtain a crude nuclei prep. All centrifugation steps were performed at 4°C at 2000 rpm. The nuclei pellet was then resuspended in the 50ul of the transposition reaction mix (25ul Tagment DNA buffer, 2.5ul TD Tn5 Transposase enzyme, 22.5ul nuclease-free water) (Illumina; 20034197). The transposition reaction was incubated at 37°C for 30 min on a thermomixer with constant gentle shaking at 1000 rpm. Immediately after transposition, DNA was purified using a Qiagen MinElute PCR Purification (Qiagen; 28004 kit) and eluted in 10mL Elution Buffer. The eluted DNA was then amplified in a reaction with 25mL NEBNext High-Fidelity 2x PCR Mastermix (NEB; M0541L) and custom 25um Nextera PCR Primers (Ad1_noMx universal primer, 0.5um Ad2.x indexing primer). PCR was performed as follows: 1 cycle of 72°C for 5min, 98°C for 30s, 5 cycles of 98°C for 10s, 63°C for 30s, 72°C for 1min. 5ul of the amplified DNA was then used to perform qPCR to determine the optimal number of additional cycles to prevent amplification saturation of DNA libraries. In all cases, either 4 or 5 additional cycles of PCR was performed at: 98°C for 10s, 63°C for 30s and 72°C for 1 min. Double size selection of amplified libraries (0.5x – left sided, 1.8x – right sided) was performed using AMPure XP beads and the DNdA was eluted in a final volume of 20ul in 0.1x TE buffer. Purified libraries were sequenced on an Illumina NextSeq 500 instrument with a 2 3 75bp read length.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics and reproducibility
All statistical analyses were performed using R (R Core Team, 2020) or Graphpad Prism. Statistical details of all experiments can be found in the relevant figure legends. RNA-Seq experiments were performed in biological triplicates. Most ChIP-Seq and ATAC-Seq experiments were performed in biological duplicates except for: H3K27ac and BRG1 ChIP-Seq in WT and SOX7KO, β-catenin ChIP-Seq in SOX17 Rescue cells and β-catenin/SOX17 ChIP-Seq in WT and 4KO 293T cells (n = 1) and NMP β-catenin ChIP-Seq (n = 3). Immunostaining and western blots were performed at least four times and representative images were used. CoIP, ChIP-qPCR experiments and reporter assays were repeated at least thrice. All differentiations and experiments were performed using cell lines maintained between passages 55–65.
Analysis of genomic data
RNA-seq
Raw reads were quality-checked using FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and if necessary, adapters were trimmed using cutadapt (Martin, 2011). Fastq files were pseudo-aligned to the hg19 reference index using salmon (Patro et al., 2017). An index of transcripts was built using default parameters (salmon index) using quasi-mapping (-quasi) and kmers of length 31 (-k 31). Relative transcript abundance was then quantified using salmon -quant using paired end fastq files and counts per transcript were obtained. The tximport package (Soneson et al., 2015)was then used for downstream analysis to convert transcript-level counts to gene-level estimates. Differential gene expression analysis was performed using DESeq2 (Love et al., 2014) using default parameters. Differentially expressed genes were defined as those with log2 fold change >|1| and adjusted FDR of p < 0.05. Transcripts were then annotated using the biomaRt (Durinck et al., 2009) package. Any genes with TPM less than 10 across all replicates were then discarded from further analysis. To perform principal component analysis, variance stabilized and transformed data from DESeq2 vst function was generated. PCA plots were visualized using plotPCA () function of DESeq2.
To identify endoderm, mesoderm or ectoderm enriched genes, we reanalyzed the following datasets: GSM1112846, GSM1112844 (RNA-Seq of day 3 ectoderm cells) and GSM1112835, GSM1112833 (RNA-Seq of day 3 mesoderm cells) and compared with our day 3 endoderm RNA-Seq data. Raw RNA-Seq data was downloaded from GEO and processed as described above, and pairwise differential gene expression analysis was performed using DESeq2 to identify genes with enriched expression in day 3 endoderm, mesoderm or ectoderm. For instance, a gene was considered to be significantly endoderm enriched if: the gene was significantly differentially expressed in endoderm over control pluripotent cells, and showed significantly enriched expression in the endoderm compared to the mesoderm and ectoderm datasets. Differential enrichment threshold: log2 fold change >|1| and adjusted FDR of p < 0.05.
ChIP-seq
Raw reads were quality-checked using FASTQC and adapters were trimmed using cutadapt. Reads were aligned to the hg19 genome using bowtie2 (Langmead and Salzberg, 2012). Unmapped and low quality (MAPQ<10) reads were discarded. Duplicates were marked using Picard (https://broadinstitute.github.io/picard/) and removed using samtools (Li et al., 2009). From the replicate datasets, a consensus set of peaks were called for each TF at each stage using HOMER (Heinz et al., 2010) getDifferentialPeaksReplicates.pl using stage-matched input samples as background and -style factor. Briefly, first tag directories were created for each target and input replicate. Peaks were quantified for both target and input tag directories and DESeq2 was then invoked to identify peaks enriched in target ChIP samples over input using a fold enrichment threshold of 1.5 and fdr of 0.1. In the absence of replicates, peak calling was performed using macs2 (Zhang et al., 2008) using –call-summits and a qvalue cutoff of 0.05. HOMER annotate-Peaks.pl was then used to annotate these peaks to their nearest gene.
Differential binding analysis was performed using the DiffBind package (Ross-Innes et al., 2012; Stark and Brown, 2021) using default parameters. Differentially bound β-catenin and ATAC peaks were identified using a fold enrichment threshold of 1.5 and adjusted -pvalue < 0.05. A genomic site was defined as both ‘SOX17’ and TCF0 bound if: a significant binding event was called for both SOX17 and at least one of the TCF/LEF TFs using the relevant input sample as background, and no statistically significant differential binding between TFs was observed. A peak was called ‘SOX17 enriched’ if: the peak was called for only SOX17 and none of the TCFs, or if SOX17 binding displayed a greater log2 fold change over input relative to all the TCFs, and SOX17 was determined to have significantly increased binding over all TCFs quantified by DiffBind and DESeq2. Similarly, a genomic site was called ‘TCF-enriched’ if a peak was called for at least one of the TCFs but not SOX17, or at least one of the TCFs was determined to have significantly increased binding over SOX17. A fold enrichment threshold of 1.5 and FDR <0.1 to identify SOX vs. TCF enriched peaks, in order to also incorporate weakly bound TCF peaks. If a binding category contained less than 500 peaks, we didn’t use it for further analysis.
ATAC-seq
FASTQ files were quality-checked using FASTQC and Nextera adapters were trimmed using cutadapt. Trimmed paired-end reads were aligned to the hg19 genome using bowtie2 and the parameters -X 2000 -very-sensitive-local. Paired end bam files were filtered for mitochondrial reads, unmapped and low-quality reads. Duplicates were marked using Picard and removed using samtools. Peaks were called on replicates using Genrich (https://github.com/jsh58/Genrich) and the parameters j -r -e -v -q 0.1.
Nucleoatac analysis
As input for nucleoatac analysis, peak files of desired categories were extended up to 2000bp across peak summits. Nucleoatac (Schep et al., 2015) was run using default parameters. For visualization and quantification of nucleosome occupancy, the occ.bedgraph files were converted to bigwigs using UCSC binary tools (https://hgdownload.cse.ucsc.edu/admin/exe/). Nucleosome occupancy scores were then computed using deeptools(Ramıŕez et al., 2016) computeMatrix and visualized using plotProfile.
Downstream data processing and visualization
To visualize ChIP-Seq and ATAC-seq data, filtered and sorted bam files were converted to bigwig files using deeptools bamCoverage with the parameters: -bS 20 –smoothLength 60 -e 200 –normalizeUsing RPGC using the effective genome size for GRCh37. Bigwig files were visualized using IGV (Thorvaldsdottir et al., 2013). Genomic algebra operations were performed using unix commands (awk, grep, sed) or using the bedtools suite (Quinlan and Hall, 2010), particularly bedtools intersect to define overlapping genomic region of interest, or bedtools merge to define a union set of genomic regions. For all quantifications, merged bam files or bigwig files of both ChIP or ATAC replicates were used.
To identify patterns in β-catenin time course ChIP-Seq datasets [Figures 1 and S1], an union of all β-catenin peaks from all days was plotted using deeptools plotHeatmap and k-means clustering was performed using kclust 8. The most predominant 5 clusters were then extracted and retained for further analysis. Heatmaps, density plots or metaplots were generated using the deeptools package by invoking the computeMatrix (-reference-point center, -a 2500, -b 2500) and then plotHeatmap or plotProfile options. Volcano plots or MA plots of differential gene expression were generated using the EnhancedVolcano (https://github.com/kevinblighe/EnhancedVolcano ) or ggpubr (https://github.com/kassambara/ggpubr) packages in R respectively. Heatmaps from RNA-Seq data was generated using the ‘pheatmap’ package.
Signal normalization (1/mapped tags/sample such that each directory contains 10 million tags) and quantification was performed on merged ChIP/ATAC tag directories by HOMER. Boxplots and violin plots of ChIP/ATAC-Seq signal quantification were generated using the ggplot2 (Wickham, 2016) package and statistically significant differences in read density between conditions was determined by ANOVA or Wilcoxon rank-sum test as appropriate in R. UpSET plots of the distribution of SOX17 or SOX2 and β-catenin co-regulated enhancers were generated using intervene (Khan and Mathelier, 2017). Data from ChIP-qPCR and reporter assays were visualized using GraphPad Prism. p-values were determined via nonparametric Mann-Whitney-U tests.
DNA-binding motif and GO enrichment analysis
The MEME-Suite of tools (Bailey et al., 2009; Machanick and Bailey, 2011) was used to perform de novo motif analysis. For motif analysis, 100bp across peak summits were extracted for each category and converted to the fasta format using bedtools getfasta. De novo motif analysis across peak sets was performed using DREME and default parameters, and motifs were identified using TOMTOM. Top enriched motifs were identified based on their E-values. E-values were calculated through DREME, where they represent the likelihood of finding similar DNA-binding motifs by chance if the input sequences were shuffled. A lower E-value represents a decreasing likelihood of observing the enrichment of that motif just by chance. GO term enrichment analysis was performed using GREAT (McLean et al., 2010) and Gene Ontology(Ashburner et al., 2000; Gene Ontology Consortium, 2021).
ADDITIONAL RESOURCES
Analysis of public data
The following public datasets were: GSM772971 (H3K4me1 ChIP-Seq in DE), GSM1112846, GSM1112844 (RNA-Seq of day 3 ectoderm cells) and GSM1112835, GSM1112833 (RNA-Seq of day 3 mesoderm cells) (Gifford et al., 2013).
Supplementary Material
Highlights.
WNT signaling activates distinct transcription programs in different cell lineages
SOX17 and SOX2 direct WNT-dependent endoderm and neuromesoderm fates in hPSCs
SOX and TCFs recruit β-catenin to different lineage-specific enhancers
SOX17 can assemble a WNT-enhanceosome complex on chromatin independent of TCFs
ACKNOWLEDGMENTS
We thank Drs. C. Cantù , S. Duncan, B. Conklin, and B. Vogelstein for cell lines and constructs, Keely Icardi and the CCHMC DNA Sequencing Core for help with ChIP-seq library preparation, CCHMC Pluripotent Stem Cell Facility and Evan Brooks for help with generating the SOX17KO line. This work was supported by NIDDK R01 DK123092 to A.M.Z, a Cincinnati Children’s CURE award, and in part by a center grant NIH P30 DK078392.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111247.
REFERENCES
- Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, et al. (2004). Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 18, 1072–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. (2009). Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). Meme suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billin AN, Thirlwell H, and Ayer DE (2000). Beta-catenin–histone deacetylase interactions regulate the transition of LEF1 from a transcriptional repressor to an activator. Mol. Cell Biol 20, 6882–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blassberg R, Patel H, Watson T, Gouti M, Metzis V, Delá s MJ, and Briscoe J (2022). Sox2 levels regulate the chromatin occupancy of WNT mediators in epiblast progenitors responsible for vertebrate body formation. Nat. Cell Biol 24, 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brafman DA, Phung C, Kumar N, and Willert K (2013). Regulation of endodermal differentiation of human embryonic stem cells through integrin-ECM interactions. Cell Death Differ. 20, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM, and Waterman ML (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol 4, a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera I, Janody F, Leeds N, Duveau F, and Treisman JE (2008). Pygopus activates Wingless target gene transcription through the mediator complex subunits Med12 and Med13. Proc. Natl. Acad. Sci. USA 105, 6644–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoglio C, Pustova I, Darzacq X, Tjian R, and Hansen AS (2020). Assessing self-interaction of mammalian nuclear proteins by Co-immunoprecipitation. Bio. Protoc 10, e3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, and Dent SYR (2014). Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet 15, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi D, Zhang W, Jia Y, Cong D, and Hu S (2019). Spalt-like transcription factor 1 (SALL1) gene expression inhibits cell proliferation and cell migration of human glioma cells through the Wnt/β-catenin signaling pathway. Med. Sci. Monit. Basic Res. 25, 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M, Orsenigo F, Bhat GP, Conze LL, Breviario F, Cunha SI, Claesson-Welsh L, Beznoussenko GV, Mironov AA, Bacigaluppi M, et al. (2019). Fine-tuning of Sox17 and canonical Wnt coordinates the permeability properties of the blood-brain barrier. Circ. Res 124, 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumpas N, Lampart F, Robinson MD, Lentini A, Nestor CE, Cantù C, and Basler K (2019). TCF/LEF dependent and independent transcriptional regulation of Wnt/β-catenin target genes. EMBO J. 38, e98873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estarás C, Benner C, and Jones KA (2015). SMADs and YAP compete to control elongation of β-catenin:LEF-1-recruited RNAPII during hESC differentiation. Mol. Cell 58, 780–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faial T, Bernardo AS, Mendjan S, Diamanti E, Ortmann D, Gentsch GE, Mascetti VL, Trotter MWB, Smith JC, and Pedersen RA (2015). Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development 142, 2121–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JB, Pulakanti K, Rao S, and Duncan SA (2017). GATA6 is essential for endoderm formation from human pluripotent stem cells. Biol. Open 6, 1084–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura N, Vacik T, Machon O, Vlcek C, Scalabrin S, Speth M, Diep D, Krauss S, and Kozmik Z (2007). Wnt-mediated down-regulation of Sp1 target genes by a transcriptional repressor Sp5. J. Biol. Chem 282, 1225–1237. [DOI] [PubMed] [Google Scholar]
- Funa NS, Schachter KA, Lerdrup M, Ekberg J, Hess K, Dietrich N, Honoré C, Hansen K, and Semb H (2015). β-catenin regulates primitive streak induction through collaborative interactions with SMAD2/SMAD3 and OCT4. Cell Stem Cell 16, 639–652. [DOI] [PubMed] [Google Scholar]
- Gammons M, and Bienz M (2018). Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol 51, 42–49. [DOI] [PubMed] [Google Scholar]
- Gao C, Xiao G, and Hu J (2014). Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerner-Mauro KN, Akiyama H, and Chen J (2020). Redundant and additive functions of the four Lef/Tcf transcription factors in lung epithelial progenitors. Proc. Natl. Acad. Sci. USA 117, 12182–12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, et al. (2013). Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 153, 1149–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti M, Tsakiridis A, Wymeersch FJ, Huang Y, Kleinjung J, Wilson V, and Briscoe J (2014). In vitro generation of neuromesodermal progenitors reveals distinct roles for Wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biol. 12, e1001937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Kim A, Li B, Ransick A, Bugacov H, Chen X, Lindström N, Brown A, Oxburgh L, Ren B, and McMahon AP (2021). A β-catenin-driven switch in TCF/LEF transcription factor binding to DNA target sites promotes commitment of mammalian nephron progenitor cells. Elife 10, e64444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagey DW, and Muhr J (2014). Sox2 acts in a dose-dependent fashion to regulate proliferation of cortical progenitors. Cell Rep. 9, 1908–1920. [DOI] [PubMed] [Google Scholar]
- Hecht A, Vleminckx K, Stemmler MP, van Roy F, and Kemler R (2000). The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 19, 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeyer K, Junghans D, Kanzler B, and Kemler R (2017). Trimethylation and acetylation of β-catenin at lysine 49 represent key elements in ESC pluripotency. Cell Rep. 18, 2815–2824. [DOI] [PubMed] [Google Scholar]
- Hou L, Srivastava Y, and Jauch R (2017). Molecular basis for the genome engagement by Sox proteins. Semin. Cell Dev. Biol 63, 2–12. [DOI] [PubMed] [Google Scholar]
- Huggins IJ, Bos T, Gaylord O, Jessen C, Lonquich B, Puranen A, Richter J, Rossdam C, Brafman D, Gaasterland T, and Willert K (2017). The WNT target SP5 negatively regulates WNT transcriptional programs in human pluripotent stem cells. Nat. Commun 8, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Nagarajan S, Woelfer M, Schoger E, Khadjeh S, Zafiriou MP, Kari V, Herting J, Pang ST, Weber T, et al. (2018). A context-specific cardiac β-catenin and GATA4 interaction influences TCF7L2 occupancy and remodels chromatin driving disease progression in the adult heart. Nucleic Acids Res. 46, 2850–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidi A, Williams AC, and Paraskeva C (2007). Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol 9, 210–217. [DOI] [PubMed] [Google Scholar]
- Kelly KF, Ng DY, Jayakumaran G, Wood GA, Koide H, and Doble BW (2011). β-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell 8, 214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, and Mathelier A (2017). Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinf. 18, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Xu X, Hecht A, and Boyer TG (2006). Mediator is a transducer of Wnt/β-catenin signaling. J. Biol. Chem 281, 14066–14075. [DOI] [PubMed] [Google Scholar]
- Koch F, Scholze M, Wittler L, Schifferl D, Sudheer S, Grote P, Timmermann B, Macura K, and Herrmann BG (2017). Antagonistic activities of Sox2 and brachyury control the fate choice of neuro-mesodermal progenitors. Dev. Cell 42, 514–526.e7. [DOI] [PubMed] [Google Scholar]
- Kondoh H, and Kamachi Y (2010). SOX-partner code for cell specification: regulatory target selection and underlying molecular mechanisms. Int. J. Biochem. Cell Biol 42, 391–399. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, and Clevers H (1997). Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/−colon carcinoma. Science 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- Kormish JD, Sinner D, and Zorn AM (2010). Interactions between SOX factors and Wnt/beta-catenin signaling in development and disease. Dev. Dyn 239, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Zu€llig S, and Basler K (2002). Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin-TCF complex. Cell 109, 47–60. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, and Fearon ER (2002). Activation of AXIN2 expression by β-catenin-T cell factor: a feedback repressor pathway regulating Wnt signaling. J. Biol. Chem 277, 21657–21665. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R 1000 Genome Project Data Processing Subgroup; 1000 Genome Project Data Processing (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Williams CE, Ruhl DA, Estevez-Silva MC, Chapman ER, Coon JJ, and Ashton RS (2015). Stem cell reports Article deterministic HOX patterning in human pluripotent stem cell-derived neuroectoderm. Stem Cell Rep. 4, 632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Ang LT, Zhang J, Kumar V, Ang J, Auyeong JQ, Lee KL, Choo SH, Lim CYY, Nichane M, et al. (2014). Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 14, 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Gao Y, Zhang Z, Cao Q, Zhang X, Zou J, and Cao Y (2015). Kdm2a/b lysine demethylases regulate canonical Wnt signaling by modulating the stability of nuclear β-catenin. Dev. Cell 33, 660–674. [DOI] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j 17, 10. [Google Scholar]
- Masuda T, and Ishitani T (2017). JB special review - Wnt signaling: biological functions and its implications in diseases: context-dependent regulation of the β-catenin transcriptional complex supports diverse functions of Wnt/β-catenin signaling. J. Biochem 161, 9–17. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers MP, Janssens DH, Meers MP, Janssens DH, and Henikoff S (2019). Pioneer factor-nucleosome binding events during article pioneer factor-nucleosome binding events during differentiation are motif encoded. Mol. Cell, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi A, Ghasemi F, Anvari K, Hassanian SM, Simab SA, Ebrahimi S, Hesari A, Forghanifard MM, Boroushaki MT, ShahidSales S, and Avan A (2017). The cross-regulation between SOX15 and Wnt signaling pathway. J. Cell. Physiol 232, 3221–3225. [DOI] [PubMed] [Google Scholar]
- Moreira S, Polena E, Gordon V, Abdulla S, Mahendram S, Cao J, Blais A, Wood GA, Dvorkin-Gheva A, and Doble BW (2017). A single TCF transcription factor, regardless of its activation capacity, is sufficient for effective trilineage differentiation of ESCs. Cell Rep. 20, 2424–2438. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Chaturvedi P, Rankin SA, Fish MB, Wlizla M, Paraiso KD, MacDonald M, Chen X, Weirauch MT, Blitz IL, et al. (2020). Sox17 and β-catenin co-occupy Wnt-responsive enhancers to govern the endoderm gene regulatory network. Elife 9, e58029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, and Schilling TF (2008). Chemokine signaling controls endodermal migration during zebrafish gastrulation. Science 322, 89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Morimoto M, Takahashi Y, Koseki H, and Saga Y (2006). Identification of Epha4 enhancer required for segmental expression and the regulation by Mesp2. Development 133, 2517–2525. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, de Paiva Alves E, Veenstra GJC, and Hoppler S (2016). Tissue- and stage-specific Wnt target gene expression is controlled subsequent to β-catenin recruitment to cis-regulatory modules. Development 143, 1914–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkis G, Tzchori I, Cohen T, Holtz A, Wier E, and Westphal H (2012). Isl1 and Ldb co-regulators of transcription are essential early determinants of mouse limb development. Dev. Dyn 241, 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LF, Kaur P, Bunnag N, Suresh J, Sung ICH, Tan QH, Gruber J, and Tolwinski NS (2019). WNT signaling in disease. Cells 8, 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2020). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing). https://www.R-project.org/.
- Ramírez F, Ryan DP, Gru€ning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Du€ndar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, and Greenleaf WJ (2015). Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 25, 1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijers J, Mokry M, Hatzis P, Cuppen E, and Clevers H (2014). Wnt-induced transcriptional activation is exclusively mediated by TCF/LEF. EMBO J. 33, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra RA, Hoverter NP, Ramirez RN, Vuong LM, Mortazavi A, Merrill BJ, Waterman ML, and Donovan PJ (2018). TCF7L1 suppresses primitive streak gene expression to support human embryonic stem cell pluripotency. Development 145, dev161075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinner D, Rankin S, Lee M, and Zorn AM (2004). Sox17 and -catenin cooperate to regulate the transcription of endodermal genes. Development 131, 3069–3080. [DOI] [PubMed] [Google Scholar]
- Sinner D, Kordich JJ, Spence JR, Opoka R, Rankin S, Lin S-CJ, Jonatan D, Zorn AM, and Wells JM (2007). Sox17 and Sox4 differentially regulate β-catenin/T-cell factor Activity and proliferation of colon carcinoma cells. Mol. Cell Biol 27, 7802–7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderholm S, and Cantù C (2021). The WNT/β-catenin dependent transcription: a tissue-specific business. WIREs Mech. Dis 13, e1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J-S, Chang C-C, Wu C-H, Dinh TK, Jan J-J, Huang K-W, Chou M-C, Shiue T-Y, Yeh K-C, Ke Y-Y, et al. (2021). A highly selective and potent CXCR4 antagonist for hepatocellular carcinoma treatment. Proc. Natl. Acad. Sci. USA 118. e2015433118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, and Brown G (2021). DiffBind: Differential Binding Analysis of ChIP-Seq Peak Data (Bioconductor). [Google Scholar]
- Thorvaldsdó ttir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompouki E, Bowman TV, Lawton LN, Fan ZP, Wu D-C, DiBiase A, Martin CS, Cech JN, Sessa AK, Leblanc JL, et al. (2011). Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 147, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li X, Wang T, Wu SP, Jeong JW, Kim TH, Young SL, Lessey BA, Lanz RB, Lydon JP, and DeMayo FJ (2018). SOX17 regulates uterine epithelial–stromal cross-talk acting via a distal enhancer upstream of Ihh. Nat. Commun 9, 4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise A, Bruser K, Elfert S, Wallmen B, Wittel Y, Wöhrle S, and Hecht A (2010). Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 38, 1964–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; )978–3-319–24277-4. https://ggplot2.tidyverse.org. [Google Scholar]
- Yano F, Kugimiya F, Ohba S, Ikeda T, Chikuda H, Ogasawara T, Ogata N, Takato T, Nakamura K, Kawaguchi H, and Chung UI (2005). The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem. Biophys. Res. Commun 333, 1300–1308. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Yang H, Tian Y, Xie Q, Lu Y, Wang Y, Su N, Dong B, Liu X, Wang C, et al. (2016). SOX7 is associated with the suppression of human glioma by HMG-box dependent regulation of Wnt/β-catenin signaling. Cancer Lett. 375, 100–107. [DOI] [PubMed] [Google Scholar]
- Zhou D, Bai F, Zhang X, Hu M, Zhao G, Zhao Z, and Liu R (2014). SOX10 is a novel oncogene in hepatocellular carcinoma through Wnt/β-catenin/TCF4 cascade. Tumour Biol. 35, 9935–9940. [DOI] [PubMed] [Google Scholar]
- Zimmerli D, Borrelli C, Jauregi-Miguel A, Sö derholm S, Brütsch S, Doumpas N, Reichmuth J, Murphy-Seiler F, Aguet M, Basler K, et al. (2020). TBX3 acts as tissue-specific component of the Wnt/β-catenin transcriptional complex. Elife 9, e58123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn AM, and Wells JM (2009). Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol 25, 221–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn AM, Barish GD, Williams BO, Lavender P, Klymkowsky MW, and Varmus HE (1999). Regulation of Wnt signaling by Sox proteins: XSox17 alpha/beta and XSox3 physically interact with beta-catenin. Mol. Cell 4, 487–498. [DOI] [PubMed] [Google Scholar]
- Heslop JA, Pournasr B, Liu JT, and Duncan SA (2021). GATA6 defines endoderm fate by controlling chromatin accessibility during differentiation of human-induced pluripotent stem cells. Cell Rep 35, 109145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data related to Figures 1 and 3 are available on Mendeley at https://doi.org/10.17632/7jxtwztwxg.1 The datasets generated during this study are available at the Gene Expression Omnibus (GEO) under the accession number GSE182842. The following public datasets were downloaded from GEO and reanalyzed: GSM772971, GSM1112846, GSM1112844, GSM1112835 and GSM1112833. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contactupon request.