

Abstract
Herein, we review a translational development plan to advance allopregnanolone to the clinic as a regenerative therapeutic for neurodegenerative diseases, in particular Alzheimer’s. Allopregnanolone, an endogenous neurosteroid that declines with age and neurodegenerative disease, was exogenously administered and assessed for safety and efficacy to promote neuro-regeneration, cognitive function and reduction of Alzheimer’s pathology. Allopregnanolone-induced neurogenesis correlated with restoration of learning and memory function in a mouse model of Alzheimer’s disease and was comparably efficacious in aged normal mice. Critical to success was a dosing and treatment regimen that was consistent with the temporal requirements of systems biology of regeneration in brain. A treatment regimen that adhered to regenerative requirements of brain was also efficacious in reducing Alzheimer’s pathology. With an optimized dosing and treatment regimen, chronic allopregnanolone administration promoted neurogenesis, oligodendrogenesis, reduced neuroinflammation and beta-amyloid burden while increasing markers of white matter generation and cholesterol homeostasis. Allopregnanolone meets three of the four drug-like physicochemical properties described by Lipinski’s rule that predict the success rate of drugs in development for clinical trials. Pharmacokinetic and pharmacodynamic outcomes, securing GMP material, development of clinically translatable formulations and acquiring regulatory approval are discussed. Investigation of allopregnanolone as a regenerative therapeutic has provided key insights into mechanistic targets for neurogenesis and disease modification, dosing requirements, optimal treatment regimen, route of administration and the appropriate formulation necessary to advance to proof of concept clinical studies to determine efficacy of allopregnanolone as a regenerative and disease modifying therapeutic for Alzheimer’s disease.
Keywords: Neurogenesis, Neurosteroid, Regenerative medicine, Treatment regimen, Pharmacokinetics, Pharmacodynamics
1. Introduction
Neurogenic mechanisms in brain are novel therapeutic targets to sustain neurological function and to prevent, delay or treat neurodegenerative diseases such as Alzheimer’s and Parkinson’s (Brinton, 2013). More than a decade of research has amassed since adult mammalian neurogenesis was detected in human brain (Eriksson et al., 1998) and has been confirmed and extensively studied in preclinical animal models. The seminal study by Eriksson et al. demonstrated unambiguously that the cell-birth marker BrdU indeed labeled adult human neurogenesis. It was recently demonstrated that a significant amount of neurogenesis occurs in the healthy aging human brain measured by a radiometric carbon dating method to birthdate neuronal and nonneuronal genomic DNA during the human lifespan (Spalding et al., 2013). The two most prominent adult neurogenic niches are the hippocampal dentate gyrus subgranular zone (SGZ) (Altman and Das, 1965; Cameron et al., 1993) and subventricular zone (SVZ) of the lateral ventricles (Altman, 1969; Luskin, 1993) reviewed by Liu and Brinton (2010). Although the evidence is less robust, data exist to support additional neurogenic niches in mammals including the cortical layers (evidence for interneurons generated after neocortical development), hypothalamus (Kokoeva et al., 2007), and cerebellum (Lee et al., 2005; Ponti et al., 2008).
Newly born dentate gyrus granule cells of the hippocampal neurogenic niche localize to the granule cell layer in the dentate gyrus and integrate into the adjacent molecular cell layer. As the new neural cells mature, dendrites sprout into the molecular cell layer to receive glutamatergic afferents from the perforant pathway of the entorhinal cortex and their axonal projections form mossy fiber synapses in the CA3 subfield to strengthen neural circuitry and function of the hippocampus. In less-described cortical neurogenesis, newly born interneurons, most destined to be GABAergic, are dispersed throughout the relatively voluminous cortical space. In the neocortex and dentate gyrus, adult neurogenic rates of replacement are similar despite differences in cell type, function, and relative numeric ratios to surrounding mature cells (Cameron and Dayer, 2008). Regenerative potential of these brain regions is possible throughout the lifespan with a neurogenic decline in aged brain (Cameron and McKay, 1999; Kuhn et al., 1996). In the SGZ, immature granule cells reach their highest level within weeks following birth in rodents; early postnatal rodents are roughly equivalent to birth maturity in humans.
Newborn granule cells are present in advanced aged mammalian brains and animal models demonstrate that the new granule cells possess intrinsic neuronal properties required for integration into the neural network. While the significance of functional neurogenesis in human AD remains unknown, new evidence demonstrates that a significant amount of neurogenesis occurs in the healthy aging human brain. Knoth et al. examined a large number of post-mortem brains that covered the lifespan range of 0–100 years. It was reported that human neurogenesis declines with age and doublecortin-positive cells decreased about tenfold from childhood to old age (Knoth et al., 2010) and the numbers roughly parallel the rate of neurogenesis in aging rodent models (Fig. 1). Recently, Spalding et al. used a 14C labeling method that relied on a serendipitous spike in atmospheric 14C levels resulting from nuclear bomb fallout (Spalding et al., 2013). The results 14 of these studies revealed that the age of the 14C-labeled DNA within adult hippocampal neurons indicate that the neurons were born during adulthood. Spalding et al. determined that each year approximately 1.75 percent of the neurons turnover within the self-renewing fraction with only a modest decline during aging. A best-fit scenario model predicted that approximately 35 percent of the hippocampal cells were cycling corresponding to slightly less than the proportion that constitute the entire dentate gyrus region. The authors estimated that the hippocampal dentate gyrus of human brain produces around 700 new neurons per day. At this rate, enough neurons could be replaced in the hippocampus to regenerate the entire hippocampal neurogenic region over the lifespan suggesting that the potential amount of neurogenesis is very significant. Interestingly, the rate of neurogenesis in adult humans was similar by comparison to the rate determined in adult rodents but the rate did not decline as steeply as is known in rodents suggesting that humans may rely on neurogenesis more during the aging process. Compared to developmental periods there are relatively few new granule neurons generated in the advanced aged rodent hippocampus yet the cells are functionally capable of survival and retain normal granule cell morphology and spine density (Morgenstern et al., 2008). Adult born neurons may also be distinctly different and possess higher plasticity and functionality than aged mature neurons. Compared to the perinatal period of rapid brain development, adult neurogenic granule cells mature at a slower rate which could be attributed to less than optimal levels of neurosteroids and other neurotrophic factors. Newly born adult rodent model neurons that survive possess appropriate synaptic density and can form afferent glutamatergic connections (Morgenstern et al., 2008). Newborn dentate granule cells have been shown to functionally integrate into the neural circuitry (van Praag et al., 2002) and to influence hippocampal-dependent processes including spatial pattern recognition (Clelland et al., 2009).
Fig. 1.
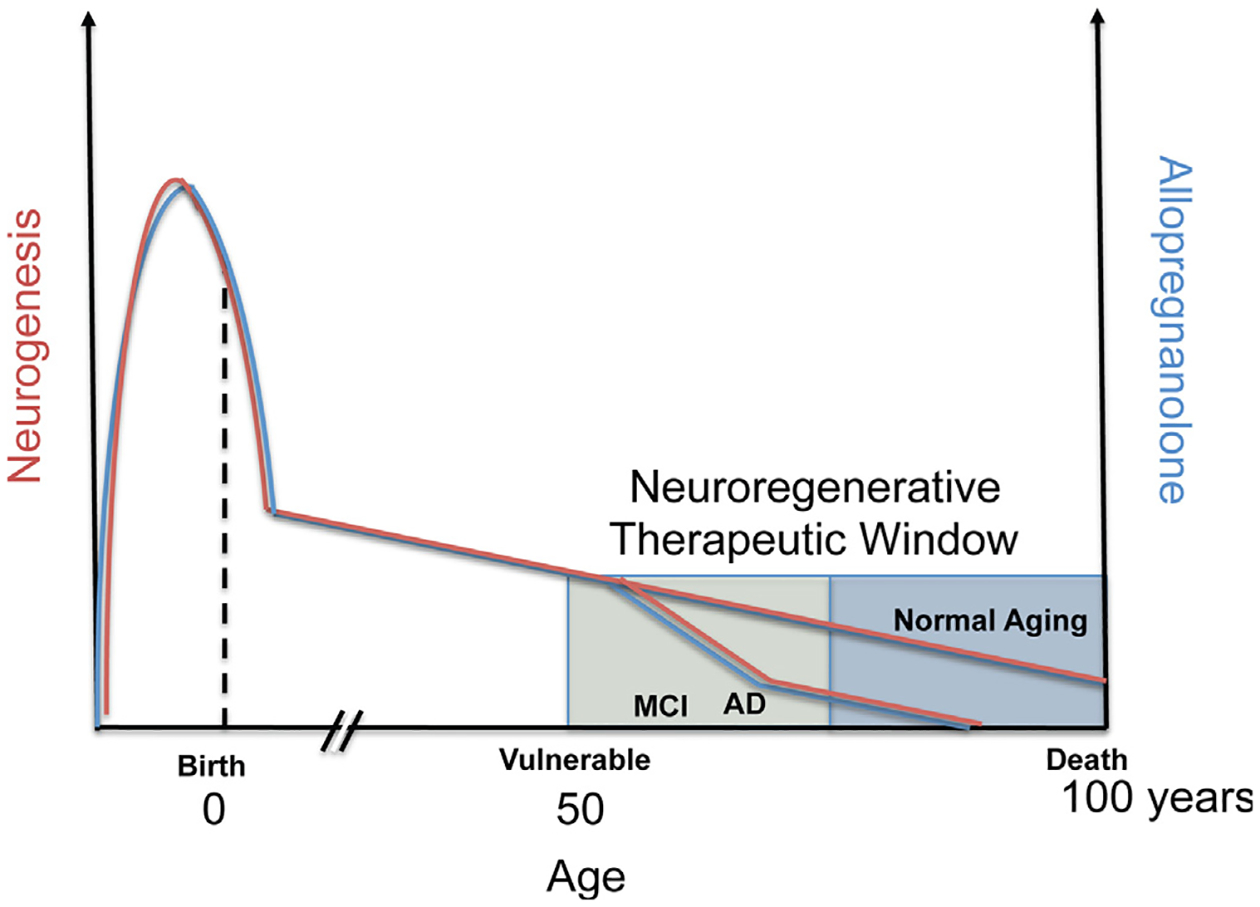
Optimal allopregnanolone (Allo) therapeutic regimen applied to therapeutic window to protect from Alzheimer’s disease (AD). Endogenous Allo (blue colored line) is produced both locally within the brain and from peripheral sources, including maternal circulation pre-natal, and post-natal and adult from the gonads and adrenal cortex in both male and female. In this generalized diagram, Allo and neurogenesis (red colored line) correlate throughout the life span and in disease. Most notable is the elevated level of Allo during fetal brain development that peaks and then sharply declines before birth (Hirst et al., 2009; Nguyen et al., 2003; Westcott et al., 2008; Luisi et al., 2000). Not depicted are the cyclic fluctuations in progesterone and closely follows progesterone levels in vivo. Also not depicted are the fluctuations in Allo during puberty. In adults, levels of neurosteroids including Allo gradually decline with advanced age and can be associated with chronological age or more specifically by what is often called endocrine age which considers the unique or categorical endocrine system status. In the presymptomatic and mild cognitive impairment (MCI) stages before AD, circulating blood and brain cortex Allo levels sharply decline correlating with onset of AD (Marx et al., 2006; Naylor et al., 2010). In most animal models of AD, the decline in neurogenesis correlates with temporal progression of AD pathology (Lazarov and Marr, 2010). Human neurogenesis declines with age and doublecortin-positive cells decreased approximately ten-fold from 0 to 100 years of age (Knoth et al., 2010) and human data roughly parallel the rate of neurogenesis in aging rodent models. Another method in which post-mortem brain genomic DNA samples containing unique radiometric signatures due to a spike in atmospheric 14C levels from global nuclear bomb fallout were used to determine neuronal and nonneuronal cell birthdates (Spalding et al., 2013). The results of these studies revealed that the age of the 14C-labeled DNA within adult hippocampal neurons were incorporated during adulthood. Each year approximately 1.75 percent of neurons turned over within the self-renewing fraction with only a modest decline during aging. A best-fit scenario model predicted that approximately 35 percent of the hippocampal cells were cycling corresponding to slightly less than the proportion that constitute the entire dentate gyrus region. Spalding and colleagues estimated that the hippocampal dentate gyrus of human brain produces around 700 new neurons per day. At this rate, enough neurons could be regenerated in the hippocampus to replace the dentate gyrus region over the human lifespan suggesting that the potential amount of neurogenesis is very substantial. Interestingly, the rate of neurogenesis in adult humans was similar by comparison to the rate determined in adult rodents but the rate did not decline as steeply as is known in rodents suggesting that humans may rely on neurogenesis more during the aging process. Chronic stress is associated with depressed responsiveness to constant levels of Allo and in correlation, chronic stress inhibits neurogenesis and exacerbates AD and loss of memory (not depicted) (Bengtsson et al., 2012, 2012). Further, neurodegeneration, a hallmark of AD negatively correlates with levels of Allo (also not depicted). The graphical representation suggests a prominent role of Allo in the neuroendocrine axis, its role both in neurogenesis and AD progression.
Glutamatergic and cholinergic synaptic inputs penetrate the SGZ of the adult dentate gyrus, yet the adult dentate gyrus SGZ retains an embryonic-like state with nearly exclusive GABAAR chloride channel depolarizing inputs influencing the local micro-environment of progenitors in the neurogenic niche (Tozuka et al., 2005). Mature neurons typically respond to GABA as an inhibitory neurotransmitter. In neural progenitor cells, the chloride ion gradient is reversed and GABA is excitatory to efflux of chloride. Depolarization of the progenitor cell activates molecular events that regulate cell proliferation. Adult-generated granule cells initially receive GABAergic input from local interneurons, isolated from extrinsic excitatory input (Overstreet Wadiche et al., 2005). GABAAR excitation initiates a cascade of events leading to calcium influx in adult neuroprogenitor cells subsequently inducing the accumulation of a neurogenic transcription factor, NeuroD (Tozuka et al., 2005). Upon GABA exposure, Type-2a cells display inward currents in SGZ (Tozuka et al., 2005). Depolarization events promote subsequent transcription factor-activated processes, mitosis, and granule cell maturation in the weeks following cell division (Overstreet-Wadiche et al., 2006).
2. Allopregnanolone (Allo) and neurogenesis
2.1. Allopregnanolone in brain development, aging and Alzheimer’s disease (AD)
Humans are naturally exposed to Allo and other neurosteroids throughout life. During the third trimester of human pregnancy endogenous levels of Allo in humans can naturally reach 150–200 nmol/L for both mother and fetus (Fig. 1) (Luisi et al., 2000). The placenta is both a barrier and a major source of Allo and other neurosteroids during gestation. During embryonic development, neuroepithelial cells proliferate in response to neurogenic cues, often from contact with the vascular niche, to form the embryonic brain (Shen et al., 2004). Neuroepithelial cells then develop the ventricular zone in the embryonic brain and give rise to mitotic radial glial cells that can differentiate into either glial cells or neurons (Noctor et al., 2001). Asymmetric radial glial cell expansion of the cortical layers could be the factor that allowed humans to possess remarkably evolved cortical layers required for human cognitive function (Kriegstein et al., 2006). In uteroplacental deficiency or very premature infancy slower brain development can result because of early withdrawal of the critical neurosteroid milieu. The preterm brain is also protected by de novo synthesis of neurosteroids in rat and sheep fetal stress models such as growth restriction or induced-hypoxia increased Allo levels for neuroprotective benefits (Hirst et al., 2009; Trotter et al., 1999). Neural progenitors have been isolated in vitro from post-mortem 11-week post-natal and adult human brain demonstrating that these progenitors are present in adults (Palmer et al., 2001). The long-term consequences of early neurosteroid deprivation remain to be studied in brain development (Saria et al., 2010) and could be associated with increased risk for cognitive impairment.
At low concentrations, neurosteroids bind to GABAAR at sites that differ from GABA, benzodiazepines, ethanol, and barbiturate binding sites and can act as positive or negative modulators of GABAAR function (Gee et al., 1987, 1988). Allopregnanolone (Allo; 3α-hydroxy-5α-pregnan-20-one) (also known as AP, APα, or THP) is a potent positive allosteric activator of the GABAAR channels whereby at nanomolar concentrations enhances the action of GABA at GABAAR and at higher concentrations directly activates GABAAR. Allo binds to two transmembrane sites of the hetero-pentameric GABAAR (Hosie et al., 2006). GABA and neurosteroid transmembrane binding sites each occur twice per channel complex. GABAAR binding sites are conserved in all receptor complex subtypes with the general subunit stoichiometry 2α:2β:1γ. The γ subunit in a subset of extrasynaptic GABAAR channel complexes can be replaced by the neurosteroid-sensitive δ subunit (Meldrum and Rogawski, 2007). The αβγ and αβδ receptor composites are pharmacologically distinct. GABA plays a key role in generation of the spontaneous network activity of immature dentate granule cells (Owens and Kriegstein, 2002; Sipila et al., 2004). In neuroprogenitor cells, GABAergic depolarization of the uniquely reversed membrane potential underlies the trophic actions of Allo. Adult dentate granule cells, devoid of the α1 subunit, are also subjected to tonic GABAergic signaling via δ subunit containing GABAARs mediated by surrounding synaptic boutons of local interneurons (Overstreet Wadiche et al., 2005). By electron microscopy, membrane bound GABAARs have been observed on BrdU co-labeled adult hippocampal SGZ progenitor cells (Mayo et al., 2005). GABAergic stimulation of neural progenitor cells elicits an efflux of chloride and a concomitant influx of calcium that contributes to the induction of cell signaling events leading to gene transcription of mitotic genes and down-regulation of anti-mitotic genes (Wang et al., 2005). GABAergic signaling likewise controls proliferation of adult progenitor cells within the SVZ neurogenic niche (Liu et al., 2005).
Allo and other trophic factors were diminished in both blood and brain of AD patients compared to age-matched controls (Fig. 2) (Marx et al., 2006; Naylor et al., 2010; Weill-Engerer et al., 2002). An early indicator of AD is the loss of episodic and semantic memory (Perry et al., 2000). AD diagnostic imaging studies using volumetric MRI revealed a decreased hippocampal volume due to atrophy of gray matter, i.e. neurodegeneration in amnestic subtype mild cognitive impairment in the progression to AD (Whitwell et al., 2007). Neuropathological hallmarks of AD include extracellular and intracellular deposition of β-amyloid (Aβ) protein, neurofibrillary tangles and neurodegeneration. Several studies have reported decreased neurogenesis in AD mouse models (Lazarov and Marr, 2010).
Fig. 2.
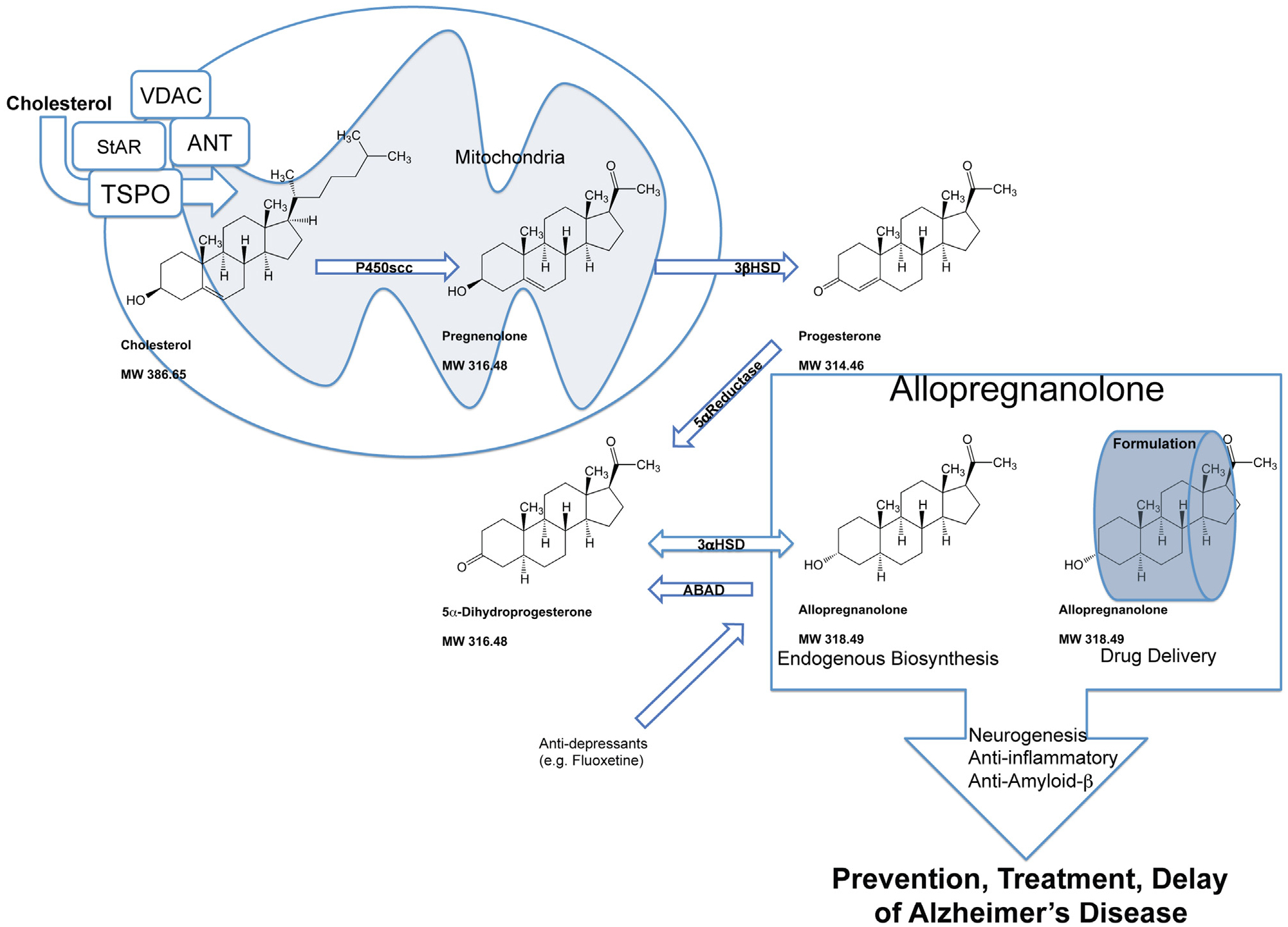
Neurosteroid biosynthesis is a multi-step enzymatic pathway. Cholesterol homeostasis, recruitment to the mitochondrial compartment, enzymatic reduction and transport to neural and glial cells or their precursor cells require regulation of multiple mechanisms critical to Alzheimer’s disease treatment. Allo mechanisms of action promote neurogenesis, oligodendrogenesis, and on a systems-level inhibit excess inflammation and β-amyloidogenesis. In the central and peripheral nervous systems, neurosteroid synthesis occurs in myelinating glial cells, astrocytes, and several neuronal cell types including neural progenitors. Cholesterol is supplied to these cell types and presented to the mitochondria. The mitochondrial membrane translocator protein (TSPO) controls the uptake of cholesterol and the synthesis of neuroactive steroids (Rupprecht et al., 2010). TSPO-associated proteins form a cholesterol transport pore in the mitochondrial inner membrane that include the steroidogenic acute regulatory protein (StAR), voltage-dependent anion channel protein (VDAC), and adenine nucleotide transporter protein (ANT). The mitochondrial pore transports cholesterol to the mitochondrial matrix to be converted into pregnenolone by the cytochrome P450 side-chain cleavage (CYP450scc) enzyme (Liu et al., 2006). Pregnenolone diffuses to the cytosol and then is converted to progesterone by the 3β-hydroxysteroid dehydrogenase (3β-HSD) enzyme. Peripherally derived progesterone and Allo cross the blood-brain barrier to also contribute to the neurosteroid concentration. Allo is synthesized from progesterone in two enzymatic steps by 5α-reductase (5α-R) type-I and 3α-hydroxysteroid dehydrogenase (3α-HSD) in the brain de novo (Mellon, 2007; Mellon et al., 2001). The rate-limiting step in neurosteroidogenesis is the reduction of progesterone to 5α-dihydroprogesterone (5α-DHP) by 5α-R. Subsequently, 3α-HSD catalyzes conversion of 5α-DHP into Allo. Amyloid beta-binding alcohol dehydrogenase or (also known as ABAD; SCHAD; 17βHSD10) is an enzyme that associates with mitochondria and facilitates back conversion of Allo to 5α-DHP (He et al., 2005; Yang et al., 2005). Additionally, anti-depressants such as fluoxetine are pro-neurogenic and have been shown to increase Allo production in the brain (Malberg et al., 2000; Uzunova et al., 2004, 2006). Endogenous Allo or optimal therapeutic dose of Allo with proper drug delivery strategy promote neurogenesis, oligodendrogenesis, and on a systems-level inhibit excess inflammation and β-amyloidogenesis.
Despite substantial evidence in AD animal models that demonstrate reduced neurogenesis with degree of AD pathology, data in human AD brain have yet to definitively demonstrate an AD-related decline in neural proliferation. In a cohort of senile patients, doublecortin, a marker for new neurons was increased and the investigators claimed that neurogenesis was increased as a compensatory mechanism of neurodegeneration (Jin et al., 2004). These unexpected findings have been disputed and other studies have demonstrated that in presenile human AD brain, most cell proliferation could be attributed to glial and vasculature-associated changes and found no evidence for altered neurogenesis in the dentate gyrus (Boekhoorn et al., 2006). Another explanation suggests that in AD brains a subpopulation of dysfunctional neurons undergo aberrant entry into the cell cycle that could lead to false positives in searches for neurogenic cell proliferation in AD brains. The aberrant cells replicate their genomes but fail to divide leading to apoptotic mechanisms instead of neurogenesis (Busser et al., 1998; Herrup, 2010). Recently, it was demonstrated that in humans, hippocampal neurogenesis occurs throughout adulthood and only modestly tapers with advanced age (Spalding et al., 2013). In future studies, the method of carbon dating used by Frisén and colleagues to retrospectively birthdate neuronal nuclei could also be used to determine the level of neurogenesis in AD victims. Thus the potential for regenerative therapeutics in AD remains an active area of investigation.
2.2. Neurosteroidogenesis
Neuroactive steroids are endogenous molecules that are present in the central nervous system and the entire peripheral system as the gonads and adrenal glands also produce neurosteroids. In the central and peripheral nervous systems, neurosteroid synthesis occurs in myelinating glial cells, astrocytes, and several neuronal cell types. Cholesterol, transported from ApoE containing LDL particles, is supplied to these cell types and presented to the mitochondria (Fig. 2). The 18 kDa outer mitochondrial membrane translocator protein (TSPO) in steroidogenic cells, controls the uptake of cholesterol and thus the synthesis of neuroactive steroids (Rupprecht et al., 2010). Cholesterol binding protein TSPO associates with other proteins that include the 30 kDa steroidogenic acute regulatory protein (StAR), 32 kDa voltage-dependent anion channel protein (VDAC), and 30 kDa adenine nucleotide transporter protein (ANT) to form a macromolecular signaling complex to transport cholesterol through the mitochondrial inner membrane to the mitochondrial matrix. TSPO regulates cholesterol compartmentalization in the mitochondria and in addition to cholesterol trafficking, could be important for mitochondrial biogenesis and thus mitochondrial function.
Activated by several different neurosteroids, cAMP dependent mechanisms then modulate protein kinase C epsilon (PKCε) to increase TSPO expression (Batarseh and Papadopoulos, 2010). Calcium ion induced nuclear translocation of ERK requires protein kinase A (PKA) activation. ERK-PKA crosstalk was found to be important for calcium-stimulated CREB-dependent transcription (Impey et al., 1998). Calcium entry also stimulates calmodulin dependent protein kinases II/IV to activate CREB genes important for neurogenesis and learning and memory. Activated PKCε translocates to the outer mitochondrial membrane, to interact with the TSPO-associated proteins VDAC and ANT.
Within the mitochondrial matrix, transported cholesterol is converted into the neurosteroid pregnenolone by the cytochrome P450 side-chain cleavage (CYP450scc) enzyme (Liu et al., 2006) (Fig. 2). Pregnenolone diffuses through the mitochondrial membrane whereby its conversion to progesterone is facilitated by the 3β-hydroxysteroid dehydrogenase (3β-HSD) enzyme in the cytosol. Progesterone is converted by enzymatic reactions to its metabolites including Allo in the brain and periphery. Allo is synthesized in the brain from progesterone by the sequential enzymatic steps of 5α-reductase (5α-R) type-I and 3α-hydroxysteroid dehydrogenase (3α-HSD) (Mellon, 2007; Mellon et al., 2001). The rate-limiting step in neurosteroidogenesis is the unidirectional reduction of progesterone to 5α-dihydroprogesterone (5α-DHP) by 5α-R. Subsequently, 3α-HSD catalyzes conversion of 5α-DHP into Allo. Both 5α-R and 3α-HSD enzymes are responsible for Allo synthesis and have been found to functionally express in pluripotent progenitor cells (Melcangi et al., 1996). This neuroactive steroid synthesis and degradation system is also importantly influenced by so-called endocrine disrupting chemicals that mimic endogenous neurosteroids such as phytochemicals and synthetic pollutants (Melcangi and Panzica, 2009). Amyloid-beta binding alcohol dehydrogenase or ABAD (aka ERAB; 17βHSD; SCHAD) is an enzyme that associates with mitochondria and has many enzymatic functions including retro-conversion of Allo to 5α-DHP (Yang et al., 2005). Intracellular oxidation of Allo via ABAD appears to maintain the normal function of GABAergic neurons (He et al., 2005). ABAD was abnormally overexpressed in activated astrocytes and enzymatic levels were high in AD brains (He et al., 2005). It is plausible that elevated expression of ABAD leads to the observed decrease in cortical Allo observed in AD. Our previous data show that neurosteroids can lower ABAD load (Chen et al., 2011). Hippocampus and cortex regions display specific expression patterns of progesterone converting enzymes important for their unique demand for neurosteroids (Mellon, 2007; Mellon et al., 2001).
The GABAergic system and its interrelation with the stress response controls the neurogenic niches in brain and presents unique molecular targets for neurosteroids and their rationale-based chemical structure derivatives (Kaminski et al., 2004). Stress has been shown to be an important determinant of neurogenic status. High levels of glucocorticoids, key mediators of the stress response, were correlated with a decline in granule cell proliferation and thus neurogenic potential (Cameron and McKay, 2001). In addition, chronic stress was shown to cause memory impairment in rodents (de Quervain et al., 1998) and humans (de Quervain et al., 2000) and genetic variations in the glucocorticoid system associate with AD risk (de Quervain et al., 2004). Chronically elevated levels of Allo that occur under severe chronic stress conditions are associated with down-regulation and malfunction of GABAAR and can cause memory impairment. Risk exposure assessments of chronic and acute exposure to neurosteroids can be estimated based on our current knowledge of the neuroendocrine system in normal and disease states.
3. Regenerative neurosteroid: allopregnanolone (Allo)
3.1. Allo efficacy in promoting neurogenesis
Our studies have demonstrated a correlation between Allo-induced neural progenitor cell survival and improved memory function in the triple transgenic mouse model of AD (3xTgAD) (Brinton, 2013; Chen et al., 2011; Irwin et al., 2011; Singh et al., 2011; Wang et al., 2010). The 3xTgAD mouse was developed by overexpression of Swedish mutant APP, mutant P301L Tau in the homozygous mutant of presenilin 1 (M146V) knock-in mouse and age-dependently develop cortical and hippocampal tangle-like pathology, intraneuronal Aβ deposition, extraneuronal Aβ deposition, and neurological deficits (Oddo et al., 2003a, 2003b). Consistent with the human AD brain neurosteroid profile, basal concentration of Allo in blood plasma (0.47 ± 0.88 ng/ml) was significantly lower (p < 0.05) than in cortex (10.36 ± 1.43 ng/g) in young adult male non-transgenic (nonTg) mice, indicating a higher brain accumulation, attributed to locally synthesized Allo in specific brain regions as required for synaptic function. 3xTgAD mice exhibited a lower basal level of Allo in the cerebral cortex (3xTgAD, 6.49 ± 2.02 ng/g versus nonTg, 10.36 ± 1.43 ng/g), suggesting either impairment of upstream Allo enzymatic production (Fig. 1) or accelerated Allo metabolism in 3xTgAD mice brain (Wang et al., 2010). Within the SGZ and SVZ in male and female 3xTgAD mice a decline in neurogenesis correlated with age related AD-like pathology progression (Brinton and Wang, 2006; Rodriguez et al., 2008, 2009; Wang et al., 2010). Our studies demonstrated that Allo promoted neurogenesis in the hippocampal SGZ to reverse learning and memory deficits. Neural progenitor cell proliferation and subsequent cell survival were determined by analysis of BrdU incorporation into the mitotic cells labeled shortly after exposure to acute Allo. Triple-immunolabeling coronal sections verified the phenotype of in vivo newly formed BrdU-positive cells in mouse hippocampus that were stereologically analyzed and derived from 3-month-old 3xTgAD mice treated with a single subcutaneous injection of 10 mg/kg Allo (Wang et al., 2010). Confocal microscopy identified BrdU-positive cells colocalized with DCX labeled young immature neurons alone or together with NeuN a marker for mature neurons. Neural progenitor cell survival was further confirmed by immunolabeling coronal sections derived from Allo (subcutaneous, 10 mg/kg) treated 3xTgAD mouse dentate gyrus 22 days post-Allo treatment and post-behavioral analyses. Analyses of Allo-treated mouse brains revealed BrdU-positive cells located deep within the granular cell layer, indicating the migration of newly formed cells from the SGZ to the granule cell layer (Wang et al., 2010). These in vivo data indicated that newly formed cells, generated following Allo treatment, expressed a neuronal phenotype.
3.2. Allo efficacy in promoting learning and memory
Previously we assessed associative learning and memory function using the hippocampal-dependent trace eye-blink conditioning paradigm. In 3xTgAD mice, 3-months of age, the basal level of BrdU-positive cells in the SGZ of 3xTgAD mice was significantly lower relative to non-transgenic (nonTg) mice, despite the lack of evident AD pathology (Wang et al., 2010). Allo significantly increased, in a dose-dependent manner, BrdU-positive cells in the SGZ of 3xTgAD mice and restored SGZ proliferation to normal magnitude of nonTg mice (Wang et al., 2010). Coinciding with deficits in adult neural progenitor proliferation, 3xTgAD mice exhibited deficits in learning and memory. Allo reversed the cognitive deficits to restore learning and memory performance to the level of normal nonTg mice (Wang et al., 2010). In 3-month-old 3xTgAD mice, Allo increased proliferation and promoted the survival of newly born hippocampal dentate granule cells (Wang et al., 2010). Neural progenitor cell numbers significantly correlated with Allo-induced memory performance. Deficits in neurogenesis were detected in this genetic model of AD prior to immunodetectable Aβ. Comparable to the 3-month mice, Allo increased proliferative activity, promoted the survival of newly born hippocampal dentate granule cells, and restored cognitive function in 6- and 9-month-old 3xTgAD mice while having no significant impact on their age-matched normal nonTg counterparts (Singh et al., 2011). BrdU-positive neural progenitor cell survival was assessed after 3 weeks, following a single exposure to Allo (10 mg/kg). During the 3-week period after neural stimulation by a single dose of Allo, trace conditioning, a hippocampal-dependent associative learning and memory task, was performed. The 3-week period allowed granule cells sufficient time to proliferate, migrate to the border between the hilus and the granule cell layer and incorporate into the existing neural network. Thus we found that Allo significantly increased survival of BrdU-positive cells and recovered hippocampal-dependent cognition in 6-, and 9-month-old 3xTgAD mice, in the presence of intraneuronal Aβ. It is important to state that Allo was ineffective after development of extraneuronal Aβ plaques that appear in 12-month-old 3xTgAD mice. Remarkably, cognition was restored to maximum by the first day of trace eyeblink conditioning, only 1 week following a single exposure to Allo in pre-plaque mice. Hippocampal-dependent associative learning accomplished by repeated trials of an auditory tone followed by a mildly aversive shock stimulus, was sustained throughout behavioral training. Allo efficacy promoted learning and memory function in 3xTgAD mice to the extent that 100% increase in magnitude was observed in learning at the start of training compared to the age-matched vehicle-treated group. The remarkable increase in associative learning was comparable to the maximal normal nonTg mouse performance. In nonTg at 12-months of age, we observed an upward trend toward Allo-enhanced learning that did not reach significance. Single dose Allo-treatment to 15-month-old nonTg mice revealed a therapeutic benefit (Singh et al., 2011). A decline in Allo has been measured in blood samples of 40-year-old healthy males and may suggest that a decline in Allo precedes many age-related diseases (Genazzani et al., 1998). The therapeutic benefit found in aged nonTg mice suggests that at least in males, Allo therapy could reverse an age- and gender-related neurosteroid decline and thus maintain or improve cognitive abilities. Our preclinical findings in the AD mouse model provided evidence that Allo-promoted survival of newly generated neural cells and paralleled restoration of cognitive performance in the pre-plaque phase of AD pathology and in late-stage normal aging.
3.3. Allo efficacy in preventing the development of and reducing severity of Alzheimer’s pathology
Increasing evidence indicates that altered cholesterol metabolism is linked to AD pathology (Brinton, 2013; Mellon et al., 2008; Schumacher et al., 2004). In addition to the mechanism of action whereby Allo induces neurogenesis through excitation of GABAAR chloride channels in neural progenitor cells (Brinton, 2013), Allo regulates cholesterol homeostasis via mechanism that influences the Liver-X-receptor (LXR) and its associated pregnane-X-receptor (PXR) system (Chen et al., 2011). LXR is a nuclear hormone receptor abundant in the brain and acts as a molecular sensor of cholesterol levels and initiates cholesterol clearance (Jakobsson et al., 2012; Whitney et al., 2002). Loss of either LXRα or LXRβ subtype expression exacerbated AD-related pathology in APP/PS1 double transgenic mice (Zelcer et al., 2007). Loss of LXR has been shown to repress cortical neurogenesis particularly during late-embryonic stage development of layer II/III (Fan et al., 2008). LXR activation increases cholesterol efflux through increased ABCA1 and ApoE expression, and prevents overactivation of γ-secretase and overproduction of Aβ (Jiang et al., 2008; Shenoy et al., 2004; Whitney et al., 2002). LXR activation improved cognitive function in multiple mouse models of amyloidogenesis (Donkin et al., 2010; Leduc et al., 2010a; Schultz et al., 2000; Whitney et al., 2002; Xiong et al., 2008; Yang et al., 2006). LXRs are recruited to ABCA1 gene promoter regions and ApoE expression to decrease Aβ plaque formation and increase Aβ clearance (Koldamova et al., 2010) through phagocytosis by microglia (Terwel et al., 2011). Both fibrillar Aβ (Koenigsknecht-Talboo and Landreth, 2005) and soluble Aβ (Mandrekar et al., 2009) are cleared by microglia. Further, LXRs are recruited to ABCG1 promoter in a ligand-dependent manner to alter epigenetic histone methylation allowing for a relaxed chromatin structure accessible to further gene expression and cholesterol efflux (Jakobsson et al., 2012). LXRs reduce neuroinflammation by inhibition of several inflammatory genes that were shown to include Ccl2, Ccl5, Cxcl10, Ifit1, Il1b, Lcn2, Ptgs2, Slc7a11, TNfrsf5, and Traf1 (Zelcer et al., 2007). Inflammatory cytokines reach high levels in AD and when suppressed by LXR activation, enhance phagocytic activity of microglia and thus Aβ clearance. LXR ligands activate pregnane-X-receptor (PXR) (Riddell et al., 2007). Results from our analyses indicated that in parallel with an Allo-induced increase in LXR expression in the pre-pathology condition, Allo also increased PXR expression in the pre-pathology 3xTgAD mouse brain (Chen et al., 2011). PXR activation induces cytochrome P450 3A (CYP3A) enzymes including CYP3A4 and CYP3A13 and this leads to cholesterol hydroxylation and activation of organic anion transporters for cholesterol extrusion (Sun et al., 2003). In addition to increased LXR and PXR expression, Allo treatment initiated in pre-Aβ pathology 3-month-old 3xTgAD mice treated once per week for six months displayed increased expression of 3-hydroxy-3-methyl-glutaryl-CoA-reductase (HMG-CoA-R) (Chen et al., 2011).
The increase in HMG-CoA-R is at first paradoxical as it is the rate-limiting enzyme in cholesterol synthesis. HMG-CoA reductase is also required for production of oxysterols that activate LXR and PXR-mediated gene transcription for cholesterol and lipid transport proteins (Leduc et al., 2010b). Thus, the Allo-induced increase in brain LXR and PXR leads to increased cholesterol efflux, thereby reducing γ-secretase activation by cholesterol-laden lipid rafts. Allo stimulated cholesterol efflux is a plausible mechanism for the observed reduction of 27 kDa and 56 kDa intraneuronal Aβ oligomers after 6 months of once per week treatment (Chen et al., 2011).
In vivo, brain cholesterol homeostasis and intraneuronal Aβ are tightly coupled to Allo efficacy (Chen et al., 2011). Deposition of Aβ in the extracellular compartment disconnected this coupled pathway and led to a loss of Allo efficacy in advanced stages of AD-like pathology in the 3xTgAD model. Allo significantly reduced Aβ generation in hippocampus, cortex, and amygdala, which was paralleled by decreased mitochondrial Aβ-binding-alcohol-dehydrogenase (ABAD) and reduced microglia activation assessed as reduced expression of Iba-1 (Chen et al., 2011). A reduction in ABAD is important for decreased mitochondrial dysfunction and simultaneously for decreasing the amount of Allo that is enzymatically back-converted to its precursor steroid 5αDHP. In organotypic slice cultures of rat cerebellum (Schumacher et al., 2004) Allo increased myelin basic protein and delayed demyelination in Niemann-Pick C mice (Mellon et al., 2008). Allo may stimulate oligodendrocyte progenitor cells in addition to neural progenitor cells and as a metabolite of progesterone, could be responsible for observed oligodendrogenesis with progesterone treatment (Schumacher et al., 2012). In oligodendrocytes, the myelin marker CNPase was increased by once per week Allo, indicating myelinating capabilities in this mouse model (Chen et al., 2011). In summary, in the 3xTgAD mouse model Allo promoted neural proliferation while simultaneously reducing AD pathology.
4. The optimal allopregnanolone treatment regimen
4.1. Allo in vivo` once per week promotes neurogenesis and reduces AD pathology
To further advance efforts to assess the preclinical efficacy of Allo for AD, our group designed long-term treatment studies. The studies were designed to test Allo using the same age of enrollment (3xTgAD male 3-months of age; prior to overt intraneuronal Aβ), efficacious dose of 10 mg/kg via subcutaneous route of administration, matching our previous studies. In addition to neurogenic efficacy, the long-term studies were extended to determine the disease modifying effects afforded by the therapeutic regimen. Once per month, once per week, and every other day treatment regimens were tested (Chen et al., 2011). Based on measured endpoints of Aβ oligomers by immunostain and immunoblot approaches, the every other day treatment regimen was maximally efficacious but failed to increase neurogenesis. The once-per-month Allo treatment was efficacious for proliferation of neural progenitor cells but not for decreased Aβ pathology. Once per month treatment regimen (Chen et al., 2011) was similar to the single exposure treatment paradigm that improved learning and memory performance after a single exposure (Singh et al., 2011). Analyses to determine the optimal treatment paradigm indicated that Allo administered once per week for six months was maximally efficacious for both neurogenic and anti-amyloidogenic endpoints. In parallel to the 3-month-old mice administered once per week Allo, we simultaneously began treatment of a 6-month-old male 3xTgAD group. When Allo was administered beginning at 6-months of age, the age at which intraneuronal plaques are apparent in this mouse model, the appearance of Aβ pathology paralleled cessation of Allo efficacy. Once intraneuronal Aβ is extracellularly localized, the efficacy of Allo is largely diminished. This suggested to us that intraneuronal Aβ accumulation is a determining factor that focuses the window of Allo therapeutic efficacy on the early stages of AD. We hypothesize that an intermittent Allo regimen will help maintain the normal rate of neurogenesis and that early clinical intervention will lead to greatest therapeutic benefit.
4.2. Chronic continuous unrelenting exposure to Allo opposes therapeutic efficacy
Bengtsson et al. investigated the effect of chronically elevated Allo levels, mimicking physiological chronic stress levels, on cognition and brain tissue Aβ levels in female and male transgenic APPSwePSEN1ΔE9 mice (see Table 1) (Bengtsson et al., 2012). When the neurosteroid Allo was administered to young adult mice, neurotransmission and intracellular Aβ release were adversely inhibited, possibly by GABAergic mechanisms, thereby leading to increased Aβ accumulation and decrements in learning and memory function. The findings point to Allo’s biphasic effects on disease states. The results in a rapid AD progression model should not be generalized to conclude that Allo is deleterious to AD progression. More likely, the chronic continuous exposure that mimics stress levels of Allo was a treatment paradigm chosen to demonstrate that dysregulation of the mechanisms controlled by neurosteroids will further exacerbate genetically programmed AD progression and negatively impact behavior as was measured by the forced swim test. Allo was administered by continuous release at the rate of: 4.7 nmol/h = 0.0015 mg/h 24 = 0.036 mg/d and another group of mice were given a double Allo level of 9.3 nmol/h = 0.003 mg/h × 24 = 0.072 mg/d which yields 2.4 mg/kg/day (Table 1). On a single day of the continuous release regimen, Allo at the rate of 4.7 nmol/h amounts to approximately 0.036 mg/d, assuming 30 g mouse yields 1.2 mg/kg/day. This amount of Allo is approximately 8-fold less (0.036 mg vs 0.3 mg) than our single subcutaneous dose studies (Table 1) (Chen et al., 2011; Singh et al., 2011; Wang et al., 2010) wherein the mice were subcutaneously treated with a single dose: 10 mg/kg = 0.3 mg/30 g mouse, single bolus by subcutaneous injection, not daily. However, the continuous release treatment paradigm was much different pharmacokinetically and pharmacodynamically than the “treat and excrete” approach wherein the neurobiological system is stimulated and then allowed to return to baseline over the period of 1 week or longer duration. Allo plasma concentration after 24 h measured: 5.4 nmol/L, 3xTgAD vehicle and 12.6 nmol/L, 3xTgAD Allo 10 mg/kg. In nonTg mice, Allo plasma levels were 1.48 nmol/L, nonTg vehicle and 9.5 nmol/L, nonTg Allo 10 mg/kg. Allo cerebral cortex concentration after 24 h measured: 6 ng/g, 3xTgAD vehicle and 15 ng/g, 3xTgAD Allo 10 mg/kg. NonTg cerebral cortex levels measured 10 ng/g, nonTg vehicle and 22 ng/g, nonTg Allo 10 mg/kg (Wang et al., 2010). Although the levels of Allo were higher in the single pulse dose experiments, the neurobiological system was able to respond in a proactive manner to induce neurogenesis and learning and memory improvements and return to homeostasis. Comparison of intermittent bolus dosing and continuous release treatment regimens further demonstrate the importance of precise neurosteroid treatment paradigms.
Table 1.
Allopregnanolone (Allo) pre-clinical research translation in AD mouse models and existing clinical exposure data in healthy subjects. Allo blood plasma and brain concentrations and reported safety and efficacy by multiple routes of administration. Intravenous (IV) dose studies determine the pharmacokinetic properties. Assumed to be 100% bioavailable, the IV route of administration is the most quantitative, and useful for gauging alternative routes of administration. Our pre-clinical Allo dosing studies with subcutaneous (SC), transdermal (TD), intranasal (IN), and IV routes of administration that resulted in beneficial effects that decreased hallmarks of AD pathology and improved cognitive performance were tabulated alongside with other pre-clinical studies that tested long-term chronic continuous exposure to SC Allo that resulted in deleterious effects on AD pathology and long-term memory impairment. Human IV dosing is included to compare the mouse pharmacokinetic parameters to clinical Allo pharmacokinetics demonstrates the safety of Allo when blood levels are below the maximum tolerated dose indicated by sedation level. When bioavailability and species-specific allometric conversions are taken into account the pre-clinical studies that demonstrate neuroregenerative efficacy in an AD mouse model are within the safe range of dosage for humans. The AD mouse models were administered both chronic and acute dosing regimens. Allo treatment induced a significant increase in neurogenesis, with the latter regimen yielding the greater increase in neurogenesis. However, the 3/week/3 months (12/month) treatment induced a significant decrease in neurogenesis. Brain sections from 3xTgAD mice treated with Allo or vehicle were immunostained. Aβ immunoreactivity was detected and indicated that the 1/week/6 months (4/month) Allo treatment significantly decreased Aβ immunoreactivity. Efficacy of Aβ reduction in 4/month was comparable to the 12/month Allo treatment whereas the 1/month Allo treatment was ineffective at reducing Aβ immunoreactivity (Chen et al., 2011). Allo tested to mimic chronic stress levels associated with AD. In a series of studies, continuous subcutaneous osmotic pump infusion of Allo proved to be detrimental and decreased learning and memory performance while simultaneously exacerbating AD pathology markers (Bengtsson et al., 2012, 2013). Although not measured, neurogenesis possibly was inhibited in the continuous Allo exposure studies. From these tabulated studies we hypothesized that the dose and frequency of Allo are critical components of efficacy at neurogenic and pathological endpoints.
| Purpose | Species/Strain | Route | ALLO mg/kg | Vehicle | Frequency of exposure | Endpoint | Blood [ALLO] | Brain [ALLO] | Results | Safety | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre-clinical | Allo pre-clinical efficacy and route of administration development in mouse model of Alzheimer’s disease | Mouse (male) 3xTgAD: APPSwe × PS1 M146V × Tau P301L | SC | 10 | PBS/5% EtOH | Single dose | 24h | 4 ng/ml (12.5 nmol/L) C24h | 15 ng/g C24h | Increased neurogenesis | No adverse effects | Wang et al. (2010) |
| SC | 10 | PBS/5% EtOH | Single dose | 24 h time course | 34ng/ml (107nmol/L) Cmax; 0.5h Tmax | 159ng/g C0.5 h | Increased neurogenesis | No adverse effects | Irwin et al. (2013) | |||
| IV | 1.5 | 6% HβCD | Single dose | 24 h time course | 215 ng/ml (675 nmol/L) Cmax; 0.08 h Tmax | 639 ng/g Cmax | Increased CREB signaling and neurodifferentiation markers | No adverse effects other than acute mild sedation | Irwin et al. (2013) | |||
| TD | 10–50 | Various soluble formulations | Single dose | 4h; 24h | 44 ng/ml (138nmol/L) C4h | – | Increased neurogenesis | No adverse effects | Irwin et al. (2013) | |||
| IN | 3–10 | – | – | |||||||||
| SC | 10 | PBS/5% EtOH | Single dose | 3 wk | – | – | Increased neurogenesis and cell survival; improved hippocampal-dependent learning and memory | No adverse effects | Singh et al. (2011) | |||
| SC | 10 | PBS/5% EtOH | Three/wk | 3 mo | – | – | No effect on neurogenesis; decreased β-amyloid | No adverse effects | Chen et al. (2011) | |||
| SC | 10 | 20% HβCD | Once/wk | 6 mo | – | – | Increased neurogenesis and cell survival; decreased AD pathology | No serious adverse effects; acute hyperactivity followed by brief sedation | Chen et al. (2011) | |||
| Allo pre-clinical efficacy and regimen development in mouse model of Alzheimer’s disease | Mouse APPSwe × PS1ΔE9 (male and female, n = 6–11) | SC | 1.2–2.4 mg/kg/day osmotic pump | 60%SBEβCD | Continuous release | 3 mo | 1.3–2.2 ng/ml (4–7 nmol/L) Cmax wk 4 | 2.5–4.8 ng/g Cmax wk 4 | Chronic treatment for three months increased soluble Aβ1–42; impaired cognitive learning assessed one month after treatment to observe long-term effects | Accelerated Alzheimer’s pathology and memory impairment | Bengtsson et al. (2012) | |
| Mouse APPSwe × PS1ΔE9 (male and female) | SC | 2.4–4.8 mg/kg/day osmotic pump | 60%SBEβCD | Continuous release | 1 mo | – | – | Brief but chronic Allo for one month increased soluble Aβ1–40 and Aβ1–42 but was not sufficient to impair cognitive learning | Accelerated Alzheimer’s pathology | Bengtsson et al. (2013) | ||
| Mouse APPSweArc (male and female) | SC | 2.4mg/kg/day osmotic pump | 60% SBEβCD | Continuous release | 1 mo | – | – | Brief but chronic treatment for one month did not affect soluble Aβ1–40 and Aβ1–42 but impaired cognitive learning | Memory impairment | Bengtsson et al. (2013) | ||
| Clinical | Clinical trial | Human(healthy male, n = 9) Human (healthy female on oral contraceptive, n = 9) | IV | 0.09 | Albumin solution | Acute 1 h cumulative doses | During 1 h cumulative dosing | 48 ng/ml (150nmol/L) Cmax | – | Male – decreased saccadic eye movement; decrease in contentedness | No adverse effects other than self reported sedation | van Broekhoven et al. (2007) |
| 32 ng/ml (100nmol/L) Cmax | – | Female – decreased saccadic eye movement; increase in contentedness | ||||||||||
| Clinical trial | Human (healthy female, n = 10) | IV | 0.09 | Albumin solution | Acute 1 h cumulative doses | During 1 h cumulative dosing | 22 ng/ml (70nmol/L) Cmax | – | Decreased saccadic eye movement | No adverse effects other than fatigue, mild nausea | Timby et al. (2006) | |
| Clinical trial | Human (healthy female, n = 28 crossover) | IV | 0.07 | Albumin solution | Single dose | 10–55 min post-dosing | 22–45 ng/ml (70–140nmol/L) Cmax | – | Acutely impaired free verbal recall episodic memory due to mild sedation with high variability; no acute effect on semantic or working memory | No adverse effects other than self reported sedation | Kask et al. (2008) | |
| Clinical trial | Human (healthy female, n = 12; PMDD, n = 16) | IV | 0.05 | Albumin solution | Single dose | 0–20 min post-dosing | 16–22 ng/ml (50–70 nmol/L) Cmax | – | No effect on startle response or prepulse inhibition to startle | No adverse effects other than self reported sedation | Kask et al. (2009) |
Human blood levels of Allo are highest in the third trimester of pregnancy at levels up to 157 nmol/l (50 ng/ml) or higher under certain pregnancy conditions. High Allo during pregnancy is associated with drowsiness although high Allo is not associated with any severe adverse effects for either mother or fetus (Luisi et al., 2000). Post-mortem human brain Allo levels from women revealed levels in the range of 14–21 ng/g (Bixo et al., 1997). In comparison, Allo levels in human reached 150 nmol/L, 10 min after intravenous administration 0.09 mg/kg compared to baseline levels in the range of 0.4–2.4 nmol/L (van Broekhoven et al., 2007). In rodents, many studies demonstrate that stress elevates Allo in plasma and brain: Allo increased in acute stress swimming in ambient temperature water (Purdy et al., 1991); Allo increased inhalation of CO2 (Barbaccia et al., 1994, 1997); and Allo increased handling in naïve rats or foot shock in handling habituated rats (Barbaccia et al., 1997). Chronic stress decreases the baseline Allo (Serra et al., 2000) interestingly such that the ability to respond to acute stress (to elevate Allo) is maintained in chronic stressed animals (Serra et al., 2007). Stress activates the HPA axis leading to an increase in cortisol levels. Allo decreases corticotropin-releasing hormone and adrenocorticotropic hormone expression (Patchev and Almeida, 1996; Patchev et al., 1994) whereas administration of exogenous corticotropin-releasing hormone increases Allo levels (Genazzani et al., 1998). These studies strengthen the point that the dosing regimen is not trivial but rather a critical parameter of successful therapeutic intervention design. The treatment intervention point is just as critical as dosing regimen, a hard lesson learned from other once-promising AD therapeutic efforts that failed in clinical trials (Mullard, 2012). Many of the learning and memory reports associated with Allo were reported in animals and humans and it is important to understand that these studies were either acute measures of memory performance for example, participants were asked to recall a list of words within minutes of a mildly sedative Allo dose (Kask et al., 2008) or chronic treatment paradigms that mimic sink-or-swim high stress conditions in rodents (Turkmen et al., 2006). When high Allo doses or chronic continuous treatment regimens are employed, Allo acts like the benzodiazepines to impair learning and memory. Allo administered twice daily at high doses to male rats for several consecutive days decreased performance on the Morris water maze, escape latency, pathlength, and thigmotaxis and pretreatment induced a partial tolerance against acute Allo effects (Turkmen et al., 2006).
Bengtsson et al. extended their investigations of the effect of chronically elevated Allo levels (3-months elevated followed by 1-month withdrawal), on cognition and brain tissue Aβ levels in female and male transgenic APPSwePSEN1ΔE9 (Bengtsson et al., 2012). The follow-up study included both APPSwePSEN1ΔE9 (predominately elevated Aβ 1–42 levels; plaques as early as 6-months) and AβPPSweArc (predominately elevated Aβ 1–40 levels; slower plaque development beginning at 9-months) mouse models of Alzheimer’s disease with a 1-month treatment followed by 1-month washout (Bengtsson et al., 2013) (Table 1). In young AD mice, the neurosteroid Allo adversely inhibits neurotransmission and intracellular Aβ release leading to increased Aβ accumulation and decrements in learning and memory function. While expected cognitive deficits were observed in both AD mouse models compared to their non-transgenic littermates, Allo treatment effects were not observed in non-trangenic mice. The effect on the GABAergic system due Allo held chronically at a stress level followed by the washout period leading to changes in various GABA receptor subunit combinations could cause deleterious withdrawal effects as well as Aβ processing changes that could far outweigh the direct effect of chronic elevation of Allo The important findings point to Allo’s biphasic effects on disease states. Under some but not all conditions in these chronic Allo studies, the treatment paradigm further exacerbated genetically programmed AD progression and negatively impacted behavior measured by the MWM swim test. From the chronic continuous exposure studies it was difficult to conclude or generalize that prior stress levels of Allo impair memory. The treatment paradigm (1-month treatment followed by one month wash-out) was such that the direct effect of Allo remained unclear. The paradigm primarily looked at long-term effects of a continuous unrelenting Allo episode in an amyloidogenic model. It could be speculated that both amyloidogenesis was exacerbated and neurogenesis and neuronal differentiation and integration to be specific were chronically inhibited that amounted to long lasting deleterious effects. In translation to humans, the chronic treatment paradigm and in young mice with very early stage AD-like pathology would not be an ideal therapeutic approach. Data varied between mouse genders within each model, brain regions, and behavior tests highlighting the importance of each variable as they relate to neurosteroid efficacy (Bengtsson et al., 2012, 2013). The long-term consequences of a previous stressful period where Allo levels were constantly elevated and then suddenly relieved in two mouse models of genetic AD could relate to a subset of stressed or depressed human cases that progress to AD either sporadically or more relevantly to familial AD.
5. Requirements for translation of preclinical allopregnanolone outcomes into clinical Alzheimer’s disease therapy
5.1. Allopregnanolone and Lipinski’s rule of 5
Lipinski’s rule of five is a mnemonic developed by Christopher A. Lipinski and colleagues for predicting success of a drug by oral route of administration (Lipinski et al., 2001). They found that four criteria with values close to multiples of five were predictive of oral availability. While the rules are based on requirements for oral administration of a drug, they are widely applied as an initial indicator for successful drug development. The rule of five predicts that poor absorption or permeation are likely if: (1) there are more than five hydrogen donors (Allo has one); (2) the molecular weight is more than 500 Da (the molecular weight of Allo is 318.49 Da); (3) the Log P value is just over five (Allo is 5.042); and (4) there are more than 10 hydrogen acceptors (Allo has two). Allo meets three of the four Lipinski rules whereas it does not meet the requirement for a Log P value less than five, although it should be mentioned that some estimates report Allo’s Log P to be just below 5. The Log P value is an indicator of lipophilicity. Molecules with a high octanol to water partition constant are preferentially distributed to hydrophobic compartments such as lipid bilayers of cells, whereas hydrophilic molecules (low octanol to water partition coefficients) are preferentially distributed in hydrophilic compartments such as blood serum. The chemical properties of Allo are not optimal for oral dosage and would require advanced formulation techniques. However, Allo availability by parenteral and other routes with proper formulations are readily delivered from the peripheral system, penetrating through the blood brain barrier to reach the central nervous system (Table 1). A Log P value of slightly greater than five indicates that Allo preferentially distributes to lipophilic compartments, a typical feature of steroid molecules.
5.2. Allopregnanolone formulation
Determining the best formulation and route of delivery is a formidable challenge for Allo and other neurosteroids. The chemical structures of most neurosteroids are not well suited for oral delivery because of poor solubility properties in aqueous environments. Neurosteroids precipitate readily from aqueous solution and thus display poor bioavailability followed by first-pass metabolism in the digestive tract and liver. Formulation of delivery vehicle for Allo is critical, as the Allo formulation will determine its bioavailability, pharmacokinetic and pharmacodynamic properties. For example, a sterile Allo suspension dosage form could be administered subcutaneously or intramuscularly whereas only soluble, particle-free sterile-filtered products would be acceptable for intravenous administration. Simple rapid-release formulations may be fine-tuned by changing the complexation agent ratio or adding viscosity agents to improve the desired pharmacokinetic profile. The complexation ratio has limitations based on solubility properties of combination of the excipients and the neurosteroid. Solubility is a major concern for most hydrophobic neurosteroids and efforts have been made to synthesize water-soluble analogs of neurosteroids, including Allo and progesterone with the goal to maintain structure-activity relationships (MacNevin et al., 2009). Excipients with prior regulatory approval are most desirable for initial formulations since they are more likely to gain regulatory acceptance in new formulations (Stella and He, 2008) whereas new excipients are required to undergo their own extensive quality and safety assessments. Use of existing excipients will, however, often limit patent strategy.
Complexing agents are substances that allow for polar-based solvents, ideally water, to complex with otherwise water-insoluble neurosteroids to enhance solubility and bioavailability. The most common complexing agents for Allo are cyclodextrins that ideally form 1:1 ratios with neurosteroids in aqueous solution (Fig. 3) (Stella and He, 2008). Clinical and translational scientists should note that cyclodextrins when delivered in vivo could form inclusion complexes with other small molecules including other drugs that may be present in the circulation or endogenous molecules including peripheral endogenous steroids and cholesterol. It is generally thought that cyclodextrins with high molecular weight cyclic saccharide structures do not penetrate the blood brain barrier when administered in reasonable quantities sufficient for drug delivery to CNS.
Fig. 3.
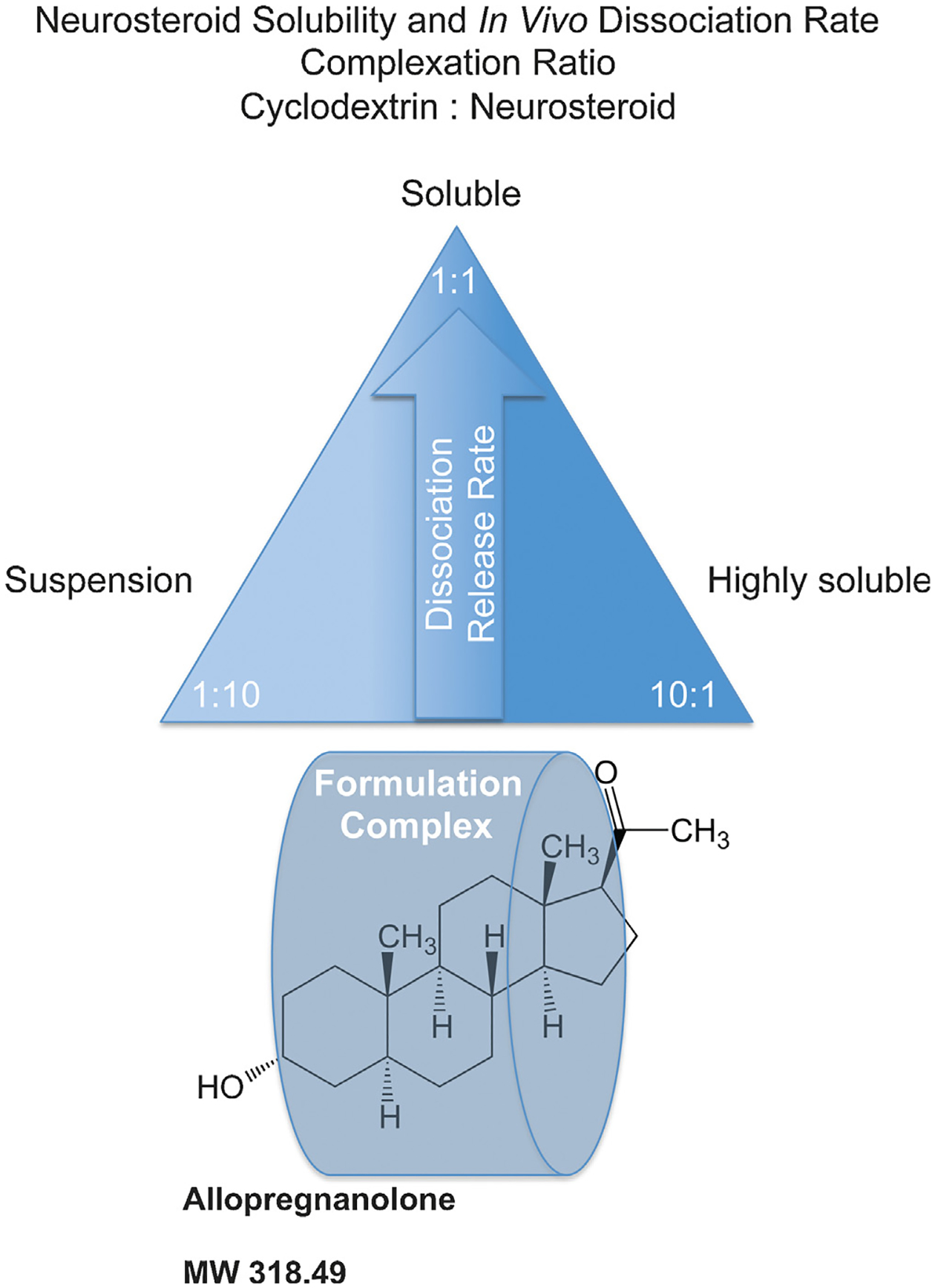
The neurosteroid release rate pyramid describes the relationship between release rate of active neurosteroid and aqueous cyclodextrin vehicle. Molar complexation ratios greatly alter the release rate or dissolution profile of neurosteroids including allopregnanolone (Allo) as illustrated by the release rate pyramid. Typical cyclodextrin carrier formulations utilize the cyclodextrin such as hydroxypropyl beta-cyclodextrin, at 5–30% in aqueous solution such as water or normal saline with a maximal achievable cyclodextrin concentration between 45–60% weight to volume. Generally, a 1:1 stoichiometry inclusion complex of neurosteroid and a cyclodextrin exists at the apex of the pyramid hypothetically set at a threshold dose that requires maximal delivery rate of the neurosteroid. For Allo, the molar complexation ratio (suitable cyclodextrin:Allo) that is optimal for release rate is closer to 1:2. The same Allo dose would exhibit a slower release rate with a cyclodextrin:Allo of 1:10 complexation ratio since most of the Allo would be suspended in a depot formulation and less accessible for release and absorption into the biological system. Likewise, a complexation ratio that possesses cyclodextrin:Allo of 10:1 at the same dose of Allo would be readily soluble in an aqueous environment similar to the 1:10 suspension formulation that sits on the opposite side of the solubility spectrum. The 10:1 formulation would also be less accessible for absorption compared to the 1:1 formulation as the Allo is held within the overly abundant empty cyclodextrin molecules governed by the binding and dissociation rate. We hypothesize that the 10:1 formulation displays a re-uptake phenomenon whereby the neurosteroid is relatively less likely to be released into the biological system and therefore slower to release and interact with the receptor targets. A desired pharmacokinetic profile can be titrated by understanding the complexation ratio and release rate to optimize a measurable biological effect such as neurogenesis, anxiolytic activity, or sedation. For Allo, ataxia or sedation can be used as a measurable biomarker of target engagement and can establish the maximally tolerated dose in relation to formulation and delivery route. The lowest possible dose to experience sedation for example at 1:1 formulation would elicit little or no effect with 1:10 or 10:1 formulation complexes.
Other formulations that are potentially viable options for neurosteroids include injectable suspension depots with standard dispersed systems, oral emulsions, or advanced nano-encapsulation technologies. Devices such as controlled release pumps theoretically could be used to cyclically deliver one or more neurosteroids to the brain although this approach remains to be investigated.
Stability of the neurosteroid formulation must be tested and often the formulation must be further developed to avoid typical stability issues such as precipitation, freeze-thaw, maintaining potency, pH fluctuation, or packaging issues. When a final Allo drug product is prepared for the regulatory approval inspection process, it is critical to consider packaging (e.g. volume, shape, cap closure system, label, materials, interaction of package with product under various conditions including time and temperature) with scale-up capability. For example, the best solvents for neurosteroids may also interact with the chemical components of typical choices for packaging such as synthetic plastics (as containers or coatings), rubber compounds, or in some cases even glass to fracture, leach or adsorb thus interfering with the intended drug product. The drug container will undergo a rigorous quality assessment by regulators. Changing any variable of the formulation requires another costly round of all-encompassing quality assessment and could require further safety and efficacy tests.
5.3. Allopregnanolone route of administration, pharmacokinetics and pharmacodynamics
From our studies (Table 1), subcutaneous and intravenous soluble cyclodextrin-based Allo and subcutaneous suspension Allo depots induced rapid neurogenic responses. Multiple formulations of Allo: (1) promoted neurogenesis with no serious adverse effects following both acute and chronic exposure at optimal intervals; (2) reduced burden of AD pathology; (3) exhibited efficacy in both male and female 3xTgAD mice; (4) reversed learning and memory deficits assessed by hippocampal dependent behavioral assays relevant to cognition in humans with AD (Wang et al., 2010; Singh et al., 2011; Chen et al., 2011; Brinton, 2013). Rapid exposure to a sub-sedative dose of Allo is sufficient to induce the Allo mechanism of neurogenic action, achievable via rapidly bioavailable intravenous or subcutaneous routes solubilized in a cyclodextrin formulation of 2-hydroxypropyl β-cyclodextrin (HβCD; Trappsol®) or sulfobutyl ether β-cyclodextrin (SBEβCD; Captisol1) a non-nephrotoxic derivative (Stella and He, 2008). Based on neurogenic regenerative efficacy determined in rodents we hypothesize that Allo will likewise induce neurogenesis in humans at risk for or affected by AD (Brinton, 2013).
Initial pharmacokinetic modeling of IV administration of Allo predicted that an intravenous infusion rate over 5 min would induce a sub-sedative peak of Allo in brain while extending the duration (area under the curve) of exposure to more closely simulate conditions generated by subcutaneous bioavailablity of cyclodextrin-solubilized Allo exposure. We chose absolute exposure doses in the mouse to correspond, factoring for allometric species conversion, to the expected intravenous dose range per average 70 kg human in order for results to best translate our preclinical results to hypothetically safe conscious-sedative clinical doses (Reagan-Shaw et al., 2008). Mice samples collected 5 min, 15 min, 30 min, 4 h, 24 h post infusion start time were used to determine Allo levels in brain and plasma to provide data for PK modeling and indication of cell cycle activation and differentiation marked by pCREB (neurogenic cell signaling) and NeuroD (developing neural cell) (Table 1). Both intravenous and subcutaneous cyclodextrin-formulated Allo had similar dose-dependent sedation profiles and elimination occurred prior to 4 h. Our preclinical data indicate that the regenerative effect of Allo occurred at sub-sedative doses given in a regimen consistent with the time course for regeneration – i.e. maximum exposure of once per week – not daily as is typical for most therapeutics (Brinton, 2013). Allo efficacy via rapidly bioavailable routes is the result of rapid calcium signaling to activate CREB regulated genes to ultimately stimulate endogenous neurogenesis and subsequent neural cell survival to enhance memory function. Phospho-CREB upregulation (Irwin et al., 2013) was induced by a rise in Allo concentration in blood and brain following IV dose of Allo 1.5 mg/kg. Pharmacokinetic analyses (Table 1) indicated that Allo doses that were mild-sedative (less than 10 min ataxia) or sub-sedative (no change in motor activity) were sufficient to activate neurogenic mechanisms by multiple routes and formulations including IV (1.5 mg/kg) and SC (10 mg/kg suspension). PK parameters of Allo in mouse plasma and brain cortex were determined by LC-MS/MS after mild-sedative IV (Allo 1.5 mg/kg) or sub-sedative administration in aged adult male nonTg mice. The Cmax reached in plasma was 215 ng/ml (675 nmol/l) with a corresponding Tmax of 5 min. Brain exposure to Allo was reached a Cmax of 639 ng/g with a corresponding Tmax of 5 min in mice IV 1.5 mg/kg. During the initial phase of distribution the plasma half-life t1/2,1 was 12.6 min followed by t1/2,z of 5.1 h for the elimination phase. The Allo pharmacokinetics in mouse models were comparable to humans, with distribution phase ranging in humans between t1/2,1 of 18.9 ± 5.3 min and 43.9 ± 7.3 min followed by elimination phase half-life t1/2,z of 4.4 ± 1.7 h treated with IV Allo cumulative dosing over 90 min (van Broekhoven et al., 2007; Timby et al., 2006). Thirty minutes after 1.5 mg/kg IV administration in the mouse, the plasma Allo concentration was 51 ng/ml (160 nmol/l) and 192 ng/g in brain cortex, comparable by 30 min post-treatment with our previously tested subcutaneous formulation (Allo 10 mg/kg s.c.) that reached 34 ng/ml (107 nmol/l) in plasma and 159 ng/g in brain cortex within 30 min. Although the peak exposure briefly spiked with IV, the mouse brain and blood exposure levels of IV (1.5 mg/kg) and suspension SC (10 mg/kg) and were nearly equal at 30 min and neurogenic effect was comparable thereby bridging IV and subcutaneous formulation and Allo exposure.
In a previous clinical study, Allo volume of distribution, elimination half-life, and the area under the curve (AUC) adjusted for body weight did not differ between men and women (van Broekhoven et al., 2007). Following dosing of Allo 0.09 mg/kg (three cumulative doses of 0.015, 0.03, 0.045 mg/kg within 1 h) fully bioavailable intravenous administration, maximum blood levels of 48 ng/ml (150 nmol/L) in men and 32 ng/ml (100 nmol/L) in women were achieved (Table 1) (van Broekhoven et al., 2007). Mean levels of endogenous plasma Allo ranged from 0.13 to 0.76 ng/ml (0.4–2.4 nmol/L) at baseline. Clinically, a dose range of 0.5–6 mg is equivalent to 0.007–0.086 mg/kg IV for a 70 kg (154 lb) adult. Within the sub-sedative to mild-sedative dose range, Allo in cyclodextrin-based formulations approximates pharmacokinetically with previously published human experiences with albumin-based formulation summarized in Table 1. Previous intravenous studies found minimal risk of adverse effects other than mild self-reported sedation in a subset of participants. The clinical plan and safety surveillance should be discussed in the US, at pre-investigational new drug application (pre-IND) meetings (21 CFR 312.82), end-of-phase 1 meetings (21 CFR 312.82), end-of-phase 2 and pre-phase 3 meetings (21 CFR 312.47), pre-new drug application license application meetings (21 CFR 312.47) with the United States Food and Drug Administration (FDA), or equivalent regulatory agencies in other countries.
Selection of starting dose is a critical first step in translating drug discovery to early clinical development. Clinical subcutaneous dose range estimates can be determined in part by computer simulations to predict pharmacokinetic and pharmacodynamic profiles using data from animal studies and known cases of exposure to the neurosteroid regarded as previous human experience. Such was the case with clinical intravenous Allo from Bäckström’s group (0.03–0.09 Allo mg/kg body weight) (Kask et al., 2008, 2009; Timby et al., 2006; van Broekhoven et al., 2007) (Table 1) to serve as a reference point for future clinical pharmacokinetic studies. In silico pharmacokinetic models can validated and used to predict the plasma and brain concentrations of Allo resulting from low doses with no predicted pharmacodynamic response up to higher doses with defined maximal tolerated side effects in the proposed study population. Computer simulations are used to predict the total neurosteroid blood and brain compartment exposure and pharmacodynamic responses such as changes in saccadic eye movement as indicator of sedation to more accurately estimate the safe dosage range.
Our preclinical studies with Allo predict that the optimal Allo treatment regimen will be a once per week parenteral, intravenous or subcutaneous administration (Fig. 4; Table 1). Again referring to clinical studies by Bäckström’s group at Umeå University, Sweden, Allo is safe with short-lived, mild self-reported drowsiness. Following dosing of Allo 0.09 mg/kg (three cumulative doses of 0.015, 0.03, 0.045 mg/kg within 1 h) fully bioavailable intravenous administration, van Broekhoven et al. measured maximum blood levels of 150 nmol/L (48 ng/ml) in men and 100 nmol/L (32 ng/ml) in women (Table 1) (van Broekhoven et al., 2007). Mean blood levels of Allo were found to be higher in men compared to women at baseline (men, 2.4 nmol/L or 0.76 ng/ml versus women, 0.4 nmol/L or 0.13 ng/ml) and after each of three doses of intravenous Allo indicating possible sex differences in absorption and metabolism. However, other pharmacokinetic parameters when adjusted for body weight did not differ between the young healthy men and women in the clinical study (van Broekhoven et al., 2007).
Fig. 4.
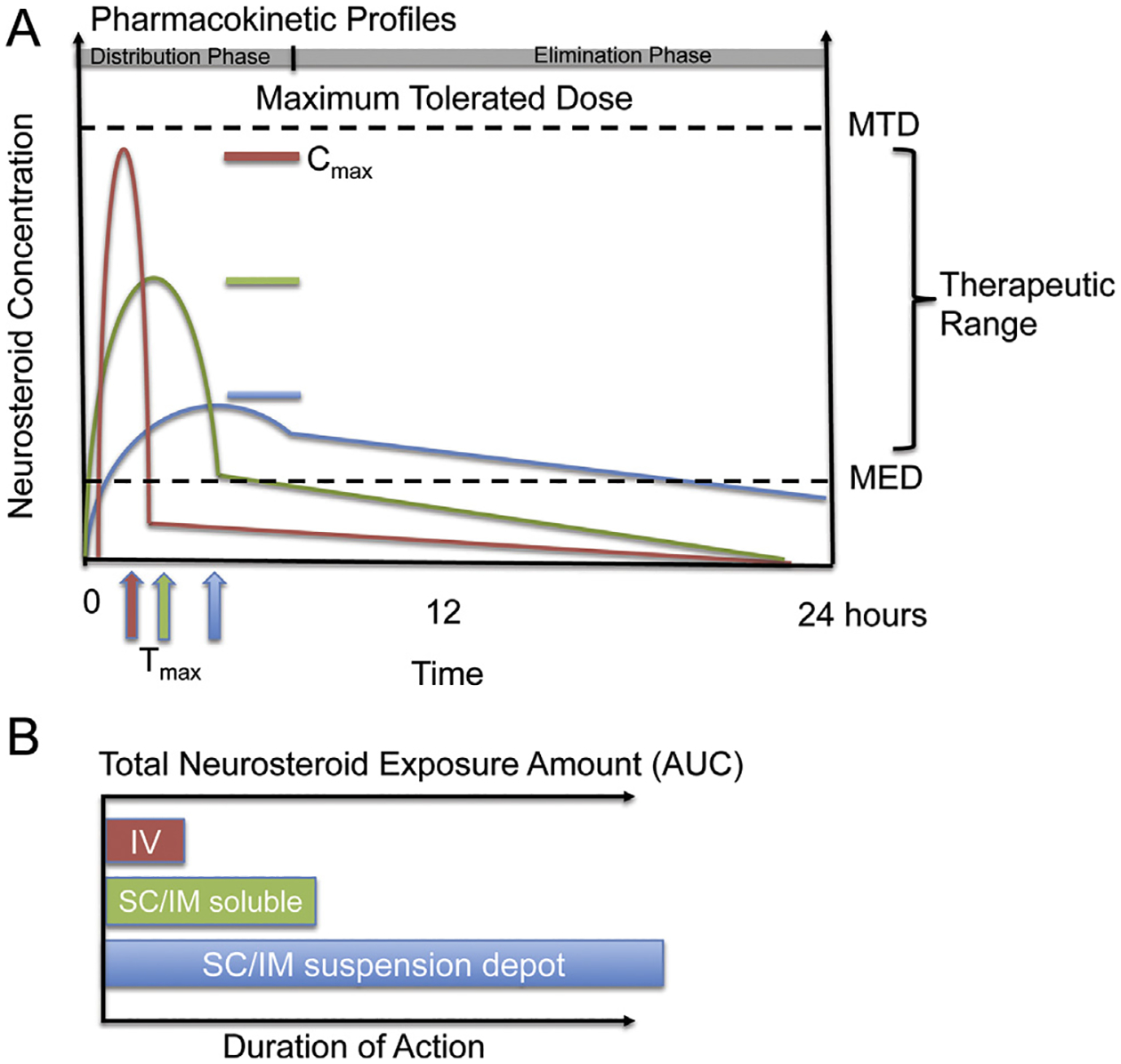
Neurosteroid pharmacokinetic profiles with consideration of formulation and route of administration. A. Pharmacokinetic profiles in blood plasma are generalized within the therapeutic range between the maximum tolerated dose (MTD) and the minimal effective dose (MED) to highlight the importance of formulation release rates and maximum exposure levels following a single equal neurosteroid bolus infusion dose. Neurosteroids readily cross the blood–brain barrier after absorption into the blood stream. The profiles: (1) Intravenous (IV) soluble dose (red colored line) – rapid release rate to reach Tmax; relatively highest concentration Cmax, rapid elimination rate; optimal for rapid cell signaling and non-genomic mechanisms within minutes and a 5–30 min distribution phase and rapid clearance. (2) Subcutaneous (SC)/intramuscular (IM) soluble dose (green colored line) – medium release; medium elimination; optimal for rapid cell signaling and non-genomic mechanisms; activates some slow mechanisms in distribution phase of 10–60 min range. (3) SC/IM suspension dose (blue colored line) – slow release; slow elimination; less optimal for rapid cell signaling, optimal for mechanisms that require slow or prolonged activation. (B) Intravenous (IV) soluble dose (red colored bar) is plotted on dual axes as the systemic amount of neurosteroid exposure or area under the curve (AUC) required to activate therapeutically relevant mechanisms and the duration of action. For example, a short burst of the neurosteroid Allo reaches MTD defined by level of conscious-sedation with relatively low exposure AUC via a bolus IV infusion of fully soluble neurosteroid. SC/IM soluble dose (green colored bar) depicts the AUC of neurosteroid that could be required for longer duration of action compared to IV. With soluble SC/IM injection, relatively less total AUC is required to activate the biological system compared to a hypothetical SC/IM suspension dose (blue bar) with relatively low solubility and slow release rate and minimal peak level at a given dose. Depending on the desired biological effect, each route has advantages and disadvantages that should be considered when developing a therapeutic strategy.
Allo pharmacokinetics and pharmacodynamics have not been studied in AD trials on men and post-menopausal women age approximately 60–80 years old. Possible differences in metabolism and clearance of Allo, unique to AD subjects, will require careful monitoring. However, clinical studies of intravenous Allo in young adult males and females have proven useful to understand the pharmacokinetics and pharmacodynamics including sedation measured by saccadic eye movement and memory tests as well as blood level differences in metabolism (Table 1). Clinically, Allo has only been studied intravenously but has not been studied by alternative routes of administration such as subcutaneously or intramuscularly in rapid soluble release or slow depot form in depot forms that would have very different pharmacodynamics and pharmacokinetic profiles that may be required to achieve the desired pharmacokinetic profile for efficacy (Fig. 4).
5.4. Safety requirements to conduct AD clinical trials
Toxicology studies are required by regulatory agencies prior to acute and chronic clinical trials with the Allo drug product. The required final Good Laboratory Practices (GLP) studies conducted using drug product generated under Good Manufacturing Practices (cGMP) will include acute and chronic studies in rodents and non-rodent species to meet or exceed the duration of the clinical trial (Steinmetz and Spack, 2009). Preclinical safety studies should seek regulatory oversight by the FDA. Useful guidance from the FDA or other regulatory agencies typically follows the International Conference on Harmonization guidelines. Meeting with the appropriate regulatory agency is crucial prior to developing an IND application. Both IND approval in the US, or its equivalent in other countries, and a complete review by the Institutional Review Board or Ethics Committee are necessary to move a preclinical development program to early clinical trial stages. In some cases where existing human exposure data is available, the neurosteroid, as is the case with Allo, may be considered safe at blood concentrations at or below levels that have been reported to occur endogenously (Luisi et al., 2000) thus guiding the toxicology study recommendations.
Several clinical studies have detected vascular edema with clinical dosing of anti-amyloid therapies (Salloway et al., 2009; Sperling et al., 2011, 2012) and have alarmed regulatory agencies to screen new drug candidates in pre-clinical stages. If the drug product affects beta-amyloid mechanisms for example, nonGLP preclinical studies may be required to include assessment of associated risk for cerebral angiopathies or microhemorrhages in an appropriate animal model. Microhemorrhage risk assessment studies require advanced age animals, 20 months old in AD mice for example, and present real challenges because of the age related attrition rates observed in AD mouse models (Pfeifer et al., 2002; Racke et al., 2005). A positive control must be included in the study to demonstrate accelerated microhemorrhage development (Demattos et al., 2012; Racke et al., 2005). These toxicology studies are important to demonstrate minimal additional risk of the drug product and therapeutic regimen in a late stage AD model and need not be aimed solely at finding an efficacious outcome on reduction of AD pathology.
5.5. Chemistry, manufacturing, and controls (CMC) and patent considerations
To develop Allo or other neurosteroids as a potential therapeutic, there are several critical manufacturing issues to carefully consider. Neurosteroids are often only available commercially in small quantities and most are manufactured for nonclinical research use only. To overcome this technical barrier, one must obtain a source of active ingredient in sufficient quantity and most importantly, manufactured under current Good Manufacturing Practice guidelines (cGMP) to conduct required preclinical toxicology and clinical trial studies. In addition to the active pharmaceutical ingredient (Allo) a cGMP batch of inactive ingredients (excipients) must be obtained. The choice of excipients is not a moot point with regards to neurosteroid formulation (refer back to Figs. 3 and 4). The precise formulation must be decided upon including concentration of stock solution and final drug product. The combination of Allo plus excipient in a drug formulation may or may not be proprietary. It is important to understand the scope of formulation patents and method of use patents at this step. Once decisions regarding dose, formulation, and treatment regimen have been carefully planned, the next step is to manufacture the cGMP drug product. The cGMP Allo product will then undergo a series of analytic tests. In addition, the proper storage conditions must be in place to maintain cGMP status of the product components and the final product. The final drug product is prepared in batches and the container is selected to minimize risk of contamination, decomposition by drug or vehicle over time and must remain sterile. Sterility testing and stability testing of the stock concentration and final drug product are required to conduct further studies. As part of an IND (investigational new drug) application, a well-defined plan for manufacturing, delivery and regulatory monitoring must be approved by national regulatory agencies, FDA for the US and the European Medicines Agency (EMA) for Europe.
5.6. Alzheimer’s therapeutic landscape and global unmet medical need
Currently approved AD drugs offer only transient benefits for symptoms of AD. There are no treatments developed to delay or reverse the progression of AD. The FDA and EMA have not approved any new drugs for Alzheimer’s disease in the past decade. Of the five FDA-approved AD drugs, four are cholinesterase inhibitors and one is an N-methyl-d-aspartate receptor antagonist. It should be noted that none of the approved AD drugs target the brain’s regenerative system. In several animal models, the regenerative system does improve cognitive function and is thus a viable candidate mechanism for AD therapeutic development. Other plausible mechanistic AD targets may not target the disease soon enough as recent clinical trials have demonstrated. Phase 3 clinical trials have failed to therapeutically modify AD via the inhibition of β-amyloid cascade targets (Mullard, 2012; Karran et al., 2011). Passive immunotherapy Anti-Aβ antibodies, delivered as human recombinant antibodies have had many challenges to overcome and thus far have resulted in lack of efficacy in human trials (Mullard, 2012). Preclinical studies in AD mouse models have demonstrated efficacy of anti-Aβ antibodies. However, Aβ42 immunization clinical trials for example have not shown appreciable clinical efficacy. Several studies have shown that anti-Aβ antibodies increase the risk for micro-hemorrhages or cerebral vascular risks in animal models and humans (Pfeifer et al., 2002; Racke et al., 2005; Salloway et al., 2009; Sperling et al., 2011, 2012). Treatment approaches to AD must be reevaluated to target the endogenous regenerative system before rather than after the neurobiological system is unresponsive to therapy. The amyloid hypothesis has been tried at the highest level and failed to demonstrate appreciable efficacy. Lessons can be learned from the costly failed AD trials (Brinton, 2013; Mullard, 2012). From a scientific standpoint, it is ineffective to evaluate therapeutic efficacy in victims that have reached the later stages of AD, when irreversible damage (beta-amyloid plaques, tauopathic tangles, and neurodegeneration) has occurred. To modify disease progression, innovative therapies such as targeting neuroregeneration should be applied early to the appropriately responsive cohorts to intervene with cognitive impairment and AD progression.
Expansion and aging of the global population is projected to increase the number of persons affected by Alzheimer’s worldwide. Current estimates of dementia indicate that there were 36 million people living with dementia worldwide in 2010, increasing to 66 million by 2030 and 115 million by 2050 (World Health Organization and Alzheimer’s Disease International, 2012). According to estimates from the World Health Organization Dementia report there were 7.7 million new cases of dementia in the year 2010, or one new case every 4 s which is three times as many as HIV/AIDS (2.6 million per year). Based on assumptions that that incidence will increase in parallel with prevalence, as global aging is driving both incidence and prevalence, the incidence is projected to increase to 24.6 million new cases annually by 2050. Based on the projected incidence over this time frame, in less than 40 years there will be 646 million new cases in addition to the current 36 million cases of Alzheimer’s disease. Summing both the current and projected number of persons living with predicts that within the next four decades 682 million people will live with dementia. To put these numbers into perspective, within 40 years the number of 682 million persons living with dementia will exceed the current population of the US and Canada (542 million) combined or nearly equal the population of Europe (738 million) (Alzheimer’s Disease International, 2012). In the United States an epidemic of Alzheimer’s disease exists where 5 million persons with Alzheimer’s currently live with the disease two-thirds of whom are women (Alzheimer’s Association, 2013; Brinton, 2008, 2009; Mosconi et al., 2007, 2009). Projections of incidence and prevalence will not change unless there is a cure or a treatment that delays the onset or progression of the disease.
6. Concluding remarks
Preclinical and translational research evidence support therapeutic development of allopregnanolone for neurodegenerative diseases including Alzheimer’s. The dose and treatment regimen of Allo must be tailored to a route of administration and pharmacokinetic profile required for neurogenesis and AD pathology reduction. Allo induces both rapid neural progenitor cell cycle gene expression (Wang et al., 2005) and induces key regulators of cholesterol homeostasis (Chen et al., 2011) while reducing toxic amyloid-beta and neuroinflammation. In a pre-clinical mouse model, intermittent treatment regimens begun prior to and during the early stages of AD pathology significantly increased the regenerative response in brain while also reducing burden of pathology in an AD mouse model and remarkably improved cognitive function measured by associative learning and memory tasks (Singh et al., 2011; Wang et al., 2010). In addition to the dosing frequency, intervention with disease progression at early stages with neurosteroid supplementation is critical. The magnitude of AD pathology and remaining neurogenic capacity at initiation of Allo treatment may influence the efficacy endpoints of the clinical trial.
Translational efforts should focus on both the pharmacodynamic effectiveness and minimized toxicological risks in both rodent and non-rodent species to predict the clinical response to the intended disease population. A clinical plan and safety surveillance program should be discussed with regulatory agencies as early as possible in the process of neurosteroid therapeutic development. Regulatory agencies on many levels offer valuable recommendations toward successful therapeutic development programs. Therapeutic development lessons can be learned from previous successful and unsuccessful drug development efforts. Several recent Alzheimer’s clinical trials have provided critical insights into the important parameters required to minimize failure (Mullard, 2012). A remaining translational and clinical development challenge is to establish neuroimaging indicators of neurogenesis and oligodendrogenesis comparable to those developed to monitor disease progression or rate of decline (e.g. white matter hyperintensity, cerebral blood flow, glucose metabolism, and amyloid-related imaging abnormalities (Salloway et al., 2009; Sperling et al., 2011, 2012). In conclusion, translational research required for development of Allo as a regenerative therapeutic has provided key insights into mechanistic targets for both neurogenesis and disease modification, dosing requirements, optimal treatment regimen, route of administration and the appropriate formulation necessary to advance to proof of concept clinical studies to determine efficacy of Allo as a regenerative and disease modifying therapeutic for Alzheimer’s disease.
Acknowledgments
The work that led to this Review was supported by grants from the NIH National Institute on Aging (U01 AG031115), the Alzheimer Drug Discovery Foundation, the Kenneth T. and Eileen L. Norris Foundation, and The California Institute for Regenerative Medicine (DR2-05410) to R. D. Brinton. The invaluable contributions of M. Rogawski and G. Bauer of the University of California at Davis; J. Wang of the University of Mississippi, and S. Chen of the University of Southern California to our discovery and translational research endeavors described herein are gratefully acknowledged. We extend special gratitude to K. Steinmetz of SRI, International for her contributions to our preclinical toxicology development efforts.
Abbreviations:
- ANT
adenine nucleotide transporter protein
- Allo
allopregnanolone
- AD
Alzheimer’s disease
- ABAD
amyloid-beta binding alcohol dehydrogenase
- AUC
area under the curve
- Aβ
β-amyloid
- CMC
chemistry, manufacturing, and controls
- CYP3A
cytochrome P450 3A
- CYP450scc
cytochrome P450 side-chain cleavage
- EMA
European Medicines Agency
- FDA
United States Food and Drug Administration
- GLP
Good Laboratory Practices
- cGMP
Good Manufacturing Practices
- IM
intramuscular
- IN
intranasal
- IV
intravenous
- IND
investigational new drug
- HβCD
2-hydroxypropyl β-cyclodextrin
- HMG-CoA-R
3-hydroxy-3-methyl-glutaryl-CoA-reductase
- 3β-HSD
3β-hydroxysteroid dehydrogenase
- 5α-R
5α-reductase
- 3α-HSD
3α-hydroxysteroid dehydrogenase
- 5α-DHP
5α-dihydroprogesterone
- LXR
liver-X-receptor
- MED
minimal effective dose
- MTD
maximum tolerated dose
- nonTg
non-transgenic
- PXR
pregnane-X-receptor
- PKA
protein kinase A
- PKCε
protein kinase C epsilon
- StAR
steroidogenic acute regulatory protein
- SBEβCD
sulfobutyl ether β-cyclodextrin
- SGZ
subgranular zone
- SVZ
subventricular zone
- SC
subcutaneous
- TD
transdermal
- TSPO
translocator protein
- 3xTgAD
triple transgenic mouse model of AD
- VDAC
voltage-dependent anion channel protein.
Footnotes
Disclosure statement
Patents pending on allopregnanolone as a therapeutic for mild cognitive impairment and Alzheimer’s disease.
References
- Altman J, 1969. Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J. Comp. Neurol 137, 433–457. [DOI] [PubMed] [Google Scholar]
- Altman J, Das GD, 1965. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol 124, 319–335. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association, 2013. Alzheimer’s Disease Facts and Figures. [DOI] [PubMed]
- Alzheimer’s Disease International, 2012. World Alzheimer Report 2012: Overcoming the Stigma of Dementia
- Barbaccia ML, Roscetti G, Trabucchi M, Cuccheddu T, Concas A, Biggio G, 1994. Neurosteroids in the brain of handling-habituated and naive rats: effect of CO2 inhalation. Eur. J. Pharmacol 261, 317–320. [DOI] [PubMed] [Google Scholar]
- Barbaccia ML, Roscetti G, Trabucchi M, Purdy RH, Mostallino MC, Concas A, Biggio G, 1997. The effects of inhibitors of GABAergic transmission and stress on brain and plasma allopregnanolone concentrations. Br. J. Pharmacol 120, 1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batarseh A, Papadopoulos V, 2010. Regulation of translocator protein 18 kDa (TSPO) expression in health and disease states. Mol. Cell. Endocrinol 327, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson SK, Johansson M, Backstrom T, Nitsch RM, Wang M, 2013. Brief but chronic increase in allopregnanolone cause accelerated AD pathology differently in two mouse models. Curr. Alzheimer Res 10, 38–47. [DOI] [PubMed] [Google Scholar]
- Bengtsson SK, Johansson M, Backstrom T, Wang M, 2012. Chronic allopregnanolone treatment accelerates Alzheimer’s disease development in AbetaPP(S-we)PSEN1(DeltaE9) mice. J. Alzheimers Dis 31, 71–84. [DOI] [PubMed] [Google Scholar]
- Bixo M, Andersson A, Winblad B, Purdy RH, Backstrom T, 1997. Progesterone, 5alpha-pregnane-3,20-dione and 3alpha-hydroxy-5alpha-pregnane-20-one in specific regions of the human female brain in different endocrine states. Brain Res 764, 173–178. [DOI] [PubMed] [Google Scholar]
- Boekhoorn K, Joels M, Lucassen PJ, 2006. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol. Dis 24, 1–14. [DOI] [PubMed] [Google Scholar]
- Brinton RD, 2008. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci 31, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD, 2009. Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci 30, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD, 2013. Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat. Rev. Endocrinol 9, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD, Wang JM, 2006. Preclinical analyses of the therapeutic potential of allopregnanolone to promote neurogenesis in vitro and in vivo in transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res 3, 11–17. [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K, 1998. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J. Neurosci.: Off. J. Soc. Neurosci 18, 2801–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, Dayer AG, 2008. New interneurons in the adult neocortex: small, sparse, but significant? Biol. Psychiatry 63, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, McKay RD, 1999. Restoring production of hippocampal neurons in old age. Nat. Neurosci 2, 894–897. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay RD, 2001. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comp. Neurol 435, 406–417. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Woolley CS, McEwen BS, Gould E, 1993. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56, 337–344. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang JM, Irwin RW, Yao J, Liu L, Brinton RD, 2011. Allopregnanolone promotes regeneration and reduces beta-amyloid burden in a preclinical model of Alzheimer’s disease. PLoS One 6, e24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD Jr., Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, Bussey TJ, 2009. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 325, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Quervain DJ, Poirier R, Wollmer MA, Grimaldi LM, Tsolaki M, Streffer JR, Hock C, Nitsch RM, Mohajeri MH, Papassotiropoulos A, 2004. Glucocorticoid-related genetic susceptibility for Alzheimer’s disease. Hum. Mol. Genet 13, 47–52. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Roozendaal B, McGaugh JL, 1998. Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394, 787–790. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Roozendaal B, Nitsch RM, McGaugh JL, Hock C, 2000. Acute cortisone administration impairs retrieval of long-term declarative memory in humans. Nat. Neurosci 3, 313–314. [DOI] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML, 2012. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron 76, 908–920. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL, 2010. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver-X-receptor agonist GW3965 on object recognition memory and amyloid burden in APP/PS1 mice. J. Biol. Chem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH, 1998. Neurogenesis in the adult human hippocampus. Nat. Med 4, 1313–1317. [DOI] [PubMed] [Google Scholar]
- Fan X, Kim HJ, Bouton D, Warner M, Gustafsson JA, 2008. Expression of liver X receptor beta is essential for formation of superficial cortical layers and migration of later-born neurons. Proc. Natl. Acad. Sci. U.S.A 105, 13445–13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee KW, Bolger MB, Brinton RE, Coirini H, McEwen BS, 1988. Steroid modulation of the chloride ionophore in rat brain: structure-activity requirements, regional dependence and mechanism of action. J. Pharmacol. Exp. Ther 246, 803–812. [PubMed] [Google Scholar]
- Gee KW, Chang WC, Brinton RE, McEwen BS, 1987. GABA-dependent modulation of the Cl ionophore by steroids in rat brain. Eur. J. Pharmacol 136, 419–423. [DOI] [PubMed] [Google Scholar]
- Genazzani AR, Petraglia F, Bernardi F, Casarosa E, Salvestroni C, Tonetti A, Nappi RE, Luisi S, Palumbo M, Purdy RH, Luisi M, 1998. Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J. Clin. Endocrinol. Metab 83, 2099–2103. [DOI] [PubMed] [Google Scholar]
- He XY, Wegiel J, Yang SY, 2005. Intracellular oxidation of allopregnanolone by human brain type 10 17beta-hydroxysteroid dehydrogenase. Brain Res 1040, 29–35. [DOI] [PubMed] [Google Scholar]
- Herrup K, 2010. The involvement of cell cycle events in the pathogenesis of Alzheimer’s disease. Alzheimers Res. Ther 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst JJ, Walker DW, Yawno T, Palliser HK, 2009. Stress in pregnancy: a role for neuroactive steroids in protecting the fetal and neonatal brain. Dev. Neurosci 31, 363–377. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG, 2006. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR, 1998. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21, 869–883. [DOI] [PubMed] [Google Scholar]
- Irwin RW, Solinsky CM, Chen S, Brinton RD, 2013. Preclinical Safety and Efficacy of Allopregnanolone for Alzheimer’s Disease Therapy. Society for Neuroscience Annual Meeting Abstracts [Google Scholar]
- Irwin RW, Wang JM, Chen S, Brinton RD, 2011. Neuroregenerative mechanisms of allopregnanolone in Alzheimer’s disease. Front. Endocrinol. (Lausanne) 2, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson T, Treuter E, Gustafsson JA, Steffensen KR, 2012. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol. Sci 33, 394–404. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE, 2008. ApoE promotes the proteolytic degradation of Abeta. Neuron 58, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA, 2004. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A 101, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski RM, Livingood MR, Rogawski MA, 2004. Allopregnanolone analogs that positively modulate GABA receptors protect against partial seizures induced by 6-Hz electrical stimulation in mice. Epilepsia 45, 864–867. [DOI] [PubMed] [Google Scholar]
- Karran E, Mercken M, Strooper BD, 2011. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov 10, 698–712. [DOI] [PubMed] [Google Scholar]
- Kask K, Backstrom T, Nilsson LG, Sundstrom-Poromaa I, 2008. Allopregnanolone impairs episodic memory in healthy women. Psychopharmacology (Berl) 199, 161–168. [DOI] [PubMed] [Google Scholar]
- Kask K, Backstrom T, Lundgren P, Sundstrom Poromaa I, 2009. Allopregnanolone has no effect on startle response and prepulse inhibition of startle response in patients with premenstrual dysphoric disorder or healthy controls. Pharmacol. Biochem. Behav 92, 608–613. [DOI] [PubMed] [Google Scholar]
- Knoth R, Singec I, Ditter M, Pantazis G, Capetian P, Meyer RP, Horvat V, Volk B, Kempermann G, 2010. Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS One 5, e8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE, 2005. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci.: Off. J. Soc. Neurosci 25, 8240–8249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoeva MV, Yin H, Flier JS, 2007. Evidence for constitutive neural cell proliferation in the adult murine hypothalamus. J. Comp. Neurol 505, 209–220. [DOI] [PubMed] [Google Scholar]
- Koldamova R, Fitz NF, Lefterov I, 2010. The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim. Biophys. Acta 1801, 824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Noctor S, Martinez-Cerdeno V, 2006. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci 7, 883–890. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH, 1996. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci 16, 2027–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Marr RA, 2010. Neurogenesis and Alzheimer’s disease: at the crossroads. Exp. Neurol 223, 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc V, Jasmin-Belanger S, Poirier J, 2010a. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med [DOI] [PubMed] [Google Scholar]
- Leduc V, Jasmin-Belanger S, Poirier J, 2010b. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med 16, 469–477. [DOI] [PubMed] [Google Scholar]
- Lee A, Kessler JD, Read TA, Kaiser C, Corbeil D, Huttner WB, Johnson JE, Wechsler-Reya RJ, 2005. Isolation of neural stem cells from the postnatal cerebellum. Nat. Neurosci 8, 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Liu J, Rone MB, Papadopoulos V, 2006. Protein–protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J. Biol. Chem 281, 38879–38893. [DOI] [PubMed] [Google Scholar]
- Liu L, Brinton RD, 2010. Gonadal hormones, neurosteroids and clinical progestins as neurogenic regenerative agents: therapeutic implications. In: Gravanis AG, Mellon SH (Eds.), Hormones in Neurodegeneration, Neuroprotection, and Neurogenesis Wiley-Blackwell, Hoboken, NJ, pp. 281–303. [Google Scholar]
- Liu X, Wang Q, Haydar TF, Bordey A, 2005. Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci 8, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisi S, Petraglia F, Benedetto C, Nappi RE, Bernardi F, Fadalti M, Reis FM, Luisi M, Genazzani AR, 2000. Serum allopregnanolone levels in pregnant women: changes during pregnancy, at delivery, and in hypertensive patients. J. Clin. Endocrinol. Metab 85, 2429–2433. [DOI] [PubMed] [Google Scholar]
- Luskin MB, 1993. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron 11, 173–189. [DOI] [PubMed] [Google Scholar]
- MacNevin CJ, Atif F, Sayeed I, Stein DG, Liotta DC, 2009. Development and screening of water-soluble analogues of progesterone and allopregnanolone in models of brain injury. J. Med. Chem 52, 6012–6023. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS, 2000. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci.: Off. J. Soc. Neurosci 20, 9104–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE, 2009. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci.: Off. J. Soc. Neurosci 29, 4252–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx CE, Trost WT, Shampine LJ, Stevens RD, Hulette CM, Steffens DC, Ervin JF, Butterfield MI, Blazer DG, Massing MW, Lieberman JA, 2006. The neurosteroid allopregnanolone is reduced in prefrontal cortex in Alzheimer’s disease. Biol. Psychiatry 60, 1287–1294. [DOI] [PubMed] [Google Scholar]
- Mayo W, Lemaire V, Malaterre J, Rodriguez JJ, Cayre M, Stewart MG, Kharouby M, Rougon G, Le Moal M, Piazza PV, Abrous DN, 2005. Pregnenolone sulfate enhances neurogenesis and PSA-NCAM in young and aged hippocampus. Neurobiol. Aging 26, 103–114. [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Froelichsthal P, Martini L, Vescovi AL, 1996. Steroid metabolizing enzymes in pluripotential progenitor central nervous system cells: effect of differentiation and maturation. Neuroscience 72, 467–475. [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Panzica G, 2009. Neuroactive steroids: an update of their roles in central and peripheral nervous system. Psychoneuroendocrinology 34 (Suppl. 1) S1–S8. [DOI] [PubMed] [Google Scholar]
- Meldrum BS, Rogawski MA, 2007. Molecular targets for antiepileptic drug development. Neurotherapeutics 4, 18–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellon SH, 2007. Neurosteroid regulation of central nervous system development. Pharmacol. Ther 116, 107–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellon SH, Gong W, Schonemann MD, 2008. Endogenous and synthetic neurosteroids in treatment of Niemann-Pick Type C disease. Brain Res. Rev 57, 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellon SH, Griffin LD, Compagnone NA, 2001. Biosynthesis and action of neurosteroids. Brain Res. Brain Res. Rev 37, 3–12. [DOI] [PubMed] [Google Scholar]
- Morgenstern NA, Lombardi G, Schinder AF, 2008. Newborn granule cells in the ageing dentate gyrus. J. Physiol 586, 3751–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, Tsui W, De Santi S, de Leon MJ, 2007. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad. Sci. U.S.A 104, 19067–19072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Mistur R, Switalski R, Brys M, Glodzik L, Rich K, Pirraglia E, Tsui W, De Santi S, de Leon MJ, 2009. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 72, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A, 2012. Sting of Alzheimer’s failures offset by upcoming prevention trials. Nat. Rev. Drug. Discov 11, 657–660. [DOI] [PubMed] [Google Scholar]
- Naylor JC, Kilts JD, Hulette CM, Steffens DC, Blazer DG, Ervin JF, Strauss JL, Allen TB, Massing MW, Payne VM, Youssef NA, Shampine LJ, Marx CE, 2010. Allopregnanolone levels are reduced in temporal cortex in patients with Alzheimer’s disease compared to cognitively intact control subjects. Biochim. Biophys. Acta 1801, 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PN, Billiards SS, Walker DW, Hirst JJ, 2003. Changes in 5alpha-pregnane steroids and neurosteroidogenic enzyme expression in the perinatal sheep. Pediatr. Res 53, 956–964. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR, 2001. Neurons derived from radial glial cells establish radial units in neocortex. Nature 409, 714–720. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM, 2003a. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 24, 1063–1070. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM, 2003b. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. [DOI] [PubMed] [Google Scholar]
- Overstreet Wadiche L, Bromberg DA, Bensen AL, Westbrook GL, 2005. GABAergic signaling to newborn neurons in dentate gyrus. J. Neurophysiol 94, 4528–4532. [DOI] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Bensen AL, Westbrook GL, 2006. Delayed development of adult-generated granule cells in dentate gyrus. J. Neurosci.: Off. J. Soc. Neurosci 26, 2326–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR, 2002. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci 3, 715–727. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Schwartz PH, Taupin P, Kaspar B, Stein SA, Gage FH, 2001. Cell culture. Progenitor cells from human brain after death. Nature 411, 42–43. [DOI] [PubMed] [Google Scholar]
- Patchev VK, Almeida OF, 1996. Gonadal steroids exert facilitating and buffering effects on glucocorticoid-mediated transcriptional regulation of corticotropin-releasing hormone and corticosteroid receptor genes in rat brain. J. Neurosci 16, 7077–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patchev VK, Shoaib M, Holsboer F, Almeida OF, 1994. The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormone-induced anxiety and alters the release and gene expression of corticotropin-releasing hormone in the rat hypothalamus. Neuroscience 62, 265–271. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Watson P, Hodges JR, 2000. The nature and staging of attention dysfunction in early (minimal and mild) Alzheimer’s disease: relationship to episodic and semantic memory impairment. Neuropsychologia 38, 252–271. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M, 2002. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 298, 1379. [DOI] [PubMed] [Google Scholar]
- Ponti G, Peretto P, Bonfanti L, 2008. Genesis of neuronal and glial progenitors in the cerebellar cortex of peripuberal and adult rabbits. PLoS One 3, e2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy RH, Morrow AL, Moore PH Jr., Paul SM, 1991. Stress-induced elevations of gamma-aminobutyric acid type A receptor-active steroids in the rat brain. Proc. Natl. Acad. Sci. U.S.A 88, 4553–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB, 2005. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J. Neurosci.: Off. J. Soc. Neurosci 25, 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N, 2008. Dose translation from animal to human studies revisited. FASEB J 22, 659–661. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, Reinhart PH, Jacobsen JS, 2007. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell. Neurosci 34, 621–628. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, Verkhratsky A, 2008. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS One 3, e2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Jones VC, Verkhratsky A, 2009. Impaired cell proliferation in the subventricular zone in an Alzheimer’s disease model. Neuroreport 20, 907–912. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M, 2010. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov 9, 971–988. [DOI] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M, 2009. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73, 2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saria S, Rajani AK, Gould J, Koller D, Penn AA, 2010. Integration of early physiological responses predicts later illness severity in preterm infants. Sci. Transl. Med 2, 48ra65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B, 2000. Role of LXRs in control of lipogenesis. Genes Dev 14, 2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M, Guennoun R, Robert F, Carelli C, Gago N, Ghoumari A, Gonzalez Deniselle MC, Gonzalez SL, Ibanez C, Labombarda F, Coirini H, Baulieu EE, De Nicola AF, 2004. Local synthesis and dual actions of progesterone in the nervous system: neuroprotection and myelination. Growth Horm. IGF Res 14 Suppl A, S18–S33. [DOI] [PubMed] [Google Scholar]
- Schumacher M, Hussain R, Gago N, Oudinet JP, Mattern C, Ghoumari AM, 2012. Progesterone synthesis in the nervous system: implications for myelination and myelin repair. Front Neurosci 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra M, Littera M, Pisu MG, Muggironi M, Purdy RH, Biggio G, 2000. Steroidogenesis in rat brain induced by short- and long-term administration of carbamazepine. Neuropharmacology 39, 2448–2456. [DOI] [PubMed] [Google Scholar]
- Serra M, Sanna E, Mostallino MC, Biggio G, 2007. Social isolation stress and neuroactive steroids. Eur. Neuropsychopharmacol 17, 1–11. [DOI] [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S, 2004. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 304, 1338–1340. [DOI] [PubMed] [Google Scholar]
- Shenoy SD, Spencer TA, Mercer-Haines NA, Alipour M, Gargano MD, Runge-Morris M, Kocarek TA, 2004. CYP3A induction by liver x receptor ligands in primary cultured rat and mouse hepatocytes is mediated by the pregnane X receptor. Drug Metab. Dispos 32, 66–71. [DOI] [PubMed] [Google Scholar]
- Singh C, Liu L, Wang JM, Irwin RW, Yao J, Chen S, Henry S, Thompson RF, Brinton RD, 2011. Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol. Aging [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipila S, Huttu K, Voipio J, Kaila K, 2004. GABA uptake via GABA transporter-1 modulates GABAergic transmission in the immature hippocampus. J. Neurosci.: Off. J. Soc. Neurosci 24, 5877–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, Possnert G, Mash DC, Druid H, Frisen J, 2013. Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M, 2012. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 11, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR Jr., Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ, 2011. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dementia: J. Alzheimer’s Assoc 7, 367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz KL, Spack EG, 2009. The basics of preclinical drug development for neurodegenerative disease indications. BMC Neurol 9 Suppl. 1, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella VJ, He Q, 2008. Cyclodextrins. Toxicol. Pathol 36, 30–42. [DOI] [PubMed] [Google Scholar]
- Sun Y, Yao J, Kim TW, Tall AR, 2003. Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J. Biol. Chem 278, 27688–27694. [DOI] [PubMed] [Google Scholar]
- Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JA, Holtzman DM, Heneka MT, 2011. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J. Neurosci.: Off. J. Soc. Neurosci 31, 7049–7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timby E, Balgard M, Nyberg S, Spigset O, Andersson A, Porankiewicz-Asplund J, Purdy RH, Zhu D, Backstrom T, Poromaa IS, 2006. Pharmacokinetic and behavioral effects of allopregnanolone in healthy women. Psychopharmacology (Berl) 186, 414–424. [DOI] [PubMed] [Google Scholar]
- Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T, 2005. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron 47, 803–815. [DOI] [PubMed] [Google Scholar]
- Trotter A, Maier L, Grill H-J, Kohn T, Heckmann M, Pohlandt F, 1999. Effects of Postnatal Estradiol and Progesterone Replacement in Extremely Preterm Infants. Journal of Clinical Endocrinology & Metabolism 84, 4531–4535. [DOI] [PubMed] [Google Scholar]
- Turkmen S, Lofgren M, Birzniece V, Backstrom T, Johansson IM, 2006. Tolerance development to Morris water maze test impairments induced by acute allopregnanolone. Neuroscience 139, 651–659. [DOI] [PubMed] [Google Scholar]
- Uzunova V, Sampson L, Uzunov DP, 2006. Relevance of endogenous 3alpha-reduced neurosteroids to depression and antidepressant action. Psychopharmacology 186, 351–361. [DOI] [PubMed] [Google Scholar]
- Uzunova V, Wrynn AS, Kinnunen A, Ceci M, Kohler C, Uzunov DP, 2004. Chronic antidepressants reverse cerebrocortical allopregnanolone decline in the olfactory-bulbectomized rat. Eur. J. Pharmacol 486, 31–34. [DOI] [PubMed] [Google Scholar]
- van Broekhoven F, Backstrom T, van Luijtelaar G, Buitelaar JK, Smits P, Verkes RJ, 2007. Effects of allopregnanolone on sedation in men, and in women on oral contraceptives. Psychoneuroendocrinology 32, 555–564. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH, 2002. Functional neurogenesis in the adult hippocampus. Nature 415, 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Johnston PB, Ball BG, Brinton RD, 2005. The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J. Neurosci 25, 4706–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson RF, Brinton RD, 2010. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A 107, 6498–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill-Engerer S, David JP, Sazdovitch V, Liere P, Eychenne B, Pianos A, Schumacher M, Delacourte A, Baulieu EE, Akwa Y, 2002. Neurosteroid quantification in human brain regions: comparison between Alzheimer’s and nondemented patients. J. Clin. Endocrinol. Metab 87, 5138–5143. [DOI] [PubMed] [Google Scholar]
- Westcott KT, Hirst JJ, Ciurej I, Walker DW, Wlodek ME, 2008. Brain allopregnanolone in the fetal and postnatal rat in response to uteroplacental insufficiency. Neuroendocrinology 88, 287–292. [DOI] [PubMed] [Google Scholar]
- Whitney KD, Watson MA, Collins JL, Benson WG, Stone TM, Numerick MJ, Tippin TK, Wilson JG, Winegar DA, Kliewer SA, 2002. Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol. Endocrinol 16, 1378–1385. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR Jr., 2007. 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain 130, 1777–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization and Alzheimer’s Disease International, 2012. Dementia: a Public Health Priority
- Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, Sue LI, Woulfe J, Xu H, Stanimirovic DB, Zhang W, 2008. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis 29, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, He XY, Schulz H, 2005. Multiple functions of type 10 17beta-hydro-xysteroid dehydrogenase. Trends Endocrinol. Metab 16, 167–175. [DOI] [PubMed] [Google Scholar]
- Yang Y, Varvel NH, Lamb BT, Herrup K, 2006. Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J. Neurosci.: Off. J. Soc. Neurosci 26, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P, 2007. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc. Natl. Acad. Sci. U.S.A 104, 10601–10606. [DOI] [PMC free article] [PubMed] [Google Scholar]