

Abstract
Cellular stress has been a key factor in the development of cardiovascular diseases. Major types of cellular stress such as mitochondrial stress, endoplasmic reticulum stress, hypoxia, and replicative stress have been implicated in clinical complications of cardiac patients. The heart is the central regulator of the body by supplying oxygenated blood throughout the system. Impairment of cellular function could lead to heart failure, myocardial infarction, ischemia, and even stroke. Understanding the effect of these distinct types of cellular stress on cardiac function is crucial for the scientific community to understand and develop novel therapeutic approaches. This review will comprehensively explain the different mechanisms of cellular stress and the most recent findings related to stress-induced cardiac dysfunction.
Keywords: Mitochondria, Endoplasmic reticulum, stress, Unfolded protein response, cardiovascular diseases, hypoxia
1. Introduction
Cell degeneration and dysfunction are among the primary causes of numerous cardiovascular diseases, leading to tissue and cumulative organ collapse and decline. Such a phenomenon is even more prominent as we age. Cardiovascular diseases (CVDs) are a common co-morbidity of ageing-associated diseases (Suzman et al., 2015). The heart is the central regulator of the body, which must pump constantly to meet daily needs. In pathological conditions, the heart faces multiple cellular stresses such as mitochondrial stress, endoplasmic reticulum stress, replicative stress, and hypoxia. But the defence system undertakes countermeasures to neutralize the stress and restore the balance. However, at a certain point, excessive stress contributes to cardiac dysfunction. Knowledge of cellular stress is imperative for the successful development of a drug specific to the disease condition. Considering the importance of stress dynamics in pathology, this review focus on the mechanism of action of different organelle molecular stress response. There are unique molecular mechanisms in each organelle, but all these mechanisms interlink with one another to maintain body homeostasis. The mitochondria provide a vital source of cellular energy: adenosine triphosphate (ATP), nicotinamide adenine dinucleotide phosphate (NADPH), and NADP (Morales et al., 2020), meeting the cell’s daily demands. But impairment of mitochondria function causes an imbalance in reactive oxygen species (ROS) which results in premature apoptosis and activates other cellular responses. One of the active responses is endoplasmic reticular (ER) stress, which leads to the accumulation of misfolded and unfolded protein, leading to the activation of the unfolded protein response (UPR) pathway and autophagy, impairing cardiac contraction and relaxation. Cells undergo a spectrum of stress and are exposed to a multitude of stress-inducing elements which cause alterations in cellular functioning. In response to stress, cells generate countermeasures to maintain cellular balance. For example, when quality control mechanisms, like autophagy, are activated, damaged organelles (such as mitochondria) are cleared and new organelles are synthesized (Fulda et al., 2010). However, prolonged stress impairs the repair mechanisms of the cell and leads to the activation of cell death mechanisms (such as apoptosis, necrosis, etc.) (Jurivich and Zhou, 2007). Similarly, cells also face pathological stress conditions such as a lack of oxygen. Oxygen is essential for the heart to function properly, a decrease in normal oxygen supply (normoxia) is known as hypoxia. Under hypoxic conditions, the heart is kept under constant pressure to maintain the normal body environment, which further increases cellular stress in cardiac cells. All these processes lead to cell senescence or the development of tumorigenic properties. A cell’s fate is also determined by the crosstalk between stress response pathways.
2. Mitochondrial stress
Mitochondria is the powerhouse of the cell: it is capable of migrating to other organelles, with compatible bioenergetics and morphology to subsidize the target organ (Adaniya et al., 2019). Mitochondria are regulated by fusion and fission. Fusion is an event where different mitochondrial compositions are mixed, thereby alleviating the mitochondrial damage, whereas, fission is the fragmentation of mitochondria to distribute the mitochondria contents evenly (Losón et al., 2013). As the heart is a continuous pumping organ, it requires huge energy demand, relying on mitochondrial bioenergetics. They cover one-third of the total cell volume and contribute to the 6–7 kg ATP/day (Morales et al., 2020). Cardiac mitochondria are segregated into three main types based on their subcellular locations: perinuclear mitochondria, subsarcolemmal mitochondria, and intermyofibrillar mitochondria (Jhun et al., 2018). The mitochondrial dynamics are maintained by multiple cellular factors, fission, for example, is regulated by dynamin-related protein-1 (Drp-1), mitochondrial fission protein (Fis1), mitochondrial fission factor (Mff), and dynamins proteins 49 and 51 (MiD49 and MiD51). Meanwhile, fusion is regulated mainly by mitofusin: Mfn1 and Mfn2, and optic atrophy 1 (Opa1) (Losón et al., 2013). Impairment of fusion and fission is known to induce cardiac dysfunctions such as heart failure, ischemia, and myocardial infarction (MI). Recently, it was identified that Mfn-2 and AMP-activated protein kinase (AMPK) are key regulators of autophagy in response to mitochondrial stress [9]. Mitochondrial stress triggers a cascade of responses such as mitochondrial fission, autophagy, and more importantly, accumulation of mitochondrial-associated ER membrane (MAM). The latter is essential for the regulation of autophagy and mitochondrial division because it triggers the translocation of AMPK from the cytosol to the MAM and the mitochondrion where it interacts directly with Mfn2 [9] (Figure 1).
Figure 1. Mitochondrial dynamics and stress response.
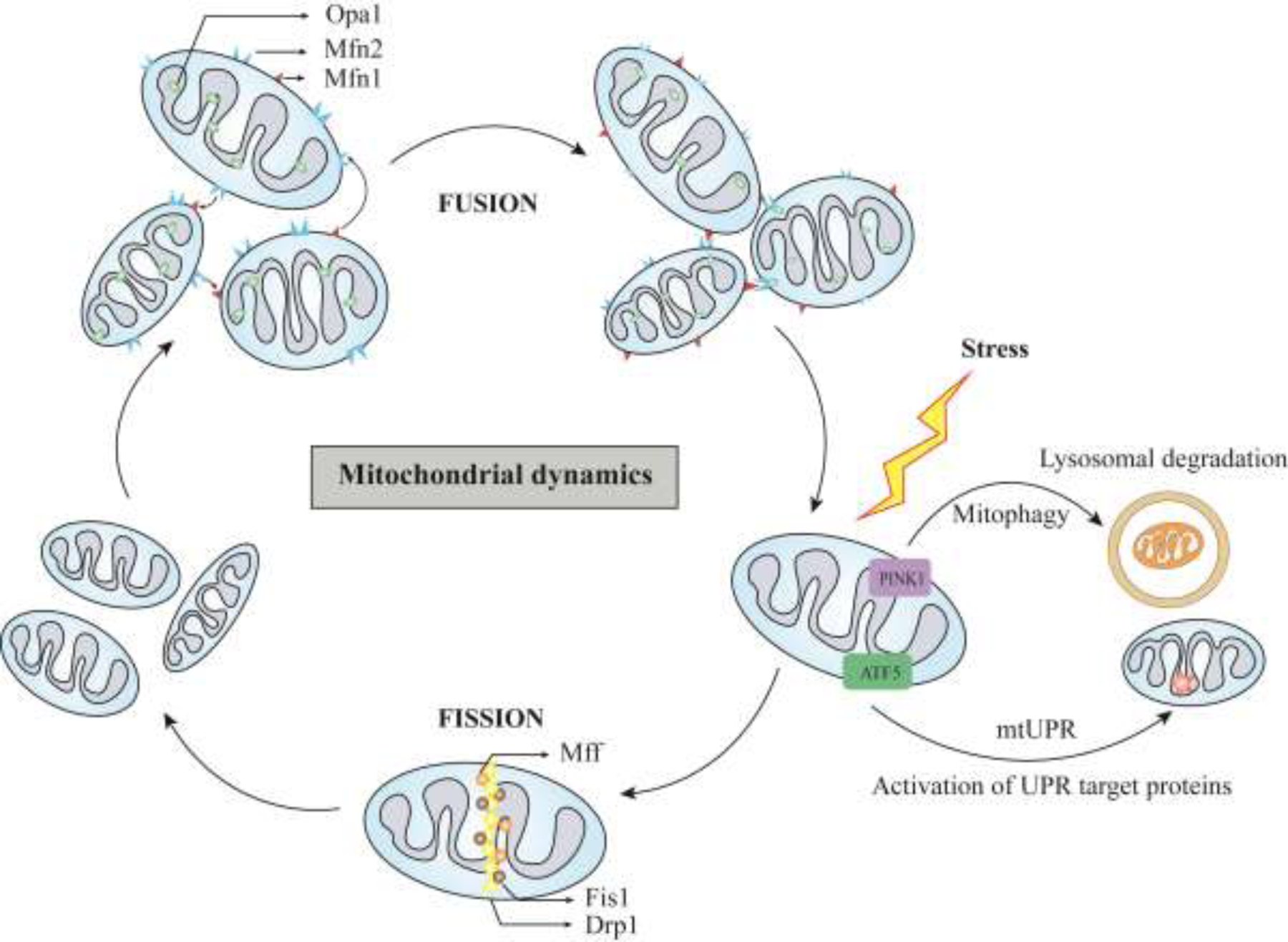
Mitochondria dynamics is regulated by balanced fusion and fission. Fusion is regulated by Mfn1, Mfn2 and Opa1. Fission is regulated by Drp1, Fis1, and Mff. Imbalance in this homeostasis results in induction of mtUPR and mitophagy. Mfn1- Mitofusin-1; Mfn2- Mitofusin-2; Opa1- Optic atrophy type 1; Drp1- Dynamin-related Protein 1; Fis1-Fission-1; Mff-Mitochondrial Fission Factor; mtUPR- Mitochondrial Unfolded protein response
2.1. Drp-1 as a key mediator of mitochondrial fission
Drp-1 has been shown to play a critical role in mitochondrial fission. A recent study found that Drp-1 deficient HeLa cells expressed altered respiratory function and changes in mitochondrial nucleoid morphology which suggests that mitochondrial fission is required to maintain proper respiratory activity and morphology of mitochondrial nucleoids in human cells (Ota et al., 2020). Drp-1 recruitment is modulated by MiD49, MiD51, Mff, and Fis1 [7]. Recently, it was found that Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy (Haileselassie et al., 2019; Losón et al., 2013). Additionally, Drp-1 is regulated by several post-translational modifications: ubiquitination, phosphorylation, SUMOlytion, palmitoylation, S-nitrosylation, and O-GlcNAcylation; these modifications play an important role in mitochondrial dynamics and are linked to cardiovascular disease pathogenesis (Jin et al., 2021). Ubiquitination of Drp-1 by mitochondrial ubiquitin ligase MITOL/MARCH5 or by Parkin targets the damaged mitochondria for proteasomal degradation; this process, for example, plays an important role in mitochondrial transport and interaction with ER (Nagashima et al., 2014; Qi et al., 2019). Deficiencies of these regulating factors are associated with increased activity of Drp-1, leading to ROS imbalance, and resulting in cardiovascular pathological conditions (Nagashima et al., 2014). Drp-1 knockout models have upregulated IL-1, IFN-γ, and TNF-α resulting in defective efferocytosis leading to atherosclerosis (Hu et al., 2020).
2.2. Mitophagy
Mitophagy is vital for the proper functioning of the cell, it is a quality control mechanism that eliminates defective mitochondria after damage or stress through the autophagosome-lysosome pathway (Onishi et al., 2021). Mitophagy is specific to defective mitochondria groups, thereby eliminating only detrimental units. Failure of the mitochondria to cope with the structural instability will activate certain autophagy-related factors such as proteases, chaperones, and ubiquitin-proteosomes to reinstate homeostasis (Braun and Westermann, 2017). Furthermore, the accumulation of unfolded proteins in mitochondria triggers the transcriptional activation of mitochondrial proteases and chaperones. There are two mechanisms by which mitophagy occurs, one dependent and another independent of autophagic factors. The autophagic-independent mechanism is modulated mainly through an outer mitochondrial membrane (OMM)-localized mitophagy receptors and proteolysis (mediated by ubiquitin ligases such as Parkin and MARCH5). In OMM receptor-mediated mitophagy, the presence of transmembrane receptors such as NIX, BNIP3, and FUNDC1 results in the formation of a bridge between the LC3-interaction region (LIR) motif of these receptors and the LC3 ligand present on the OMM. A recent study found that dysregulation of FUNDC1 destabilized cardiomyocytes during high-fat consumption (Pei et al., 2021). An example of indirect mitophagy regulation is BMAL1, a cardiac rhythm gene involved in monitoring the cardiac cycle. It regulates BNIP3 by binding to its promoter region, thereby indirectly controlling mitophagy (Li et al., 2020). On the other hand, proteolysis-mediated mitophagy is activated by the accumulation of PINK1 (PTEN-induced putative kinase protein 1), which is known to activate E3 ubiquitin ligase Parkin. Parkin targets OMM-related proteins using PINK1-generated phosphoubiquitin, which is feedbacked as a substrate for PINK1. The long non-coding RNA H19 prevents superfluous mitophagy by downregulating PINK1. H19-deficient cells showed low PINK1 expression, causing dysfunctional cardiomyocytes via excess mitophagy (Wang, S.H. et al., 2021). Drp1 also plays a crucial role in regulating mitophagy by interrupting the PINK1 pathway and inducing apoptosis in senescent cells (Wei et al., 2021). Alternatively, mitophagy can be regulated independently of autophagic factors. Though this pathway is not well explored, it was reported that mitochondria can be eliminated by lysosomes through early Rab5+ve endosomes. In obesity-induced cardiac failure, overexpression of Rab9 protected the heart from high-fat consumption by promoting mitophagy (Tong et al., 2021). Additionally, the inactivation of mitophagy was reported to cause diabetic cardiomyopathy. One of the main results of doxorubicin (DOX)-induced cardiotoxicity is the fragmentation of mitochondria and impaired mitophagy. Mitophagy also poses a promising target for DOX-induced cardiomyopathy, as mice treated with DOX, showed reduced expression of Rubicon, a negative regulator of autophagy (Liu, X. et al., 2019).
2.3. The Mitochondrial unfolded protein response (mtUPR) pathway
Under stress conditions, mitochondrial unfolded protein response (mtUPR) acts as a last resort to neutralize the affecters and reinstate the homeostasis of the system. Additionally, mtUPR is tightly regulated and helps prevent any aberrant misregulation by directing the activation of autophagy/mitophagy to eliminate the dysfunctional mitochondria. However, prolonged activation of mtUPR has detrimental effects on cellular metabolism. Furthermore, mtUPR activation has been implicated in various pathological conditions such as heart failure, neurological diseases, and cancer. Initially, mtUPR was discovered in mammalian cells and it was reported that when mitochondria face certain specific stressors, mtUPR promotes the upregulation of mitochondrial chaperones; this up regulation does not affect the other organelles’ functions. Later, Zhao et al. (2002) displayed the same effects by inducing the accumulation of the misfolded proteins in the mitochondria, which lead to the confirmation of mtUPR existence in mitochondria. mtUPR is activated under various stress conditions which include deletions in mtDNA, and overexpression of misfolded protein via activation of transcriptional activation of nucleus DNA to the expression of mitochondrial chaperones. This activation of nucleus-localized DNA occurs by retrograde mode of transport of transcriptional factors from the mitochondria to the nucleus. The accumulation of truncated proteins is the most effective mode of mtUPR activation, which triggers the expression of mitochondrial chaperones such as DNAJ, Hsp60, and Hsp10. Failure of these chaperones will disrupt the protein trafficking and produces powerful mtUPR responses. For example, the inhibition of the oxidative phosphorylation pathway (OXPHOS) by retenone causes significant increases in UPR activation, which in turn dysregulate the OXPHOS transcriptional factors.
2.3.1. Regulation of mtUPR
The mechanism underlying mtUPR stress response is densely regulated. mtDNA deletion causes upregulation of mitochondrial chaperones such as hsp60, and hsp10. In addition, the C/EBP homologous protein (CHOP) further activates DNAJ, ClpP, and LonP1 within the UPR cascade. Moreover, activation transcription factor 5 (ATF5) modulates the mtUR cascade which carries two factors, the mitochondrial targeting sequence (MTS) and the nuclear localization sequence (NLS). Under healthy conditions, ATF5 is localized in mitochondria; in stress conditions, ATF5 translocates to the nucleus to regulate the proteases and chaperone transcription levels. In addition to the above-mentioned factors, ATF4 is also implicated in mtUPR activation. Compared with ER stress factors, activation of all CHOP, ATF4, and ATF5, requires recruitment of ISR (integrated stress response). ISR is an evolutionarily conserved pathway, activated by eIF2α kinases namely PRKR-like endoplasmic reticulum kinase (PERK), double-stranded RNA-dependent protein kinase PKR, General Control Nonderepressible 2 (GCN2), and heme-regulated eukaryotic translation initiation factor 2α kinase (HRI) (Wek et al., 2006). The eIF2α kinase can significantly reduce global protein synthesis while enhancing the generation of selective genes such as ATF4, assisting in cell recovery. However, the severity of the cell stress leads to the induction of cell death. The dephosphorylation of eIF2α kinase can abort the ISR and return to normal protein synthesis. In addition, kinase response varies based on the stress stimulus such as amino acid depletion (Pakos-Zebrucka et al., 2016). Though various stress elements activate eIF2α phosphorylation, the outcome varies based on the context. The decisive result of ISR depends on the duration and severity of stress, other than its nature of it. As such prolonged ISR is considered to culminate in cell death whereas, short-term ISR are meant to establish a pro-survival response and reinstate homeostasis (Dey et al., 2010). Various studies have reported on how ISR is diversely regulated by CHOP, ATF4, and ATF5 (Michel et al., 2015; Sasaki et al., 2020). For instance, altered expression of mtDNA causes reduced expression of ISR-responsive genes which depend on the transcriptional regulator ATF4 (Michel et al., 2015). However, further studies are required to establish strong relationships between ISR, mtUPR, and mitochondrial stress.
2.3.2. The connection between mtUPR and mitophagy
Mitophagy and mtUPR are two damage responses that balance homeostasis in the mitochondria. The process of activation for both responses shares common factors that are essential in expressing the downstream target. For example, perturbations in ROS level, accumulation of misfolded/unfolded proteins, and mutations in the mtDNA trigger both mtUPR and mitophagy (Smyrnias, 2021). Although some studies state the independence of mitophagy and other mitochondrial stress responses (Chen et al., 2021), some of the signaling is common to both pathways, for instance, excess mitophagy activates mtUPR in HNC cells (Kang et al., 2021). Remarkedly, inhibition of mtUPR partially distorted the mitophagy response; it has been shown that mitophagy regulates myocardial stress via mtUPR responses (Wang, Y. et al., 2021). One primary example of a shared factor for activation of both mitophagy and mtUPR especially the SKN-1 gene, which senses an imbalance in ROS levels. SKN-1 activates mitophagy in a retrospective manner (Palikaras et al., 2015), whereas, it has also been shown to interact with ATF5 (ATFS-1, C.elegans homologue), thereby inducing the mtUPR pathway (Nargund et al., 2012).
2.3.3. Clinical significance of mtUPR in cardiac dysfunction
Mitochondria is the main provider of the daily energy requirements for heart contraction and relaxation. These organelles are the main source of ROS production, which act as signaling molecules, and control systemic communication. However, an imbalance in ROS causes impairment of normalcy, which triggers the homeostasis mechanism (Dietl and Maack, 2017). In cardiomyocytes, activation of mtUPR is reported to have beneficial effects by alleviating the deteriorating effects of mitochondrial dysfunction and improving contractile failure (Smyrnias et al., 2019). Furthermore, mtUPR is also activated by hemodynamic overload and neurohumoral stress, which protect against cardiac dysfunction (Smyrnias et al., 2019). ATF5 is a key mammalian modulator of mtUPR, which has been proved to enable cardioprotection (Wang, Y.T. et al., 2019). Most of the treatments available for cardiac diseases act by blocking neurohumoral hyperactivation (such as angiotensin-receptors blockers, and angiotensin-converting enzyme inhibitors) to alleviate cardiac stress. Examining the activation of mtUPR under neurohumoral overload inhibitors will help to understand its systemic regulation of cardiac stress (Bozi et al., 2019). LonP1 protease is a multi-functional enzyme that regulates mitochondrial proteostasis and prevents cellular stress. Under hypoxic conditions, LonP1 protease regulates ROS levels, providing cardioprotection (Kuo et al., 2015). Furthermore, LonP1 mitigates cardiac injury by adjusting the mitochondrial bioenergetics and it adapts to maintain homeostasis by reducing the complex one composition and activity (Venkatesh et al., 2019). Interestingly, LonP1 is also indirectly involved in controlling ferroptosis (Wang et al., 2020). Being ubiquitous in mitochondria, oxidative post-translational modifications impair the electron transport chain, mitochondrial respiration, and left ventricular dysfunction (Hoshino et al., 2014). Mitochondrial dysfunction has also been linked to ER stress; however, the main mechanism of interaction has not yet been elucidated. For example, mtDNA damage induced eIF2α phosphorylation, which activated integrated stress response (ISR) genes, implicating activation of ISR by ATF4 rather than by mtUPR induction (Sasaki et al., 2020). Mitochondrial dysfunction activates the ISR arm of UPR focusing mainly on ATF4. The interaction of ATF4 with CHOP is a well-studied interplay in the UPR pathway which results in negative regulation of ATF4-dependent genes (Su and Kilberg, 2008). Activation of ATF4 upregulates several genes involved in autophagy: Atg3, Atg5, Atg7, Atg10, Atg12, Atg12, Atg16, Becn1, Gabarap, Gabarap12, Map1lc3b and Sqstm1 upon amino acid deprivation (B’Chir et al., 2013). The regulation of autophagy genes depends on the ATF4 and CHOP signaling, which is decided by the ratio of CHOP and ATF4 bound to specific promoter cis-elements. Therefore, ATF4 acts as a fine tuner of autophagic response based on cellular requirements (B’Chir et al., 2013). A study by Kaspar et al. (2021) group showed that ATF4 acts as an attenuator diminish prolonged ISR signaling, where CHOP-C/EBPβ interaction modulates the ATF4 expression. Thus, prevention of overactivation of ATF4 during mitochondrial stress delays cardiomyopathy. Moreover, the upstream targets of ATF4 also affect stress responses such as HRI which plays vital in the OMA1-DELE1-HRI pathway. Repression of OMA1 is proved in vivo to be protective against heart failure conditions under mitochondrial dysfunction which is essential in the cleavage of DELE1 which modulates HRI kinase activity (Guo et al., 2020). Blockage of OMA1-DELE1-HRI is beneficial in certain stress conditions, on the other hand detrimental. Inhibition of this pathway also repressed cytosolic Hsp70 and displayed a protective effect under mitochondrial stress. Furthermore, a similar correlation was confirmed by the absence of mitochondrial protease and chaperone alteration (such as Hsp60 and LonP1) (Evinova et al., 2022). Contrastingly, the loss of other mitochondrial protease CLPP ameliorates cardiac injury by inducing the mtUPR pathway (Seiferling et al., 2016). Similarly, upregulation of Hsp60 results in cardiomyocyte cell death (Lin et al., 2007). Surprisingly, mtProhibitin depletion activates mtUPR, through lipid modulation and autophagy instead of mitophagy resulting in extending the lifespan of mammals (de la Cruz-Ruiz et al., 2021). Choline improved the cardiac condition by promoting mtUPR via the AMPK-SIRT3 pathway (Xu et al., 2019). Thus, mtUPR acts as a promising therapeutic target for treating CVD.
2.4. Correlation between mitochondrial stress and cardiac dysfunction
Mitochondria are overly sensitive organelles; they could be easily damaged by a lack of nutrients or oxygen supply. However, their adaptation mechanisms allow them to eliminate the damaged mitochondria to maintain proper mitochondrial function in the cell. Proper mitochondrial function is vital for many organs of high energy consumption such as the heart. When the mitochondrial function is impaired in cardiac cells, there is enhanced production of reactive oxygen species, uncoupling of the electron transport chain, depletion of cell ATP stock, and many other disruptions that eventually lead to extensive cell damage and apoptosis. Usually, mitophagy will restore normal cell function but when this process is dysfunctional, the cardiomyocytes cannot withstand the increased oxidative stress derived from mitophagy dysfunction and this will lead to apoptosis. Altered mitochondrial function and clearance will lead to cardiovascular disease due to enhanced ROS formation and decreased ATP levels in cardiomyocytes. Below, we will discuss other ways mitochondrial dysfunction can lead to cardiovascular disease (Figure 2).
Figure 2. Summary of mitochondria mediated cardiovascular pathologies.
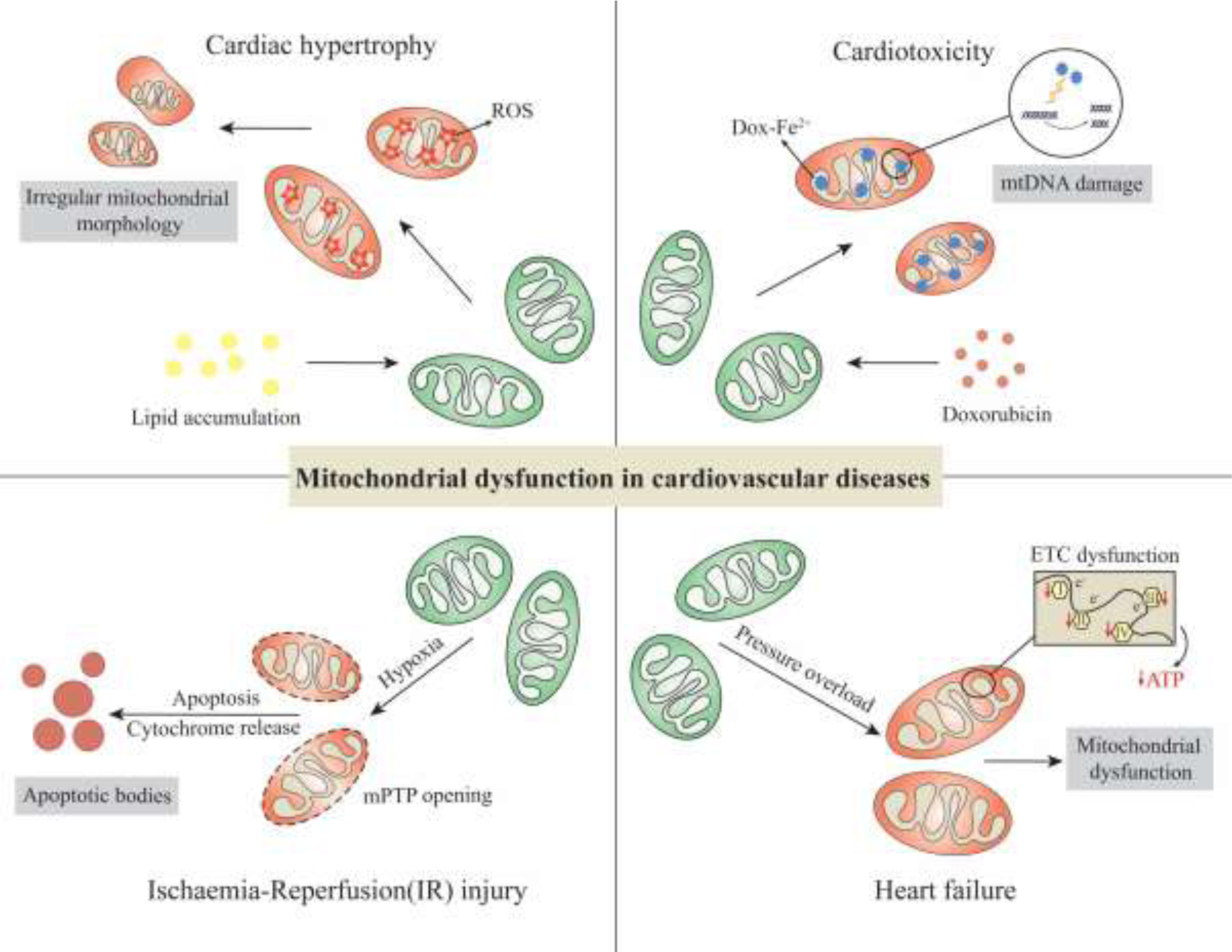
Any interruptions in mitochondrial network causes imbalance in homeostasis. In cardiac hypertrophy, increased lipid accumulation causes, accumulation of ROS in the mitochondria, disrupting mitochondrial morphology. In Cardiotoxicity, formation of damages Doxorubicin-Fe2+ the mtDNA. Under low oxygen condition, it causes release of cytochrome C and results in apoptosis. In heart failure condition, pressure overload induces ETC impairment and results in mitochondrial dysfunction.
2.4.1. Proteolytic processing of OPA1
The proteolytic processing of dynamin-like GTPase OPA1, in the inner mitochondrial membrane, is a pivotal step in the regulation of mitochondrial dynamics. There are two mitochondrial proteases, YMEL1 and OMA1, that mediate the cleavage of OPA1 from long (L-OPA10) to the short form (OPA1-S) which is essential for mitochondrial fission. Under cellular stress, OPA1 is cleaved to S-OPA1, inhibiting fusion, and causing mitochondrial fragmentation, resulting in dilated cardiomyopathy and heart failure (Wai et al., 2015). Contrastingly, mtUPR induces fusion, instead of fission and upregulates mitochondrial biogenesis elements. Furthermore, FUNDC1-mediated mitophagy induces mtDNA fragmentation and induces mtUPR during myocardial ischemic injury (Ji et al., 2022). Several studies are required to delineate the coupling of mitophagy and mtUPR to design highly specific targeted therapy.
2.4.2. Lipid peroxidation
Lipid oxidation-mediated cardiac dysfunction has not been well investigated. Lipid overload is a primary cause of heart-related diseases, inhibiting glucose utilization and resulting in lipotoxicity (Wende and Abel, 2010). Increased fatty acid utilization causes metabolic inflexibility through the Randle cycle and suppresses glucose oxidation (Goodpaster and Sparks, 2017). Lipid oxidation is the main commodity for cardiac functioning. At the prenatal stage, the heart relies on maternal lactate and glucose supply as the main source of energy. However, during perinatal development, cardiomyocytes shift to lipid oxidation as a central energy source (Makinde et al., 1998). During this process, mitochondria undergo dire conformal change, both shape and volume, accompanied by a change in cellular enzyme composition (LCAD, MCAD, CPT1, and ACSL1) to adapt to energy requirements (Lopaschuk et al., 1994). Post-translational modification of Drp-1 plays an essential role in cardiac maturation after birth. Extended exposure to lipids causes enhanced ROS production, morphological changes, increased mitochondrial respiration and promotes mitochondrial fission (Tsushima et al., 2018). Furthermore, the accumulation of ceramide caused the upregulation of mitochondrial dynamic factors such as OPA1, MFF, and mitophagic factors such PINK1. In addition, insulin dependent Akt regulation is limited in cells with ceramide accumulation, triggering protein phosphatase 2A (PP2A) activation (Bekhite et al., 2021).
2.4.3. Doxorubicin cardiotoxicity
Cancer treatment is associated with multiple layers of chemotherapeutic treatment to eliminate tumor growth. However, chemotherapy has been associated with damage to several organs. Doxorubicin (dox), an anthracycline agent, is a primary anti-cancer drug for a spectrum of cancer diseases: breast, ovarian, gastric, acute lymphoblastic leukaemia, neuroblastoma, small cell lung, bladder, thyroid, osteogenic bone tumors, Wilm’s tumor, Hodgkin’s and cutaneous T cell lymphoma (Sritharan and Sivalingam, 2021). However, doxorubicin-mediated anti-cancer therapy is associated with increased heart failure because of upregulated ROS environment (Varricchi et al., 2018). DOX-mediated cardiotoxicity is caused because of the accumulation of DOX in mitochondria. It forms an iron complex, which causes increased ROS production (Ichikawa et al., 2014). Furthermore, downregulated glutathione peroxidase 4 and increased expression of lipid oxidation via the Dox-Fe2+ reaction culminate in the condition of toxicity (Tadokoro et al., 2020). Loss of ubiquitin E3 ligase TRIM21, which negatively regulates the p62-Keap1-Nrf2 - antioxidant pathway, improved the mortality of mice and protected against DOX-induced cardiotoxicity (Hou et al., 2021). Dox treatment caused non-heme deposition through heme breakdown by Nrf-2 mediated pathway (Fang et al., 2019). Supportively, Ferrostatin and iron chelation have alleviated heart failure (Fang et al., 2019; Tadokoro et al., 2020).
3. ENDOPLASMIC RETICULUM STRESS
Post-translational modification of protein takes place in the endoplasmic reticulum (ER), where proteins are folded into the proper three-dimensional structure and destined for different cellular locations. The ER is the main server of the cell organization and takes part in the secretory pathway, cellular storage of Ca2+, cellular communication, protein folding, and translocation (Sun et al., 2021). Interruptions in ER functioning activate the unfolded protein response (UPR) and ER-associated degradation (ERAD) pathway, which neutralizes the cause of ER stress to promote homeostasis. Inefficacy of these mechanisms leads to activation of cell death, and apoptosis, eliminating the cell (Hwang and Qi, 2018). The heart being the constant worker of the body requires a constant flow of energy; impairment in the cardiac function such as ischemia, cardiomyopathy, heart failure, or atherosclerosis will impose increased demand on the ER protein folding unity, which induces ER stress. The induction of ER stress has been a negative regulator of pathological conditions in the heart, which increases the chance of heart failure (Zhou et al., 2021).
3.1. Crosstalk between ERAD and other signaling pathways
The ER is an oxidative environment, stocked with enzymes like protein disulfide isomerase (PDI), which enhances the formation of a disulfide bond, thereby folding into proper protein structure (Trevelin and Lopes, 2015). Any disruptions in the folding will cause the retention of misfolded and unfolded proteins in the ER, which will activate ER stress, resulting in apoptosis (Delbrel et al., 2018). ERAD is a checkpoint mechanism, that monitors protein folding in the ER, and maintains homeostasis, for example, ERAD can sense alterations in lipid biogenesis, which can either upregulate/downregulate the expression of enzymes involved in the process (Lemberg and Strisovsky, 2021). The misfolded/unfolded protein sensing mechanism of ERAD involves multiple stages: detection, translocation, and elimination (Lemberg and Strisovsky, 2021). Briefly, ERAD detects any unfolded proteins by N-glycans motif in the misfolded proteins with the help of EDEM (ER degradation-enhancing α-mannosidase-like protein) and lectin chaperones. Later, the misfolded proteins are translocated to the cytosol by the Hrd1-Sel1l-ERAD axis for proteasomal degradation (Li et al., 2010).
The ER also regulates the flow of Ca2+ in the cytoplasm, ER, and extracellular matrix. The Ca2+ required for the contraction and relaxation of cardiac muscle regulates the opening and closing of the ER membrane. In addition, sarcoplasmic/ER Ca2+ ATPase 2a (SERCA2a) also controls the Ca2+ balance that is required for proper heart functioning (Saheki and De Camilli, 2017). The misaligned nature of Ca2+ circulation in CVD promotes the activation of proteases, lipases, and nucleases. Stromal interaction molecule 1(STIM1) is a molecular Ca2+ detector that regulates cardiac metabolism. A previous study showed that knockout of STIM1 causes abrogated mitochondria, erratic lipid accumulation, and dilated cardiomyopathy (Collins et al., 2019). Overall, ER is involved in regulating protein, lipid, and Ca2+ balance in cardiac tissues. Additionally, junctions between ER and other organelles have been crucial for effective cellular communication. For instance, the lack of Golgi membrane tethering protein, At CASP which is responsible for forming a junction between ER and Golgi to eliminate ceramide, will cause an accumulation of toxic lipids in ER (Gillingham and Munro, 2016; Osterrieder et al., 2017). Furthermore, ER trafficking is also associated with mitochondria, by the presence of MAM (Mitochondrial associated ER Membrane), which regulates the signaling molecule Ca2+, as well as the translocation of lipid molecules vital for promoting cell survival (Sasi et al., 2020). In post-mitotic cells like cardiomyocytes, it is essential to monitor calcium overloading (Whelan et al., 2010). A study by Beretta et al. (2020) displayed the pro-survival mechanism involved in the generation of ROS-generating protein Nox4. Under stress conditions, Nox4 is recruited at the MAM site and amplifies Akt-mediated phosphorylation of lnsP3R (which is a major channel for calcium release from ER), preventing calcium flux and thereby inhibiting necrosis. Heart exposed to ischemia-reperfusion condition activates Nox4 mediated mechanism to reduce the infract size. Various proteins carry the MAM domain, such as the FUN14 domain-containing protein 1, which interacts with the IP3 receptor, modulating the Ca2+ signaling in diabetic heart conditions (Wu et al., 2019). Accumulation of Ca2+ has a two-edged effect on the system: Excess Ca2+ promotes autophagy (Wang and Kaufman, 2016) and parallelly it also blocks autophagy by inducing calpain during ischemic reperfusion (IR) injury (Lu et al., 2020). A study showed that blocking the transposon has potential therapeutic benefits to restore cardiac performance, diastolic Ca2+ flow, and recovery of contraction ability in myocardium-stunned hearts (Mariángelo et al., 2022). Furthermore, in IR injury, ferroptosis seems to augment the effect of ER stress; overexpression of miR-133a and H2S treatment attenuated ER stress-mediated cell death and cardiomyocyte motility (Ren et al., 2019). Chen et al. (2020) reported circDLGAP4, a circular RNA, to be a mediator of ER stress, which regulates the expression of HECT domain E3 ubiquitin-protein ligase 1 (HECTD1) and mediates IR injury.
3.2. Unfolded protein response (UPR) pathway
The unfolded protein response maintains the cellular proteins in a balanced state. It functions in either two ways: increasing or limiting the protein folding capacity of ER (Wang and Kaufman, 2012). Three vital signaling components are involved in this mechanism: IRE1α (inositol-requiring enzyme 1α), ATF6(activating transcription factor 6), and PERK (pancreatic endoplasmic reticulum kinase), which help ER to propagate its actions (Ron and Walter, 2007). The common element between all three of the signaling components is the ER luminal domain, which can sense the critical concentration of misfolded protein in the ER. When the protein requirement is high, UPR tries to increase the folding machinery by increasing ER capacity, transcription of ER chaperones, and biogenesis of ER components. Each UPR sensor has a unique mechanism to execute its response. IRE1α has two enzymatic actions at the cytoplasmic tail: The serine/threonine kinase domain and the endoribonuclease domain (RNase) (Tirasophon et al., 1998; Wang et al., 1998). In presence of unfolded proteins, IRE1α is trans-auto phosphorylated and gains RNase activity, which leads to the splicing of XBP1(X-box protein1) mRNA (Calfon et al., 2002; Korennykh and Walter, 2012). Then, the active XBP1 translocates to the nucleus and upregulates the proteins associated with increasing ER size and function like MANF (Mesencephalic astrocyte-derived neurotrophic factor) to protect from ER stress (Wang et al., 2018; Yoshida et al., 2001). Additionally, ATF6 collaborates with XBP1 by translocating to the Golgi and gains active form by cleaving at site-1 and site-2 proteases, increasing the ER size (Yamamoto et al., 2007). After cardiac arrest, during recovery, the UPR pathway is activated in various organs to restore the damage. Activation of ATF6 arms improved neuronal function and triggered protein removal pathways. This showed the prosurvival capabilities of UPR-mediated ATF6 activation after cardiac arrest (Shen et al., 2021). Contrastingly, PERK slows protein translation by inactivating eukaryotic translation initiation factor 2α (eIF2α) to clear the unfolded proteins (Bertolotti et al., 2000; Harding et al., 1999). However, this stalling is detrimental: hyperactivation of PERK upregulates CHOP (C/EBP-homologous protein) which inhibits the Bcl2 expression, resulting in cell death (Marciniak et al., 2004; McCullough et al., 2001). Extended ER stress results in pressure overload in cardiomyocytes triggering CHOP signaling, deletion of CHOP, improved cardiomyocyte resistance to cardiac remodeling, and cardiac dysfunction. Recently, Al-Yacoub et al. (2021) reported the existence of CHOP-mediated non-canonical pathways in FBXO32-associated cardiovascular diseases. FBXO32 is a muscle-specific E3 ubiquitin ligase, which regulates Akt and calcineurin A dependent cardiac hypertrophy mice model (Li et al., 2004; Li et al., 2007). Likewise, exposure to chronic ER stress enables IRE1α to gain off-target RNA substrate by oligomerization, resulting in the degradation of ER elements (Han et al., 2009; Hollien and Weissman, 2006). Furthermore, the aberrant splicing of IRE1α limits some microRNAs, which leads to the upregulation of proapoptotic factors such as TXNIP (thioredoxin-interacting protein), which promotes Caspase1 mediated cell death (Lerner et al., 2012) (Figure 3).
Figure 3. ER stress mechanism.
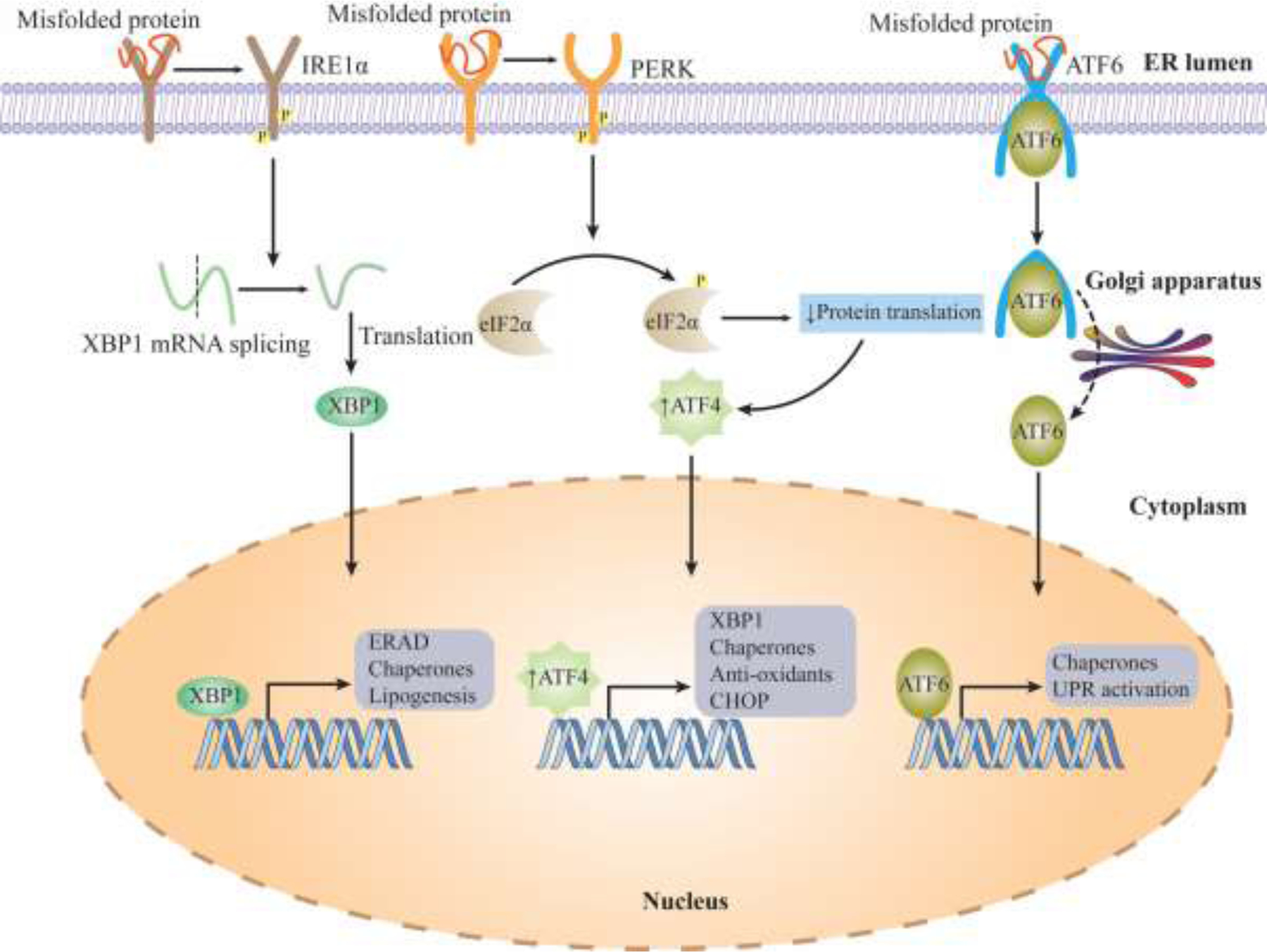
Unfolded Protein Response (UPR) maintains the cellular homeostasis. When misfolded proteins accumulate, they activate three major mechanisms: IRE1α, PERK, ATF6. These proteins translocate to the nucleus and upregulate the proteins responsible for increased functioning in the ER. Failure to eliminate the accumulation of misfolded proteins, triggers apoptosis.
The ER stress response also protects against cardiac dysfunction, for example, cardiac-specific kinase, Pak2 is a stress-responsive protein that protects from apoptosis. Pak2 is localized near the ER membrane; it is activated by PI3K (phosphatidylinositol 3 kinases)-Akt, AMPK, and PERK-ERK1-ERK2(extracellular signal-regulated kinase 1) to inhibit cell death (Binder et al., 2019). However, in ischemic reperfusion conditions, PAK2 is downregulated, inducing ROS generation, Ca2+ overload, and activation of caspase 12 resulting in apoptosis, implicating the protective role of PAK2 in cardiac disease conditions (Wang, S. et al., 2019). Furthermore, Pak2 also regulates Nrf2 in a stressed heart; deletion of Pak2 upregulated Nrf2 expression. Pak2 attenuates the ER-mediated Nrf2 activation by inducing the XBP1-Hrd1 axis (Binder et al., 2022). The UPR pathway interlinks with mitochondria-mediated cell death. Lowering of the mitochondrial permeability transition pore (mPTP) causes the release of cytochrome C, which activates the caspase cascade, leading to intrinsic apoptosis. The mPTP is regulated by the distribution ratio of proapoptotic (such as Bax, Bak) to antiapoptotic factors (bcl2, mcl1, Bcl-xl) (Shi et al., 2021). The terminal UPR has been shown to interact with at least six BH3-only proteins (Bim, Bik, Bid, Bad, Noxa, and Puma) (Arystarkhova et al., 2021; Li et al., 2006; López et al., 2017; Puthalakath et al., 2007; Rodriguez et al., 2012; Upton et al., 2008). In addition, UPR is correlated with the biogenesis of mitochondria, it is involved in the control of mitophagy and mitochondrial integrity. For instance, IRE1α has a scaffolding role for Ca2+ transport into the mitochondria through its non-canonical path in MAM (Malli and Graier, 2019). Furthermore, UPR also monitors the major elements of mitochondrial biogenesis such as PGC1α, Nrf1, TFAM, citrate synthetase, and TFEB (Transcription factor EB), altered expression of these genes leads to heart failure (He et al., 2020; Pan et al., 2022; Prola et al., 2019; Xu et al., 2020; Zhang et al., 2021).
3.3. ER stress and autophagy
Autophagy is another adaptive measure in ER to remove the surplus accumulation of misfolded/unfolded protein, termed reticulophagy. Precisely, uncontrolled deposition of proteins, activates auto-ubiquitination of E3 ubiquitin ligase, TRIM13 (tripartite motif-containing protein 13), which binds with p62 (or sequestosome1), autophagy regulator (Ji et al., 2019). Simultaneously, the arginine transferase enzyme attaches arginine residues to the N terminal of ER chaperones, causing destabilization of ER-resident chaperones such as Bip (Ji et al., 2019). This unstable N-terminal, known as N-degron, binds with p62, forms an oligomer of TRIM13 and p62, and causes induction of autophagy by recruiting an LC3B autophagy adaptor (Ji et al., 2020). In ER stress, induction of autophagy exerts cytoprotective effects, for example, PERK signaling during ER stress, recruits autophagy-related protein 12 (ATG12) and activates the ATG12-ATG16-ATG5 axis, which converts LC3-I to LC3-II form, and promotes cardioprotection (Hsieh et al., 2020; Kouroku et al., 2007). Autophagy is a double-edged sword, as ER stress prolongs, it activates the apoptotic pathway, instead of recovering the ER. In some pathological cases, ER stress impairs autophagy, disrupts the clearance of protein accumulation, and promotes the disease condition (Rashid et al., 2015).
4. Hypoxia
Hypoxia is a condition of low oxygen in the body or a region of the body. Hypoxia-inducing factor (HIF) is the fundamental factor in promoting hypoxia during oxygen deprivation (Wang and Semenza, 1995). In normoxia, HIF is prevented from translocating to the nucleus by proteasomal degradation. HIF is highly regulated by prolyl-4-hydroxylases (PHDs), which hydroxylate the proline residues in HIF-1α; asparagine residues are hydroxylated by Factors Inhibiting Hypoxia (FIH) (Majmundar et al., 2010). These hydroxylated residues are recognized by Von Hippel–Lindau protein (pVHL), an E3 ubiquitin ligase, a signaling enzyme for proteasomal degradation (Ivan et al., 2001). However, under hypoxia conditions, pVHL and FIH expressions are repressed, promoting HIF-1α translocation. In the nucleus, HIF-1α binds with HIF-1β forming a complex and interacting with Hypoxia Responsive Elements (HRE), thereby promoting hypoxia (Jaakkola et al., 2001). Apart from HIF-1, there are HIF-2α and HIF-3α. Similar to HIF-1α, HIF-2α is also stabilized during hypoxia by forming a dimer with ART(aryl hydrocarbon receptor nuclear translocator), transactivating the VEGF (Vascular endothelial growth factor) and erythropoietin promoter, implicating a vital role in promoting vascular development (Ozaki, 2007). The exact role of HIF-3α during hypoxia is still unknown, however, it dimerizes with HIF-1β despite its lack of the c-terminal transactivation domain, thereby negatively regulating the transcription of HIF-1α and HIF-2α (Knutson et al., 2021) (Figure 4).
Figure 4. Pictorial representation of Hypoxia mechanism.
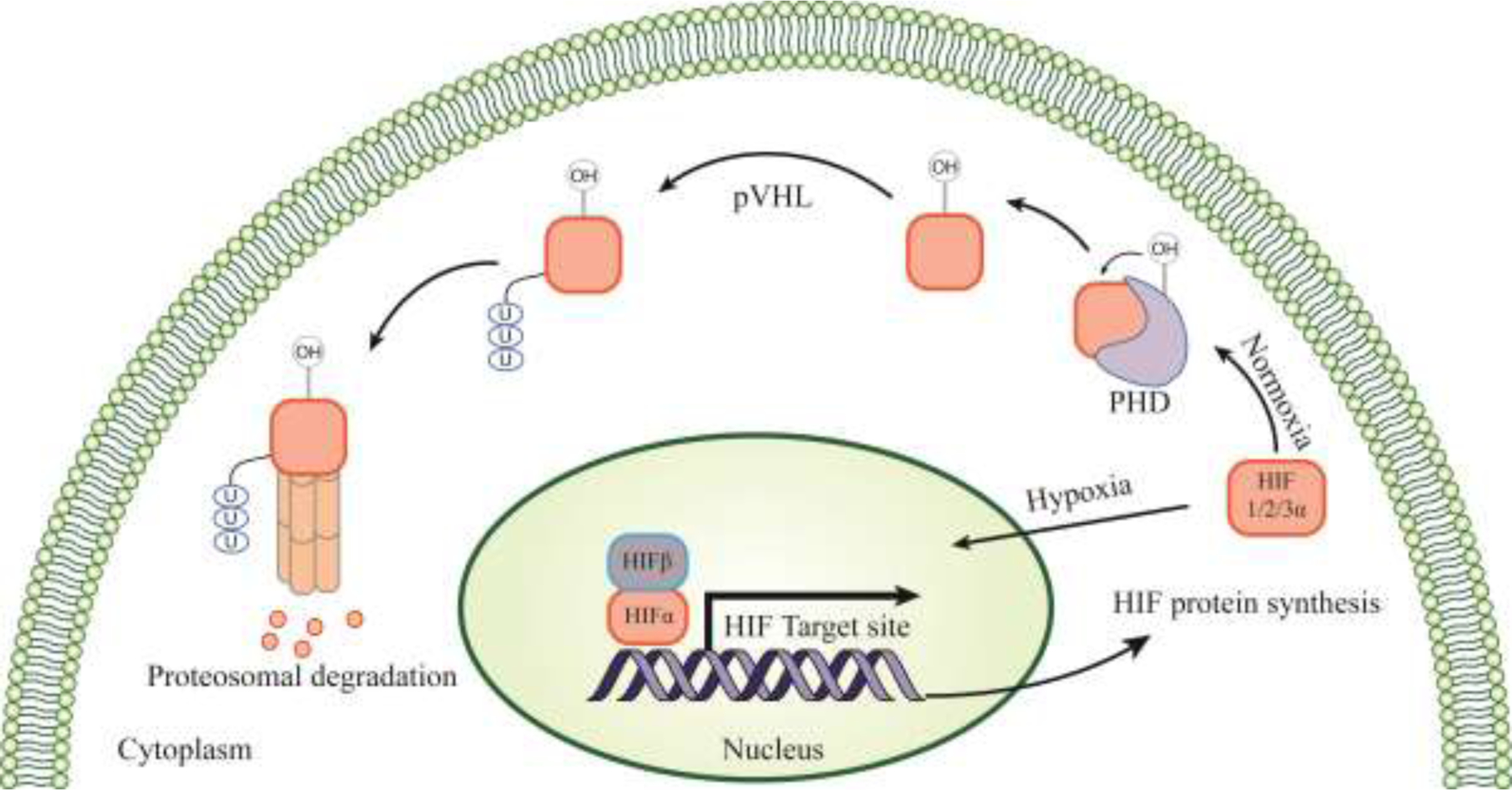
Under Normoxia, Hif protein is eliminated by proteasomal mechanism with the help of PHD and pVHL enzymes. However, depletion of oxygen leads to downregulation of PHD and pVHL, results in translocation of Hif to nucleus and forms complex with Hif1β. This complex promotes the transcription of Hif protein. pVHL- Von Hippel-Lindau protein; PHD- Prolyl-4-hydroxylases
4.1. Metabolic switch alternation in hypoxia
The human body mainly relies on two major sources of energy, glucose, and fatty acids. Around 96% percent of ATP production depends on fatty acids and glucose, but, oxidation of fatty acids produces the highest ATP yield (Su et al., 2021). However, the utilization of minimal oxygen, which is required for glucose oxidation, yields a prominent amount of ATP compared to fatty acids (Su et al., 2021). This variation in energy dependence correlates with the geographical location of ethnic groups such as Tibetans, Sherpas in Nepal, and the Andeans (Holden et al., 1995). People located at higher altitudes rely on glucose as their main source of energy and tend to suffer from severe cardiovascular disease, due to the presence of a constant hypoxia environment. Compared to the high landers, low landers showed increased utilization of fatty acids as their metabolic fuel (Holden et al., 1995). Two chief regulators that are associated with modulating the hypoxic environment are peroxisome proliferator-activated receptor-α (PPAR-α) and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α); both are downregulated under hypoxic conditions (Cole et al., 2016). High altitude residents: Tibetans and Sherpas have shown PPAR-α lacking genetic variants among the population (Ge et al., 2012). PPAR-α is found to be downregulated by the elevated expression of HIF1α. Additionally, PPAR-α regulates PDK2 and PDK4 factors that are involved in glucose oxidation (Cole et al., 2016; Mansor et al., 2016). Furthermore, PPAR-δ, an isoform of PPAR also regulates HIF-1α, by restoring vascular homogenesis during post-ischemia in a ligand-dependent manner (Wu et al., 2022). PGC-1α is a regulator of PPAR-α, it is involved in mitochondrial biogenesis, and seems to function erratically in the left and right ventricles in the heart during hypoxic conditions (Dorn et al., 2015). For instance, 3-week hypoxia-exposed both the left and right ventricle in a rat model showed downregulated PGC-1α (Ramjiawan et al., 2013). However, in a 4-week hypoxia model, the left ventricle displayed no notable difference, but in the right ventricle, PGC-1α expression was downregulated, and PGC-1β was upregulated with increased mitochondrial respiration (Ferri et al., 2018). Under a hypoxic environment, the energy consumption by either of the ventricles (left and right) varies; the muscles respond differently to the same hypoxic exposure. For example, in a study of rats exposed to 10% oxygen, the left ventricle relied on pyruvate for maximum energy utilization and its maximal mass-specific respiration and mitochondrial-specific respiration were significantly increased after hypoxia exposure when compared to normal conditions. On the other hand, the right ventricle’s maximal mass-specific and mitochondrial-specific respiration did not show any significant differences between hypoxia and normal conditions (Ferri et al., 2018). This study demonstrated the energy reliance of the heart on glucose-dependent oxidation under hypoxic conditions and it also demonstrated that muscles respond differently to hypoxia. The changes were correlated with the alterations in the electron transport chain (ETC) complex: I, II, IV (Heather et al., 2012). Coenzyme Q (CoQ) is an important structural component of ETC has been associated with potential antioxidant capabilities and acts as a vital electron carrier (Zozina et al., 2018). In hypoxia, the mRNA stabilization which is associated with the CoQ is interfered with, causing reduced ATP synthesis, increased cell death and leading heart failure (Liu, Y. et al., 2019). Supplementation of CoQ, improved antioxidant properties in high altitude inhabitants (Biuomy et al., 2020).
A study by Cho et al. (2013) lead to the finding of PGC-1 and ERR-induced Regulator in Muscle 1 (PERM1), a novel effector of PGC-1 and ERR, enriched in skeletal muscle and cardiomyocytes. Perm1 regulates the mitochondrial biogenesis in cardiomyocytes, thereby monitoring the energy metabolism required for the heart. PERM1 protected the cardiomyocytes against hypoxic conditions and promoted mitochondrial biogenesis (Cho et al., 2021). Insulin resistance development in diabetic hearts, impairs HIF-1α mediated signaling, disabling the metabolic switch from fatty acid to glucose, resulting in cardiac dysfunction (Su et al., 2021). Factors such as obesity and hyperglycemia, are notably associated with CVD in high landers (Zheng et al., 2019). The development of insulin resistance is a common characteristic observed in intermittent hypoxia and congenital heart disease (Ip et al., 2002; Niwa, 2019). In addition to glucose and fatty acids, ketone poses another prime source of energy; energy generation from ketone is comparable to glucose and fatty acid utilization (Zozina et al., 2018). A recent study showed that elevated levels of β-OHB, the main constituent of ketone bodies, are harmful to cardiomyocytes exposed to hypoxic/ischemic conditions, however, inhibition of HIF-1α by Roxadustat administration alleviated the effects of β-OHB by inducing a metabolic shift toward glycolysis (Ma et al., 2021). Their results suggest that increased ketone body supply in cardiomyocytes under hypoxic conditions exacerbates cardiac dysfunction.
4.2. Induction of mitophagy in Hypoxia
The heart is more sensitive to oxygen-deprived conditions like hypoxia. Most of the high energy demand of the heart is supplemented by mitochondrial OXPHOS. In heart failure, excess demand for energy will compromise the OXPHOS activity causing depletion of ATP synthesis and augmenting the cardiac injury. Under ischemia, a low oxygen-deficient condition that causes cardiac damage, the Ca2+ flux and the electrochemical gradient are impaired. Excessive autophagy in the ischemia-reperfusion models reflects the degradation of vital organelles that are complementing the energy supply. Hypoxia also induces receptor-mediated-mitophagy. For instance, FUNDC1 is known to cause mitophagy in response to hypoxia, by dephosphorylation of PGAM5. This circuit is blocked by the Bcl2L1/Bcl-xL binding to PGAM5, inhibiting the dephosphorylation. Polo-like kinase 1(PLK1) is known to cause mitophagy; it suppresses ischemia-induced cardiac injury by FUNDC1-dependent pathway resulting in mitophagy activation. In a PLK1 overexpression model, upregulated mitophagy and attenuated hypoxia were observed (Mao et al., 2021). Conditions like ischemia and starvation trigger a phenomenon known as autosis, a cell death process mediated by ion channels, characterized by increased formation of autophagosomes (Liu et al., 2013).
4.3. Hypoxia in CVD
Ischemia occurs due to the narrowing of the coronary artery or ruptures of plaques. This leads to an impoverished oxygen supply causing irregular ATP synthesis, increased ROS generation, and cardiac dysfunction. Ischemia injury occurs during heart transplantation, surgery, or angioplasty (Lee et al., 2019). The two critical causes of ischemia-reperfusion injury are Ca2+ overloading and subsequent ROS accumulation, lowering the mitochondrial permeability transition pore (mPTP). Low mPTP will result in the release of cytochrome C into the cytoplasm leading to cell death by activation of the caspase cascade (Abdelwahid et al., 2017). Interestingly, periodical exposure to hypoxia such as Chronic Continuous Normobaric Hypoxia (CNH) and short-term intermittent hypoxia (IH) causes acclimatization, developing resistance toward hypoxia (Chang et al., 2019; Prokudina et al., 2019). In a recent study, rat hearts under CNH conditions showed reduced infarct size in response to ischemia and reperfusion (Lishmanov et al., 2017; Maslov et al., 2013; Neckar et al., 2003). This cardiac protection was characterized by developing MPTP resistance towards Ca2+ loading, however, blockage of opioid receptors completely terminates the MPTP resistance effects (Lishmanov et al., 2017).
Similarly, IH also known as cyclic hypoxia involves the exposure of cells to cycles of hypoxia/reoxygenation. These short cycles of hypoxia are sufficient for the occurrence of the metabolic switch and non-oxidative energy consumption. One of the consequences of transient normoxia in hypoxic cells is the accumulation of ROS, due to the inability of mitochondria to activate the ROS-scavenging pathway. Furthermore, restored oxygen availability will lead to rapid HIF-α degradation and the hypoxic stress response necessary for survival will be compromised exposing the cells to oxidative stress (Guan et al., 2019). IH leads to obstructive sleep apnea, which is associated with intermittent loss of airflow in sleep. Several studies point out that IH is characterized by hypertension, stroke, and MI (Marin et al., 2005). Contrastingly, IH also improves cardioprotection by regulating the vital signaling molecule, ROS. This perpetual exposure is enabling cardioprotection, however, the mechanism underlying this protection is yet to be explored. This cardioprotection is attained by upregulating -antioxidant factors, mainly superoxide dismutase (SOD) and glutathione peroxidase (GPx) and preventing the mPTP opening(Aguilar et al., 2018). Furthermore, this protection is enabled by maintaining Ca2+ homeostasis. This adaptation is associated with the elevation of the O-linked N-acetylglucosamine protein, activating the glucose-6-phosphate dehydrogenase (G6PDH). The activation of G6PDH results in homeostasis by regulating NADPH/NADP+ and GSH/GSS implicating control of anti-oxidant properties(Ou et al., 2021). HIF1α exerts its protective effect by generating adenosine molecules and promoting adenosine receptors which are proven to display anti-inflammatory effects (Eltzschig et al., 2003; Kong et al., 2006). Consequently, hypoxia upregulates HIF2α, an isoform of HIF that promote cardioprotection by inducing its target site amphiregulin (AREG) (Koeppen et al., 2018). Lee, et. Al., (Lee et al., 2020) reported that HIF2α protects from myocardial ischemia by induction of epidermal growth signaling receptor ERBB1. Furthermore, deletion of HIF2α resulted in increased infarct size, implicating the HIF2α’s role in ensuring cardio-specific protection. HIF2α shows cardioprotective action by regulating IL6 through PI3K/Akt axis and STAT3 pathway (Wu et al., 2021).
Recently, Hif3α was found to regulate hypoxia tolerance; knockout of HIF3α significantly reduced the hypoxia tolerance. The mechanism undermining this kind of characteristic is erythropoiesis via GATA4 (Cai et al., 2020). Alternative splicing of HIF3α results in the generation of different HIF3α variants hypothesized to have a spectrum of regulatory functions. Furthermore, HIF3α2 can directly regulate the erythropoietin (EPO) gene, by binding at the HRE element situated at the promoter of EPO (Tolonen et al., 2020). Surprisingly, HIF3α2 can also induce apoptosis by accumulating at the later stages of hypoxia. Surfeit deposition of the HIF3α2 isoform can induce the expression of DNA damage-inducible transcript 4 (DDIT4); as a consequence, caspase 3-mediated apoptosis will be activated, resulting in cell death (Jaskiewicz et al., 2022). The effects of differential expression of hypoxia isoform in cardiac disorder are summarized in Table 1 (Chen et al., 2022; Eubank et al., 2011; Ghosh et al., 2021; Hickey et al., 2010; Hnatiuk et al., 2016; Menendez-Montes et al., 2021; Sun et al., 2015; Tolonen et al., 2020).
Table-1:
Effect of differential expression of HIF isoforms
| Isoforms | Alterations | Cell line/Model/Organism | Mode of mechanism | Ref |
|---|---|---|---|---|
| Hif1α | Overexpression | peri-infarct of sheep (n=6) undergoing coronary occlusion | Reduced infarct size and improved LV systolic performance increased neovascularization | Chen et al., 2022 |
| Hif1α null mouse model | Increased HIF2Α Increased mitochondrial number Impaired embryonic glycolysis Activation of amino acid catabolism |
Eubank et al., 2011 | ||
| Knockdown | H9c2 cells exposed to high glucose | Downregulated angiopoietin-like protein 2, alleviating the hypoxia injury by promoting nrf2 and HO-1 expression | Ghosh et al., 2021 | |
| Hif2α | Overexpression | 46C ESC cell line | Promotes cardiomyogenesis by wnt/β-catenin pathway | Hickey et al., 2010 |
| Knockdown | VHLR200W (Chuvash Polycythemia)mouse model | Normalized erythropoietin and endothelin level suppressed both the polycythemia and pulmonary hypertension partial protection against vascular remodeling, haemorrhage, and edema |
Hnatiuk et al., 2016
Menendez-Montes et al., 2021 |
|
| HIF-1αflox/flox/LysMcre knockout mice | Inhibited soluble form of the VEGF receptor-1 (sVEGFR-1) production in a hypoxic condition Production of VEGF was unaffected |
Sun et al., 2015 | ||
| Hif3α | Overexpression | - | - | |
| Knockdown | Hep3B hepatoma cells | Downregulation of EPO gene | Tolonen et al., 2020 | |
| Hif3α2 | Overexpression | Hep3B hepatoma cells SK-N-AS cells | Promotes expression of erythropoietin gene (EPO), BMP6, PTX3 | Tolonen et al., 2020 |
| Knockdown | - | - | - |
Future perspectives and conclusion
Response to cell stress is a vital element in deciding cell fate. For temporal stress, a systemic response will try to eliminate the damaged organelles and reinstate them to their original form. However, protracted stress often leads to either cell death or senescence. Because stress pathways are dynamic, understanding their mutual crosstalk will aid in understanding the molecular mechanisms underlying them. Nonetheless, despite the emergence of new findings demonstrating their dynamic nature, several interconnections between these stress pathways remain largely unknown. Furthermore, research in this area has been restricted to in-vitro culture-based systems. Novel approaches to developing complex model systems involving 3D culture systems such as endothelial cells and CMCs should be initiated (Bartoszewska and Collawn, 2020; Bartoszewska et al., 2022). Understanding the mechanism triggering these responses will lay a stronger foundation to develop potential therapeutics for the existing pathophysiology. Another limitation in this field is that the results do not account for the pathways’ genome-wide consequences, as activation of PERK and IRE1 leads to transcription and translation of multiple genes. This is possible by incorporating next-generation sequencing and single-cell sequencing. Although challenging, incorporating this will significantly improve our understanding of the crosstalk.
A large portion of hypoxia-related research has concentrated on exposure for a specific amount of time. However, because physiological oxygen demands vary with each cell in our body, our experiments should be tailored to account for IH. Another issue is the use of hypoxia mimics, which merely degrade FIH. Such compounds do not shed light on the molecular complexities of these pathways. So, the results obtained should be substantiated by limited oxygen availability(Bartoszewska and Collawn, 2020; Bartoszewska et al., 2022). Since mitochondria are the central source of energy, it is highly regulated. However, several regulatory mechanisms are still unknown or not explored. For instance, the LC3-independent mode of mitophagy is still not completely resolved, though it contributes to the lysosomal way of mitochondrial removal. Abnormal stress responses are frequently reported in most disease conditions, they exacerbate the patient’s state of condition and diminish their lifespan. Therefore, considerable studies are required to clear out the perplexity of the underlying stress mechanism to obtain a lucid view of these stress elements.
Acknowledgments
Johnson Rajasingh reports financial support was provided by The University of Tennessee Health Science Center. Johnson Rajasingh reports financial support was provided by National Institutes of Health. Johnson Rajasingh reports financial support was provided by American Heart Association.
FUNDING INFORMATION
This work was supported in part, by American Heart Association Transformational Project Award 20TPA35490215 and National Institute of Health R01 grant HL141345 to JR.
ABBREVIATIONS
- MI
Myocardial infraction
- Drp-1
Dynamin related protein-1
- Dox
Doxorubicin
- mtUPR
Mitochondrial unfolded protein response
- HGPS
Hutchinson-Gilford progeria syndrome
- ER
Endoplasmic reticulum
- ERAD
Endoplasmic reticulum associated degradation
- UPR-
Unfolded protein response
- CVDs
Cardiovascular diseases
- ROS
Reactive oxygen species
- iCM
Inner cell mass
- VEGF
Vascular endothelial growth factor
- iNOS
Induced nitric oxide synthetase
- IL
Interleukin
- DNA
Deoxyribonucleic acid
- RNA
Ribonucleic Acid
- LncRNA
Long non-coding RNA
- MALAT1
Metastasis associated lung adenocarcinoma transcript 1
- PI3K
Phosphoinositide 3-kinases
- Akt
Ak strain transforming/Protein kinase B
- FUS1
Fused in sarcoma
- Mff
Mitochondrial fission factor
- MiD
Mitochondrial dynamics protein
- OPA1
Optic atrophy-1
- Mfn
Mitofusin
- MAM
Mitochondiral associated membrane
- AMP
Adenosine monophosphate
- AMPK
AMP-activated protein kinase
- SUMO
Small ubiquitin-like Modifier
- OMM
Outer mitochondrial membrane
- IFN-γ
Interferon-gamma
- FGF
Fibroblast growth factor
- TNF-α
Tumor necrosis factor-alpha
- LCAD
Long-chain acyl-CoA dehydrogenase
- MCAD
Medium-chain acyl-CoA dehydrogenase
- CPT-1
Carnitine palmitoyltransferase 1
- ACSL-1
Acyl-CoA synthetase-1
- PTEN
Phosphatase and tensin homolog
- PINK1
PTEN-induced kinase 1
- PP2A
Protein phosphatase 2A
- TRIM21
Tripartite motif containing-2
- p62k
protein 62 kinase
- KEAP1
Kelch like ECH associated protein 1
- Nrf-2
Nuclear respiratory factor 2
- BNIP3
BCL2 interacting protein 3
- LC3
Microtubule-associated protein light chain 3
- OXPHOS
Oxidative phosphorylation
- CHOP
CCAAT-enhancer-binding protein homologous protein
- CLPP
Caseinolytic mitochondrial matrix peptidase proteolytic subunit
- LONP1
Lon protease-1
- ATFS-1
Activating transcription factor associated with stress—1
- MTS
mitochondrial targeting sequence
- NLS
Nuclear localization signals
- ISR
Integrated stress response
- PERK
RNA-dependent protein kinase (PKR)-like ER kinase
- PKR
Protein kinase RNA
- GCN2
General controlled nonrepressed-2
- HRI
Heme-regulated inhibitor
- eIF2α
Eukaryotic initiation factor-2α
- mt
Mitochondira
- SIRT
Sirtuin
- SKN-1
Skinhead-1
- PDI
Protein disulfide isomerase
- EDEM
ERAD-enhancing α-mannosidase-like proteins
- SERCA
Sarco/endoplasmic reticulum Ca2+-ATPase
- STIM1
Stromal interaction molecule 1
- IR
Ischemic- reperfusion
- XBP-1
X-box binding protein-1
- MANF
Mesencephalic Astrocyte-Derived Neurotrophic Factor
- Bcl2
B-cell lymphoma 2
- TXNIP
Thioredoxin-interacting protein
- Pak2
p21-activated kinase-2
- ERK
Extracellular signal-regulated kinase
- mPTP
Mitochondrial permeability transition pore
- TFEB
Transcription factor EB
- PGC-1α
Peroxisome proliferator-activated receptor-gamma coactivator-1α
- Bip
Binding immunoglobin protein
- FIH
Factor inhibiting HIF
- HIF
Hypoxia inducing factor
- PHD
Prolyl-4-hydroxylases
- pVHL
Von Hippel–Lindau protein
- ART
Aryl hydrocarbon receptor nuclear translocator
- ATP
Adenosine triphosphate
- CoQ
Coenzyme Q
- ETC
electron transport chain
- PPARα
Peroxisome proliferator activated receptor-α
- PERM1
PGC-1 and ERR-induced Regulator in Muscle 1
- ERR
Estrogen related receptor
- CNH
Chronic Continuous Normobaric Hypoxia
- IH
Short-term intermittent hypoxia
- SOD
Superoxide dismutase
- GPx-
Glutathione peroxidase
- G6PDH
Glucose-6-phosphate dehydrogenase
- AREG
Amphiregulin
- EPO
Erythropoietin
- DDIT4
DNA damage-inducible transcript 3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit author statement
Thannavan Ulaganathan: conceptualization and literature collection, original draft preparation; Selene Perales: Original draft preparation, reviewing, and editing; Saiprahalad Mani: conceptualization and literature collection; Boula A Baskhairoun: conceptualization and literature collection, Johnson Rajasingh: Supervision, Writing-Reviewing and Editing.
Declaration of interests
☒ The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
REFERENCES
- Abdelwahid E, Stulpinas A, Kalvelyte A, 2017. Effective Agents Targeting the Mitochondria and Apoptosis to Protect the Heart. Curr Pharm Des 23(8), 1153–1166. [DOI] [PubMed] [Google Scholar]
- Adaniya SM, J, O.U., Cypress MW, Kusakari Y, Jhun BS, 2019. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am J Physiol Cell Physiol 316(5), C583–c604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar M, González-Candia A, Rodríguez J, Carrasco-Pozo C, Cañas D, García-Herrera C, Herrera EA, Castillo RL, 2018. Mechanisms of Cardiovascular Protection Associated with Intermittent Hypobaric Hypoxia Exposure in a Rat Model: Role of Oxidative Stress. Int J Mol Sci 19(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Yacoub N, Colak D, Mahmoud SA, Hammonds M, Muhammed K, Al-Harazi O, Assiri AM, Al-Buraiki J, Al-Habeeb W, Poizat C, 2021. Mutation in FBXO32 causes dilated cardiomyopathy through up-regulation of ER-stress mediated apoptosis. Commun Biol 4(1), 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arystarkhova E, Ozelius LJ, Brashear A, Sweadner KJ, 2021. Misfolding, altered membrane distributions, and the unfolded protein response contribute to pathogenicity differences in Na,K-ATPase ATP1A3 mutations. J Biol Chem 296, 100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A, 2013. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41(16), 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoszewska S, Collawn JF, 2020. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol Biol Lett 25, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoszewska S, Collawn JF, Bartoszewski R, 2022. The Role of the Hypoxia-Related Unfolded Protein Response (UPR) in the Tumor Microenvironment. Cancers (Basel) 14(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekhite M, González-Delgado A, Hübner S, Haxhikadrija P, Kretzschmar T, Müller T, Wu JMF, Bekfani T, Franz M, Wartenberg M, Gräler M, Greber B, Schulze PC, 2021. The role of ceramide accumulation in human induced pluripotent stem cell-derived cardiomyocytes on mitochondrial oxidative stress and mitophagy. Free Radic Biol Med 167, 66–80. [DOI] [PubMed] [Google Scholar]
- Beretta M, Santos CX, Molenaar C, Hafstad AD, Miller CC, Revazian A, Betteridge K, Schroder K, Streckfuss-Bomeke K, Doroshow JH, Fleck RA, Su TP, Belousov VV, Parsons M, Shah AM, 2020. Nox4 regulates InsP(3) receptor-dependent Ca(2+) release into mitochondria to promote cell survival. EMBO J 39(19), e103530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D, 2000. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2(6), 326–332. [DOI] [PubMed] [Google Scholar]
- Binder P, Nguyen B, Collins L, Zi M, Liu W, Christou F, Luo X, Hille SS, Frey N, Cartwright EJ, Chernoff J, Müller OJ, Guan K, Wang X, 2022. Pak2 Regulation of Nrf2 Serves as a Novel Signaling Nexus Linking ER Stress Response and Oxidative Stress in the Heart. Front Cardiovasc Med 9, 851419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder P, Wang S, Radu M, Zin M, Collins L, Khan S, Li Y, Sekeres K, Humphreys N, Swanton E, Reid A, Pu F, Oceandy D, Guan K, Hille SS, Frey N, Müller OJ, Cartwright EJ, Chernoff J, Wang X, Liu W, 2019. Pak2 as a Novel Therapeutic Target for Cardioprotective Endoplasmic Reticulum Stress Response. Circ Res 124(5), 696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biuomy AR, Oraby FSH, Khalifa EA, El-Sherif HA, Hussein J, Abdel-Latif Y, 2020. Hypoxia-induced oxidative stress in high altitude population: impact of coenzyme Q10 supplementation. J Complement Integr Med 18(3), 621–626. [DOI] [PubMed] [Google Scholar]
- Bozi LHM, Campos JC, Gross ER, Ferreira JCB, 2019. Mitochondrial Unfolded Protein Response (UPR(mt)) Activation in Cardiac Diseases: Opportunities and Challenges. J Am Coll Cardiol 74(7), 1011–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun RJ, Westermann B, 2017. With the Help of MOM: Mitochondrial Contributions to Cellular Quality Control. Trends Cell Biol 27(6), 441–452. [DOI] [PubMed] [Google Scholar]
- Cai X, Zhou Z, Zhu J, Liao Q, Zhang D, Liu X, Wang J, Ouyang G, Xiao W, 2020. Zebrafish Hif3alpha modulates erythropoiesis via regulation of gata1 to facilitate hypoxia tolerance. Development 147(22). [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D, 2002. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415(6867), 92–96. [DOI] [PubMed] [Google Scholar]
- Chang J-C, Lien C-F, Lee W-S, Chang H-R, Hsu Y-C, Luo Y-P, Jeng J-R, Hsieh J-C, Yang K-T, 2019. Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca(2+) Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species. Cells 8(6), 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Luo W, Zhang W, Chu H, Wang J, Dai X, Cheng Y, Zhu T, Chao J, 2020. circDLPAG4/HECTD1 mediates ischaemia/reperfusion injury in endothelial cells via ER stress. RNA Biol 17(2), 240–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LT, Lin CT, Lin LY, Hsu JM, Wu YC, Pan CL, 2021. Neuronal mitochondrial dynamics coordinate systemic mitochondrial morphology and stress response to confer pathogen resistance in C. elegans. Dev Cell 56(12), 1770–1785 e1712. [DOI] [PubMed] [Google Scholar]
- Chen W, Wang J, Wang X, Chang P, Liang M, 2022. Knockdown of hypoxia-inducible factor 1-alpha (HIF1α) interferes with angiopoietin-like protein 2 (ANGPTL2) to attenuate high glucose-triggered hypoxia/reoxygenation injury in cardiomyocytes. Bioengineered 13(1), 1476–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Hazen BC, Russell AP, Kralli A, 2013. Peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1)- and estrogen-related receptor (ERR)-induced regulator in muscle 1 (Perm1) is a tissue-specific regulator of oxidative capacity in skeletal muscle cells. J Biol Chem 288(35), 25207–25218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Tachibana S, Lam K, Arita Y, Khosrowjerdi S, Zhang O, Liang A, Li R, Andreyev A, Murphy AN, Ross RS, 2021. Perm1 promotes cardiomyocyte mitochondrial biogenesis and protects against hypoxia/reoxygenation-induced damage in mice. J Biol Chem 297(1), 100825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MA, Abd Jamil AH, Heather LC, Murray AJ, Sutton ER, Slingo M, Sebag-Montefiore L, Tan SC, Aksentijević D, Gildea OS, Stuckey DJ, Yeoh KK, Carr CA, Evans RD, Aasum E, Schofield CJ, Ratcliffe PJ, Neubauer S, Robbins PA, Clarke K, 2016. On the pivotal role of PPARα in adaptation of the heart to hypoxia and why fat in the diet increases hypoxic injury. Faseb j 30(8), 2684–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins HE, Pat BM, Zou L, Litovsky SH, Wende AR, Young ME, Chatham JC, 2019. Novel role of the ER/SR Ca(2+) sensor STIM1 in the regulation of cardiac metabolism. Am J Physiol Heart Circ Physiol 316(5), H1014–h1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz-Ruiz P, Hernando-Rodriguez B, Perez-Jimenez MM, Rodriguez-Palero MJ, Martinez-Bueno MD, Pla A, Gatsi R, Artal-Sanz M, 2021. Prohibitin depletion extends lifespan of a TORC2/SGK-1 mutant through autophagy and the mitochondrial UPR. Aging Cell 20(5), e13359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbrel E, Soumare A, Naguez A, Label R, Bernard O, Bruhat A, Fafournoux P, Tremblais G, Marchant D, Gille T, Bernaudin JF, Callard P, Kambouchner M, Martinod E, Valeyre D, Uzunhan Y, Planès C, Boncoeur E, 2018. HIF-1α triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci Rep 8(1), 17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC, 2010. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem 285(43), 33165–33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietl A, Maack C, 2017. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr Heart Fail Rep 14(4), 338–349. [DOI] [PubMed] [Google Scholar]
- Dorn GW 2nd, Vega RB, Kelly DP, 2015. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29(19), 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP, 2003. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med 198(5), 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubank TD, Roda JM, Liu H, O’Neil T, Marsh CB, 2011. Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood 117(1), 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evinova A, Hatokova Z, Tatarkova Z, Brodnanova M, Dibdiakova K, Racay P, 2022. Endoplasmic reticulum stress induces mitochondrial dysfunction but not mitochondrial unfolded protein response in SH-SY5Y cells. Mol Cell Biochem 477(3), 965–975. [DOI] [PubMed] [Google Scholar]
- Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F, 2019. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116(7), 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Panariti A, Miserocchi G, Rocchetti M, Buoli Comani G, Rivolta I, Bishop DJ, 2018. Tissue specificity of mitochondrial adaptations in rats after 4 weeks of normobaric hypoxia. European Journal of Applied Physiology 118(8), 1641–1652. [DOI] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O, Samali A, 2010. Cellular Stress Responses: Cell Survival and Cell Death. International Journal of Cell Biology 2010, 214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge RL, Simonson TS, Cooksey RC, Tanna U, Qin G, Huff CD, Witherspoon DJ, Xing J, Zhengzhong B, Prchal JT, Jorde LB, McClain DA, 2012. Metabolic insight into mechanisms of high-altitude adaptation in Tibetans. Mol Genet Metab 106(2), 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh MC, Zhang DL, Ollivierre WH, Noguchi A, Springer DA, Linehan WM, Rouault TA, 2021. Therapeutic inhibition of HIF-2α reverses polycythemia and pulmonary hypertension in murine models of human diseases. Blood 137(18), 2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham AK, Munro S, 2016. Finding the Golgi: Golgin Coiled-Coil Proteins Show the Way. Trends Cell Biol 26(6), 399–408. [DOI] [PubMed] [Google Scholar]
- Goodpaster BH, Sparks LM, 2017. Metabolic Flexibility in Health and Disease. Cell Metab 25(5), 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan G, Yang L, Huang W, Zhang J, Zhang P, Yu H, Liu S, Gu X, 2019. Mechanism of interactions between endoplasmic reticulum stress and autophagy in hypoxia/reoxygenation‑induced injury of H9c2 cardiomyocytes. Molecular Medicine Reports 20(1), 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M, 2020. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579(7799), 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haileselassie B, Mukherjee R, Joshi AU, Napier BA, Massis LM, Ostberg NP, Queliconi BB, Monack D, Bernstein D, Mochly-Rosen D, 2019. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J Mol Cell Cardiol 130, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR, 2009. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138(3), 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D, 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397(6716), 271–274. [DOI] [PubMed] [Google Scholar]
- He F, Ru X, Wen T, 2020. NRF2, a Transcription Factor for Stress Response and Beyond. Int J Mol Sci 21(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heather LC, Cole MA, Tan JJ, Ambrose LJ, Pope S, Abd-Jamil AH, Carter EE, Dodd MS, Yeoh KK, Schofield CJ, Clarke K, 2012. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol 107(3), 268. [DOI] [PubMed] [Google Scholar]
- Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, Yu QC, Solomides CC, Morrisey EE, Khurana TS, Christofidou-Solomidou M, Simon MC, 2010. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest 120(3), 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnatiuk AP, Ong SG, Olea FD, Locatelli P, Riegler J, Lee WH, Jen CH, De Lorenzi A, Giménez CS, Laguens R, Wu JC, Crottogini A, 2016. Allogeneic Mesenchymal Stromal Cells Overexpressing Mutant Human Hypoxia-Inducible Factor 1-α (HIF1-α) in an Ovine Model of Acute Myocardial Infarction. J Am Heart Assoc 5(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JE, Stone CK, Clark CM, Brown WD, Nickles RJ, Stanley C, Hochachka PW, 1995. Enhanced cardiac metabolism of plasma glucose in high-altitude natives: adaptation against chronic hypoxia. J Appl Physiol (1985) 79(1), 222–228. [DOI] [PubMed] [Google Scholar]
- Hollien J, Weissman JS, 2006. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313(5783), 104–107. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Okawa Y, Ariyoshi M, Kaimoto S, Uchihashi M, Fukai K, Iwai-Kanai E, Matoba S, 2014. Oxidative post-translational modifications develop LONP1 dysfunction in pressure overload heart failure. Circ Heart Fail 7(3), 500–509. [DOI] [PubMed] [Google Scholar]
- Hou K, Shen J, Yan J, Zhai C, Zhang J, Pan JA, Zhang Y, Jiang Y, Wang Y, Lin RZ, Cong H, Gao S, Zong WX, 2021. Loss of TRIM21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. EBioMedicine 69, 103456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CL, Huang HS, Chen KC, Saka T, Chiang CY, Chung LWK, Sung SY, 2020. A Novel Salicylanilide Derivative Induces Autophagy Cell Death in Castration-Resistant Prostate Cancer via ER Stress-Activated PERK Signaling Pathway. Mol Cancer Ther 19(1), 101–111. [DOI] [PubMed] [Google Scholar]
- Hu Q, Zhang H, Gutierrez Cortes N, Wu D, Wang P, Zhang J, Mattison JA, Smith E, Bettcher LF, Wang M, Lakatta EG, Sheu SS, Wang W, 2020. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ Res 126(4), 456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Qi L, 2018. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem Sci 43(8), 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H, 2014. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 124(2), 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS, 2002. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med 165(5), 670–676. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr., 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292(5516), 464–468. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ, 2001. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292(5516), 468–472. [DOI] [PubMed] [Google Scholar]
- Jaskiewicz M, Moszynska A, Serocki M, Króliczewski J, Bartoszewska S, Collawn JF, Bartoszewski R, 2022. Hypoxia-inducible factor (HIF)-3a2 serves as an endothelial cell fate executor during chronic hypoxia. Excli j 21, 454–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhun BS, O-Uchi J, Adaniya SM, Cypress MW, Yoon Y, 2018. Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology. Antioxidants 7(12), 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji CH, Kim HY, Heo AJ, Lee MJ, Park DY, Kim DH, Kim BY, Kwon YT, 2020. Regulation of reticulophagy by the N-degron pathway. Autophagy 16(2), 373–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji CH, Kim HY, Heo AJ, Lee SH, Lee MJ, Kim SB, Srinivasrao G, Mun SR, Cha-Molstad H, Ciechanover A, Choi CY, Lee HG, Kim BY, Kwon YT, 2019. The N-Degron Pathway Mediates ER-phagy. Mol Cell 75(5), 1058–1072.e1059. [DOI] [PubMed] [Google Scholar]
- Ji H, Wang J, Muid D, Song W, Jiang Y, Zhou H, 2022. FUNDC1 activates the mitochondrial unfolded protein response to preserve mitochondrial quality control in cardiac ischemia/reperfusion injury. Cell Signal 92, 110249. [DOI] [PubMed] [Google Scholar]
- Jin JY, Wei XX, Zhi XL, Wang XH, Meng D, 2021. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin 42(5), 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurivich DA, Zhou X, 2007. Stress: Physiological, in: Birren JE (Ed.) Encyclopedia of Gerontology (Second Edition). Elsevier, New York, pp. 559–565. [Google Scholar]
- Kang SU, Kim DH, Lee YS, Huang M, Byeon HK, Lee SH, Baek SJ, Kim CH, 2021. DIM-C-pPhtBu induces lysosomal dysfunction and unfolded protein response - mediated cell death via excessive mitophagy. Cancer Lett 504, 23–36. [DOI] [PubMed] [Google Scholar]
- Kaspar S, Oertlin C, Szczepanowska K, Kukat A, Senft K, Lucas C, Brodesser S, Hatzoglou M, Larsson O, Topisirovic I, Trifunovic A, 2021. Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci Adv 7(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson AK, Williams AL, Boisvert WA, Shohet RV, 2021. HIF in the heart: development, metabolism, ischemia, and atherosclerosis. J Clin Invest 131(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen M, Lee JW, Seo SW, Brodsky KS, Kreth S, Yang IV, Buttrick PM, Eckle T, Eltzschig HK, 2018. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat Commun 9(1), 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP, 2006. HIF-dependent induction of adenosine A2B receptor in hypoxia. Faseb j 20(13), 2242–2250. [DOI] [PubMed] [Google Scholar]
- Korennykh A, Walter P, 2012. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol 28, 251–277. [DOI] [PubMed] [Google Scholar]
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T, 2007. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14(2), 230–239. [DOI] [PubMed] [Google Scholar]
- Kuo CY, Chiu YC, Lee AY, Hwang TL, 2015. Mitochondrial Lon protease controls ROS-dependent apoptosis in cardiomyocyte under hypoxia. Mitochondrion 23, 7–16. [DOI] [PubMed] [Google Scholar]
- Lee JW, Ko J, Ju C, Eltzschig HK, 2019. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med 51(6), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Koeppen M, Seo SW, Bowser JL, Yuan X, Li J, Sibilia M, Ambardekar AV, Zhang X, Eckle T, Yoo SH, Eltzschig HK, 2020. Transcription-independent Induction of ERBB1 through Hypoxia-inducible Factor 2A Provides Cardioprotection during Ischemia and Reperfusion. Anesthesiology 132(4), 763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Strisovsky K, 2021. Maintenance of organellar protein homeostasis by ER-associated degradation and related mechanisms. Mol Cell 81(12), 2507–2519. [DOI] [PubMed] [Google Scholar]
- Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR, 2012. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16(2), 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Li X, Huang J, Xu C, Liang Q, Ren K, Bai A, Lu C, Qian R, Sun N, 2020. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell 11(9), 661–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C, 2004. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest 114(8), 1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C, 2007. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest 117(11), 3211–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee B, Lee AS, 2006. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 281(11), 7260–7270. [DOI] [PubMed] [Google Scholar]
- Li S, Spooner RA, Allen SC, Guise CP, Ladds G, Schnöder T, Schmitt MJ, Lord JM, Roberts LM, 2010. Folding-competent and folding-defective forms of ricin A chain have different fates after retrotranslocation from the endoplasmic reticulum. Mol Biol Cell 21(15), 2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, Torre-Amione G, Knowlton AA, 2007. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol 293(4), H2238–2247. [DOI] [PubMed] [Google Scholar]
- Lishmanov YB, Naryzhnaya NV, Tsibul’nikov SY, Wang H, Maslov LN, 2017. Role of ATP-Sensitive K+ Channels in Myocardial Infarct Size-Limiting Effect of Chronic Continuous Normobaric Hypoxia. Bulletin of Experimental Biology and Medicine 163(1), 22–24. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang S, An L, Wu J, Hu X, Lai S, Mazhar H, Zou Y, He L, Zhu H, 2019. Loss of Rubicon ameliorates doxorubicin-induced cardiotoxicity through enhancement of mitochondrial quality. Int J Cardiol 296, 129–135. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B, 2013. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 110(51), 20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Xing J, Li Y, Luo Q, Su Z, Zhang X, Zhang H, 2019. Chronic hypoxia-induced Cirbp hypermethylation attenuates hypothermic cardioprotection via down-regulation of ubiquinone biosynthesis. Sci Transl Med 11(489). [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Witters LA, Itoi T, Barr R, Barr A, 1994. Acetyl-CoA carboxylase involvement in the rapid maturation of fatty acid oxidation in the newborn rabbit heart. J Biol Chem 269(41), 25871–25878. [PubMed] [Google Scholar]
- López I, Tournillon AS, Prado Martins R, Karakostis K, Malbert-Colas L, Nylander K, Fåhraeus R, 2017. p53-mediated suppression of BiP triggers BIK-induced apoptosis during prolonged endoplasmic reticulum stress. Cell Death Differ 24(10), 1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losón OC, Song Z, Chen H, Chan DC, 2013. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24(5), 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H-T, Feng R-Q, Tang J-K, Zhou J-J, Gao F, Ren J, 2020. CaMKII/calpain interaction mediates ischemia/reperfusion injury in isolated rat hearts. Cell Death & Disease 11(5), 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Dong Z, Liu J, Ma L, Sun X, Gao R, Pan L, Zhang J, A, D., An J, Hu K, Sun A, Ge J, 2021. β-Hydroxybutyrate Exacerbates Hypoxic Injury by Inhibiting HIF-1α-Dependent Glycolysis in Cardiomyocytes-Adding Fuel to the Fire? Cardiovasc Drugs Ther [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC, 2010. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40(2), 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinde AO, Kantor PF, Lopaschuk GD, 1998. Maturation of fatty acid and carbohydrate metabolism in the newborn heart. Mol Cell Biochem 188(1–2), 49–56. [PubMed] [Google Scholar]
- Malli R, Graier WF, 2019. IRE1α modulates ER and mitochondria crosstalk. Nat Cell Biol 21(6), 667–668. [DOI] [PubMed] [Google Scholar]
- Mansor LS, Mehta K, Aksentijevic D, Carr CA, Lund T, Cole MA, Le Page L, Sousa Fialho M.d.L., Shattock MJ, Aasum E, Clarke K, Tyler DJ, Heather LC, 2016. Increased oxidative metabolism following hypoxia in the type 2 diabetic heart, despite normal hypoxia signaling and metabolic adaptation. The Journal of physiology 594(2), 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao S, Tian S, Luo X, Zhou M, Cao Z, Li J, 2021. Overexpression of PLK1 relieved the myocardial ischemia-reperfusion injury of rats through inducing the mitophagy and regulating the p-AMPK/FUNDC1 axis. Bioengineered 12(1), 2676–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D, 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18(24), 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariángelo JIE, Valverde CA, Vittone L, Said M, Mundiña-Weilenmann C, 2022. Pharmacological inhibition of translocon is sufficient to alleviate endoplasmic reticulum stress and improve Ca(2+) handling and contractile recovery of stunned myocardium. Eur J Pharmacol 914, 174665. [DOI] [PubMed] [Google Scholar]
- Marin JM, Carrizo SJ, Vicente E, Agusti AG, 2005. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365(9464), 1046–1053. [DOI] [PubMed] [Google Scholar]
- Maslov LN, Naryzhnaia NV, Tsibulnikov SY, Kolar F, Zhang Y, Wang H, Gusakova AM, Lishmanov YB, 2013. Role of endogenous opioid peptides in the infarct size-limiting effect of adaptation to chronic continuous hypoxia. Life Sci 93(9–11), 373–379. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ, 2001. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21(4), 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez-Montes I, Escobar B, Gomez MJ, Albendea-Gomez T, Palacios B, Bonzon-Kulichenko E, Izquierdo-Garcia JL, Alonso AV, Ferrarini A, Jimenez-Borreguero LJ, Ruiz-Cabello J, Vázquez J, Martin-Puig S, 2021. Activation of amino acid metabolic program in cardiac HIF1-alpha-deficient mice. iScience 24(2), 102124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel S, Canonne M, Arnould T, Renard P, 2015. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 21, 58–68. [DOI] [PubMed] [Google Scholar]
- Morales PE, Arias-Durán C, Ávalos-Guajardo Y, Aedo G, Verdejo HE, Parra V, Lavandero S, 2020. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med 71, 100822. [DOI] [PubMed] [Google Scholar]
- Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S, 2014. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem 155(5), 273–279. [DOI] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM, 2012. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337(6094), 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckar J, Szarszoi O, Herget J, Ostadal B, Kolár F, 2003. Cardioprotective effect of chronic hypoxia is blunted by concomitant hypercapnia. Physiological research / Academia Scientiarum Bohemoslovaca 52, 171–175. [PubMed] [Google Scholar]
- Niwa K, 2019. Metabolic Syndrome in Adult Congenital Heart Disease. Korean Circ J 49(8), 691–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K, 2021. Molecular mechanisms and physiological functions of mitophagy. Embo j 40(3), e104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterrieder A, Sparkes IA, Botchway SW, Ward A, Ketelaar T, de Ruijter N, Hawes C, 2017. Stacks off tracks: a role for the golgin AtCASP in plant endoplasmic reticulum-Golgi apparatus tethering. J Exp Bot 68(13), 3339–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota A, Ishihara T, Ishihara N, 2020. Mitochondrial nucleoid morphology and respiratory function are altered in Drp1-deficient HeLa cells. The Journal of Biochemistry 167(3), 287–294. [DOI] [PubMed] [Google Scholar]
- Ou W, Liang Y, Qin Y, Wu W, Xie M, Zhang Y, Zhang Y, Ji L, Yu H, Li T, 2021. Hypoxic acclimation improves cardiac redox homeostasis and protects heart against ischemia-reperfusion injury through upregulation of O-GlcNAcylation. Redox biology 43, 101994–101994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki CK, 2007. Endothelium-Intrinsic Requirement for Hif-2α During Vascular DevelopmentDuan L-J, Zhang-Benoit Y, Fong GH (Univ of Connecticut, Farmington) Circulation 111:2227–2232, 2005§. Yearbook of Vascular Surgery 2007, 35–36. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM, 2016. The integrated stress response. EMBO Rep 17(10), 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N, 2015. Coupling mitogenesis and mitophagy for longevity. Autophagy 11(8), 1428–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Hu Z, Shao Z, Ning Y, 2022. Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) overexpression alleviates endoplasmic reticulum stress after acute kidney injury. Ren Fail 44(1), 358–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Z, Liu Y, Liu S, Jin W, Luo Y, Sun M, Duan Y, Ajoolabady A, Sowers JR, Fang Y, Cao F, Xu H, Bi Y, Wang S, Ren J, 2021. FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metabolism 122, 154840. [DOI] [PubMed] [Google Scholar]
- Prokudina ES, Naryzhnaya NV, Mukhomedzyanov AV, Gorbunov AS, Zhang Y, Jaggi AS, Tsibulnikov SY, Nesterov EA, Lishmanov YB, Suleiman MS, Oeltgen PR, Maslov LN, 2019. Effect of Chronic Continuous Normobaric Hypoxia on Functional State of Cardiac Mitochondria and Tolerance of Isolated Rat Heart to Ischemia and Reperfusion: Role of µ and δ2 Opioid Receptors. Physiological Research, 909–920. [DOI] [PubMed] [Google Scholar]
- Prola A, Nichtova Z, Pires Da Silva J, Piquereau J, Monceaux K, Guilbert A, Gressette M, Ventura-Clapier R, Garnier A, Zahradnik I, Novotova M, Lemaire C, 2019. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc Res 115(2), 328–342. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A, 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129(7), 1337–1349. [DOI] [PubMed] [Google Scholar]
- Qi Z, Huang Z, Xie F, Chen L, 2019. Dynamin-related protein 1: A critical protein in the pathogenesis of neural system dysfunctions and neurodegenerative diseases. J Cell Physiol 234(7), 10032–10046. [DOI] [PubMed] [Google Scholar]
- Ramjiawan A, Bagchi RA, Blant A, Albak L, Cavasin MA, Horn TR, McKinsey TA, Czubryt MP, 2013. Roles of histone deacetylation and AMP kinase in regulation of cardiomyocyte PGC-1α gene expression in hypoxia. Am J Physiol Cell Physiol 304(11), C1064–1072. [DOI] [PubMed] [Google Scholar]
- Rashid H-O, Yadav RK, Kim H-R, Chae H-J, 2015. ER stress: Autophagy induction, inhibition and selection. Autophagy 11(11), 1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L, Wang Q, Chen Y, Ma Y, Wang D, 2019. Involvement of MicroRNA-133a in the Protective Effect of Hydrogen Sulfide against Ischemia/Reperfusion-Induced Endoplasmic Reticulum Stress and Cardiomyocyte Apoptosis. Pharmacology 103(1–2), 1–9. [DOI] [PubMed] [Google Scholar]
- Rodriguez DA, Zamorano S, Lisbona F, Rojas-Rivera D, Urra H, Cubillos-Ruiz JR, Armisen R, Henriquez DR, Cheng EH, Letek M, Vaisar T, Irrazabal T, Gonzalez-Billault C, Letai A, Pimentel-Muiños FX, Kroemer G, Hetz C, 2012. BH3-only proteins are part of a regulatory network that control the sustained signaling of the unfolded protein response sensor IRE1α. Embo j 31(10), 2322–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P, 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7), 519–529. [DOI] [PubMed] [Google Scholar]
- Saheki Y, De Camilli P, 2017. Endoplasmic Reticulum-Plasma Membrane Contact Sites. Annu Rev Biochem 86, 659–684. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Uchiumi T, Toshima T, Yagi M, Do Y, Hirai H, Igami K, Gotoh K, Kang D, 2020. Mitochondrial translation inhibition triggers ATF4 activation, leading to integrated stress response but not to mitochondrial unfolded protein response. Biosci Rep 40(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasi USS, Ganapathy S, Palayyan SR, Gopal RK, 2020. Mitochondria Associated Membranes (MAMs): Emerging Drug Targets for Diabetes. Curr Med Chem 27(20), 3362–3385. [DOI] [PubMed] [Google Scholar]
- Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, Konig T, Kukat A, Trifunovic A, 2016. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep 17(7), 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Li R, Yu S, Zhao Q, Wang Z, Sheng H, Yang W, 2021. Activation of the ATF6 (Activating Transcription Factor 6) Signaling Pathway in Neurons Improves Outcome After Cardiac Arrest in Mice. J Am Heart Assoc 10(12), e020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Tang H, Ai W, Zeng Q, Yang H, Zhu F, Wei Y, Feng R, Wen L, Pu P, He Q, 2021. Schisandrin B Antagonizes Cardiotoxicity Induced by Pirarubicin by Inhibiting Mitochondrial Permeability Transition Pore (mPTP) Opening and Decreasing Cardiomyocyte Apoptosis. Front Pharmacol 12, 733805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyrnias I, 2021. The mitochondrial unfolded protein response and its diverse roles in cellular stress. Int J Biochem Cell Biol 133, 105934. [DOI] [PubMed] [Google Scholar]
- Smyrnias I, Gray SP, Okonko DO, Sawyer G, Zoccarato A, Catibog N, López B, González A, Ravassa S, Díez J, Shah AM, 2019. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. Journal of the American College of Cardiology 73(14), 1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sritharan S, Sivalingam N, 2021. A comprehensive review on time-tested anticancer drug doxorubicin. Life Sci 278, 119527. [DOI] [PubMed] [Google Scholar]
- Su N, Kilberg MS, 2008. C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J Biol Chem 283(50), 35106–35117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Liu Y, Zhang H, 2021. Adaptive Cardiac Metabolism Under Chronic Hypoxia: Mechanism and Clinical Implications. Front Cell Dev Biol 9, 625524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Tang X, Guo Y, Hu J, 2021. Endoplasmic reticulum composition and form: Proteins in and out. Curr Opin Cell Biol 71, 1–6. [DOI] [PubMed] [Google Scholar]
- Sun X, Pang L, Shi M, Huang J, Wang Y, 2015. HIF2α induces cardiomyogenesis via Wnt/β-catenin signaling in mouse embryonic stem cells. J Transl Med 13, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzman R, Beard JR, Boerma T, Chatterji S, 2015. Health in an ageing world—what do we know? The Lancet 385(9967), 484–486. [DOI] [PubMed] [Google Scholar]
- Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI, Imai H, Tsutsui H, 2020. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ, 1998. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev 12(12), 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolonen JP, Heikkilä M, Malinen M, Lee HM, Palvimo JJ, Wei GH, Myllyharju J, 2020. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Cell Mol Life Sci 77(18), 3627–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J, 2021. Alternative Mitophagy Protects the Heart Against Obesity-Associated Cardiomyopathy. Circ Res 129(12), 1105–1121. [DOI] [PubMed] [Google Scholar]
- Trevelin SC, Lopes LR, 2015. Protein disulfide isomerase and Nox: new partners in redox signaling. Curr Pharm Des 21(41), 5951–5963. [DOI] [PubMed] [Google Scholar]
- Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, McGlauflin R, Kenny HC, Zhang Y, Souvenir R, Hu XX, Sloan CL, Pereira RO, Lira VA, Spitzer KW, Sharp TL, Shoghi KI, Sparagna GC, Rog-Zielinska EA, Kohl P, Khalimonchuk O, Schaffer JE, Abel ED, 2018. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ Res 122(1), 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JP, Austgen K, Nishino M, Coakley KM, Hagen A, Han D, Papa FR, Oakes SA, 2008. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cell Biol 28(12), 3943–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varricchi G, Ameri P, Cadeddu C, Ghigo A, Madonna R, Marone G, Mercurio V, Monte I, Novo G, Parrella P, Pirozzi F, Pecoraro A, Spallarossa P, Zito C, Mercuro G, Pagliaro P, Tocchetti CG, 2018. Antineoplastic Drug-Induced Cardiotoxicity: A Redox Perspective. Front Physiol 9, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Li M, Saito T, Tong M, Rashed E, Mareedu S, Zhai P, Barcena C, Lopez-Otin C, Yehia G, Sadoshima J, Suzuki CK, 2019. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J Mol Cell Cardiol 128, 38–50. [DOI] [PubMed] [Google Scholar]
- Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T, 2015. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350(6265), aad0116. [DOI] [PubMed] [Google Scholar]
- Wang D, Hou C, Cao Y, Cheng Q, Zhang L, Li H, Feng L, Shen Y, 2018. XBP1 activation enhances MANF expression via binding to endoplasmic reticulum stress response elements within MANF promoter region in hepatitis B. Int J Biochem Cell Biol 99, 140–146. [DOI] [PubMed] [Google Scholar]
- Wang GL, Semenza GL, 1995. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 270(3), 1230–1237. [DOI] [PubMed] [Google Scholar]
- Wang H, Liu C, Zhao Y, Zhang W, Xu K, Li D, Zhou Y, Li H, Xiao G, Lu B, Gao G, 2020. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem Biophys Res Commun 522(4), 1063–1068. [DOI] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ, 2016. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529(7586), 326–335. [DOI] [PubMed] [Google Scholar]
- Wang S, Bian W, Zhen J, Zhao L, Chen W, 2019. Melatonin-Mediated Pak2 Activation Reduces Cardiomyocyte Death Through Suppressing Hypoxia Reoxygenation Injury-Induced Endoplasmic Reticulum Stress. J Cardiovasc Pharmacol 74(1), 20–29. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ, 2012. The impact of the unfolded protein response on human disease. J Cell Biol 197(7), 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Zhu XL, Wang F, Chen SX, Chen ZT, Qiu Q, Liu WH, Wu MX, Deng BQ, Xie Y, Mai JT, Yang Y, Wang JF, Zhang HF, Chen YX, 2021. LncRNA H19 governs mitophagy and restores mitochondrial respiration in the heart through Pink1/Parkin signaling during obesity. Cell Death Dis 12(6), 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D, 1998. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. Embo j 17(19), 5708–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Jasper H, Toan S, Muid D, Chang X, Zhou H, 2021. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol 45, 102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Lim Y, McCall MN, Huang KT, Haynes CM, Nehrke K, Brookes PS, 2019. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am J Physiol Heart Circ Physiol 317(2), H472–H478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Wu YE, Wang W, Zhang S, Liu D, Liu H, 2021. Decreased dynamin-related protein 1-related mitophagy induces myocardial apoptosis in the aging heart. Acta Biochim Biophys Sin (Shanghai) 53(10), 1354–1366. [DOI] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, Anthony TG, 2006. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 34(Pt 1), 7–11. [DOI] [PubMed] [Google Scholar]
- Wende AR, Abel ED, 2010. Lipotoxicity in the heart. Biochim Biophys Acta 1801(3), 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan RS, Kaplinskiy V, Kitsis RN, 2010. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 72, 19–44. [DOI] [PubMed] [Google Scholar]
- Wu J-W, Hu H, Li D, Ma L-K, 2021. Hypoxia-inducible factor 2-alpha-dependent induction of IL-6 protects the heart from ischemia/reperfusion injury. Aging 13(3), 3443–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, Mao X, Huang K, Xie Z, Zou MH, 2019. Hyperglycemia-Driven Inhibition of AMP-Activated Protein Kinase α2 Induces Diabetic Cardiomyopathy by Promoting Mitochondria-Associated Endoplasmic Reticulum Membranes In Vivo. Circulation 139(16), 1913–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Tang X, Lee S, Hong H, Cao X, Lau CW, Liu B, Chawla A, Ma RCW, Huang Y, Lui KO, Tian XY, 2022. Endothelial PPARδ facilitates the post-ischemic vascular repair through interaction with HIF1α. Theranostics 12(4), 1855–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HX, Cui SM, Zhang YM, Ren J, 2020. Mitochondrial Ca(2+) regulation in the etiology of heart failure: physiological and pathophysiological implications. Acta Pharmacol Sin 41(10), 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Xue RQ, Lu Y, Yong SY, Wu Q, Cui YL, Zuo XT, Yu XJ, Zhao M, Zang WJ, 2019. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc Res 115(3), 530–545. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K, 2007. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 13(3), 365–376. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K, 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107(7), 881–891. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Qian Q, Li M, Shao F, Ding WX, Lira VA, Chen SX, Sebag SC, Hotamisligil GS, Cao H, Yang L, 2021. The unfolded protein response regulates hepatic autophagy by sXBP1-mediated activation of TFEB. Autophagy 17(8), 1841–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ, 2002. A mitochondrial specific stress response in mammalian cells. Embo j 21(17), 4411–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Chen Z, Zhang L, Wang X, Dong Y, Wang J, Shao L, Tian Y, Wang Z, Zhao T, Fan G, Jia X, Gao R, Guo Y, Tong Y, Chen Y, Duan P, Li N, Suo F, Cao M, He L, Kang Y, Li S, Zhang Z, 2019. Metabolic Risk Factors and Left Ventricular Diastolic Function in Middle‑Aged Chinese Living in the Tibetan Plateau. Journal of the American Heart Association 8(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Murugan DD, Khan H, Huang Y, Cheang WS, 2021. Roles and Therapeutic Implications of Endoplasmic Reticulum Stress and Oxidative Stress in Cardiovascular Diseases. Antioxidants (Basel) 10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zozina VI, Covantev S, Goroshko OA, Krasnykh LM, Kukes VG, 2018. Coenzyme Q10 in Cardiovascular and Metabolic Diseases: Current State of the Problem. Curr Cardiol Rev 14(3), 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]