

Abstract
The human genome is folded into a multi-level 3D structure that controls many nuclear functions including gene expression. Recently, alterations in 3D genome organization were associated with several genetic diseases and cancer. As a consequence, experimental approaches are now being developed to modify the global 3D genome organization and that of specific loci. Here, we discuss emerging experimental approaches of 3D genome editing that may prove useful in biomedicine.
Keywords: genetic diseases, cancer, 3D genome, epigenome editing, therapy
Graphical abstract
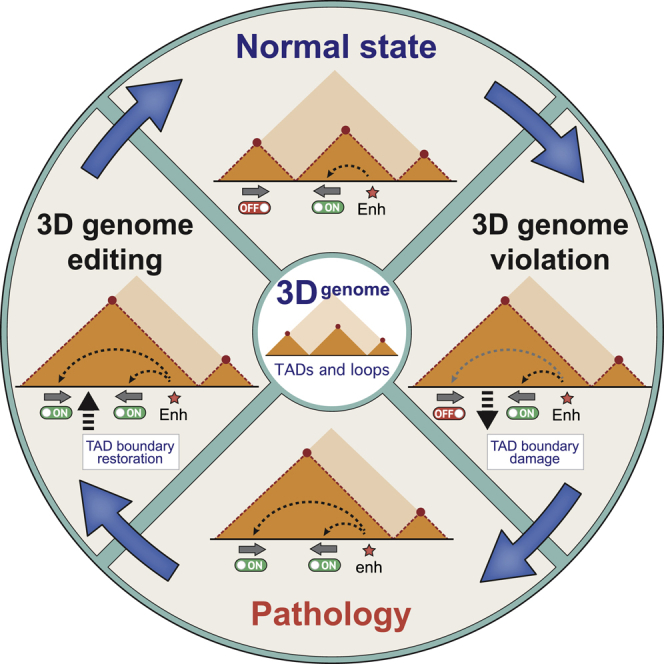
The genome is folded into a multi-level 3D structure that controls many nuclear functions, including gene expression. Alterations in 3D genome organization are linked to genetic diseases and cancer. We discuss new approaches to modify the 3D genome organization that may prove useful in biomedicine.
Introduction
The interphase genome is folded in a highly ordered manner, essential both for DNA compaction and for the regulation of various intranuclear processes. Each chromosome occupies a restricted volume in the nucleus, a chromosome territory (CT). In mammals, large chromosomes and chromosomes with low gene density tend to localize at the nuclear periphery, whereas smaller chromosomes with high gene density are located more centrally.1,2 CTs have a spongy internal structure and are composed of bulk chromatin masses penetrated by the channels of the “interchromatin compartment,” a dynamically organized system of cavities serving for the diffusion of nucleoplasm components.3
Within CTs, active and repressed genomic regions are spatially segregated into A and B compartments formed by local cis as well as distant cis and trans interactions (Figure 1A).4 A compartments are early-replicating gene-rich and typically highly transcribed regions enriched in active histone marks such as H3K36me3, H3K27ac, and H3K4me1–3. In contrast, B compartments contain late-replicating transcriptionally silenced regions enriched with nucleolus- and lamina-associated domains (NADs and LADs, respectively) marked with H3K9me2 and H3K9me3.5,6,7,8 Compartment partitioning strongly correlates with the transcription profile and thus is highly cell-type specific,9,10 whereas the degree of compartmentalization might vary significantly within a cell population.11,12
Figure 1.

Different levels of 3D genome organization
(A) Top panel: contact maps of 3D genome structures; bottom panel: graphical representation of the corresponding structures. (B) Summary of different levels of 3D genome organization.
The compartments are further divided into subcompartments, distinguished by the patterns of different histone modifications, and further into topologically associated domains (TADs) with a high CCCTC-binding factor (CTCF)/cohesin occupancy at their borders13,14 and representing globular structures with a remarkable cell-to-cell variability in their 3D shape and folding density.15,16 TADs serve as “warehouses” for genes, gene loci, and their regulatory systems,17 delimiting the areas of enhancer action (Figure 1B).18,19 Consequently, genes within a TAD are often co-regulated20; this is achieved by looping between enhancers/locus control regions and promoters.21,22 Molecular details of the loop formation mechanisms are still not fully understood, but cohesin-driven extrusion23,24 and liquid-liquid phase separation25,26 are the most consistent models. The CTCF-cohesin complex preferentially determines strong long-range interactions including contacts between TAD borders,8 while the short-range enhancer-promoter and promoter-promoter interactions are also maintained through the Mediator complex and various transcription factors (TFs)27 in cooperation with transcription machinery.28 Complex contact patterns within TADs are manifested in non-structured hierarchical nucleosome assemblies such as clutches29 and nanodomains.30 Clutches are relatively small nucleosome agglomerates (about 2–20 nucleosomes/clutch) whose density and size strongly depend on the level of histone acetylation. This implies that clutches are formed by weak transient electrostatic interactions between nucleosomes. A group of clutches constitutes a nanodomain. Nanodomains are distributed throughout the nucleus, but their concentration increases near the nuclear periphery. Nanodomain structures are preserved upon CTCF and cohesin degradation, and they seem to be formed through liquid-liquid phase separation.30,31
In sum, TADs represent cornerstone structural and functional units of the 3D genome. These units contribute to multiple cellular processes including stem cell differentiation, limb growth and development, epidermal-mesenchymal transition, and cellular senescence.9,19,32,33,34 Changes in the normal profile of TADs can lead to pathologies. Below, we highlight the role of TAD rearrangement and disruption of TAD borders in the development of severe pathologies and discuss 3D genome editing techniques.
3D organization of the genome and pathologies
Over the past several years, a number of diseases have been associated with 3D genome structure abnormalities (Figure 2). Pathological changes in loop and TAD profiles are caused by structural variations (SVs; rearrangements of 50 nucleotides or more in length: insertions, duplications, deletions, etc.), single-nucleotide polymorphisms (SNPs), large chromosomal rearrangements, viral/transposon DNA integrations, and epigenetic factors. These genome and epigenome perturbations could affect or eliminate TAD boundaries and violate distant regulatory interactions that result in transcription dysregulation and manifest in development of pathologies.
Figure 2.

TAD-centric view on the 3D genome organization and disease
Note that some disorders may be causing different rearrangements: autosomal-dominant retinitis pigmentosa, F-syndrome (acropectorovertebral dysgenesis), acute myeloid leukemia, medulloblastoma, brachydactyly, polydactyly.
Deletions
Deletions are among the most frequent SVs in the human genome.35 Extended deletions eliminating CTCF-marked TAD boundaries result in aberrant activation of proto-oncogenes TAL1 and LMO2 by distal enhancers in T cell acute lymphoid leukemia (T-ALL).36 A 600-kb deletion eliminating a TAD boundary results in interactions between unrelated strong enhancers and the LMNB1 promoter, causing LMNB1 overexpression and myelin degeneration in autosomal-dominant adult-onset demyelinating leukodystrophy (ADLD).37 In the developing human limb bud, deletion of an entire TAD including boundaries within the 6p22.3 locus correlates with activation of the ID4 gene by enhancers from the neighboring TAD. This results in the development of mesomelic dysplasia with hypoplastic tibia and fibula.38
Deletions also cause the Liebenberg syndrome, a limb malformation due to dysregulation of the PITX1 expression: the forelimbs develop into the hindlimbs. PITX1 controls the normal development of the hindlimbs where its expression is governed by the interaction with the Pen enhancer. In the forelimb buds, PITX1 is not expressed due to spatial isolation from the Pen by a nearby insulator. In Liebenberg syndrome, multiple deletions eliminate this insulator, allowing the Pen enhancer to activate PITX1 and leading to abnormal formation of the forelimb bones and the kneecap near the elbow.32,39
An SV-affecting CTCF binding site (CBS) is associated with facioscapulohumeral muscular dystrophy (FSHD). This disease is caused by abnormal expression of the DUX4 gene and potentially some other genes, including FRG1. In healthy muscles, enhancers regulating DUX4 and FRG1 are physically separated from their target genes by an FR-MAR insulator.40,41,42 Massive deletion, including several copies of the DUX4 gene and/or hypomethylation of the locus, cause redistribution of loop contacts. A decrease in the FR-MAR insulator activity leads to FRG1 and DUX4 upregulation and muscle pathology.43
Duplications
Duplications also affect the TAD profile and/or intra-TAD spatial interactions. The SOX9/KCNJ2/KCNJ16 locus contains genes coding for the developmental regulator SOX9 and potassium channels KCNJ2/KCNJ16, located in adjacent TADs. Duplications within the SOX9 TAD result in female-to-male sex reversal. In the same locus, duplications encompassing the TAD boundary and the entire KCNJ2 gene result in the formation of a new TAD, where KCNJ2 is upregulated by SOX2-specific enhancers. This leads to the limb malformations with aplasia of nails and short digits known as Cooks syndrome.44 In addition to duplications, a translocation involving the SOX9/KCNJ2/KCNJ16 locus leads to the Snijders Blok-Campeau syndrome, which is characterized by intellectual disability, speech problems, and distinctive facial features.45
Inversions
Large-scale inversions lead to branchiooculofacial syndrome (BOFS), which is characterized by skin, face, and eye defects of varying severity. This pathology occurs when the normal expression of the neural crest regulator TFAP2A is impaired. Usually, it is a result of a partial gene deletion; however, a recent study46 describes a patient with an 89-Mb inversion that does not affect TFAP2A, per se, but compromises its expression. This inversion disrupts the TFAP2A-containing TADs and the interaction between the TFAP2A gene and a group of its enhancers.
Multiple rearrangements
Numerous SVs lead to the emergence of new TADs within the YPEL2/LINC01476 locus leading to dysregulation of gene expression in autosomal-dominant retinitis pigmentosa (adRP).47 In some loci, large-scale SVs cause a variety of distinct pathologies. The canonical example is the WNT6/IHH/EPHA4/PAX3 locus, where different deletions, inversions, and duplications result in limb malformation. Some of the intergenic SVs induce F syndrome (acropectorovertebral dysgenesis), a rare inherited skeletal disorder characterized by the fusion of the thumb and index finger. In particular, F syndrome is caused by an inversion, which leaves the TAD boundary intact but relocates an enhancer from the neighboring TAD to a close vicinity of the WNT6 gene, promoting its overexpression. Other duplications and deletions affecting TAD structure within the regions cause brachydactyly and polydactyly.48,49
Different SVs affecting the 3D genome can lead to a phenomenon called “enhancer hijacking” where enhancers activate genes that are not their normal targets. For example, an SV in the vicinity of the GFI1B gene leads to GFI1B interaction with distal superenhancers and overexpression in medulloblastoma.50 In acute myeloid leukemia (AML), multiple rearrangements affect more than 40 cancer-related loci.51 These mutations are represented by translocations, deletions, and inversions; SVs lead to the formation of new loops with “hijacking” of an enhancer or a silencer in 27 of these loci.
SNPs
SNPs affect the genome topology by mutations in the binding sites of architectural proteins or tissue-specific TFs.
In the human 3p21.2 locus, the SNP rs2535629 has a strong association with schizophrenia. rs2535629 (A/G) is located within the CBS, which resides in a repressor element in the seventh intron of the ITIH3 gene. The presence of this SV prevents CTCF binding to CBS and also changes the expression profiles of nearby genes: it downregulates GLT8D1 and SFMBT1 and upregulates NEK4. The SFMBT1 gene product is involved in the regulation of proliferation and differentiation of nerve stem cells, the formation of dendritic spines, and the proper functioning of neural synapse. Its downregulation is associated with schizophrenia development.52
SNP rs7903146 (CT/TT) is present in the enhancer of the TCF7L2 locus and promotes the formation of the gene-enhancer contact.53 This interaction leads to an increase in the TCF7L2 expression with a subsequent decrease in insulin secretion, possibly contributing to the development of type 2 diabetes.54
STR expansion and viral DNA integration
Expansion of DNA repeats and viral DNA integration can also affect the profile of spatial interactions between remote genomic elements. Short tandem repeats (STRs) alter TAD boundaries by modulating CTCF binding.55 In several pathological models, STRs accumulate at TAD boundaries, increasing the density of CpG islands, which are often hypermethylated in pathologies. One example is the FMR1 gene, whose repression leads to fragile X chromosome syndrome (Martin-Bell syndrome). STR accumulation at the boundary of encompassing TADs promotes DNA hypermethylation followed by a decrease of CTCF binding and significant alteration in the enhancer landscape of the locus. These lead to the loss of the TAD boundary and FMR1 repression.56
Integration of a primate-specific human endogenous retrovirus subfamily H (HERV-H) transposon establishes TAD boundaries in the genome of human pluripotent stem cells.57 In this case, active viral transcription creates a TAD boundary at the site of the HERV-H integration. This is in line with recent observations showing that active transcription constitutes a barrier for the cohesin-driven extrusion.58 In contrast to HERV-H, insertion of the human T-lymphotropic virus HTLV-1 establishes de novo loops due to the presence of a CBS within the viral genome. This results in abnormal host gene transcription not only in loci proximal to the integration site but also more than 300-kb away.59 The same was observed for the bovine leukemia virus (BLV) carrying several CBSs involved in the formation of new chromatin loops with the host chromosome loci after the provirus integration.60
Epigenetic factors
Abnormal DNA methylation is also involved in violation of the 3D genome via suppression of CTCF binding. This decreases insulation at TAD boundaries, allowing aberrant enhancer-promoter interactions that may affect disease-related genes. In human glioma, mutations in the IDH gene result in an increased genome-wide DNA methylation including that in the PDGFRA oncogene locus. Methylation of the 5′-flanking insulator serving as a TAD boundary in this locus results in a loss of CTCF binding and perturbs local interaction patterns; this drives an abnormal activation of the PDGFRA gene by a distal enhancer.61 In some cases, abnormal methylation at CBSs could be driven by external factors, such as drug use. For example, cocaine addiction results in a pathogenic looping within the IRXА locus in brain neurons due to DNA hypomethylation at a set of CBSs.62 Recent studies reveal that besides abnormal methylation and CBS mutations, some other mechanisms could impact chromatin looping. In T-ALL, disappearance of the TAD boundary between MYC and a group of enhancers in the neighbor TAD leads to MYC overexpression.63 Interestingly, in this case, a loss of CTCF binding is not caused by CBS mutations or methylation and is accompanied by decreased chromatin accessibility. Thus some additional factors may influence CTCF binding. One potential candidate is Jpx non-coding RNA, which regulates CTCF binding at a subset of developmentally sensitive loci by competitive inhibition.64
Finally, at a whole-nucleus scale, contacts between non-homologous chromosomes can also lead to oncogenic chromosomal translocations. This disrupts local regulatory 3D interaction networks and alters cellular transcription programs. A few examples include interactions of chromosomes 8 and 14 or 11 and 14 in B lymphocytes leading to Burkitt or mantle cell lymphomas, respectively; chromosomes 12 and 16 in adipocytes leading to liposarcoma; and chromosomes 5 and 6 in hepatocytes leading to hepatocarcinoma.65,66,67,68,69
Together, alterations in the genome 3D organization are quite common in cancer and developmental disorders. Screening revealed that 7.3% of balanced chromosomal abnormalities disrupt TADs at known syndromic loci70; 14% of SVs affect TAD boundaries and lead to remarkable changes in expression of nearby genes in cancers.71 The ability of naturally occurring SVs to affect 3D genome and cause socially significant diseases imposes the development and further clinical validation of 3D genome editing technologies applicable for treating patients.
3D genome editing
Since alterations in the chromatin contact profiles are associated with many pathologies, development of 3D genome editing methods is on the frontlines of biomedicine. The existing approaches largely rely on the use of either native architectural proteins such as CTCF or artificial looping proteins (Figure 3).
Figure 3.

Strategies for 3D genome editing
As highlighted above, CTCF is a master regulator of the mammalian interphase genome folding72 and thus is a predominant target for 3D genome engineering. CBS deletion/insertion/inversion and point mutations are streamlined paths for the precise control of CTCF binding within genome regulatory elements and TAD boundaries/loop anchors.73,74 In several loci, such manipulations alter loop profile and result in changes of gene expression that could be potentially used as a strategy for the cell-type-specific transcription reprogramming in patients.75 However, genomic DNA editing possesses risks of chromosome rearrangements that should be considered while designing clinically relevant applications.76
In the case of de novo TAD formation or excessive CTCF binding, depletion of CTCF from particular sites in chromatin can be accomplished through epigenetic modifications of CBS to inhibit CTCF binding without changes in the primary DNA sequence. In this case, chimeric proteins consisting of catalytically deficient (“dead” Cas9 [dCas9]) fused with the Krüppel-associated box (KRAB), an H3K9me3-catalyzing repressor (dCas9-KRAB), or DNA-methyltransferases DNMT3A and DNMT3A3L were shown to be effective inhibitors of CTCF binding when precisely targeted to the CBS.77,78
Another approach being developed for clinical applications is the usage of small molecules interfering with the CTCF binding. Treatment of cells with the anti-cancer agent curaxin (CBL0137) leads to a partial depletion of CTCF from chromatin and compromises enhancer-promoter contacts.79 On the other hand, DNA methylation interfering with the CTCF binding could be eliminated by cell treatment with 5-aminosalicylic acid (5-ASA). In the case of AML cells, treatment with 5-ASA restores a normal pattern of CTCF-dependent insulation in chromatin and effectively suppresses the interactions between a set of enhancers and oncogenes.51
Other perspective molecules for the 3D genome manipulations are bromodomain and extra-terminal motif inhibitors (BETis). BET proteins are widespread transcription regulators often associated with architectural proteins. For instance, BET protein bromodomain-containing protein 4 (BRD4) binds to CTCF-associated Yin Yang 1 (YY1) factor throughout the genome including at TAD boundaries. Pan-BETi (Apa20, JQ1, IBET762) treatment weakens BRD4-YY1 binding, removes BRD4 from chromatin, and causes chromatin decondensation.80 BETi JQ1 treatment suppresses cohesin and CTCF binding to the Kaposi’s sarcoma-associated herpesvirus (KSHV) genome. This results in the loss of looping between latent and lytic control regions of the viral chromosome and virus transition to a lytic state.81
Together, this opens an avenue for the rational in silico design of inhibitors of DNA binding and competitors for the protein-protein interactions for the factors involved in the 3D genome maintenance.
Since some diseases are associated with the decrease of CTCF occupancy at particular CBSs,82 stabilization of CTCF binding at such loci is a strategy to be considered. Several post-translational modifications are essential for the CTCF insulator and barrier activities.83 Indeed, mutations in the CTCF region subjected to poly(ADP-ribosyl)ation by poly(ADP-ribose) polymerase 1 (PARP1) compromise cohesin enrichment at CBSs, e.g., interfere with cohesion-/CTCF-dependent loop formation.84 In the Epstein-Barr virus genome, PARP1 acts to stabilize CTCF binding at particular sites.85 Thus, inducible recruitment of PARP1 to certain CBSs could be a tool for the stabilization of selective CTCF binding to these sites. Other post-translational modifications of CTCF such as SUMOylation and phosphorylation86,87 are also of interest for 3D genome manipulation in both genome-wide and locus-specific manners. Together, these examples illustrate that epigenome targeting and inducible CTCF post-translational modifications could potentially serve as a proxy for 3D genome editing.
A number of diseases are characterized by a complete loss of CBSs at critical regulatory elements due to deletions and other SVs.88,89,90 In such cases, recruitment of CTCF by an unrelated DNA-binding module and restoration of the original loop profile might be a potential treatment strategy. One example is a dCas9-mediated CTCF recruitment enforced by the coupling with SunTag technology, allowing recruitment of multiple CTCF molecules to the same binding site.91 This method has been tested in the TFF locus associated with breast, lung, and colon cancers92 and demonstrated a potential utility for 3D genome engineering in multi-gene disease-associated loci.
Some pathologies are accompanied by a total loss of CTCF expression followed by genome-wide alterations of chromatin 3D structure. Consequently, insertion of a functional CTCF gene could restore an original loop pattern of the affected loci. For example, breast cancer could be inhibited by the increased CTCF expression; the CTCF gene is frequently deleted in this type of cancer, and this negatively affects the survival rate of patients at late stages.93 CTCF gene insertion by pseudoviruses also slows down the cancer cell division and/or migration, as well as metastasis in the lungs and brain, and affects the expression of almost 130 genes.
An alternative approach for manipulation of the 3D genome architecture relies on the expression of chimeric proteins containing DNA-binding modules (zinc finger [ZF]),transcription activator-like effector [TALE], dCas9) fused with units forming homo- or heterodimers, such as dimerization domains of mammalian TFs, e.g., the self-associating domain of the Ldb1 protein.94 These chimeric proteins form relatively stable dimers and are suitable for the formation of constant distant interactions in chromatin. These contacts can be made reversible via inducible polymerization. One example is the chromatin loop reorganization using CRISPR-dCas9 (CLOuD9) system, where PYL1 and ABI1 fused with two different dCas9 modules interact with each other in the presence of abscisic acid (ABA). Application of the CLOuD9 system to the Oct4 locus associated with various types of cancers demonstrated that recruitment of these fusion proteins to the Oct4 promoter and its distal enhancer induced loop formation and upregulation of Oct4 after ABA addition.95 A similar approach, light-activated dynamic looping (LADL), utilizes co-expression of cryptochrome 2 (CRY2) and N-truncated CRY-interacting basic-helix-loop-helix protein 1 (CIBN) fused to dCas9. Exposure of cells to 470-nm blue light induces dimerization of CRY2 and heteromerization of CRY2 and CIBN. As a result, loci targeted by dCas9-CIBN form a transient loop. In a proof-of-concept study, LADL-induced looping between Zfp462 and the Klf4 superenhancer was successfully used to increase the Zfp462 expression in mouse embryonic stem cells.96
Concluding remarks
Genome-wide association studies revealed a number of genomic SVs associated with the development of various diseases. Most of these SVs are located outside genes and their regulatory modules. Consequently, the mechanical links between disease-associated SVs and regulation of genome activity remained obscure. Recent results discussed here argue that many of the disease-associated SVs affect 3D genome organization. This raises the issue of the need for 3D genome editing. Several approaches for such editing have been proposed and tested on cell cultures. The question is whether any of the developed strategies have the prospect of practical application. In the case of cancer, the straightforward strategy is to kill a cancer cell if it can be recognized and targeted rather than to try to correct anything in this cell. The only possible application here is to use low-molecular-weight agents (e.g., curaxins) that affect all cells with some preference to cancer cells. More interesting opportunities for practical applications of 3D genome editing arise in cases where it is necessary to deliver to the organism its own normal cells, which will exist alongside corrupted ones or replace them. This strategy assumes that damaged cells are taken from the patient, manipulated in the laboratory, and returned to the patient’s body. In the future, this approach could be useful for the treatment of a number of diseases of the hematopoietic and endocrine organs, as well as some types of muscular dystrophies.
However, similarly to genome editing, 3D genome manipulations could have off-target effects. In particular, targeted recruitment of the full-length CTCF or any other natural architectural protein to an ectopic site requires its overexpression in a cell. This could increase the abundance of the overexpressed factor at endogenous binding sites affecting their contact profile genome-wide. Usage of truncated forms of architectural proteins lacking natural DNA-binding domains and/or weak promoters for the expression cassettes potentially solve this problem. Further, binding by an artificial module such as dCas9 may change the chromatin residence time of the recruited protein, which, in turn, may affect the looping strength and nucleosome/epigenetic profiles in a vicinity of the binding site.97 Thus, a sophisticated design of chimeric proteins and comprehensive analysis of epigenetic and transcription profiles within the edited locus and in its neighborhood are prerequisites for the development of clinically relevant applications.
Acknowledgments
This work was supported by the Russian Science Foundation (21-64-00001) and by the Russian Ministry of Science and Higher Education (075-15-2021-1062) to S.V.U. and S.V.R. and from the AFM (CTCFSHD) and the IDB RAS Government basic research programs (0088-2022-0007 and 0088-2022-0016) to Y.V.
Author contributions
S.V.R. and Y.V. conceptualized the manuscript. E.A.T., S.V.U., and A.K. wrote the draft. S.V.U. prepared the figures. All authors made corrections to the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Boyle S., Gilchrist S., Bridger J.M., Mahy N.L., Ellis J.A., Bickmore W.A. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 2001;10:211–219. doi: 10.1093/hmg/10.3.211. [DOI] [PubMed] [Google Scholar]
- 2.Crosetto N., Bienko M. Radial organization in the mammalian nucleus. Front. Genet. 2020;11:33. doi: 10.3389/fgene.2020.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albiez H., Cremer M., Tiberi C., Vecchio L., Schermelleh L., Dittrich S., Küpper K., Joffe B., Thormeyer T., von Hase J., et al. Chromatin domains and the interchromatin compartment form structurally defined and functionally interacting nuclear networks. Chromosome Res. 2006;14:707–733. doi: 10.1007/s10577-006-1086-x. [DOI] [PubMed] [Google Scholar]
- 4.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dillinger S., Straub T., Németh A. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS ONE. 2017;12:e0178821. doi: 10.1371/journal.pone.0178821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W., van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 7.Nichols M.H., Corces V.G. Principles of 3D compartmentalization of the human genome. Cell Rep. 2021;35:109330. doi: 10.1016/j.celrep.2021.109330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S., Aiden E.L. A three-dimensional map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W., et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winick-Ng W., Kukalev A., Harabula I., Zea-Redondo L., Szabó D., Meijer M., Serebreni L., Zhang Y., Bianco S., Chiariello A.M., et al. Cell-type specialization is encoded by specific chromatin topologies. Nature. 2021;599:684–691. doi: 10.1038/s41586-021-04081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bintu B., Mateo L.J., Su J.-H., Sinnott-Armstrong N.A., Parker M., Kinrot S., Yamaya K., Boettiger A.N., Zhuang X. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science. 2018;362:eaau1783. doi: 10.1126/science.aau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H.-J., Yardımcı G.G., Bonora G., Ramani V., Liu J., Qiu R., Lee C., Hesson J., Ware C.B., Shendure J., et al. Capturing cell type-specific chromatin compartment patterns by applying topic modeling to single-cell Hi-C data. Plos Comput. Biol. 2020;16:e1008173. doi: 10.1371/journal.pcbi.1008173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gómez-Marín C., Tena J.J., Acemel R.D., López-Mayorga M., Naranjo S., de la Calle-Mustienes E., Maeso I., Beccari L., Aneas I., Vielmas E., et al. Evolutionary comparison reveals that diverging CTCF sites are signatures of ancestral topological associating domains borders. Proc. Natl. Acad. Sci. USA. 2015;112:7542–7547. doi: 10.1073/pnas.1505463112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merkenschlager M., Nora E.P. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genomics Hum. Genet. 2016;17:17–43. doi: 10.1146/annurev-genom-083115-022339. [DOI] [PubMed] [Google Scholar]
- 15.Stevens T.J., Lando D., Basu S., Atkinson L.P., Cao Y., Lee S.F., Leeb M., Wohlfahrt K.J., Boucher W., O’Shaughnessy-Kirwan A., et al. 3D structure of individual mammalian genomes studied by single cell Hi-C. Nature. 2017;544:59–64. doi: 10.1038/nature21429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ulianov S.V., Zakharova V.V., Galitsyna A.A., Kos P.I., Polovnikov K.E., Flyamer I.M., Mikhaleva E.A., Khrameeva E.E., Germini D., Logacheva M.D., et al. Order and stochasticity in the folding of individual Drosophila genomes. Nat. Commun. 2021;12:41. doi: 10.1038/s41467-020-20292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Razin S.V., Gavrilov A.A., Vassetzky Y.S., Ulianov S.V. Topologically-associating domains: gene warehouses adapted to serve transcriptional regulation. Transcription. 2016;7:84–90. doi: 10.1080/21541264.2016.1181489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nora E.P., Goloborodko A., Valton A.-L., Gibcus J.H., Uebersohn A., Abdennur N., Dekker J., Mirny L.A., Bruneau B.G. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169:930–944.e22. doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang Q.Y., Tan T.Z., Sundararajan V., Chiu Y.-C., Chee E.Y.W., Chung V.Y., Choolani M.A., Huang R.Y.-J. 3D genome organization in the epithelial-mesenchymal transition spectrum. Genome Biol. 2022;23:121. doi: 10.1186/s13059-022-02687-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon J.R., Gorkin D.U., Ren B. Chromatin domains: the unit of chromosome organization. Mol. Cell. 2016;62:668–680. doi: 10.1016/j.molcel.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amândio A.R., Lopez-Delisle L., Bolt C.C., Mascrez B., Duboule D. A complex regulatory landscape involved in the development of mammalian external genitals. eLife. 2020;9:e52962. doi: 10.7554/eLife.52962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonev B., Mendelson Cohen N., Szabo Q., Fritsch L., Papadopoulos G.L., Lubling Y., Xu X., Lv X., Hugnot J.-P., Tanay A., Cavalli G. Multiscale 3D genome rewiring during mouse neural development. Cell. 2017;171:557–572.e24. doi: 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bauer B.W., Davidson I.F., Canena D., Wutz G., Tang W., Litos G., Horn S., Hinterdorfer P., Peters J.-M. Cohesin mediates DNA loop extrusion by a “swing and clamp” mechanism. Cell. 2021;184:5448–5464.e22. doi: 10.1016/j.cell.2021.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davidson I.F., Bauer B., Goetz D., Tang W., Wutz G., Peters J.-M. DNA loop extrusion by human cohesin. Science. 2019;366:1338–1345. doi: 10.1126/science.aaz3418. [DOI] [PubMed] [Google Scholar]
- 25.Hnisz D., Shrinivas K., Young R.A., Chakraborty A.K., Sharp P.A. A phase separation model predicts key features of transcriptional control. Cell. 2017;169:13–23. doi: 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razin S.V., Gavrilov A.A. The role of liquid-liquid phase separation in the compartmentalization of cell nucleus and spatial genome organization. Biochemistry. 2020;85:643–650. doi: 10.1134/S0006297920060012. [DOI] [PubMed] [Google Scholar]
- 27.Krietenstein N., Abraham S., Venev S.V., Abdennur N., Gibcus J., Hsieh T.-H.S., Parsi K.M., Yang L., Maehr R., Mirny L.A., et al. Ultrastructural details of mammalian chromosome architecture. Mol. Cell. 2020;78:554–565.e7. doi: 10.1016/j.molcel.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh T.-H.S., Cattoglio C., Slobodyanyuk E., Hansen A.S., Rando O.J., Tjian R., Darzacq X. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol. Cell. 2020;78:539–553.e8. doi: 10.1016/j.molcel.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricci M.A., Manzo C., García-Parajo M.F., Lakadamyali M., Cosma M.P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160:1145–1158. doi: 10.1016/j.cell.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 30.Szabo Q., Donjon A., Jerković I., Papadopoulos G.L., Cheutin T., Bonev B., Nora E.P., Bruneau B.G., Bantignies F., Cavalli G. Regulation of single-cell genome organization into TADs and chromatin nanodomains. Nat. Genet. 2020;52:1151–1157. doi: 10.1038/s41588-020-00716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulianov S.V., Velichko A.K., Magnitov M.D., Luzhin A.V., Golov A.K., Ovsyannikova N., Kireev I.I., Gavrikov A.S., Mishin A.S., Garaev A.K., et al. Suppression of liquid–liquid phase separation by 1,6-hexanediol partially compromises the 3D genome organization in living cells. Nucleic Acids Res. 2021;49:10524–10541. doi: 10.1093/nar/gkab249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kragesteen B.K., Spielmann M., Paliou C., Heinrich V., Schöpflin R., Esposito A., Annunziatella C., Bianco S., Chiariello A.M., Jerković I., et al. Dynamic 3D chromatin architecture contributes to enhancer specificity and limb morphogenesis. Nat. Genet. 2018;50:1463–1473. doi: 10.1038/s41588-018-0221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodríguez-Carballo E., Lopez-Delisle L., Willemin A., Beccari L., Gitto S., Mascrez B., Duboule D. Chromatin topology and the timing of enhancer function at the HoxD locus. Proc. Natl. Acad. Sci. USA. 2020;117:31231–31241. doi: 10.1073/pnas.2015083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Criscione S.W., Teo Y.V., Neretti N. The chromatin landscape of cellular senescence. Trends Genet. 2016;32:751–761. doi: 10.1016/j.tig.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.Y., et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hnisz D., Weintraub A.S., Day D.S., Valton A.-L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A., et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giorgio E., Robyr D., Spielmann M., Ferrero E., Di Gregorio E., Imperiale D., Vaula G., Stamoulis G., Santoni F., Atzori C., et al. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD) Hum. Mol. Genet. 2015;24:3143–3154. doi: 10.1093/hmg/ddv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flöttmann R., Wagner J., Kobus K., Curry C.J., Savarirayan R., Nishimura G., Yasui N., Spranger J., Van Esch H., Lyons M.J., et al. Microdeletions on 6p22.3 are associated with mesomelic dysplasia Savarirayan type. J. Med. Genet. 2015;52:476–483. doi: 10.1136/jmedgenet-2015-103108. [DOI] [PubMed] [Google Scholar]
- 39.Kragesteen B.K., Brancati F., Digilio M.C., Mundlos S., Spielmann M. H2AFY promoter deletion causes PITX1 endoactivation and Liebenberg syndrome. J. Med. Genet. 2019;56:246–251. doi: 10.1136/jmedgenet-2018-105793. [DOI] [PubMed] [Google Scholar]
- 40.Himeda C.L., Jones T.I., Jones P.L. Facioscapulohumeral muscular dystrophy as a model for epigenetic regulation and disease. Antioxid. Redox Signal. 2015;22:1463–1482. doi: 10.1089/ars.2014.6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrov A., Pirozhkova I., Carnac G., Laoudj D., Lipinski M., Vassetzky Y.S. Chromatin loop domain organization within the 4q35 locus in facioscapulohumeral dystrophy patients versus normal human myoblasts. Proc. Natl. Acad. Sci. USA. 2006;103:6982–6987. doi: 10.1073/pnas.0511235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petrov A., Allinne J., Pirozhkova I., Laoudj D., Lipinski M., Vassetzky Y.S. A nuclear matrix attachment site in the 4q35 locus has an enhancer-blocking activity in vivo: implications for the facio-scapulo-humeral dystrophy. Genome Res. 2008;18:39–45. doi: 10.1101/gr.6620908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karpukhina A., Tiukacheva E., Dib C., Vassetzky Y.S. Control of DUX4 expression in facioscapulohumeral muscular dystrophy and cancer. Trends Mol. Med. 2021;27:588–601. doi: 10.1016/j.molmed.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Franke M., Ibrahim D.M., Andrey G., Schwarzer W., Heinrich V., Schöpflin R., Kraft K., Kempfer R., Jerković I., Chan W.-L., et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 2016;538:265–269. doi: 10.1038/nature19800. [DOI] [PubMed] [Google Scholar]
- 45.Melo U.S., Schöpflin R., Acuna-Hidalgo R., Mensah M.A., Fischer-Zirnsak B., Holtgrewe M., Klever M.-K., Türkmen S., Heinrich V., Pluym I.D., et al. Hi-C identifies complex genomic rearrangements and TAD-shuffling in developmental diseases. Am. J. Hum. Genet. 2020;106:872–884. doi: 10.1016/j.ajhg.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laugsch M., Bartusel M., Rehimi R., Alirzayeva H., Karaolidou A., Crispatzu G., Zentis P., Nikolic M., Bleckwehl T., Kolovos P., et al. Modeling the pathological long-range regulatory effects of human structural variation with patient-specific hiPSCs. Cell Stem Cell. 2019;24:736–752.e12. doi: 10.1016/j.stem.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 47.de Bruijn S.E., Fiorentino A., Ottaviani D., Fanucchi S., Melo U.S., Corral-Serrano J.C., Mulders T., Georgiou M., Rivolta C., Pontikos N., et al. Structural variants create new topological-associated domains and ectopic retinal enhancer-gene contact in dominant retinitis pigmentosa. Am. J. Hum. Genet. 2020;107:802–814. doi: 10.1016/j.ajhg.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lupiáñez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R., et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lupiáñez D.G., Spielmann M., Mundlos S. Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet. 2016;32:225–237. doi: 10.1016/j.tig.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Northcott P.A., Lee C., Zichner T., Stütz A.M., Erkek S., Kawauchi D., Shih D.J.H., Hovestadt V., Zapatka M., Sturm D., et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu J., Song F., Lyu H., Kobayashi M., Zhang B., Zhao Z., Hou Y., Wang X., Luan Y., Jia B., et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature. 2022;611:387–398. doi: 10.1038/s41586-022-05365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y., Ma C., Li S., Wang J., Li W., Yang Y., Li X., Liu J., Yang J., Liu Y., et al. Regulatory variant rs2535629 in ITIH3 intron confers schizophrenia risk by regulating CTCF binding and SFMBT1 expression. Adv. Sci. 2022;9:e2104786. doi: 10.1002/advs.202104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miguel-Escalada I., Bonàs-Guarch S., Cebola I., Ponsa-Cobas J., Mendieta-Esteban J., Atla G., Javierre B.M., Rolando D.M.Y., Farabella I., Morgan C.C., et al. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat. Genet. 2019;51:1137–1148. doi: 10.1038/s41588-019-0457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyssenko V., Lupi R., Marchetti P., Del Guerra S., Orho-Melander M., Almgren P., Sjögren M., Ling C., Eriksson K.-F., Lethagen A.L., et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Invest. 2007;117:2155–2163. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ibrahim D.M., Mundlos S. Three-dimensional chromatin in disease: what holds us together and what drives us apart? Curr. Opin. Cell Biol. 2020;64:1–9. doi: 10.1016/j.ceb.2020.01.003. [DOI] [PubMed] [Google Scholar]
- 56.Sun J.H., Zhou L., Emerson D.J., Phyo S.A., Titus K.R., Gong W., Gilgenast T.G., Beagan J.A., Davidson B.L., Tassone F., Phillips-Cremins J.E. Disease-associated short tandem repeats co-localize with chromatin domain boundaries. Cell. 2018;175:224–238.e15. doi: 10.1016/j.cell.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y., Li T., Preissl S., Amaral M.L., Grinstein J.D., Farah E.N., Destici E., Qiu Y., Hu R., Lee A.Y., et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 2019;51:1380–1388. doi: 10.1038/s41588-019-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brandão H.B., Paul P., van den Berg A.A., Rudner D.Z., Wang X., Mirny L.A. RNA polymerases as moving barriers to condensin loop extrusion. Proc. Natl. Acad. Sci. USA. 2019;116:20489–20499. doi: 10.1073/pnas.1907009116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Melamed A., Yaguchi H., Miura M., Witkover A., Fitzgerald T.W., Birney E., Bangham C.R. The human leukemia virus HTLV-1 alters the structure and transcription of host chromatin in cis. eLife. 2018;7:e36245. doi: 10.7554/eLife.36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bellefroid M., Rodari A., Galais M., Krijger P.H.L., Tjalsma S.J.D., Nestola L., Plant E., Vos E.S.M., Cristinelli S., Van Driessche B., et al. Role of the cellular factor CTCF in the regulation of bovine leukemia virus latency and three-dimensional chromatin organization. Nucleic Acids Res. 2022;50:3190–3202. doi: 10.1093/nar/gkac107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flavahan W.A., Drier Y., Liau B.B., Gillespie S.M., Venteicher A.S., Stemmer-Rachamimov A.O., Suvà M.L., Bernstein B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaillancourt K., Yang J., Chen G.G., Yerko V., Théroux J.F., Aouabed Z., Lopez A., Thibeault K.C., Calipari E.S., Labonté B., et al. Cocaine-related DNA methylation in caudate neurons alters 3D chromatin structure of the IRXA gene cluster. Mol. Psychiatry. 2021;26:3134–3151. doi: 10.1038/s41380-020-00909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kloetgen A., Thandapani P., Ntziachristos P., Ghebrechristos Y., Nomikou S., Lazaris C., Chen X., Hu H., Bakogianni S., Wang J., et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 2020;52:388–400. doi: 10.1038/s41588-020-0602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oh H.J., Aguilar R., Kesner B., Lee H.-G., Kriz A.J., Chu H.-P., Lee J.T. Jpx RNA regulates CTCF anchor site selection and formation of chromosome loops. Cell. 2021;184:6157–6173.e24. doi: 10.1016/j.cell.2021.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allinne J., Pichugin A., Iarovaia O., Klibi M., Barat A., Zlotek-Zlotkiewicz E., Markozashvili D., Petrova N., Camara-Clayette V., Ioudinkova E., et al. Perinucleolar relocalization and nucleolin as crucial events in the transcriptional activation of key genes in mantle cell lymphoma. Blood. 2014;123:2044–2053. doi: 10.1182/blood-2013-06-510511. [DOI] [PubMed] [Google Scholar]
- 66.Germini D., Tsfasman T., Klibi M., El-Amine R., Pichugin A., Iarovaia O.V., Bilhou-Nabera C., Subra F., Bou Saada Y., Sukhanova A., et al. HIV Tat induces a prolonged MYC relocalization next to IGH in circulating B-cells. Leukemia. 2017;31:2515–2522. doi: 10.1038/leu.2017.106. [DOI] [PubMed] [Google Scholar]
- 67.Kuroda M., Tanabe H., Yoshida K., Oikawa K., Saito A., Kiyuna T., Mizusawa H., Mukai K. Alteration of chromosome positioning during adipocyte differentiation. J. Cell Sci. 2004;117:5897–5903. doi: 10.1242/jcs.01508. [DOI] [PubMed] [Google Scholar]
- 68.Parada L.A., McQueen P.G., Misteli T. Tissue-specific spatial organization of genomes. Genome Biol. 2004;5:R44. doi: 10.1186/gb-2004-5-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roix J.J., McQueen P.G., Munson P.J., Parada L.A., Misteli T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 2003;34:287–291. doi: 10.1038/ng1177. [DOI] [PubMed] [Google Scholar]
- 70.Redin C., Brand H., Collins R.L., Kammin T., Mitchell E., Hodge J.C., Hanscom C., Pillalamarri V., Seabra C.M., Abbott M.-A., et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat. Genet. 2017;49:36–45. doi: 10.1038/ng.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akdemir K.C., Le V.T., Chandran S., Li Y., Verhaak R.G., Beroukhim R., Campbell P.J., Chin L., Dixon J.R., Futreal P.A., et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 2020;52:294–305. doi: 10.1038/s41588-019-0564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Ruiten M.S., Rowland B.D. On the choreography of genome folding: a grand pas de deux of cohesin and CTCF. Curr. Opin. Cell Biol. 2021;70:84–90. doi: 10.1016/j.ceb.2020.12.001. [DOI] [PubMed] [Google Scholar]
- 73.Narendra V., Bulajić M., Dekker J., Mazzoni E.O., Reinberg D. CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev. 2016;30:2657–2662. doi: 10.1101/gad.288324.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Willemin A., Lopez-Delisle L., Bolt C.C., Gadolini M.-L., Duboule D., Rodriguez-Carballo E. Induction of a chromatin boundary in vivo upon insertion of a TAD border. Plos Genet. 2021;17:e1009691. doi: 10.1371/journal.pgen.1009691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gong W., Liu Y., Qu H., Liu A., Sun P., Wang X. The effect of CTCF binding sites destruction by CRISPR/Cas9 on transcription of metallothionein gene family in liver hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2019;510:530–538. doi: 10.1016/j.bbrc.2019.01.107. [DOI] [PubMed] [Google Scholar]
- 76.Samuelson C., Radtke S., Zhu H., Llewellyn M., Fields E., Cook S., Huang M.-L.W., Jerome K.R., Kiem H.-P., Humbert O. Multiplex CRISPR/Cas9 genome editing in hematopoietic stem cells for fetal hemoglobin reinduction generates chromosomal translocations. Mol. Ther. Methods Clin. Dev. 2021;23:507–523. doi: 10.1016/j.omtm.2021.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu X.S., Wu H., Ji X., Stelzer Y., Wu X., Czauderna S., Shu J., Dadon D., Young R.A., Jaenisch R. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–247.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tarjan D.R., Flavahan W.A., Bernstein B.E. Epigenome editing strategies for the functional annotation of CTCF insulators. Nat. Commun. 2019;10:4258. doi: 10.1038/s41467-019-12166-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kantidze O.L., Luzhin A.V., Nizovtseva E.V., Safina A., Valieva M.E., Golov A.K., Velichko A.K., Lyubitelev A.V., Feofanov A.V., Gurova K.V., et al. The anti-cancer drugs curaxins target spatial genome organization. Nat. Commun. 2019;10:1441. doi: 10.1038/s41467-019-09500-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsujikawa L.M., Kharenko O.A., Stotz S.C., Rakai B.D., Sarsons C.D., Gilham D., Wasiak S., Fu L., Sweeney M., Johansson J.O., et al. Breaking boundaries: Pan BETi disrupt 3D chromatin structure, BD2-selective BETi are strictly epigenetic transcriptional regulators. Biomed. Pharmacother. 2022;152:113230. doi: 10.1016/j.biopha.2022.113230. [DOI] [PubMed] [Google Scholar]
- 81.Chen H.-S., De Leo A., Wang Z., Kerekovic A., Hills R., Lieberman P.M. BET-inhibitors disrupt Rad21-dependent conformational control of KSHV latency. PLoS Pathog. 2017;13:e1006100. doi: 10.1371/journal.ppat.1006100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miyata K., Imai Y., Hori S., Nishio M., Loo T.M., Okada R., Yang L., Nakadai T., Maruyama R., Fujii R., et al. Pericentromeric noncoding RNA changes DNA binding of CTCF and inflammatory gene expression in senescence and cancer. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2025647118. e2025647118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pavlaki I., Docquier F., Chernukhin I., Kita G., Gretton S., Clarkson C.T., Teif V.B., Klenova E. Poly(ADP-ribosyl)ation associated changes in CTCF-chromatin binding and gene expression in breast cells. Biochim. Biophys. Acta Gene Regul. Mech. 2018;1861:718–730. doi: 10.1016/j.bbagrm.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pugacheva E.M., Kubo N., Loukinov D., Tajmul M., Kang S., Kovalchuk A.L., Strunnikov A.V., Zentner G.E., Ren B., Lobanenkov V.V. CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. Proc. Natl. Acad. Sci. USA. 2020;117:2020–2031. doi: 10.1073/pnas.1911708117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lupey-Green L.N., Caruso L.B., Madzo J., Martin K.A., Tan Y., Hulse M., Tempera I. PARP1 stabilizes CTCF binding and chromatin structure to Maintain epstein-Barr virus latency type. J. Virol. 2018;92:007555-18–e818. doi: 10.1128/JVI.00755-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kitchen N.S., Schoenherr C.J. Sumoylation modulates a domain in CTCF that activates transcription and decondenses chromatin. J. Cell. Biochem. 2010;111:665–675. doi: 10.1002/jcb.22751. [DOI] [PubMed] [Google Scholar]
- 87.Luo H., Yu Q., Liu Y., Tang M., Liang M., Zhang D., Xiao T.S., Wu L., Tan M., Ruan Y., et al. LATS kinase-mediated CTCF phosphorylation and selective loss of genomic binding. Sci. Adv. 2020;6:eaaw4651. doi: 10.1126/sciadv.aaw4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dahlqvist J., Fulco C.P., Ray J.P., Liechti T., de Boer C.G., Lieb D.J., Eisenhaure T.M., Engreitz J.M., Roederer M., Hacohen N. Systematic identification of genomic elements that regulate FCGR2A expression and harbor variants linked with autoimmune disease. Hum. Mol. Genet. 2022;31:1946–1961. doi: 10.1093/hmg/ddab372. ddab372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo Y.A., Chang M.M., Huang W., Ooi W.F., Xing M., Tan P., Skanderup A.J. Mutation hotspots at CTCF binding sites coupled to chromosomal instability in gastrointestinal cancers. Nat. Commun. 2018;9:1520. doi: 10.1038/s41467-018-03828-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ushiki A., Zhang Y., Xiong C., Zhao J., Georgakopoulos-Soares I., Kane L., Jamieson K., Bamshad M.J., Nickerson D.A., University of Washington Center for Mendelian Genomics, et al. Deletion of CTCF sites in the SHH locus alters enhancer-promoter interactions and leads to acheiropodia. Nat. Commun. 2021;12:2282. doi: 10.1038/s41467-021-22470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oh S., Shao J., Mitra J., Xiong F., D’Antonio M., Wang R., Garcia-Bassets I., Ma Q., Zhu X., Lee J.-H., et al. Enhancer release and retargeting activates disease-susceptibility genes. Nature. 2021;595:735–740. doi: 10.1038/s41586-021-03577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu Q., Chen M.Y., He C.Y., Sun L.P., Yuan Y. Promoter polymorphisms in trefoil factor 2 and trefoil factor 3 genes and susceptibility to gastric cancer and atrophic gastritis among Chinese population. Gene. 2013;529:104–112. doi: 10.1016/j.gene.2013.07.070. [DOI] [PubMed] [Google Scholar]
- 93.Duan J., Bao C., Xie Y., Guo H., Liu Y., Li J., Liu R., Li P., Bai J., Yan Y., et al. Targeted core-shell nanoparticles for precise CTCF gene insert in treatment of metastatic breast cancer. Bioact. Mater. 2022;11:1–14. doi: 10.1016/j.bioactmat.2021.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deng W., Rupon J.W., Krivega I., Breda L., Motta I., Jahn K.S., Reik A., Gregory P.D., Rivella S., Dean A., Blobel G.A. Reactivation of developmentally silenced Globin genes by forced chromatin looping. Cell. 2014;158:849–860. doi: 10.1016/j.cell.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morgan S.L., Mariano N.C., Bermudez A., Arruda N.L., Wu F., Luo Y., Shankar G., Jia L., Chen H., Hu J.-F., et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat. Commun. 2017;8:15993. doi: 10.1038/ncomms15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kim J.H., Rege M., Valeri J., Dunagin M.C., Metzger A., Titus K.R., Gilgenast T.G., Gong W., Beagan J.A., Raj A., Phillips-Cremins J.E. LADL: light-activated dynamic looping for endogenous gene expression control. Nat. Methods. 2019;16:633–639. doi: 10.1038/s41592-019-0436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Soochit W., Sleutels F., Stik G., Bartkuhn M., Basu S., Hernandez S.C., Merzouk S., Vidal E., Boers R., Boers J., et al. CTCF chromatin residence time controls three-dimensional genome organization, gene expression and DNA methylation in pluripotent cells. Nat. Cell Biol. 2021;23:881–893. doi: 10.1038/s41556-021-00722-w. [DOI] [PubMed] [Google Scholar]