

SUMMARY
The parasite Cryptosporidium hominis is a leading cause of the diarrheal disease cryptosporidiosis, whose incidence in the United States has increased since 2005. Here, we show that the newly emerged and hyper-transmissible subtype IfA12G1R5 is now dominant in the United States. In a comparative analysis of 127 newly sequenced and 95 published C. hominis genomes, IfA12G1R5 isolates from the United States place into three of the 14 clusters (Pop6, Pop13, and Pop14), indicating that this subtype has multiple ancestral origins. Pop6 (IfA12G1R5a) has an East Africa origin and has recombined with autochthonous subtypes after its arrival. Pop13 (IfA12G1R5b) is imported from Europe, where it has recombined with the prevalent local subtype, whereas Pop14 (IfA12G1R5c) is a progeny of secondary recombination between Pop6 and Pop13. Selective sweeps in invasion-associated genes have accompanied the emergence of the dominant Pop14. These observations offer insights into the emergence and evolution of hyper-transmissible pathogens.
Graphical Abstract
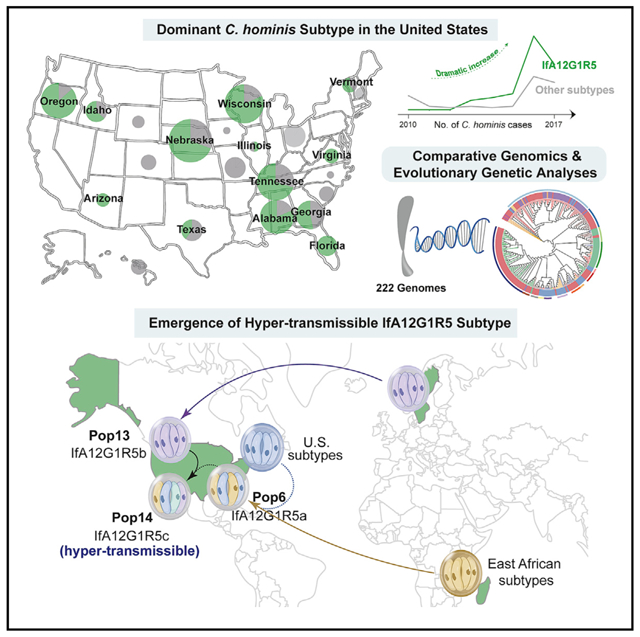
In brief
A newly emerged Cryptosporidium hominis subtype is associated with increased incidence of cryptosporidiosis in the United States. Huang et al. use comparative genomics to trace this subtype’s evolutionary history involving multiple imports and secondary recombination. Adaptive selection in invasion-associated genes has led to the dominance of one of the three variants.
INTRODUCTION
Cryptosporidiosis is a major cause of diarrhea and diarrhea-associated deaths in children in low- and middle-income countries and waterborne diseases in high-income nations, including the United States.1 Research on the pathogen Cryptosporidium has therefore attacked major attention recently.2 In the United States, because of its nationally notifiable disease status, cryptosporidiosis has been under surveillance since the massive waterborne outbreak in Milwaukee in 1993, which caused illness in 403,000 people.3,4 For a long time, the reported incidence of human cryptosporidiosis in the United States had been approximately 1 case per 100,000 persons. Since 2005, however, there has been a substantial increase in the incidence of cryptosporidiosis; the factors contributing to this increase are not fully clear.5
Cryptosporidium hominis (C. hominis) is an anthroponotic species and the dominant cause of human cryptosporidiosis in most areas.6 It is responsible for most outbreaks of cryptosporidiosis in the United States and European countries.7,8 Thus far, over 10 C. hominis subtype families have been identified based on sequence analysis of the 60-kDa glycoprotein (gp60) gene, with Ia, Ib, Id, Ie, and If being the most common ones.6 Among them, the IbA10G2 subtype is widely distributed in both low- and high-income countries and is the dominant subtype responsible for C. hominis-associated outbreaks in Europe.8
In the United States, IbA10G2 was the dominant C. hominis subtype for outbreaks in early years, including the massive 1993 Milwaukee outbreak.9 In 2005, a previously undetected C. hominis subtype, IaA28R4, appeared in the United States. By 2007, it was identified in a multistate outbreak and the majority of sporadic cases.10 This subtype largely disappeared in the United States within a few years and appears to be replaced by IfA12G1R5, which is frequently seen in outbreaks and sporadic cases since 2013.7 IfA12G1R5 has recently become a common C. hominis subtype in Australia and New Zealand.11,12 The genetic factors involved in the alternation of C. hominis subtypes and emergence of the hyper-transmissible subtype IfA12G1R5 in the United States are poorly understood.
In this study, we have acquired whole-genome sequencing (WGS) data from 127 C. hominis isolates collected from the United States, Spain, and China in recent years and conducted comparative genomics and evolutionary genetic analyses of the data together with 95 published ones to understand the evolution of C. hominis and the emergence of IfA12G1R5 in the United States. Results of the analyses indicate that multiple introductions and genetic recombination events and the subsequent adaptive selection have led to the emergence of the hyper-transmissible subtype.
RESULTS
IfA12G1R5 has become the most frequently detected C. hominis subtype in the United States
Among the 1,075 Cryptosporidium-positive stool samples submitted by public health laboratories (Figure S1A), C. hominis was identified in 368 samples. Sequence analysis of the gp60 gene indicated the presence of 21 subtypes in six subtype families, including Ia, Ib, Id, Ie, If, and Ig (Figure 1A). During this period, IfA12G1R5 appeared first in 2013 and became the dominant subtype in the United States ever since (Figures 1B and 1C). This subtype was detected in 14 of 23 states that submitted C. hominis samples during 2010–2017 (Figure 1D).
Figure 1. Distribution of Cryptosporidium spp. and subtypes in recent years (2010–2017) in the United States.
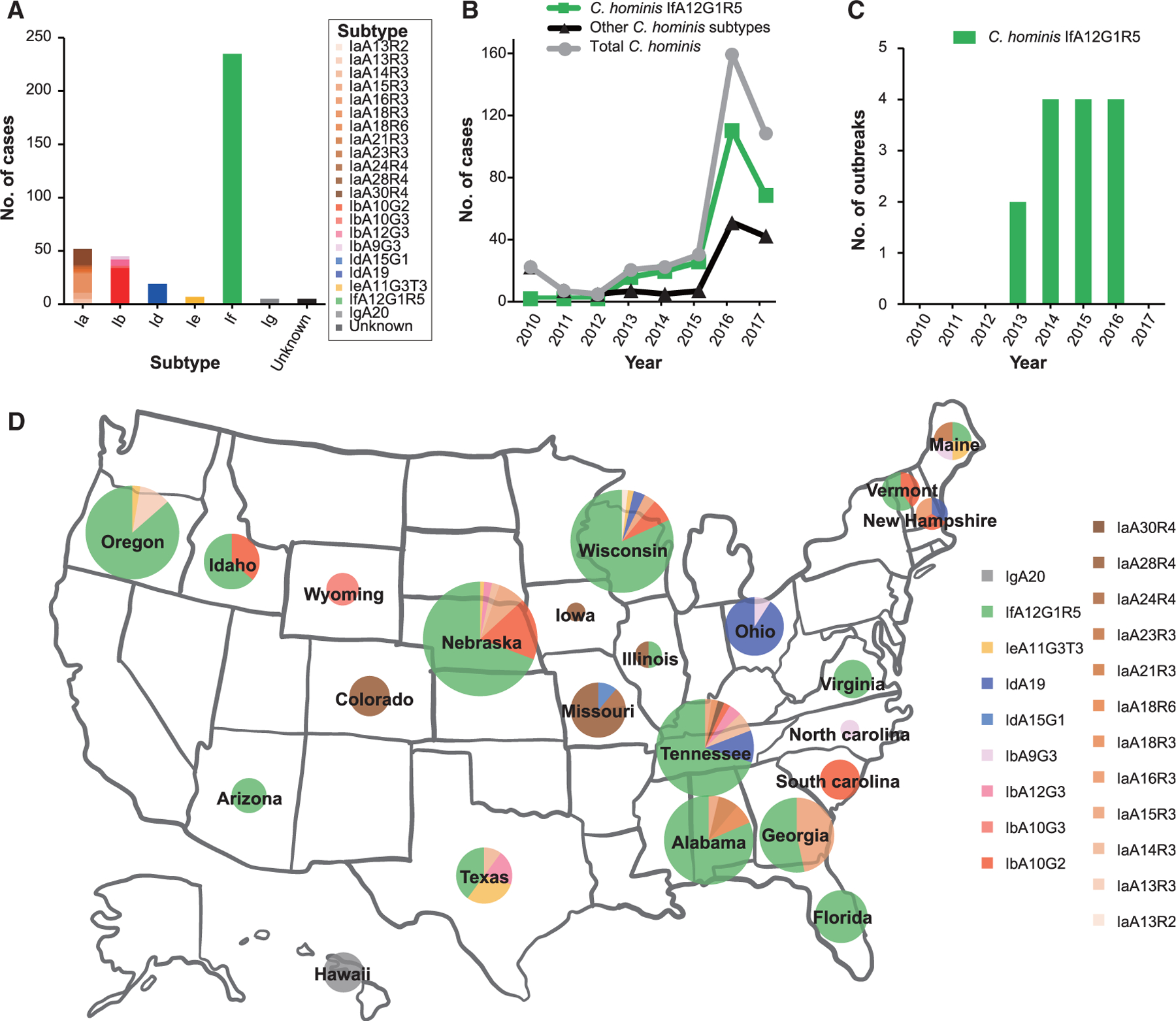
(A) Number of cases of C. hominis subtype families and subtypes in outbreak and sporadic cases in the United States.
(B) Number of cases of C. hominis subtypes in outbreak and sporadic cases in the United States by year.
(C) C. hominis IfA12G1R5 identified in outbreak cases in the United States.
(D) Wide occurrence of IfA12G1R5 across the United States.
Distribution of C. hominis isolates used in comparative genomics analyses
Diversity and phylogenetic relationship of C. hominis were examined at the whole-genome level. We used 249 C. hominis genomes in the initial analysis, including 146 and 103 genomes newly sequenced and downloaded from the Sequence Read Archive (SRA) database, respectively. Genomes with one of following characteristics were excluded from further analyses: sequencing depth below 5, genome coverages below 90%, no gp60 sequences, two or more types of gp60 or 18S rRNA sequences, and genome length over 9.1 Mb. After the removal of low-quality genomes, 222 were included, of which 127 were from this study and 95 from public databases (Figure S1; Table S1). The WGS datasets were from six continents. Among the six subtype families, Ib, Ia, and If subtype families were well represented (Figure 2A). Most If isolates (63 of 66), however, were from North America.
Figure 2. Population subdivision within Cryptosporidium hominis and the formation of three populations of IfA12G1R5.
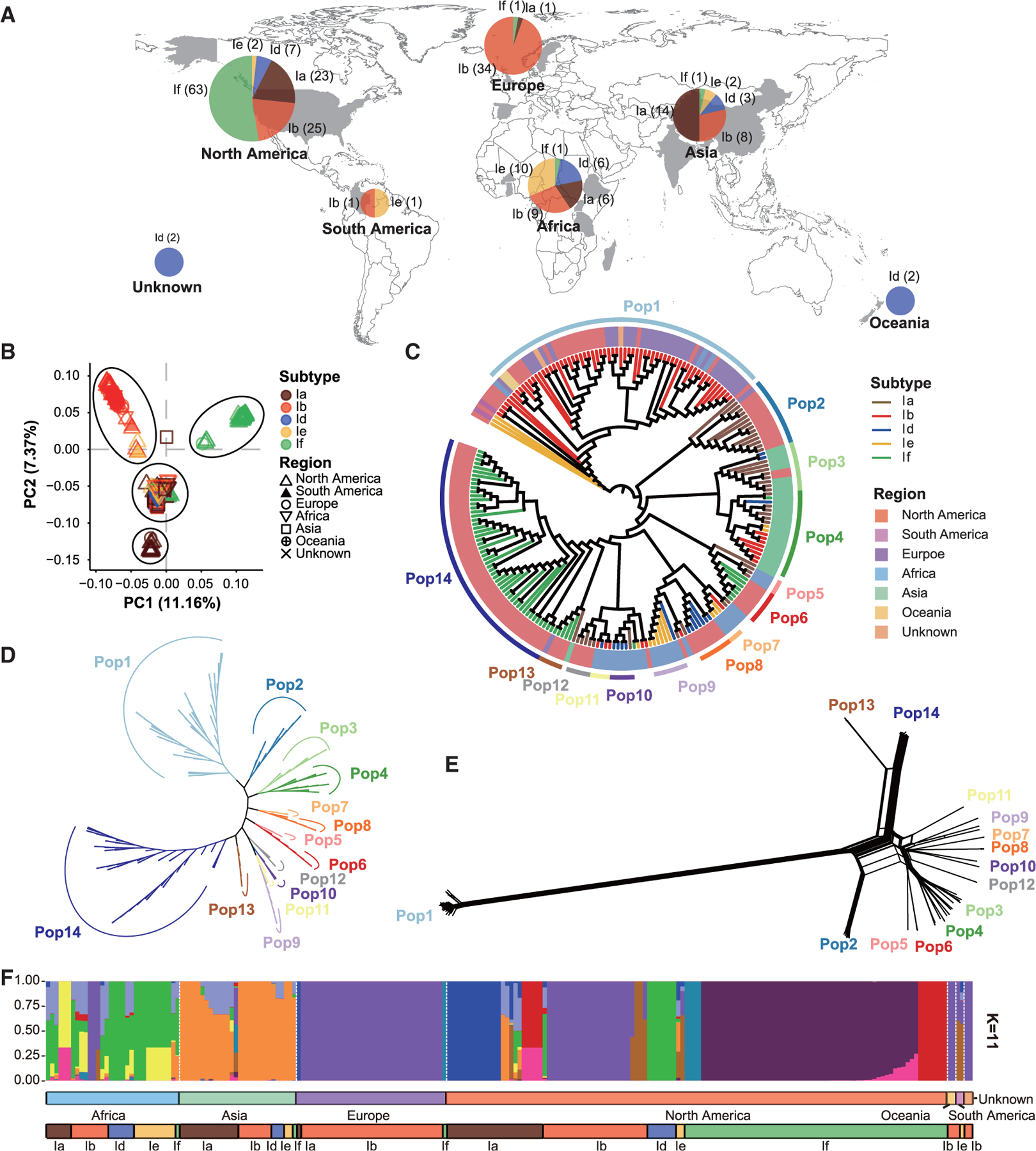
Isolates are colored according to their subtype families (A–C and F), geographical origins (C), and populations (C–E). (A) Geographical origins of 222 samples used in this study. Subtypes Ia, Ib, Id, Ie, and If are represented in brown, red, blue, yellow, and green, respectively. (B) Principal-component analysis (PCA) of C. hominis isolates based on pruned SNPs, in which PC1 and PC2 account for variability among isolates. The colors of the symbols represent the C. hominis subtypes (colored the same as in A). The shapes show the sample sources. (C) Phylogenetic analysis inferred by maximum likelihood (ML) using 12,736 wgSNPs. The circular tree is shown ignoring the branch length, and the branch colors represent different subtypes (colored the same as in A). The background colors of the sample names represent the sample sources. These C. hominis isolates formed 14 clades. (D) Phylogenetic analysis of 9,394 wgSNPs among 199 isolates in the 14 populations. The unrooted ML tree was constructed using the transversion model and gamma distribution (TVM + G) implemented. The colors of the branches represent the populations (colored the same as in C). (E) A phylogenetic network of the 199 isolates based on 9,394 wgSNPs. The parallel edges in the network are suggestive of gene flows among isolates. (F) STRUCTURE plot representing the percentage of shared ancestry among the C. hominis metapopulation (for K = 11).
Population structure of C. hominis
We determined population structure of C. hominis using principal-component analysis (PCA), maximum likelihood (ML), phylogenetic network, and STRUCTURE analyses of the WGS data. There were 12,736 SNPs among the 222 genomes. In the PCA analysis of the SNPs, the genomes formed 4 major clusters, with most Ia, Ib, and If isolates forming their own clusters. Some isolates, including most from Africa and Asia, however, formed the fourth cluster between the other three major clusters (Figure 2B). In agreement with these findings, the C. hominis genomes formed 14 clades organized in the four major clusters in the ML analysis (Figures 2C, 2D, and S2A). Most Ib isolates collected from multiple continents over a long period of time (2004–2018), all of the IbA10G2 subtype, formed one super-clade, which was distinct from the other subtypes. Except for IbA10G2 isolates, other US isolates formed six clades largely segregated by gp60 subtype. In contrast, the Dhaka (Asian) isolates of diverse gp60 subtypes formed two sister clades, whereas the African isolates formed five clades mainly segregated by country origin. This differed significantly from the ML analysis of the gp60 sequences from these isolates, which expectedly formed clusters by subtype family (Figure S2B).
Gene flow among C. hominis populations
As expected, four major clusters of 14 populations were seen in phylogenetic network analysis of the whole-genome SNPs (wgSNPs) data. The presence of parallel edges between some of the populations supported the occurrence of gene flows among isolates, especially between Pop13 and Pop14 and between Pop2 and Pop6 from the United States (Figure 2E). In addition, the STRUCTURE plot showed several more homogeneous populations, including all IbA10G2 isolates from Europe, North America, and Africa and most IfA12G1R5 isolates from North America. In contrast, genome admixture was seen in other isolates from Africa, Asia, and North America, affirming the occurrence of genetic recombination among some subtypes (Figures 2F and S3A).
Formation of three populations within IfA12G1R5
In the phylogenetic and the PCA analyses, the 63 IfA12G1R5 isolates from the United States formed three clades, Pop6, Pop13, and Pop14 (Figures 2C and 3A). Among them, Pop6 and Pop14 isolates were collected during 2014–2017, whereas Pop13 isolates were collected from 2016 to 2017 (Figure 3B). Pop14 showed the widest geographic distribution, being found in eight states. In contrast, the seven Pop6 isolates were collected from Idaho, Maine, and Nebraska, whereas the four Pop13 isolates were collected from Oregon and Alabama. However, most of the states with Pop6 and Pop13 also had Pop14 (Figure 3C). The IfA12G1R5 isolate from Sweden was placed in Pop13, indicating that it is closely related to some US isolates. The genomes of Pop13 and Pop14 showed the most identity. In contrast, genomes of Pop6 were more similar to Pop2-Pop12 (particularly Pop5) than to Pop13 and Pop14 (Figures 3D, S3B, and S3C). Between Pop13 and Pop14, Pop14 had more sequence identity to the other populations. These findings suggest that the three populations (Pop6, Pop13, and Pop14) of IfA12G1R5 from the United States had different ancestral origins.
Figure 3. Origin of Pop6 (IfA12G1R5a).
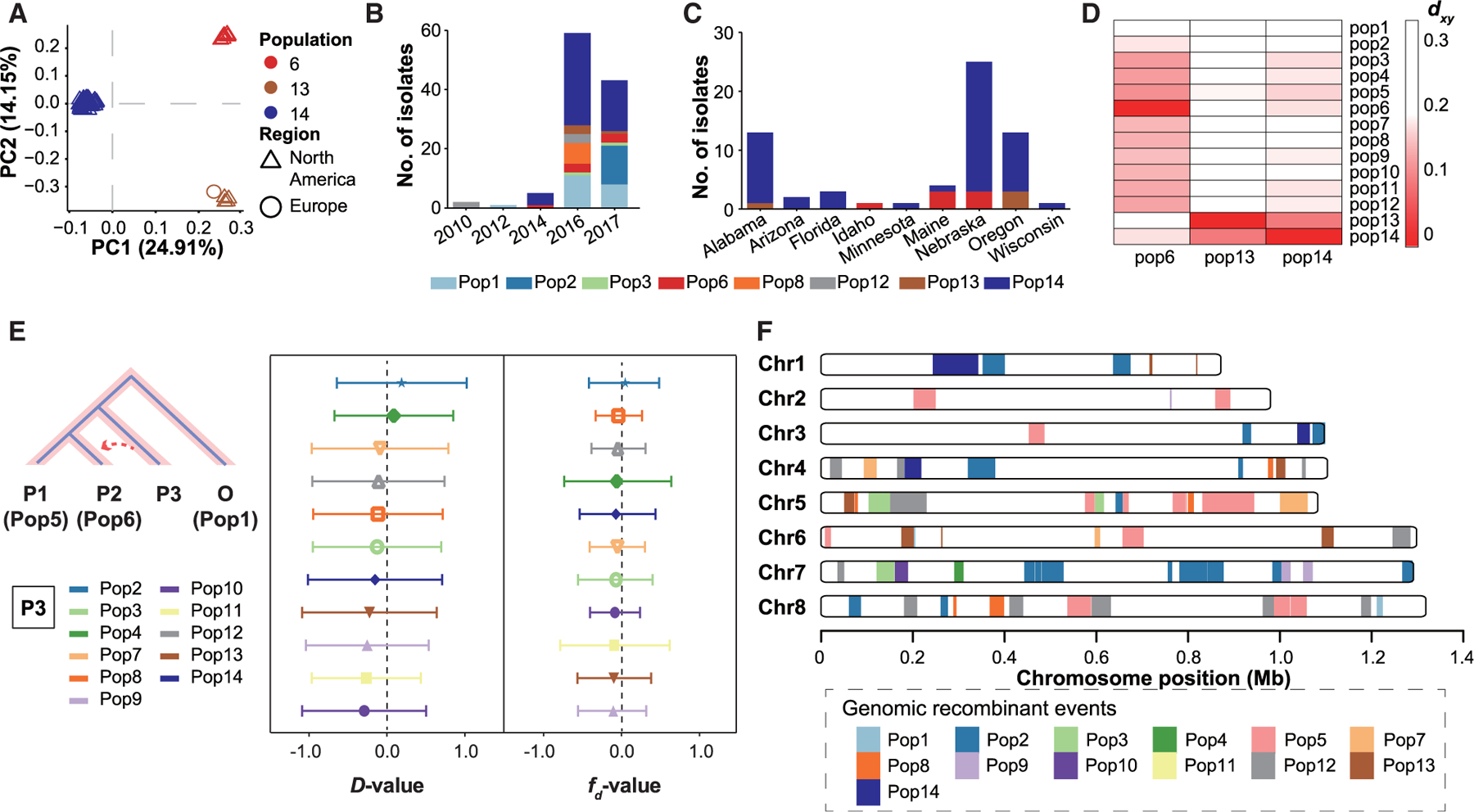
Populations of the C. hominis isolates are named and colored the same as in Figure 2C.
(A) PCA of C. hominis isolates in Pop6, Pop13, and Pop14 based on 2,255 wgSNPs. The colors of the symbols represent the C. hominis populations (Pop6, Pop13, and Pop14), whereas the shapes represent the sources of isolates (North America and Europe).
(B) Number of C. hominis isolates collected from the United States in the present study by year.
(C) Number of IfA12G1R5 cases in Pop6, Pop13, and Pop14 by state and variant.
(D) Mean absolute divergence (dxy) between three populations of IfA12G1R5 and other populations.
(E) D and fd statistics calculated using 100-kb windows and 10-kb steps. Gene flows were simulated from Pop2, 3, 4, 7, 8, 9, 10, 11, 12, 13, and 14 to Pop6.
(F) Distribution of introgressed regions in the genomes of Pop6 based on the genetic differentiation (Fst) value of 0 between Pop6 and the other populations across the eight chromosomes.
East African origin of Pop6 (IfA12G1R5a)
In phylogenetic analysis, Pop6 formed a sister clade with Pop5 (isolates from East Africa), indicating high nucleotide identity between each other (Figures 2C and 3D). As other C. hominis isolates from Asian and African countries formed country-specific clusters, Pop5 and Pop6 probably have similar origins. This is also supported by the lack of the cgd2_4380 gene in Pop6, which is present in most genomes from Europe (Pop1 and Pop13) and some genomes from North America (Pop1, Pop8, and Pop13) but largely absent from genomes from Africa (Pop5, Pop7, and Pop9–11) (Table S2). The results of phylogenetic network, ABBA-BABA test (D-statistics), and modified f-statistic (fd) analyses all showed the presence of gene flows from Pop2 (Ia subtypes in the United States) to Pop6 (Figures 2E, 3E, and 3F). In addition, in network analysis of linkage disequilibrium (LD) blocks across the genomes, there was different clustering of populations among regions of the eight chromosomes (Figures S4A and S4B). This suggests the occurrence of multiple sequence introgression events within Pop6 at the whole-genome level. For example, a large-linked region in chromosome 2 (nt 199,090–303,437) has high sequence identity between Pop5 and Pop6 (Figure S4A). In contrast, Pop6 has high sequence identity to Pop2 in several regions within chromosome 7 (nt 337,784–537,673) (Figure S4B). In genetic differentiation (Fst) analysis, Pop2 (several Ia subtypes in the United States), Pop5 (IaA14R3 subtype in Madagascar), and Pop12 (IaA28R4 subtype in the United States) were the top three populations with the largest sequence contributions to Pop6 genomes, 5.4%, 5.2%, and 3.6%, respectively (Figures 3F and S4C).
European origin of Pop13 (IfA12G1R5b)
The IfA12G1R5 isolate obtained from Sweden in 2013 was placed in Pop13 with isolates collected from the United States during 2016–2017 (Figures 2C and 3B, and Table S1). The patient traveled to Denmark several weeks prior to the infection.13 To further identify the relationship between the European isolate and the three US IfA12G1R5 populations, we undertook identity-by-descent (IBD) analysis of the populations (Figure 4A). The results indicated that the shared IBDs between the European isolate and other isolates in Pop13 (mean IBD sharing fraction over 99%) were much higher than those between the European isolate and Pop14 (mean IBD sharing fraction over 61%). The latter were comparable with those shared between Pop13 and Pop14 (Figure 4A). In nucleotide diversity (Pi) analysis of the Swedish isolate and US isolates from Pop13, the Pi values of 863 (94.7%) windows were 0 due to sequence identity. In contrast, 301 (33.0%) and 578 (63.4%) windows had Pi values of 0 between the Swedish isolate and Pop6 (p = 0.00) or Pop14 (p = 0.03), respectively (Figure 4B). In the absolute divergence (dxy) analysis, low dxy values were seen between the Swedish isolate and Pop13 in most regions across the eight chromosomes (Figure 4C). These data indicate that the European isolate is genetically related to Pop13.
Figure 4. Origin of Pop13 (IfA12G1R5b).
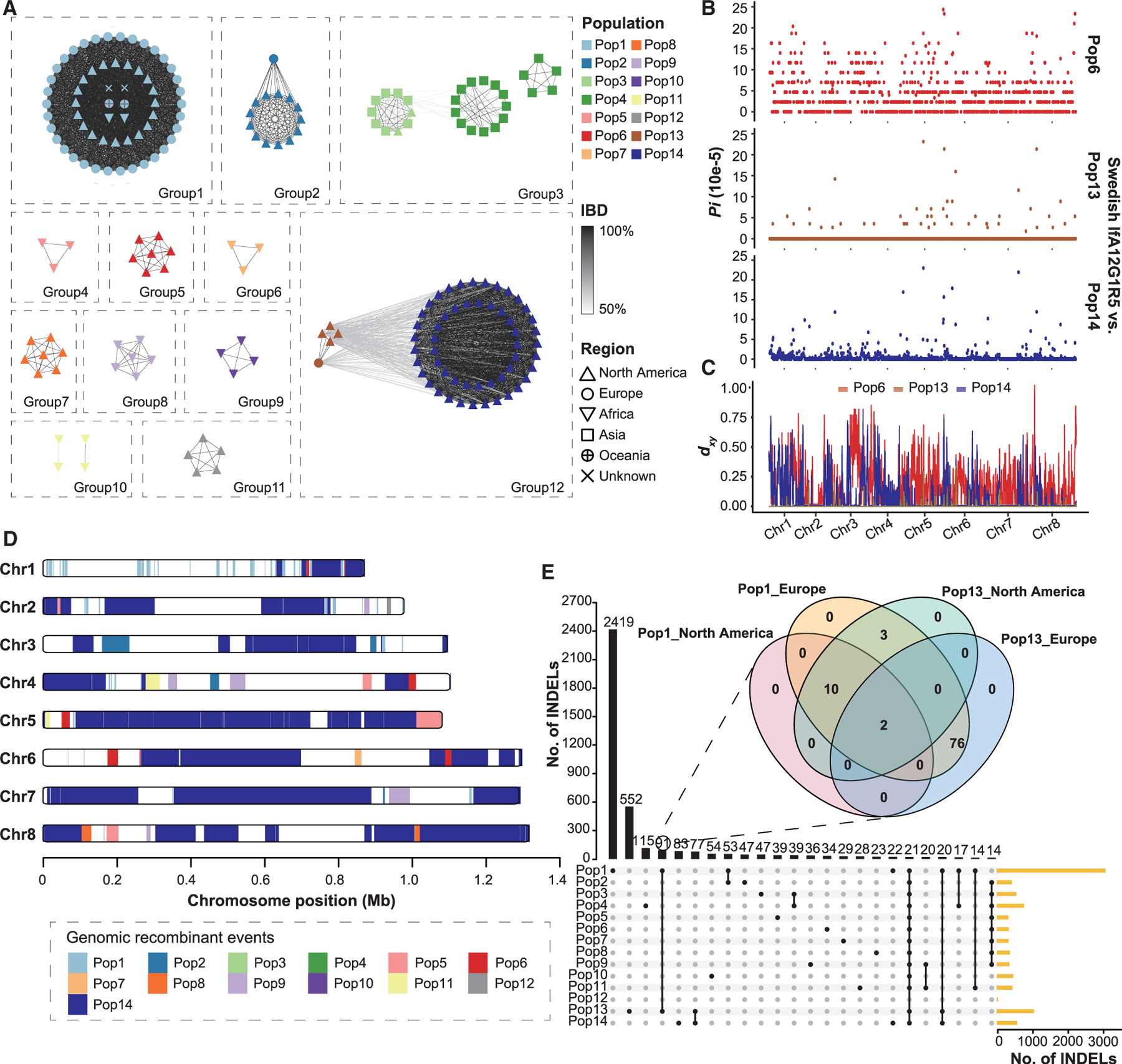
(A) Relatedness network for pairs of isolates inferred using identity-by-descent (IBD) analysis. Nodes represent isolates, and their colors and shapes represent the populations and the geographic origins. Edges between two nodes indicate IBD sharing. All edges with IBD higher than the threshold (mean value + SD) are shown.
(B) Nucleotide diversity (Pi) between Swedish and US isolates of the IfA12G1R5 using a 10-kb window, showing genetic similarity of the Swedish IfA12G1R5 to Pop13.
(C) Absolute divergence (dxy) between the Swedish IfA12G1R5 isolate and the three IfA12G1R5 populations (Pop6, Pop13, and Pop14) across the eight chromosomes.
(D) Distribution of introgressed regions in the genomes of Pop13 based on the Fst value of 0 between Pop13 and the other populations across the eight chromosomes.
(E) Relationships of insertion and deletion (INDEL) sites among 14 populations. The venn diagram in the format of overlapping circles shows the relationships among Pop1 and Pop13 isolates from both North America and Europe, whereas Upset plot shows the relationships of INDELs among the 14 populations.
One Chinese isolate (IaA18R4) appeared to group with the North American Pop13 (IfA12G1R5) in the phylogenetic analysis but formed a deep branch (Figure 2C). In Pi analysis of the Asian isolate and the three IfA12G1R5 groups, the Pi values of 345 (37.9%), 294 (32.3%), and 179 (19.6%) windows were 0 due to sequence identity between the Asian isolate and Pop13, Pop14, and Pop6, respectively (Figure S5A). In addition, the dxy values between the Asian isolate and the three other groups were far above 0 in most regions across the eight chromosomes (Figure S5B). These data indicate that the Asian isolate is divergent from others, although it shares sequences at some genetic loci with the US IfA12G1R5 isolates.
In addition to the close relationship between Pop13 and Pop14, TreeMix analysis detected significant signatures of sequence introgression from Pop1, Pop5, and Pop9 to Pop13 (Figure S4D). In multiple sequence alignment and phylogenetic analyses of sequences, the topology of chromosome 1 was different from that of other chromosomes, with Pop1 and Pop13 being clustered together and having an almost identical SNP pattern (Figure S4E). The Fst analysis suggested that about 2.1% of the Pop13 genomes were identical to Pop1. Therefore, Pop1 appears to be the main source of sequence introgression in Pop13, and the genetic introgression has occurred mostly in chromosome 1 (Figures 4D and S4F). In addition, we attempted to identify the geographic location of the sequence introgression through comparisons of insertion and deletions (INDELs), which evolve much fast than SNPs. The data showed that Pop13 isolates from North America shared 3 INDELs with Pop1 isolates from Europe but none with Pop1 isolates from North America (Figure 4E). These results suggest that the introgression event between Pop13 and Pop1 likely happened in Europe. Almost all genomes from Europe and a few from North America contain the cgd2_4380 gene. More importantly, Pop13 is the only IfA12G1R5 population that has this gene, supporting the European origin of Pop13 (Table S2).
Secondary recombination led to the formation of Pop14 (IfA12G1R5c)
In the IBD analysis, the C. hominis isolates formed 12 groups (groups 1–12), largely corresponding to the 14 populations with two exceptions. One included both Pop14 and Pop13, supporting their similar ancestral origin (mean IBD sharing fraction over 63%) (Figure 4A). Phylogenetic topology weighting across the IfA12G1R5 genomes of Pop6, Pop13, and Pop14 confirmed the genetic relatedness of Pop13 and Pop14, with the average weighting of the two as sister populations (topo3) accounting for >60% of the genome (Figure 5A). The presence of other topologies (topo1 and topo2) indicates the occurrence of genetic recombination among the three populations. This was mostly seen in chromosomes 1–4, with sequence introgressions from Pop6 to Pop14 (Figure 5B). The dxy values between Pop14 and Pop13 were the lowest in most regions across the eight chromosomes (Figure S6A). However, low dxy values were seen between Pop14 and Pop6 in a large region (from 24 to 32 kb) of chromosome 1 (Figure 5C). Phylogenetic analysis of sequences of the region yielded a topology different from that of the genome or chromosome 1, with Pop6 and Pop14 clustered together (Figures 2D, S6B, and S6C). Results of the Fst analysis further confirmed the contribution of Pop13 and Pop6 to the formation of the Pop14 genomes, for about 48.6% and 2.2% sequences, respectively, (Figures 5D and S6D). The results suggest that the recombination between Pop6 and Pop13 has led to the formation of Pop14 (Figure 5E).
Figure 5. Formation of Pop14 (IfA12G1R5c).
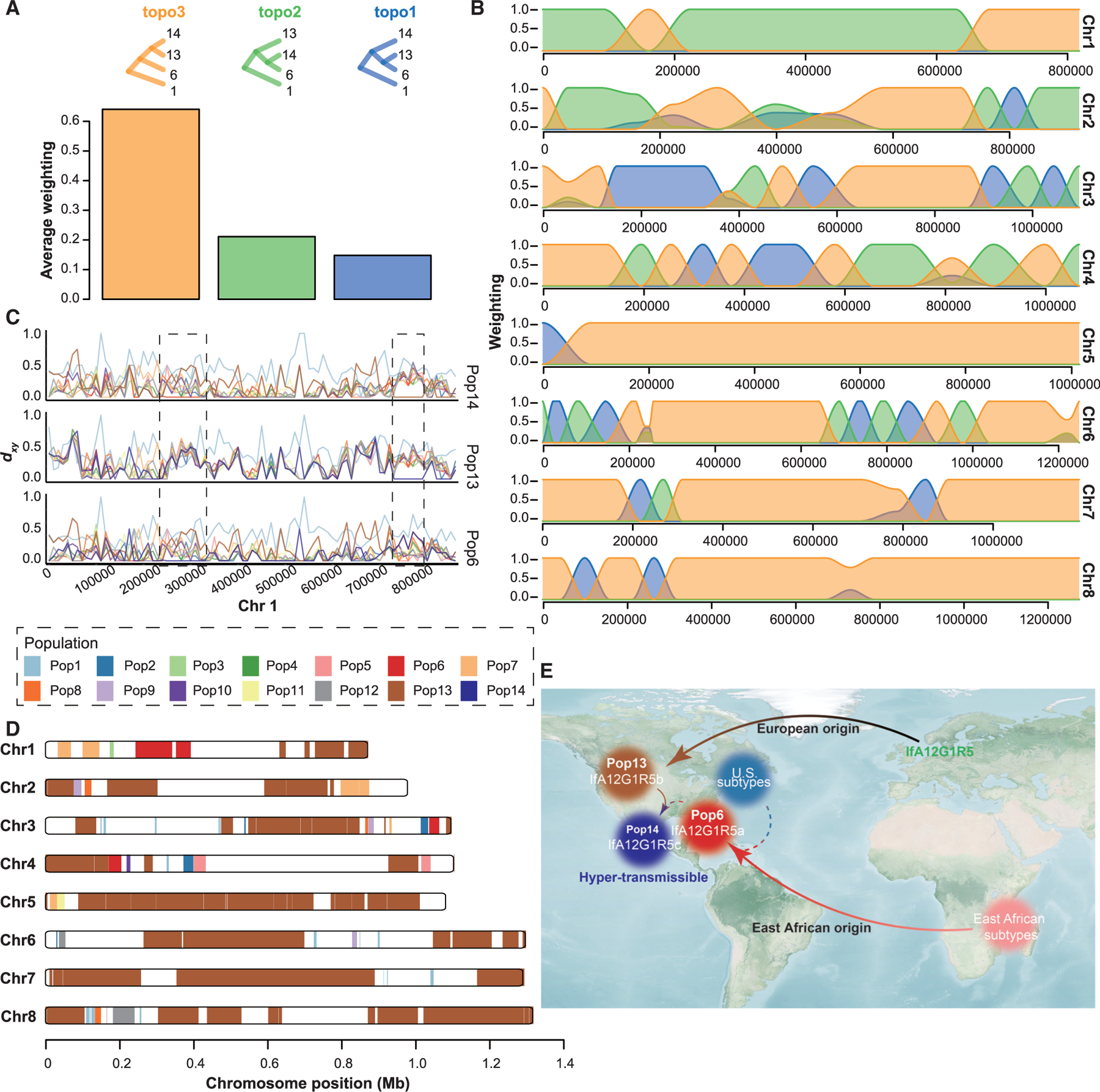
(A) Genome-wide distribution of phylogenetic relationships among the three IfA12G1R5 populations (Pop6, Pop13, and Pop14) based on 50-SNP sliding window with Pop1 as the outgroup. The top panel shows all possible topologies, while the bottom panel shows the genome-wide average weighting of each topology.
(B) Distribution of topology weightings across eight chromosomes (colors as in A).
(C) Absolute divergence for paired comparisons of the three populations of IfA12G1R5 and other populations of C. hominis isolates using 10-kb sliding windows across chromosome 1. The first box shows the introgression of Pop6 sequences in Pop14, whereas the second box shows sequence identity between Pop13 and Pop14 in the region.
(D) Distribution of introgressed regions in the genomes of Pop14 based on the Fst value of 0 between Pop14 and the other populations across the eight chromosomes.
(E) Summary of the evolutionary history of IfA12G1R5 in the United States. The import of C. hominis IfA12G1R5 from Africa and Europe is shown with the solid arrow, whereas the putative genetic recombination is shown with the dashed arrow.
Adaptive selection led to the dominance of Pop14 (IfA12G1R5c)
To investigate whether the current dominance of Pop14 was the result of adaptive selection, a selective sweep analysis was performed on the genomes of Pop13 and Pop14, which are closely related but differ in transmissibility. Pop14 had lower polymorphism than Pop13 (median Pipop14/Pipop13 = 0.4) (Figure 6A), reflecting its higher homogeneity. We detected four large genomic regions with strong selective sweep signals in Pop14 (Figure 6B). The selected regions exhibited significantly lower Pi ratios and Tajima’s D values and higher Fst values (p = 3 × 10−8, 5 × 10−15, and 2 × 10−6, respectively, by Mann-Whitney U test) (Figures 6C–6E). These data indicate that the genomes of Pop14 have gone through selective sweeps, resulting in higher transmissibility of the IfA12G1R5c variant. The four selected regions altogether contained 26 protein-encoding genes. Except for the region in chromosome 3, most these genes encoded hypothetical proteins (Table S3). However, two regions contained genes (cgd6_40 and cgd8_700) encoding invasion-associated mucin-like glycoproteins.
Figure 6. Presence of strong selective sweep signals in genomes of the hyper-transmissible Pop14 (IfA12G1R5c).
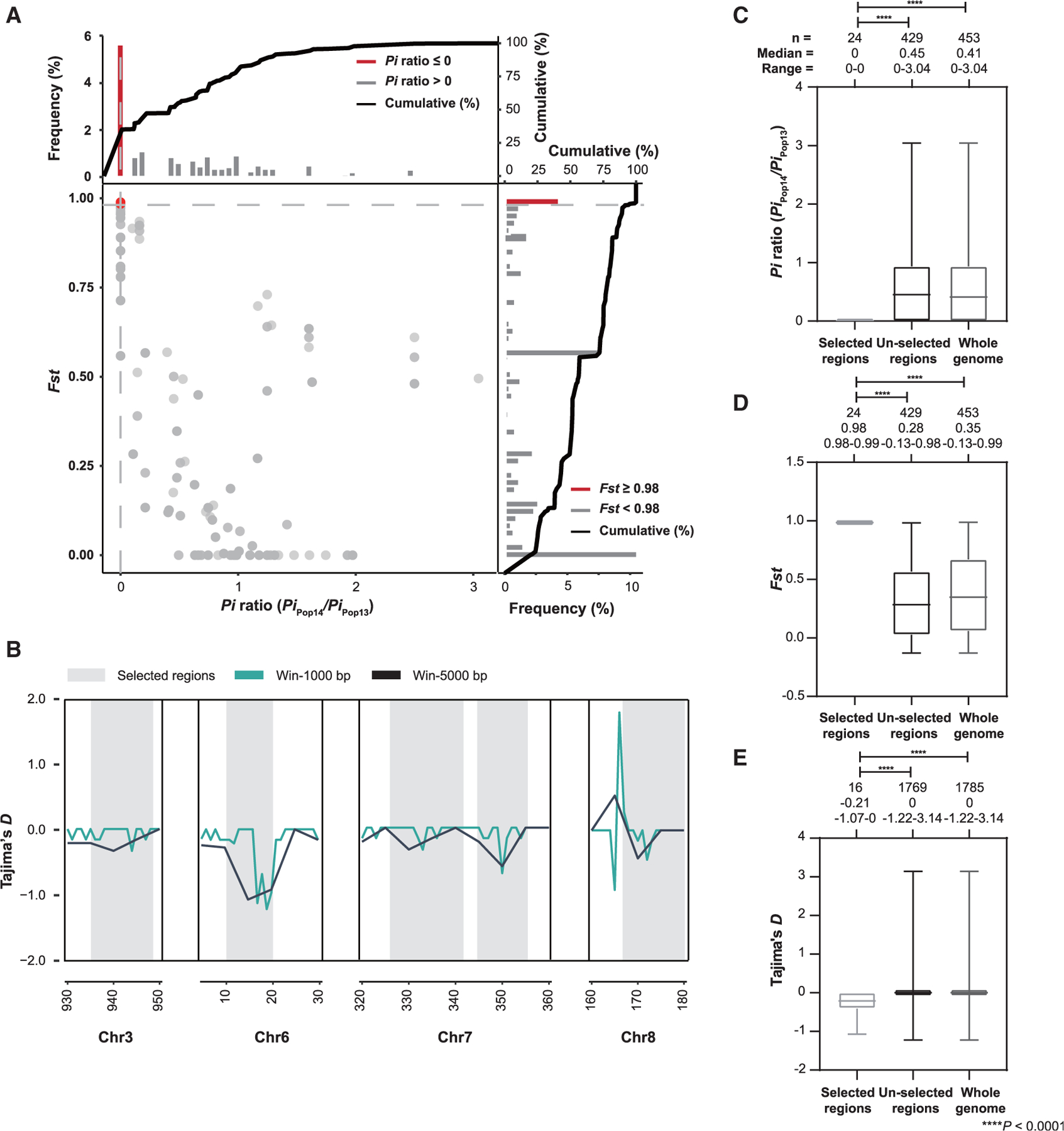
(A) Distribution of Pi ratios (Pipop14/Pipop13) and Fst values calculated using 10-kb sliding windows in 1-kb steps. Data points located to the left of the left vertical dashed lines (corresponding to the 5% left tails of the empirical Pi ratio distribution, where the Pi ratios were 0), and above the horizontal dashed line (the 5% right tail of the empirical Fst distribution, where Fst was 0.98) were identified as selective sweep signals for Pop14 (red points).
(B) Four genetic regions (in gray) with selective sweeps in Pop14 based on Tajima’s D analysis using 1- and 5-kb sliding window.
(C–E) Boxplot of Pi ratios (Pipop14/Pipop13) (C), Fst values (D), and Tajima’s D (E) for regions of Pop13 and Pop14 that have undergone selective sweeps in comparison with the un-selected regions and the whole genome. The boxes denote the interquartile ranges between the first and third quartiles and the line inside denotes the median, whereas the whiskers denote the lowest and highest values. The statistical significance was assessed using the Mann-Whitney U test. p < 0.0001 for selected regions versus un-selected regions and the whole genome in Pi ratios, Fst values, and Tajima’s D.
DISCUSSION
Data from the study indicate that IfA12G1R5 has become a dominant C. hominis subtype in the United States and the hyper-transmissible subtype has a complicated evolutionary history. In comparative genomics and population genetic analyses of 222 isolates from diverse areas, C. hominis genomes clustered mainly according to the country origins of isolates. Among them, isolates of the IfA12G1R5 subtype were placed in three of the 14 populations, suggesting that they have different ancestral origins. Further analyses indicated that IfA12G1R5 was initially imported into the United States from two sources (East Africa and Europe) but had gone through subsequent genetic recombination with each other and local subtypes. In addition to the sequence introgression, natural selection in several genomic regions containing genes encoding invasion-associated proteins might have played significant roles in shaping the evolution of IfA12G1R5 in the United States.
Accompanying the dramatic increase in incidence of cryptosporidiosis, IfA12G1R5 has become a dominant C. hominis subtype in the United States. In the present study, epidemiological data show that IfA12G1R5 is frequently seen in outbreaks and sporadic cases during 2013–2017. In the United States, C. hominis infection is mainly linked to recreational water usage and day care attendance.14,15 In contrast, there were no major differences in the transmission of other enteric diseases during the study period according to surveillance data on foodborne illnesses. The incidence of some bacterial pathogens increased in 2016, but this was attributed to the increased use of culture-independent diagnostic tests.16
C. hominis isolates of different origins appear to have different population genetic structures. In our phylogenomic analysis of the data, C. hominis genomes have shown isolation-by-distance. This contradicts the finding in one recent study, which indicated that C. hominis encompassed mainly two lineages, one of European and American isolates and the other of African and Asian isolates.17 In previous comparative genomics studies conducted in Asia (Bangladesh) and Africa (Gabon, Ghana, Madagascar, and Tanzania), C. hominis isolates also clustered mainly by country of origin irrespective of their gp60 subtypes.18,19 This is probably due to gene flow among isolates within countries and between neighboring areas, producing distinct lineages.19 The results of previous multilocus sequence analyses had also indicated the presence of geographical segregation within C. hominis and suggested that Cryptosporidium parasites have population structure depending on the transmission intensity.20 In the present study, the data generated indicate that the population structure of C. hominis in low- and middle-income countries with high transmission intensity differs from that in high-income countries with low transmission intensity.
The two dominant C. hominis subtypes in industrialized nations, IfA12G1R5 and IbA10G2, have very different population genetics. Between the two, IbA10G2 is almost the only C. hominis subtype in European countries and most frequently detected subtype for cryptosporidiosis outbreaks in the United States prior to 2005.7,8 In the present study, IbA10G2 isolates from different countries showed high genetic identity, indicating the subtype mostly has a simple ancestral origin. This confirms the result of a recent analysis of 114 C. hominis genomes, which has named IbA10G2 as C. hominis aquapotentis and other subtypes as C. hominis based on the genetic differences between the two groups.21 In contrast, the IfA12G1R5 subtype, which is now the dominant C. hominis subtype for sporadic cases and outbreaks in the United States, has three variants with mosaic genomes. Therefore, IfA12G1R5 in the United Sates is a heterogeneous subtype and has multiple origins.
Among the three IfA12G1R5 variants, Pop6 (IfA12G1R5a) appears to have an East African origin and has gone through genetic recombination with US subtypes after its arrival in the United States. Phylogenomic evidence shows that Pop6 is a sister clade of Pop5 (isolates from East Africa). As C. hominis largely forms country-specific clades,18,19 Pop6 might have originated from East Africa. Indeed, the If subtype family, including the IfA12G1R5 subtype, is common in East Africa and rare elsewhere.22 Therefore, the earlier and common occurrence of IfA12G1R5 in Africa suggests that the subtype in the United States could have derived from the area. In addition, we detected sequence introgression from locally circulating Ia subtypes (Pop2) in the United States into Pop6. Thus, although Pop6 originated from East Africa, it went through recombination with US subtypes. The occurrence of genetic recombination is facilitated by the presence of multiple C. hominis subtype families within the United States.7 Recently, genetic recombination has been shown to play an important role of shaping the population structure of C. parvum isolates.23–25
In contrast, the variant Pop13 (IfA12G1R5b) appears to be initially introduced into the United States from Europe. In Europe, IfA12G1R5 was first identified in the United Kingdom26 but has since been detected in Denmark, Germany, Ireland, Sweden, and the Netherlands.27 The earlier occurrence of IfA12G1R5 in Europe suggests it could be the origin of this subtype in the United States. We also detected haploblocks of IbA10G2 from Europe in the Pop13 genomes. This indicates that before Pop13 was imported into the United States from Europe, it went through recombination with IbA10G2 subtype there. This is not surprising as IbA10G2 is the dominant subtype in European countries.8
Genetic recombination between the two IfA12G1R5 variants is probably responsible for the emergence of hyper-transmissible Pop14 (IfA12G1R5c) in the United States. Among the three IfA12G1R5 variants, Pop14 is a relative of Pop13 and has some sequence introgression from Pop6. Therefore, the hyper-transmissible Pop14 variant is probably a progeny of recombination of Pop6 and Pop13 after their import into the United States. Previously, multilocus sequence typing of isolates indicated that genetic recombination could have played a role in the emergence of the IaA28R4 subtype of C. hominis in the United States.10 Genetic recombination has also been identified in IbA10G2 in Peru.28 The occurrence of genetic recombination in IaA28R4 and IbA10G2 subtypes in the United States has been confirmed by comparative genomics analysis of a small number of isolates.29
In addition to genetic recombination, natural selection probably plays an important role in the evolution of IfA12G1R5 in the United States. Selective sweeps were detected in several regions in the genomes of the dominant Pop14 variant, encoding secretory proteins. These genes are considered secreted pathogenesis determinants in Cryptosporidium spp.30 Of particular interest is selective sweeps were observed in chromosomes 6 and 8 around two mucin-like glycoproteins. Mucin glycoproteins play critical roles in sporozoite invasion.31 Therefore, post-recombination selective sweep could have contributed to the emergence of the hyper-transmissible Pop14.
Prior to the present study, our understanding of the evolution of C. hominis has been hampered by the lack of WGS data. Previously, less than 100 high-quality WGS data of C. hominis are available in public databases. They were mostly collected from Europe, Africa, and Asia (all from Dhaka, Bangladesh). In this study, we acquired WGS data from 127 C. hominis isolates collected mostly from the United States, filling a major data gap in WGS data from the Western Hemisphere. Nevertheless, we still lack comparable data from Oceania, where IfA12G1R5 is emerging.12 Although the emergence of this subtype there is more recent than in the United States, more systematic collection and analysis of isolates from this area are needed to improve the understanding of the transmission of this emerging C. hominis subtype.
In conclusion, the recently emerged IfA12G1R5 subtype in the United States has a complex evolutionary history, with two imports from East Africa and Europe and subsequent genetic recombination with each other and local subtypes. This has led to the formation of three variants of the subtype in the United States. Adaptive selection at invasion-associated loci in the genomes has eventually led to the dominance of one hyper-transmissible variant, Pop14 (IfA12G1R5c). The results of this study shed light on the understanding of the evolution of C. hominis and mechanisms for the emergence of hyper-transmissible subtypes. They demonstrate an urgent need for the implementation of molecular surveillance systems to monitor the global dispersal of IfA12G1R5 and other hyper-transmissible subtypes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lihua Xiao (lxiao1961@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All sequence data have been deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra/) and are publicly available as of the date of publication. The accession number is listed in the key resources table. Information on the samples used in this study and summary statistics of whole genome sequencing data have been deposited at Mendeley. The DOI is listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains (parasite strains) | ||
| Cryptosporidium hominis (12146 isolates) | This paper | N/A |
| Cryptosporidium hominis (103 isolates) | SRA | https://www.ncbi.nlm.nih.gov/sra/ |
| Critical commercial assays | ||
| FastDNA SPIN Kit for Soil | MP Biomedicals | Cat#116560–200 |
| Q5 Hot Start High-Fidelity 2X master mix | New England Biosciences | Cat#M0494S |
| Dynabeads Anti-Cryptosporidium kit | Invitrogen | Cat#73011 |
| Qiagen DNeasy Blood & Tissue Kit | QIAGEN | Cat#69504 |
| QIAamp DNA Mini kit | QIAGEN | Cat#51304 |
| REPLI-g Midi Kit | QIAGEN | Cat#150043 |
| Oligonucleotides | ||
| Primer: C. hominis 18S rRNA | Xiao et al.32 | N/A |
| Primer: C. hominis gp60 | Alves et al.33 | N/A |
| Software and algorithms | ||
| CLC Genomics Workbench | QIAGEN | https://digitalinsights.qiagen.com |
| SPAdes v3.1 | St. Petersburg State University | http://cab.spbu.ru/software/spades/ |
| Blastn v2.10.1 | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| MUSCLE v3.8.31 | EMBL-EBI | https://www.ebi.ac.uk/ |
| RAxML-NG v1.0.0 | Kozlov et al.34 | https://github.com/amkozlov/raxml-ng |
| jModelTest v2.1.10 | Posada35 | https://github.com/ddarriba/jmodeltest2/releases |
| BWA-MEM v0.7.17 | Li and Durbin36 | https://github.com/lh3/bwa |
| SAMtools v1.7 | SAMtools | http://samtools.sourceforge.net/ |
| BCFtools v1.12 | SAMtools | https://samtools.github.io/bcftools/ |
| GATK4 | Broad Institute | https://gatk.broadinstitute.org/hc/en-us |
| SnpEff | Pablo Cingolani | https://pcingola.github.io/SnpEff/ |
| Vcftools 0.1.16 | Vcftools | https://vcftools.github.io/index.html |
| PLINK v1.90 | PLINK | http://zzz.bwh.harvard.edu/plink/ |
| LDAK v5.1 | Speed et al.37 | https://dougspeed.com/ldak/ |
| ggplot2 | R | https://www.rdocumentation.org/packages/ggplot2/versions/3.3.5 |
| Structure v.2.3.4 | Stanford University | https://web.stanford.edu/group/pritchardlab/structure.html |
| Pophelper v2.3.1 | R | http://www.royfrancis.com/pophelper/articles/index.html |
| DnaSP v6.12.03 | Rozas et al.38 | http://www.ub.edu/dnasp/ |
| POPart v1.7 | Allan Wilson Centre | http://popart.otago.ac.nz |
| hmmIBD v2.0.4 | Schaffner et al.39 | https://github.com/glipsnort/hmmIBD |
| Cytoscape v3.9.1 | National Institute of General mMedical Sciences | https://cytoscape.org/ |
| SplitsTree5 | Huson and Bryant40 | https://github.com/husonlab/splitstree5 |
| Phyml v3.3 | Guindon et al.41 | http://www.atgc-montpellier.fr/phyml/ |
| Twist.py | Github | https://github.com/simonhmartin/twisst |
| PlotTwist | Github | https://github.com/JoachimGoedhart/PlotTwist |
| TreeMix v1.13 | Institut Pasteur | https://bitbucket.org/nygcresearch/treemix/ |
| Genomics_general | Github | https://github.com/simonhmartin/genomics_general |
| Other | ||
| Sequence data (146 isolates) | This paper | BioProject:PRJNA821705 |
| Information on samples | This paper | Table S1 and Mendeley Data: https://doi.org/10.17632/g6tr57tb2c.1 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cryptosporidium strains
A total of 1,075 Cryptosporidium-positive samples submitted by state public health laboratories during 2010–2017 as part of cryptosporidiosis surveillance were used in examining the occurrence of C. hominis IfA12G1R5 subtype in the United States. These were mostly stored in Cary-Blair transport medium, 2.5 % potassium dichromate solution, or unpreserved at 4 °C. Information on isolates sequenced is presented in Table S1.
The study was done with delinked residual diagnostic samples under the Human Subjects Protocol No. 990115 “Use of residual human specimens for the determination of frequency of genotypes or sub-types of pathogenic parasites”, which was approved by the Institutional Review Board of the Centers for Disease Control and Prevention (CDC). They were submitted to CDC by local public health laboratories as part of technical assistance to the investigations of cryptosporidiosis cases and outbreak surveillance.
METHOD DETAILS
Cryptosporidium hominis subtyping
Genomic DNA was extracted from these samples using the FastDNA SPIN Kit for Soil (MP Biomedicals, Solon, USA). Cryptosporidium spp. in the DNA preparations were genotyped and subtyped by PCR and sequence analyses of the 18S rRNA and gp60 genes.32,33
Whole-genome sequencing
C. hominis oocysts were purified from 131 surveillance samples from the United States described above, 14 samples from Spain, and one sample from China using immunomagnetic separation (Dynabeads anti-Cryptosporidium, ThermoFisher, United States). After five freeze-thaw cycles, DNA was extracted from the purified oocysts using the QIAamp DNA minikit (Qiagen, United States) and sequenced on an Illumina HiSeq 2500 (Illumina, San Diego, CA, United States) using the 250-bp paired-end approach as described.29
A total of 103 sets of WGS data of C. hominis were retrieved from the SRA database of the NCBI (https://www.ncbi.nlm.nih.gov/sra/). They were from published studies.13,17–19,29,42–46 Some basic information on the WGS data is shown in Table S1.
Genome assembly and molecular characterization
Sequence reads of all samples were trimmed for adapter sequences and poor sequence quality (phred-score < 25), and assembled de novo using CLC Genomics Workbench with a word size of 63 and bubble size of 400. In addition, genomes were assembled using SPAdes 3.1 (http://cab.spbu.ru/software/spades/) with Kmer of 63 and the careful mode. The assemblies were aligned and sorted with published reference genome of C. hominis 30976 using Mauve 2.3.1 for assessment of the final genome length and gene insertions and deletions among isolates.
The 18S rRNA genes and the gp60 genes were extracted from genomes using Blastn 2.10.1+ (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The genomes were assigned to subtype families and subtypes using the established nomenclature.7 Genomes with no gp60 sequences and mixed sequence types of the 18S rRNA or gp60 gene were excluded from further analyses. These sequences were aligned using MUSCLE v3.8.31 (https://www.ebi.ac.uk/). A ML tree was reconstructed from the sequence alignment using RAxML-NG v1.0.0,34 with the model of general time reversible and proportion of invariable sites (GTR + I) and 1,000 bootstrap replicates. The substitution model was selected using jModelTest v2.1.10 based on values from the Akaike Information Criterion.35
Variant analysis
The reads were trimmed and mapped to the published reference genome of C. hominis 30976 (of IaA28R4 subtype) from the United States29 using the BWA-MEM v0.7.1736 and procedures described in a previous publication.47 The genome coverage and sequence depth were estimated using the mpileup algorithm of SAMtools v1.7 (http://samtools.sourceforge.net/), with genomes with < 90% coverage and < tenfold depth being excluded from further analyses. BCFtools v1.12 (https://samtools.github.io/bcftools/) was used to call the SNPs and to generate a VCF file of sequence variants, with parameters -c 50 and -d 500. Low quality SNPs (QUAL < 30, FORMAT/DP < 3 and AVG FORMAT/DP < 25)25 were filtered out using BCFtools with only homozygous SNPs being retained. Alternatively, the bam files from BWA were used to identify SNPs and INDELs using the GATK4 HaplotypeCaller (https://gatk.broadinstitute.org/hc/en-us). The VariantFiltration in GATK4 was used to remove low quality SNPs and INDELs as recommended (https://gatk.broadinstitute.org/hc/en-us/sections/360007226631-Tutorials).
The SNPs and INDELs identified above were annotated using SnpEff (https://pcingola.github.io/SnpEff/) for variant types and genes affected. Allele frequency was calculated using Vcftools 0.1.16 (https://vcftools.github.io/index.html).
Population structure analyses
We investigated the relationships among C. hominis isolates using PCA, STRUCTURE, phylogenetic, and IBD analyses. The high-quality SNPs identified were pruned based on LD using PLINK v1.90 (http://zzz.bwh.harvard.edu/plink/). A set of unlinked sites was generated with 100-kb sliding windows. The dataset was subjected to PCA analysis using LDAK v5.1.37 The clustering among the isolates was visualized using R package ‘ggplot2’ (https://www.rdocumentation.org/packages/ggplot2/versions/3.3.5). To reduce the influence of population sizes, representative genomes of each population were selected in an additional PCA analysis. The pruned SNPs were also analyzed using Structure v.2.3.4 (https://web.stanford.edu/group/pritchardlab/structure.html), with the best number of subpopulations (K value) being calculated using Pophelper v2.3.1 (http://www.royfrancis.com/pophelper/articles/index.html). SNPs for representative isolates and by chromosome were extracted from the wgSNPs using Vcftools 0.1.16. The haplotype network of LD blocks was visualized using DnaSP v6.12.0338 and POPart v1.7 (http://popart.otago.ac.nz).
ML trees were generated from the wgSNPs using RAxM-NG with a GTR + G substitution model and 1,000 replicates of bootstrapping. The substitution model was selected using jModelTest. The IBD analysis was used to identify isolates with shared ancestry using hmmIBD v2.0.4.39 Isolates with IBD sharing greater than the mean + SD of their genomes were considered as related ones. Relatedness networks for pairs of isolates were generated using Cytoscape v3.9.1 (https://cytoscape.org/).
Assessment of gene flow among populations
The high-quality SNPs were further used in phylogenetic network analysis and topology weighting. Phylogenetic networks were generated using neighbor-net algorithm of SplitsTree5.40 ML phylogenies in 50-SNP windows across the genome were estimated using Phyml v3.341 with a GTR substitution model. Topology weighting was used to investigate the phylogenetic relationships across the genome among three populations of C. hominis isolates and an outgroup, using Twist.py (https://github.com/simonhmartin/twisst). Genome-wide average weighting of each topology and distribution of topology weightings across eight chromosomes were visualized using the R package ‘PlotTwist’ (https://github.com/JoachimGoedhart/PlotTwist).
To detect the potential gene flow among populations, the D-statistics and modified fd test with 100-kb sliding windows and 10-kb steps were performed as described.48 Three populations and an outgroup with the relationship (((P1, P2), P3), O) were used, of which P1 is closer to P2 than P3. Positive D-values and fd-values were considered as introgression signals.
To infer migration events among the populations, we used TreeMix v1.13 (https://bitbucket.org/nygcresearch/treemix/) to construct ML trees using a window size (-K) of 500 SNPs to account for LD with Pop1 as the root, migration events (-m) of 0–15, corresponding residuals, and 100 bootstrap replications. The trees were visualized using the suggested approach.49
Identification of introgressed genomic regions
To identify the introgressed genomic regions across the whole-genome, population-genetics parameters were estimated across the genome using a set of SNPs with minor allele frequency of more than 0.01. dxy between two populations was calculated in 10-kb sliding windows using popgenWindows.py in Genomics_general (https://github.com/simonhmartin/genomics_general), and visualized in line plots using the R package ‘ggplot2’. The mean dxy between populations was calculated. Fst between IfA12G1R5 and the other populations was calculated using 500-bp sliding windows with 100-bp steps and Vcftools v0.1.16, with Fst value of 0 indicating no genetic differentiation and near 1 indicating significant differentiation.50 Therefore, windows with Fst value of 0 between two populations were considered the introgressed regions, with the adjacent windows being merged as concatenated introgressed regions. Pi was used to measure the degree of variability in a group.51 It was calculated between Swedish and U.S. isolates of the IfA12G1R5 using the Vcftools and a 10-kb sliding window.
Identification of selective sweeps
Regions with signatures of selective sweeps in the evolution of IfA12G1R5 were identified by calculating Pi ratios, Fst values, and Tajima’s D. Using Vcftools, a sliding-window approach (10-kb windows in 1-kb steps) was applied to quantify Pi in Pop13 and Pop14 and Fst between Pop13 and Pop14. Windows with low or high Pi ratios (the 20% left and right tails, where the Pi ratios were 0 and 1.3, respectively) and high Fst values (the 20% right tail, where Fst was 0.8) were considered regions with strong selective sweep signals.52 In addition, Tajima’s D values were calculated using 1-kb and 5-kb sliding windows across the genome, with windows with Tajima’s D values < 0 as candidate selective sweep regions.53 The selective sweep regions were integrated, and the genes involved were annotated using Blast v2.10.1 (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using the R package unless otherwise specified. A Mann Whitney U test was used when comparing the mean of two groups.
Supplementary Material
Highlights.
Newly emerged IfA12G1R5 has become the dominant C. hominis subtype in the United States
IfA12G1R5 originates from subtypes in East Africa and Europe
IfA12G1R5 has gone through genetic recombination with local US subtypes
Natural selection played an additional role in shaping the evolution of IfA12G1R5
ACKNOWLEDGMENTS
This work was supported in part by the Guangdong Major Project of Basic and Applied Basic Research (2020B0301030007), the National Natural Science Foundation of China (31820103014, 32150710530, and U1901208), 111 Project (D20008), Innovation Team Project of Guangdong Universities (2019K CXTD001), and the Advanced Molecular Detection Program of the US Centers for Disease Control and Prevention. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2022.11.013.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Checkley W, White AC Jr., Jaganath D, Arrowood MJ, Chalmers RM, Chen XM, Fayer R, Griffiths JK, Guerrant RL, Hedstrom L, et al. (2015). A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for Cryptosporidium. Lancet Infect. Dis 15, 85–94. 10.1016/S1473-3099(14)70772-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guérin A, and Striepen B (2020). The biology of the intestinal intracellular parasite Cryptosporidium. Cell Host Microbe 28, 509–515. 10.1016/j.chom.2020.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Collier SA, Deng L, Adam EA, Benedict KM, Beshearse EM, Blackstock AJ, Bruce BB, Derado G, Edens C, Fullerton KE, et al. (2021). Estimate of burden and direct healthcare cost of infectious waterborne disease in the United States. Emerg. Infect. Dis 27, 140–149. 10.3201/eid2701.190676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mac Kenzie WR, Hoxie NJ, Proctor ME, Gradus MS, Blair KA, Peterson DE, Kazmierczak JJ, Addiss DG, Fox KR, and Rose JB (1994). A massive outbreak in Milwaukee of Cryptosporidium infection transmitted through the public water supply. N. Engl. J. Med 331, 161–167. 10.1056/NEJM199407213310304. [DOI] [PubMed] [Google Scholar]
- 5.Painter JE, Gargano JW, Yoder JS, Collier SA, and Hlavsa MC (2016). Evolving epidemiology of reported cryptosporidiosis cases in the United States, 1995–2012. Epidemiol. Infect 144, 1792–1802. 10.1017/S0950268815003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng Y, Ryan UM, and Xiao L (2018). Genetic diversity and population structure of Cryptosporidium. Trends Parasitol. 34, 997–1011. 10.1016/j.pt.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Xiao L, and Feng Y (2017). Molecular epidemiologic tools for waterborne pathogens Cryptosporidium spp. and Giardia duodenalis. Food Waterborne Parasitol. 8–9, 14–32. 10.1016/j.fawpar.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cacciò SM, and Chalmers RM (2016). Human cryptosporidiosis in Europe. Clin. Microbiol. Infect 22, 471–480. 10.1016/j.cmi.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 9.Zhou L, Singh A, Jiang J, and Xiao L (2003). Molecular surveillance of Cryptosporidium spp. in raw wastewater in Milwaukee: implications for understanding outbreak occurrence and transmission dynamics. J. Clin. Microbiol 41, 5254–5257. 10.1128/JCM.41.11.5254-5257.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng Y, Tiao N, Li N, Hlavsa M, and Xiao L (2014). Multilocus sequence typing of an emerging Cryptosporidium hominis subtype in the United States. J. Clin. Microbiol 52, 524–530. 10.1128/JCM.02973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braima K, Zahedi A, Oskam C, Reid S, Pingault N, Xiao L, and Ryan U (2019). Retrospective analysis of Cryptosporidium species in Western Australian human populations (2015–2018), and emergence of the C. hominis IfA12G1R5 subtype. Infect. Genet. Evol 73, 306–313. 10.1016/j.meegid.2019.05.018. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-R JC, Pita AB, Velathanthiri N, French NP, and Hayman DTS (2020). Species and genotypes causing human cryptosporidiosis in New Zealand. Parasitol. Res 119, 2317–2326. 10.1007/s00436-020-06729-w. [DOI] [PubMed] [Google Scholar]
- 13.Sikora P, Andersson S, Winiecka-Krusnell J, Hallström B, Alsmark C, Troell K, Beser J, and Arrighi RB (2017). Genomic variation in IbA10G2 and other patient-derived Cryptosporidium hominis subtypes. J. Clin. Microbiol 55, 844–858. 10.1128/JCM.01798-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hlavsa MC, Roellig DM, Seabolt MH, Kahler AM, Murphy JL, McKitt TK, Geeter EF, Dawsey R, Davidson SL, Kim TN, et al. (2017). Using molecular characterization to support investigations of aquatic facility-associated outbreaks of cryptosporidiosis – Alabama, Arizona, and Ohio, 2016. MMWR Morb. Mortal. Wkly. Rep 66, 493–497. 10.15585/mmwr.mm6619a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loeck BK, Pedati C, Iwen PC, McCutchen E, Roellig DM, Hlavsa MC, Fullerton K, Safranek T, and Carlson AV (2020). Genotyping and subtyping Cryptosporidium to identify risk factors and transmission patterns – Nebraska, 2015–2017. MMWR Morb. Mortal. Wkly. Rep 69, 335–338. 10.15585/mmwr.mm6912a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marder EP, Cieslak PR, Cronquist AB, Dunn J, Lathrop S, Rabatsky-Ehr T, Ryan P, Smith K, Tobin-D’Angelo M, Vugia DJ, et al. (2017). Incidence and trends of infections with pathogens transmitted commonly through food and the effect of increasing use of culture-independent diagnostic tests on surveillance – foodborne diseases active surveillance network, 10 U.S. Sites, 2013–2016. MMWR Morb. Mortal. Wkly. Rep 66, 397–403. 10.15585/mmwr.mm6615a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cabarcas F, Galvan-Diaz AL, Arias-Agudelo LM, García-Montoya GM, Daza JM, and Alzate JF (2021). Cryptosporidium hominis phylogenomic analysis reveals separate lineages with continental segregation. Front. Genet 12, 740940. 10.3389/fgene.2021.740940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilchrist CA, Cotton JA, Burkey C, Arju T, Gilmartin A, Lin Y, Ahmed E, Steiner K, Alam M, Ahmed S, et al. (2018). Genetic diversity of Cryptosporidium hominis in a Bangladeshi community as revealed by whole-genome sequencing. J. Infect. Dis 218, 259–264. 10.1093/infdis/jiy121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tichkule S, Jex AR, van Oosterhout C, Sannella AR, Krumkamp R, Aldrich C, Maiga-Ascofare O, Dekker D, Lamshöft M, Mbwana J, et al. (2021). Comparative genomics revealed adaptive admixture in Cryptosporidium hominis in Africa. Microb. Genom 7, mgen000493. 10.1099/mgen.0.000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanriverdi S, Grinberg A, Chalmers RM, Hunter PR, Petrovic Z, Akiyoshi DE, London E, Zhang L, Tzipori S, Tumwine JK, and Widmer G (2008). Inferences about the global population structures of Cryptosporidium parvum and Cryptosporidium hominis. Appl. Environ. Microbiol 74, 7227–7234. 10.1128/AEM.01576-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tichkule S, Cacciò SM, Robinson G, Chalmers RM, Mueller I, Emery-Corbin SJ, Eibach D, Tyler KM, van Oosterhout C, and Jex AR (2022). Global population genomics of two subspecies of Cryptosporidium hominis during 500 years of evolution. Mol. Biol. Evol 39, msac056. 10.1093/molbev/msac056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krumkamp R, Aldrich C, Maiga-Ascofare O, Mbwana J, Rakotoz andrindrainy N, Borrmann S, Caccio SM, Rakotozandrindrainy R, Adegnika AA, Lusingu JPA, et al. (2021). Transmission of Cryptosporidium species among human and animal local contact networks in Sub-Saharan Africa: a multicountry study. Clin. Infect. Dis 72, 1358–1366. 10.1093/cid/ciaa223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T, Guo Y, Roellig DM, Li N, Santín M, Lombard J, Kváč M, Naguib D, Zhang Z, Feng Y, and Xiao L (2022). Sympatric recombination in zoonotic Cryptosporidium leads to emergence of populations with modified host preference. Mol. Biol. Evol 39, msac150. 10.1093/molbev/msac150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corsi GI, Tichkule S, Sannella AR, Vatta P, Asnicar F, Segata N, Jex AR, van Oosterhout C, and Cacciò SM (2022). Recent genetic exchanges and admixture shape the genome and population structure of the zoonotic pathogen Cryptosporidium parvum. Mol. Ecol 10.1111/mec.16556. [DOI] [PubMed] [Google Scholar]
- 25.Nader JL, Mathers TC, Ward BJ, Pachebat JA, Swain MT, Robinson G, Chalmers RM, Hunter PR, van Oosterhout C, and Tyler KM (2019). Evolutionary genomics of anthroponosis in Cryptosporidium. Nat. Microbiol 4, 826–836. 10.1038/s41564-019-0377-x. [DOI] [PubMed] [Google Scholar]
- 26.Chalmers RM, Hadfield SJ, Jackson CJ, Elwin K, Xiao L, and Hunter P (2008). Geographic linkage and variation in Cryptosporidium hominis. Emerg. Infect. Dis 14, 496–498. 10.3201/eid1403.071320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebbad M, Winiecka-Krusnell J, Stensvold CR, and Beser J (2021). High diversity of Cryptosporidium species and subtypes identified in cryptosporidiosis acquired in Sweden and abroad. Pathogens 10, 523. 10.3390/pathogens10050523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li N, Xiao L, Cama VA, Ortega Y, Gilman RH, Guo M, and Feng Y (2013). Genetic recombination and Cryptosporidium hominis virulent subtype IbA10G2. Emerg. Infect. Dis 19, 1573–1582. 10.3201/eid1910.121361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Y, Tang K, Rowe LA, Li N, Roellig DM, Knipe K, Frace M, Yang C, Feng Y, and Xiao L (2015). Comparative genomic analysis reveals occurrence of genetic recombination in virulent Cryptosporidium hominis subtypes and telomeric gene duplications in Cryptosporidium parvum. BMC Genomics 16, 320. 10.1186/s12864-015-1517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Z, Li N, Guo Y, Feng Y, and Xiao L (2020). Comparative genomic analysis of three intestinal species reveals reductions in secreted pathogenesis determinants in bovine-specific and non-pathogenic Cryptosporidium species. Microb. Genom 6, e000379. 10.1099/mgen.0.000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ludington JG, and Ward HD (2016). The Cryptosporidium parvum C-type lectin CpClec mediates infection of intestinal epithelial cells via interactions with sulfated proteoglycans. Infect. Immun 84, 1593–1602. 10.1128/IAI.01410-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao L, Singh A, Limor J, Graczyk TK, Gradus S, and Lal A (2001). Molecular characterization of Cryptosporidium oocysts in samples of raw surface water and wastewater. Appl. Environ. Microbiol 67, 1097–1101. 10.1128/AEM.67.3.1097-1101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alves M, Xiao L, Sulaiman I, Lal AA, Matos O, and Antunes F (2003). Subgenotype analysis of Cryptosporidium isolates from humans, cattle, and zoo ruminants in Portugal. J. Clin. Microbiol 41, 2744–2747. 10.1128/JCM.41.6.2744-2747.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozlov AM, Darriba D, Flouri T, Morel B, and Stamatakis A (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Posada D (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol 25, 1253–1256. 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 36.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Speed D, Holmes J, and Balding DJ (2020). Evaluating and improving heritability models using summary statistics. Nat. Genet 52, 458–462. 10.1038/s41588-020-0600-y. [DOI] [PubMed] [Google Scholar]
- 38.Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, and Sánchez-Gracia A (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol 34, 3299–3302. 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- 39.Schaffner SF, Taylor AR, Wong W, Wirth DF, and Neafsey DE (2018). hmmIBD: software to infer pairwise identity by descent between haploid genotypes. Malar. J 17, 196. 10.1186/s12936-018-2349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huson DH, and Bryant D (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol 23, 254–267. 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 41.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, and Gascuel O (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol 59, 307–321. 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 42.Isaza JP, Galván AL, Polanco V, Huang B, Matveyev AV, Serrano MG, Manque P, Buck GA, and Alzate JF (2015). Revisiting the reference genomes of human pathogenic Cryptosporidium species: reannotation of C. parvum Iowa and a new C. hominis reference. Sci. Rep 5, 16324. 10.1038/srep16324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hadfield SJ, Pachebat JA, Swain MT, Robinson G, Cameron SJ, Alexander J, Hegarty MJ, Elwin K, and Chalmers RM (2015). Generation of whole genome sequences of new Cryptosporidium hominis and Cryptosporidium parvum isolates directly from stool samples. BMC Genomics 16, 650. 10.1186/s12864-015-1805-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amid C, Pakseresht N, Silvester N, Jayathilaka S, Lund O, Dynovski LD, Pataki BÁ, Visontai D, Xavier BB, Alako BTF, et al. (2019). The COMPARE data hubs. Database (Oxford) 2019, baz136. 10.1093/database/baz136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arias-Agudelo LM, Garcia-Montoya G, Cabarcas F, Galvan-Diaz AL, and Alzate JF (2020). Comparative genomic analysis of the principal Cryptosporidium species that infect humans. PeerJ 8, e10478. 10.7717/peerj.10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knox MA, Garcia-R JC, and Hayman DTS (2021). Draft genome assemblies of two Cryptosporidium hominis isolates from New Zealand. Microbiol. Resour. Announc 10, e0036321. 10.1128/MRA.00363-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Y, Li N, Roellig DM, Kelley A, Liu G, Amer S, Tang K, Zhang L, and Xiao L (2017). Comparative genomic analysis of the IId subtype family of Cryptosporidium parvum. Int. J. Parasitol 47, 281–290. 10.1016/j.ijpara.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin SH, Davey JW, and Jiggins CD (2015). Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evol 32, 244–257. 10.1093/molbev/msu269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reich D, Thangaraj K, Patterson N, Price AL, and Singh L (2009). Reconstructing Indian population history. Nature 461, 489–494. 10.1038/nature08365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin T, Zhu G, Zhang J, Xu X, Yu Q, Zheng Z, Zhang Z, Lun Y, Li S, Wang X, et al. (2014). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet 46, 1220–1226. 10.1038/ng.3117. [DOI] [PubMed] [Google Scholar]
- 51.Tajima F (1983). Evolutionary relationship of DNA sequences in finite populations. Genetics 105, 437–460. 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li M, Tian S, Jin L, Zhou G, Li Y, Zhang Y, Wang T, Yeung CK, Chen L, Ma J, et al. (2013). Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet 45, 1431–1438. 10.1038/ng.2811. [DOI] [PubMed] [Google Scholar]
- 53.Luo X, Li H, Wu Z, Yao W, Zhao P, Cao D, Yu H, Li K, Poudel K, Zhao D, et al. (2020). The pomegranate (Punica granatum L.) draft genome dissects genetic divergence between soft- and hard-seeded cultivars. Plant Biotechnol. J 18, 955–968. 10.1111/pbi.13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data have been deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra/) and are publicly available as of the date of publication. The accession number is listed in the key resources table. Information on the samples used in this study and summary statistics of whole genome sequencing data have been deposited at Mendeley. The DOI is listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains (parasite strains) | ||
| Cryptosporidium hominis (12146 isolates) | This paper | N/A |
| Cryptosporidium hominis (103 isolates) | SRA | https://www.ncbi.nlm.nih.gov/sra/ |
| Critical commercial assays | ||
| FastDNA SPIN Kit for Soil | MP Biomedicals | Cat#116560–200 |
| Q5 Hot Start High-Fidelity 2X master mix | New England Biosciences | Cat#M0494S |
| Dynabeads Anti-Cryptosporidium kit | Invitrogen | Cat#73011 |
| Qiagen DNeasy Blood & Tissue Kit | QIAGEN | Cat#69504 |
| QIAamp DNA Mini kit | QIAGEN | Cat#51304 |
| REPLI-g Midi Kit | QIAGEN | Cat#150043 |
| Oligonucleotides | ||
| Primer: C. hominis 18S rRNA | Xiao et al.32 | N/A |
| Primer: C. hominis gp60 | Alves et al.33 | N/A |
| Software and algorithms | ||
| CLC Genomics Workbench | QIAGEN | https://digitalinsights.qiagen.com |
| SPAdes v3.1 | St. Petersburg State University | http://cab.spbu.ru/software/spades/ |
| Blastn v2.10.1 | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| MUSCLE v3.8.31 | EMBL-EBI | https://www.ebi.ac.uk/ |
| RAxML-NG v1.0.0 | Kozlov et al.34 | https://github.com/amkozlov/raxml-ng |
| jModelTest v2.1.10 | Posada35 | https://github.com/ddarriba/jmodeltest2/releases |
| BWA-MEM v0.7.17 | Li and Durbin36 | https://github.com/lh3/bwa |
| SAMtools v1.7 | SAMtools | http://samtools.sourceforge.net/ |
| BCFtools v1.12 | SAMtools | https://samtools.github.io/bcftools/ |
| GATK4 | Broad Institute | https://gatk.broadinstitute.org/hc/en-us |
| SnpEff | Pablo Cingolani | https://pcingola.github.io/SnpEff/ |
| Vcftools 0.1.16 | Vcftools | https://vcftools.github.io/index.html |
| PLINK v1.90 | PLINK | http://zzz.bwh.harvard.edu/plink/ |
| LDAK v5.1 | Speed et al.37 | https://dougspeed.com/ldak/ |
| ggplot2 | R | https://www.rdocumentation.org/packages/ggplot2/versions/3.3.5 |
| Structure v.2.3.4 | Stanford University | https://web.stanford.edu/group/pritchardlab/structure.html |
| Pophelper v2.3.1 | R | http://www.royfrancis.com/pophelper/articles/index.html |
| DnaSP v6.12.03 | Rozas et al.38 | http://www.ub.edu/dnasp/ |
| POPart v1.7 | Allan Wilson Centre | http://popart.otago.ac.nz |
| hmmIBD v2.0.4 | Schaffner et al.39 | https://github.com/glipsnort/hmmIBD |
| Cytoscape v3.9.1 | National Institute of General mMedical Sciences | https://cytoscape.org/ |
| SplitsTree5 | Huson and Bryant40 | https://github.com/husonlab/splitstree5 |
| Phyml v3.3 | Guindon et al.41 | http://www.atgc-montpellier.fr/phyml/ |
| Twist.py | Github | https://github.com/simonhmartin/twisst |
| PlotTwist | Github | https://github.com/JoachimGoedhart/PlotTwist |
| TreeMix v1.13 | Institut Pasteur | https://bitbucket.org/nygcresearch/treemix/ |
| Genomics_general | Github | https://github.com/simonhmartin/genomics_general |
| Other | ||
| Sequence data (146 isolates) | This paper | BioProject:PRJNA821705 |
| Information on samples | This paper | Table S1 and Mendeley Data: https://doi.org/10.17632/g6tr57tb2c.1 |