


Abstract
Methylation of CpG dinucleotides is correlated with transcriptional repression of genes, including imprinted genes. In the case of the imprinted H19 gene, a 2 kb imprinting control region (ICR) is subject to differential methylation, as it is methylated only on the silenced paternal allele. This region has previously been shown to act as a silencer element at the endogenous locus. The proteins that bind at the H19 differentially methylated domain (DMD) and mediate transcriptional silencing have yet to be identified, although a family of proteins containing a methyl-CpG-binding domain (MBD), of which MeCP2 is the best characterised, are obvious candidates. MeCP2 can bind to a single methylated CpG dinucleotide through its MBD and also contains a transcriptional repression domain (TRD). The TRD interacts with Sin3a and histone deacetylases (HDACs) in vivo, forming a repressive complex. Here we show that MeCP2 is recruited to the H19 DMD in vivo and can silence a reporter gene regulated by the H19 DMD in a methylation-dependent manner. This repression can be alleviated by deletion of the TRD from MeCP2 or by inhibition of HDAC activity. These data indicate that transcriptional silencing from the H19 ICR involves recruitment of MeCP2 and presumably an associated protein complex with deacetylase activity. This complex may also be recruited to the ICR in vivo, resulting in a compact, repressive chromatin structure capable of silencing the paternal H19 allele.
INTRODUCTION
Genomic imprinting is responsible for the parental allele-specific expression of a subset of genes in the mammalian genome. Silencing of one of the parental alleles requires DNA methylation (1) and chromatin structural modifications (2–5). A strong correlation exists between CpG methylation and transcriptional repression at a number of genes (6). However, the precise mechanisms by which these epigenetic modifications induce transcriptional repression at imprinted loci are poorly understood. In the case of the H19 gene the paternally inherited allele is silenced. A 2 kb CpG-rich differentially methylated domain (DMD) upstream of the H19 gene is essential for repression and a 1.2 kb region within the DMD functions specifically as a silencer element on the methylated paternal chromosome (7,8). The proteins that bind at the H19 DMD and mediate this silencing have yet to be identified, although a family of proteins containing a conserved methyl-CpG-binding domain (MBD) (9,10), of which MeCP2 (11,12) is the best characterised, are obvious candidates.
MeCP2 can bind to a single methylated CpG dinucleotide through its MBD and interacts with Sin3a and histone deacetylases (HDACs) through a transcriptional repression domain (TRD) in vivo, forming a repressive complex (13,14). The TRD is also capable of interacting with TFIIB, a component of the basal transcription machinery (15,16), suggesting that it may also be capable of causing repression independently of HDAC activity.
In mice, MeCP2 is particularly abundant in the brain (17) and mutations in MeCP2 have recently been shown to contribute to Rett syndrome in humans (6,18–21), an X-linked neurodevelopmental disorder that is one of the most common causes of mental retardation in females (22). These mutations affect the ability of the protein to repress transcription either by impairing selectivity for methylated DNA binding or by decreasing the stability of the protein in vivo (23,24). MeCP2 is required for normal post-natal neurological development in mice (25,26). However, its role in regulating gene expression earlier in development is unknown and this role may be difficult to establish accurately because of possible functional redundancy amongst the MBD family members (27).
In this study we have investigated whether MeCP2 mediates transcriptional repression from the H19 DMD. Using chromatin immunoprecipitation (ChIP) with an anti-MeCP2 antibody, coupled with PCR amplification of the immunoprecipitated DNA, we show that MeCP2 is recruited to the H19 DMD in vivo. Using a HeLa cell transfection assay we also demonstrate that transcriptional repression from the DMD is mediated by MeCP2 in a methylation-dependent manner and requires the TRD of the protein. Transcriptional repression can be almost completely alleviated by inhibition of HDAC activity. These data demonstrate that in vivo silencing of the paternal H19 allele from the DMD involves DNA methylation and the possible recruitment, through MeCP2 and Sin3a, of a HDAC, which presumably acts to ensure an inactive repressed chromatin state.
MATERIALS AND METHODS
HeLa cell transfection reporter assay
Regions from the mouse H19 DMD were amplified by PCR and cloned into the MluI site in the pGL3-Promoter vector (Promega), upstream of an SV40 promoter driving a firefly luciferase reporter gene, and sequenced to confirm integrity and orientation. The following PCR primers were used: full-length DMD, 4S (5′-TGCCTACAGTTCCCGAATCACC-3′) and 2AS (5′-CGGCATCGTCTGTCCATTTAGC-3′); 5′-DMD, 4S and 2.8AS (5′-GCTTTTCTGCTTTCTGGCATCG-3′); 3′-DMD, 2.8S (5′-AGCCGTTGTGAGTGGAAAGACC-3′) and 2AS; mid DMD, 3.1S (5′-AACCGCCAACAAGAAAGTCTGG-3′) and 2.6AS (5′-GCTTCGGACATTGCTGTGGG-3′); down DMD, 2.6S (5′-AAGCCGCTATGCCTCAGTGG-3′) and 2AS.
An aliquot of 2 µg of these DNA constructs was transfected into HeLa cells which were cultured for 20 h. The cells were lysed and luciferase levels were measured. The firefly luciferase values were normalised against 0.2 µg co-transfected Renilla luciferase reporter gene driven by a thymidine kinase (TK-Renilla) promoter, as described in the Dual-Luciferase Reporter Assay System protocol (Promega). Each construct was tested in triplicate in each experiment, and the experiments were repeated a minimum of three times. The constructs were either unmethylated or fully methylated at all CpGs by incubation with SssI methylase (NEB) in the presence of 160 µM S-adenosylmethionine in NEB buffer 2 (50 mM NaCl, 10 mM Tris–HCl pH 7.9, 10 mM MgCl2, 1 mM DTT) at 37°C for 16 h. Cells transfected with TK-Renilla alone demonstrated no firefly luciferase activity.
To test the effect of MeCP2 expression, constructs were generated containing full-length human MeCP2 or MeCP2 lacking the TRD (residues 207–310), designated MeCP2 ΔTRD, cloned between the KpnI and XhoI sites of the mammalian expression vector pcDNAI (Invitrogen). The inserts were generated by PCR from the plasmid Actin5C.MeCP2 (a gift of S. Kudo). The Rett mutation, R306C, was introduced into the MeCP2 construct using the QuikChange mutagenesis kit (Stratagene). All constructs were checked by automated DNA sequencing. The Ezh1 expression construct (pKW2T/Ezh1#10) consists of a CMV promoter, the Ezh1 open reading frame and SV40 poly(A) tail and was a gift from T. Jenuwein.
Anti-MeCP2 antibody
An antiserum was raised in rabbits against the His-tagged recombinant methyl-binding domain of rat MeCP2 (residues 78–164) expressed in Escherichia coli from plasmid pET6HMBD, which was a gift from A. Bird. In western blots of rat liver and brain nuclei, used at 1 in 1000 dilution, it recognised a single band with an apparent Mr of ∼68 000 in an SDS–12% polyacrylamide gel. (The actual Mr of rat MeCP2 is 53 014, but it migrates anomalously; authentic recombinant MeCP2 with a 20 residue N-terminal tag, whose actual Mr is 55 022 migrates with an apparent Mr of ∼70 000.)
Chromatin immunoprecipitation assay
Crosslinking, sonication and immunoprecipitation. Samples of 400 A260 units of nuclei (28) isolated from adult or neonate mouse liver were resuspended in 5 ml of 0.32 mM sucrose, 1 mM MgCl2, 1 mM phenylmethylsulphonyl chloride. The nuclei were crosslinked with 1% formaldehyde (added from a 38% stock; Sigma) at room temperature with gentle shaking for 15 min and the reaction was then quenched with 125 mM glycine (added from a 2.5 M stock). After 5 min further incubation at room temperature the nuclei were collected by centrifugation (5000 r.p.m. for 5 min at 4°C in a Sorvall SS34 rotor), washed twice in ice-cold buffer (2 mM potassium phosphate, pH 7.4, 0.15 M NaCl) and resuspended in 0.25 ml lysis buffer (50 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% v/v Triton X-100, 0.1% w/v sodium deoxycholate) supplemented with protease inhibitors (0.24 mg/ml aminoethylbenzenesulphonyl fluoride, 1 µg/ml leupeptin, 1 µg/ml aproteinin, 1.56 mg/ml benzamidine, 20 µg/ml tosyl-l-chloroketone; all from Sigma). The nuclei were sonicated in an ice–water bath to shear the chromatin with three 8 s pulses of 2.5 kV with a rest interval of 20 s; the resulting average DNA size was ∼0.6 kb (range ∼0.25–2.0 kb). The lysate was cleared by centrifugation (twice for 5 min at 13 000 r.p.m. at 4°C in a microfuge).
An aliquot of 1.25 µl of anti-MeCP2 antiserum (or 1.25 µl of preimmune serum) was added to 100 µl of the formaldehyde-crosslinked chromatin and the samples (and crosslinked chromatin alone) were mixed on a rotator for 2 h at 4°C. Sepharose–protein A beads (15 µl) (Pharmacia) were added and the samples mixed for a further 2 h at 4°C. The beads, with bound chromatin fragments, were collected by centrifugation (1 min at 13 000 r.p.m. in a microfuge at 4°C); the unbound fraction was retained for analysis by western blotting (see below). The beads were washed successively, each for 5 min on a rotator, with 1 ml lysis buffer (see above), 1 ml wash buffer 1 (50 mM HEPES pH 7.5, 500 mM NaCl, 1 mM EDTA, 1% v/v Triton X-100, 0.1% w/v sodium deoxycholate), 1 ml wash buffer 2 (10 mM Tris–HCl pH 8.0, 0.5% v/v NP40, 0.5% w/v sodium deoxycholate, 1 mM EDTA, 0.14 M LiCl2) and finally 1 ml TE (10 mM Tris–HCl pH 8.0, 1 mM EDTA). The final wash was retained for western analysis. The washed beads were resuspended in 60 µl elution buffer (50 mM Tris–HCl pH 8.0, 1 mM EDTA, 1% w/v SDS) and incubated at 65°C for 10 min to release bound (immunoprecipitated) chromatin fragments. The beads were then removed by centrifugation (10 min at 13 000 r.p.m. in a microfuge). An aliquot of 20 µl of the supernatant was retained for DNA extraction.
To 20 µl of eluted crosslinked chromatin (or input sonicated chromatin) was added 110 µl TE containing 1% SDS and the samples were incubated at 65°C for 12 h to reverse the protein–DNA crosslinks. They were then allowed to cool to room temperature and deproteinised by digestion with 100 µg proteinase K in the presence of 2 µg glycogen at 37°C for 1 h. To this was added 25 µl of 5 M LiCl2 and the DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1 v/v) and ethanol precipitated. ‘Input’ DNA samples were resuspended in 50 µl TE and ‘immunoprecipitated’ DNA samples were resuspended in 250 µl TE for the PCR (see below).
Crosslinked proteins present in 1/25th of the input chromatin, the fraction not bound to beads, the final TE wash from beads and the fraction bound by beads were released from DNA by heating at 90°C for 10 min in elution buffer (50 mM Tris–HCl pH 8.0, 1 mM EDTA, 1% w/v SDS), collected by precipitation with 25% (final) trichloroacetic acid, washed with acetone/10 mM HCl followed by acetone and then analysed by western blotting (using anti-MeCP2 and preimmune serum) after electrophoresis in an SDS–12% polyacrylamide gel. Bound MeCP2 was detected with horseradish peroxidase-linked secondary antibody (donkey anti-rabbit; Amersham Pharmacia) coupled to the enhanced chemiluminescence detection system (Amersham Pharmacia).
Polymerase chain reaction. The sequences of the PCR primers used to amplify selected genes in the ChIP assay were as follows: mouse H19 3.1S (5′-AACCGCCAACAAGAAAGTCTGG-3′); mouse H19 2.6AS (5′-GCTTCGGACATTGCTGTGGG-3′); rat H19 forward (5′-CCCGGTATTGGAATCCAC-3′); rat H19 reverse (5′-GAAATGCATGTGTCCTGCCCTCC-3′); GAPDH forward (5′-CATTAACGTCAACTACATGG-3′); GAPDH reverse (5′-TGATGACCAGCTTCCCATTCTCAGC-3′).
The primers for the mouse H19 DMD amplify a region –2.6 to –3.1 kb upstream of the transcription start site. The primers for rat GAPDH amplify a 100 bp region at the start of the coding region of this gene and the rat H19 primers a 100 bp region –2 to –2.1 kb upstream of the transcription start site. The number of cycles of PCR required to give a linear response was determined for each primer set and these conditions were used to amplify the DNA immunoprecipitated in the ChIP assay, using 2.5 U Taq polymerase (Promega). The programme consisted of a hot start of 95°C for 5 min followed by 24–28 cycles of a denaturing step (95°C for 30 s), an annealing step (50°C for 30 s) and an elongation step (72°C for 30 s), followed by a final elongation step of 72°C for 10 min. Controls with no DNA or genomic DNA were included. The PCR products were analysed in an 8% polyacrylamide–Tris acetate/EDTA gel which was stained with 0.5 µg/ml ethidium bromide and viewed on a UV transilluminator.
RESULTS
MeCP2-mediated silencing from methylated H19 DMD
In order to test whether MeCP2 mediates transcriptional repression from the H19 DMD we used a HeLa cell transfection assay. The aim of this in vitro study was to analyse the direct effect of MeCP2 on transcriptional regulation from the H19 DMD, which may not be possible with targeted mutations in vivo due to functional redundancy amongst the MBD-containing proteins (27).
The full-length mouse H19 DMD was cloned upstream of an SV40 promoter-driven firefly luciferase reporter gene (Fig. 1A) and transfected into HeLa cells, which lack endogenous MeCP2 (29). When unmethylated this construct (designated H19 DMD) gives ∼2-fold repression over the corresponding construct containing the promoter alone (Fig. 1B). Methylation of the H19 DMD (H19 DMDm) caused a significant increase in repression, which can only be partially attributed to direct methylation-mediated silencing of the SV40 promoter (Fig. 1B). Co-transfection of a human MeCP2-expressing vector with the methylated promoter sequence alone did not mediate further significant silencing (Fig. 1B, SV40 promoterm + MeCP2). This observation is consistent with the continued repression seen from a methylated SV40 promoter in MeCP2–/– cells, just as in wild-type cells (26), and suggests that MeCP2 does not mediate significant repression on the promoter alone. In contrast, co-transfection of the MeCP2-expressing vector with the methylated H19 DMD reporter construct caused an ∼5.5-fold increase in repression of the reporter gene (Fig. 1B), corresponding to ∼200-fold repression over the unmethylated SV40 promoter alone. Significantly, MeCP2-mediated silencing is dependent on methylation of the H19 DMD, since no repression occurred in the absence of DNA methylation. The repression is also dose dependent, as shown by proportionate reductions in repression when 200, 100 and 50 ng of the MeCP2 vector, respectively, were co-transfected (Fig. 1B).
Figure 1.

MeCP2 represses transcription from the methylated H19 DMD in a HeLa cell transfection assay. (A) Schematic representation of the H19 gene and 4 kb upstream sequence. Individual methylation-sensitive CfoI restriction enzyme sites in the H19 promoter and upstream region are shown as unmethylated on the maternal chromosome (open circles, above line) or methylated on the paternal chromosome (filled circles, below line). Vertical arrows indicate DNase I hypersensitive sites, which are clustered on the maternal chromosome. The lower part of the figure shows the 1.2 kb silencer element (white box), 2 kb differentially methylated domain (grey box) and the sub-regions tested in the cell transfection assay (see text). The number of CpG dinucleotides and the mean separation (in bp) of CpGs in the regions tested are indicated in parentheses. B, BamHI; Bs, BspEI; H, HindIII; R, EcoRI. (B) Repression of transfected firefly luciferase reporter constructs in HeLa cells. Regions from the mouse H19 DMD, amplified by PCR, were cloned into the MluI site in the pGL3-Promoter vector (Promega), upstream of an SV40 promoter driving a firefly luciferase reporter gene, and sequenced to confirm integrity and orientation. The SV40 promoter alone and the H19 differentially methylated domain + SV40 promoter (H19 DMD) constructs were tested unmethylated and methylated (m). A MeCP2-expressing vector (200, 100 or 50 ng) was transfected as indicated. Light emission values were normalised with respect to the value for the unmethylated SV40 promoter alone, taken as 1. Error bars show calculated SEM values.
To examine the specificity of the MeCP2-mediated silencing we also tested the effect of Ezh1, a member of the Polycomb group of heterochromatin proteins, which has a role in gene silencing (30). Co-transfection of a mouse Ezh1 expression construct with either the unmethylated or methylated H19 DMD construct caused no additional repression (data not shown). We also examined MeCP2-mediated repression from four sub-regions of the H19 DMD (Fig. 1A). In contrast to the full-length DMD, none of the methylated sub-regions exhibited significant repression when MeCP2 was co-transfected in this assay (data not shown), despite the approximately equivalent density of CpGs between all the sub-regions and full-length DMD (1 CpG dinucleotide every 30–41 nt; see Fig. 1A). This observation is in agreement with our previous in vivo studies, where methylation of a 1.1 kb segment from the 5′-region of the DMD alone is not sufficient for silencing of H19 in many tissues (7), suggesting that the total number of CpGs is important for repression. It is also possible that the size of the region from the DMD, or its spacing relative to the SV40 promoter, are important for repression, as the sub-regions are all smaller than the full-length DMD.
Inhibition of HDAC activity relieves H19 DMD-mediated repression
To determine whether MeCP2-mediated repression at the H19 DMD requires the TRD of MeCP2, a deletion mutant of MeCP2 (ΔTRD), lacking the TRD, was tested in the transfection assay. The ΔTRD mutant had no repressive activity (Fig. 2A). A mutation (R306C, very near the C-terminal end of the TRD) found in some Rett syndrome patients (20) did not disrupt repression from the H19 DMD (Fig. 3A), in agreement with another study in which effects on transcription were tested by injection into Xenopus oocytes (24). The TRD of MeCP2 has been shown to interact with the co-repressor molecule Sin3a and HDACs (13,14). To test whether the recruitment of HDACs to the H19 DMD is responsible for repression from this control region in this assay we analysed expression of the transfected reporter construct in the presence of co-transfected MeCP2-expressing vector and trichostatin A (TSA), a known inhibitor of HDAC activity (31). TSA had no effect on expression of the unmethylated H19 DMD–SV40–luc reporter construct (Fig. 2B). However, repression of the methylated reporter construct in the presence of MeCP2 was alleviated by >95% by TSA (Fig. 2B), suggesting that histone deactylation is the predominant mechanism of repression from the H19 DMD.
Figure 2.
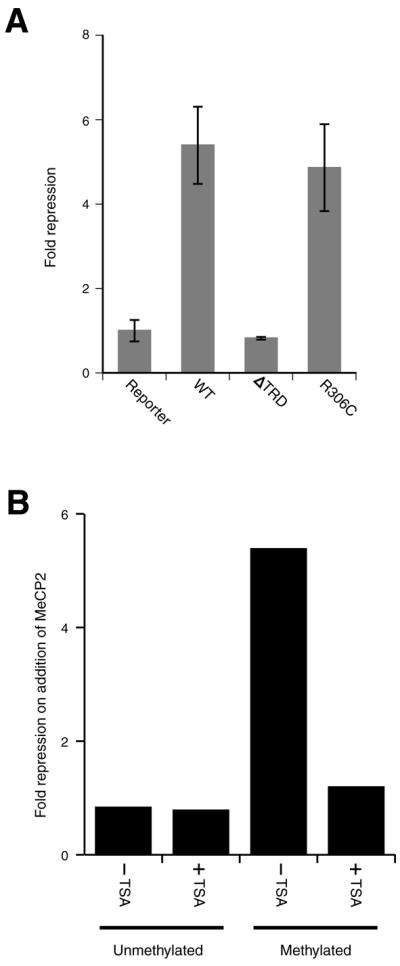
MeCP2-mediated repression is linked to HDAC recruitment. (A) Comparison of repression mediated by wild-type (WT), TRD-deleted (ΔTRD) and R306C mutant MeCP2 expression vectors. Constructs were generated containing full-length human MeCP2, MeCP2 lacking the transcriptional repression domain (residues 207–310), designated MeCP2 ΔTRD, or the Rett mutation MeCP2 (R306C). Light emission values were normalised with respect to the value for the methylated H19 DMD–SV40 promoter–firefly luciferase construct. Error bars show calculated SEM values. (B) The effect of TSA on repression of transfected constructs. Repression, on addition of an MeCP2-expressing vector, of an unmethylated or methylated H19–SV40 promoter–firefly luciferase reporter construct in the absence (–TSA) or presence (+TSA) of 100 mM TSA is shown. TSA alleviates >95% of the repression observed for the methylated construct in the absence of TSA.
Figure 3.

MeCP2 is associated with the H19 ICR in vivo. (A) The primers for mouse H19 DMD amplify a region –3.1 to –2.6 kb upstream of the transcription start site (mid DMD PCR primers). (B) PCR amplification of DNA co-immunoprecipitated with the MeCP2 antibody from mouse adult and neonatal liver chromatin. Formaldehyde-crosslinked, sonicated chromatin fragments were immunoprecipitated with anti-MeCP2 antiserum and Sepharose–protein A beads or with preimmune serum. Protein–DNA crosslinks in the chromatin immunoprecipitated with both preimmune (as a control) and MeCP2 antisera were reversed and the 527 bp region from the H19 DMD was amplified by PCR and in parallel amplified from input DNA (not immunoprecipitated). MeCP2 was detected in the bound fraction in both adult and neonatal liver chromatin. The number of cycles of PCR required to give a linear response was determined. N, no DNA; I, input; P, preimmune; B, bound (MeCP2 serum-precipitated DNA); M, size markers (pBR322/MspI). (C and D) Model of methylation-dependent repression from the H19 ICR. (C) At the paternal H19 allele, DNA methylation (filled circles) at the DMD is recognised by MeCP2. MeCP2 is capable of recruiting Sin3a and HDACs via its TRD (dark grey ellipse). HDACs act locally to deacetylate the tails of histones proximal to H19, resulting in chromatin compaction and silencing of the H19 gene. (D) At the maternal H19 allele the absence of DNA methylation (open circles) at the DMD prevents recruitment of the MeCP2/Sin3a/HDAC complex. The histones in the region therefore remain acetylated and the chromatin is accessible to factors necessary for transcription of the H19 gene.
MeCP2 is associated with the H19 differentially methylated domain in vivo
To investigate whether endogenous MeCP2 was recruited to the H19 DMD in vivo, we used ChIP (32,33) with an anti-MeCP2 antibody. The antibody we raised specifically detects MeCP2 in adult rat liver nuclei, where a single band is observed at ∼68 kDa, corresponding to MeCP2 (which migrates anomalously slowly, Mr ∼53 kDa), and in formaldehyde-crosslinked and sonicated chromatin (data not shown). MeCP2 is immunoprecipitated by the antiserum, but not by the preimmune serum. In experiments on both adult and neonatal mouse liver, a 0.5 kb region from the H19 DMD (Fig. 3A) could be amplified from the immunoprecipitated DNA (after reversal of the crosslinks), showing that MeCP2 was recruited to this region in both tissues (Fig. 3B). In neonatal liver, H19 is predominantly expressed only from the maternal allele and silenced on the paternal allele (7). The association of MeCP2 therefore correlates both with general repression in adult tissue and with repression in a tissue in which H19 is imprinted. MeCP2 can also be detected by ChIP at the region upstream of the H19 gene in rat liver and brain, but not at the ubiquitously expressed GAPDH gene in these tissues (data not shown).
DISCUSSION
At the mouse H19/Igf2 genomic locus the DMD upstream of H19 acts as a cis-regulatory imprinting control region (ICR). The activity of the DMD is modified depending on its methylation status. On the maternal chromosome it is unmethylated and is proposed to act as an insulator, capable of disrupting the interaction of downstream enhancers with the Igf2 promoter (34–37). Conversely, on the paternal chromosome the methylated DMD contains a silencer element responsible for repression of H19 (7,37). Here we show that repression from this silencer element is mediated by recruitment of MeCP2 and possibly an associated protein complex containing HDACs, capable of modifying chromatin structure.
Recent studies have revealed that in the H19/Igf2 imprinted domain it is only in the DMD/promoter region of the H19 gene that there is a correlation between DNA methylation, histone H4 hypoacetylation and transcriptional repression (3). Our data demonstrate that MeCP2 is recruited to the H19 DMD in vivo. This factor can facilitate silencing of a reporter gene from this cis-acting ICR in a transfection assay in vitro. DNA methylation is required for MeCP2-mediated repression and this repression is therefore likely to occur in vivo only on the methylated paternal H19 allele (Fig. 3C). Sub-regions of the methylated DMD are not sufficient to mediate additional repression when MeCP2 is co-expressed. As the 3′-DMD fragment we tested overlaps extensively with the silencer region identified by deletion in vivo (see Fig. 1A), this suggests that transcriptional repression at the endogenous H19 locus may be mediated by other trans-acting factors in addition to MeCP2. Different members of the MBD-containing family of proteins are obvious candidates for this role. Indeed, the existing genetic evidence points to some level of functional redundancy amongst these factors in vivo (27). While our study does not address the role of MeCP2-mediated repression from the H19 ICR directly on the endogenous H19 promoter, we have previously shown that deletion of the MeCP2-binding region at the DMD in vivo results in reactivation of endogenous H19 expression (7). In addition, transgenic studies have demonstrated that the silencer region from the DMD is capable of repressing transcription both from the H19 promoter (8) and heterologous promoters (38), indicating that the repression is not promoter specific but may be a consequence of promoter proximity to the H19 ICR silencer.
The silencing mediated by MeCP2 via the full-length H19 DMD in the transfection assay can be almost completely alleviated by inhibition of HDAC activity, suggesting that deacetylases play a major role in facilitating repression. The targets of this repressive deacetylation activity at the endogenous locus are likely to include the tails of histones located within or close to the H19 ICR on the paternal allele (3,5; Fig. 3C). It is known that at the endogenous H19 locus the level of acetylation on histones H3 and H4 is important for gene regulation, as the silenced paternal allele is hypoacetylated and the expressed maternal allele is hyperacetylated (5). In our model, the absence of DNA methylation on the maternal H19 allele would prevent binding of the repressive complex, facilitating acetylation of histones and potentially allowing the recruitment of factors necessary for transcriptional activation of the gene (Fig. 3D). The existence of differentially methylated genomic regions at a number of genes exhibiting allele-specific expression will make it of interest to examine whether similar mechanisms of transcriptional regulation are employed at other imprinted loci.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Teca Galvao for providing the recombinant MBD of MeCP2 used to raise an antiserum, Prof. S. Kudo for the human MeCP2 clone and Esther Bae, Katharine Arney and Michael Levine for helpful comments on the manuscript. R.A.D. acknowledges receipt of a Wellcome Trust Prize Studentship and Fellowship and C.J.G of a Medical Research Council Studentship. This work was supported by grants from the Wellcome Trust to M.A.S. and J.O.T.
REFERENCES
- 1.Li E., Beard,C. and Jaenisch,R. (1993) Role of DNA methylation in genomic imprinting. Nature, 366, 362–365. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson-Smith A.C., Sasaki,H., Cattanach,B.M. and Surani,M.A. (1993) Parental-origin-specific modification of the mouse H19 gene. Nature, 362, 751–755. [DOI] [PubMed] [Google Scholar]
- 3.Grandjean V., O’Neill,L., Sado,T., Turner,B. and Ferguson-Smith,A. (2001) Relationship between DNA methylation, histone H4 acetylation and gene expression in the mouse imprinted Igf2-H19 domain. FEBS Lett., 488, 165–169. [DOI] [PubMed] [Google Scholar]
- 4.Khosla S., Aitchison,A., Gregory,R., Allen,N.D. and Feil,R. (1999) Parental allele-specific chromatin configuration in a boundary-imprinting-control element upstream of the mouse H19 gene. Mol. Cell. Biol., 19, 2556–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pedone P.V., Pikaart,M.J., Cerrato,F., Vernucci,M., Ungaro,P., Bruni,C.B. and Riccio,A. (1999) Role of histone acetylation and DNA methylation in the maintenance of the imprinted expression of the H19 and Igf2 genes. FEBS Lett., 458, 45–50. [DOI] [PubMed] [Google Scholar]
- 6.Robertson K.D. and Wolffe,A.P. (2000) DNA methylation in health and disease. Nature Rev. Genet., 1, 11–19. [DOI] [PubMed] [Google Scholar]
- 7.Drewell R.A., Brenton,J.D., Ainscough,J.F., Barton,S.C., Hilton,K.J., Arney,K.L., Dandolo,L. and Surani,M.A. (2000) Deletion of a silencer element disrupts H19 imprinting independently of a DNA methylation epigenetic switch. Development, 127, 3419–3428. [DOI] [PubMed] [Google Scholar]
- 8.Brenton J.D., Drewell,R.A., Viville,S., Hilton,K.J., Barton,S.C., Ainscough,J.F.-X. and Surani,M.A. (1999) A silencer element identified in Drosophila is required for imprinting of H19 reporter transgenes in mice. Proc. Natl Acad. Sci. USA, 96, 9242–9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cross S.H., Meehan,R.R., Nan,X.S. and Bird,A. (1997) A component of the transcriptional repressor MeCP1 shares a motif with DNA methyltransferase and HRX proteins. Nature Genet., 16, 256–259. [DOI] [PubMed] [Google Scholar]
- 10.Hendrich B., Abbott,C., McQueen,H., Chambers,D., Cross,S. and Bird,A. (1999) Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3 and Mbd4 genes. Mamm. Genome, 10, 906–912. [DOI] [PubMed] [Google Scholar]
- 11.Lewis J.D., Meehan,R.R., Henzel,W.J., Maurer-Fogy,I., Jeppesen,P., Klein,F. and Bird,A. (1992) Purification, sequence and cellular localisation of a novel chromosomal protein that binds to methylated DNA. Cell, 69, 905–914. [DOI] [PubMed] [Google Scholar]
- 12.Meehan R., Lewis,J. and Bird,A. (1992) Characterisation of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res., 20, 5085–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones P.L., Veenstra,G.J., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger,N., Strouboulis,J. and Wolffe,A.P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- 14.Nan X., Ng,H.H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenman,R.N. and Bird,A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex [see comments]. Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 15.Kaludov N.K. and Wolffe,A.P. (2000) MeCP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res., 28, 1921–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu F., Thiesen,J. and Stratling,W.H. (2000) Histone deacetylase-independent transcriptional repression by methyl-CpG-binding protein 2. Nucleic Acids Res., 28, 2201–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tate P., Skarnes,W. and Bird,A. (1996) The methyl-CpG binding protein MeCP2 is essential for embryonic development in the mouse. Nature Genet., 12, 205–208. [DOI] [PubMed] [Google Scholar]
- 18.Amir R.E., Van den Veyver,I.B., Wan,M., Tran,C.Q., Francke,U. and Zoghbi,H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2 [see comments]. Nature Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 19.Amir R.E., Van den Veyver,I.B., Schultz,R., Malicki,D.M., Tran,C.Q., Dahle,E.J., Philippi,A., Timar,L., Percy,A.K., Motil,K.J. et al. (2000) Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann. Neurol., 47, 670–679. [PubMed] [Google Scholar]
- 20.Van den Veyver I.B. and Zoghbi,H.Y. (2000) Methyl-CpG-binding protein 2 mutations in Rett syndrome. Curr. Opin. Genet. Dev., 10, 275–279. [DOI] [PubMed] [Google Scholar]
- 21.Wan M., Zhao,K., Lee,S.S.J. and Francke,U. (2001) MECP2 truncating mutations cause histone H4 hyperacetylation in Rett syndrome. Hum. Mol. Genet., 10, 1085–1092. [DOI] [PubMed] [Google Scholar]
- 22.Rett A. (1966) [On an unusual brain atrophy syndrome in hyperammonemia in childhood]. Wien. Med. Wochenschr., 116, 723–726. [PubMed] [Google Scholar]
- 23.Ballestar E., Yusufzai,T.M. and Wolffe,A.P. (2000) Effects of rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry, 39, 7100–7106. [DOI] [PubMed] [Google Scholar]
- 24.Yusufzai T.M. and Wolffe,A.P. (2000) Functional consequences of Rett Syndrome mutations on human MeCP2. Nucleic Acids Res., 28, 4172–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen R.Z., Akbarian,S., Tudor,M. and Jaenisch,R. (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nature Genet., 27, 327–331. [DOI] [PubMed] [Google Scholar]
- 26.Guy J., Hendrich,B., Holmes,M., Martin,J.E. and Bird,A. (2001) A mouse MeCP2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature Genet., 27, 322–326. [DOI] [PubMed] [Google Scholar]
- 27.Hendrich B., Guy,J., Ramsahoye,B., Wilson,V.A. and Bird,A. (2001) Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev., 15, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas J.O. (1998) Isolation and fractionation of chromatin and linker histones. In Gould,H.J. (ed.), Chromatin. Oxford University Press, Oxford, UK, pp. 1–34.
- 29.Ng H.H., Zhang,Y., Hendrich,B., Johnson,C.A., Turner,B.M., Erdjument-Bromage,H., Tempst,P., Reinberg,D. and Bird,A. (1999) MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex [see comments]. Nature Genet., 23, 58–61. [DOI] [PubMed] [Google Scholar]
- 30.Laible G., Wolf,A., Dorn,R., Reuter,G., Nislow,C., Lebersorger,A., Popkin,D., Pillus,L. and Jenuwein,T. (1997) Mammalian homologues of the Polycomb-group gene Enhancer of zeste mediate gene silencing in Drosophila heterochromatin and at S.cerevisiae telomeres. EMBO J., 16, 3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida M., Kijima,M., Akita,M. and Beppu,T. (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem., 265, 17174–17179. [PubMed] [Google Scholar]
- 32.Hecht A., Strahl-Bolsinger,S. and Grunstein,M. (1999) Mapping DNA interaction sites of chromosomal proteins. Crosslinking studies in yeast. Methods Mol. Biol., 119, 469–479. [DOI] [PubMed] [Google Scholar]
- 33.Strutt H. and Paro,R. (1999) Mapping DNA target sites of chromatin proteins in vivo by formaldehyde crosslinking. Methods Mol. Biol., 119, 455–467. [DOI] [PubMed] [Google Scholar]
- 34.Bell A.C. and Felsenfeld,G. (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature, 405, 482–485. [DOI] [PubMed] [Google Scholar]
- 35.Hark A.T., Schoenherr,C.J., Katz,D.J., Ingram,R.S., Levorse,J.M. and Tilghman,S.M. (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature, 405, 486–489. [DOI] [PubMed] [Google Scholar]
- 36.Kanduri C., Pant,V., Loukinov,D., Pugacheva,E., Qi,C.F., Wolffe,A., Ohlsson,R. and Lobanenkov,V.V. (2000) Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol., 10, 853–856. [DOI] [PubMed] [Google Scholar]
- 37.Thorvaldsen J.L., Duran,K.L. and Bartolomei,M.S. (1998) Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev., 12, 3693–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyko F., Brenton,J.D., Surani,M.A. and Paro,R. (1997) An imprinting element from the mouse H19 locus functions as a silencer in Drosophila. Nature Genet., 16, 171–173. [DOI] [PubMed] [Google Scholar]