

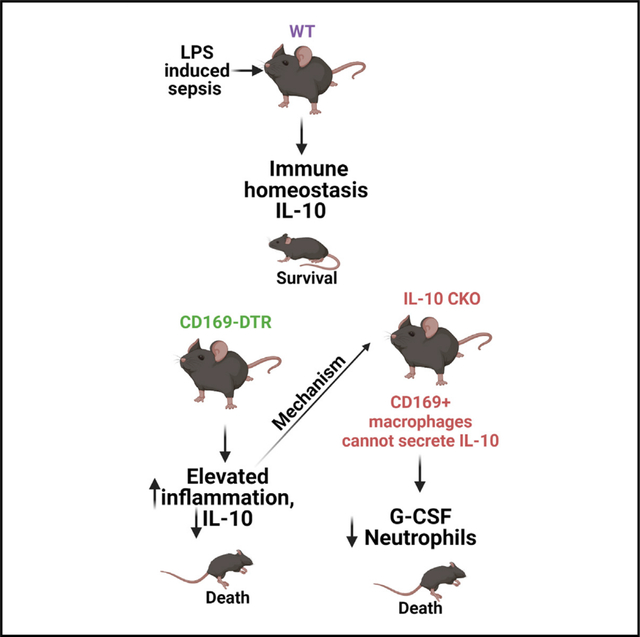
SUMMARY
Macrophages facilitate critical functions in regulating pathogen clearance and immune homeostasis in tissues. The remarkable functional diversity exhibited by macrophage subsets is dependent on tissue environment and the nature of the pathological insult. Our current knowledge of the mechanisms that regulate the multifaceted counter-inflammatory responses mediated by macrophages remains incomplete. Here, we report that CD169+ macrophage subsets are necessary for protection under excessive inflammatory conditions. We show that in the absence of these macrophages, even under mild septic conditions, mice fail to survive and exhibit increased production of inflammatory cytokines. Mechanistically, CD169+ macrophages control inflammatory responses via interleukin-10 (IL-10), as CD169+ macrophage-specific deletion of IL-10 was lethal during septic conditions, and recombinant IL-10 treatment reduced lipopolysaccharide (LPS)-induced lethality in mice lacking CD169+ macrophages. Collectively, our findings show a pivotal homeostatic role for CD169+ macrophages and suggest they may serve as an important target for therapy under damaging inflammatory conditions.
In brief
Yeung et al. investigate the mechanisms that regulate the multifaceted counter-inflammatory responses mediated by macrophages under inflammatory conditions. They show that the CD169+ macrophage subset is necessary for host survival even during mild septic conditions. The mechanism by which these macrophages impart the pivotal anti-inflammatory function is by secreting IL-10.
Graphical Abstract
INTRODUCTION
Tissue-resident macrophages are myeloid cells that are distributed in virtually all organs and play critical roles in maintaining immune and tissue homeostasis under steady-state or inflammatory conditions.1–4 The CD169+ subset of macrophages is located in secondary lymphoid tissues and nonlymphoid tissues.5–9 In lymphoid tissues, these macrophages are positioned strategically to serve as gatekeepers for phagocytosis and clearance of invading pathogens from the blood and lymph and to promote erythropoiesis in the bone marrow.6,10–13
Macrophages exhibit remarkable functional diversity; for example, in the lungs, CD169+ alveolar macrophages (AMs) provide pro-inflammatory cues and assist in pathogen clearance, while CD169+ interstitial macrophages play an immunoregulatory role following viral infection.14 Similarly, CD169+ macrophages in the spleen respond robustly to Listeria infection by polarizing to a pro-inflammatory state and help clear the bacteria. Depleting these macrophages resulted in dramatically higher bacterial burden and spread to other organs.13 Moreover, depleting these subsets of macrophages in the colon alleviated colonic inflammation following dextran sodium sulfate (DSS) administration.5 However, it remains to be determined if the same populations of macrophages can polarize differently depending on the location, context, and inflammatory cue. Considering the robust inflammatory response to Listeria infection by CD169+ splenic macrophages, we reasoned that depleting these macrophages during an overt inflammatory condition such as toxin-induced sepsis may serve to be therapeutic and protective for the host.
Sepsis ranks among the top 10 causes of death worldwide and leading causes of death (1 in every 2 min) in hospitals in the United States, with more than 1.6 million individuals diagnosed, and poses an economic burden of more than $24 billion each year.15 Currently, there are no specific therapies for sepsis; nonetheless, current therapeutic management of sepsis consists of resuscitative therapy (i.e., oxygen and intravenous fluids) to keep vital organs functional and antibiotics to combat infections with patient prognosis contingent on early detection.16 Sepsis involves complex interactions between pathogens and the host immune cells resulting in a systemic hyper inflammatory state.17 These interactions are crucial for clearing infection; however, they can also result in unchecked inflammatory response characterized by excessive production of cytokines/chemokines and recruitment of immune cells culminating in severe organ damage. A considerable amount of work to date has focused on understanding the immune signaling pathways and bulk immune cell populations during sepsis.18–22 However, our knowledge of the precise counter-regulatory mechanisms that are important for preventing immunopathology during sepsis is limited. Particularly, the extent to which tissue-resident macrophage subsets control immunoregulatory responses during sepsis remains unknown. Thus, it is critical to determine the precise cellular subsets that uniquely contribute to the multifaceted counter-inflammatory responses in vivo.
Here, we used the well-established lipopolysaccharide (LPS) and cecal slurry infusion models of sepsis to investigate the role of the CD169+ macrophage subset in mediating anti-inflammatory countermeasures. Contrary to our aforementioned hypothesis, we show that the selective depletion of CD169+ macrophages in fact results in hyper susceptibility to LPS− and cecal slurry-induced septic shock. Upon LPS and cecal slurry challenge, CD169+ macrophage-depleted mice exhibited overt inflammatory cytokine production and impaired interleukin-10 (IL-10) production. Importantly, we show that IL-10 production exclusively by CD169+ macrophages is critical for mediating protection against LPS shock. And this protection afforded by CD169+ macrophages was dependent of Syk signaling but independent of MyD88. Taken together, our data demonstrate a certain macrophage subset that, in one instance (i.e., Listeria infection), performs pro-inflammatory function of clearing bacteria and yet provide critical immunoregulatory cues to maintain immune homeostasis during uncontrolled inflammatory conditions such as toxin mediated sepsis.
RESULTS
CD169+ macrophages closely associate with LPS and regulate LPS clearance from tissues
In our previous study, we showed that CD169+ splenic macrophages are the initial cells that interact with blood-borne bacteria, thus we began our study by determining whether CD169+ macrophages also associate with LPS.13 We treated wild-type (WT) C57Bl/6 mice with 200 μg purified fluorescein isothiocyanate (FITC)-conjugated LPS intraperitoneally. Confocal microscopy of immunostained spleen sections revealed that as early as 1.5 h, LPS closely associated with the marginal zone CD169+ macrophages, and this interaction continued as late as 12 h after LPS treatment (Figure 1A). Since CD169+ macrophages closely associated with the LPS, we next wanted to determine if, in the absence of these cells, the amount of LPS would be elevated in the circulation or in the tissues. To this end, we utilized the limulus amebocyte lysate (LAL) assay, and we found that in the absence of CD169+ macrophages, there was in fact similar level of detectable LPS in circulation (Figure 1B). However, using fluorescently tagged low-dose LPS (75 μg), we observed that in the CD169-diphtheria toxin receptor (DTR) mouse model, which allows for temporal depletion of CD169+ macrophages upon the administration of DT, there was notably higher level of LPS detected in the spleen compared with WT mice (Figure 1C). Furthermore, we found that compared with WT mice, CD169-DTR mice exhibited significantly more LPS staining in the spleen (Figure 1D) over time following LPS treatment, which was preferentially localized with the F4/80 + red pulp macrophages (Figure 1E). In contrast, in WT animals LPS was preferentially localized to the CD169+ marginal zone macrophages (Figure 1F). This suggested that in the absence of CD169+ macrophages, the half-life of LPS within the spleen was increased, while LPS levels in circulation are not affected significantly. These data also suggest that an increase in association of LPS with F4/80+ red pulp macrophages in the absence of CD169+ splenic macrophages may be pathogenic during a septic response.
Figure 1. CD169+ macrophages closely associate with LPS and regulate LPS clearance from tissue.
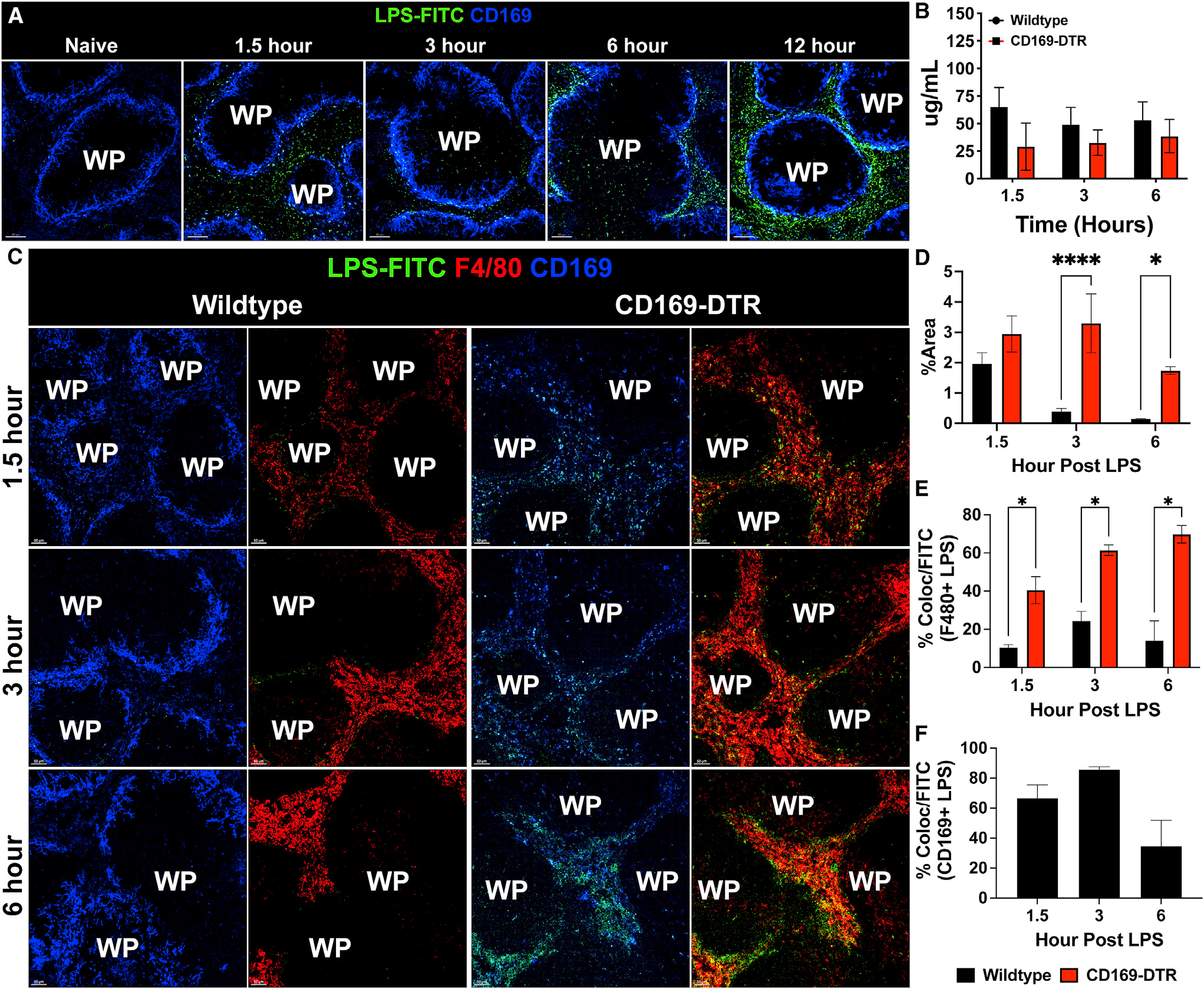
(A) Confocal imaging of splenic sections immunostained with CD169 (blue) and FITC-LPS (green) at various time points from naive to 12 h post 200 μg LPS treatment. Scale bar: 80 um.
(B) LAL assay to quantify free LPS in serum.
(C) Immunofluorescence staining in the spleen from WT (left) and CD169-DTR (right) mice at 1.5 (top), 3 (middle), and 6 (bottom) h post 75 μg LPS treatment. Tissue were stained for CD169 (blue), F4/80 (red), and LPS (green). Scale bar: 50 um
(D) Percentage of area of the spleen occupied by LPS at the indicated time after LPS treatment between WT (black) and CD169-DTR (red) mice.
(E) Percentage of LPS colocalized with F4/80+ macrophages at the indicated time points after LPS treatment between WT (black) and CD169-DTR (red) mice.
(F) Percentage of LPS colocalized with CD169+ macrophages at the indicated time points after LPS treatment in WT mice.
The images and graphs are representative of 2–4 independent experiments. WP, white pulp. **p < 0.0, *p < 0.001, p < 0.01. n = 3–5/group ad of 2 independent experiments.
CD169+ macrophages are required for the host survival during sepsis
Given the observation that CD169+ macrophages interact with and mediate the clearance of LPS, we next utilized two established model of sepsis to understand the physiological role of CD169+ macrophages during septic conditions. To this end, we used the CD169-DTR mice to specifically deplete CD169-expressing macrophage subsets.13,14,23 We found that depletion of CD169+ macrophages made mice highly susceptible to septic shock with higher doses (125 μg LPS) or even with extremely low-dose LPS (25 μg LPS) treatment (Figures 2A–2C). Compared with WT, mice the CD169-depleted mice exhibited all the signs of disease associated with septic shock, including lethargy, reduced motor activity, social withdrawal, and, ultimately, increased mortality. To validate the hypothesis that CD169+ macrophages are critical for regulating the inflammatory response during septic shock, we utilized an alternative model of sepsis induced by the intraperitoneal administration of cecal slurry, which has been shown to be an excellent and reliable alternative to the cecal ligation and puncture model of sepsis.24 Briefly, cecal slurry was prepared from C57Bl/6 WT mice as described previously,24 and CD169+ macrophage-depleted and control mice were injected intraperitoneally with a sublethal dose of cecal slurry preparation and monitored for survival. We found that CD169-DTR mice are significantly more susceptible to cecal slurry-induced septic shock compared with WT mice by day 2 post injection (Figure 2D).
Figure 2. CD169+ macrophages are required for the host survival during sepsis.
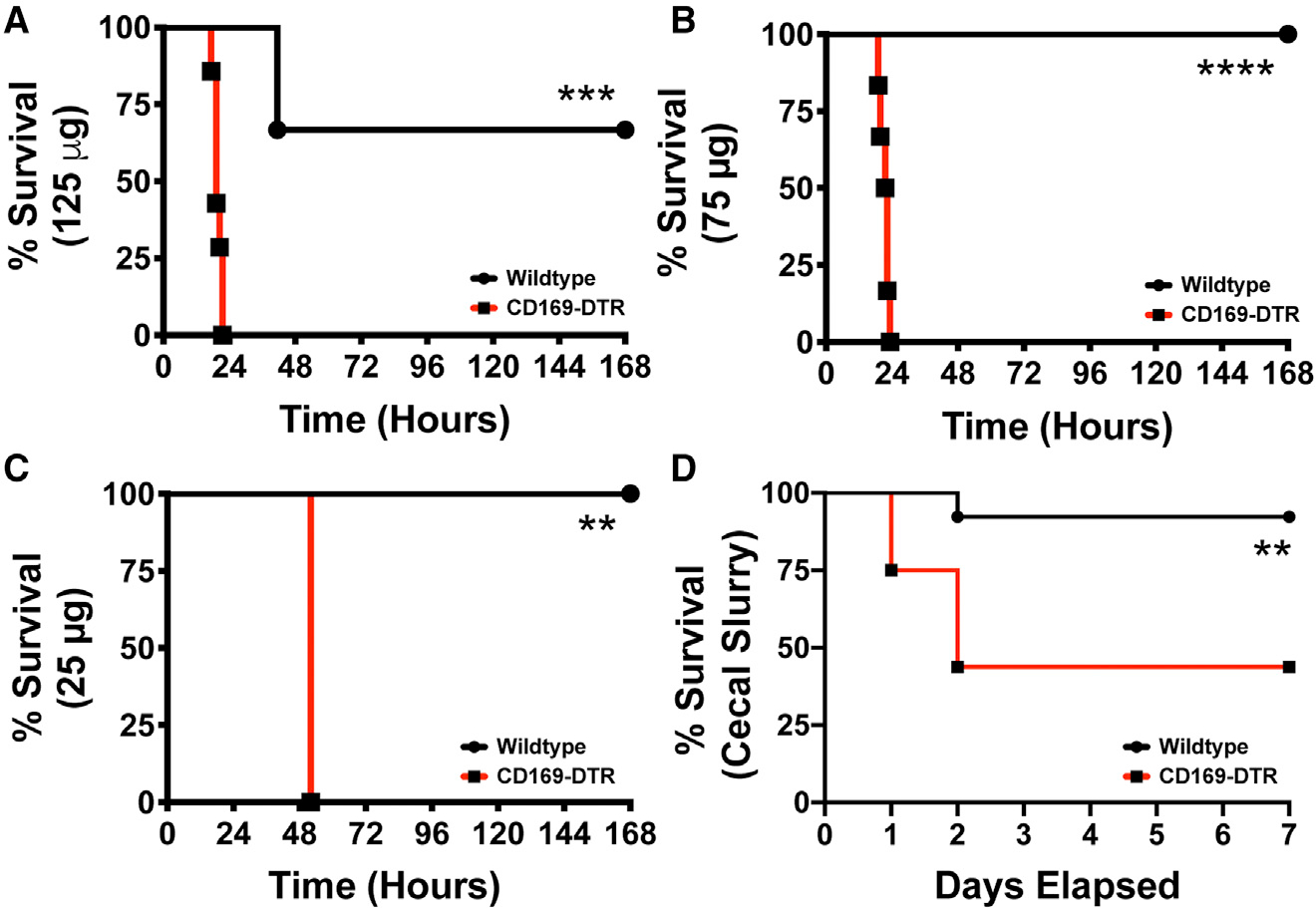
(A–C) WT (black) vs. CD169-DTR (red) mice challenged with Escherichia coli O111:B4 phenol LPS (A) 125, (B) 75, and (C) 25 μg O111:B4 Phenol LPS.
(D) Survival curve of WT (black) and CD169-DTR (red) mice following 150 μL cecal slurry injection. **p < 0.01, ***p < 0.001, ****p < 0.0001. n = 3–5/group and of 2 independent experiments.
CD169+ macrophages in the liver can also contribute to systemic inflammation25; however, only multiple systemic DT treatments results in depletion of these cells in the liver.13 Nevertheless, we next determined if only splenic CD169+ macrophages were critical for LPS sensitivity in the CD169-DTR mice. We treated WT and CD169-DTR mice with DT intraperitoneally or intravenously followed by intraperitoneal LPS injection (Figure S1). We found that the intravenous DT treatment, which primarily results in depletion of CD169+ macrophages in the spleen and not liver (as previously shown), resulted in equal susceptibility compared with intraperitoneal depletion (Figure S1).13 This suggests that splenic-resident CD169+ macrophages have a critical role in regulating systemic LPS susceptibility independently of the liver.
CD169+ macrophages control inflammatory cytokine responses during endotoxemia
We next sought to understand the immunological dysregulation in the absence of CD169+ macrophages in vivo. We analyzed inflammatory cytokines in the serum at 1.5, 3, 6, and 12 h post LPS treatment in WT and CD169-DTR mice. We found that in the absence of CD169+ macrophages, the mice exhibited elevated production of pro-inflammatory cytokine/chemokine characterized by increase in IL-6, tumor necrosis factor α (TNF-α), IL-12(p70), CCL2, CCL3, CCL4, and CCL5 (Figures 3A and 3B) production. Interestingly, we also observed impaired production of anti-inflammatory cytokines IL-10 and IL-12(p40) (Figure 3C), with transient changes in IL-1β, IL-4, and G-CSF (Figures 3A and 3D). This suggested that splenic CD169+ macrophages are critical for controlling the cytokine storm elicited by LPS. Analysis of several other cytokines and chemokines showed that majority of them were slightly higher when the macrophages were depleted after LPS treatment with the exception of IL-18 (Figure S2). As expected, IL-18, which is known to be produced by CD169+ macrophages in lymph nodes, was reduced in CD169 DTR mice.26 Flow cytometric analysis revealed that in the absence of CD169+ macrophages, there was a significant increase in neutrophil infiltration in the peritoneal cavity and spleen at 6 h post LPS treatment (Figures 3E and 3G). We observed a significant increase in Ly6C+ monocytes and natural killer (NK) cells in the peritoneal cavity and blood at 3 and 6 h post LPS treatment (Figures 3E and 3F). Interestingly, we also observed a loss of peritoneal CD45+ large cells upon LPS stimulation (Figure 3E). This suggests that in the absence of CD169+ macrophages, the increase in chemokines observed results in a rapid recruitment of inflammatory immune cells that are promoting inflammation and tissue damage.
Figure 3. CD169-DTR mice exhibit increased pro-inflammatory cytokines and chemokines, impaired anti-inflammatory cytokines, and infiltration of pro-inflammatory cells in the peritoneal cavity, spleen, and circulation.
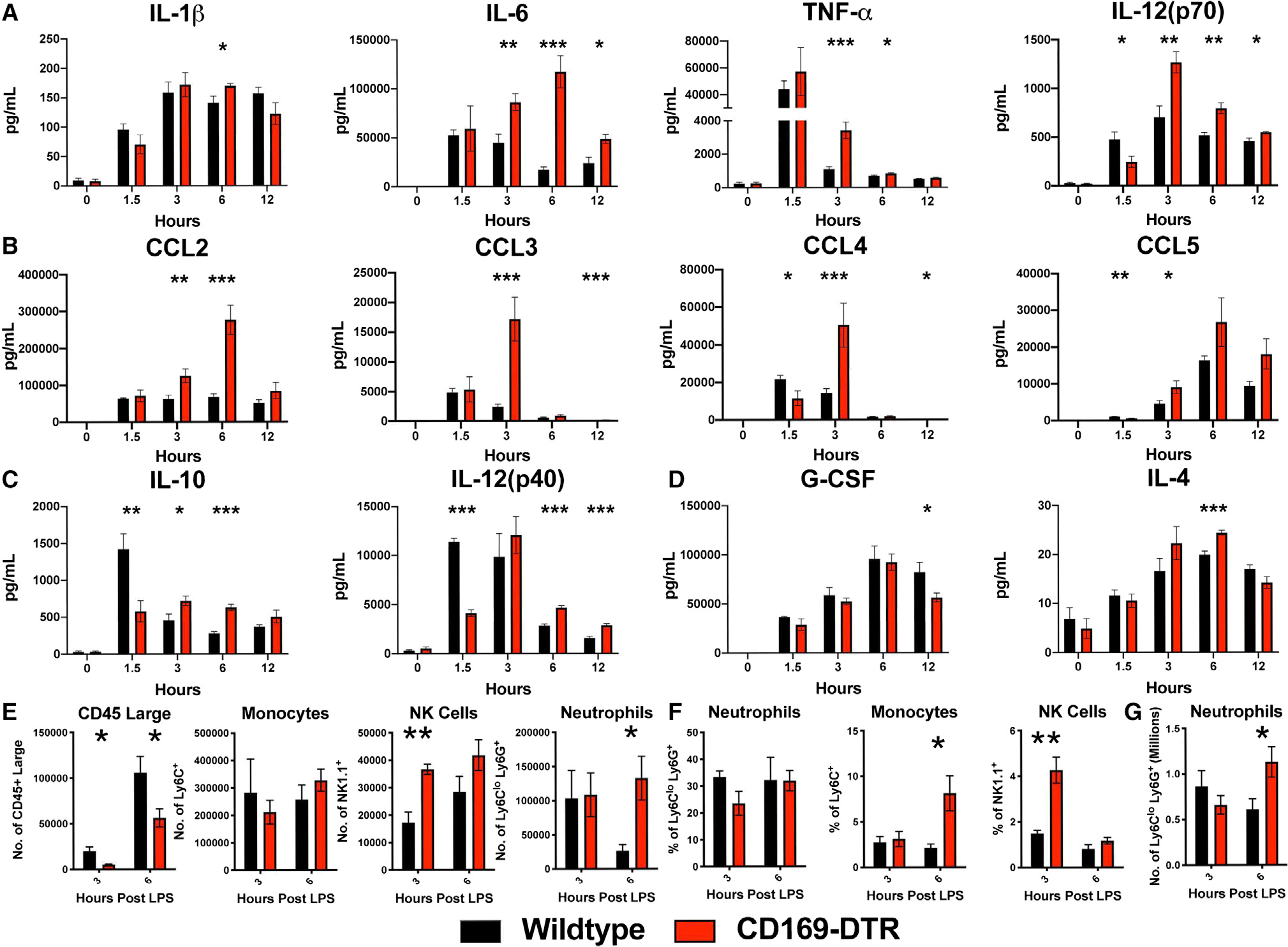
(A–D) Cytokines and chemokines levels between WT (black) and CD169-DTR (red) mice challenged with 75 μg LPS from E. coli O111:B4 intraperitoneally (i.p.).
(A) Pro-inflammatory cytokines (left to right) IL-1β, IL-6, TNF-α, and IL-12(p70), (B) chemokines (left to right) CCL2, CCL3, CCL4, and CCL5, and (C) anti-inflammatory cytokines IL-10 and IL-12(p40) and (D) G-CSF and IL-4.
(E–G) Time course cellularity of (E) peritoneal exudate cells (PECs), (F) blood, and (G) spleen of WT (black) and CD169-DTR (red) mice 3 and 6 h post treatment with 75 μg LPS from E. coli O111:B4 i.p.
*p < 0.05, **p < 0.01, ***p < 0.001. n = 3–5/group and of 2 independent experiments.
IL-6 neutralization fails to ameliorate, and neutrophil depletion exacerbates, the inflammatory response under septic conditions
We next began exploring the mechanisms responsible for the lethality observed in CD169+ macrophage-depleted mice during endotoxin shock. As shown above, we observed a significant increase in pro-inflammatory cytokine IL-6 in CD169-DTR mice early after LPS treatment. Thus, we next aimed to determine whether neutralization of IL-6 would be sufficient to rescue mice depleted of CD169+ macrophages during LPS-induced septic shock. To address this, we treated WT and CD169-DTR mice 2 h post LPS treatment with either anti-IL-6 or isotype control antibody intraperitoneally and monitored the mice for survival (Figure 4A). IL-6 neutralization failed to rescue CD169-DTR mice from septic shock following LPS treatment (Figure 4B). Additionally, we observed an increase neutrophil infiltration in the spleen following LPS treatment. Since unrestrained neutrophil response can lead to overt immunopathology, we next aimed to determine whether neutrophil depletion would be sufficient to rescue the CD169+ macrophage-depleted mice from septic shock. We administered either anti-Ly6G depleting antibody or isotype control 1 h post LPS treatment (Figure 4C). Temporal depletion of neutrophils in fact exasperated LPS-induced inflammatory disease in CD169-DTR mice (Figure 4D). These results suggest that neither overproduction of IL-6 or increased neutrophil recruitment was responsible for endotoxin-induced lethality in the absence of CD169+ macrophages.
Figure 4. Neutrophil depletion or IL-6 neutralization fails to rescue CD169-DTR susceptibility to LPS.
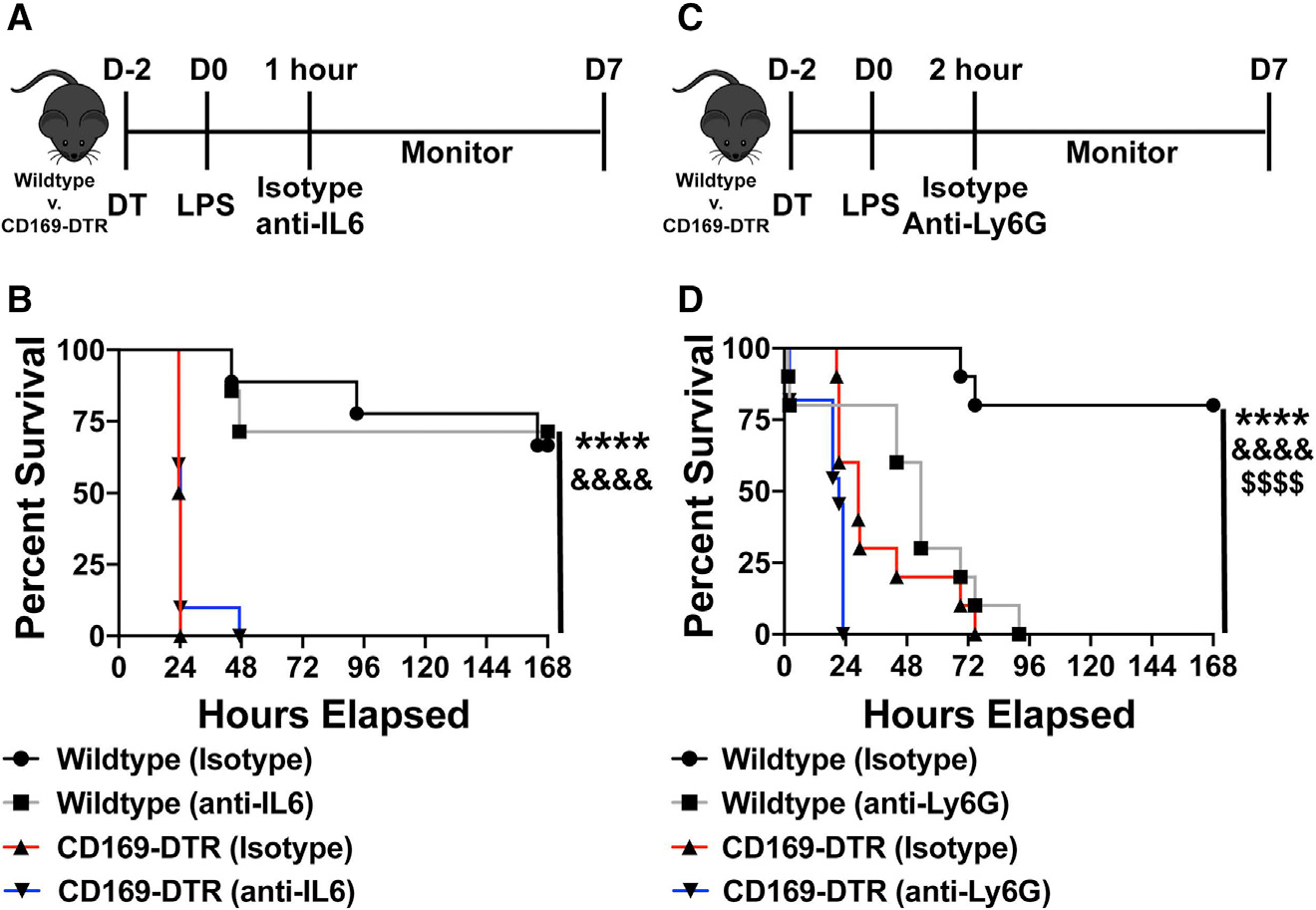
(A) Experimental timeline of IL-6 neutralization following LPS challenge.
(B) Survival curve between WT and CD169-DTR mice given isotype control or anti-IL-6 antibody.
(C) Experimental timeline of neutrophil depletion following LPS challenge.
(D) Survival curve between WT and CD169-DTR mice given isotype control or anti-Ly6G antibody.
****p < 0.0001, &&&&p < 0.0001, $$$$p < 0.0001. n = 3–5/group and of 2 independent experiments.
CD169+ macrophages actively produce IL-10
We observed that IL-10 production was significantly reduced at 1.5 h after LPS treatment in mice depleted of CD169+ macrophages. This suggested that CD169+ macrophages might be a source of early IL-10, which may be an important component of the counter response to the inflammatory cytokine storm during septic shock. Moreover, macrophages can produce baseline IL-10 independent of regulatory T cells,27–30 and even under inflammatory conditions, macrophages are an early source of IL-10.29,30 However, which macrophage subset is the main source of IL-10 during septic shock is not known. Thus, to determine if CD169+ macrophages are capable of producing IL-10 during homeostatic conditions and after the onset of septic shock, we utilized the IL-10 GFP mice (VeRT-X mouse model). The VeRT-X IL-10-GFP mouse to interrogate IL-10 production is a useful tool; however, since there are caveats associated with this model,31,32 we only used these mice initially for the purposes of determining if CD169+ macrophages contribute to early IL-10 production following LPS treatment. First, we conducted flow cytometric analysis of IL-10 GFP splenocytes at 0 (naive), 1, and 3 h post LPS stimulation. We found that even during homeostatic conditions, approximately 20%–30% of CD169+ macrophages secreted IL-10 or 40%–60% of IL-10+ myeloid cells are CD169+ macrophages (Figures 5A and 5B). Additionally, at 1 and 3 h following LPS treatment, nearly 40% of CD169+ macrophages remained GFP+ (Figure 5B). These data suggested that among other splenic cells, CD169+ macrophages are a major contributor of IL-10 production early after LPS treatment.
Figure 5. CD169+ macrophages actively produce IL-10, and recombinant IL-10 is sufficient to rescue CD169-DTR sensitivity to LPS.
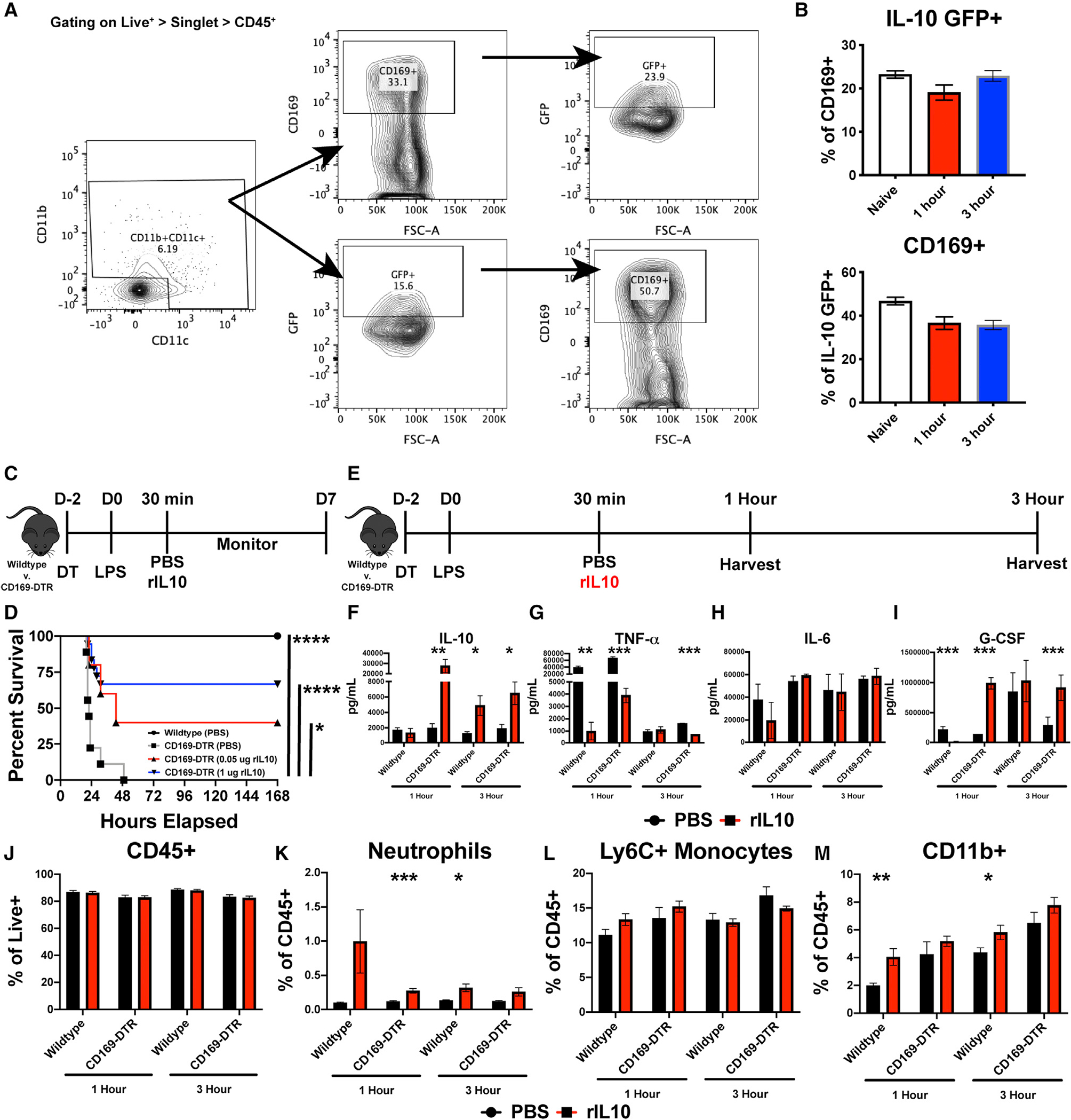
(A) Flow cytometry gating strategy from IL-10-GFP mice.
(B) Percentage of CD169+ cells that are IL-10 GFP+ and IL-10 GFP+ cells that are CD169+ (top) and percentage of IL-10 GFP+ cells that are CD169+ (bottom).
(C) Experimental timeline of recombinant IL-10 (rIL-10) treatment following LPS challenge.
(D) Survival curve between WT and CD169-DTR mice following 75 μg LPS and dose-dependent rIL-10 treatment.
(E) Experimental timeline of organ harvest at 1 and 3 h post LPS stimulation.
(F–I) Cytokine levels following 1 and 3 h post LPS and 1 μg rIL-10, (F) IL-10, (G) TNF-α, (H) IL-6, and (I) G-CSF.
(J–M) Flow cytometric analysis following 1 and 3 h post LPS and 1 μg rIL-10, (J) CD45+, (K) neutrophils, (L) Ly6C + monocytes, and (M) CD11b+ myeloid cells.
Scale bar: 80 μm.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n = 3–5/group and of 2 independent experiments.
CD169+ macrophages rein in inflammatory responses and lethality during endotoxemia by producing IL-10
Next, we determined whether recombinant IL-10 treatment would rescue CD169-DTR mice from endotoxin-induced shock. WT or CD169-DTR mice were either treated with PBS or recombinant IL-10 30 min post LPS treatment (Figure 5C). Strikingly, administration of recombinant IL-10 was sufficient to rescue majority of the CD169+ macrophage-depleted mice from LPS-induced septic shock (Figure 5D). To understand through which mechanism recombinant IL-10 (rIL-10) is able to rescue CD169-DTR mice from LPS-induced septic shock, we analyzed the serum and spleen at 1 and 3 h post LPS treatment for cytokines and chemokines, flow cytometry, and confocal microscopy (Figure 5E). In line with administrating rIL-10, we observed an increase in IL-10 levels in the serum, along with decrease in TNF-α and, interestingly, an increase in G-CSF production but no change in IL-6 levels (Figures 5F–5I). Flow cytometric analysis revealed an increase in neutrophils and CD11b+ cells but no change in total immune cells and monocytes (Figures 5J–5M).
Since IL-10 treatment resulted in the striking rescue of LPS-treated CD169-DTR mice, we next determined if CD169+ macrophage intrinsic IL-10 production is a critical regulatory mechanism that prevents disease associated with septic shock. Thus, we generated mice in which only CD169+ macrophages fail to produce IL-10 by crossing CD169-Cre mice with IL-10flox/flox (CD169-IL-10 conditional knockout [CKO] mice; Figures 6A–6D). We found that upon sublethal administration of LPS, CD169-IL-10 CKO mice exhibited similar susceptibility to LPS shock compared with CD169-DTR mice (Figure 6B). To understand the increased susceptibility in CD169-IL-10 CKO mice from LPS-induced septic shock, we analyzed the serum and spleen at 1 h post LPS treatment for cytokines and chemokines and flow cytometry (Figures 6C and 6D). Consistent with our previous finding of CD169+ macrophages being primary producers of IL-10 (Figure 2C), CD169-IL-10 CKO mice exhibit impaired IL-10 production. Additionally, we observed impaired G-CSF levels in CD169-IL-10 CKO mice compared with control, and no changes were observed in TNF-α or IL-6 (Figure 6C). Flow cytometric analysis revealed decreased neutrophils and CD169+ macrophages but no change in monocytes and F4/80+ macrophages (Figure 6D). Taken together, these results suggest that during homeostatic and acute inflammatory conditions, CD169+ macrophages are a major source of IL-10, and IL-10 production by CD169+ macrophages is an essential regulatory mechanism by which the host is protected during septic shock.
Figure 6. IL-10 production by CD169+ macrophages is required for LPS-induced sepsis protection.
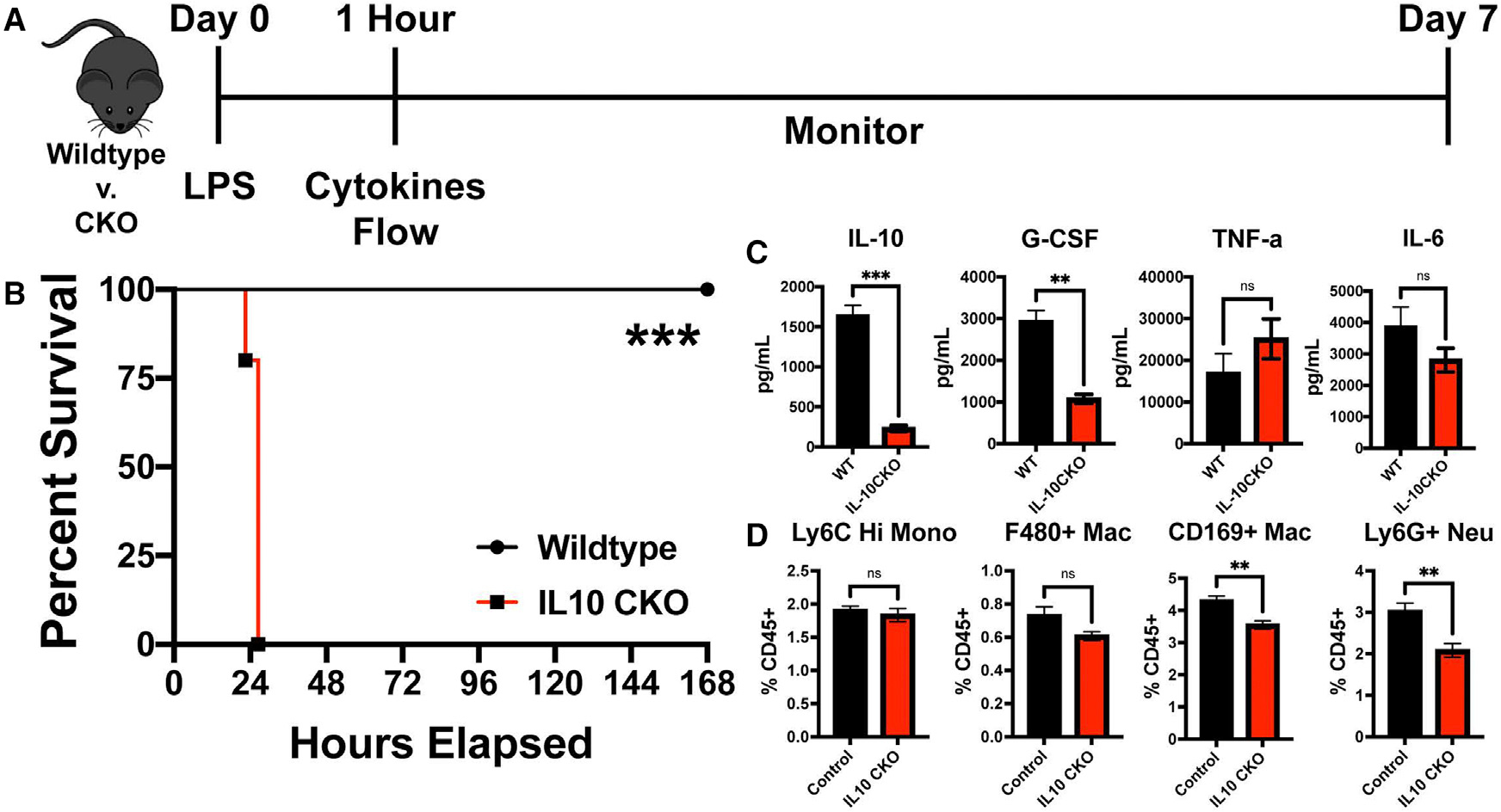
(A) Experimental timeline of LPS challenge of control vs. CD169-IL-10 CKO (IL-10 CKO).
(B) Survival curve between control and IL-10 CKO mice following 75 μg LPS.
(C) (Left to right) IL-10, G-CSF, TNF-α, and IL-6 cytokine levels following 1 h post LPS between control vs. IL-10 CKO.
(D) (Left to right) Splenic Ly6C Hi monocytes, F4/80+ macrophages, CD169+ macrophages, and Ly6G+ neutrophil flow cytometric analysis following 1 h post LPS between control vs. IL-10 CKO mice.
**p < 0.01, ***p < 0.001, ns, not significant. n = 3–5/group and of 2 independent experiments.
DISCUSSION
Macrophage subsets are remarkably versatile and pleiotropic cells that can perform diverse functions such as pathogen clearance and yet maintain immune homeostasis in the tissues in which they reside. The remarkable functional diversity exhibited by different macrophage subsets is largely dependent on the tissue environment and the broad spectrum of the pathological insult. Our current knowledge of the mechanisms that regulate the multifaceted counter-inflammatory responses mediated by macrophages remains incomplete. One such inflammatory condition that is an outcome of uncontrolled bacterial replication that can lead to toxin-mediated excessive inflammation is sepsis. The onset of sepsis induces a complex immunopathological immune response characterized by, but not limited to, increased myeloid cell expansion and cytokine storm that often overwhelms the counter-immunoregulatory measures resulting in tissue damage and morbidity. Although our knowledge of the mechanisms that regulate unrestrained inflammatory pathways in vivo such as sepsis have improved, we know less about the protective counter-regulatory immune mechanisms orchestrated by macrophages in vivo. Increasing our understanding of the regulatory mechanisms that can restrain the damaging effects of the unchecked inflammation under conditions such as sepsis will open new avenues for therapies against myriad of inflammatory disorders. Here, we demonstrated a critical immune-regulatory pathway mediated by a subset of CD169+ macrophages that is necessary for protecting the host during sepsis/bacteremia by secreting IL-10. Macrophages play important roles in the maintenance of immune and tissue homeostasis under steady-state and inflammatory conditions.1–4,13,14,33,34 Growing evidence shows that there is a complex division of labor with respect to the diverse function of macrophage subsets in vivo.5,13,14,27,33,35,36 Although recent studies have begun investigating macrophage function during sepsis in vivo, our understanding of the precise roles these cells play in response to toxins such as LPS is largely based on in vitro studies utilizing bone marrow-derived macrophages or immortalized macrophage cell lines (RAW 264.7, THP-1, etc).34,37–39 Furthermore, in vivo studies have heavily relied on global KO mouse strains (Toll-like receptor 4 [TLR4] KO) or non-specific phagocyte depletion systems (clodronate liposomes) to study macrophage responses during inflammatory conditions.18–22,40 Although these studies have provided important insights into the mechanisms important for pathogen recognition and signaling cascades, our understanding of the subset of macrophages and the underlying mechanisms that these cells use to regulate the damaging inflammation in vivo remains limited
In the current study, we investigated how a particular subset of tissue-resident macrophages (CD169+) respond under septic conditions. We previously showed that the splenic CD169+ macrophages are in fact robustly inflammatory following Listeria infection and are critical for eliminating bacteria.13 Moreover, we recently reported the unique ability of a macrophage subset in the lungs (nerve- and airway-associated interstitial macrophage [NAM]) with similar morphological and phenotypic attributes (as the splenic CD169+ macrophages) to exhibit robust immunoregulatory properties.14 Thus, we sought to study whether splenic CD169+ macrophages serve a protective or pathogenic role under septic conditions13,14
Depletion of CD169+ macrophages resulted in lethality within the first 24 h following LPS challenge as low as 25–75 μg/mouse. Although LPS is a highly utilized model of studying septic shock, to validate our phenotype, we also found that CD169-DTR mice were more susceptible to cecal slurry-induced septic shock. These results suggest that CD169+ macrophages regulate not only LPS-induced shock but polymicrobial-induced septic shock. Previous studies have suggested that liver Kupffer cells were the primary cells to respond to free LPS and its clearance41,42; however, depletion of splenic CD169+ macrophages through intravenous DT treatment resulted in deaths of all animals compared with WT mice. This was not due to depletion of macrophages (i.e., Kupffer cells) in the liver since we had previously shown that that a single DT dose administered intravenously did not deplete CD169+ macrophages or other macrophage subsets in the liver.13,43 Accordingly, we found equal susceptibility to LPS in mice that either received DT intraperitoneally or intravenously. These data suggest that splenic CD169+ macrophages are critical for regulating LPS-induced inflammation. However, it is possible that in addition to the effects we observed in the spleen, lack of CD169+ macrophages and the concomitant increase in LPS in other tissues such as the lungs may also contribute to the increase in inflammation observed in CD169-DTR mice. In the current study, we have used the spleen as a surrogate tissue to interrogate the mechanisms since we have observed that the spleen is primary organ where LPS accumulates early after treatment. LPS is known to be a potent inducer of both pro-inflammatory and anti-inflammatory cytokines and chemokine production.21,44,45 Although, we observed a dramatic increase in pro-inflammatory cytokines and chemokine production, we also found that in the absence of CD169+ macrophages, there was an impairment in early IL-10 production. This suggested that CD169+ macrophages may be a key producer of IL-10, which is consistent with our findings of a population of lung-resident CD169+ macrophage subset (NAMs) that are heavily anti-inflammatory (M2) skewed.14 We also observed little difference in the amount of LPS we detected in the blood of CD169-DTR mice when compared with WT mice. Given this result, we reasoned that CD169+ macrophages in the spleen may serve to uptake LPS for clearance and shield other inflammatory cells from responding to LPS. Indeed, our microscopy studies showed that there was elevated and protracted LPS retention in the spleen in CD169-DTR mice, which likely contributed to the increased pro-inflammatory cytokine storm observed in the absence of CD169+ macrophages.
Immunotherapy such as recombinant cytokine treatment, cytokine blockade/neutralization, or cellular depletion have been shown to be effective to treat overall survival in mouse sepsis studies.40,46–48 IL-6 is a pivotal pro-inflammatory cytokine that is secreted by monocytes and macrophages following TLR activation.49 It has previously been reported that either anti-IL-6 or anti-IL-6R antibody administration following either cecal ligation puncture or LPS-induced septic shock could improve overall survival.40,50 Although we observed a dramatic increase in IL-6 production in the absence of CD169+ macrophages, anti-IL-6 antibody treatment failed to rescue CD169-DTR mice from susceptibility to LPS-induced septic shock. This suggests that although IL-6 neutralization is sufficient to rescue sepsis phenotype in WT mice, it is insufficient in a situation in which CD169+ macrophages are absent. However, although IL-6 neutralization is insufficient, this does not eliminate the potential of using IL-6R neutralization to inhibit trans-presentation of IL-6 by immune and non-immune cells.51 Since IL-6 blockage failed to rescue mice from LPS-induced shock and death, we next considered the possibility that increased neutrophil response was leading to organ damage and failure. Although a previous study showed that neutrophils are in fact protective during LPS-induced septic conditions,52 the dramatic increase in neutrophil numbers in CD169-DTR mice raised the possibility that the ideal protective threshold of the neutrophil response may have been crossed in the absence of CD169+ macrophages, leading to overt damaging inflammation. However, we found that neutrophil depletion, even in the absence of CD169+ macrophages, in fact resulted in an increase susceptibility to septic shock. The precise protective role of neutrophils under septic conditions remains poorly understood; however, neutrophil-derived myeloperoxidase52,53 or secretion of immunosuppressive cytokines such IL-1054 may play an important role. Since IL-6 neutralization or neutrophil depletion failed to alleviate septic shock-induced lethality, we turned our attention to IL-10. Recent studies have shown that IL-10 sensing by macrophages is important for preventing damaging inflammation in several different contexts.27,33,34 However, we observed a significant reduction in IL-10 production early after LPS treatment. IL-10 is a critical anti-inflammatory cytokine that was originally thought to be produced only by regulatory T cells; however, in recent years, macrophages have been shown to produce IL-10 as well.14,27–30,55,56 Indeed, we found that nearly 50% of the immune cells that were producing IL-10 early after LPS challenge were CD169+ macrophages in the spleen, and we were able to rescue lethality due to septic shock by treating CD169-DTR mice with rIL-10 in a dose-dependent manner. We also found that this rescue phenotype is associated with a reduction in TNF-α and IL-6 and an increase in G-CSF, which results in an increase neutrophil response. Finally, the critical evidence that IL-10 production exclusively by CD169+ macrophages is a major protective regulatory mechanism during skeptic shock conditions was revealed by our CD169-IL-10 conditionally deficient mice that responded similarly to LPS treatment as the CD169-DTR mice. Interestingly, IL-10 protein production in CD169-IL-10 conditionally deficient mice was dramatically reduced, which suggested that the majority of the IL-10 in the serum was being produced by CD169+ macrophages. This result did not completely agree with the results we obtained with the IL-10 reporter mice in which we observed that nearly 50% of the cells capable of producing IL-10 were cells other than CD169+ macrophages. This discrepancy may be related to the inherent caveats of using cytokine reporter mice that may not report cytokine protein production with high fidelity.32 The downstream targets of IL-10 are still unknown, although our data showing a reduction in inflammatory cytokine production such as IL-6 and TNF-α and an increase in regulatory cytokines such as G-CSF provide some clues. The scale of observed systemic changes in cytokine production in CD169-DTR mice is too large to be exclusively associated with the absence of CD169+ macrophages. It is likely that in the absence of these macrophages, LPS is sensed by other myeloid cell subsets in peripheral tissues such as the F4/80+ red pulp macrophages, resulting in dramatic alteration in cytokine production that is pathogenic to the animals during LPS-induced shock. Although our study thoroughly explores the remarkable functional diversity of resident macrophages in regulating the inflammatory responses during septic conditions and implicates the important role of the CD169+ macrophage subset in providing the regulatory cues by secreting IL-10, the precise mechanisms by which IL-10 provides protection during sepsis need to be investigated further.
As noted above in our previous study, we demonstrated that splenic CD169+ macrophages polarize toward a potent inflammatory cell type in order to clear Listeria infection. However, in the current study, we observed that the same macrophages serve in a completely different way and provide critical immunoregulatory function. Thus, it appears that resident macrophages in different tissues respond uniquely depending on the context. On one hand, during a pathogenic bacterial infection, splenic CD169+ macrophages become highly inflammatory and are critical in eliminating the bacteria; the very same macrophages during septic conditions provide critical immunoregulatory function and protect the host from inflammatory damage. Thus, the specific nature of the inflammatory stimuli can serve as an important rheostat for regulating macrophage polarization in vivo.
In conclusion, this study reveals the pivotal immunoregulatory role of CD169+ macrophages during sepsis by the secretion of IL-10, thereby providing new therapeutic targets for treating sepsis. Additionally, these results have important implications for other inflammatory disorders such as acute respiratory distress syndrome induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and the potential for targeting specific macrophage subsets and/or IL-10 treatment as a therapeutic strategy that should be explored in future studies.
Limitations of the study
In this study, we have used the spleen as a surrogate for determining the mechanisms utilized by CD169+ macrophages in the marginal zone. However, we were not able to definitively explore the potential role of other CD169+ macrophage subsets in tissues such as the lungs in regulating the inflammatory response during septic conditions. Although our study demonstrates the remarkable functional diversity of resident macrophages in regulating the inflammatory responses during septic conditions and implicates the important role of CD169+ macrophage subset in providing the regulatory cues by secreting IL-10, the precise mechanisms by which IL-10 provides protection during sepsis still remain unknown. Furthermore, in this study, we observed an important role of neutrophils in providing protection against LPS-induced septic shock; however, the precise mechanisms by which neutrophils protect mice from death during septic shock remain to be determined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kamal M. Khanna (kamal.khanna@nyulangone.org).
Materials availability
Mouse lines generated in this study are available from the lead contact upon request (subject to their availability at the time of request).
Data and code availability
All data reported in the paper are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Female C57Bl/6 mice were purchased from Charles River (Frederick, MD). IL-10 F/F mice were kindly provided by Dr. Joseph Sun (Weill Cornell Medicine). All mice listed above, including Siglec1DTR/+ (CD169-DTR) and Siglec1Cre/+ (CD169-Cre) mice, were bred and genotyped at New York University. All the mouse strains used were on a C57BL/6J background between 8 and 15 weeks of age at the start of the experiment. All mice were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited facility. Animals were bred and maintained with food and water ad libitum under a 12-h dark/light cycle in a specific-pathogen free facility at New York University. All animal experiments were performed in accordance with regulatory guidelines and standards set by the Institutional Animal Care and Use Committee of New York University and UConn Health. All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of New York University or UConn Health and were performed in accordance to guidelines from the National Institutes of Health (NIH), the Animal Welfare Act, and U.S. Federal Law.
METHODS DETAILS
Diphtheria toxin administration
Diphtheria toxin (DT, Sigma Aldrich, St. Louis, MO, USA) was prepared in endotoxin free PBS and administered intraperitoneal (i.p.) or intravenously (i.v.) (40 ng/g body weight) day −2 for depletion studies.
LPS treatment
Lipopolysaccharide was prepared in sterile endotoxin free water and 25–400 μg per 200 μL endotoxin free PBS was administered i.p.
Serum collection
Blood serum was collected and isolated using BD Microtainer serum separator tube (Becton Dickinson and Co, Franklin Lakes, NJ, USA) at timepoints listed in experiments.
Cecal slurry shock
Cecal slurry stock was isolated as described.24,45,57 Briefly, whole cecal content dissection from in-house C57Bl/6 WT mice were weighed and resuspended in 0.5 mL 1xPBS/100 mg of cecal content. Slurry was vortexed until homogeneous and filtered through a 100 μm filter. Equal volumes of 30% glycerol/PBS were added and glycerol stocks were aliquoted and frozen down in −8°C until needed. Cecal Slurry shock was conducted by quick thawing cecal slurry aliquot at 37°C and 150 μL of slurry was injected intraperitoneally.
LAL assay
Chromo-Limulus Amebocyte Lysate (LAL) Assay (Associates of Cape Cod, Inc, East Falmouth, MA) was conducted per manufacturer guidelines.
Cytokine and chemokine analysis
Blood serum was quantified through Mouse Cytokine 23-plex array analysis (Bio-Rad, Hercules, CA, USA) for cytokine and chemokine protein levels.
Recombinant mouse IL-10 (rmIL-10)
Recombinant mouse IL-10 (Cat: 417-ML-025, R&D Systems, Minneapolis, MN, USA) was reconstituted in sterile endotoxin-free PBS at 100 μg/mL. Mice received either 1 μg or 0.05 μg in 200 μL of endotoxin-free PBS intraperitoneally 30 min post LPS stimulation.
Neutrophil depletion
Anti-mouse Ly6G depleting antibody (Cat: BE0075–1, Clone: 1A8, BioXCell, West Lebanon, NH, USA) or rat IgG2a isotype control (Cat: BE0089, Clone: 2A3, BioXCell, West Lebanon, NH, USA) was given at 750 μg/mouse intraperitoneally 1 h post LPS stimulation.
Mouse anti-IL-6 blocking
Mouse anti-IL-6 blocking antibody (Cat: BE0046, Clone: MP5–20F3, BioXCell, West Lebanon, NH, USA) or rat IgG1 isotype control (Cat: BE0088, Clone: HRPN, BioXCell, West Lebanon, NH, USA) was given at 1 mg/mouse intraperitoneally 2 h post LPS stimulation.
Tissue preparation for immunofluorescence and confocal microscopy
Tissue were fixed in paraformaldehyde, lysine, and sodium periodate buffer (PLP, 0.05 M phosphate buffer, 0.1M L-lysine, p.H. 7.4, 2 mg/mL NaIO4, and 10 mg/mL paraformaldehyde) overnight at 4°C followed by 30% sucrose overnight at 4°C and subsequent embedding in OCT media. 20 μm frozen tissue sections were sectioned using either a Leica CM3050S or Leica CM1520 cryostat (Leica Biosystems Inc, Buffalo Grove, IL, USA). FcR blocked with anti-CD16/32 Fc block antibody (clone 93, Biolegend, San Diego, CA, USA) diluted in 1xphosphate buffer solution (PBS) containing 2% goat serum and 2% fetal bovine serum (FBS) for 1 h at room temperature. Sections were stained with F4/80-PE (clone BM8, Biolegend, San Diego, CA, USA) and CD169-ef660 (clone Ser-4, Invitrogen, Hampton, NH, USA) that were diluted in PBS containing 2% goat serum and 2% FBS for 1 h at room temperature. For the intracellular staining, all the antibodies including the Fc block were diluted in PBS containing 2% goat serum, 2% FBS, and 0.05% Triton X-. Sections were also washed with the same buffer. Sections were cover slipped using Immun-mount mounting medium (Fisher Scientific, Hampton, NH, USA) and Cover Glasses with a 0.13 to 0.17 mm thickness (Fisher Scientific, Hampton, NH, USA). Fluorescence was detected with a Zeiss LSM 880 confocal microscope (Carl Zeiss, Oberkochen, Germany) equipped with 405, 488, 514, 561, 594, and 633 nm solid-state laser lines, a 32-channel spectral detector (409–695 nm), and 10×0.3, 20x PlanApochromat 0.8, 40x, and 6331.40 objectives. Zen Black (Carl Zeiss, Oberkochen, Germany) software suite was used for data collection. The imaging data were processed and analyzed using Imaris software version 8.3.1 (Bitplane USA; Oxford Instruments, Concord, MA, USA).
Image analysis by FIJI ImageJ and imaris
Single color images were analyzed using ImageJ FIJI software version 2.1.0/1.53c (NIH, Bethesda, MD, USA). where images were converted to 8-bit to establish grey-scale. Next, LPS staining threshold was established, normalized, measured, and percent area was recorded. For LPS colocalized CD169+ macrophages images were analyzed using Imaris software version 8.3.1 (Bitplane USA; Oxford Instruments, Concord, MA, USA). Briefly. CD169+ macrophage or F4/80 + macrophages and LPS-FITC channels generated a new “colocalized” channel which represents CD169+ FITC + or F4/80 + FITC + channel. Next, surface renders were constructed of LPS-FITC, CD169+FITIC+, and F4/80 + FITC + channel. We recorded the sum volume and present the data as % colocalized/FITC which represents LPS colocalized with CD169+ macrophages or F4/80 + macrophages.
Single cell suspension
Organs was extracted and placed in harvest buffer (RPMI supplemented with 5% fetal bovine serum) until cells were subsequently isolated into single cell suspension using organ specific protocols. Splenic tissue was digested in Collagenase composed of RPMI-1640 media containing 5% fetal bovine serum (FBS), 100 U/mL collagenase IV, 1mM HEPES, 5mM glutamine, 1mM CaCl2, 1 mM MgCl2, and 1–50 μg/mL DNase I for 30 min at 37°C. Following incubation, splenic tissue was crushed through a 70 μm filter into a single cell suspension and treated with Tris-buffered ammonium chloride (TAC, 20.6 g/L Trizma HCl (Sigma-Aldrich, St. Louis, MO, USA), 8.3 g/L NH4Cl (Fisher Scientific, Hampton, NH, USA)). Cells were spun down at 1500 RPM for 5 min at 4°C and the supernatant was discarded and cells were resuspended for counting and staining. Peritoneal Exudate Cells Using scissor and forceps the outer skin of the peritoneum was cut and gently pull apart to expose the inner skin lining the peritoneal cavity. 5 mL of ice-cold PBS + 5% FBS was injected into the peritoneal cavity using a 27-gauge needle. After injection, the peritoneum was gently massaged to dislodge any attached cells. Peritoneal PBS solution was collected using the same syringe and deposited into a 15 mL falcon tube and kept on ice. Remaining fluid was collected by opening the peritoneal cavity and pipetting remaining fluid with a transfer pipette. Cells were spun down at 1500 RPM for 5 min at 4°C and the supernatant was discarded and cells were resuspended for counting and staining. Blood was collected via cardiac puncture or retro-orbital bled and red blood cells were lysed in Tris-buffered ammonium chloride (TAC, 20.6 g/L Trizma HCl (Sigma-Aldrich, St. Louis, MO, USA), 8.3 g/L NH4Cl (Fisher Scientific, Hampton, NH, USA)). Cells were spun down at 1500 RPM for 5 min at 4°C and the supernatant was discarded and cells were resuspended for counting and staining.
Flow cytometry
Cells were resuspended in FACS buffer (1xPBS (Gibco, Waltham, MA, USA), 5% FBS, and 0.5% sodium azide). Fc receptors were blocked with anti-CD16/32 Fc block antibody (Biolegend, San Diego, CA, USA) and stained with Live Dead AF350 NHS Ester (Thermofisher, Hampton, NH, USA), CD11c-APC-ef780 (clone N418, Thermofisher, Hampton, NH, USA), CD169-ef660 (clone Ser-4, Thermofisher, Hampton, NH, USA), Ly6G-PerCP-Cy5.5 (clone 1A8, Biolegend, San Diego, CA, USA), CD11b-BV711 (clone M1/70, Biolegend, San Diego, CA, USA), Ly6C (clone HK1.4, Biolegend, San Diego, CA, USA), B220-AF700 (clone RA3–6B2, Biolegend, San Diego, CA, USA), F4/80-BV650 (clone BM8, Biolegend, San Diego, CA, USA), CX3CR1-BV605 (clone SA011F11, Biolegend, San Diego, CA, USA), MHCII-Pacific Blue (clone M5/114.15.2, Biolegend, San Diego, CA, USA), CD64-PE Dazzle 594 (clone X54–5/7.1, Biolegend, San Diego, CA, USA), NK1.1-PE-Cy7 (clone PK136, Biolegend, San Diego, CA, USA), CCR2-PE (R&D Systems, Minneapolis, MN, USA), CD8a-BUV737 (clone 53-6-7, BD Bioscience, Franklin Lakes, NJ, USA), CD45-BUV661 (clone 30-F11, BD Bioscience, Franklin Lakes, NJ, USA), and CD4-Pacific Orange (clone RM4–5, Invitrogen, Hampton, NH, USA) for 20 min in 4°C. Cells were fixed with 2% PFA for 20 min in 4°C and resuspended in FACS buffer. Cells suspension processed on Becton-Dickinson LSRII or Bio-Rad ZE5 Yeti instrument and data analyzed in FlowJo software. For cell sorting, lung tissue was treated in the same way without fixation.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was performed using Prism 7 GraphPad Software (La Jolla, CA). For experiments comparing 2 groups, two-tail Student t-test was used to determine statistical significance. For survival studies, Log rank Mantel-Cox test and Gehan-Breslow-Wilcoxon test for extra weight for early time points was performed. For bacterial quantification experiments non-Gaussian distribution was assumed and Mann-Whitney test was performed. For experiments comparing three or more groups, one-way ANOVA, Bonferroni test was used to determine statistical significance for sample groups that assume Gaussian distribution or Kruskal-Wallis H test with sample groups that assume non-Gaussian distribution. Statistical significance is determined as *p < 0.05, **p < 0.01, ***p < 0.001 and N.S. for not significant. Data are represented as mean ± SEM, unless otherwise specified.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| AF350 NHS Ester (Live/Dead) | ThermoFisher | Catalog # A10168 |
| Rat Anti-Mouse CD16/32 (clone 93) | Biolegend | Catalog # 101302 |
| Armenian Hamster anti-Mouse CD11c, PE (clone N418) | Biolegend | Catalog # 117308 |
| Rat anti-Mouse CD169, ef660 (clone Ser-4) | Invitrogen | Catalog # 50–5755-82 |
| Rat anti-Mouse F4/80, PE (clone BM8) | Biolegend | Catalog #123110 |
| Streptavidin, Alexa Fluor 488, Conjugate | Invitrogen | Catalog #S11223 |
| Rat anti-Mouse CD45, BUV661 (clone 30-F11) | BD Bioscience | Catalog # 565079 |
| Rat anti-Mouse CD8a, BUV737 (clone 53–6.7) | BD Bioscience | Catalog # 564297 |
| Rat anti-Mouse Ly6G, PerCP-Cy5.5 (clone 1A8) | Biolegend | Catalog # 127616 |
| Rat anti-Mouse CCR2, PE | R&D Systems | Catalog # FAB5538P |
| Rat anti-Mouse NK1.1, PE-Cy7 (clone PK136) | Biolegend | Catalog # 108714 |
| Rat ant-Mouse CD64, PE-Dazzle 594 (clone X54–5/7.1) | Biolegend | Catalog # 139320 |
| Rat anti-Mouse MHCII, Pacific Blue (clone M5/114.15.2) | Biolegend | Catalog # 107620 |
| Rat anti-Mouse CD4, Pacific Orange (clone RM4–5) | Invitrogen | Catalog # MCD0430 |
| Rat anti-Mouse CX3CR1, BV605 (clone SA011F11) | Biolegend | Catalog # 149027 |
| Rat anti-Mouse F4/80, BV650 (clone BM8) | Biolegend | Catalog # 123149 |
| Rat anti-Mouse CD11b, BV711 (clone M1/70) | Biolegend | Catalog # 101241 |
| Rat anti-Mouse Ly6C, BV786 (clone HK1.4) | Biolegend | Catalog # 128041 |
| Rat anti-Mouse B220, AF700 (clone RA3–6B2) | Biolegend | Catalog # 103232 |
| Rat anti-Mouse CD11c, APC-eF780 (clone N418) | Invitrogen | Catalog # 47–0114-82 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Triton X-100 | Fisher Scientific | Catalog # BP151–100 |
| 32% Paraformaldehyde | EMS Inc | Catalog # 15714S |
| Sodium Periodate | Sigma Aldrich | Catalog #311448 |
| Shandon Immu-Mount | Fisher Scientific | Catalog # 9990402 |
| Normal Goat Serum | Millipore | Catalog # NS02L |
| Fetal Calf Serum | Gibco | Catalog # 16000044 |
| RPMI 1640 with L-Glutamine | Lonza | Catalog # BE12–702F |
| DNase I | Sigma Aldrich | Catalog # 10104159001 |
| Collagenase IV | Gibco | Catalog # 17–104-019 |
| Gibco Pen/Strep | ThermoFisher | Catalog # 15140122 |
| CaCl2 | Fisher Scientific | Catalog # C79 |
| MgCl2 | Fisher Scientific | Catalog # M35 |
| Gentamicin | Gibco | Catalog # 15750–060 |
| Disodium Phosphate (Na2HPO4) | Sigma Aldrich | Catalog # S3264 |
| Monosodium Phosphate (H2NaO4P) | Sigma Aldrich | Catalog # S3139 |
| L-Lysine HCl | Sigma Aldrich | Catalog # 4400 |
| Sucrose | Sigma Aldrich | Catalog #S8501 |
| IxPBS | Gibco | Catalog # 10010023 |
| Endotoxin-Free Ultra Pure Water | Millipore Sigma | Catalog # TMS-011-A |
| Endotoxin-Free Dulbecco's PBS (1X) (w/o Ca2+ & Mg++) | Millipore Sigma | Catalog #TMS-012-A |
| Phenol LPS - E.coli O111:B4 | Sigma Aldrich | Catalog # L2630 |
| LPS-FITC - E.coli O111.B4 | Sigma Aldrich | Catalog # F3665 |
| Diphtheria toxin | Sigma Aldrich | Catalog # D0564 |
| Recombinant mouse IL-10 | R&D Systems | Catalog # 417-ML-025 |
| Anti-mouse Ly6G depleting antibody (1A8) | BioXCell | Catalog # BE0075–1 |
| Rat IgG2a isotype control (2A3) | BioXCell | Catalog # BE0089 |
| Anti-mouse anti-IL-6 blocking antibody (MP5–20F3) | BioXCell | Catalog # BE0046 |
| Rat IgG1 isotype control (HRPN) | BioXCell | Catalog # BE0088 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Bio-Plex Pro mouse cytokine 23-plex Assay | Bio-Rad | Catalog # M60009rdpd |
| Chromo-LAL | ACCIUSA | Catalog # C0031–5 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: CD169-DTR: B6; 129-Siglec1 < tm1(HBEGF)Mtka> | New York University: Originally from Makoto Tanaka | RRID:IMSR_NM-KO-201894 |
| Mouse: C57BL/6-Siglec1 <tm1(cre)Mtka> | New York University: Originally from Makoto Tanaka | RRID:IMSR_RBRC06239 |
| Mouse: C57Bl/6NCrl | New York University: Originally from Charles Rivers | RRID:IMSR_CRL:027 |
| Mouse: IL-10flox/flox | New York University: Originally from Joseph Sun | RRID:IMSR_NM-CKO-200003 |
| Mouse: B6(cg)-Il10tm1.1Karp/J | New York University: Originally from The Jackson Laboratory | RRID:IMSR_JAX:014530 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo (Versions 10.7.1) | Becton Dickinson & Company | RRID:SCR_008520 |
| Prism (Version 8) | Graphpad | RRID:SCR_002798 |
| Illustrator CC2020 | Adobe | RRID:SCR_010279 |
| ImageJ - FIJI (Version 2.1.0/1.53c | NIH | RRID:SCR_002285 |
| Imaris (Version 9.0) | Bitplane | RRID:SCR_007370 |
| Zen Digital Imaging for Light Microscopy, Zen 2012 | Zeiss | RRID:SCR_013672 |
| Everest Software (Version 2.3) | Bio-Rad | Bio-rad.com |
| BD FACSDiva (Version 8.01) | Becton Dickinson & Company | RRID:SCR_001456 |
Highlights.
CD169+ macrophages provide critical regulatory cues to maintain immune homeostasis
CD169+ macrophages provide counter-inflammatory responses during sepsis by secreting IL-10
The type of inflammatory stimuli serves as a rheostat for regulating macrophage polarization
ACKNOWLEDGMENTS
The authors would like to acknowledge Xuanmai Pham and Quynh Pham for technical assistance. We acknowledge NYU Langone Health Microscopy Laboratory, which is partially funded by NYU Cancer Center support grant NCI P30CA016087, for assistance and equipment support. The graphical abstract was drawn with Biorender software. This study was funded by 1R01AI143861 and 3R01AI143861-02S1 to K.M.K.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112171.
REFERENCES
- 1.Amit I, Winter DR, and Jung S (2016). The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat. Immunol. 17, 18–25. 10.1038/ni.3325. [DOI] [PubMed] [Google Scholar]
- 2.Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, and Lecuit M (2015). Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 42, 145–158. [DOI] [PubMed] [Google Scholar]
- 3.Glass CK, and Natoli G (2016). Molecular control of activation and priming in macrophages. Nat. Immunol. 17, 26–33. 10.1038/ni.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kierdorf K, Prinz M, Geissmann F, and Gomez Perdiguero E (2015). Development and function of tissue resident macrophages in mice. Semin. Immunol. 27, 369–378. 10.1016/j.smim.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asano K, Takahashi N, Ushiki M, Monya M, Aihara F, Kuboki E, Moriyama S, Iida M, Kitamura H, Qiu CH, et al. (2015). Intestinal CD169(+) macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat. Commun. 6, 7802. 10.1038/ncomms8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, Pinho S, Leboeuf M, Noizat C, van Rooijen N, et al. (2013). CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 19, 429–436. 10.1038/nm.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.den Haan JMM, and Kraal G (2012). Innate immune functions of macrophage subpopulations in the spleen. J. Innate Immun. 4, 437–445. 10.1159/000335216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez-Pomares L, and Gordon S (2012). CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol. 33, 66–70. [DOI] [PubMed] [Google Scholar]
- 9.O’Neill ASG, van den Berg TK, and Mullen GED (2013). Sialoadhesin - a macrophage-restricted marker of immunoregulation and inflammation. Immunology 138, 198–207. 10.1111/imm.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aichele P, Zinke J, Grode L, Schwendener RA, Kaufmann SHE, and Seiler P (2003). Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J. Immunol. 171, 1148–1155. 10.4049/jimmunol.171.3.1148. [DOI] [PubMed] [Google Scholar]
- 11.Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, and von Andrian UH (2010). Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature 465, 1079–1083. 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, et al. (2007). Subcapsular sinus macrophages in lymph nodes clear lymphborne viruses and present them to antiviral B cells. Nature 450, 110–114. 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 13.Perez OA, Yeung ST, Vera-Licona P, Romagnoli PA, Samji T, Ural BB, Maher L, Tanaka M, and Khanna KM (2017). CD169(+) macrophages orchestrate innate immune responses by regulating bacterial localization in the spleen. Sci. Immunol. 2, eaah5520. 10.1126/sciimmunol.aah5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ural BB, Yeung ST, Damani-Yokota P, Devlin JC, de Vries M, Vera-Licona P, Samji T, Sawai CM, Jang G, Perez OA, et al. (2020). Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci. Immunol. 5, eaax8756. 10.1126/sciimmunol.aax8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin GS, Mannino DM, Eaton S, and Moss M (2003). The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348, 1546–1554. 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 16.Seymour CW, Gesten F, Prescott HC, Friedrich ME, Iwashyna TJ, Phillips GS, Lemeshow S, Osborn T, Terry KM, and Levy MM (2017). Time to treatment and mortality during mandated emergency Care for sepsis. N. Engl. J. Med. 376, 2235–2244. 10.1056/NEJMoa1703058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hotchkiss RS, Tinsley KW, and Karl IE (2003). Role of apoptotic cell death in sepsis. Scand. J. Infect. Dis. 35, 585–592. 10.1080/00365540310015692. [DOI] [PubMed] [Google Scholar]
- 18.Dauphinee SM, and Karsan A (2006). Lipopolysaccharide signaling in endothelial cells. Lab. Invest. 86, 9–22. 10.1038/labinvest.3700366. [DOI] [PubMed] [Google Scholar]
- 19.Shen XF, Cao K, Jiang JP, Guan WX, and Du JF (2017). Neutrophil dysregulation during sepsis: an overview and update. J. Cell Mol. Med. 21, 1687–1697. 10.1111/jcmm.13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiche L, Forel JM, Thomas G, Farnarier C, Vely F, Bléry M, Papazian L, and Vivier E (2011). The role of natural killer cells in sepsis. J. Biomed. Biotechnol. 2011, 986491. 10.1155/2011/986491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russo AJ, Behl B, Banerjee I, and Rathinam VAK (2018). Emerging insights into noncanonical inflammasome recognition of microbes. J. Mol. Biol. 430, 207–216. 10.1016/j.jmb.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller SI, Ernst RK, and Bader MW (2005). LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 3, 36–46. 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 23.Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, Kanagawa O, Fujii SI, and Tanaka M (2011). CD169-Positive macrophages dominate antitumor immunity by crosspresenting Dead cell-associated antigens. Immunity 34, 85–95. [DOI] [PubMed] [Google Scholar]
- 24.Starr ME, Steele AM, Saito M, Hacker BJ, Evers BM, and Saito H (2014). A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PLoS One 9, e115705. 10.1371/journal.pone.0115705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta P, Lai SM, Sheng J, Tetlak P, Balachander A, Claser C, Renia L, Karjalainen K, and Ruedl C (2016). Tissue-resident CD169(+) macrophages form a crucial front line against plasmodium infection. Cell Rep. 16, 1749–1761. 10.1016/j.celrep.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Kastenmüller W, Torabi-Parizi P, Subramanian N, Läammermann T, and Germain RN (2012). A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 150, 1235–1248. 10.1016/j.cell.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, Brenner O, Krauthgamer R, Varol C, Müller W, and Jung S (2014). Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 40, 720–733. 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Schridde A, Bain CC, Mayer JU, Montgomery J, Pollet E, Denecke B, Milling SWF, Jenkins SJ, Dalod M, Henri S, et al. (2017). Tissuespecific differentiation of colonic macrophages requires TGFbeta receptor-mediated signaling. Mucosal Immunol. 10, 1387–1399. 10.1038/mi.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Müller W, Sparwasser T, Förster R, and Pabst O (2011). Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246. 10.1016/j.immuni.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 30.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, and Kronenberg M (2009). Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 10, 1178–1184. 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bouabe H (2012). Cytokine reporter mice: the special case of IL-10. Scand. J. Immunol. 75, 553–567. 10.1111/j.1365-3083.2012.02695.x. [DOI] [PubMed] [Google Scholar]
- 32.Özkan M, Eskiocak YC, and Wingender G (2021). The IL-10GFP (VeRT-X) mouse strain is not suitable for the detection of IL-10 production by granulocytes during lung inflammation. PLoS One 16, e0247895. 10.1371/journal.pone.0247895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shemer A, Scheyltjens I, Frumer GR, Kim JS, Grozovski J, Ayanaw S, Dassa B, Van Hove H, Chappell-Maor L, Boura-Halfon S, et al. (2020). Interleukin-10 prevents pathological microglia hyperactivation following peripheral endotoxin challenge. Immunity 53, 1033–1049.e7. 10.1016/j.immuni.2020.09.018. [DOI] [PubMed] [Google Scholar]
- 34.Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. 10.1126/science.aal3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q, Wang D, Hao S, Han X, Xia Y, Li X, Chen Y, Tanaka M, and Qiu CH (2017). CD169 expressing macrophage, a key subset in mesenteric lymph nodes promotes mucosal inflammation in dextran sulfate sodium-induced colitis. Front. Immunol. 8, 669. 10.3389/fimmu.2017.00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bosshart H, and Heinzelmann M (2016). THP-1 cells as a model for human monocytes. Ann. Transl. Med. 4, 438. 10.21037/atm.2016.08.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taciak B, Bia1asek M., Braniewska A., Sas Z., Sawicka., Kiraga Ł., Rygiel T, and Król M. (2018). Evaluation of phenotypic and functional stability of RAW 264.7 cell line through serial passages. PLoS One 13, e0198943. 10.1371/journal.pone.0198943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hagar JA, Powell DA, Aachoui Y, Ernst RK, and Miao EA (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Honda SI, Sato K, Totsuka N, Fujiyama S, Fujimoto M, Miyake K, Nakahashi-Oda C, Tahara-Hanaoka S, Shibuya K, and Shibuya A (2016). Marginal zone B cells exacerbate endotoxic shock via interleukin-6 secretion induced by Fcalpha/muR-coupled TLR4 signalling. Nat. Commun. 7, 11498. 10.1038/ncomms11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao Z, Mates JM, Cheplowitz AM, Hammer LP, Maiseyeu A, Phillips GS, Wewers MD, Rajaram MVS, Robinson JM, Anderson CL, and Ganesan LP (2016). Blood-borne lipopolysaccharide is rapidly eliminated by liver sinusoidal endothelial cells via high-density lipoprotein. J. Immunol. 197, 2390–2399. 10.4049/jimmunol.1600702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shao B, Munford RS, Kitchens R, and Varley AW (2012). Hepatic uptake and deacylation of the LPS in bloodborne LPS-lipoprotein complexes. Innate Immun. 18, 825–833. 10.1177/1753425912442431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, and Tanaka M (2007). Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J. Clin. Invest. 117, 2268–2278. 10.1172/JCI31990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gnauck A, Lentle RG, and Kruger MC (2016). The characteristics and function of bacterial lipopolysaccharides and their endotoxic potential in humans. Int. Rev. Immunol. 35, 189–218. 10.3109/08830185.2015.1087518. [DOI] [PubMed] [Google Scholar]
- 45.Gonnert FA, Recknagel P, Seidel M, Jbeily N, Dahlke K, Bockmeyer CL, Winning J, Lösche W, Claus RA, and Bauer M (2011). Characteristics of clinical sepsis reflected in a reliable and reproducible rodent sepsis model. J. Surg. Res. 170, e123–e134. 10.1016/j.jss.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 46.Pajkrt D, Camoglio L, Tiel-van Buul MC, de Bruin K, Cutler DL, Affrime MB, Rikken G, van der Poll T, ten Cate JW, and van Deventer SJ (1997). Attenuation of proinflammatory response by recombinant human IL-10 in human endotoxemia: effect of timing of recombinant human IL-10 administration. J. Immunol. 158, 3971–3977. [PubMed] [Google Scholar]
- 47.Pajkrt D, van der Poll T, Levi M, Cutler DL, Affrime MB, van den Ende A, ten Cate JW, and van Deventer SJ (1997). Interleukin-10 inhibits activation of coagulation and fibrinolysis during human endotoxemia. Blood 89, 2701–2705. [PubMed] [Google Scholar]
- 48.Gérard C, Bruyns C, Marchant A, Abramowicz D, Vandenabeele P, Delvaux A, Fiers W, Goldman M, and Velu T (1993). Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J. Exp. Med. 177, 547–550. 10.1084/jem.177.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garbers C, Heink S, Korn T, and Rose-John S (2018). Interleukin-6: designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 17, 395–412. 10.1038/nrd.2018.45. [DOI] [PubMed] [Google Scholar]
- 50.Riedemann NC, Neff TA, Guo RF, Bernacki KD, Laudes IJ, Sarma JV, Lambris JD, and Ward PA (2003). Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J. Immunol. 170, 503–507. 10.4049/jimmunol.170.1.503. [DOI] [PubMed] [Google Scholar]
- 51.Moore JB, and June CH (2020). Cytokine release syndrome in severe COVID-19. Science 368, 473–474. 10.1126/science.abb8925. [DOI] [PubMed] [Google Scholar]
- 52.Reber LL, Gillis CM, Starkl P, Jönsson F, Sibilano R, Marichal T, Gaudenzio N, Bérard M, Rogalla S, Contag CH, et al. (2017). Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J. Exp. Med. 214, 1249–1258. 10.1084/jem.20161238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ioannou M, Hoving D, Aramburu IV, Temkin MI, De Vasconcelos NM, Tsourouktsoglou TD, Wang Q, Boeing S, Goldstone R, Vernardis S, et al. (2022). Microbe capture by splenic macrophages triggers sepsis via T cell-death-dependent neutrophil lifespan shortening. Nat. Commun. 13, 4658. 10.1038/s41467-022-32320-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.González LA, Melo-González F, Sebastián VP, Vallejos OP, Noguera LP, Suazo ID, Schultz BM, Manosalva AH, Peñaloza HF, Soto JA, et al. (2021). Characterization of the anti-inflammatory capacity of IL-10-producing neutrophils in response to Streptococcus pneumoniae infection. Front. Immunol. 12, 638917. 10.3389/fimmu.2021.638917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pils MC, Pisano F, Fasnacht N, Heinrich JM, Groebe L, Schippers A, Rozell B, Jack RS, and Müller W (2010). Monocytes/macrophages and/or neutrophils are the target of IL-10 in the LPS endotoxemia model. Eur. J. Immunol. 40, 443–448. 10.1002/eji.200939592. [DOI] [PubMed] [Google Scholar]
- 56.Siewe L, Bollati-Fogolin M, Wickenhauser C, Krieg T, Müller W, and Roers A (2006). Interleukin-10 derived from macrophages and/or neutrophils regulates the inflammatory response to LPS but not the response to CpG DNA. Eur. J. Immunol. 36, 3248–3255. 10.1002/eji.200636012. [DOI] [PubMed] [Google Scholar]
- 57.Consoli DC, Jesse JJ, Klimo KR, Tienda AA, Putz ND, Bastarache JA, and Harrison FE (2020). A cecal slurry mouse model of sepsis leads to acute consumption of vitamin C in the brain. Nutrients 12, 911. 10.3390/nu12040911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in the paper are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.