

Abstract
Tyrosyl-DNA phosphodiesterase-1 (Tdp1) is the only known enzyme to remove tyrosine from complexes in which the amino acid is linked to the 3′-end of DNA fragments. Such complexes can be produced following DNA processing by topoisomerase I, and recent studies in yeast have demonstrated the importance of TDP1 for cell survival following topoisomerase I-mediated DNA damage. In the present study, we used synthetic oligodeoxynucleotide–peptide conjugates (nucleopeptides) and recombinant yeast Tdp1 to investigate the molecular determinants for Tdp1 activity. We find that Tdp1 can process nucleopeptides with up to 13 amino acid residues but is poorly active with a 70 kDa fragment of topoisomerase I covalently linked to a suicide DNA substrate. Furthermore, Tdp1 was more effective with nucleopeptides with one to four amino acids than 15 amino acids. Tdp1 was also more effective with nucleopeptides containing 15 nt than with homolog nucleopeptides containing 4 nt. These results suggest that DNA binding contributes to the activity of Tdp1 and that Tdp1 would be most effective after topoisomerase I has been proteolyzed in vivo.
INTRODUCTION
DNA topoisomerase I is a ubiquitous enzyme which relieves DNA torsional stress. It is active during transcription, replication and chromatin changes (1,2). Eukaryotic and viral topoisomerases I are related to site-specific recombinases (also referred to as tyrosine recombinases) (1,3,4). They introduce transient DNA single-strand breaks by forming a catalytic intermediate in which a covalent bond is generated between an enzyme tyrosine residue (Tyr723 for human topoisomerase I) and the 3′-end of the broken DNA (Fig. 1A). After DNA relaxation, the break is resealed by nucleophilic attack of the 5′-end of the broken DNA, thereby liberating the intact topoisomerase I, which can then carry out additional catalytic cycles (1,2).
Figure 1.

Schematic representation of topoisomerase I cleavage intermediates and processing by Tdp1 and PNKP. (A) Reversible topoisomerase I (top1) cleavage complex. (B) Tdp1 hydrolyzes the phosphotyrosyl bond. The resulting DNA break bears a 3′-phosphate and 5′-hydroxyl. PNKP (such as T4 PNKP) has two activities: removal of the phosphate at the 3′-end and phosphorylation of the 5′-end.
A number of DNA modifications have been shown to interfere with the religation reaction and to generate irreversible topoisomerase I–DNA complexes, which are commonly referred to as ‘suicide complexes’ (5). These modifications include DNA strand breaks (6–8), carcinogenic adducts (9,10), mismatches and abasic sites (11). The anticancer drug camptothecin and its clinically used derivatives also stabilize topoisomerase I cleavage complexes and collision of replication forks with these reversible cleavage complexes converts the cleavage complexes into replication-mediated DNA double-strand breaks and topoisomerase I covalent complexes (12–15).
An enzymatic activity (16) and a gene product (17) whose function is to selectively remove 3′-phosphotyrosyl adducts were discovered recently. The gene has been named tyrosyl-DNA phosphodiesterase-1 (TDP1) and orthologs have been found in a wide variety of eukaryotic species, including yeast and human cells (17). Tdp1 is a member of the phospholipase D superfamily (18). TDP1 null mutants in the budding yeast Saccharomyces cerevisiae are hypersensitive to a mutant topoisomerase I that induces high levels of cleavage complexes (19) and to camptothecin in the absence of Rad9 (17), indicating that Tdp1 acts to remove topoisomerase I–DNA complexes. Figure 1B shows the reactions carried out by Tdp1. Recent biochemical studies performed with oligodeoxynucleotides containing a 3′-linked tyrosine demonstrated that Tdp1 is markedly more active on substrates where the scissile (tyrosyl–DNA) bond is located in a tailed duplex rather than in a nicked duplex DNA segment (19), which is consistent with the possibility that Tdp1 repairs suicide topoisomerase I complexes, such as the replication-mediated double-strand breaks generated by replication fork collisions (15).
The aim of the present study was to further define the biochemical requirements for Tdp1 activity using synthetic nucleopeptides. We investigated the influence of the size of the polypeptide chain and of the length of the oligonucleotide on Tdp1 activity.
MATERIALS AND METHODS
Fmoc-l-amino acids and Boc-l-amino acids (where Fmoc is 9-fluorenylmethoxycarbonyl and Boc is t-butoxycarbonyl) and p-methylbenzhydrylamine resin were obtained from Bachem (Bubendorf, Switzerland) and Novabiochem (Bad Soden, Germany). Boc-Thr(Ac)-OH (where Ac is acetyl) and Boc-Arg(Fmoc)2-OH were obtained as previously described (20). The 3′-phosphoramidite derivatives of 4,4′-dimethoxytrityl (DMT)-dABz (where Bz is benzoyl), DMT-dCBz, DMT-dGiBu (where iBu is isobutyryl) and DMT-T were purchased from Glen Research Corp. (Sterling, VA). Solid phase syntheses were performed manually in a polypropylene syringe fitted with a polyethylene disc (peptides) or in a 380B Applied Biosystems synthesizer (oligonucleotide fragments). Reversed phase HPLC analyses and purifications were performed on either a Nucleosil C18 column (250 × 4.6 mm, 10 µm, 1 ml/min) or a Kromasil C18 column (250 × 10 mm, 10 µm, 3 ml/min) using a linear gradient of 0.01 M aqueous ammonium acetate and acetonitrile (ACN)/H2O (1:1). Octadecylsyloxane-Vydac resin was used for purification by reversed phase MPLC. Electrophoreses were run under denaturing conditions (7 M urea) on 20% polyacrylamide gels, at 500–750 V for 3–4 h. On the analytical scale, nucleopeptides were detected by reaction with Stains-all dye (Sigma, St Louis, MO). In preparative gels, products were visualized by UV-shadowing and isolated by the crush-and-soak method (21). The target products were desalted by elution through Sep-Pak cartridges (Phase Separations Ltd, Flintshire, UK). Amino acid analyses were performed on a Beckman System 6300 analyzer (Fullerton, CA). Hydrolyses of peptide resins were carried out in 12 M HCl/propionic acid (1:1) either at 150°C for 60–90 min or at 110°C for 24 h (serine and threonine are to some extent degraded under these hydrolysis conditions; a crystal of phenol was added to reduce tyrosine degradation; asparagine and glutamine are converted to aspartic and glutamic acid, respectively). Digestions with snake venom phosphodiesterase (EC 3.1.4.1; Sigma) and alkaline phosphatase (EC 3.1.3.1; Sigma) were carried out on 0.5–1.0 OD260 units of nucleopeptides. The digestion mixtures were analyzed by reversed phase HPLC (10 min 5% B, linear gradient from 5 to 30% B over 10 min). Matrix assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometric analysis was carried out using a Voyager-DE RP instrument (Perseptive Biosystems, Foster City, CA). Calculated mass values for neutral compounds (M) are indicated in all cases. Synthetic oligonucleotides and 3′-tyrosyl-oligonucleotide H-Tyr(p3′-dTTTCAGAAAATCTAG)-OH (N15Y) were purchased from Midland Certified Reagents Co. (Midland, TX). Purified human topoisomerase was obtained as described previously (22). Saccharomyces cerevisiae Tdp1 was bacterially expressed as an N-terminal his-tagged fusion as described (15). Tdp1 protein was purified with a Novagen His-Bind purification kit according to the instructions (Novagen, Madison, WI).
Preparation of nucleopeptides
Incorporation of the internal reference amino acid (leucine or phenylalanine) onto p-methylbenzhydrylamine resin (f = 0.5–0.6 mmol/g) was carried out by reaction with 0.5 equivalents of both the Fmoc-amino acid derivative and N,N′-dicyclohexylcarbodiimide (DCC) (15 min) in order to achieve partial acylation of polymeric amine groups. The substitution degree was determined on an aliquot by UV spectrophotometric quantitation of the amount of N-(9-fluorenylmethyl)piperidine formed upon deprotection with piperidine and, if suitable for oligonucleotide synthesis (23), unreacted amine groups were capped by reaction with Ac2O and N-ethyl-N,N-diisopropylamine (DIEA) (substitution degree values ranged from 0.17 to 0.19 mmol/g). The Fmoc group was eliminated and the bifunctional linker N-2-(9-hydroxymethylfluorenyl) succinamic acid (24,25) coupled in the presence of DCC (3 equiv of each reagent, 2–4 h). Hydroxyl groups were esterified by reaction with the C-terminal Boc-amino acid and DCC (10 equiv) in the presence of 0.5 equiv 4-N,N-dimethylaminopyridine for 2 h. The coupling yield was determined from the relative proportion [C-terminal residue]/[internal reference amino acid] after acid hydrolysis and amino acid analysis (typical values 0.91–0.95) and unreacted hydroxyl groups were capped by reaction with acetic anhydride and pyridine.
The remaining amino acids of the peptide sequence were incorporated following standard procedures for solid phase peptide assembly (Boc removal by reaction twice with 30% trifluoroacetic acid in dichloromethane for 15 min, neutralization with 5% DIEA in dichloromethane for 3 + 7 min and coupling with 3 equiv Boc-amino acid and DCC for 1–1.5 h), with addition of an equimolar amount of 1-hydroxybenzotriazole in incorporation of the arginine derivative. To prevent diketopiperazine formation, the Boc group of Boc-Asp(OFm)-Pro resin (where Fm is 9-fluorenylmethyl) was removed as usual and Boc-leucine (5 equiv) was incorporated onto the trifluoroacetate salt of the dipeptide resin by reaction with 7-azabenzotriazol-1-yloxytris(pyrrolidino)phosphonium-hexafluorophosphate (5 equiv) and DIEA (10 equiv). Acylation of the N-terminal amine group was carried out by reaction with acetic anhydride, when required, otherwise the N-terminal amino acid was incorporated as the Fmoc derivative.
Oligonucleotide elongation at the tyrosine side chain of peptide resins (20) was carried out following standard phosphite triester procedures with minor modifications that render this methodology compatible with the use of polystyrene supports (23). Aliquots of 0.2 M solutions of commercially available 3′-phosphoramidite derivatives [5′-DMT-N-P(OCNE)NiPr2, N = T, ABz, CBz or GiBu; where CNE is 2-cyanoethyl] in anhydrous dichloromethane and 0.8 M solutions of tetrazole in anhydrous tetrahydrofuran were used for the coupling step (15 min). Phosphite to phosphate oxidation was accomplished with 1 M t-BuOOH treatment. Coupling yields for incorporation of the first nucleoside derivative typically ranged between 90 and 99% and the average coupling yield was 97–98%. The 5′-DMT group was removed from the nucleopeptide resin.
Nucleopeptides were deprotected and detached from the solid support by overnight treatment with concentrated aqueous ammonia/dioxane (1:1) at 55°C.
H-Ile-Ala-Leu-Gly-Thr-Ser-Lys-Leu-Asn-Tyr(p3′-dTTTCAGAAAATCTAG)-Leu-Asp-Pro-OH (N15P13). Internal reference amino acid: Phe. Amino acid analysis of peptide resin: Asp 2.15, Thr 0.65, Ser 0.33, Gly 0.87, Ala 0.95, Ile 0.60, Leu 1.97, Tyr 1.09, Lys 1.02, Pro 1.04. Purification: preparative polyacrylamide gel electrophoresis (PAGE) and reversed phase HPLC, linear gradient from 5 to 35% B over 30 min. Nucleoside composition: dC 3.05, dG 2.04, dT 4.23, dA 5.77. Calculated mass (M): 6038.54; MALDI-TOF mass spectrometry (MALDI-TOF MS) [negative mode, linear, trihydroxyacetophenone (THAP)]: m/z 6036.3, [M-H]–.
Ac-Tyr(p3 ′-dTTTCAGAAAATCTAG)-Ala-Phe-Gly-OH (N15P4). Internal reference amino acid: Leu. Amino acid analysis of peptide resin: Gly 1.02, Ala 0.99, Tyr 1.00, Phe 0.99. Purification: preparative PAGE. Nucleoside composition: dC 2.43, dG 1.92, dT 4.87, dA 5.78. Calculated mass (M): 5132.99; MALDI-TOF MS (negative mode, linear, THAP): m/z 5135.1, [M-H]–.
H-Ile-Ala-Leu-Gly-Thr-Ser-Lys-Leu-Asn-Tyr(p3 ′-dACGT)-Leu-Asp-Pro-OH (N4P13). Internal reference amino acid: Phe. Amino acid analysis of peptide resin: Asp 2.15, Thr 0.65, Ser 0.33, Gly 0.87, Ala 0.95, Ile 0.60, Leu 1.97, Tyr 1.09, Lys 1.02, Pro 1.04. Purification: reversed phase HPLC, linear gradient from 5 to 35% B over 30 min. Nucleoside composition: dC 1.25, dG 1.19, dT 1.06, dA 0.51. Calculated mass (M): 2638.97; MALDI-TOF MS (negative mode, reflectron, THAP): m/z 2639.1, [M-H]–.
Ac-Tyr(p3 ′-dACGT)-Ala-Phe-Gly-OH (N4P4). Internal reference amino acid: Leu. Amino acid analysis of peptide resin: Gly 1.02, Ala 0.99, Tyr 1.00, Phe 0.99. Purification: reversed phase (C18, Vydac) medium pressure liquid chromatography, A = 0.05 M ammonium acetate, B = ACN/H2O (1:1), gradient from 0 to 30% B, 600 ml of each solvent. Nucleoside composition: dC 0.99, dG 1.08, dT 0.97, dA 0.96. Calculated mass (M): 1733.41; MALDI-TOF MS (negative mode, reflectron, THAP): m/z 1732.8, [M-H]–.
Ac-Tyr(p3 ′-dTTTCAGAAAATCTAG)-Leu-Asp-Pro-Arg-Ile-Thr-Val-OH (N15R8). Internal reference amino acid: Phe. Amino acid analysis of peptide resin: Asp 1.00, Thr 0.39, Val 1.65, Ile 0.82, Leu 1.01, Tyr 0.99, Arg 1.06, Pro 0.84. Purification: preparative PAGE. Nucleoside composition: dC 2.12, dG 2.05, dT 4.95, dA 5.92. Calculated mass (M): 5652.32; MALDI-TOF MS (negative mode, lineal, THAP): m/z 5651.6, [M-H]–.
Oligonucleotide and nucleopeptide radiolabeling
Oligonucleotides and nucleopeptides were 5′-end-labeled using [γ-32P]ATP and T4 polynucleotide kinase (T4 PKP). Typically, a 20 µl reaction mixture containing 60 µCi [γH32P]ATP (6000 Ci/mmol; DuPont NEN, Boston, MA), 5 U T4 polynucleotide kinase (Gibco BRL, Rockville, MD), 10 pmol oligonucleotide or nucleopeptide, 70 mM Tris–HCl, pH 7.6, 10 mM MgCl2, 100 mM KCl, 1 mM 2-mercaptoethanol was incubated at 37°C for 1 h, 1 µl of 0.5 M ethylendiaminotetraacetic acid (EDTA) was added and the mixture heated to 70°C for 10 min and passed over a QuickSpin G-25 column (Boehringer Mannheim, Mannheim, Germany).
Tdp1 reactions
Typically, 12 µl of reaction mixtures containing 1 pmol 5′-32P-nucleopeptide, 11 ng Tdp1, 50 mM Tris–HCl, pH 8.0, 80 mM KCl, 2 mM EDTA, 1 mM dithiothreitol (DTT), 40 µg/ml bovine serum albumin (BSA) were incubated at 30°C for 12 min. Reactions were stopped by addition of 36 µl of Maxam–Gilbert loading buffer (80% v/v formamide, 10 mM NaOH, 1 mM EDTA, 1% w/v xylene cyanol, 1% w/v bromophenol blue). Control oligonucleotide or nucleopeptide samples were prepared in the same way, except that Tdp1 was replaced by an equal volume of dilution buffer for Tdp1 (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 5 mM Na2EDTA, 5 mM DTT, 10% glycerol, 500 µg/ml BSA).
Time–course kinetics of Tdp1 reactions
For each substrate, a 12 µl reaction mixture was prepared as described above and incubated at 30°C. Time t = 0 was defined as the moment when Tdp1 enzyme was added to the mixture. Aliquots of 1.5 µl were withdrawn after 0.33, 0.67, 1, 3, 5, 10 and 30 min and reactions were stopped by adding 4.3 µl of Maxam–Gilbert loading buffer.
Determination of the Michaelis–Menten kinetic parameters
For each substrate, 0.25 pmol radiolabeled material was mixed with 0, 2, 10, 20 and 35 pmol unlabeled material to yield 6 µl reaction mixtures with final substrate concentrations of 0.042, 1.71, 3.38 and 5.88 µM, respectively. Following incubation with 5.5 ng Tdp1 at 30°C for 1 min (N15P4 and N15Y), 3 min (N15P13 and N4P4) or 6 min (N4P13), 1.5 µl aliquots were withdrawn and reactions were stopped with 4.3 µl of Maxam–Gilbert loading buffer. For each substrate the experiments were performed independently three times and the data obtained were fitted to the Briggs–Haldane approximation of the Michaelis–Menten equation and represented in a Lineweaver–Burk, or double reciprocal, plot to yield estimates of Michaelis constant Km and maximum velocity Vmax (26,27). The value for Vmax was converted to a catalytic constant or turnover number kcat by dividing by the concentration of enzyme used in the experiments (14.72 nM). The specificity constant kcat/Km was also calculated. In all cases, the standard deviation was calculated.
Preparation of the topoisomerase I–14mer suicide complex
An 18mer oligonucleotide was 5′-end-labeled as previously described and hybridized to a complementary 36mer strand (11). The 5′-OH end of the lower 36mer strand was then phosphorylated to avoid its recombination with the upper 3′-OH strand (28). The covalent topoisomerase I–DNA complex was formed as described (11), in a reaction mixture containing 50 fmol DNA duplex, 5 ng purified human topoisomerase I, 10 mM Tris–HCl, pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 0.2 mM DTT and 15 µg/ml BSA, and the mixture was incubated at 25°C for 2 h.
Dephosphorylation reactions with T4 polynucleotide kinase phosphatase (T4 PNKP)
After incubation with Tdp1, the reaction mixtures were brought to 10 mM MgCl2, supplemented with 15 U of T4 PNKP and incubated at 37°C for 20 min. Reactions were stopped by adding 36 µl of Maxam–Gilbert loading buffer.
Gel electrophoresis analysis
The reaction samples (typically 5 µl) were analyzed by electrophoresis through 20% polyacrylamide gels containing 7.0 M urea in Tris-boric acid–EDTA buffer (K·D Medical Inc., Columbia, MD) on a dedicated height sequencer (52 × 33 cm) from CBS Scientific (Del Mar, CA). Autoradiographs were scanned with a phosphorimager from Molecular Dynamics (Sunnyvale, CA) in order to quantify the intensity of each band.
RESULTS AND DISCUSSION
Recognition of nucleopeptides as Tdp1 substrates and determination of the structure of the reaction product
To obtain some insight into the structural preferences of Tdp1, we synthesized nucleopeptides that mimic the topoisomerase I–DNA covalent complex, with phosphodiester linkages between the side chain hydroxyl group of a tyrosine residue and the 3′-end of an oligonucleotide chain (Fig. 2). The nucleopeptide sequences combine peptides and oligonucleotides of different lengths and compositions. The topoisomerase I amino acid sequence around the catalytic tyrosine residue (Tyr723 for human topoisomerase 1) is reproduced in nucleopeptides with the P13 and R8 peptide chains (13). The N15 oligonucleotide moiety is derived from the piece of DNA that remains linked to the protein after the cleavage reaction (10,11,28).
Figure 2.

Nucleopeptides used as Tdp1 substrates. (Insert) Structure of the tyrosyl–3′-nucleoside phosphodiester bond in the nucleopeptides.
In order to assess whether nucleopeptides can be processed by Tdp1, in a first set of experiments the closest models of covalent complexes, nucleopeptides N15P13 and N15R8, were incubated with the enzyme and aliquots of the reaction mixtures were analyzed by gel electrophoresis. As shown in Figure 3 (lanes 3 and 6), treatment of both nucleopeptides with Tdp1 yielded a common and unique product with a slightly faster electrophoretic mobility than the control 15mer oligonucleotide, suggesting that the product was the 3′-phosphorylated oligonucleotide. This interpretation was supported by the fact that when Tdp1 reactions were followed by a treatment with T4 PNKP (Fig. 3, lanes 4 and 7), the products coincided with the control 15mer.
Figure 3.
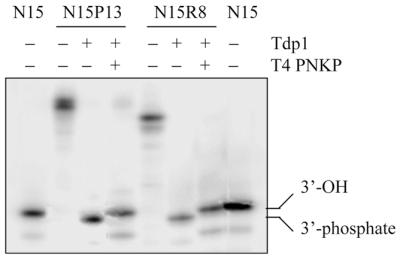
Reaction of Tdp1 with nucleopeptides N15P13 and N15R8. Controls: 15mer oligonucleotide (lanes 1 and 8) and nucleopeptides N15P13 (lane 2) and N15R8 (lane 5). All products were 5′-32P-radiolabeled. Incubation of 1 pmol nucleopeptide with 11 ng Tdp1 at 30°C for 30 min, followed by reaction with 15 U T4 PNKP at 37°C for 20 min yielded the 15mer oligonucleotides N15P13 (lane 4) and N15R8 (lane 7), whereas the reaction of N15P13 and N15R8 (1 pmol) with Tdp1 (11 ng) resulted in formation of a product with a slightly faster mobility (lanes 3 and 6, respectively). As a result, Tdp1 cleavage of the nucleopeptides generates the corresponding 3′-phosphate oligonucleotide and treatment with T4 PNKP removes the 3′-end phosphate.
Phosphodiesterase activity was further evaluated with N15P13. It was found that 95% of N15P13 was transformed into the corresponding 3′-phosphorylated oligonucleotide after incubation with 11 ng Tdp1 at 30°C for 30 min, but that phosphodiesterase activity persisted at lower enzyme concentrations. Thus, using 2.2 ng and 440 and 73 pg Tdp1 we observed transformation of 83, 44 and 8% of N15P13, respectively. In the subsequent experiments, we invariably used 11 ng Tdp1 per µmol substrate.
Time–course kinetics of nucleopeptide processing by Tdp1
To study the determinants of the tyrosine–phosphate cleavage reaction, in particular the influence of the size of the peptide and oligonucleotide fragments on recognition and processing by the enzyme, we carried out a time–course kinetic study of the hydrolysis of nucleopeptides N15P13, N15P4, N4P13, N4P4 and N15Y by Tdp1. Analysis of the relative intensities of substrate and product bands allowed a graphical representation of the percentage of processing as a function of time for each substrate obtained (Fig. 4).
Figure 4.

Kinetics of Tdp1 processing of nucleopeptides N15P13, N15P4, N4P13, N4P4 and N15Y. (A) An aliquot of 1 pmol of each nucleopeptide (5′-32P-radiolabeled) was incubated at 30°C with 11 ng Tdp1. Aliquots were taken at 0.33, 0.67, 1, 3, 5, 10 and 30 min and, after addition of a 3-fold excess of Maxam–Gilbert loading buffer to stop the reactions, mixtures were analyzed by gel electrophoresis. (B) Representation of the percentage of Tdp1 cleavage as a function of time.
We observed a general trend in the behavior of the enzyme depending on the structure of the substrate. Processing by Tdp1 appeared most effective with substrates consisting of long oligonucleotides and short peptides, as inferred from the relative percentages of hydrolyzed nucleopeptide. N15P13 was better processed than N4P13 and N15P4 was better processed than N4P4 when the peptide length was kept constant. On the other hand, when the oligonucleotide length was kept constant, N15P4 was better processed than N15P13 and N4P4 was better processed than N4P13. Thus, ranking of the four nucleopeptides according to the efficiency of reaction with Tdp1 would be as follows: N15P4 > N15P13 ≈ N4P4 > N4P13. Processing of nucleopeptides N15P13 and N4P4 was probably similar due to compensation of the influences of the oligonucleotide and peptide fragments, respectively. The products with a 15mer oligonucleotide and the shortest peptides (nucleopeptide N15P4 and 3′-tyrosine-oligonucleotide N15Y) seemed to have quite similar behaviors.
To support the previously observed qualitative trends, the Michaelis–Menten parameters of the Tdp1–nucleopeptide reactions were determined (Table 1). The Michaelis constant Km is the substrate concentration at which the velocity is half-maximal. Km does not indicate how tightly a substrate binds to the active site of the enzyme, but may be related to the ability of the enzyme to recognize the substrate. In our case, the Km values observed for the different substrates yielded the following: N15P4 < N15P13 < N15Y < N4P13 < N4P4. The long oligonucleotide-containing substrates appeared to have smaller Km values than the nucleopeptides containing short oligonucleotides, which indicates that the oligonucleotide moiety could help Tdp1 in recognizing the substrate as suitable. This observation confirms the preference for long oligonucleotide-containing substrates observed in the time–course kinetics and suggests the presence of an oligonucleotide-binding site on the enzyme.
Table 1.
| Substrate | Vmax (µM s–1) | Km (µM) | kcat (min–1) | kcat/Km (µM–1 min–1) |
|---|---|---|---|---|
| N15P13 | 0.222 ± 0.060 | 0.71 ± 0.24 | 15.08 ± 4.09 | 21.66 ± 1.40 |
| N15P4 | 0.814 ± 0.082 | 0.61 ± 0.23 | 55.27 ± 5.57 | 100.18 ± 38.63 |
| N4P13 | 0.074 ± 0.011 | 1.39 ± 0.39 | 5.06 ± 0.76 | 3.95 ± 1.77 |
| N4P4 | 0.319 ± 0.092 | 1.67 ± 0.84 | 21.67 ± 6.30 | 13.97 ± 3.64 |
| N15Y | 0.670 ± 0.141 | 0.97 ± 0.19 | 45.53 ± 9.58 | 55.85 ± 21.22 |
In our experiments, the enzyme concentration was the same for all the substrates and, as a consequence, the maximum velocity Vmax was directly proportional to the catalytic constant, or turnover number, kcat (see Materials and Methods). kcat is the maximum number of molecules of substrate that could be converted into product per second per active site. Because the maximum rate is obtained at high substrate concentrations, when all the active sites are occupied with substrate, kcat is a measure of how rapidly an enzyme can operate once the active site is filled. The kcat values determined for the different substrates were: N4P13 < N15P13 < N4P4 < N15Y < N15P4. Thus, Tdp1 appears to process substrates bearing long oligonucleotides (N4P13 < N15P13 and N4P4 < N15P4) and substrates bearing short peptides (N15P13 < N15P4 and N4P13 < N4P4) more efficiently. These results are consistent with the general trends observed in the time–course kinetics. Should the difference between the N15P4 and N15Y kcat values be significant, this would mean that a minimal size of the peptide fragment is a requirement for efficient enzyme action, either for the recognition or the processing step. In any case, the influence of factors such as amino acid composition and structural properties cannot be excluded based on the present evidence.
The absolute values of kcat observed for this kind of substrate (0.08–1.0 molecules per s) appear quite small when compared with the values described for other enzymes, such as acetylcholinesterase (kcat = 14 000) and chymotrypsin (kcat = 100) (27,29) for example. Nevertheless, two points should be taken into account. On the one hand, we have determined the kinetic parameters for synthetic substrates that reproduce the covalent topoisomerase I–DNA complexes and not the kinetic parameters of native complexes in cellular medium, which may be different. On the other hand, small values of kcat are not necessarily opposed to efficient action of the enzyme in the cell, since Tdp1 would be expected to process dead-end topoisomerase I–DNA complexes, allowing recycling of the DNA without interfering with the normal catalytic cycle of the topoisomerase.
Finally, we calculated the ratio kcat/Km, which is referred to as the specificity constant and provides a measure of how rapidly the enzyme can work at low substrate concentrations. This constant gives a quite realistic description of enzyme efficiency since, under physiological conditions, enzymes do not usually operate at saturating concentrations. In our case, the kcat/Km values observed for the different substrates afforded the following classification: N4P13 < N4P4 < N15P13 < N15Y < N15P4. Once more, Tdp1 appeared to process substrates containing long oligonucleotides (since N4P13 < N15P13 and N4P4 < N15P4) and short peptides (as N15P13 < N15P4 and N4P13 < N4P4) more efficiently.
Processing of the topoisomerase I–14mer suicide complex by Tdp1
Besides using nucleopeptides as a tool to evaluate Tdp1 preferences, we considered the possibility of studying the processing of a suicide complex made of human topoisomerase I covalently linked to a 14mer oligonucleotide (Fig. 5A and B) (11,22,28). For these experiments we used a 68 kDa topoisomerase I polypeptide truncated at its N-terminus and purified from insect cells infected with a baculovirus construct (22). As with the nucleopeptides, we treated the topoisomerase I–14mer suicide complex with Tdp1 and withdrew aliquots at different times. Progressive appearance of a new band corresponding to the liberated 3′-phosphorylated 14mer oligonucleotide was observed (Fig. 5C). The bands corresponded to the expected 3′-phosphate 14mer single-stranded products as we carried out electrophoresis under denaturing conditions so as to avoid possible aggregation phenomena, of either the protein–protein or protein–nucleic acid type. The relative intensities of the 14mer bands allowed us to infer that after 30 min Tdp1 had processed only ∼9% of the suicide complex (Fig. 5D). This result confirms the difficulty of Tdp1 in processing substrates with long peptides and short oligonucleotides. The suicide complex can be viewed as an extreme case of nucleopeptide, with a very long peptide fragment, the topoisomerase I polypeptide, and a relatively short oligonucleotide. In the gel we also observed an invariable band corresponding to the excess starting 18mer oligonucleotide, as well as bands corresponding to the deletion 17, 16, 15 and 14mer oligonucleotides which accompanied the 18mer used in this experiment. The poor activity of Tdp1 against the topoisomerase I suicide substrate contrasts with the efficiency of Tdp1 for DNA fragments linked to short peptides, which suggests that Tdp1 acts after the topoisomerase I covalent complex has been processed in vivo (14). Consistently, proteolysis of topoisomerase I has been observed in cells treated with camptothecin (30) and correlated with cellular resistance (31). Whether PNKP further processes the DNA ends resulting from Tdp1 action remains unknown. Our recent observation that the 5′-hydroxyl ends of replication-mediated DNA breaks produced by camptothecin in human colon carcinoma cells become 5′-phosphorylated in vivo (15) would be consistent with this hypothesis (schematized in Fig. 1B).
Figure 5.

Structure of the DNA duplex used as substrate for (A) human topoisomerase I and (B) of the topoisomerase I–14mer oligonucleotide suicide complex. Tdp1 processing of the topoisomerase I–14mer oligonucleotide suicide complex: (C) PAGE analysis and (D) graphical representation of suicide complex processing as a function of time.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Drs Jeffrey J. Pouliot and Howard Nash (LMB, NIMH) for providing us with yeast Tdp1 and valuable suggestions. This work was supported by funds from the Spanish MEC (grant PB97-0941-C02-01) and the Generalitat de Catalunya (1998SGR-1, 2000SGR-18 and Centre de Referència de Biotecnologia). Laurent Debéthune was a recipient of a fellowship of the Generalitat de Catalunya.
REFERENCES
- 1.Wang J.C. (1996) DNA topoisomerases. Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- 2.Champoux J.J. (2001) DNA topoisomerases: structure, function and mechanism. Annu. Rev. Biochem., 70, 369–413. [DOI] [PubMed] [Google Scholar]
- 3.Wigley D.B. (1998) Teaching a new dog old tricks? Curr. Biol., 6, 543–548. [DOI] [PubMed] [Google Scholar]
- 4.Cheng C., Kussie,P., Pavletich,N. and Shuman,S. (1998) Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell, 92, 841–850. [DOI] [PubMed] [Google Scholar]
- 5.Pourquier P. and Pommier,Y. (2001) Topoisomerase I-mediated DNA damage. Adv. Cancer Res., 80, 189–216. [DOI] [PubMed] [Google Scholar]
- 6.Shuman S. (1992) Two classes of DNA end-joining reactions catalyzed by vaccinia topoisomerase I. J. Biol. Chem., 267, 16755–16758. [PubMed] [Google Scholar]
- 7.Svejstrup J.Q., Christiansen,K., Gromova,I.I, Andersen,A.H. and Westergaard,O. (1991) New technique for uncoupling the cleavage and religation reactions of eukaryotic topoisomerase I. The mode of action of camptothecin at a specific recognition site. J. Mol. Biol., 222, 669–678. [DOI] [PubMed] [Google Scholar]
- 8.Pourquier P., Pilon,A., Kohlhagen,G., Mazumder,A., Sharma,A. and Pommier,Y. (1997) Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps: importance of DNA end phosphorylation and camptothecin effects. J. Biol. Chem., 272, 26441–26447. [DOI] [PubMed] [Google Scholar]
- 9.Pommier Y., Kohlhagen,G., Pourquier,P., Sayer,J.M., Kroth,H. and Jerina,D.M. (2000) Benzo[a]pyrene epoxide adducts in DNA are potent inhibitors of a normal topoisomerase I cleavage site and powerful inducers of other topoisomerase I cleavages. Proc. Natl Acad. Sci. USA, 97, 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pommier Y., Kohlhagen,G., Laco,G.S., Sayer,J.M., Kroth,H. and Jerina,D.M. (2000) Position-specific trapping of topoisomerase I-DNA cleavage complexes by the intercalated benzo[a]pyrene diol epoxide adducts at the 6-amino group of adenine. Proc. Natl Acad. Sci. USA, 97, 10739–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pourquier P., Ueng,L.-M., Kohlhagen,G., Mazumder,A., Gupta,M., Kohn,K.W. and Pommier,Y. (1997) Effects of uracil incorporation, DNA mismatches and abasic sites on cleavage and religation activities of mammalian topoisomerase I. J. Biol. Chem., 272, 7792–7796. [DOI] [PubMed] [Google Scholar]
- 12.Chen A.Y. and Liu,L.F. (1994) DNA topoisomerases: essential enzymes and lethal targets. Annu. Rev. Pharmacol. Toxicol., 94, 194–218. [DOI] [PubMed] [Google Scholar]
- 13.Gupta M., Fujimori,A. and Pommier,Y. (1995) Eukaryotic DNA topoisomerases I. Biochim. Biophys. Acta, 1262, 1–14. [DOI] [PubMed] [Google Scholar]
- 14.Pommier Y., Pourquier,P., Urasaki,Y., Wu,J. and Laco,G. (1999) Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resistance Update, 2, 307–318. [DOI] [PubMed] [Google Scholar]
- 15.Strumberg D., Pilon,A.A., Smith,M., Hickey,R., Malkas,L. and Pommier,Y. (2000) Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol., 20, 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang S.-W., Burgin,A.B., Huizenga,B.N., Robertson,C.A., Yao,K.C. and Nash,H.A. (1996) A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl Acad. Sci. USA, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pouliot J.J., Yao,K.C., Robertson,C.A. and Nash,H.A. (1999) Yeast gene for a Tyr-DNA phosphodiesterase that repairs topo I covalent complexes. Science, 286, 552–555. [DOI] [PubMed] [Google Scholar]
- 18.Interthal H., Pouliot,J.J. and Champoux,J.J. (2001) The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl Acad. Sci. USA, 98, 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pouliot J.J., Robertson,C.A. and Nash,H.A. (2001) Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells, 6, 677–687. [DOI] [PubMed] [Google Scholar]
- 20.Robles J., Beltrán,M., Marchán,V., Pérez,Y., Travesset,I., Pedroso,E. and Grandas,A. (1999) Towards nucleopeptides containing any trifunctional amino acid. Tetrahedron, 55, 13251–13264. [Google Scholar]
- 21.Efcavitch J.W. (1990) The electrophoresis of synthetic oligonucleotides. In Dickwood,D. and Hames,B.D. (eds), Gel Electrophoresis of Nucleic Acids. A Practical Approach, 2nd edn. IRL Press, Oxford, pp. 125–149. [Google Scholar]
- 22.Pourquier P., Takebayashi,Y., Urasaki,Y., Gioffre,C., Kohlhagen,G. and Pommier,Y. (2000) Induction of topoisomerase I cleavage complexes by 1-β-D-arabinofuranosylcytosine (Ara-C) in vitro and in ara-C-treated cells. Proc. Natl Acad. Sci. USA, 97, 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montserrat F.X., Grandas,A., Eritja,R. and Pedroso,E. (1994) Criteria for the economic large scale solid-phase synthesis of oligonucleotides. Tetrahedron, 50, 2617–2622. [Google Scholar]
- 24.Rabanal F., Giralt,E. and Albericio,F. (1992) A new fluorene-derived anchor for the solid-phase synthesis of protected peptides. Tetrahedron Lett., 33, 1775–1778. [Google Scholar]
- 25.Albericio F., Cruz,M., Debéthune,L., Eritja,R., Giralt,E., Grandas,A., Marchán,V., Pastor,J.J., Pedroso,E., Rabanal,F. et al. (2001) An improved synthesis of N-[(9-hydroxymethyl)-2-fluorenyl]succinamic acid (HMFS), a versatile handle for the solid-phase synthesis of biomolecules. Synth. Commun., 31, 225–232. [Google Scholar]
- 26.Cormish-Bowden A. (1995) Fundamentals in Enzyme Kinetics. Portland Press, London, UK.
- 27.Zubay G. (1998) Enzyme kinetics. In Brown (ed.), Biochemistry. Dubuque, IA, pp. 158–176.
- 28.Pommier Y., Jenkins,J., Kohlhagen,G. and Leteurtre,F. (1995) DNA recombinase activity of eukaryotic DNA topoisomerase I; effects of camptothecin and other inhibitors. Mutat. Res., 337, 135–145. [DOI] [PubMed] [Google Scholar]
- 29.Boyer P.D. (1971) The Enzymes, 3rd Edn. Academic Press, New York, NY.
- 30.Beidler D.R. and Cheng,Y.-C. (1995) Camptothecin induction of a time- and concentration-dependent decrease of topoisomerase I and its implication in camptothecin activity. Mol Pharmacol., 47, 907–914. [PubMed] [Google Scholar]
- 31.Desai S.D., Li,T.K., Rodriguez-Bauman,A., Rubin,E.H. and Liu,L.F. (2001) Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer Res., 61, 5926–5932. [PubMed] [Google Scholar]