

Abstract
While a number of methods exist to investigate CRISPR off-target (OT) editing, few have been compared head-to-head in primary cells after clinically relevant editing processes. Therefore, we compared in silico tools (COSMID, CCTop, and Cas-OFFinder) and empirical methods (CHANGE-Seq, CIRCLE-Seq, DISCOVER-Seq, GUIDE-Seq, and SITE-Seq) after ex vivo hematopoietic stem and progenitor cell (HSPC) editing. We performed editing using 11 different gRNAs complexed with Cas9 protein (high-fidelity [HiFi] or wild-type versions), then performed targeted next-generation sequencing of nominated OT sites identified by in silico and empirical methods. We identified an average of less than one OT site per guide RNA (gRNA) and all OT sites generated using HiFi Cas9 and a 20-nt gRNA were identified by all OT detection methods with the exception of SITE-seq. This resulted in high sensitivity for the majority of OT nomination tools and COSMID, DISCOVER-Seq, and GUIDE-Seq attained the highest positive predictive value (PPV). We found that empirical methods did not identify OT sites that were not also identified by bioinformatic methods. This study supports that refined bioinformatic algorithms could be developed that maintain both high sensitivity and PPV, thereby enabling more efficient identification of potential OT sites without compromising a thorough examination for any given gRNA.
Keywords: CRISPR/Cas9, genome editing, hematopoietic stem cells, genotoxicity, DNA damage repair, next-generation sequencing
Graphical abstract
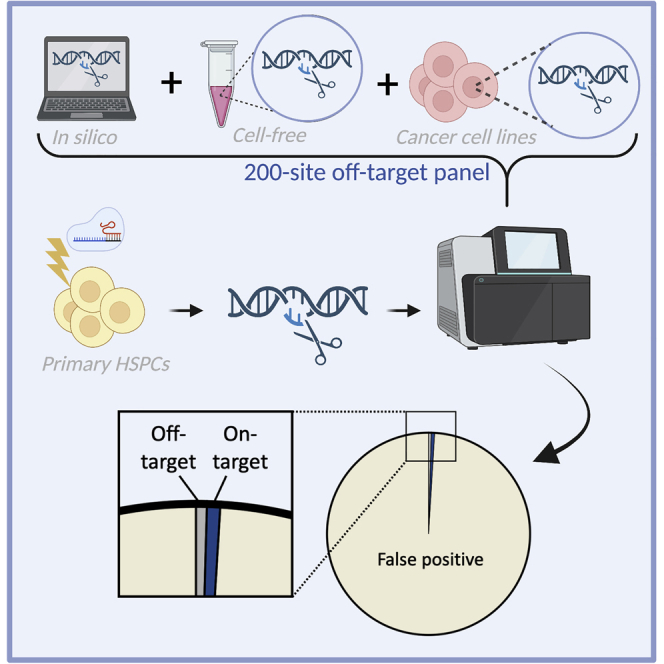
Cromer et al. investigated the landscape of CRISPR OT activity after a clinically relevant genome editing process. In this study, we found that OT activity in human primary HSPCs is exceedingly rare and that virtually all sites are found by available OT detection methods.
Introduction
CRISPR-Cas9 technology enables precision engineering of the genome with single base-pair resolution.1 The site specificity of this system is imparted by a guide RNA (gRNA)—typically containing a 20 nucleotide (nt) spacer sequence—which couples with the Cas9 nuclease allowing it to scan the genome for a suitable protospacer adjacent motif (PAM), then bind and cleave sequences with homology to the gRNA. While the location of the highest cleavage activity often occurs at the intended (on-target) site with perfect homology to the gRNA, activity at sites with lower degrees of homology may also occur.2,3,4
After a DNA double-strand break (DSB) at both on-target and off-target (OT) sites, the cell’s endogenous DNA repair machinery will either resolve the break using the non-homologous end-joining (NHEJ) pathway, which can result in inserted or deleted base pairs (indels) adjacent to the break site, or using homologous recombination where the sister chromosome or exogenous DNA donor is used as a repair template. If these DNA repair pathways are not successful, the cell will undergo cell-cycle arrest because of the presence of an unresolved DSB.5 In addition to these outcomes of DSB resolution, lower frequency events may also occur such as translocations, inversions, large deletions,6 or chromothripsis.7
While several CRISPR-based therapies have entered the clinic,8,9 there is an ongoing debate about which methodologies are most effective at determining the location and frequency of potentially deleterious Cas9 OT activity. Currently, a range of tools and workflows have been developed to identify possible OT sites in the human genome. The first of these—in silico-based (bioinformatic) tools—use the specific gRNA as input to return a list of potential OT sites with varying degrees of homology to the gRNA that may be screened for activity. While cleavage by the Cas9:gRNA ribonucleoprotein (RNP) complex is largely homology dependent, there is concern that purely homology-based prediction tools may miss some sites harboring bona fide OT activity. Additionally, the computational approaches to identify homology vary between in silico tools, leading to discrepancies in the sites identified. Furthermore, these computational tools primarily search a consensus reference genome and are thus unable to account for genetic variation that could lead to differential Cas9 activity across patients.
As the CRISPR field matured, wet laboratory-based empirical approaches were developed to identify DSBs regardless of gRNA homology. Examples of these methods include CIRCLE-Seq,10 GUIDE-Seq,11 and SITE-Seq,12 all of which tag or enrich for DSBs after the delivery of Cas9 and gRNA (Table S1). These studies reported a large number of sites with a range of OT activity, some of which were missed by homology-based in silico prediction tools. While alarming, each of these empirical methods identified OT sites following Cas9:gRNA delivery to cell-free genomic DNA or immortalized cancer cell lines, which are known to harbor polyploidy, aneuploidy, tumorigenic single nucleotide polymorphisms, and dysfunctional DNA damage repair and response mechanisms.13,14,15 In addition, the typically rapid doubling time of immortalized lines may impact OT activity profiles by providing excess unwound genomic DNA substrate on which Cas9 may bind and cleave. Similar to bioinformatic methods, the empirical methods are usually performed on a single cell line or genomic DNA from a single source and thus also do not fully account for genomic variation across patients. The cell lines used are aneuploid with acquisition of multiple new mutations and structural variants as well, also making them different from intact diploid genomes.
In clinical ex vivo editing, Cas9:gRNA RNP is delivered transiently to live, primary cells with functional DNA damage repair processes.16 To improve the specificity of this process, high-fidelity (HiFi) variants of Cas9 (e.g., HiFi Cas9) have been developed17,18 and are being incorporated into the latest translational efforts.19,20,21 Because this clinical workflow departs significantly from the context in which the original empirical methods were conducted, there is a need to compare the performance of both in silico and empirical detection tools to determine whether true OT sites are currently being overlooked in the clinic. The ideal OT detection method should not only display high sensitivity (i.e., capturing the majority of sites of unintended OT editing activity), but have high positive predictive value as well (i.e., reporting as few false positive OT sites as possible).
To determine the performance of the existing methods, we designed a study to compare in silico-based and empirical methods in their ability to predict Cas9 OT activity using transient delivery of HiFi Cas9:gRNA RNP to primary human hematopoietic stem and progenitor cells (HSPCs) ex vivo. First, we selected a series of 11 gRNAs previously investigated in the literature with a range of predicted activity at OT sites. From there, we compared the similarity and differences between OT sites nominated by various methods and interrogated sites that were both unique, as well as those that were largely overlapping across methods for evidence of OT editing in the ex vivo HSPC system. To gain insight into the impact of the type of Cas9 used and length of gRNA spacer on OT activity, we compared wild-type (WT) Cas9 with HiFi Cas9 in two conditions as well as an 18-nt spacer gRNA to the standard length 20-nt gRNA in one of the conditions. After targeted deep sequencing, we then classified sites as true or false positives and evaluated the capability of these various methods to successfully recover bona fide OT sites. In doing so, this work provides clarity on how to successfully produce genome editing safety data with a focus on HiFi ex vivo genome editing systems.
Results
Development of next-generation sequencing panels to compare performance of in silico and empirical OT prediction methods
To compare the performance of both in silico and empirical OT prediction methods, we chose to edit primary human CD34+-purified HSPCs using 11 different gRNAs previously reported in the literature (Table 1). We selected gRNAs based on disease relevance and/or inclusion in prior studies, including many from the original empirical OT detection publications (Table S1). To maximize the number of detectable OT sites, we chose guides with a range of expected OT activity (Figure S1A). COSMID likely had the fewest predicted OT sites due to the tool’s more stringent mismatch criteria (three mismatches tolerated vs. five for CCTop), as well as application of a cutoff score that limited reporting of low likelihood sites (a scoring factor absent in both CCTop and Cas-OFFinder). The distribution of selected guides had predicted OT scores (higher = less predicted OT activity) ranging from 79 to 0 according to IDT’s CRISPR-Cas9 Guide RNA Design Checker tool, with a median predicted OT score of 21 (Figure S1B).
Table 1.
Summary of Cas9 gRNAs
| Target | Guide sequence (PAM) Citation |
Coordinates feature | CHAN. -Seq |
CIRC. -Seq |
DISC. -Seq |
GUID. -Seq |
SITE -Seq |
|---|---|---|---|---|---|---|---|
| AAVS1 | GGGGCCACTAGGGACAGGAT(NGG) Cameron et al.12 Lazzarotto et al.22 |
chr19:55115749-71 intron 1 |
X | X | |||
| AR | GTTGGAGCATCTGAGTCCAG(NGG) Vakulskas et al.17 |
chrX:67545905-27 exon 1 |
X | ||||
| CD33 | (GA)GTCAGTGACGGTACAGGA(NGG) Kim et al.23 |
chr19:51225259-81 exon 2 |
X | ||||
| CTNNB1 | TAAAGGCAATCCTGAGGAAG(NGG) Gehrke et al.24 |
chr3:41224656-78 exon 2 |
X | ||||
| EMX1 | GAGTCCGAGCAGAAGAAGAA(NGG) Tsai et al.11; Tsai et al.10 |
chr2:72933853-75 3′ UTR |
X | X | |||
| FANCF | GGAATCCCTTCTGCAGCACC(NGG) Tsai et al.11; Cameron et al.12; Tsai et al.10 |
chr11:22625786-808 exon 1 |
X | X | X | ||
| GRHPR | GATCCTCTTGTCCACGTGGT(NGG) Wienert et al.25 |
chr9:37424940-62 exon 2 |
|||||
| HBB | CTTGCCCCACAGGGCAGTAA(NGG) Tsai et al.10; Wienert et al., 202025 |
chr11:5226968-90 exon 1 |
X | X | |||
| HBG | CTTGTCAAGGCTATTGGTCA(NGG) Métais et al., 201926 |
chr11:5254880-902 intron 1 |
X | ||||
| HPRT | AATTATGGGGATTACTAGGA(NGG) Vakulskas et al.17 |
chrX:134498209-31 intron 6 |
X | ||||
| VEGFA | GGGTGGGGGGAGTTTGCTCC(NGG) Tsai et al.11; Cameron et al.12; Tsai et al.10 |
chr6:43769554-76 5′ of TSS |
X | X | X |
Characteristics of the gRNAs included in this study, including the gene name, coordinates (genome build hg38), and gene feature targeted by the gRNA. Presence of an "X" denotes inclusion in prior studies corresponding to the various empirical OT detection methods. Absence of an "X" indicates that data was unavailable for a given gRNA.
After choosing gRNAs for this study, we then used three different in silico prediction tools—COSMID,27 CCTop,28 and Cas-OFFinder29—to identify potential OT sites for each of the guide sequences. These were chosen because they are publicly available, allowed interrogation of any given gRNA sequences (including those outside of exons), and their prior use in the literature.17,30,31,32 At the same time, we compiled all previously published OT data for the 11 gRNAs generated by empirical methods. These data were then used to establish custom 200-site panels for each gRNA that we interrogated using a rhAmpSeq-based NGS workflow—a standard method for identifying editing at potential OT sites (Figure 1A).20
Figure 1.

Experimental design and sequencing summary
(A) Experimental design. In silico prediction programs (COSMID, CCTop, and Cas-OFFinder) were used to call potential OT sites for each gRNA. These sites were then overlapped with published data using empirical methods to discover high-likelihood sites of OT activity for each gRNA. Panels of 200 candidate sites were compiled and synthesized, which included those with high concordance across prediction methods as well as top-ranked sites called by individual methods. Concurrently, CD34+ HSPCs from three donors were edited via electroporation of Cas9 protein complexed with each respective gRNA and gDNA was harvested 2 days after editing from mock and edited treatments. Libraries were prepared from gDNA, applied to the Illumina MiSeq platform, and NGS data were analyzed using a bioinformatic pipeline. (B) Each dot depicts number of OT sites found for each gRNA by each discovery method. (C) Coverage across all sites on panel for each gRNA (edited and mock treatments averaged at each site for all three HSPC donors). The middle line represents median, and box extends from 25th to 75th percentiles. Whiskers extend from 10th to 90th percentiles. Sequencing was performed in two separate rounds. All treatments were performed using HiFi Cas9 with 20-nt gRNA unless otherwise noted in parentheses. Dotted line represents 5,000× coverage. (D) Each dots depicts read coverage at each individual OT site for a single donor. Shown on base 10 logarithmic scale.
We found that both in silico and empirical methods nominated a widely variable number of OT sites (Figure 1B). While some tools returned a feasible number of nominated sites for follow-up screening (e.g., DISCOVER-Seq,25 GUIDE-Seq, and COSMID found an average and standard deviation of 2.0 ± 0.0, 22.6 ± 9.1, and 66.4 ± 32.2 sites per gRNA, respectively), others returned much larger lists of possible OT sites that would be time and cost intensive to screen by current methods (e.g., SITE-Seq, CHANGE-Seq,22 and Cas-OFFinder found an average and standard deviation of 893.7 ± 695.9, 1365.0 ± 0.0, and 1418.4 ± 353.6 sites per gRNA, respectively). In designing 200-site panels for each gRNA, the total number of sites nominated by each method was directly correlated with representation on each panel (i.e., more sites called by a given method resulted in greater representation on each panel) (Figure S1C).
In the 200-site panels, we prioritized nominated sites that had the greatest degree of overlap across all methods, as well as OT sites found by the empirical methods to have the highest likelihood of indels that were not identified by any in silico tool (Figure S1D; Extended Data). We hypothesized that these two categories would allow us to identify the most likely loci with OT activity and determine whether specific empirical methods were capturing sites of bona fide activity that may have been missed by other prediction tools. Not surprisingly, the tools that identified the greatest number of candidate OT sites also had the greatest degree of overlap with other detection methods (Figure S1E; note highest values in rows for Cas-OFFinder and SITE-Seq). The on-target editing site was also included in the rhAmpSeq panel.
NGS panels reveal high on-target and low OT editing profiles across all gRNAs
After designing the panels, we edited primary human CD34+ HSPCs by transiently delivering Cas9 RNP by electroporation—a workflow that has already been used to successfully treat patients suffering from β-thalassemia and sickle cell disease in the clinic.9 We then harvested genomic DNA, prepared sequencing libraries, and sequenced each gRNA-specific panel on an Illumina MiSeq. We achieved an average coverage of 19,516 reads over all sites across all treatments, with 80.6% of sites exceeding our ideal coverage threshold of 5,000 reads to enable detection of low-frequency editing events (Figure 1C). We also observed a high degree of consistency for read counts at each site across donors and treatments (Figure 1D). While the majority of sites were sequenced at high depth, regions that were prone to low coverage had consistently low read counts across all donors and treatments. We observed no apparent decrease in read coverage in edited vs. mock treatments.
After sequencing, we processed NGS data using CRISPAltRations (https://www.idtdna.com/pages/tools/rhampseq-crispr-analysis-tool).33 We observed a high frequency of on-target indels across all gRNAs (median of 74.0% across all donors at all target loci) (Figure 2A), and indel frequencies were highly consistent across donors. Despite efficient on-target editing, we found that the majority of OT sites displayed virtually no editing after subtraction of mock background from the edited treatments at each site (Figures 2B and S2). In fact, 94.8% of candidate OT sites (2,499 of 2,635 total sites) displayed less than 0.1% indels after subtracting the mock background (Figure 2C).
Figure 2.

Summary of on-target and OT editing
(A) On-target activity of each sgRNA determined by inclusion in rhAmpSeq NGS panel. All treatments were performed using HiFi Cas9 with 20-nt gRNA unless otherwise noted in parentheses. N = 3 separate HSPC donors per treatment. (B) Each dot depicts percent adjusted indels (Edit-Mock) for each donor at the on-target as well as each OT site in each category. Numbers above dots indicate the total number of sites in each category. (C) Each dot depicts the average percent indels (Edit-Mock) across donors at each site on panel in AAVS1-HiFi treatment (including the on-target site). The dotted line depicts 0.1% adjusted indel detection threshold after Mock is subtracted from Edited treatments. All sites are shown before filtering. (D) Each dot depicts the percent indels averaged across donors for each site on panel with average coverage or more than 5,000×. Top left quadrant indicates indels more than 0.5% indels in Edit treatment and less than 0.4% indels in Mock treatment. Blue dots represent on-target indels and orange dots represent classified OT sites. Shown on base 10 logarithmic scale. (E) Each dot depicts the percent indels (Edit-Mock) for each donor at the on-target as well as each OT site that remained after filtering using HiFi Cas9 and 20-nt spacers. The solid bars depict the median percent indels for all three HSPC donors. The dotted line depicts 0.1% adjusted indel detection threshold after Mock is subtracted from Edited treatments.
We then applied a binary classification method used previously to determine if a nominated OT site is edited within a limit of detection of 0.5% indels given certain experimental bounds.33,34 We found that the majority of sites probed met our coverage criteria of more than 5,000× (2,118 of 2,635 total sites) (Figure 2C). However, 99.5% of sites with adequate coverage displayed indels frequencies below our limit of detection (2,107 of 2,118 total sites)—yielding only 11 total OT sites for classification (Figure 2C). Importantly, all 11 sites were found to have p values of less than 0.05 by Fisher’s exact test. This extremely low frequency of OT sites is also evident when plotting percent indels in edited treatments vs. percent indels in mock treatments (Figure 2D). The majority of indels skewed toward deletions rather than insertions (Figure S3), and indel spectrums were unique to each gRNA in a manner highly consistent across HSPC donors. Remarkably, the total number of on-target editing events exceeded the total number of classified OT editing events across all conditions (14 on-target vs. 11 OT sites) (Figure 2E). We also found that only 5 of the 11 OT sites occurred when using HiFi Cas9 and a standard 20-nt gRNA.
Comparison with WT Cas9 reveals that HiFi version dramatically decreases OT editing
When editing with HiFi Cas9 and a 20-nt gRNA, we found that the majority of gRNAs tested in this study showed no evidence of OT activity (i.e., 7/11 gRNAs tested using HiFi Cas9 and 20-nt spacer elicited no detectable OT events) (Figure 2E). Furthermore, 4 of the 11 bona fide OT sites identified using non-standard conditions (three from WT Cas9 treatments and one from truncated gRNA treatment) had no measurable indels when using HiFi Cas9 with a 20-nt gRNA. Compared with WT Cas9, the use of HiFi decreased the total number of detectable OT events in the AAVS1 treatment from four to two total OTs. We also found that HiFi Cas9 dramatically decreased the frequency of OT editing at the top OT site by an average of 36.8-fold compared with WT Cas9 using AAVS1 and HBB gRNAs, without compromising on-target editing frequency (Figures 3A and 3B). Interestingly, we found that OT-2 in the AAVS1 HiFi Cas9 treatment did not meet our criteria for classification in the WT Cas9 treatment. Given that this site was the closest of all OTs to our limit of detection (0.26% indels in the AAVS1 HiFi treatment), it is likely that this is either a false positive in the HiFi condition or a false negative in the WT condition.
Figure 3.

OT activity across comparative treatments
(A) Each dot depicts % indels averaged across donors for each site on panel with average coverage of more than 5,000×. The top and bottom panels represent treatments with HiFi and WT Cas9, respectively. Top left quadrant indicates indels of more than 0.5% indels in Edit treatment and less than 0.4% indels in Mock treatment. Blue dots represent on-target indels and orange dots represent classified OT sites. Shown on base 10 logarithmic scale. (B) Same as above, but with top and bottom panel representing treatments with 18-nt truncated and 20-nt CD33 gRNAs, respectively. (C) Each dot depicts the percent adjusted indels (Edit-Mock) for each donor at the on-target as well as each OT site that remained post-filtering. Top and bottom panel represent treatments with WT and HiFi Cas9, respectively. The solid bars depict the median percent indels for all three HSPC donors. Dotted line depicts 0.1% adjusted indel detection threshold after Mock is subtracted from Edited treatments. Note: OT1s for AAVS1 and HBB are at the same locus between WT and HiFi treatments. All other sites are at different genomic loci. (D) Same as above, but with the top and bottom panel representing treatments with 18-nt truncated and 20-nt CD33 gRNAs, respectively. Note: OT1s between 18-nt and 20-nt gRNA treatments are at different genomic loci.
In our analysis the previously reported 18-nt spacer gRNA targeting CD3323 seemed to decrease OT activity; however, it is difficult to determine whether this is a function of the truncated gRNA being more specific or simply possessing less activity altogether (indicated by the lower on-target editing frequency of the shorter gRNA, an average of 31.2% vs. 83.8%) (Figures 3C and 3D). In these comparative treatments, we observed a high degree of consistency in the indel spectrums generated that seemed to be more dependent on the core gRNA sequence rather than the type of Cas9 or length of the spacer (Figure S3).
OT sites for all selected gRNAs are called by most methods
Of the 11 true OT sites identified, we found that all were called by at least one in silico prediction method, but no method successfully called all 11 sites (Figure 4A). However, under conditions of HiFi Cas9 and 20-nt gRNA, all 5 true OT sites were found by all in silico prediction methods. We also found that empirical methods reliably captured true OT activity as well, with only a single OT editing event missed by SITE-Seq in the AAVS1 treatment (which also happened to be the site with the lowest adjusted indel frequency [0.26%] and as discussed above could be a false positive). At the outset of this work, we sought to test the claims made by the developers of the empirical methods that these methods identified clinically relevant OT sites that were not identified by bioinformatic methods, despite the fact that the empirical methods used immortalized cell lines or ratios of Cas9 to DNA that would be supra-pharmacologic to levels attained in cells. For the seven gRNA conditions with no detectable OT activity, all methods therefore reported no false negatives (Figure 4B). Further investigation of our classified OT sites indicated that tolerance of gRNA mismatches increases with corresponding distance from the PAM site (Figure 4C). We also did not observe any true OT sites that disrupted the NGG PAM used by SpCas9. Heavy dependence on the core guide sequence as well as an intact PAM is well documented.35
Figure 4.

Summary of OT activity
(A) The characteristics of all true-positive and false-negative OT sites identified in this study, including gene name, coordinates, and gene feature targeted by the gRNA. The percent of indels was calculated as the percent of indels in Cas9-treated conditions, minus the background percent indels in Mock controls (averaged across all three donors for a given OT site). The green boxes denote successful prediction for a particular OT site by a particular detection tool. The orange boxes indicate a false negative for a particular OT site for a given detection tool. The gray boxes indicate that data was unavailable for a given gRNA and method. (B) All gRNAs that had no confirmed OT sites, and methods that therefore yielded no false negatives (indicated by green boxes). The gray boxes indicate that data were unavailable for a given gRNA and method. (C) Percent mismatches at each position of the gRNA for 6 total OT sites in 11 HiFi Cas9 conditions and 5 total OT sites in 2 WT Cas9 conditions. Individual values and linear trendlines are plotted for each condition. Sites edited in both WT and HiFi treatments were only counted once in the “Total” grouping.
We next sought to compare the performance of different OT detection tools by quantifying both sensitivity and positive predictive value (PPV) using our panel of sites that had sufficient coverage to perform binary editing classification (>5,000× coverage). For the sake of calculating the PPV, any variants that were not highly ranked enough by a tool’s criteria to make our 200-target panels were considered a false positive. Because of the low number of true OT sites identified and the fact that most of these were captured by the majority of detection tools, we observed high sensitivity across all methods (Figure 5A). When editing under standard conditions (HiFi Cas9 and 20-nt spacer gRNA), the average sensitivity across all methods was 0.98, and all in silico methods attained a sensitivity of 1.0. However, when sites were edited under more promiscuous, non-standard conditions (WT Cas9 or truncated gRNA), the average sensitivity decreased to 0.82. All empirical detection methods except SITE-Seq had a sensitivity of 1.0 for all guides that the method was performed on. SITE-Seq had a decreased sensitivity of 0.5 at the AAVS1 locus. However, there was a greater difference among methods when quantifying PPV (Figure 5B). With the exception of DISCOVER-Seq, which had a PPV of 0.5 (although the data were only available for the HBB gRNA and only nominated two sites), all other methods had PPVs of less than 0.05 for all gRNAs tested. In other words, each method called far more false positives than true positives, although that could be expected because of the exceedingly low rate of OT activity generated by the ex vivo editing workflow.
Figure 5.

Performance of OT discovery methods
(A) Each dot depicts sensitivity for each gRNA for each discovery method. White dots indicate results derived from non-standard (i.e., WT Cas9 or truncated gRNA) conditions. Note: sensitivity was unable to be calculated in treatments where no OT sites were found. (B) Each dot depicts PPV for each gRNA for each discovery method. All sites not on panel are assumed to be false positives. White dots indicate results derived from non-standard conditions. Note: PPV was not plotted for treatments where no OT sites were found. (C) Each dots depicts the COSMID score for all candidate OT sites for all gRNAs. True positives and corresponding indel frequencies are shown by dotted lines (WT indel frequency underlined). Orange, white, and yellow dots indicate OTs generated by HiFi, WT, and both HiFi and WT Cas9, respectively. (D) For each OT site—rank ordered left to right from high COSMID score to low along the x-axis—COSMID score and adjusted indel percent (Edit-Mock) is plotted in standard and non-standard treatments. (E) Each dot depicts the score assigned to a single DISCOVER-Seq OT site after editing using HBB gRNA. True positive and corresponding indel frequency is shown by dotted line (WT indel frequency underlined). The yellow dot indicates OT generated by WT and HiFi Cas9. (F) Each dot depicts the number of reads covering a single GUIDE-Seq OT site following editing with AR, CTNNB1, EMX1, FANCF, HPRT, and VEGFA gRNAs. True positive and corresponding indel frequency is shown by dotted line. The orange dot indicates OT generated by HiFi Cas9. (G) Pie chart summarizing proportion of on-target, OT, and false positives across all treatments for sites achieving sufficient coverage depth. False positives are defined as putative OT sites on panel that met coverage threshold that were not found to have OT activity.
When plotting all calls by COSMID (736 across all 11 intended gRNA cut sites), true OT sites were given likelihood-of-activity scores at a median in the 88th percentile (a lower score indicates a greater likelihood of OT activity) (Figure 5C). While the vast majority of sites called did not reach our OT classification threshold for activity (727/736 sites called), we found that a lower COSMID score both increased likelihood of activity as well as indel frequency should OT cleavage occur (Figure 5D). In fact, the two top scores given to any OT site displayed activity, even with HiFi Cas9. We next plotted cumulative detection frequency as a function of COSMID score and found that 75% of all bona fide OT sites generated by standard conditions would have been found by interrogating the top 21% of all COSMID-nominated sites (148 total sites across all 11 gRNAs) (Figure S4A). However, to achieve 100% detection, sequencing would have to have extended to the top 51% of all nominated sites (370 total sites across all 11 gRNAs).
While CCTop and Cas-OFFinder did not provide OT activity likelihood scores, we were able to group them into bins for number of mismatches and bulge sites from the target sequence (Figures S4B and S4C). For both methods, a greater number of sites were nominated as similarity to the gRNA decreased, and bona fide OT sites were clustered in bins with higher degrees of homology to the intended target. In assessing performance of empirical methods, for DISCOVER-Seq and GUIDE-Seq—the two empirical methods with the highest degree of sensitivity and PPV—both true OT events occurred at the top-ranked call (determined by read count) by each method (Figures 5E and 5F). Even among the empirical methods with a lower PPV, true OT sites generally ranked at or near the top for CHANGE-Seq, CIRCLE-Seq, and SITE-Seq (Figures S4D–S4G).
True OT sites predominantly map to non-coding regions of the genome
Overall, we found more than two orders of magnitude more false positives than true OT sites within our NGS panels (Figure 5G). Even though we also found more on-target activity than OT activity (11 on-target events vs. 9 OT events [combining standard and non-standard conditions]), we nevertheless sought to determine whether these true OT sites are likely to cause genotoxicity or oncogenic expansion. Of bona fide OT sites for our selected gRNAs, five of nine resided in intergenic regions of the genome, the effects of which remain difficult to interpret (Figures 4A and 5G). Three of nine bona fide OT sites resided in intronic regions of genes, indels which do not typically disrupt exon splicing or gene expression.36 Only a single OT site was found in the exon of a gene, SIGLEC6, which was caused by the CD33 gRNA. The indel spectrum created by the CD33 gRNA is consistent with a frameshift 1-bp insertion, which would likely knock out this gene (Figure S3). However, the detected indel frequency was less than 1% and knockdown of this gene is not known to impart oncogenic potential.37 To better understand the overall likelihood that OTs will disrupt coding regions of genes, we performed an in silico aggregated search for all possible on-target gRNAs targeting 19,222 human genes using the IDT CRISPR-Cas9 Guide RNA Design Checker tool and investigated the relative frequency that exons are nominated OTs. From this we found that an average of 7.2% ± 2.9% of predicted OTs for gRNAs targeting a gene are annotated within exonic regions (Figure S5). This is comparable with our experimental findings here that one of nine unique bona fide OTs target an exon. Exons only make up approximately 3% of the total genomic content, so this is enriched approximately more than 2-fold compared with what could be expected if genomic regions were selected at random. We have no definitive explanation, but possible hypotheses include (1) a greater degree of similarity among coding regions because of gene duplication events and shared features common across genes (such as start and stop codons, splice donor, and acceptor sites, etc.) or (2) a possible enrichment for NGG PAM sites in coding regions because C to A conversions by deamination of methyl-C may be selected against in coding regions to protect the function of the protein for which it codes.
Discussion
In this study, we investigated the efficacy of different OT nomination tools to successfully identify sites with a high likelihood of Cas9 activity to inform genome editing safety investigations. When editing HSPCs in a HiFi ex vivo genome editing workflow, we found that, of 2,061 potential OT sites identified by both in silico and empirical methods from 11 different previously identified targets, there were only 9 sites with detectable indels when the Cas9 nuclease is delivered as an RNP into repair-competent healthy human HSPCs. Importantly, these numbers only include sites on our NGS panels that reached sufficient coverage depth and does not count the greater number of lower ranked sites called by each method that were not included on our panels. In addition, supporting previously published data,17 the use of HiFi Cas9 resulted in a more than 30-fold decrease in the frequency of OT indels without a decrease in on-target activity. These results demonstrate that the CRISPR-Cas9 system can be used with very high specificity. We also found less than one detectable OT event per gRNA to a limit of detection of 0.5% indels across a set of guides with highly variable predicted OT activity. Our analysis revealed that existing OT detection tools, both in silico prediction and empirical methods, identify the majority of detectable OT events for the guides interrogated, especially using HiFi genome editing conditions (HiFi Cas9 delivered as an RNP). Notably, all of the bioinformatic programs identified all true OT sites when editing with HiFi Cas9.
We believe that our results contrast with prior findings for several reasons. First, we delivered Cas9 to primary cells rather than immortalized cell lines that often harbor gross chromosomal abnormalities (polyploidy, aneuploidy, translocations, etc.) with dysfunctional DNA damage and nucleic acid delivery-sensing responses.13,14,15 Furthermore, the delivery of Cas9 to living cells likely serves as a better model for Cas9 binding and cleavage because of the presence of vast regions of inaccessible chromatin,38 active DNA damage repair mechanisms, and a pharmacologic rather than supra-pharmacologic ratio of RNP to genomic DNA. These biological phenomena are likely why CHANGE-Seq, CIRCLE-Seq, and SITE-Seq—methods relying on delivery of Cas9 to cell-free genomic DNA—attained the lowest PPVs of all empirical methods tested (Figure 4B). This may be why our study identified so few bona fide OT sites—because we investigated OT activity in healthy donor primary cells rather than immortalized cell lines or cell-free DNA. Considering this fact, we would expect the PPV to also be higher across detection methods when using cell lines because of the increased number of true-positive OT sites. Additionally, unlike prior studies that relied on a pre-existing pool of cells with oncogenic mutations,39,40 by conducting our experiments in primary HSPCs that are not likely to harbor such aberrations,41 it is not surprising that this study found no bona fide OT sites residing in exons of known tumor suppressors or oncogenes. This was further confirmed by a recent publication that found no evidence that Cas9 introduced or enriched for oncogenic mutations after ex vivo editing in primary HSPCs.41 Taken together, our study highlights the importance of evaluating Cas9 OT detection tool performance in the appropriate application-specific context (e.g., HiFi nuclease, clinically relevant cell type) where epigenetic factors and DNA damage repair mechanisms can impact the appearance of unwanted OT editing.
Collectively, our findings reinforce the importance of using HiFi variants of Cas9 to dramatically reduce the frequency of unintended editing events. Since HiFi Cas917 decreases editing at OT sites without compromising on-target editing, it is also likely that the risk of translocations or inversions between the on-target and OT sites are decreased accordingly and has been shown in one example.20 Furthermore, other HiFi Cas9 enzymes as well as gRNA innovations have been reported18,42,43 and continued efforts attempt to increase activity and specificity of Cas9 via protein engineering. This may be especially important as genome editing technology begins to be adapted for in vivo delivery.8
While this study did not detect a large number of OT events, the experimental workflow primarily allowed us to capture the most common unintended genomic event after Cas9 editing—small, site-specific indels at sites other than the targeted locus. Other abnormalities such as translocations, inversions, large deletions,6 or chromothripsis would have likely been overlooked by our methodology. In fact, chromothripsis has even been documented as a consequence of on-target Cas9 cleavage,7 a phenomenon our experimental workflow cannot capture accurately. Several strategies have been developed to identify and quantify the frequency of genomic rearrangements following CRISPR editing. These include No-Amp long-range sequencing protocols that avoid PCR and use Cas9 to enrich for the sequence of interest44 as well as protocols which use a sequence at the on-target site as bait followed by NGS and bioinformatics to identify the prey.45,46,47,48,49 However, because these abnormalities are reported to occur at relatively low frequencies, limited read depth (especially in the absence of PCR amplification) can limit the ability to detect these events. Therefore, further studies are needed to better understand, and possibly prevent, creation and enrichment of cells harboring large-scale genomic disruptions following CRISPR-based editing.
Our study provides a comparison of OT detection tools in the context of genome editing HSPCs using HiFi nucleases. These tools approach the problem of finding Cas9-generated OT edits from two different directions—a priori or a posteriori. The a priori in silico prediction tools operate under the assumption that Cas9 binding and cleavage requires the presence of the PAM and homology to the gRNA—a concept derived from a number of prior studies35,50—while a posteriori empirical methods identify OT sites experimentally. However, OT sites nominated by empirical methods are often so numerous that filtering based on homology may be used as a quality control step.51 Therefore, it is reasonable to ask, if filtering based on homology to the intended cut site is recommended during processing putative OT sites identified by empirical methods, then how do we interpret sites that pass coverage metrics but display no homology to the gRNA? Despite deliberate inclusion of OT sites on NGS panels that were nominated only by empirical tools, we found none of these sites to harbor detectable levels of OT editing, leading to the conclusion that minimal Cas9 activity occurs at sites lacking homology to the gRNA. Overall, this highlights the importance of applying appropriate filters to NGS data that was either generated by targeted sequencing of in silico-predicted OT sites or by one of the empirical methods. This was a critical component of our workflow as well, using coverage depth filters to ensure we could detect indels down to 0.5% frequency as well as unedited background subtraction to ensure that OT events were due to Cas9 activity rather than misalignment, mismapping, or errors introduced by PCR amplification.
While the in silico-based prediction of OT sites requires minimal time, effort, and expertise, empirical methods are comparably cumbersome to establish in the laboratory and require the development of specialized skills to be used robustly and reproducibly. Although datasets for empirical methods were not available for many of the guides used in this study, we found no OT sites validated under HiFi editing conditions that were identified solely by empirical methods. In light of this, we believe the simplest possible method for high-confidence capture of OT sites (at least when editing primary HSPCs with HiFi nucleases) is to perform targeted NGS on the highest-ranked 55% of COSMID calls. Such a workflow would have captured all true variants that occurred after editing with HiFi Cas9 in this study, yielding an average of 37 sites per gRNA for follow-up sequencing (which is less than the number of sites typically screened after translational ex vivo editing).21,52,53,54 One caveat to this strategy is that although in silico methods seem to be capable of capturing most true OT events, current tools rely on homology to a consensus genome and, therefore, would not take into account patient-specific variation, which has been shown to impact Cas9 activity.41 The empirical methods, however, are also limited by not being performed on a diverse array of genomes and are usually also performed on a single genome. As a solution, both in silico and empirical methods could be performed on diverse genomes, especially in silico methods as the catalog of sequenced human genomes continually grows. Therefore, we hope our results prompt the development of improved tools that take into account the large amounts of new data that have been generated since their development.
Taken together, our results provide a comparative analysis of the relative performance of currently available OT detection methods. While no one method emerged as ideal for detecting all OT events without a high frequency of false positives, certain methods had a better profile of sensitivity and positive prediction value than others, particularly using a HiFi system. COSMID, for example, is an easily accessible online program that requires no user expertise and identified all bona fide OT sites generated by the HiFi system, while also having the highest positive prediction frequency of in silico programs investigated. All of the bioinformatic tools evaluated are several years old, however, and it is likely that improved tools using machine learning or other techniques could be developed in the future using datasets like this and others.35 In conclusion, the efficient characterization of genomic outcomes of Cas9 activity, both intended and not, will help to ensure that genome editing-based therapies are developed and delivered in a way that maximizes safety and minimizes the risk of genotoxic or oligoclonal expansion events while facilitating translational development of these potentially curative therapies.
Materials and methods
Acquisition of HSPCs
Primary human HSPCs were sourced from fresh umbilical cord blood (generously provided by the Binns Family program for Cord Blood Research) under protocol 33818, which was approved and renewed annually by the National Heart, Lung, and Blood Institute Institutional Review Board committee. All patients provided informed consent for the study. Patient information was de-identified before laboratory experiments—we, therefore, are unable to make a statement speaking to sex or ethnicity of participants. Donors were not aware of the research purpose or compensated for their participation. Consent forms provided express permission to publish de-identified genetic information.
Ex vivo culturing of HSPCs
CD34+ HSPCs were bead-enriched using Human CD34 Microbead Kits (Mitenyi Biotec, Inc., Bergisch Gladbach, Germany) according to manufacturer’s protocol and cultured at 1 × 105 cells/mL in CellGenix GMP SCGM serum-free base media (Sartorius CellGenix GmbH, Freiburg, Germany) supplemented with stem cell factor (SCF) (100 ng/mL), thrombopoietin (100 ng/mL), FLT3-ligand (100 ng/mL), IL-6 (100 ng/mL), UM171 (35 nM), 20 mg/mL streptomycin, and 20 U/mL penicillin.
Genome editing of HSPCs
Chemically modified gRNAs used to edit HSPCs were purchased from Synthego (Menlo Park, CA). The gRNA modifications added were the 2′-O-methyl-3′-phosphorothioate at the three terminal nucleotides of the 5′ and 3′ ends.38 All Cas9 protein (both Alt-R S.p. HiFi and WT Cas9 nuclease) was purchased from IDT, Inc. (Coralville, IA). HSPCs were cultured in vitro for 2 d and then edited as follows: RNPs were complexed at a Cas9:gRNA molar ratio of 1:2.5 at 25°C for 10 min before electroporation, HSPCs were resuspended in P3 buffer (Lonza, Basel, Switzerland) with complexed RNPs, and electroporated using the Lonza 4D Nucleofector (program DZ-100). Cells were plated at 1 × 105 cells/mL after electroporation in the cytokine-supplemented media described above.
rhAmpSeq panel design
The primary goal of rhAmpSeq panel design was to assemble a list of 200 sites with either a high degree of overlap across methods (representing sites of high likelihood of OT activity) or those that were found only by one or more empirical methods (representing sites that can inform whether purely homology-based prediction is missing true OT sites). To assemble these panels, we took virtually all sites with a high degree of overlap. Then for lower overlap samples, we established a ranking system that allowed us to prioritize OT sites that were nominated by one or several methods. For COSMID, we used the internal score that is generated as output (lower score indicates higher likelihood of activity). For CCTop and Cas-OFFinder, where scores were not generated, we prioritized sites with the fewest mismatches and bulges. For DISCOVER-Seq, we were able to include both predicted OT locations on our panel. And for all other empirical methods, we prioritized the sites with the highest read counts from the highest concentration Cas9 conditions.
rhAmpSeq library preparation
On-target and OT editing rates in HSPCs were measured by amplicon-based NGS. Genomic DNA from edited and unedited HSPC samples was harvested 48 h after editing using QuickExtract DNA Extraction Solution according to manufacturer’s recommendations (Lucigen Corp., Teddington, UK) and diluted to 4.55 ng/μL in IDTE pH 8.0 (IDT, Inc.). Amplicon libraries were generated using target-specific rhAmpSeq primer panels (as described above) with 4× rhAmpSeq library mix 1 and 50 ng gDNA input. The following cycling conditions were used for PCR 1: 95°C for 10 min; (95°C for 15 s; 61°C for 8 min) × 14 cycles; 99.5°C for 15 min; 4°C hold. Target-specific amplicon libraries from PCR 1 were diluted 20-fold in nuclease-free water and were subsequently tagged with P5 and P7 Illumina adapter primers with dual unique indices via a second round of PCR using 4× rhAmpSeq library mix 2. The following cycling conditions were used for PCR 2: 95°C for 3 min; (95°C for 15 s; 60°C for 30 s; 72°C for 30 s) × 24 cycles; 72°C for 1 min; 4°C hold. Libraries were purified using the AMPure XP system (Beckman Coulter, Pasadena, CA), and quantified using Qubit 1× dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA) or quantitative PCR before loading onto the Illumina MiSeq or NextSeq 500/550 platform (Illumina, San Diego, CA). Paired-end, 150-bp reads were sequenced using V2 or V2.5 chemistry.
Data analysis
NGS data were analyzed and CRISPR editing quantified using CRISPAltRations with a default window parameter for Cas9 (8 bp).33 The following filter was used to determine sites that had sufficient data and bounds to be binarily classified as “Edited” or “Not Edited”: (1) greater than 5,000× read depth; (2) greater than 0.5% indels in the “Edited” treatment; and (3) less than 0.4% indels in the “Mock” treatment. These cutoffs were based on average values across Mock and Edited samples for all three HSPC donors. We also ensured that each OT site remaining after filtering had at least one donor that met all coverage and indel frequency criteria on their own. To quantify sensitivity and PPV, the following values were determined for all treatment conditions, using the following definitions: (1) true positives = all sites called by a given method that showed detectable OT activity; (2) false positives = all sites called by a given method that met coverage metrics but remained below detection; and (3) false negatives = all true OT sites missed by a given method (see Extended Data). Sensitivity was then defined as the number of true positives captured as a percentage of all true positives and false negatives. The PPV was defined as the number of true positives captured as a percentage of all true positives and false positives. To predict the specificity of previously published gRNAs, all sequences were submitted to the IDT gRNA design checker (https://www.idtdna.com/site/order/designtool/index/CRISPR_SEQUENCE) and OT scores recorded.
Statistical analyses
Statistical analysis for binary classification of editing was performed on the treated/edited samples compared with the untreated/unedited samples using a Fisher’s exact test (p < 0.05) using set parameters for read depth (>5,000×) and percent NHEJ (>0.5% in treated samples, <0.4% in untreated samples) based on previously published methods.33
Acknowledgments
This project was supported by the Food and Drug Administration (FDA) of the US Department of Health and Human Services (HHS) as part of a financial assistance award (Center of Excellence in Regulatory Science and Innovation grant to University of California, San Francisco [UCSF] and Stanford University, U01FD005978) totaling $75,000 with 100% funded by the FDA/HHS. The contents are those of the authors and do not necessarily represent the official views of, nor an endorsement by, FDA/HHS, or the US government. Products and tools supplied by IDT are for research use only and not intended for diagnostic or therapeutic purposes. Purchaser and/or user are solely responsible for all decisions regarding the use of these products and any associated regulatory or legal obligations.
Author contributions
M.K.C. and M.H.P. supervised the project. M.K.C. and M.H.P. designed experiments. M.K.C., K.R.M., G.R.R., K.M., J.P.H., G.K., C.A.V., and M.A.B. carried out experiments. M.K.C. wrote the manuscript.
Declaration of interests
The authors of this study also wish to declare the following conflicts of interest: M.H.P. is on the Board of Directors of Graphite Bio. M.H.P. serves on the SAB of Allogene Tx and is an advisor to Versant Ventures. M.H.P. and M.K.C. have equity in Graphite Bio. M.H.P. has equity in CRISPR Tx. G.R.R., K.M., G.K., C.A.V., and M.A.B .are employees of IDT, Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.02.011.
Supplemental information
Data availability
High-throughput sequencing data generated for all rhAmpSeq panels will be uploaded to the NCBI Sequence Read Archive submission. The filtered data for all figures in this study are provided in the Supplementary Information/Source Data file.
References
- 1.Porteus M.H. A new class of medicines through DNA editing. N. Engl. J. Med. 2019;380:947–959. doi: 10.1056/NEJMra1800729. [DOI] [PubMed] [Google Scholar]
- 2.Bao X.R., Pan Y., Lee C.M., Davis T.H., Bao G. Tools for experimental and computational analyses of off-target editing by programmable nucleases. Nat. Protoc. 2021;16:10–26. doi: 10.1038/s41596-020-00431-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shrivastav M., de Haro L.P., Nickoloff J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 6.Park S.H., Cao M., Pan Y., Davis T.H., Saxena L., Deshmukh H., Fu Y., Treangen T., Sheehan V.A., Bao G. Comprehensive analysis and accurate quantification of unintended large gene modifications induced by CRISPR-Cas9 gene editing. Sci. Adv. 2022;8:eabo7676. doi: 10.1126/sciadv.abo7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.-Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O’Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 9.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 10.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron P., Fuller C.K., Donohoue P.D., Jones B.N., Thompson M.S., Carter M.M., Gradia S., Vidal B., Garner E., Slorach E.M., et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods. 2017;14:600–606. doi: 10.1038/nmeth.4284. [DOI] [PubMed] [Google Scholar]
- 13.Passerini V., Ozeri-Galai E., de Pagter M.S., Donnelly N., Schmalbrock S., Kloosterman W.P., Kerem B., Storchová Z. The presence of extra chromosomes leads to genomic instability. Nat. Commun. 2016;7:10754. doi: 10.1038/ncomms10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshihara M., Oguchi A., Murakawa Y. Genomic instability of iPSCs and challenges in their clinical applications. Adv. Exp. Med. Biol. 2019;1201:23–47. doi: 10.1007/978-3-030-31206-0_2. [DOI] [PubMed] [Google Scholar]
- 15.Mittelman D., Wilson J.H. The fractured genome of HeLa cells. Genome Biol. 2013;14:111. doi: 10.1186/gb-2013-14-4-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bravo J.P.K., Liu M.-S., Hibshman G.N., Dangerfield T.L., Jung K., McCool R.S., Johnson K.A., Taylor D.W. Structural basis for mismatch surveillance by CRISPR-Cas9. Nature. 2022;603:343–347. doi: 10.1038/s41586-022-04470-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodwin M., Lee E., Lakshmanan U., Shipp S., Froessl L., Barzaghi F., Passerini L., Narula M., Sheikali A., Lee C.M., et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci. Adv. 2020;6:eaaz0571. doi: 10.1126/sciadv.aaz0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lattanzi A., Camarena J., Lahiri P., Segal H., Srifa W., Vakulskas C.A., Frock R.L., Kenrick J., Lee C., Talbott N., et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021;13:eabf2444. doi: 10.1126/scitranslmed.abf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cromer M.K., Camarena J., Martin R.M., Lesch B.J., Vakulskas C.A., Bode N.M., Kurgan G., Collingwood M.A., Rettig G.R., Behlke M.A., et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021;27:677–687. doi: 10.1038/s41591-021-01284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazzarotto C.R., Malinin N.L., Li Y., Zhang R., Yang Y., Lee G., Cowley E., He Y., Lan X., Jividen K., et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR-Cas9 genome-wide activity. Nat. Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim M.Y., Yu K.-R., Kenderian S.S., Ruella M., Chen S., Shin T.-H., Aljanahi A.A., Schreeder D., Klichinsky M., Shestova O., et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. 2018;173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gehrke J.M., Cervantes O., Clement M.K., Wu Y., Zeng J., Bauer D.E., Pinello L., Joung J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 2018;38:977–982. doi: 10.1038/nbt.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wienert B., Wyman S.K., Richardson C.D., Yeh C.D., Akcakaya P., Porritt M.J., Morlock M., Vu J.T., Kazane K.R., Watry H.L., et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science. 2019;364:286–289. doi: 10.1126/science.aav9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Métais J.Y., Doerfler P.A., Mayuranathan T., Bauer D.E., Fowler S.C., Hsieh M.M., Katta V., Keriwala S., Lazzarotto C.R., Luk K., et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3:3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: a web-based tool for identifying and validating CRISPR/cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobson L., Reményi I., Tusnády G.E. CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res. 2015;43:W408–W412. doi: 10.1093/nar/gkv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae S., Park J., Kim J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin R.M., Ikeda K., Cromer M.K., Uchida N., Nishimura T., Romano R., Tong A.J., Lemgart V.T., Camarena J., Pavel-Dinu M., et al. Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell. 2019;24:821–828.e5. doi: 10.1016/j.stem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Kang S.-H., Lee W.J., An J.-H., Lee J.-H., Kim Y.-H., Kim H., Oh Y., Park Y.-H., Jin Y.B., Jun B.-H., et al. Prediction-based highly sensitive CRISPR off-target validation using target-specific DNA enrichment. Nat. Commun. 2020;11:3596. doi: 10.1038/s41467-020-17418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labuhn M., Adams F.F., Ng M., Knoess S., Schambach A., Charpentier E.M., Schwarzer A., Mateo J.L., Klusmann J.-H., Heckl D. Refined sgRNA efficacy prediction improves large- and small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 2018;46:1375–1385. doi: 10.1093/nar/gkx1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurgan G., Turk R., Li H., Roberts N., Rettig G.R., Jacobi A.M., Tso L., Sturgeon M., Mertens M., Noten R., et al. CRISPAltRations: a validated cloud-based approach for interrogation of double-strand break repair mediated by CRISPR genome editing. Mol. Ther. Methods Clin. Dev. 2021;21:478–491. doi: 10.1016/j.omtm.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kath J., Du W., Pruene A., Braun T., Thommandru B., Turk R., Sturgeon M.L., Kurgan G.L., Amini L., Stein M., et al. Pharmacological interventions enhance virus-free generation of TRAC-replaced CAR T cells. Mol. Ther. Methods Clin. Dev. 2022;25:311–330. doi: 10.1016/j.omtm.2022.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyle E.A., Becker W.R., Bai H.B., Chen J.S., Doudna J.A., Greenleaf W.J. Quantification of Cas9 binding and cleavage across diverse guide sequences maps landscapes of target engagement. Sci. Adv. 2021;7:eabe5496. doi: 10.1126/sciadv.abe5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong H., Ceballos C.C., Massengill C.I., Muniak M.A., Ma L., Qin M., Petrie S.K., Mao T. High-fidelity, efficient, and reversible labeling of endogenous proteins using CRISPR-based designer exon insertion. Elife. 2021;10:e64911. doi: 10.7554/eLife.64911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang J., Peng H., Shaffer B.C., Baskar S., Wecken I.C., Cyr M.G., Martinez G.J., Soden J., Freeth J., Wiestner A., et al. Siglec-6 on chronic lymphocytic leukemia cells is a target for post-allogeneic hematopoietic stem cell transplantation antibodies. Cancer Immunol. Res. 2018;6:1008–1013. doi: 10.1158/2326-6066.CIR-18-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horlbeck M.A., Witkowsky L.B., Guglielmi B., Replogle J.M., Gilbert L.A., Villalta J.E., Torigoe S.E., Tjian R., Weissman J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife. 2016;5:e12677. doi: 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C., et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 40.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 41.Cromer M.K., Barsan V.V., Jaeger E., Wang M., Hampton J.P., Chen F., Kennedy D., Xiao J., Khrebtukova I., Granat A., et al. Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat. Commun. 2022;13:4724. doi: 10.1038/s41467-022-32233-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kulcsár P.I., Tálas A., Ligeti Z., Krausz S.L., Welker E. SuperFi-Cas9 exhibits remarkable fidelity but severely reduced activity yet works effectively with ABE8e. Nat. Commun. 2022;13:6858. doi: 10.1038/s41467-022-34527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riesenberg S., Helmbrecht N., Kanis P., Maricic T., Pääbo S. Improved gRNA secondary structures allow editing of target sites resistant to CRISPR-Cas9 cleavage. Nat. Commun. 2022;13:489. doi: 10.1038/s41467-022-28137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang S.-Q., Liu P., Smith J.L., Mintzer E., Maitland S., Dong X., Yang Q., Lee J., Haynes C.M., Zhu L.J., et al. Genome-wide detection of CRISPR editing in vivo using GUIDE-tag. Nat. Commun. 2022;13:437. doi: 10.1038/s41467-022-28135-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin J., Liu M., Liu Y., Wu J., Gan T., Zhang W., Li Y., Zhou Y., Hu J. Optimizing genome editing strategy by primer-extension-mediated sequencing. Cell Discov. 2019;5:18. doi: 10.1038/s41421-019-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turchiano G., Andrieux G., Klermund J., Blattner G., Pennucci V., El Gaz M., Monaco G., Poddar S., Mussolino C., Cornu T.I., et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021;28:1136–1147.e5. doi: 10.1016/j.stem.2021.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Zheng Z., Liebers M., Zhelyazkova B., Cao Y., Panditi D., Lynch K.D., Chen J., Robinson H.E., Shim H.S., Chmielecki J., et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014;20:1479–1484. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 48.Hu J., Meyers R.M., Dong J., Panchakshari R.A., Alt F.W., Frock R.L. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 2016;11:853–871. doi: 10.1038/nprot.2016.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giannoukos G., Ciulla D.M., Marco E., Abdulkerim H.S., Barrera L.A., Bothmer A., Dhanapal V., Gloskowski S.W., Jayaram H., Maeder M.L., et al. UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genomics. 2018;19:212. doi: 10.1186/s12864-018-4561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu B.X.H., Hansen L.L., Artiles K.L., Nonet M.L., Fire A.Z. Landscape of target:guide homology effects on Cas9-mediated cleavage. Nucleic Acids Res. 2014;42:13778–13787. doi: 10.1093/nar/gku1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaudhari H.G., Penterman J., Whitton H.J., Spencer S.J., Flanagan N., Lei Zhang M.C., Huang E., Khedkar A.S., Toomey J.M., Shearer C.A., et al. Evaluation of homology-independent CRISPR-cas9 off-target assessment methods. CRISPR J. 2020;3:440–453. doi: 10.1089/crispr.2020.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J., et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaidyanathan S., Salahudeen A.A., Sellers Z.M., Bravo D.T., Choi S.S., Batish A., Le W., Baik R., de la O S., Kaushik M.P., et al. High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell. 2020;26:161–171.e4. doi: 10.1016/j.stem.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park S.H., Lee C.M., Dever D.P., Davis T.H., Camarena J., Srifa W., Zhang Y., Paikari A., Chang A.K., Porteus M.H., et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47:7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data generated for all rhAmpSeq panels will be uploaded to the NCBI Sequence Read Archive submission. The filtered data for all figures in this study are provided in the Supplementary Information/Source Data file.