

Abstract
The ‘healthy cell bias of estrogen action’ hypothesis examines the role that regulating mitochondrial function and bioenergetics play in promoting neural health and the mechanistic crossroads that lead to divergent outcomes following estrogen exposure. Estrogen-induced signaling pathways in hippocampal and cortical neurons converge upon the mitochondria to enhance aerobic glycolysis coupled to the citric acid cycle, mitochondrial respiration and ATP generation. Convergence of estrogen-induced signaling onto mitochondria is also a point of vulnerability when activated in diseased neurons which exacerbates degeneration through increased load on dysregulated calcium homeostasis. As the continuum of neurological health progresses from healthy to unhealthy so too do the benefits of estrogen or hormone therapy. The healthy cell bias of estrogen action hypothesis provides a lens through which to assess disparities in outcomes across basic and clinical science and on which to predict outcomes of estrogen interventions for sustaining neurological health and preventing age-associated neurodegenerative diseases such as Alzheimer s.
Introduction
Decades of basic science investigation of estrogen action in the brain and subsequent observational and clinical trials have indicated the benefit of estrogen-based therapies [1–4]. Embedded among these reports were suggestions that the beneficial effects of estrogen were conditional [5–11]. Results of the widely publicized Women’s Health Initiative Memory Study (WHIMS) clinical trial drew substantial attention to just how conditional estrogen therapy (ET) and hormone therapy (HT) can be [12,13].
Analysis of the model systems used across the basic to clinical research continuum separates into two broad classes, those that use prevention interventions in healthy organisms and those that use hormone interventions in organisms with compromised neurological function [1]. Basic science analyses that led to elucidation of the neurotrophic and neuroprotective effects of estrogen and the underlying mechanisms of action typically used a prevention experimental paradigm [1]. The prevention paradigm relies on healthy neurons/brains/animals/humans as the starting foundation followed by exposure to estrogen/hormone followed by exposure to neurodegenerative insult. The prevention paradigm of basic science analyses parallels the human studies of Sherwin and colleagues, who investigated the cognitive impact of estrogen therapy in women with surgical- or pharmacological-induced menopause [11]. Observational, retrospective and prospective studies are also consistent with the outcomes of basic science analyses [1]. For the most part, the epidemiological observational data indicate reduction in risk of Alzheimer’s disease (AD) in women who began estrogen or hormone therapy at the time of the menopause [1,6,11,14] (but see Ref. [15]). The benefit seen in most observational studies and basic science analyses suggests that for the most part, the data within the observational studies were derived from women with healthy neurological status.
By contrast, studies that fall within the second class, hormone intervention in women with compromised neurological function, that is, a treatment paradigm, exhibit a mixed profile [1]. This was first evident in the results from Cache County, in which risk of AD varied with age of HT initiation and duration of use [6]. A woman’s gender-associated increase in risk disappeared entirely with more than 10 years of treatment, with most of the HT-related reduction in incidence reflecting former use. There was no effect with current hormone replacement therapy use unless duration of treatment exceeded 10 years [6]. Efficacy of ET observed in early AD treatment trials which typically lasted 1.5–2 months [16] was not sustained when ET was administered for a year or more [17,18]. In these randomized double-blind clinical trials of ET in a cohort in aged women, 72 years of age, diagnosed with Alzheimer’s disease, estrogen therapy resulted in a modest benefit of estrogen therapy in the short term (2 months) and adverse progression of disease in the long term (12 months) [17,18]. In the WHIMS cohort of women, 65–79 years of age, with no indicators of neurological disease but with variable health status, no statistically significant increase in AD risk occurred in the ET/conjugated equine estrogen (CEE) arm of the trial [12]. However, there was no benefit of ET and there was a clear decline in cognitive performance over time [12]. By contrast, the combination of CEE + medroxyprogesterone acetate (MPA) for 5 years increased the risk of developing Alzheimer’s disease by twofold [13], and when the results of the ET and HT data were combined there was a twofold increase in the risk of AD [13]. Subsequent post hoc analyses of the WHIMS data suggested that women who had reported prior hormone use had a significantly lower risk of AD disease and all-cause dementia during the WHIMS trials [19]. Collectively, the data suggest that as the continuum of neurological health progresses from healthy to unhealthy so too do the benefits of estrogen or hormone therapy [1]. If neurons are healthy at the time of estrogen exposure, their response to estrogen is beneficial for both neurological function and survival. By contrast, if neurological health is compromised, estrogen exposure over time exacerbates neurological demise. Based on the analyses reviewed herein, the hypothesis of a “healthy cell bias of estrogen action’ is proposed (see Figure 1).
Figure 1.
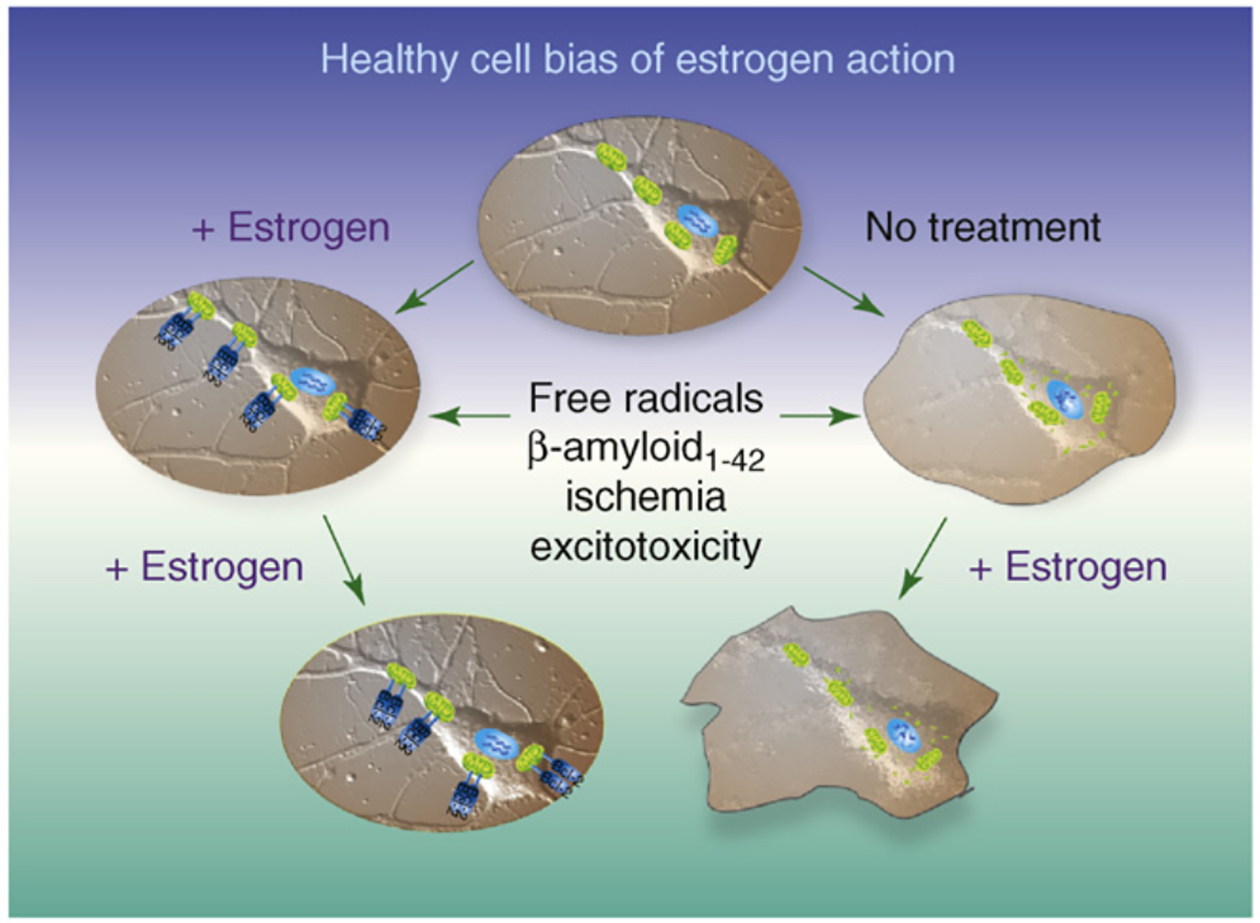
Healthy cell bias of estrogen action evidence from basic to clinical science indicates that neurons and women treated with estrogen before exposure to neurodegenerative insult prevents neural demise. In stark contrast, basic and clinical evidence further indicates that exposure to estrogen following neurodegenerative insult can result in an exacerbation of neurological demise. Estrogen regulation of calcium signaling and mitochondrial function play key roles in determining the outcome of estrogen exposure. Figure modified from Ref. [1].
Estrogen regulation of mitochondrial function: a window into understanding the healthy cell bias of estrogen action
Our investigation of estrogen regulation of mitochondrial function was stimulated by our findings that 17β-estradiol (E2) prevented dysregulation of Ca2+ homeostasis by increasing mitochondrial sequestration of Ca2+ while simultaneously sustaining mitochondrial respiration [2,20,21]. Further, we serendipitously observed years earlier that estrogens increased ATP generation in healthy hippocampal neurons and sustained ATP generation in hippocampal neurons following exposure to Aβ1–42 [22]. These findings, coupled with our increasing awareness that estrogen-induced signaling pathways converged upon the mitochondria [20,21,23,24], led us to directly investigate mitochondria as a pivotal convergence point of estrogen action in neurons (see Figure 2).
Figure 2.
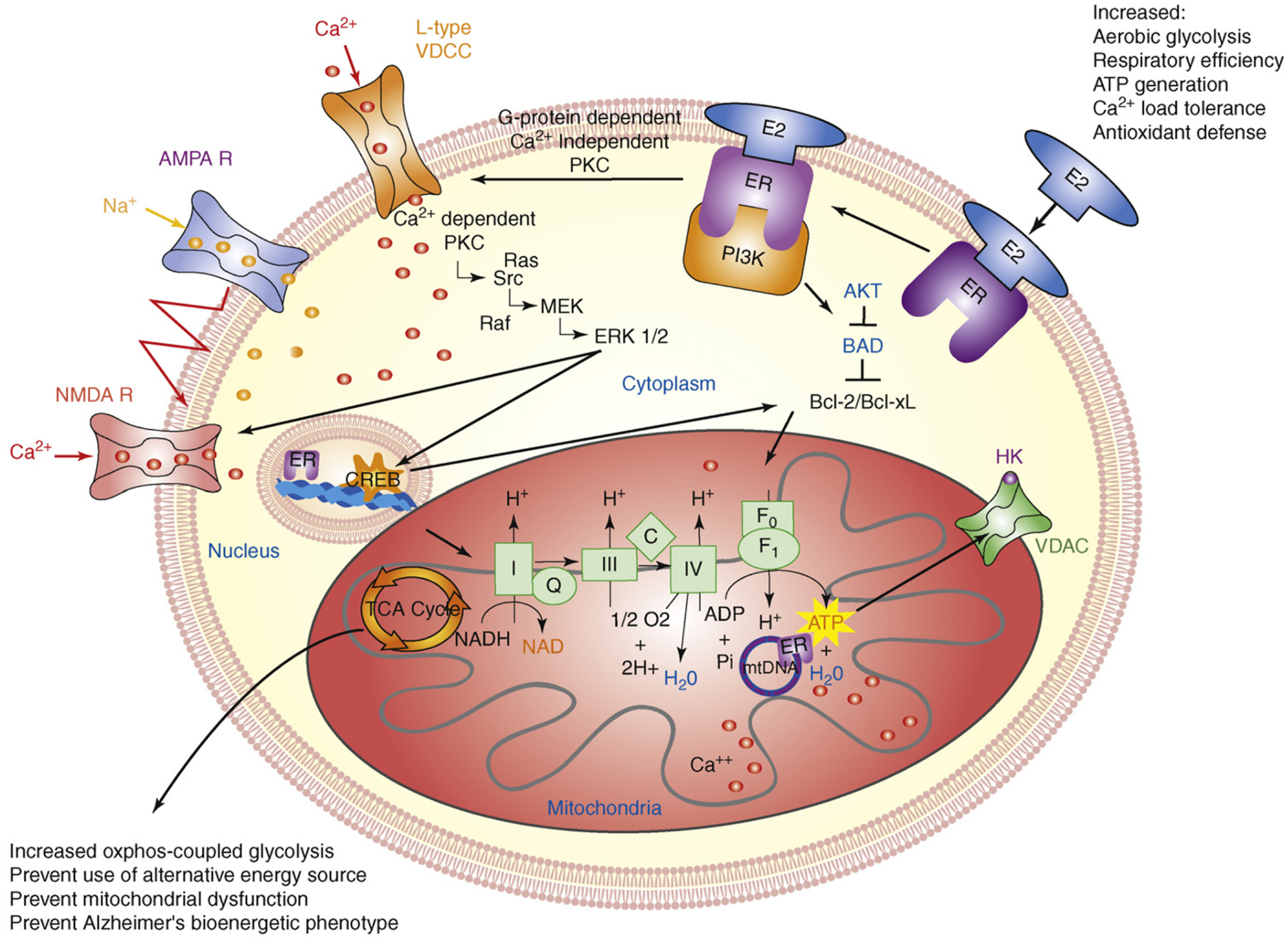
Estrogen mechanisms of action converge upon the mitochondria. Estrogen (17β-estradiol; E2) binding to a membrane-associated estrogen receptor (ER) undergoes a protein-protein interaction with the regulatory subunit of PI3K, p85, to activate the divergent but coordinated activation of the Akt and MAPk signaling cascades. These E2-induced signaling pathways in hippocampal and cortical neurons converge upon the mitochondria to enhance glucose uptake and metabolism, aerobic glycolysis and pyruvate dehydrogenase to couple aerobic glycolysis to acetyl-CoA production and tricarboxylic acid cycle (TCA) -coupled oxidative phosphorylation and ATP generation. In parallel, E2 increases antioxidant defense and antiapoptotic mechanisms. Estrogen receptors at the membrane, in mitochondria and within the nucleus are well positioned to regulate coordinated mitochondrial and nuclear gene expression required for optimal bioenergetics. Enhancing and sustaining glycolysis, aerobic metabolism and mitochondrial function would be predicted to prevent the shift to alternative fuel sources and the hypometabolism characteristic of Alzheimer’s disease. Figure modified from Ref. [2].
As a starting point, we conducted a proteomic analysis of brain mitochondria from female rats treated with E2. Results of our proteomic analyses indicated that of the 499 detected proteins, 66 proteins exhibited a twofold or greater change in expression, and of these, 28 proteins increased in expression, whereas 38 proteins were decreased relative to control [25]. The protein expression profile indicated that, overall, E2 induced marked changes in proteins involved in cellular energetics, free radical maintenance, metabolism, stress response and cell survival (see Figure 3).
Figure 3.
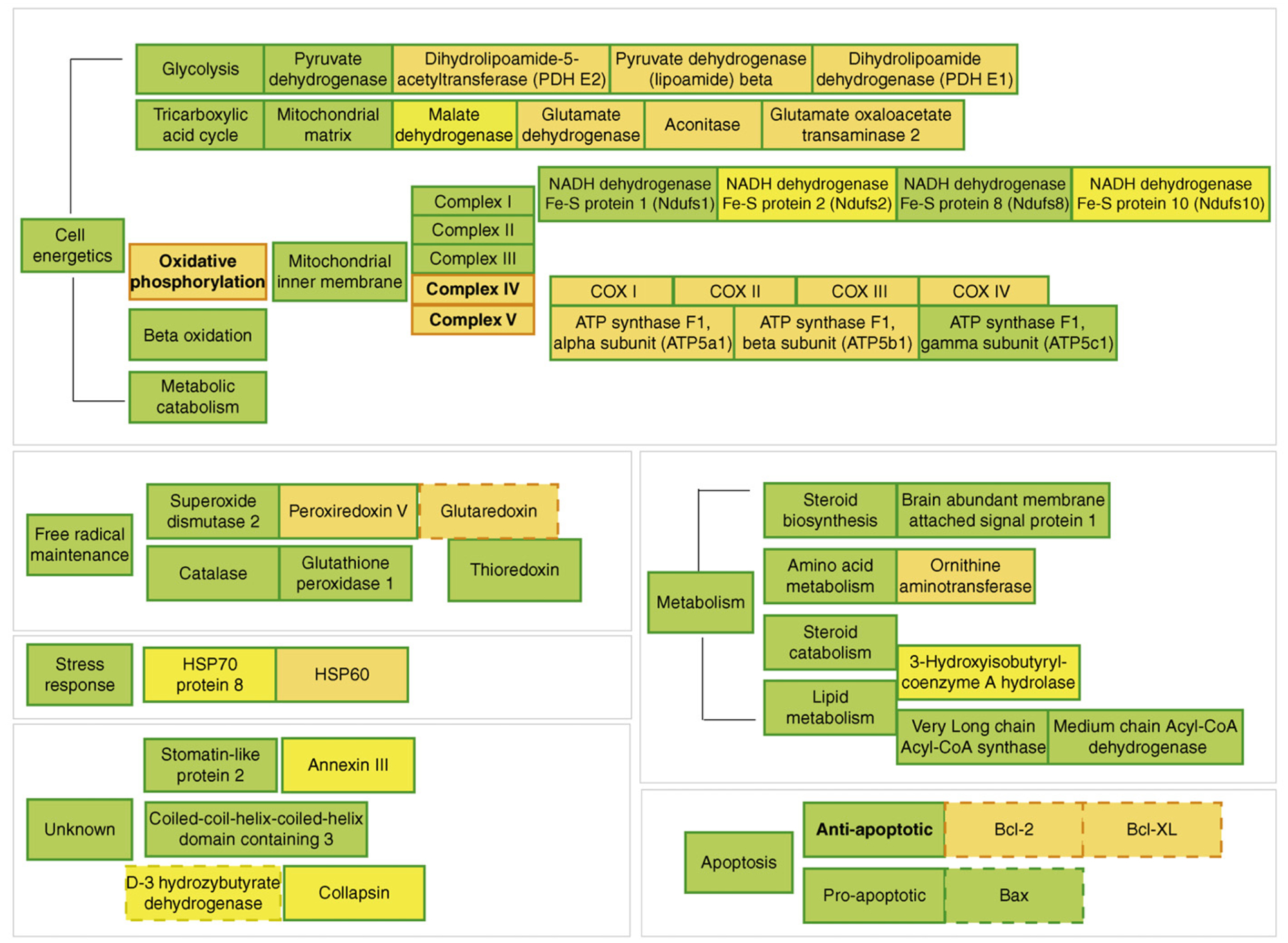
Overview of 17β-estradiol (E2) regulation of female rat brain mitoproteome in vivo. Results of the functional proteomic analysis of the brain mitoproteome were combined with a bioinformatic assessment of the brain mitoproteome regulated by E2. Proteins with known responses to E2 were separated into functional subgroups based on common mitochondrial ontology. Orange represents upregulation and yellow represents downregulation. Filled boxes are based on results of Nilsen and Irwin and colleagues [25]. Dashed boxes are derived from published literature (reviewed in Ref. [25]). Bold lettering represents altered activity. E2 significantly increased key components of the cellular energetic machinery including proteins involved in the tricarboxylic acid cycle and oxidative phosphorylation. Further, E2 increased expression of antioxidant enzymes and antiapoptotic proteins. Collectively, the data indicate a comprehensive regulation of mitochondrial function by E2 which increases key elements in the tricarboxylic acid cycle, pyruvate metabolism, mitochondrial oxidative phosphorylation, respiratory efficiency and ATP generation while reducing free radical leak and oxidative damage.
E2 increased protein expression and activity of key metabolic enzymes including pyruvate dehydrogenase, aconitase and ATP synthase. Illustrative of changes in metabolic enzymes, E2 increased expression of multiple subunits of the pyruvate dehydrogenase (PDH) enzyme complex. PDH is a key regulatory enzyme complex linking the glycolytic metabolism to the citric acid cycle by transforming pyruvate into acetyl-CoA. In the brain, PDH is further responsible for directing acetyl-CoA to either the tricarboxylic acid cycle (TCA; also known as the citric acid cycle) or to acetylcholine synthesis (reviewed in Ref. [25]). Given the role PDH plays in linking glycolytic metabolism to the citric acid cycle, a commensurate change in glycolytic enzymes would be anticipated. E2 increases activity of the key cytosolic glycolytic enzymes hexokinase, phosphofructokinase and phosphoglycerate kinase in rodent brain [26]. Together, these findings indicate that E2 promotes enhanced utilization of glucose, the main energy source for the brain (see Figure 4).
Figure 4.
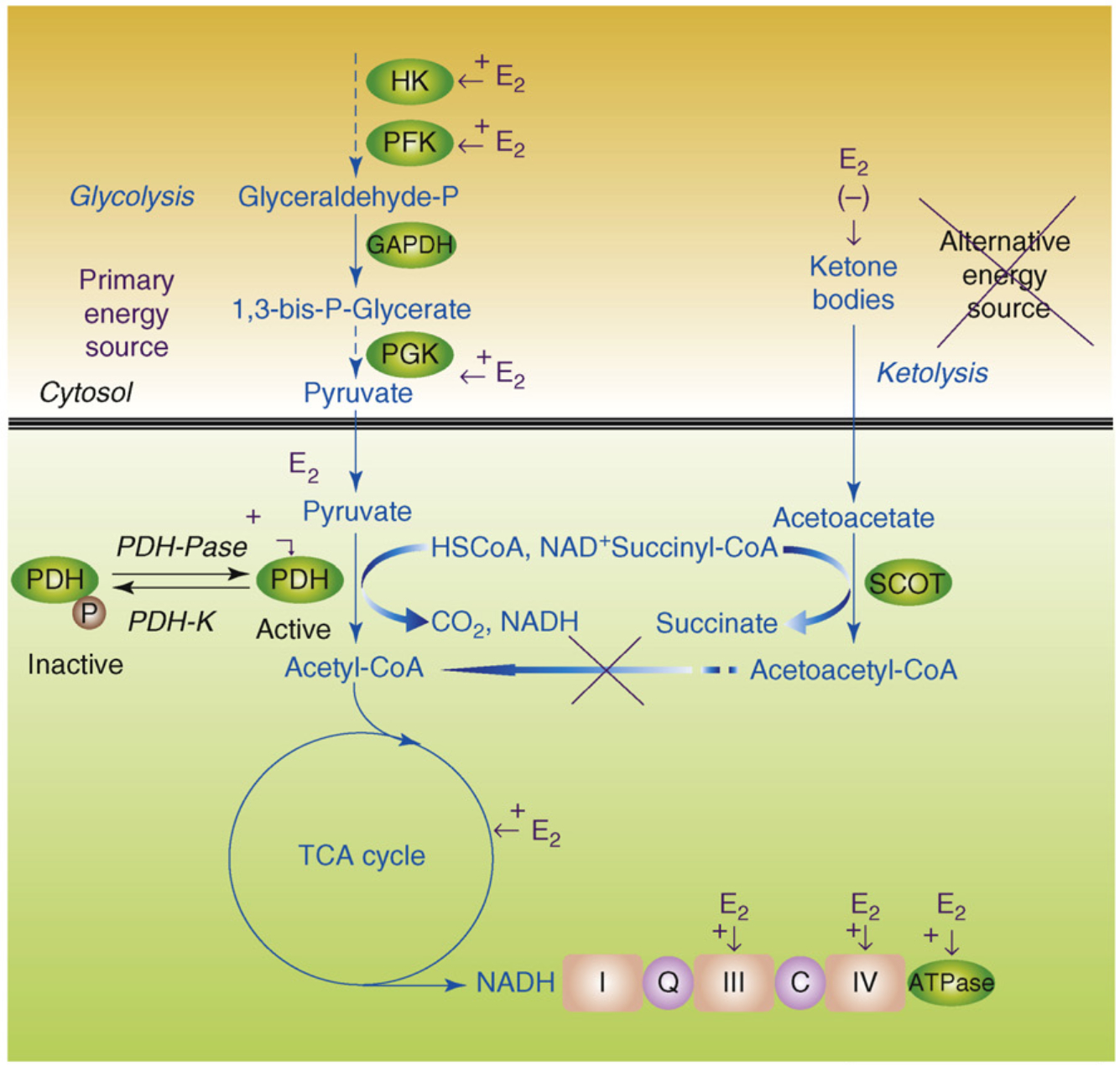
Estrogen (E2) promotes glycolysis and glycolytic-coupled tricarboxylic acid cycle (TCA) function, mitochondrial respiration and ATP generation to prevent a switch to ketone bodies as an alternative fuel source. E2 increases key enzymes in the glycolytic pathway to promote generation of pyruvate and its conversion by pyruvate dehydrogenase (PDH) to acetyl-CoA to initiate and sustain the TCA cycle. Under metabolically challenging conditions (i.e. starvation, aging and neurodegeneration), neurons can utilize acetyl-CoA generated from ketone body metabolism (ketolysis), produced by the liver or under conditions of starvation in neighboring glial cells. This latter pathway is much less efficient and can inhibit residual glycolysis. In AD, there is a generalized shift toward use of an alternative fuel, ketone bodies, and away from glycolytic energy production. Estrogen enhances glucose uptake into the brain glycolytic/pyruvate/acetyl-CoA pathway to generate electrons required for oxidative phosphorylation and ATP generation. Collectively, estrogen enhancement of glucose metabolism and aerobic glycolysis promotes and sustains utilization of glucose as the primary fuel source of the brain, thereby preventing the shift to alternative fuels such as ketone bodies which is characteristic of Alzheimer’s disease.
Estrogen further increased expression and activity of proteins required for oxidative phosphorylation electron transfer, a result that was consistent with a coordinated response that optimizes glucose metabolism in the brain [25]. E2 induced a significant increase of both protein expression and activity of complex IV subunits I–IV [25], consistent with previous reports [27,28]. The E2-induced increase is particularly relevant, given that reduction in complex IV is an early marker of Alzheimer’s disease [29,30]. E2 also increased expression of ATP synthase F1α and F1β, which is consistent with the increase in proteins required for mitochondrial respiration and with our previous report of estrogen-induced increases in ATP levels in primary neuronal cultures [22,25]. The importance of estrogen regulation of mitochondrial function is further heightened by the recent report by Mosconi and colleagues, who found a relationship between maternal history of AD and reduction in glucose metabolic rate in the brain [31]. This finding is of particular interest, as mitochondrial DNA is maternally inherited [32], and suggests a relationship between the maternally inherited mitochondrial genome which, among other genes, encodes for subunits 1,2 and 3 of complex IV, brain metabolism and risk for AD.
E2-induced enhancement of energetic efficiency was paralleled by an increase in free radical defense systems. Increased expression of peroxiredoxin-V is consistent with the well-documented antioxidant effects of estrogens, including increased glutaredoxin expression and MnSOD [20,25]. Free radical balance is maintained by reduction of the superoxide anion to hydrogen peroxidase by superoxide dimutases, with the resulting hydrogen peroxide then being removed by various peroxidases, including peroxiredoxin-V [33]. Reduction in reactive oxygen species contributes to neuroprotection and can reduce the overall stress response. In this context, we identified significant alterations in the expression of two mitochondrial heat-shock proteins, Hsp70 and Hsp60, which are important in the correct import of nascent proteins to the mitochondrial matrix. Many components of the mitochondrial bioenergetic network are vulnerable to oxidative stress, which can impair mitochondrial and cellular function as well as increase apoptotic vulnerability [29,30]. Damaged electron transport chain complexes compromise ATP synthesis and accelerate the generation of free radicals, which could cause or exacerbate neuronal degeneration [29,30]. Estrogen-induced increase in antioxidants, reduction in free radicals and substantially lower oxidative damage to mitochondrial DNA have been posited by Vina and colleagues to be a major contributor to the greater longevity of females relative to males [34].
Remarkably, E2 regulation of mitochondrial function in neural tissue is closely paralleled in the vasculature [35]. In vascular endothelium, chronic estrogen treatment increased mitochondrial capacity for oxidative phosphorylation while simultaneously decreasing production of reactive oxygen species. In contrast to the emerging data regarding ERβ regulation of neural mitochondrial function [36], E2 regulation of mitochondrial function in cerebral blood vessels is mediated by ERα [37]. Estrogen regulation of mitochondrial function in both neural and vascular tissue has functional importance for coordinated responses between neural activity and vascular integrity on the one hand and sustaining neural viability on the other.
E2 regulation of both mitochondrial- and nuclear-encoded gene products requires coordinated control of mitochondrial- and nuclear-encoded gene transcription [25]. Within neurons, estrogen receptors (ER) have been detected in the nucleus, associated with the plasma membrane and within mitochondria [36,38–40]. The simultaneous labeling of membrane, mitochondrial and nuclear ERs within the same neuron and/or glial cell remains a challenge. Although the mechanisms whereby ERs coordinate the complex signaling pathway between the three main compartments, membrane, mitochondria and nucleus, remain to be determined, it is striking that ERs are perfectly positioned to coordinate events at the membrane with events in the mitochondria and nucleus.
Estrogen regulation of glucose metabolism: how sweet it is
If E2 is inducing enhanced coupling between glycolysis and the TCA cycle, then one would expect to observe increased glucose transport and metabolism in the brain. Earlier work from the Simpkins group demonstrated that E2 increased expression of glucose transporter subunits and increased glucose transport in blood-brain barrier endothelium [36]. Later work by Bondy and colleagues confirmed E2 regulation of glucose transporter proteins and that regulation of glucose transporters occurs in neurons in the non-human primate brain [41]. In the frontal cortex of ovariectomized non-human primates, E2 treatment induced two- to fourfold increases in glucose transporter proteins Glut3 and Glut4 mRNA and protein [41]. Analysis of cellular localization indicated that E2 induced a marked rise in neuronal Glut1 mRNA levels with no appreciable effect on expression of the gene encoding vascular Glutl. Collectively, these data indicate that E2 regulates critical elements required for glucose transport into the brain to sustain the energetic demands of neuronal activation [24,42].
If E2 is promoting glucose transport into the brain and into neural cells, then concomitant regulation of glycolytic enzymes would be anticipated. Evidence derived from rat brain indicates that, in vivo, E2 significantly increases glycolytic enzyme activity of hexokinase (soluble and membrane-bound), phosphofructokinase and pyruvate kinase [26]. The neuroprotective effect of E2 is mediated by the coordinated and near simultaneous activation of both the MAPK and Akt signaling pathways through activation of PI3K in hippocampal neurons (see Figure 2) [23]. Remarkably, the antiapoptotic effect of Akt is dependent upon hexokinase association with the voltage-dependent anion channel (VDAC) of mitochondria [43]. Hexokinases are known to bind to VDAC to directly couple intramitochondrial ATP synthesis to glucose metabolism (see Figure 2) [44]. Moreover, of the four hexokinase isoforms, only HKI and HKII are known to associate with mitochondria, where they associate with the mitochondrial outer membrane and bind to VDAC [43]. Although it is known that E2 activates Akt [23,45] and increases HKII activity [26], it remains to be directly demonstrated that E2 induces the association of HKII and VDAC in neural cells.
A functional impact of estrogen-induced glucose transporter protein would require a concomitant change in factors regulating glucose uptake, which in turn suggests a role for insulin or its brain homolog insulin growth factor 1 (IGF-1) and its cognate receptor IGF-1R. Bondy and colleagues found that IGF-1R mRNA was concentrated in cortical neurons in a distribution similar to Gluts 3 and 4 [41]. In non-human primate frontal cortex, E2-treated animals showed a significant increase in IGF-1 mRNA without a concomitant rise in IGF-1 receptor mRNA [41]. These investigators went on to elucidate the molecular mechanisms whereby IGF-1 regulated neuronal metabolism by demonstrating that the active, phosphorylated form of Akt/PKB was selectively colocalized with the ‘insulin-sensitive’ glucose transporter GLUT4 in IGF-1-expressing neurons. Akt is a major target of insulin signaling in the regulation of glucose transport via the facilitative glucose transporter (GLUT4) and glycogen synthesis in peripheral tissues. In parallel to these studies of glucose transport and metabolism, Garcia-Segura and colleagues have for many years demonstrated the synergistic coupling between ERs and IGF-1R [46]. Results of their analyses provide substantial evidence for the role of IGF-1, the PI3K to Akt signaling pathway and ER in estrogen-inducible neuroprotection [46]. Findings of the neuroprotective actions of the synergy between the ER and IGF-1 signaling cascades are particularly relevant to prevention of neurodegenerative diseases. Torres-Aleman and coworkers have shown that low-circulating IGF-1 in the brain is associated with greater accumulation of β amyloid (Aβ), whereas Aβ burden can be reduced by increasing serum IGF-1 [47]. The inverse relationship between serum IGF-1 and brain Aβ levels reflects the ability of IGF-1 to induce clearance of Aβ from the brain, likely by enhancing transport of Aβ carrier proteins such as albumin and transthyretin into the brain [47].
Estrogen regulation of brain metabolism in vivo: I think therefore I metabolize
If E2 is increasing glucose uptake, metabolism and utilization in the brain, then there should be evidence of increased metabolic activity in the brain following estrogen administration. As part of a 9 year study in the Baltimore Longitudinal Study of Aging, Resnick and colleagues conducted positron emission topography (PET) to assess regional cerebral blood flow in a small cohort of women who were ET users versus women who were not. Results of this analysis showed that ET users and nonusers showed significant differences in PET regional cerebral blood flow relative to activation patterns during memory tasks. ET users showed better performance on neuropsychological tests of figural and verbal memory and on some aspects of the PET activation tests [48]. In a follow-up longitudinal study of the same cohort of healthy menopausal women, Maki and Resnick [49] found that regional cerebral blood flow was increased in estrogen therapy users relative to nonusers in the hippocampus, parahippocampal gyrus and temporal lobe, regions that form a memory circuit and that are sensitive to preclinical AD [49]. Further, these investigators found that the increase in regional cerebral blood flow was associated with higher scores on a battery of cognitive tests [49]. In a separate 2 year follow-up analysis, Rasgon and colleagues detected a significant decrease in metabolism of the posterior cingulate cortex among postmenopausal women at 2 year follow-up who did not receive estrogen, whereas those women who were estrogen users did not exhibit significant metabolic change in the posterior cingulate cortex [50]. These findings that estrogen use can preserve regional cerebral metabolism and protect against metabolic decline in postmenopausal women, especially in the posterior cingulate cortex, is particularly important given that metabolism in this region of the brain declines in the earliest stages of AD [50,51].
Hypometabolism precedes cognitive decline of Alzheimer’s disease
Are these findings of E2 regulation of mitochondrial function and enhancement of aerobic glycolysis relevant to Alzheimer’s disease risk? The role of mitochondria in health and disease has long been recognized [29,52], and the evidence for mitochondrial dysfunction as a key precipitating factor in age-associated neurodegenerative diseases such as Alzheimer’s and Parkinson’s continues to mount [29,52,53]. Further, a strong link is mounting between hypometabolism and reduction in mitochondrial gene expression and function in the brain as precipitating antecedents to the cognitive deficits of Alzheimer’s [51,54–56]. The association between hypometabolism and AD is based on multiple levels of analysis and experimental paradigms that range from genomic analyses in animal models and postmortem autopsy of human brain to in vitro cell model systems to brain imaging in humans. Overall, each of these levels of analyses indicates that dysfunction in glucose metabolism, bioenergetics and mitochondrial function are consistent antecedents to development of Alzheimer pathology [30,31,51,56–61]. The decline in brain glucose metabolism and mitochondrial function can appear decades before diagnosis and thus might serve as a biomarker of AD risk as well as therapeutic target.
Circumstances that initiate the hypometabolic cascade remain to be fully determined, but one clue is the shift in the brain from glucose as the primary energetic fuel to ketone bodies. Under metabolically challenging conditions (i.e. starvation, aging and neurodegeneration), neurons can utilize acetyl-CoA generated from ketone body metabolism (ketolysis), produced by the liver or, under conditions of starvation, in neighboring glial cells. This latter pathway is much less efficient and can inhibit residual glycolysis via the Randle cycle [25]. In AD there is a generalized shift toward use of an alternative fuel, ketone bodies. This is evidenced by an observed 45 reduction in cerebral glucose utilization in AD patients [62] which is paralleled by decrease in the expression of glycolytic enzymes which are coupled to a decrease in the activity of the pyruvate dehydrogenase complex [63]. Furthermore, whereas there is a 100:0 ratio of glucose to other substrate utilization in young controls, patients with incipient AD exhibit a 2:1 ratio of glucose to other substrates compared to a ratio of 29:1 in healthy elderly controls (reviewed in Ref. [25]). The switch from glucose as the primary fuel to the alternative of ketone bodies in the AD brain has been the basis for Accera to develop Ketasyn, which is converted to ketone bodies in the liver for subsequent use by the brain. This approach capitalizes on the brain’s relative inability to utilize glucose and its dependency on ketone bodies. Efficacy of Ketasyn as a therapeutic for AD is currently in phase II clinical trials.
The ability of estrogen to sustain glucose as the primary fuel source in the brain by enhancing glucose transport, uptake and aerobic glycolysis (oxidative phosphorylation coupled to pyruvate metabolism) is likely linked to its ability to prevent age-associated metabolic decline in the brain, and thus could be a key mechanism whereby estrogen reduces the risk of AD in postmenopausal women.
Estrogen regulation of calcium homeostasis requires functional mitochondria
Calcium has long been recognized as a pivotal intracellular signal that can regulate a wide array of neural functions including synaptic plasticity, morphogenesis and neurogenesis [64]. Calcium regulation of diverse cellular processes is achieved through a tightly controlled intricate system of Ca2+ dynamics that operate across time spans ranging from milliseconds to hours and include changes in signal amplitude from low to high and transient dynamics that range from rapid on/off to unique oscillatory patterns of varying duration to a slow steady rise and descent [64]. Because Ca2+ plays such a pivotal and near-ubiquitous role in many signaling cascades, dysregulation of Ca2+ signaling and homeostasis has the potential to impact a myriad of neural responses ranging from neurogenesis to neurodegeneration. Thus, sustaining intracellular Ca2+ homeostasis is key to sustaining optimal neural function and survival.
Dysregulation of intracellular Ca2+ homeostasis [Ca2+]i has long been proposed as a fundamental mechanism in age-associated decline in brain function and a key element in the degenerative cascade leading to Alzheimer’s disease [53,65–67]. Nearly two decades ago, Khachaturian proposed a hypothesis of ‘a complex interaction between the amount of [Ca2+]i perturbation and the duration of such deregulation of Ca2+ homeostasis and that a small disturbance in Ca2+ homeostasis with a sustained increase in [Ca2+]i over a long period has similar cell injuring consequences as that produced by a large increase in [Ca2+]i over a shorter period.’ Further, ‘disruptions in energy metabolism and changes in the structure and function of membranes are the most likely antecedent events which lead to disruption of Ca2+ homeostasis’ [65]. Consistent with this prediction, we and others have found that Aβ induces a small sustained increase in [Ca2+]i that rises over a long period [10,66,68]. Gene array analyses of hippocampus from aging rodents [57], humans and Alzheimer’s victims [58] all indicate significant and profound changes in gene expression associated with Ca2+ homeostasis that are paralleled by downregulation of multiple genes required for mitochondrial energy metabolism and positively correlated with behavioral impairment [57,58,60,68]. The relationship between Ca2+ homeostasis and AD recently advanced with the finding that a polymorphism (P89L) for the CALHM1 Ca2+ channel that has homology to the NMDA receptor and which generates a large Ca2+ conductance across the plasma membrane is linked to late-onset AD [69]. This finding is consistent with the discovery many years ago by Landfield and colleagues that voltage-dependent Ca2+ channels increased with age and was associated with cognitive decline [68].
The pivotal role of Ca2+ signaling in neural function and the importance of sustaining intracellular Ca2+ homeostasis is mirrored in estrogen mechanisms of action. Through activating the PI3 kinase signaling pathway, E2 promotes influx of Ca2+ via L-type Ca2+ channels which in turns activates the Src/ERK/CREB cascade (Figure 2) [23,70,71]. Estrogen induction of this Ca2+-dependent signaling cascade leads to activation of mechanisms of learning and memory and neural defense [2,72]. Our studies of E2 regulation of intracellular Ca2+ dynamics and homeostasis originated in an attempt to resolve the paradox of dual regulation of [Ca2+]i by E2 in hippocampal neurons after nontoxic and excitotoxic glutamate exposure [73]. Analyses of [Ca2+]i dynamics between the cytosolic and mitochondrial compartments revealed that E2 caused an increase in mitochondrial sequestration of [Ca2+]i when neurons were exposed to excitotoxic glutamate, which was paralleled by attenuation of cytoplasmic [Ca2+]i [74]. E2-induced attenuation of [Ca2+]i was correlated with an increase in Bcl-2 expression, which could provide a mechanism whereby neurons are protected against deleterious effects of increased mitochondrial [Ca2+] [21,75]. Further, increased mitochondrial sequestration of Ca2+ induced by E2 protected neurons against adverse consequences of excess cytoplasmic [Ca2+]i and subsequent dysregulation of [Ca2+]i homeostasis. Despite an increased mitochondrial Ca2+ load, E2 preserved mitochondrial respiratory capacity [74].
Because these mechanistic studies were conducted in healthy neurons derived from embryonic hippocampus, we sought to determine whether E2 regulation of Ca2+ homeostasis extended to neurons derived from middle-aged and aged rodent hippocampus [64]. Results of these analyses were both striking and consistent with earlier observations. Age-associated dysregulation of [Ca2+]i homeostasis was prevented by 48 h of prior exposure to E2, a time frame consistent with E2-induced Bcl-2 expression [70,74]. Embryonic neurons exhibited the greatest capacity to sustain Ca2+ homeostasis, followed by middle-age neurons followed by neurons derived from the aged brain which had a near nonexistent capacity to maintain Ca2+ homeostasis [64]. In neurons derived from aged rat hippocampus, the first peak of [Ca2+]i was substantially greater than at other ages and the return to baseline Ca2+ rapidly dysregulated with an inability to restore [Ca2+]i following the first glutamate pulse which persisted throughout the 20 pulses. Remarkably, E2 pretreatment of aged neurons profoundly attenuated the peak [Ca2+]i rise and delayed the age-associated dysregulation of baseline [Ca2+]i, normalizing responses to those of middle-age neurons treated with E2 [64].
It is the dependency upon Ca2+ signaling and the requirement for optimal Ca2+ homeostatic mechanisms that we believe is the Achilles heel of estrogen action. In a series of experiments designed to address controversies of estrogen therapy, we conducted in vitro experiments designed to simulate the WHIMS trial in cultured neurons. We hypothesized that E2 exposure of healthy neurons in a prevention mode would promote [Ca2+]i homeostasis to prevent Aβ1–42-induced neurodegeneration, whereas E2 exposure of degenerating neurons in a treatment mode would exacerbate Aβ1–42-induced dysregulation of [Ca2+]i homeostasis [10]. Results of those analyses indicated that in a prevention mode of exposure, E2 was most effective in sustaining [Ca2+]i homeostasis when present before and during Aβ1–42 insult. By contrast, E2 treatment following Aβ1–42 exposure was ineffective in reversing Aβ-induced degeneration and exacerbated Aβ1–42-induced cell death. We further found that low E2 significantly prevented Aβ1–42-induced rise in [Ca2+]i, whereas high E2 significantly increased [Ca2+]i and did not prevent Aβ1–42-induced [Ca2+]i dysregulation [10]. Therapeutic benefit resulted only from low-dose E2 exposure before, but not following, Aβ1–42-induced neurodegeneration. Collectively, these data support a role of low-dose E2 in promoting [Ca2+]i homeostasis in healthy embryonic, middle-aged and aged neurons. Further, the data indicate that once dysregulation of [Ca2+]i homeostasis has occurred, as in the case of Aβ1–42-induced [Ca2+]i dysregulation, exposure to low-dose E2 is of no benefit and exposure to high-dose E2 is deleterious and exacerbates neural demise.
Implications for initiating estrogen therapy and for generalizing preventive strategies to treatment strategies
The healthy cell bias of estrogen action hypothesis predicts that estrogen therapy, if initiated at the time of peri- to menopause when neurological health is not yet compromised, will be of benefit, as manifested as reduced risk for age-associated neurodegenerative diseases such as Alzheimer’s and Parkinson’s. Further, E2 promotion of glycolysis and glycolytic-coupled citric acid function, mitochondrial respiration and ATP generation, and antioxidant and antiapoptotic mechanisms serves as the pivotal pathway by which estrogen sustains neurological health and defense. The reliance of this pathway on Ca2+ signaling and on mitochondrial Ca2+ buffering is an Achilles heel of estrogen action in degenerating systems in which [Ca2+]i homeostasis is dysregulated. Addition of estrogen under these conditions, although of modest benefit initially, an effect likely mediated by neurons not yet affected by the disease, adds to the dysregulation of [Ca2+]i homeostasis with predictable exacerbation of the degenerative process [10].
Major challenges for optimal estrogen and hormone therapy remain. Beyond the timing issue, the real and perceived risks of hormone therapy remain and were amplified by results from both the WHI and WHIMS trials. It is clear that many, but not all, women could potentially benefit from estrogen or hormone therapy intervention. Biomarkers to identify women appropriate for which type of hormone regimen remain largely undeveloped beyond the hot flash. Hormone therapy interventions that selectively target the benefits of estrogen while avoiding untoward risk factors remains an unmet need in women’s health.
Investigating mechanisms of estrogen action in parallel to identifying the antecedents to Alzheimer’s pathology provides insights into the basis for disparities between basic science discovery and clinical outcomes. More generally, results of these investigations raise concerns regarding applying preventive strategies to treatment modalities in the clinical realm. In the preclinical basic science domain, reliance upon healthy cellular and animal models that are acutely exposed to toxic insults as models of neurodegenerative diseases that typically develop incrementally, slowly and accumulate damage over time raises concern regarding the translational validity of these data. This is particularly true for age-associated neurodegenerative diseases in which the normal aging brain undergoes dramatic changes that are either unrelated to or are the earliest signs of neurodegenerative vulnerability [57,58,60,61,68]. Efforts to bridge these gaps in women’s neurological health are emerging and hold the promise to serve as a model for mechanistic and translational neuroscience research at the bench and the bedside [76] (http://www.nia.nih.gov/ResearchInformation/ExtramuralPrograms/NeuroscienceOfAging/NNA_Conferences/BenchtoBedside.htm).
Acknowledgements
The many contributions of the Brinton laboratory estrogen and mitochondria research team, especially Jon Nilsen, Ronald Irwin, Jia Yao, Shuhua Chen, Liqin Zhao and Ryan Hamilton are gratefully acknowledged. I also thank Enrique Cadenas for critique of this manuscript and helpful discussions. Graphic contributions by Kathy Cho, Syeda Ahmed and Karren Wong are gratefully acknowledged. Research from our laboratory and preparation of this review were supported by grants from the National Institute of Mental Health (1R01 MH67159), National Institute on Aging (P01AG026572) and the Kenneth T. and Eileen L. Norris Foundation to R.D.B.
References
- 1.Brinton RD (2005) Investigative models for determining hormone therapy-induced outcomes in brain: evidence in support of a healthy cell bias of estrogen action. Ann. N. Y. Acad. Sci 1052, 57–74 [DOI] [PubMed] [Google Scholar]
- 2.Morrison JH et al. (2006) Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J. Neurosci 26, 10332–10348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wise PM (2006) Estrogen therapy: does it help or hurt the adult and aging brain? Insights derived from animal models. Neuroscience 138, 831–835 [DOI] [PubMed] [Google Scholar]
- 4.Singh M et al. (2008) Estrogens and progesterone as neuroprotectants: what animal models teach us. Front. Biosci 13, 1083–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nilsen J and Brinton RD (2002) Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology 143, 205–212 [DOI] [PubMed] [Google Scholar]
- 6.Zandi PP et al. (2002) Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. J. Am. Med. Assoc 288, 2123–2129 [DOI] [PubMed] [Google Scholar]
- 7.Resnick SM and Henderson VW (2002) Hormone therapy and risk of Alzheimer disease: a critical time. J. Am. Med. Assoc 288, 2170–2172 [DOI] [PubMed] [Google Scholar]
- 8.Yaffe K (2003) Hormone therapy and the brain: deja vu all over again? J. Am. Med. Assoc 289, 2717–2719 [DOI] [PubMed] [Google Scholar]
- 9.Sohrabji F (2005) Estrogen: a neuroprotective or proinflammatory hormone? Emerging evidence from reproductive aging models. Ann. N. Y. Acad. Sci 1052, 75–90 [DOI] [PubMed] [Google Scholar]
- 10.Chen S et al. (2006) Dose and temporal pattern of estrogen exposure determines neuroprotective outcome in hippocampal neurons: therapeutic implications. Endocrinology 147, 5303–5313 [DOI] [PubMed] [Google Scholar]
- 11.Sherwin BB and Henry JF (2008) Brain aging modulates the neuroprotective effects of estrogen on selective aspects of cognition in women: a critical review. Front. Neuroendocrinol 29, 88–113 [DOI] [PubMed] [Google Scholar]
- 12.Shumaker SA et al. (2004) Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. J. Am. Med. Assoc 291, 2947–2958 [DOI] [PubMed] [Google Scholar]
- 13.Shumaker SA et al. (2003) Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. J. Am. Med. Assoc 289, 2651–2662 [DOI] [PubMed] [Google Scholar]
- 14.Yaffe K et al. (1998) Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. J. Am. Med. Assoc 279, 688–695 [DOI] [PubMed] [Google Scholar]
- 15.Petitti DB et al. (2008) Incidence of dementia in long-term hormone users. Am. J. Epidemiol 167, 692–700 [DOI] [PubMed] [Google Scholar]
- 16.Fillit H et al. (1986) Observations in a preliminary open trial of estradiol therapy for senile dementia-Alzheimer’s type. Psychoneuroendocrinology 11, 337–345 [DOI] [PubMed] [Google Scholar]
- 17.Mulnard RA et al. (2000) Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer’s Disease Cooperative Study. J. Am. Med. Assoc 283, 1007–1015 [DOI] [PubMed] [Google Scholar]
- 18.Henderson VW et al. (2000) Estrogen for Alzheimer’s disease in women: randomized, double-blind, placebo-controlled trial. Neurology 54, 295–301 [DOI] [PubMed] [Google Scholar]
- 19.Henderson VW et al. (2007) Prior use of hormone therapy and incident Alzheimer’s disease in the Women’s Health Initiative Memory Study. Neurology 68, A205 [Google Scholar]
- 20.Nilsen J and Brinton RD (2004) Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr. Drug Targets CNSNeurol Disord 3, 297–313 [DOI] [PubMed] [Google Scholar]
- 21.Nilsen J and Diaz Brinton R (2003) Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc. Natl. Acad. Sci. U. S. A 100, 2842–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brinton RD et al. (2000) The Women’s Health Initiative estrogen replacement therapy is neurotrophic and neuroprotective. Neurobiol. Aging 21, 475–496 [DOI] [PubMed] [Google Scholar]
- 23.Mannella P and Brinton RD (2006) Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J. Neurosci 26, 9439–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilsen J et al. (2006) Estrogen protects neuronal cells from amyloid β-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci 7, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsen J et al. (2007) Estradiol in vivo regulation of brain mitochondrial proteome. J. Neurosci 27, 14069–14077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kostanyan A and Nazaryan K (1992) Rat brain glycolysis regulation by estradiol-17 β. Biochim. Biophys. Acta 1133, 301–306 [DOI] [PubMed] [Google Scholar]
- 27.Bettini E and Maggi A (1992) Estrogen induction of cytochrome c oxidase subunit III in rat hippocampus. J. Neurochem 58, 1923–1929 [DOI] [PubMed] [Google Scholar]
- 28.Stirone C et al. (2005) Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol. Pharmacol 68, 959–965 [DOI] [PubMed] [Google Scholar]
- 29.Lin M and Beal M (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 [DOI] [PubMed] [Google Scholar]
- 30.Moreira PI et al. (2007) Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J. Neurol. Sci 257, 206–214 [DOI] [PubMed] [Google Scholar]
- 31.Mosconi L et al. (2007) Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad. Sci. U. S. A 104, 19067–19072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallace DC (2007) Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu. Rev. Biochem 76, 781–821 [DOI] [PubMed] [Google Scholar]
- 33.Cadenas E (2004) Mitochondrial free radical production and cell signaling. Mol. Aspects Med 25, 17–26 [DOI] [PubMed] [Google Scholar]
- 34.Borras C et al. (2007) Mitochondrial oxidant generation is involved in determining why females live longer than males. Front. Biosci 12, 1008–1013 [DOI] [PubMed] [Google Scholar]
- 35.Duckles SP et al. (2006) Estrogen and mitochondria: a new paradigm for vascular protection? Mol. Interv 6, 26–35 [DOI] [PubMed] [Google Scholar]
- 36.Simpkins JW et al. (2008) Estrogen actions on mitochondria – physiological and pathological implications. Mol. Cell. Endocrinol 290, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Razmara A et al. (2008) Mitochondrial effects of estrogen are mediated by ERα in brain endothelial cells. J. Pharmacol. Exp. Ther 325, 782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levin ER (2001) Cell localization, physiology, and nongenomic actions of estrogen receptors. J. Appl. Physiol 91, 1860–1867 [DOI] [PubMed] [Google Scholar]
- 39.Wu TW and Brinton RD (2004) Estrogen membrane receptor imaging coupled with estradiol activation of intracellular calcium rise and ERK activation in single neurons. Abstr. Soc. Neurosci 659, 654 [Google Scholar]
- 40.Milner TA et al. (2008) Nuclear and extranuclear estrogen binding sites in the rat forebrain and autonomic medullary areas. Endocrinology 149, 3306–3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng CM et al. (2001) Estrogen augments glucose transporter and IGF1 expression in primate cerebral cortex. FASEB J 15, 907–915 [DOI] [PubMed] [Google Scholar]
- 42.Simpkins JW and Dykens JA (2008) Mitochondrial mechanisms of estrogen neuroprotection. Brain Res. Rev 57, 421–430 [DOI] [PubMed] [Google Scholar]
- 43.Gottlob K et al. (2001) Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 15, 1406–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyamoto S et al. (2008) Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ 15, 521–529 [DOI] [PubMed] [Google Scholar]
- 45.Singh M (2001) Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine 14, 407–415 [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Segura LM et al. (2007) Estradiol, insulin-like growth factor-I and brain aging. Psychoneuroendocrinology 32 (Suppl. 1), S57–S61 [DOI] [PubMed] [Google Scholar]
- 47.Carro E et al. (2002) Serum insulin-like growth factor I regulates brain amyloid-β levels. Nat. Med 8, 1390–1397 [DOI] [PubMed] [Google Scholar]
- 48.Resnick SM et al. (1998) Effects of estrogen replacement therapy on PET cerebral blood flow and neuropsychological performance. Horm. Behav 34, 171–182 [DOI] [PubMed] [Google Scholar]
- 49.Maki PM and Resnick SM (2000) Longitudinal effects of estrogen replacement therapy on PET cerebral blood flow and cognition. Neurobiol. Aging 21, 373–383 [DOI] [PubMed] [Google Scholar]
- 50.Rasgon NL et al. (2005) Estrogen use and brain metabolic change in postmenopausal women. Neurobiol. Aging 26, 229–235 [DOI] [PubMed] [Google Scholar]
- 51.Liang WS et al. (2008) Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U. S. A 105, 4441–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet 39, 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martins IJ et al. (2006) Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol. Psychiatry 11, 721–736 [DOI] [PubMed] [Google Scholar]
- 55.Reiman EM et al. (2008) Cholesterol-related genetic risk scores are associated with hypometabolism in Alzheimer’s-affected brain regions. Neuroimage 40, 1214–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosconi L et al. (2008) Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J. Nucl. Med 49, 390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blalock EM et al. (2003) Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J. Neurosci 23, 3807–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blalock EM et al. (2004) Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U. S. A 101, 2173–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reiman EM et al. (2004) Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. U. S. A 101, 284–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rowe WB et al. (2007) Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J. Neurosci 27, 3098–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller JA et al. (2008) A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J. Neurosci 28, 1410–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ishii K and Minoshima S (2005) PET is better than perfusion SPECT for early diagnosis of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 32, 1463–1465 [DOI] [PubMed] [Google Scholar]
- 63.Blass J et al. (2000) Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann. N. Y. Acad. Sci 903, 204–221 [DOI] [PubMed] [Google Scholar]
- 64.Brewer GJ et al. (2006) Prevention of age-related dysregulation of calcium dynamics by estrogen in neurons. Neurobiol. Aging 27, 306–317 [DOI] [PubMed] [Google Scholar]
- 65.Khachaturian ZS (1989) Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N. Y. Acad. Sci 568, 1–4 [DOI] [PubMed] [Google Scholar]
- 66.LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci 3, 862–872 [DOI] [PubMed] [Google Scholar]
- 67.Landfield P (1987) ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol. Aging 8, 346–347 [DOI] [PubMed] [Google Scholar]
- 68.Toescu EC et al. (2004) Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci 27, 614–620 [DOI] [PubMed] [Google Scholar]
- 69.Dreses-Werringloer U et al. (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell 133, 1149–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu TW et al. (2005) 17β-Estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience 135, 59–72 [DOI] [PubMed] [Google Scholar]
- 71.Zhao L et al. (2005) 17β-Estradiol induces Ca2+ influx, dendritic and nuclear Ca2+ rise and subsequent cyclic AMP response element-binding protein activation in hippocampal neurons: a potential initiation mechanism for estrogen neurotrophism. Neuroscience 132, 299–311 [DOI] [PubMed] [Google Scholar]
- 72.Brinton RD (2001) Cellular and molecular mechanisms of estrogen regulation of memory function and neuroprotection against Alzheimer’s disease: recent insights and remaining challenges. Learn. Mem 8, 121–133 [DOI] [PubMed] [Google Scholar]
- 73.Nilsen J et al. (2002) Dual action of estrogen on glutamate-induced calcium signaling: mechanisms requiring interaction between estrogen receptors and src/mitogen activated protein kinase pathway. Brain Res 930, 216–234 [DOI] [PubMed] [Google Scholar]
- 74.Nilsen J and Brinton RD (2003) Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc. Natl. Acad. Sci. U. S. A 100, 2842–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murphy AN et al. (1996) Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc. Natl. Acad. Sci. U. S. A 93, 9893–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Asthana S et al. Frontiers proposal. National Institute on Aging “bench to bedside: estrogen as a case study”. Age Omaha) (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]