

Abstract
Tauopathies including Alzheimer's disease, are characterized by progressive cognitive decline, neurodegeneration, and intraneuronal aggregates comprised largely of the axonal protein Tau. It has been unclear whether cognitive deficits are a consequence of aggregate accumulation thought to compromise neuronal health and eventually lead to neurodegeneration. We use the Drosophila tauopathy model and mixed-sex populations to reveal an adult onset pan-neuronal Tau accumulation-dependent decline in learning efficacy and a specific defect in protein synthesis-dependent memory (PSD-M), but not in its protein synthesis-independent variant. We demonstrate that these neuroplasticity defects are reversible on suppression of new transgenic human Tau expression and surprisingly correlate with an increase in Tau aggregates. Inhibition of aggregate formation via acute oral administration of methylene blue results in re-emergence of deficient memory in animals with suppressed human Tau (hTau)0N4R expression. Significantly, aggregate inhibition results in PSD-M deficits in hTau0N3R-expressing animals, which present elevated aggregates and normal memory if untreated with methylene blue. Moreover, methylene blue–dependent hTau0N4R aggregate suppression within adult mushroom body neurons also resulted in emergence of memory deficits. Therefore, deficient PSD-M on human Tau expression in the Drosophila CNS is not a consequence of toxicity and neuronal loss because it is reversible. Furthermore, PSD-M deficits do not result from aggregate accumulation, which appears permissive, if not protective of processes underlying this memory variant.
SIGNIFICANCE STATEMENT Intraneuronal Tau aggregate accumulation has been proposed to underlie the cognitive decline and eventual neurotoxicity that characterizes the neurodegenerative dementias known as tauopathies. However, we show in three experimental settings that Tau aggregates in the Drosophila CNS do not impair but rather appear to facilitate processes underlying protein synthesis-dependent memory within affected neurons.
Keywords: Drosophila, memory, methylene blue, tau, tau aggregation, tauopathies
Introduction
Tauopathies involve dysregulation of the essential neuronal microtubule-associated protein Tau and are the most widespread neurodegenerative dementias including Alzheimer's disease (AD) and Pick's disease, among others (Spillantini and Goedert, 1998; Lee et al., 2001; Delacourte, 2005; Zhang et al., 2022). There are six Tau isoforms in the human CNS arising by alternative splicing of a single transcript (Andreadis et al., 1995; Arendt et al., 2016; Zhang et al., 2022) and are engaged in multiple intraneuronal processes including axonal microtubule stability and function (Wang and Mandelkow, 2016; Sotiropoulos et al., 2017).
Although the initiating mechanisms remain largely elusive, pathogenic transformation of physiological Tau isoforms is characterized by their hyperphosphorylation and eventual aggregate formation (Alonso et al., 2001; Cowan and Mudher, 2013; Arendt et al., 2016). This has led to hypotheses positing that aggregates act as gain-of-function mutations (Trojanowski and Lee, 2005), obstructing housekeeping or neuroplasticity mechanisms and mediate neuronal dysfunction, toxicity, and neurodegeneration (Arendt et al., 2016; Wang and Mandelkow, 2016; Zhang et al., 2022). However, the contribution of aggregates, such as the characteristic neurofibrillary tangles (NFTs) in neuronal dysfunction and neurodegeneration, has been questioned (Spires-Jones et al., 2009, 2011; Wang and Mandelkow, 2016). Typically, NFT formation is preceded by cognitive deficits (Andorfer et al., 2005), and their presence generally does not correlate with cognitive deficits in mouse tauopathy models (Santacruz et al., 2005; Sydow et al., 2011; Van der Jeugd et al., 2012). In Drosophila, pharmacological or genetic inhibition of hyperphosphorylation, which reverses Tau-mediated dysfunction, is reported to be accompanied by increased Tau aggregation (Cowan et al., 2015). Furthermore, inhibition of Tau aggregation in clinical trials did not benefit AD patients or those with the behavioral variant of frontotemporal dementia (Wischik et al., 1996, 2015; Gauthier et al., 2016; Shiells et al., 2020). Therefore, although larger Tau aggregates such NFTs may eventually mediate neuronal death and underlie neurodegeneration, they appear unlikely to be causal of neuronal dysfunction and initial cognitive deficits.
Tau is proposed to form extended β-sheet amyloid-like filamentous inclusions with structures characterizing distinct tauopathies (Shi et al., 2021) via a stepwise mechanism involving a number of apparent intermediates. Pathologically hyperphosphorylated Tau is thought to form oligomers such as dimers and trimers that act as intermediates and promote formation of larger globular oligomers, which aggregate further adopting β-sheet conformations to yield filaments and eventually NFTs (Sahara et al., 2007, 2008; Patterson et al., 2011; Kaniyappan et al., 2017). Small oligomers, comprising a few to a dozen monomers, are thought to be soluble, whereas larger insoluble ones are referred to as granular tau oligomers (GTOs; Cowan et al., 2015). Significantly, the small oligomers have been linked to neuronal dysfunction and synaptotoxicity (Kaniyappan et al., 2017), whereas the larger ones form in conditions associated with suppression of these phenotypes (Cowan et al., 2015).
We aimed to determine whether Tau aggregation underlies cognitive deficits capitalizing on the genetic facility of a Drosophila tauopathy model (Papanikolopoulou and Skoulakis, 2011; Giong et al., 2021). Human Tau isoform-encoding transgenes expressed in the adult Drosophila CNS result in isoform and time-dependent deficits in associative learning (Mershin et al., 2004; Kosmidis et al., 2010; Papanikolopoulou and Skoulakis, 2015; Sealey et al., 2017; Keramidis et al., 2020) and memory (Prifti et al., 2021). The exquisite spatiotemporal regulation of transgene expression in this system (McGuire et al., 2004a,b) provides precise description of Tau pathogenic modifications ostensibly underlying learning deficits (Papanikolopoulou and Skoulakis, 2015) and the formation of high-molecular-weight aggregates (Cowan and Mudher, 2013; Papanikolopoulou and Skoulakis, 2015; Sealey et al., 2017). Using regulated spatiotemporal expression in the fly CNS of two human Tau isoforms, one known to precipitate learning defects and another that does not (Sealey et al., 2017), we ask whether the presence of aggregates correlates with memory deficits.
Materials and Methods
Drosophila culture and strains
Drosophila crosses were set up en masse in standard wheat-flour-sugar food supplemented with soy flour and CaCl2 and cultured at 18°C at 50–70% humidity in a 12 h light/dark cycle unless noted otherwise. Adult-specific pan-neuronal and panmushroom body transgene expression was achieved using the ElavC155-Gal4; Tub-Gal80ts (ElavGal4;Gal80ts; Papanikolopoulou and Skoulakis, 2015) or LeoMB-Gal4; Tub-Gal80ts (LeoGal4;Gal80ts; Papanikolopoulou et al., 2019), respectively. The fly line carrying UAS-htau0N4R (human Tau 0N4R) was a gift from Mel Feany (Harvard Medical School) and UAS-hTau0N3R of Dr. Amrit Mudher (University of Southampton). The generation of UAS-hTau0N4Ra1 transgene has been described previously (Keramidis et al., 2020). The bacterial plasmid pGEX-5× expressing the hTau0N4R isoform was a gift from Martin Chow (University of Kentucky). The cDNA was subcloned into pUASattB vector (Bischof et al., 2007) as a BglII/XbaI fragment. The sequence of the construct was confirmed by dsDNA sequencing (Vienna BioCenter). Transgenic flies were generated by phiC31-mediated transgenesis by BestGene. DNAs were injected into genomic landing site 53B2 and ZH-86Fb on the second (0N4Ra1) and third (0N4Ra2) chromosomes, respectively (Bloomington Drosophila Stock Center #9736 and #24749, respectively). The double-Tau transgene strain (0N4R2a) was constructed by standard genetic crosses of the above transgenes (0N4Ra1 and 0N4Ra2). All initial fly strains were backcrossed into the resident Cantonized w1118 control background for six generations.
Drug feeding
Adult flies were collected and maintained on a standard food supplement with methylene blue (MetBlu, Sigma-Aldrich) in the concentrations indicated. Flies were transferred to fresh vials every 2 d.
Life span determination
Flies accumulating hTau0N4R or hTau0N3R under ElavC155-Gal4; Tub-Gal80ts were raised at 18°C along with control driver heterozygotes. Groups of 20 young male flies (1–3 d old) were collected and maintained at the transgene-expression permissive temperature of 30°C until they expired. Flies were transferred to fresh vials every 3 d. For the drug experiments, flies were transferred to fresh food supplemented with methylene blue every 2 d. At least 300 flies were assessed per genotype.
Behavioral analyses
Animals expressing UAS-hTau0N4R or UAS-hTau0N3R under the control of the ElavC155-Gal4; Tub-Gal80ts or LeoMB-Gal4; Tub-Gal80ts drivers were raised at 18°C. On eclosion they were collected in fresh bottles or vials, and transgene expression was induced by placing them at 30°C for 6 or 12 d. For expression reversal experiments, pan-neuronal transgene expression was allowed for 12 d at 30°C as before, but it was followed by 10 d of maintaining the flies at 18°C as described in the text, and flies were transferred to fresh vials with or without methylene blue every 2 d. Flies on methylene blue for behavioral testing were transferred to fresh vials without the drug for 1 h before conditioning commenced.
All associative learning and memory experiments were performed under dim red light at 25°C and 70–75% humidity in a genotype-balanced manner. All genotypes involved in an experiment were tested per day. Olfactory aversive conditioning was performed as previously described (Keramidis et al., 2020) using the aversive odors benzaldehyde (BNZ) and 3-octanol (OCT) diluted in isopropyl myristate (Fluka; 6% v/v for BNZ and 50% v/v for OCT) as conditioned stimuli (CS+ and CS−) with 90 V electric shocks as unconditioned stimuli (US). One hour before training flies were transferred to fresh food vials. To assess immediate memory (learning), a group of 50–70 flies were tested immediately after a single training cycle consisting of the CS+ odor for 40 s paired with eight 90 V shocks, 30 s air, and CS− odor for 40 s without shock and then 30 s of air. To assess immediate performance (learning) after a five-round Extended Conditioning (5X Immediate), flies were tested immediately after five training cycles each consisting of the CS+ odor for 60 s paired with 12 90 V shocks, 30 s air, and CS− odor for 60 s without shock and then 30 s of air, with 15 min rest intervals between rounds. For 24 h memory after Spaced Conditioning (PSD-M) flies were submitted to 12 US/CS pairings per round and five such training cycles with a 15 min rest interval between cycles as above, but they were kept at 18°C for 24 h before testing. For 24 h memory after Massed Conditioning [protein synthesis-independent memory (PSI-M)], flies were submitted to 12 US/CS pairings per round and five such rounds of training, without the 15 min inter-round interval. The flies were also kept at 18°C until tested 24 h later. In all above experiments, two groups of animals of the same genotype were trained simultaneously with the CS+ and CS− odors switched. Both groups of flies were tested in a T-maze apparatus being allowed to choose between the two odors for 90 s. A performance index (PI) was calculated as described before (Keramidis et al., 2020) and represents n = 1.
RNA extraction and RT-PCR
Total RNA was extracted using TRIzol Reagent (Sigma-Aldrich) following instructions from the manufacturer. RT reaction was conducted using SuperScript II Reverse Transcriptase (Invitrogen), and 1 μg cDNA from each RT reaction was then subjected to PCR using the following conditions: 95°C for 10 min, followed by 28 cycles of 95°C for 60 s, 62°C for 40 s, and 72°C for 60 s. A final extension step at 72°C for 10 min was performed, and the PCR products were analyzed by agarose gel electrophoresis. The ribosomal gene rp49 was used as a normalizer. The primers used were the following: Tau forward, 5'-CCCGCACCCCGTCCCTTCC-3′; Tau reverse, 5'-GATCTCCGCCCCGTGGTCTGTCTT-3′; rp49 forward, 5'- GATCGTGAAGAAGCGCAC-3′; and rp49-reverse, 5'-CTTCTTGAATCCGGTGGG-3′. Quantification was performed using ImageJ software.
Western blot and antibodies
Total Tau levels in three to six adult female heads were determined by homogenization in 1× Laemmli buffer (50 mm Tris, pH 6.8, 5% 2-mercaptoethanol, 2% SDS, 10% glycerol, and 0.01% bromophenol blue), boiling for 5 min at 95°C, centrifugation for 5 min at 11,000 × g and separation in 10% SDS-acrylamide gels. Proteins were transferred to PVDF membranes and probed with mouse monoclonal anti-Tau (5A6, Developmental Studies Hybridoma Bank) at a 1:1000 dilution. Anti-syntaxin (Syx) primary antibody (8C3, Developmental Studies Hybridoma Bank) at a 1:3000 dilution was used to normalize sample loading. HRP-conjugated secondary antibodies were applied at 1:5000, the signal was detected by chemiluminescence (Immobilon Crescendo, Millipore) and quantified by densitometry with the Image Lab 5.2 program (Bio-Rad).
Tau solubility assay
For the extraction of insoluble Tau species with SDS, adult fly heads were homogenized in TBS/sucrose buffer (50 mm Tris HCl, pH 7.4, 175 mm NaCl, 1 m sucrose, and 5 mm EDTA, supplemented with protease and phosphatase inhibitors) as described in (Sealey et al., 2017; Prifti et al., 2021). The samples were then spun for 2 min at 1000 × g, and the supernatant was centrifuged at 200,000 × g for 2 h at 4°C. The resulting supernatant was regarded as the soluble fraction, and the pellet was resuspended in 5% SDS/TBS (50 mm Tris HCl, pH 7.4, 175 mm NaCl, 5% SDS) and centrifuged at 200,000 × g for 2 h at 25°C. The supernatants were collected as the SDS-soluble, aqueous-insoluble fraction. All samples were diluted in 2× Laemmli buffer and boiled for 5 min at 95°C. Equivalent volumes were loaded and analyzed by immunoblotting.
Atomic force microscopy
To extract the insoluble Tau fraction enriched for filaments and excluding GTOs, 50 adult fly heads were homogenized in TBS/sucrose (50 mm Tris HCl, pH 7.4, 175 mm NaCl, 1 m sucrose, 5 mm EDTA, and protease inhibitor cocktail) as described in Sealey et al. (2017) and Prifti et al. (2021). The samples were then spun for 2 min at 1000 × g, and the supernatant was centrifuged at 100,000 × g for 30 min at 4°C. The resulting supernatant included the aqueous soluble fraction and monomeric Tau, NS1. The pellet was resuspended at room temperature in 5% SDS/TBS buffer and spun at 100,000 × g for 30 min at 25°C. The resulting NP1 pellet was washed three times with water to remove residual SDS and resuspended in 1× PBS. The pellet sample was placed in a freshly cleaved 10 mm mica disk (Agar Scientific) and incubated at room temperature for 5 min to allow absorbing. Samples were rinsed four times with ultrapure water and dried with compressed air. Samples were imaged in air with a digital multimode Nanoscope IV AFM operating in tapping mode with an aluminum-coated noncontact/Tapping mode probe with a resonance frequency of 320 kHz and force constant of 42N/m (Pointprobe NHCR, NanoWorld). Representative images were taken at random points on the sample with a scan rate of 1 Hz to 2 Hz. The acquired images were processed by WSxM software.
Experimental design and statistical analyses
For all experiments, controls and experimental genotypes were tested in the same session in a balanced design. Genotypes were trained and tested in a random order. Performance indexes in behavioral experiments were analyzed parametrically with the JMP 7.1 statistical software package (SAS) and plotted using GraphPad Prism 9.5 software. Following an initial positive ANOVA, the means were compared with the control with planned multiple comparisons using the least squares means (LSM) approach or with Dunnett's tests as indicated. Survival curves were compared at each assessment day using Wilcoxon/Kruskal–Wallis tests. The means and SEMs from each genotype for the days with significant differences were compared using Steel's test with control tests. Quantification of all Western blots was performed by densitometry. Tau levels were normalized using the Syx as a loading control and are shown as a ratio of their mean ±SEM values relative to respective levels of the control genotype, which was set to one. The means were compared following an initial positive ANOVA, using Dunnett's tests relative to the designated control. All statistical details are presented in the text and the relevant tables.
Results
Deficient protein-synthesis-dependent memory on hTau0N4R accumulation in the adult CNS
Deficient associative learning was reported to emerge in a time-dependent manner after 12 d of pan-neuronal adult-specific expression of hTau0N4R (Papanikolopoulou and Skoulakis, 2015; Sealey et al., 2017). As before (Keramidis et al., 2020), we used the well-established negatively reinforced olfactory conditioning assay to assess learning and consolidated memory forms (Tully et al., 1994). Learning was normal after 6 d of hTau0N4R expression (Fig. 1A; ANOVA, F(5,74) = 17.6063, p = 3.3 × 10−11; subsequent LSM planned comparisons with both control strains, 6 d, p = 0.2303 and p = 0.7165, respectively), but a strong learning deficit emerged by day 12 (Fig. 1A; LSM planned comparisons with both controls, 12 d, p < 0.0001 from both). This verified independently the previously reported (Papanikolopoulou and Skoulakis, 2015) time-dependent manifestation of neuronal dysfunction in this Drosophila tauopathy model. To determine whether deficits in consolidated memory emerge with the same time dependence, performance was assessed 24 h post-training with five rounds of Spaced Training, known to yield PSD-M (Tully et al., 1994). PSD-M appeared intact for 6 d of hTau expression (Fig. 1B; ANOVA, F(5,67) = 10.433, p = 2.7 × 10−7; subsequent LSM planned comparisons with both controls, 6 d, p = 0.8911 and p = 0.3287, respectively). However, a robust deficit was evident after 12 d of hTau0N4R expression (Fig. 1B; LSM planned comparisons with both controls, 12 d, p = 0.0025 and p = 0.0089, respectively).
Figure 1.

Deficient associative learning and PSD-M emerge in a time-dependent manner on hTau0N4R expression in the adult CNS. Bars represent the mean PI and ± SEM for the number of indicated experimental replicates (n). Stars indicate significant differences. All statistical details are presented in the statistics table (Table 4). Black bars represent the experimental strains and open bars the controls as indicated. A, Immediate Performance after one round of standard conditioning (Learning) of animals accumulating pan-neuronally the hTau0N4R isoform for 6 and 12 d compared with that of driver and transgene heterozygotes; n ≥ 12 for all genotypes. B, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of animals accumulating pan-neuronally hTau0N4R for 6 and 12 d compared with that of driver and transgene heterozygotes; n ≥ 11 for all genotypes. C, Immediate Performance after Extended Conditioning (5X) of flies accumulating pan-neuronally hTau0N4R for 12 d compared with that of driver and transgene heterozygotes; n ≥ 12 for all genotypes. D, Twenty-four-hour Massed Conditioning (PSI-M) memory of flies accumulating pan-neuronally hTau0N4R for 12 d compared with that of driver and transgene heterozygotes; n ≥ 12 for all genotypes. E, Representative Western blots from head lysates of flies pan-neuronally accumulating hTau0N4R for 12 d compared with similar lysates from hTau0N4Ra1, hTau0N4Ra2, and the double transgenic strain hTau0N4R2a, probed with the 5A6 anti-Tau antibody. Syx levels in the lysates were used as quantification normalizer. Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM values relative to respective levels in flies accumulating hTau0N4R, which was set to one; n ≥ 4 for all genotypes. F, Performance immediately after one round of standard conditioning (Learning) animals accumulating pan-neuronally hTau0N4R from the double transgenic hTau0N4R2a strain for 12 d and heterozygous controls; n ≥ 12 for all genotypes. G, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of animals accumulating pan-neuronally hTau0N4R from the double transgenic hTau0N4R2a for 12 d and heterozygous controls; n ≥ 13 for all genotypes. H, Immediate Performance after Extended Conditioning (5X) of flies accumulating pan-neuronally hTau0N4R from the double transgenic hTau0N4R2a strain for 12 d and heterozygous controls; n ≥ 9 for all genotypes. I, Twenty-four-hour Massed Conditioning (PSI-M) memory of flies accumulating pan-neuronally hTau0N4R from the double transgenic hTau0N4R2a for 12 d and heterozygous controls; n ≥ 11 for all genotypes.
These robust learning and memory deficits raised the question of whether the 12-day accumulation of pathologically hyperphosphorylated hTau0N4R (Papanikolopoulou and Skoulakis, 2015), affects processes underlying neuronal dysfunction specifically, or the deficits are consequent of nonspecific neurotoxicity. To probe whether flies after 12 d of hTau0N4R expression are learning competent, immediate performance (learning) after a five-round Extended Conditioning of 12 CS/US pairings each (Gouzi et al., 2018) was assessed. This conditioning regime yielded identical learning for hTau0N4R-accumulating animals and controls (Fig. 1C; ANOVA, F(2,40) = 3.136, p = 0.0549). Therefore, although hTau0N4R accumulation in the adult Drosophila CNS compromises learning, the deficit can be rescued by overconditioning, suggesting that it results from a compromised learning rate as reported before for Drosophila mutants (Moressis et al., 2009), rather than ability to learn consistent with neuronal loss.
In addition, the Massed Conditioning-elicited PSI-M (Tully et al., 1994), was not affected after 12 d of hTau0N4R accumulation (Fig. 1D; ANOVA, F(2,47) = 3.202, p = 0.0501). Because of the two consolidated memory types, PSD-M is preferentially compromised, and hTau0N4R accumulation appears to impair translation in affected neurons, in accord with previous suggestions (Papanikolopoulou et al., 2019), but spares the translation-independent PSI-M. It appears then that adult CNS-limited hTau0N4R accumulation compromises specific plasticity processes and behavioral outputs, arguing against the impairments resulting from neurotoxicity and neuronal death, which would likely affect neuroplasticity rather indiscriminately.
To verify these surprising results, two independent hTau0N4R-encoding transgenes (0N4Ra1 and 0N4Ra2) on different chromosomal sites (attp9A and attp86F) were generated. However, expression of both of these site-specific inserted transgenes was low, and they were combined in a double transgenic strain 0N4R2a to approximate hTau levels yielded by the single 0N4R transgene (Wittmann et al., 2001; Fig. 1E; ANOVA, F(3,18) = 135.648, p = 4.3 × 10−11; subsequent LSM planned comparisons with ElavGal4;Gal80ts>0N4R, p = 4.9 × 10−11, p = 2.9 × 10−11 and p = 2.4 × 10−5, respectively). Consistent with the results above (Fig. 1A), adult specific pan-neuronal expression of hTau0N4R2a for 12 d resulted in impaired learning on a single round of eight CS/US pairings (Fig. 1F; ANOVA, F(2,35) = 143.048, p = 5.5 × 10−17; subsequent LSM planned comparisons with both controls, p = 2.5 × 10−8 and p = 9.5 × 10−18, respectively), which, however, was eliminated on Extended Conditioning (Fig. 1H; ANOVA, F(2,27) = 3.119, p = 0.062). Nevertheless, this spaced conditioning regime resulted in impaired PSD-M (Fig. 1G; ANOVA, F(2,42) = 13.829, p = 2.7 × 10−5; subsequent LSM planned comparisons with both controls, p = 0.0001 and p = 1.9 × 10−5) but left PSΙ-Μ intact (Fig. 1I; ANOVA, F(2,34) = 2.963, p = 0.0659). These results confirm with an independent transgenic strain that adult-specific pan-neuronal hTau0N4R accumulation results in impaired, but not abolished, associative learning and specific attenuation of PSD-M.
Tau insoluble aggregate accumulation correlates with reversal of the PSD-M deficit
Because the effects of hTau accumulation on neuroplasticity appeared specific to PSD-M, and even learning deficits were ameliorated with overtraining, we hypothesized that the CNS is unlikely to have sustained extensive neurodegenerative damage. If the fly CNS was not damaged, then repressing expression of the hTau transgene would reduce the hTau0N4R load, which could attenuate the neuroplasticity deficits as in vertebrate models expressing the frontotemporal dementia and parkinsonism (FTDP)-linked mutant hTau0N4R (Santacruz et al., 2005; Sydow et al., 2011; Van der Jeugd et al., 2012). To that end, adult-specific pan-neuronal hTau0N4R transgene expression was permitted for 12 d at 30°C as before (Papanikolopoulou and Skoulakis, 2015; Fig. 1), but it was followed by 10 d of maintaining the flies at 18°C, the nonpermissive temperature for transgene expression (McGuire et al., 2004b). Another group of flies of the identical genotype were maintained as adults for 10 d at 18°C and then switched to transgene-inducing 30°C for 12 d (Fig. 2A). Therefore, in the two groups of genotypically identical and of similar age animals, hTau0N4R is either repressed for 10 d following 12 d of expression (OFF), or it is expressed for 12 d (ON) after 10 d of repression. Transgene expression levels under these conditions were assessed on day 22 after adult emergence and revealed (Fig. 2B; ANOVA, F(1,13) = 99.548, p = 3.7 × 10−7) at least a 50% reduction in htau0N4R transcripts on transgene repression (OFF), relative to its expression under permissive conditions (ON). In contrast, protein levels remained equivalent if not somewhat elevated under transgene transcriptional repression conditions (Fig. 2C; ANOVA, F(1,12) = 1.012, p = 0.3327), indicating that the hTau0N4R protein is rather stable in the fly CNS.
Figure 2.

Reversal of the PSD-M deficit is correlated with Tau insoluble aggregate accumulation. A, A schematic of the hTau0N4R transgene repression (OFF) and expression protocol conditions (ON). The two groups of genotypically identical and of similar age animals hTau0N4R are either repressed for 10 d of maintaining the flies at 18°C, following 12 d of expression (OFF), or expressed for 12 d at 30°C (ON) after maintaining the adults flies for 10 d at 18°C. B, Representative RT-PCR of Tau mRNA levels in flies with either repressed (OFF) or pan-neuronally expressing hTau0N4R (ON). The rp49 RNA levels served as an internal reference and as a normalization control for the quantifications. The normalized level of hTau0N4R (ON) for each quantification was fixed to one. Error bars indicate mean ± SEM relative mRNA levels at the OFF condition relative to that of the ON condition. The star indicates significant differences from the control; n = 7 determinations for both conditions. C, Representative Western blots from head lysates of flies accumulating hTau0N4R pan-neuronally for 12 d (ON) compared with similar lysates from flies with hTau0N4R transgene repression (OFF) probed with the 5A6 anti-Tau antibody. The level of Syx in the lysates was used as control for quantifications. For the quantification, Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM values relative to the respective levels under ON conditions; n = 6 independent blots for both conditions. D, Representative Western blot of soluble fractions of head lysates under expression (ON) or repression (OFF) conditions probed with the 5A6 anti-Tau antibody. Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM relative to respective levels in flies accumulating pan-neuronally hTau0N4R or hTau0N4R2a for 12 d, which were set to one. n ≥ 5 for hTau0N4R and n ≥ 6 for hTau0N4R2a, n = 6 independent blots. E, Representative Western blot of insoluble fractions of head lysates under expression (ON) or repression (OFF) conditions probed with the 5A6 anti-Tau antibody. Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM relative to respective levels in flies accumulating pan-neuronally hTau0N4R or hTau0N4R2a for 12 d, which were set to one. n ≥ 5 for hTau0N4R and n ≥ 4 for hTau0N4R2a independent blots. F, G, Bars represent the mean PIs and ± SEM for the number of indicated experimental replicates (n). Stars indicate significant differences. Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of animals accumulating pan-neuronally hTau0N4R (F) or hTau0N4R2a (G) for 12 d at 30°C (ON, gray bars) compared with driver and transgene heterozygotes (open bars) and animals with repressed transgenes (black bars); n ≥ 9 for F and n ≥ 10 for G. H, Mean PIs and standard SEMs for 24 h Spaced Conditioning memory (PSD-M) performance of control animals kept either for 12 d at 30°C (gray bar) after 10 d as adults at 18°C or 10 d at 18°C following 12 d at 30°C (black bar); n = 8 for both groups. Statistical details on Table 4.
Sustained accumulation of hTau in the fly (Cowan et al., 2015; Papanikolopoulou and Skoulakis, 2015) or vertebrate CNS (Santacruz et al., 2005; Wang and Mandelkow, 2016) results in turnover-resistant aggregate formation. Therefore, we aimed to determine whether the apparently stable levels of hTau0N4R protein under transcriptional attenuation result from aggregate accumulation. Total head lysate proteins from flies with the 0N4R and 0N4R2a transgenes transcriptionally active for 12 d (ON), or inactive for 10 d (OFF), were fractionated, and hTau0N4R levels were quantified in the soluble and insoluble fractions. Interestingly, soluble hTau0N4R levels remained unchanged, if not somewhat decreased, regardless of whether the 0N4R and 0N4R2a transgenes were ON or OFF (Fig. 2D; ANOVA, F(1,11) = 0.145, p = 0.711 for hTau0N4R and F(1,13) = 4.262, p = 0.061 for hTau0N4R2a, respectively). However, insoluble hTau was elevated when the transgenes were transcriptionally inactive (Fig. 2E; ANOVA, F(1,11) = 9.191, p = 0.0126 for and F(1,9) = 11.556, p = 0.0094 for hTau0N4R2a, respectively). Therefore, aggregates accumulate in the fly CNS, ostensibly formed from pre-existing soluble hTau, and likely account for the apparently stable levels of the protein even after 10 d without new transgene transcription (Fig. 2B).
Importantly, silencing transgene transcription (OFF) for 10 d after 12 d of expression, resulted in recovery of the PSD-M deficit compared with the significantly attenuated memory of animals expressing hTau0N4R (ON). For hTau0N4R (Fig. 2F), ANOVA, F(3,39) = 12.466, p = 9.6 × 10−6; subsequent LSM planned comparisons with ElavG4;Gal80ts>0N4R (OFF) and ElavG4;Gal80ts>0N4R (ON), p = 0.0015; whereas in comparison with w1118>0N4R, p = 0.0099. Conversely, for hTau0N4R2a (Fig. 2G), ANOVA, F(3,43) = 17.761, p = 1.5 × 10−7; subsequent LSM planned comparisons with ElavG4;Gal80ts>0N4R2a (OFF) and ElavG4;Gal80ts>0N4R2a (ON), p = 0.002; whereas in comparison with w1118>0N4R2a, p = 7.1 × 10−5. Moreover, PSD-M was not affected by the temperature switching regimes in ElavG4;Gal80ts>w1118 controls (Fig. 2H; ANOVA, F(1,15) = 0.018, p = 0.8959), indicating that the differences in PSD-M in the experimental animals are not a consequence of the experimental manipulations.
These results are consistent with the notion that neuronal dysfunction manifested as memory deficits is not consequent of irreversibly damaged or degenerating CNS neurons but rather of reversibly impaired processes essential for PSD-M. Considering that transcriptional silencing of the transgenes elevates insoluble hTau, the results suggest that such aggregates not only do not precipitate neuronal dysfunction but may in fact suppress or prevent it. The deficient PSD-M could then be mediated by newly translated, hence largely soluble, hTau0N4R expected in the CNS of flies expressing the transgenes for 12 d (ON).
Blocking hTau0N4R insoluble aggregate formation results in defective PSD-M
Is it hTau0N4R aggregate accumulation that suppresses the PSD-M deficit or reduction of soluble protein on transcriptional silencing of the transgene? To differentiate between these two alternatives, we aimed to prevent hTau insoluble aggregate formation or induce their decomposition under transgene silencing conditions. To that end, flies expressing hTau0N4R for 12 d at 30°C were switched to the nonpermissive 18°C in the presence of a range of concentrations of the nonneuroleptic phenothiazine MetBlu. The drug has been experimentally shown to bind to the repeat domains of hTau and inhibit hTau-hTau interactions essential for formation of insoluble aggregates (Hosokawa et al., 2012) and paired helical filaments (Wischik et al., 1996). As the 0N4R and 0N4R2a transgenes yielded identical results in all experiments detailed above, to reduce redundancy, we used only the original randomly inserted hTau0N4R transgene (Wittmann et al., 2001) for all subsequent experiments unless specified otherwise.
Initially we used the control genotype ElavGal4;Gal80ts heterozygotes to determine the toxicity range of MetBlu at 30°C, where we typically assay the longevity of hTau0N4R-expressing animals (Papanikolopoulou and Skoulakis, 2015; Keramidis et al., 2020). MetBlu in the food media at the range of 10 to 250 μm did not affect survival significantly, but at 500 μm, it reduced the date that 50% of the population was expired (50% attrition date; Keramidis et al., 2020) by 16 d and at 1 mm by 22 d (Fig. 3A, Table 1). Conversely, 10–100 μm of the drug did not change the 50% attrition date of hTau0N4R-expressing flies relative to untreated ones but reduced it by 5 d relative to controls. The 50% attrition at 500 μm and 1 mm MetBlu were shortened by 15 d and 17 d, respectively, relative to untreated animals (Fig. 3B, Table 2). Therefore, in agreement with prior reports (Gillman, 2011), MetBlu precipitates significant concentration-dependent toxicity above 250 μm at 30°C, and this was more pronounced for hTau0N4R-expressing flies over the range of the experiment, where the 50% attrition date for these flies at 30°C was shortened by 13 d relative to their untreated siblings (Fig. 3B, Table 2).
Figure 3.
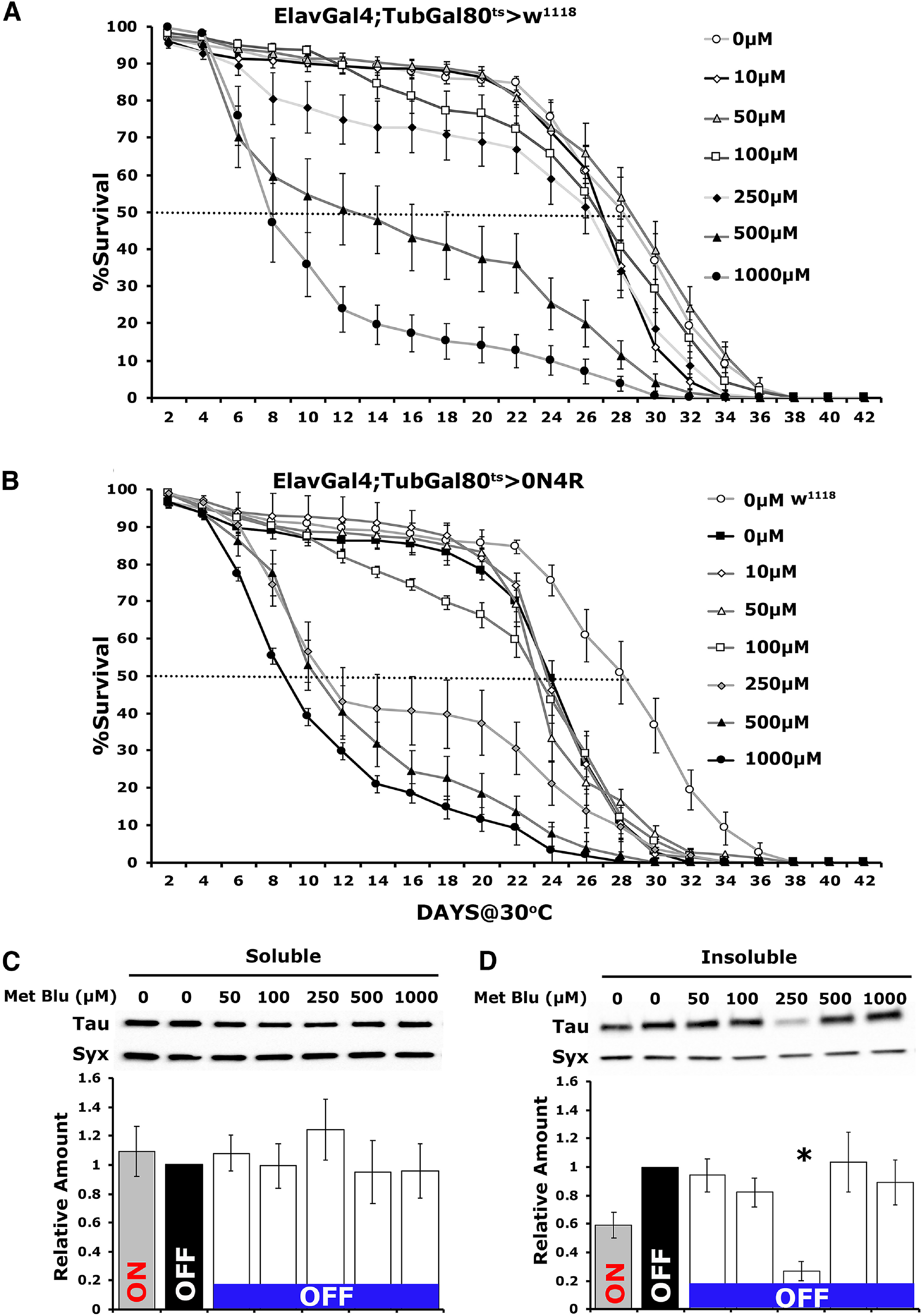
Methylene blue prevents insoluble hTau0N4R aggregate formation at a specific concentration. A, B, Survival curves of untreated and treated with different concentrations of MetBlu driver heterozygote control (A) and animals accumulating pan-neuronally hTau0N4R at 30°C (B). The data represent the mean ± SEM from two independent experiments with at least 300 flies assessed per genotype. Right, The different concentrations of MetBlu. The dotted lines indicate the 50% attrition levels. Statistical details are provided in Table 1 and 2. C, D, Representative Western blots of soluble (C) and insoluble (D) fractions generated from adult flies untreated or treated with different concentrations of MetBlu probed with 5A6 anti-Tau antibody. hTau0N4R was either expressed for 12 d (ON) or is repressed for 10 d following 12 d of expression (OFF). To determine the effect of the drug on hTau0N4R insoluble aggregate formation, flies were shifted onto food containing MetBlu ranging from 50 to 1000 μm at 18°C to silence the transgene for 10 d (OFF). The different concentrations of MetBlu used are indicated above each bar. The level of Syx was used as control for quantifications. The normalized level of hTau0N4R (OFF condition, untreated) for each quantification was fixed to one. Error bars indicate mean ± SEM relative to respective levels in flies that exist under transgene transcriptional silencing conditions. The star indicates significant differences from the control genotype; n ≥ 5 for C and n ≥ 6 independent blots for D.
Table 1.
Survival statistics for control heterozygotes kept on the indicated concentrations of MetBlu at 30°C
| Wilcoxon/Kruskal–Wallis |
Means comparison (Steel test with control) |
||||
|---|---|---|---|---|---|
| Day | χ2, (df, count) | p > χ2 | Genotype (μm MetBlu) | z | p |
| 2 | 17.015 (6,17) | 0.0092 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | |||||
| Elav;G80ts> + 500 | 0.4029 | 0.9975 | |||
| Elav;G80ts> + 1000 | 2.9594 | 0.0161 | |||
| 6 | 8.044 (6,17) | 0.2349 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | |||||
| Elav;G80ts> + 500 | |||||
| Elav;G80ts> + 1000 | |||||
| 10 | 44.54 (6,17) | <0.0001 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | |||||
| Elav;G80ts> + 500 | −2.9765 | 0.0153 | |||
| Elav;G80ts> + 1000 | −4.9758 | <0.0001 | |||
| 14 | 60.578 (6,17) | <0.0001 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | −2.6753 | 0.0372 | |||
| Elav;G80ts> + 500 | −3.8591 | 0.0007 | |||
| Elav;G80ts> + 1000 | −4.9943 | <0.0001 | |||
| 18 | 63.859 (6,17) | <0.0001 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | −2.4139 | 0.075 | |||
| Elav;G80ts> + 500 | −4.2418 | 0.0001 | |||
| Elav;G80ts> + 1000 | −5.0056 | <0.0001 | |||
| 22 | 58.397 (6,17) | <0.0001 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | −1.8873 | 0.2439 | |||
| Elav;G80ts> + 500 | −4.3894 | <0.0001 | |||
| Elav;G80ts> + 1000 | −4.9321 | <0.0001 | |||
| 26 | 43.698 (6,17) | <0.0001 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | −1.0555 | 0.7870 | |||
| Elav;G80ts> + 500 | −3.4394 | 0.0032 | |||
| Elav;G80ts> + 1000 | −4.4177 | <0.0001 | |||
| 30 | 39.271 (6,17) | <0.0001 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | −2.1832 | 0.3149 | |||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | −2.1833 | 0.1287 | |||
| Elav;G80ts> + 500 | −3.6036 | 0.0018 | |||
| Elav;G80ts> + 1000 | −4.1500 | 0.0002 | |||
| 34 | 26.7302 (6,17) | 0.0002 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | −2.6180 | 0.0436 | |||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | −2.0723 | 0.1642 | |||
| Elav;G80ts> + 500 | −2.1460 | 0.1398 | |||
| Elav;G80ts> + 1000 | −2.6181 | 0.0436 | |||
| 38 | 0.0000 (6,17) | 1.0000 | Elav;G80ts> + 0 | ||
| Elav;G80ts> + 10 | |||||
| Elav;G80ts> + 50 | |||||
| Elav;G80ts> + 100 | |||||
| Elav;G80ts> + 250 | |||||
| Elav;G80ts> + 500 | |||||
| Elav;G80ts> + 1000 | |||||
Survival results from all the independent determinations were compared with Wilcoxon/Kruskal–Wallis tests for the indicated days. If a positive (χ2) outcome, the means from each genotype for the days with significant differences were compared using the Steel with control tests, whose z ratio and p values are shown. Significant differences from controls are shown in boldface.
Table 2.
Survival statistics for flies expressing hTau0N4R kept on the indicated concentrations of MetBlu at 30°C
| Wilcoxon/Kruskal–Wallis |
Means comparison (Steel test with control) |
||||
|---|---|---|---|---|---|
| Day | χ2, (df, count) | p > χ2 | Genotype (μm MetBlu) | z | p |
| 2 | 13.5961 (6,17) | 0.0834 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | |||||
| Elav;G80ts>0N4R 250 | |||||
| Elav;G80ts>0N4R 500 | |||||
| Elav;G80ts>0N4R 1000 | |||||
| 6 | 18.4831 (6,17) | 0.0051 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | |||||
| Elav;G80ts>0N4R 250 | |||||
| Elav;G80ts>0N4R 500 | |||||
| Elav;G80ts>0N4R 1000 | −2.1034 | 0.1536 | |||
| 10 | 65.901 (6,17) | <0.0001 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | |||||
| Elav;G80ts>0N4R 250 | −3.0928 | 0.0106 | |||
| Elav;G80ts>0N4R 500 | −3.8612 | 0.0006 | |||
| Elav;G80ts>0N4R 1000 | −4.8594 | <0.0001 | |||
| 14 | 76.510 (6,17) | <0.0001 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | −1.7623 | 0.3028 | |||
| Elav;G80ts>0N4R 250 | −3.7252 | 0.0011 | |||
| Elav;G80ts>0N4R 500 | −4.9123 | <0.0001 | |||
| Elav;G80ts>0N4R 1000 | −4.8496 | <0.0001 | |||
| 18 | 73.901 (6,17) | <0.0001 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | −1.5947 | 0.4017 | |||
| Elav;G80ts>0N4R 250 | −3.4581 | 0.0030 | |||
| Elav;G80ts>0N4R 500 | −4.8533 | <0.0001 | |||
| Elav;G80ts>0N4R 1000 | −4.9939 | <0.0001 | |||
| 22 | 70.196 (6,17) | <0.0001 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | −0.7294 | 0.9497 | |||
| Elav;G80ts>0N4R 250 | −3.6844 | 0.0013 | |||
| Elav;G80ts>0N4R 500 | −4.8786 | <0.0001 | |||
| Elav;G80ts>0N4R 1000 | −4.9903 | <0.0001 | |||
| 26 | 30.019 (6,17) | <0.0001 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | |||||
| Elav;G80ts>0N4R 250 | −1.5003 | 0.4648 | |||
| Elav;G80ts>0N4R 500 | −2.9561 | 0.0163 | |||
| Elav;G80ts>0N4R 1000 | −3.6461 | 0.0015 | |||
| 30 | 15.404 (6,17) | 0.0173 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | |||||
| Elav;G80ts>0N4R 250 | −0.9155 | 0.8626 | |||
| Elav;G80ts>0N4R 500 | −2.2021 | 0.1204 | |||
| Elav;G80ts>0N4R 1000 | −2.2018 | 0.1204 | |||
| 34 | 5.1170 (6,17) | 0.5289 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | |||||
| Elav;G80ts>0N4R 250 | |||||
| Elav;G80ts>0N4R 500 | |||||
| Elav;G80ts>0N4R 1000 | |||||
| 38 | 0.0000 (6,17) | 1.0000 | Elav;G80ts>0N4R 0 | ||
| Elav;G80ts>0N4R 10 | |||||
| Elav;G80ts>0N4R 50 | |||||
| Elav;G80ts>0N4R 100 | |||||
| Elav;G80ts>0N4R 250 | |||||
| Elav;G80ts>0N4R 500 | |||||
| Elav;G80ts>0N4R 1000 | |||||
| Elav;G80ts>0N4R 1000 | |||||
Survival results from all the independent determinations were compared with Wilcoxon/Kruskal–Wallis tests for the indicated days. If a positive (χ2) outcome, the means from each genotype for the days with significant differences were compared using the Steel with control tests, whose z ratio and p values are shown. Significant differences from controls are shown in boldface.
To determine the effect of the drug on the steady-state levels of hTau0N4R insoluble aggregates, flies expressing the transgene for 12 d were shifted to 18°C to silence transcription, and for these 10 d, were offered food containing MetBlu ranging from 50 to 1000 μm. Head lysates from these animals were fractionated, and the amount of hTau in the soluble and insoluble fractions was quantified relative to animals kept on normal food for the same period (0). Soluble hTau levels were not significantly affected by any concentration of MetBlu, but were somewhat, yet not significantly, elevated at 250 μm (Fig. 3C; ANOVA, F(6,40) = 0.323, p = 0.9204). Importantly, insoluble hTau0N4R levels were not significantly different from controls at any MetBlu concentration except at 250 μm, where they were significantly reduced (Fig. 3D; ANOVA, F(6,44) = 4.142, p = 0.0027). Subsequent comparisons with ElavGal4;Gal80ts>0N4R OFF revealed a significant effect of 250 μm MetBlu (p = 0.0008). The reason for this sharp optimum in the MetBlu concentration leading to insoluble aggregate reduction is unclear but has been consistent over a number of technical and biological experimental repeats.
Importantly, the elevated lethality of hTau0N4R-expressing flies on 250 μm MetBlu (Fig. 3B) was not apparent over the 10 d these animals were treated at 18°C, with typical survival rates over 98% (Fig. 4A; ANOVA, F(19 299) = 1.1663, p = 0.042). This agrees with previous suggestions (Schirmer et al., 2011) that the toxicity of the drug is likely dependent on the metabolic rate. For the poikilothermic Drosophila, metabolism is expected much higher at 30°C than 18°C, and it is most likely reflected on the lack of significant differences from controls at the lower temperature.
Figure 4.

Preventing hTau0N4R insoluble aggregate formation results in defective PSD-M under transgene transcriptional silencing conditions. A, Survival histogram for hTau0N4R animals kept under transgene silencing conditions (OFF, 18°C) but on MetBlu for 10 d compared with driver heterozygotes. The survival rates were over 98% in these conditions for all genotypes independent of drug administration. The data represent the mean ± SEM from two independent experiments with at least 300 flies assessed per genotype. B–G, Bars represent the mean PIs and ± SEM for the number of indicated experimental replicates (n). Stars indicate significant differences. Statistical details appear on Table 4. B, Immediate Performance after Extended Conditioning (5X) of hTau0N4R-expressing flies kept for 10 d in the OFF condition in the absence (0 μm) or presence of 250 μm MetBlu; n ≥ 7 per condition. C, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies kept for 10 d in the OFF condition in the absence (0 μm) or presence of 250 μm MetBlu; n ≥ 13 per condition. D, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of control animals kept for 10 d in the OFF condition in the absence (0 μm) or presence of 250 μm MetBlu; n ≥ 11 per condition. E, Twenty-four-hour Massed Conditioning (PSI-M) memory of hTau0N4R-expressing flies kept for 10 d in the OFF condition in the absence (0 μm) or presence of 250 μm MetBlu; n ≥ 14 per condition. F, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies kept for 10 d in the OFF condition in the absence (0 μm) or presence of 500 μm MetBlu; n ≥ 7 per condition. G, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of control animals kept for 10 d in the OFF condition in the absence (0 μm) or presence of 500 μm MetBlu; n = 12 per condition.
Significantly, immediate memory after Extended Conditioning of hTau0N4R-expressing animals treated for 10 d with 250 μm MetBlu was not significantly different from untreated flies of the same genotype (Fig. 4B; ANOVA, F(1,15) = 0.138, p = 0.7154). However, treated animals presented a significant reduction in 24 h PSD-M relative to untreated ones (Fig. 4C; ANOVA, F(1,27) = 10.435, p = 0.0033), but feeding control animals 250 μm MetBlu for 10 d did not impair PSD-M relative to that of their untreated siblings (Fig. 4D; ANOVA, F(1,22) = 0.201, p = 0.6584). Similarly, PSI-M was not affected in treated hTau0N4R-expressing flies (Fig. 4E; ANOVA, F(1,29) = 0.0016, p = 0.9681). Therefore, under these conditions, the drug does not appear to precipitate nonspecific dysfunction in the neurons or mechanisms underlying PSD-M.
In support of this interpretation and disfavoring the notion of differential MetBlu-mediated dysfunction in hTau0N4R-expressing flies, treatment with 500 μm of the drug, which does not appear to affect hTau aggregates (Fig. 3D), did not attenuate PSD-M in hTau0N4R-expressing animals (Fig. 4F; ANOVA, F(1,14) = 2.056, p = 0.1752), or in controls (Fig. 4G; ANOVA, F(1,23) = 0.701, p = 0.4115). Therefore, the relative elevation of aggregates on silencing hTau0N4R transcription likely accounts for the resultant reversal of PSD-M deficits (Fig. 2F,G). The collective results strongly argue that although hTau0N4R aggregates are benign, or protective, the smaller apparently soluble protein species are deleterious to processes requisite for PSD-M.
Efficient PSD-M in hTau0N3R-expressing flies correlates with elevated aggregates and is reversible with MetBlu
Unlike for hTau0N4R expressing flies, associative learning and PSD-M are normal in animals expressing the hTau0N3R isoform even after 12 d of transgene induction (Sealey et al., 2017). Quantification of insoluble hTau0N3R in head lysates revealed a nearly sixfold elevation over aggregates in lysates from hTau0N4R animals after 12 d at 30°C (Fig. 5A; ANOVA, F(1,9) = 51.036, p = 9.8 × 10−5). Considering the results above, this difference led to the hypothesis that the reported lack of learning and memory defects in hTau0N3R-expressing animals is a consequence of the elevated steady-state aggregates. To address this hypothesis, hTau0N3R-expressing animals were subjected to MetBlu-mediated aggregation inhibition for the 12 d transgene was actively transcribed posteclosion.
Figure 5.

Blocking hTau0N3R insoluble aggregate formation results in defective PSD-M. A, Representative Western blots of insoluble fractions generated from adult heads, following pan-neuronal expression of hTau0N4R and hTau0N3R transgenes for 12 d at 30°C, probed with the 5A6 anti-Tau antibody. The level of Syx was used as control for quantifications. The normalized level of hTau0N4R for each quantification was fixed to one. Error bars indicate mean ± SEM of insoluble hTau levels in flies that express hTau0N3Rover that of the hTau0N4R. The star indicates significant differences from that in hTau0N4R-expressing lysates; n ≥ 4 independent blots. B, Representative Western blots of soluble and insoluble fractions generated from adult heads, following pan-neuronal hTau0N3R expression for 12 d at 30°C in flies kept on different concentrations of MetBlu (0, 10, 50 and 100 μm), as indicated, probed with the 5A6 antibody., The level of Syx was used as loading control. For the quantification, Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM values relative to respective levels in untreated flies accumulating hTau0N3R, which were set to one. The star indicates significant differences from the untreated with MetBlu animals; n ≥ 4 for soluble and n > 12 for Insoluble independent blots. C, Survival histogram of animals of the indicated genotype untreated or treated with 50 μm MetBlu at 30°C compared with driver heterozygotes. MetBlu at 50 μm did not affect survival of driver heterozygotes. The data represent the mean ± SEM from two independent experiments with at least 300 flies assessed per genotype. Statistical details in the statistics table. The star indicates significant differences from the control genotype on the respective day. D–K, Bars represent the mean PIs and SEM for the number of indicated experimental replicates (n). Stars indicate significant differences. Statistical details are presented on Table 4. D, Immediate Performance after Extended Conditioning (5X) of hTau0N3R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 50 μm MetBlu; n ≥ 11 per condition. E, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N3R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 50 μm MetBlu; n ≥ 14 per condition. F, Twenty-four-hour Massed Conditioning (PSI-M) memory of hTau0N3R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 50 μm MetBlu; n ≥ 10 per condition. G, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of control animals kept for 12 d in the ON condition in the absence (0 μm) or presence of 50 μm MetBlu; n ≥ 14 per condition. H, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N3R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 10 μm MetBlu; n ≥ 13 per condition. I, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N3R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 100 μm MetBlu; n ≥ 12 per condition. J, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 50 μm MetBlu; n = 12 per condition. K, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 100 μm MetBlu; n ≥ 11 per condition.
As reported before (Sealey et al., 2017), hTau0N3R-expressing animals presented significantly reduced survival at 30°C, and this premature mortality was exaggerated by MetBlu at concentrations higher than 100 μm (Table 3), likely because of enhanced metabolism at the higher temperature (Schirmer et al., 2011). Treatment with the less toxic MetBlu concentrations over the 12 d of hTau0N3R expression in adults did not affect significantly the levels of soluble hTau0N3R (Fig. 5B; ANOVA, F(3,15) = 0.495, p = 0.6927). However, the levels of insoluble hTau0N3R were significantly different on 50 μm MetBlu and appeared reduced on the other concentrations assayed as well (Fig. 5B; ANOVA, F(3,48) = 3.013, p = 0.0397; subsequent comparisons with untreated p = 0.0044). As expected, survival of hTau0N3R-expressing flies on 50 μm MetBlu was reduced by the 12th day at 30°C, but not earlier (Fig. 5C; ANOVA, F(23 407) = 8.534, p = 5.1 × 10−23; subsequent planned comparisons, 12 d treated ElavGal4;Gal80ts heterozygotes vs treated ElavGal4;Gal80ts>0N3R: p = 2.4 × 10−6, but p = 0.0002 for the same comparison at 8 d and p = 0.0271 at 6 d). Further survival reduction by MetBlu suggests that toxicity is not affected by insoluble hTau0N3R accumulation, but rather results from the newly translated upon transgene induction soluble protein, or the accumulation of oligomeric species because of MetBlu-mediated aggregation inhibition.
Table 3.
Survival statistics for flies expressing hTau0N3R kept on the indicated concentrations of MetBlu at 30°C
| Wilcoxon/Kruskal–Wallis |
Means comparison (Steel test with control) |
||||
|---|---|---|---|---|---|
| Day | χ2, (df, count) | p > χ2 | Genotype (μm MetBlu) | z | p |
| 2 | 14,6455 (6,17) | 0.0232 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | |||||
| Elav;G80ts>0N3R 50 | |||||
| Elav;G80ts>0N3R 100 | −0.6696 | 0.9657 | |||
| Elav;G80ts>0N3R 250 | −2.5453 | 0.0530 | |||
| Elav;G80ts>0N3R 500 | |||||
| Elav;G80ts>0N3R 1000 | |||||
| 6 | 83,7625 (6,17) | <0.0001 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | |||||
| Elav;G80ts>0N3R 50 | |||||
| Elav;G80ts>0N3R 100 | −0.7908 | 0.9280 | |||
| Elav;G80ts>0N3R 250 | −4.5530 | <0.0001 | |||
| Elav;G80ts>0N3R 500 | −4.6673 | <0.0001 | |||
| Elav;G80ts>0N3R 1000 | −4.9777 | <0.0001 | |||
| 10 | 99.929 (6,17) | <0.0001 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | |||||
| Elav;G80ts>0N3R 50 | −0.8090 | 0.9208 | |||
| Elav;G80ts>0N3R 100 | −3.9799 | 0.0004 | |||
| Elav;G80ts>0N3R 250 | −5.1601 | <0.0001 | |||
| Elav;G80ts>0N3R 500 | −5.2584 | <0.0001 | |||
| Elav;G80ts>0N3R 1000 | −5.2050 | <0.0001 | |||
| 14 | 104.714 (6,17) | <0.0001 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | 0.2637 | 0.9998 | |||
| Elav;G80ts>0N3R 50 | −3.1629 | 0.0084 | |||
| Elav;G80ts>0N3R 100 | −4.9986 | <0.0001 | |||
| Elav;G80ts>0N3R 250 | −5.3209 | <0.0001 | |||
| Elav;G80ts>0N3R 500 | −5.2584 | <0.0001 | |||
| Elav;G80ts>0N3R 1000 | −5.3209 | <0.0001 | |||
| 18 | 108.364 (6,17) | <0.0001 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | −2.3846 | 0.0799 | |||
| Elav;G80ts>0N3R 50 | −4.9932 | <0.0001 | |||
| Elav;G80ts>0N3R 100 | −5.1725 | <0.0001 | |||
| Elav;G80ts>0N3R 250 | −5.3345 | <0.0001 | |||
| Elav;G80ts>0N3R 500 | −5.2715 | <0.0001 | |||
| Elav;G80ts>0N3R 1000 | −5.3345 | <0.0001 | |||
| 22 | 90.179 (6,17) | <0.0001 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | −1.9251 | 0.2222 | |||
| Elav;G80ts>0N3R 50 | −3.7813 | 0.0009 | |||
| Elav;G80ts>0N3R 100 | −4.9195 | <0.0001 | |||
| Elav;G80ts>0N3R 250 | −5.0693 | <0.0001 | |||
| Elav;G80ts>0N3R 500 | −5.0693 | <0.0001 | |||
| Elav;G80ts>0N3R 1000 | −5.0609 | <0.0001 | |||
| 26 | 21.133 (6,17) | 0.0017 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | |||||
| Elav;G80ts>0N3R 50 | |||||
| Elav;G80ts>0N3R 100 | |||||
| Elav;G80ts>0N3R 250 | |||||
| Elav;G80ts>0N3R 500 | −1.3926 | 0.5432 | |||
| Elav;G80ts>0N3R 1000 | −1.3926 | 0.5432 | |||
| 30 | 6.000 (6,17) | 0.4232 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | |||||
| Elav;G80ts>0N3R 50 | |||||
| Elav;G80ts>0N3R 100 | |||||
| Elav;G80ts>0N3R 250 | |||||
| Elav;G80ts>0N3R 500 | |||||
| Elav;G80ts>0N3R 1000 | |||||
| 34 | 0.000 (6,17) | 1.0000 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | |||||
| Elav;G80ts>0N3R 50 | |||||
| Elav;G80ts>0N3R 100 | |||||
| Elav;G80ts>0N3R 250 | |||||
| Elav;G80ts>0N3R 500 | |||||
| Elav;G80ts>0N3R 1000 | |||||
| 38 | 0.000 (6,17) | 1.0000 | Elav;G80ts>0N3R 0 | ||
| Elav;G80ts>0N3R 10 | |||||
| Elav;G80ts>0N3R 50 | |||||
| Elav;G80ts>0N3R 100 | |||||
| Elav;G80ts>0N3R 250 | |||||
| Elav;G80ts>0N3R 500 | |||||
| Elav;G80ts>0N3R 1000 | |||||
Survival results from all the independent determinations were compared with Wilcoxon/Kruskal-Wallis tests for the indicated days. If a positive (χ2) outcome, the means from each genotype for the days with significant differences were compared using the Steel with control tests whose z ratio and p values are shown. Significant differences from controls are emphasized with bold.
Table 4.
Collective statistics table
| Genotype | Mean ± SEM | F ratio | p |
|---|---|---|---|
| Figure 1A. ANOVA F(5,74) = 17.6036, p = 3.34e-11 | |||
| w1118>0N4R (6 d) | 73.435 ± 2.034 | ||
| ElavGal4;Gal80ts>w1118 (6 d) | 70.924 ± 2.830 | 0.5485 | 0.4614 |
| ElavGal4;Gal80ts>0N4R (6 d) | 69.789 ± 2.488 | 1.4645 | 0.2303 |
| ElavGal4;Gal80ts>w1118 (6 d) | 70.924 ± 2.830 | ||
| ElavGal4;Gal80ts>0N4R (6 d) | 69.789 ± 2.488 | 0.1330 | 0.7165 |
| w1118>0N4R (12 d) | 67.671 ± 1.432 | ||
| ElavGal4;Gal80ts>w1118 (12 d) | 65.831 ± 1.020 | 0.3580 | 0.5516 |
| ElavGal4;Gal80ts>0N4R (12 d) | 50.609 ± 1.981 | 42.762 | 9.00e-9 |
| ElavGal4;Gal80ts>w1118 (12 d) | 65.831 ± 1.020 | ||
| ElavGal4;Gal80ts>0N4R (12 d) | 50.609 ± 1.981 | 24.507 | 5.03e-6 |
| Figure 1B. ANOVA F(5,67) = 10.433, p = 2.7e-7 | |||
| w1118>0N4R (6 d) | 34.306 ± 3.639 | ||
| ElavGal4;Gal80ts>w1118 (6 d) | 38.662 ± 3.074 | 1.1534 | 0.2870 |
| ElavGal4;Gal80ts>0N4R (6 d) | 38.128 ± 3.556 | 0.9688 | 0.3287 |
| ElavGal4;Gal80ts>w1118 (6 d) | 38.662 ± 3.074 | ||
| ElavGal4;Gal80ts>0N4R (6 d) | 38.128 ± 3.556 | 0.0189 | 0.8911 |
| w1118>0N4R (12 d) | 27.931 ± 1.459 | ||
| ElavGal4;Gal80ts>w1118 (12 d) | 26.506 ± 2.482 | 0.1418 | 0.7078 |
| ElavGal4;Gal80ts>0N4R (12 d) | 16.473 ± 1.656 | 9.9603 | 0.0025 |
| ElavGal4;Gal80ts>w1118 (12 d) | 26.506 ± 2.482 | ||
| ElavGal4;Gal80ts>0N4R (12 d) | 16.473 ± 1.656 | 7.2917 | 0.0089 |
| Figure 1C. ANOVA F(2,40) = 3.136, p = 0.0549 | |||
| w1118>0N4R | 65.774 ± 1.955 | ||
| ElavGal4;Gal80ts>w1118 | 68.305 ± 1.583 | 1.053 | 0.3112 |
| ElavGal4;Gal80ts>0N4R | 71.697 ± 1.567 | 6.179 | 0.0174 |
| ElavGal4;Gal80ts>w1118 | 68.305 ± 1.583 | ||
| ElavGal4;Gal80ts>0N4R | 71.697 ± 1.567 | 2.026 | 0.1628 |
| Figure 1D. ANOVA F(2,47) = 3.202, p = 0.0501 | |||
| w1118>0N4R | 17.937 ± 1.609 | ||
| ElavGal4;Gal80ts>w1118 | 27.051 ± 3.348 | 5.574 | 0.0226 |
| ElavGal4;Gal80ts>0N4R | 25.300 ± 2.854 | 3.874 | 0.0552 |
| ElavGal4;Gal80ts>w1118 | 27.051 ± 3.348 | ||
| ElavGal4;Gal80ts>0N4R | 25.300 ± 2.854 | 0.212 | 0.6477 |
| Figure 1E. ANOVA F(3,18) = 135.648, p = 4.3e-11 | |||
| ElavGal4;Gal80ts>0N4R | 1 | ||
| ElavGal4;Gal80ts>0N4Ra1 | 0.088 ± 0.0199 | 272.71 | 4.9e-11 |
| ElavGal4;Gal80ts>0N4Ra2 | 0.0545 ± 0.006 | 293.05 | 2.9e-11 |
| ElavGal4;Gal80ts>0N4R2a | 0.647 ± 0.080 | 36.188 | 2.4e-5 |
| Figure 1F. ANOVA F(2,35) = 143.048, p = 5.505e-17 | |||
| w1118>0N4R2a | 40.506 ± 1.030 | ||
| ElavGal4;Gal80ts>w1118 | 60.492 ± 1.746 | 119.56 | 1.65e-12 |
| ElavGal4;Gal80ts>0N4R2a | 27.596 ± 1.223 | 52.768 | 2.47e-8 |
| ElavGal4;Gal80ts>w1118 | 60.492 ± 1.746 | ||
| ElavGal4;Gal80ts>0N4R2a | 27.596 ± 1.223 | 282.78 | 9.49e-18 |
| Figure 1G. ANOVA F(2,42) = 13.829, p = 2.7e-5 | |||
| w1118>0N4R2a | 27.926 ± 1.731 | ||
| ElavGal4;Gal80ts>w1118 | 26.202 ± 2.079 | 0.502 | 0.4828 |
| ElavGal4;Gal80ts>0N4R2a | 16.326 ± 1.246 | 23.496 | 1.9e-5 |
| ElavGal4;Gal80ts>w1118 | 26.202 ± 2.079 | ||
| ElavGal4;Gal80ts>0N4R2a | 16.326 ± 1.246 | 17.031 | 0.0001 |
| Figure 1H. ANOVA F(2,27) = 3.119, p = 0.062 | |||
| w1118>0N4R2a | 66.526 ± 2.471 | ||
| ElavGal4;Gal80ts>w1118 | 72.845 ± 4.099 | 2.5096 | 0.1257 |
| ElavGal4;Gal80ts>0N4R2a | 62.930 ± 1.753 | 0.9146 | 0.3480 |
| ElavGal4;Gal80ts>w1118 | 72.845 ± 4.099 | ||
| ElavGal4;Gal80ts>0N4R2a | 62.930 ± 1.753 | 6.1794 | 0.0199 |
| Figure 1I. ANOVA F(2,34) = 2.963, p = 0.0659 | |||
| w1118>0N4R2a | 20.437 ± 2.228 | ||
| ElavGal4;Gal80ts>w1118 | 25.966 ± 1.669 | 5.021 | 0.0321 |
| ElavGal4;Gal80ts>0N4R2a | 25.598 ± 1.250 | 4.375 | 0.0445 |
| ElavGal4;Gal80ts>w1118 | 25.966 ± 1.669 | ||
| ElavGal4;Gal80ts>0N4R2a | 25.598 ± 1.250 | 0.0272 | 0.8701 |
| Genotype | Mean ± SEM | Dunnett's p |
|---|---|---|
| Figure 2B. ANOVA F(1,13) = 99.548, p = 3.7e-7 | ||
| ElavGal4;Gal80ts>0N4R ON | 1 | 1 |
| ElavGal4;Gal80ts>0N4R OFF | 0.544 ± 0.046 | 1.1e-8 |
| Figure 2C. ANOVA F(1,12)=1.012, p = 0.3327 | ||
| ElavGal4;Gal80ts>0N4R ON | 1 | 1 |
| ElavGal4;Gal80ts>0N4R OFF | 1.205 ± 0.114 | 0.332 |
| Figure 2D. 0N4R, ANOVA F(1,11) = 0.145, p = 0.711 0N4R2a, ANOVA F(1,13) = 4.262, p= 0.061 | ||
| ElavGal4;Gal80ts>0N4R ON | 1 | |
| ElavGal4;Gal80ts>0N4R OFF | 1.009 ± 0.107 | 0.7114 |
| ElavGal4;Gal80ts>0N4R2a ON | 1 | |
| ElavGal4;Gal80ts>0N4R2a OFF | 0.836 ± 0.046 | 0.0612 |
| Figure 2E. 0N4R, ANOVA F(1,11) = 9.191, p = 0.0126 0N4R2a, ANOVA F(1,9) = 11.556, p = 0.0094 | ||
| ElavGal4;Gal80ts>0N4R ON | 1 | |
| ElavGal4;Gal80ts>0N4R OFF | 1.656 ± 0.197 | 0.0126 |
| ElavGal4;Gal80ts>0N4R2a ON | 1 | |
| ElavGal4;Gal80ts>0N4R2a OFF | 1.879 ± 0.183 | 0.0094 |
| Genotype | Mean ± SEM | F ratio | p |
|---|---|---|---|
| Figure 2F. ANOVA F(3,39) = 12.466, p = 9.6e-6 | |||
| w1118>0N4R OFF | 30.701 ± 2.261 | ||
| ElavGal4;Gal80ts>w1118 OFF | 41.380 ± 2.698 | 11.240 | 0.0019 |
| ElavGal4;Gal80ts>0N4R OFF | 32.962 ± 2.346 | 0.504 | 0.4825 |
| ElavGal4;Gal80ts>0N4R ON | 22.034 ± 1.548 | 7.405 | 0.0099 |
| ElavGal4;Gal80ts>w1118 OFF | 41.380 ± 2.698 | ||
| ElavGal4;Gal80ts>0N4R OFF | 32.962 ± 2.346 | 6.985 | 0.0120 |
| ElavGal4;Gal80ts>0N4R ON | 22.034 ± 1.548 | 36.891 | 5.5e-7 |
| ElavGal4;Gal80ts>0N4R OFF | 32.962 ± 2.346 | ||
| ElavGal4;Gal80ts>0N4R ON | 22.034 ± 1.548 | 11.770 | 0.0015 |
| Figure 2G. ANOVA F(3,43) = 17.761, p = 1.5e-7 | |||
| w1118>0N4R2a OFF | 25.398 ± 2.722 | ||
| ElavGal4;Gal80ts>w1118 OFF | 33.028 ± 1.944 | 7.395 | 0.009 |
| ElavGal4;Gal80ts>0N4R2a OFF | 21.768 ± 0.991 | 2.061 | 0.159 |
| ElavGal4;Gal80ts>0N4R2a ON | 13.839 ± 1.765 | 19.541 | 7.1e-5 |
| ElavGal4;Gal80ts>w1118 OFF | 33.028 ± 1.944 | ||
| ElavGal4;Gal80ts>0N4R2a OFF | 21.768 ± 0.991 | 18.625 | 9.8e-5 |
| ElavGal4;Gal80ts>0N4R2a ON | 13.839 ± 1.765 | 50.777 | 1.1e-8 |
| ElavGal4;Gal80ts>0N4R2a OFF | 21.768 ± 0.991 | ||
| ElavGal4;Gal80ts>0N4R2a ON | 13.839 ± 1.765 | 10.892 | 0.002 |
| Genotype | Mean ± SEM | Dunnett's p |
|---|---|---|
| Figure 2H. ANOVA F(1,15) = 0.018, p = 0.8959 | ||
| ElavGal4;Gal80ts>w1118 OFF | 23.482 ± 1.284 | 1 |
| ElavGal4;Gal80ts>w1118 ON | 23.237 ± 1.308 | 0.8959 |
| Genotype | Mean ± SEM | F ratio | p |
|---|---|---|---|
| Figure 3C. ANOVA F(6,40) = 0.323, p = 0.9204 | |||
| ElavGal4;Gal80ts>0N4R OFF | 1 | ||
| ElavGal4;Gal80ts>0N4R ON | 1.091 ± 0.172 | 0.128 | 0.7223 |
| ElavGal4;Gal80ts>0N4R OFF 50 μm Met Blu | 1.080 ± 0.121 | 0.0998 | 0.7539 |
| ElavGal4;Gal80ts>0N4R OFF 100 μm Met Blu | 0.99345 ± 0.153 | 0.001 | 0.9796 |
| ElavGal4;Gal80ts>0N4R OFF 250 μm Met Blu | 1.245 ± 0.211 | 0.927 | 0.3425 |
| ElavGal4;Gal80ts>0N4R OFF 500 μm Met Blu | 0.952 ± 0.219 | 0.035 | 0.8521 |
| ElavGal4;Gal80ts>0N4R OFF 1000 μm Met Blu | 0.959 ± 0.186 | 0.023 | 0.8794 |
| Figure 3D. ANOVA F(6,44) = 4.142, p = 0.0027 | |||
| ElavGal4;Gal80ts>0N4R OFF | 1 | ||
| ElavGal4;Gal80ts>0N4R ON | 0.591 ± 0.090 | 5.1285 | 0.0293 |
| ElavGal4;Gal80ts>0N4R OFF 50 μm Met Blu | 0.942 ± 0.118 | 0.0839 | 0.7736 |
| ElavGal4;Gal80ts>0N4R OFF 100 μm Met Blu | 0.822 ± 0.103 | 0.7924 | 0.3789 |
| ElavGal4;Gal80ts>0N4R OFF 250 μm Met Blu | 0.269 ± 0.069 | 13.301 | 0.0008 |
| ElavGal4;Gal80ts>0N4R OFF 500 μm Met Blu | 1.034 ± 0.208 | 0.029 | 0.864 |
| ElavGal4;Gal80ts>0N4R OFF 1000 μm Met Blu | 0.892 ± 0.158 | 0.289 | 0.593 |
| Genotype | Mean ± SEM | F ratio | p |
|---|---|---|---|
| Figure 4A. ANOVA F(19,299) = 1.663 p = 0.042 | |||
| ElavGal4;Gal80ts>w1118 (2 d) | 100 ± 0 | ||
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (2 d) | 99.67 ± 0.333 | 0.245 | 0.6212 |
| ElavGal4;Gal80ts>0N4R (2 d) | 99.667 ± 0.333 | 0.245 | 0.6212 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (2 d) | 100 ± 0 | 2.7e-32 | 1 |
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (2 d) | 99.67 ± 0.333 | ||
| ElavGal4;Gal80ts>0N4R (2 d) | 99.667 ± 0.333 | 2.7e-32 | 1 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (2 d) | 100 ± 0 | 0.245 | 0.6212 |
| ElavGal4;Gal80ts>0N4R (2 d) | 99.667 ± 0.333 | ||
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (2 d) | 100 ± 0 | 0.245 | 0.6212 |
| ElavGal4;Gal80ts>w1118 (4 d) | 100 ± 0 | ||
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (4 d) | 99.33 ± 0.454 | 0.979 | 0.3233 |
| ElavGal4;Gal80ts>0N4R (4 d) | 99.33 ± 0.454 | 0.979 | 0.3233 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (4 d) | 100 ± 0 | 1.1e-31 | 1 |
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (4 d) | 99.33 ± 0.454 | ||
| ElavGal4;Gal80ts>0N4R (4 d) | 99.33 ± 0.454 | 4.2e-34 | 1 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (4 d) | 100 ± 0 | 0.979 | 0.3233 |
| ElavGal4;Gal80ts>0N4R (4 d) | 99.33 ± 0.454 | ||
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (4 d) | 100 ± 0 | 0.979 | 0.3233 |
| ElavGal4;Gal80ts>w1118 (6 d) | 100 ± 0 | ||
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (6 d) | 99 ± 0.723 | 2.203 | 0.1389 |
| ElavGal4;Gal80ts>0N4R (6 d) | 99 ± 0.534 | 2.203 | 0.1389 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (6 d) | 100 ± 0 | 2.4e-31 | 1 |
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (6 d) | 99 ± 0.723 | ||
| ElavGal4;Gal80ts>0N4R (6 d) | 99 ± 0.534 | 6.1e-32 | 1 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (6 d) | 100 ± 0 | 2.203 | 0.1389 |
| ElavGal4;Gal80ts>0N4R (6 d) | 99 ± 0.534 | ||
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (6 d) | 100 ± 0 | 2.203 | 0.1389 |
| ElavGal4;Gal80ts>w1118 (8 d) | 100 ± 0 | ||
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (8 d) | 99 ± 0.723 | 2.203 | 0.1389 |
| ElavGal4;Gal80ts>0N4R (8 d) | 98.67 ± 0.766 | 3.916 | 0.0488 |
| ElavGal4;Gal80ts>0N4R (8 d) | 98.67 ± 0.766 | ||
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (8 d) | 99 ± 0.534 | 2.203 | 0.1389 |
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (8 d) | 99 ± 0.723 | ||
| ElavGal4;Gal80ts>0N4R (8 d) | 98.67 ± 0.766 | 0.245 | 0.6212 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (8 d) | 99 ± 0.534 | 2.7e-32 | 1 |
| ElavGal4;Gal80ts>0N4R (8 d) | 98.67 ± 0.766 | ||
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (8 d) | 99 ± 0.534 | 0.245 | 0.6212 |
| ElavGal4;Gal80ts>w1118 (10 d) | 99.67 ± 0.333 | ||
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (10 d) | 99 ± 0.723 | 0.979 | 0.3233 |
| ElavGal4;Gal80ts>0N4R (10 d) | 98.33 ± 0.797 | 3.916 | 0.0488 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (10 d) | 98 ± 0.655 | 6.119 | 0.01396 |
| ElavGal4;Gal80ts>w1118 + 250 μm MetBlu (10 d) | 99 ± 0.723 | ||
| ElavGal4;Gal80ts>0N4R (10 d) | 98.33 ± 0.797 | 0.979 | 0.3233 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (10 d) | 98 ± 0.655 | 2.203 | 0.1389 |
| ElavGal4;Gal80ts>0N4R (10 d) | 98.33 ± 0.797 | ||
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu (10 d) | 98 ± 0.655 | 0.245 | 0.6212 |
| Genotype | Mean ± SEM | Dunnett's p |
|---|---|---|
| Figure 4B. ANOVA F(1,15) = 0.138, p = 0.7154 | ||
| ElavGal4;Gal80ts>0N4R | 77.210 ± 1.779 | 1 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu | 78.161 ± 1.834 | 0.7154 |
| Figure 4C. ANOVA F(1,27) = 10.435, p = 0.0033 | ||
| ElavGal4;Gal80ts>0N4R | 34.005 ± 1.815 | 1 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu | 23.804 ± 2.479 | 0.0033 |
| Figure 4D. ANOVA F(1,22) = 0.201, p = 0.6584 | ||
| W1118 | 42.675 ± 4.298 | 1 |
| W1118 250 μm MetBlu | 40.213 ± 3.297 | 0.6584 |
| Figure 4E. ANOVA F(1,29) = 0.0016, p = 0.9681 | ||
| ElavGal4;Gal80ts>0N4R | 30.585 ± 1.619 | 1 |
| ElavGal4;Gal80ts>0N4R + 250 μm MetBlu | 30.487 ± 1.817 | 0.9681 |
| Figure 4F. ANOVA F(1,14) = 2.056, p = 0.1752 | ||
| ElavGal4;Gal80ts>0N4R | 30.584 ± 3.218 | 1 |
| ElavGal4;Gal80ts>0N4R + 500 μm MetBlu | 37.413 ± 3.521 | 0.1752 |
| Figure 4G. ANOVA F(1,23) = 0.701, p = 0.4115 | ||
| W1118 | 26.104 ± 2.944 | 1 |
| W1118 500 μm MetBlu | 30.460 ± 4.290 | 0.4115 |
| Genotype | Mean ± SEM | Dunnett's p |
|---|---|---|
| Figure 5A. ANOVA F(1,9) = 51.036, p = 9.8e-5 | ||
| ElavGal4;Gal80ts>0N4R | 1 | 1 |
| ElavGal4;Gal80ts>0N3R | 6.049 ± 0.516 | 7.7e-5 |
| Genotype | Mean ± SEM | F ratio | p |
|---|---|---|---|
| Figure 5A. ANOVA F(1,9) = 51.036, p = 9.8e-5 | |||
| ElavGal4;Gal80ts>0N4R | 1 | 1 | |
| ElavGal4;Gal80ts>0N3R | 6.049 ± 0.516 | 7.7e-5 | |
|
Figure 5B. Soluble, ANOVA F(3,15) = 0.495, p = 0.6927 Insoluble, ANOVA F(3,48) = 3.013, p = 0.0397 |
|||
| Soluble | |||
| ElavGal4;Gal80ts>0N3R | 1 | ||
| ElavGal4;Gal80ts>0N3R 10 μm Met Blu | 1.042 ± 0.154 | 0.0377 | 0.8492 |
| ElavGal4;Gal80ts>0N3R 50 μm Met Blu | 1.071 ± 0.146 | 0.107 | 0.7495 |
| ElavGal4;Gal80ts>0N3R 100 μm Met Blu | 1.246 ± 0.159 | 1.280 | 0.2799 |
| Insoluble | |||
| ElavGal4;Gal80ts>0N3R | 1 | ||
| ElavGal4;Gal80ts>0N3R 10 μm Met Blu | 0.816 ± 0.115 | 1.704 | 0.1984 |
| ElavGal4;Gal80ts>0N3R 50 μm Met Blu | 0.578 ± 0.058 | 8.970 | 0.0044 |
| ElavGal4;Gal80ts>0N3R 100 μm Met Blu | 0.782 ± 0.133 | 2.484 | 0.1219 |
| Genotype | Mean ± SEM | F ratio | p |
|---|---|---|---|
| Figure 5C. ANOVA F(23,407) = 8.534, p = 5.1e-23 | |||
| ElavGal4;Gal80ts>w1118 (2 d) | 96.765 ± 0.851 | ||
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (2 d) | 97.353 ± 1.060 | 0.055 | 0.8154 |
| ElavGal4;Gal80ts>0N3R (2 d) | 98.529 ± 0.713 | 0.491 | 0.4839 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (2 d) | 98.235 ± 0.597 | 0.341 | 0.5596 |
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (2 d) | 97.353 ± 1.060 | ||
| ElavGal4;Gal80ts>0N3R (2 d) | 98.529 ± 0.713 | 0.218 | 0.6406 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (2 d) | 98.235 ± 0.597 | 0.123 | 0.7262 |
| ElavGal4;Gal80ts>0N3R (2 d) | 98.529 ± 0.713 | ||
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (2 d) | 98.235 ± 0.597 | 0.014 | 0.9071 |
| ElavGal4;Gal80ts>w1118 (4 d) | 95 ± 1.213 | ||
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (4 d) | 96.471 ± 1.407 | 0.341 | 0.5596 |
| ElavGal4;Gal80ts>0N3R (4 d) | 93.529 ± 1.647 | 0.341 | 0.5596 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (4 d) | 94.706 ± 1.740 | 0.014 | 0.9071 |
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (4 d) | 96.471 ± 1.407 | ||
| ElavGal4;Gal80ts>0N3R (4 d) | 93.529 ± 1.647 | 1.364 | 0.2436 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (4 d) | 94.706 ± 1.740 | 0.491 | 0.4839 |
| ElavGal4;Gal80ts>0N3R (4 d) | 93.529 ± 1.647 | ||
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (4 d) | 94.706 ± 1.740 | 0.218 | 0.6406 |
| ElavGal4;Gal80ts>w1118 (6 d) | 93.529 ± 1.532 | ||
| ElavGal4;Gal80ts>w1118 + 50 μΜ MetBlu (6 d) | 94.118 ± 1.5597 | 0.055 | 0.8154 |
| ElavGal4;Gal80ts>0N3R (6 d) | 89.412 ± 1.813 | 2.673 | 0.1028 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (6 d) | 88.529 ± 1.807 | 3.942 | 0.0478 |
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (6 d) | 94.118 ± 1.5597 | ||
| ElavGal4;Gal80ts>0N3R (6 d) | 89.412 ± 1.813 | 3.492 | 0.0624 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (6 d) | 88.529 ± 1.807 | 4.924 | 0.0271 |
| ElavGal4;Gal80ts>0N3R (6 d) | 89.412 ± 1.813 | ||
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (6 d) | 88.529 ± 1.807 | 0.123 | 0.7262 |
| ElavGal4;Gal80ts>w1118 (8 d) | 91.471 ± 1.471 | ||
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (8 d) | 92.941 ± 1.549 | 0.341 | 0.5596 |
| ElavGal4;Gal80ts>0N3R (8 d) | 87.353 ± 1.923 | 2.673 | 0.1028 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (8 d) | 83.529 ± 2.804 | 9.944 | 0.0017 |
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (8 d) | 92.941 ± 1.549 | ||
| ElavGal4;Gal80ts>0N3R (8 d) | 87.353 ± 1.923 | 4.924 | 0.0271 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (8 d) | 83.529 ± 2.804 | 13.968 | 0.0002 |
| ElavGal4;Gal80ts>0N3R (8 d) | 87.353 ± 1.923 | ||
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (8 d) | 83.529 ± 2.804 | 2.305 | 0.1298 |
| ElavGal4;Gal80ts>w1118 (10 d) | 90.882 ± 1.5597 | ||
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (10 d) | 91.176 ± 1.518 | 0.014 | 0.9071 |
| ElavGal4;Gal80ts>0N3R (10 d) | 86.176 ± 2.123 | 3.492 | 0.0624 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (10 d) | 82.059 ± 2.910 | 12.276 | 0.0005 |
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (10 d) | 91.176 ± 1.518 | ||
| ElavGal4;Gal80ts>0N3R (10 d) | 86.176 ± 2.123 | 3.942 | 0.0478 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (10 d) | 82.059 ± 2.910 | 13.108 | 0.0003 |
| ElavGal4;Gal80ts>0N3R (10 d) | 86.176 ± 2.123 | ||
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (10 d) | 82.059 ± 2.910 | 2.673 | 0.1028 |
| ElavGal4;Gal80ts>w1118 (12 d) | 89.412 ± 1.813 | ||
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (12 d) | 91.176 ± 1.518 | 0.491 | 0.4839 |
| ElavGal4;Gal80ts>0N3R (12 d) | 85.588 ± 2.095 | 2.305 | 0.1298 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (12 d) | 79.118 ± 3.008 | 16.709 | 5.3e-5 |
| ElavGal4;Gal80ts>w1118 + 50 μm MetBlu (12 d) | 91.176 ± 1.518 | ||
| ElavGal4;Gal80ts>0N3R (12 d) | 85.588 ± 2.095 | 4.924 | 0.0271 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (12 d) | 79.118 ± 3.008 | 22.929 | 2.4e-6 |
| ElavGal4;Gal80ts>0N3R (12 d) | 85.588 ± 2.095 | ||
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu (12 d) | 79.118 ± 3.008 | 6.602 | 0.0106 |
| Genotype | Mean ± SEM | Dunnett's p |
|---|---|---|
| Figure 5D. ANOVA F(1,23) = 0.107, p = 0.7470 | ||
| ElavGal4;Gal80ts>0N3R | 61.532 ± 2.408 | 1 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu | 62.742 ± 2.815 | 0.7470 |
| Figure 5E. ANOVA F(1,28) = 9.407, p = 0.0049 | ||
| ElavGal4;Gal80ts>0N3R | 33.772 ± 2.136 | 1 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu | 24.232 ± 2.201 | 0.0049 |
| Figure 5F. ANOVA F(1,20) = 4.120, p = 0.0566 | ||
| ElavGal4;Gal80ts>0N3R | 30.408 ± 3.872 | 1 |
| ElavGal4;Gal80ts>0N3R + 50 μm MetBlu | 36.975 ± 2.589 | 0.0566 |
| Figure 5G. ANOVA F(1,28) = 0.113, p = 0.7397 | ||
| W1118 | 23.995 ± 2.303 | 1 |
| W1118 + 50 μΜ MetBlu | 22.931 ± 2.180 | 0.7397 |
| Figure 5H. ANOVA F(1,25) = 0.007, p = 0.936 | ||
| ElavGal4;Gal80ts>0N3R | 34.074 ± 4.120 | 1 |
| ElavGal4;Gal80ts>0N3R + 10 μm MetBlu | 34.520 ± 3.649 | 0.936 |
| Figure 5I. ANOVA F(1,23) = 0.571, p = 0.458 | ||
| ElavGal4;Gal80ts>0N3R | 39.685 ± 2.389 | 1 |
| ElavGal4;Gal80ts>0N3R + 100 μm MetBlu | 37.081 ± 2.485 | 0.458 |
| Figure 5J. ANOVA F(1,23) = 7.211, p = 0.0135 | ||
| ElavGal4;Gal80ts>0N4R | 24.318 ± 3.448 | 1 |
| ElavGal4;Gal80ts>0N4R + 50 μm MetBlu | 12.492 ± 2.739 | 0.0135 |
| Figure 5K. ANOVA F(1,22) = 2.576, p = 0.1234 | ||
| ElavGal4;Gal80ts>0N4R | 27.778 ± 2.615 | 1 |
| ElavGal4;Gal80ts>0N4R + 100 μm MetBlu | 22.875 ± 1.420 | 0.1234 |
| Genotype | Mean ± SEM | Dunnett's p |
|---|---|---|
| Figure 7A. ANOVA F(1,11) = 37.416, p = 0.0001 | ||
| ElavGal4;Gal80ts>0N4R | 1 | 1 |
| LeoGal4;Gal80ts>0N4R | 14.199 ± 1.586 | 0.0001 |
| Figure 7B. ANOVA F(1,11) = 34.926, p= 0.0001 | ||
| ElavGal4;Gal80ts>0N4R2a | 1 | 1 |
| LeoGal4;Gal80ts>0N4R2a | 4.341 ± 0.671 | 0.0001 |
| Genotype | Mean ± SEM | F ratio | p |
|---|---|---|---|
| Figure 7C. ANOVA F(2,39) = 1.527, p = 0.2306 | |||
| LeoGal4;Gal80ts>W1118 | 37.573 ± 3.367 | ||
| W1118>0N4R | 31.291 ± 2.029 | 2.308 | 0.1372 |
| LeoGal4;Gal80ts>0N4R | 31.915 ± 2.564 | 2.201 | 0.1463 |
| W1118>0N4R | 31.291 ± 2.029 | ||
| LeoGal4;Gal80ts>0N4R | 31.915 ± 2.564 | 0.0234 | 0.8792 |
| Figure 7D. ANOVA F(2,39) = 2.706, p = 0.0799 | |||
| LeoGal4;Gal80ts>W1118 | 35.773 ± 3.475 | ||
| W1118>0N4R2a | 29.079 ± 2.046 | 3.2223 | 0.0808 |
| LeoGal4;Gal80ts>0N4R2a | 27.518 ± 2.306 | 4.9009 | 0.0331 |
| W1118>0N4R2a | 29.079 ± 2.046 | ||
| LeoGal4;Gal80ts>0N4R2a | 27.518 ± 2.306 | 0.1899 | 0.6655 |
| Genotype | Mean ± SEM | Dunnett's p |
|---|---|---|
|
Figure 7E. Soluble, ANOVA F(1,11) = 291.294, p = 1.0e-8 Insoluble, ANOVA F(1,11) = 49.499, p = 3.6e-5 |
||
| Soluble | ||
| ElavGal4;Gal80ts>0N4R | 1 | 1 |
| LeoGal4;Gal80ts>0N4R | 5.256 ± 0.249 | 6.9e-10 |
| Insoluble | ||
| ElavGal4;Gal80ts>0N4R | 1 | 1 |
| LeoGal4;Gal80ts>0N4R | 4.445 ± 0.447 | 3.1e-5 |
| Figure 7F. ANOVA F(1,23) = 0.719, p = 0.4797 | ||
| LeoGal4;Gal80ts>0N4R | 72.327 ± 2.333 | 1 |
| LeoGal4;Gal80ts>0N4R + 10 μm MetBlu | 69.959 ± 2.324 | 0.4797 |
| Figure 7G. ANOVA F(1,23) = 6.768, p = 0.0163 | ||
| LeoGal4;Gal80ts>0N4R | 37.588 ± 3.709 | 1 |
| LeoGal4;Gal80ts>0N4R + 10 μm MetBlu | 26.842 ± 1.816 | 0.0163 |
| Figure 7H. ANOVA F(1,23) = 6.192, p = 0.0209 | ||
| LeoGal4;Gal80ts>0N4R2a | 40.723 ± 2.455 | 1 |
| LeoGal4;Gal80ts>0N4R2a + 10 μm MetBlu | 30.747 ± 3.169 | 0.0209 |
If the elevated insoluble species are indeed responsible for the lack of PSD-M deficits after 12 d of 0N3R transgene induction as hypothesized, then memory deficits are expected to emerge on MetBlu-mediated aggregate attenuation. Therefore, hTau0N3R-expressing flies were kept on 50 μm MetBlu-containing media, which was effective at attenuating aggregates (Fig. 5B), for the 12 d of adult transgene expression. This treatment did not affect Immediate Memory after Extended Conditioning (Fig. 5D; ANOVA, F(1,23) = 0.107, p = 0.7470) compared with untreated congenic animals. However, PSD-M (Fig. 5E) was significantly reduced (ANOVA, F(1,28) = 9.407, p = 0.0049) by 50 μm MetBlu treatment, whereas PSI-M remained unaffected (Fig. 5F; ANOVA, F(1,20) = 4.120, p = 0.0566). The specificity of the impairment only for PSD-M suggests that the deficit is unlikely the result of nonspecific drug toxicity. To ascertain this, control animals were kept on 50 μm MetBlu for 12 d at 30°C, which does not have an impact on their survival (Fig. 3A) nor their PSD-M performance relative to that of untreated flies (Fig. 5G; ANOVA, F(1,28) = 0.113, p = 0.7397). This provides independent validation that the deficit in hTau0N3R-expressing flies on 50 μm MetBlu treatment is not a consequence of drug toxicity. Furthermore, PSD-M was not affected in hTau0N3R-expressing flies kept on 10 μm (Fig. 5H; ANOVA, F(1,25) = 0.007, p = 0.936), or 100 μm MetBlu (Fig. 5I; ANOVA, F(1,23) = 0.571, p = 0.458), conditions that do not significantly reduce aggregates (Fig. 5B).
Therefore, memory deficits emerge in hTau0N3R-expressing animals only under conditions that attenuate aggregate formation, independently confirming that aggregation of this hTau isoform is also not inhibitory and may in fact be permissive to PSD-M. Accordingly, the deficient PSD-M presented by hTau0N4R-expressing animals kept at 30°C for 12 d was further significantly decreased if these animals were simultaneously kept at 50 μm (Fig. 5J; ANOVA, F(1,23) = 7.211, p = 0.0135), but was not affected if flies were kept on 100 μm MetBlu (Fig. 5K; ANOVA, F(1,22) = 2.576, p = 0.1234), both concentrations that do not affect their survival (Fig. 3B) but only the former inhibiting aggregation. Therefore, inhibiting insoluble aggregate formation of two different hTau isoforms in the Drosophila CNS results in specific PSD-M deficits.
The size and abundance of aggregates in their CNS correlate with the memory deficits in hTau0N4R and hTau0N3R-expressing animals
To independently verify the results supporting the notion that hTau aggregation does not impair but rather may be permissive to processes required for PSD-M formation, storage, or recall, insoluble Tau species were recovered from adult head lysates as detailed before (Cowan et al., 2015; Sealey et al., 2017), placed on mica disks, and their sizes and configurations examined under AFM are summarized in Figure 6.
Figure 6.

Aggregate accumulation in the CNS of hTau0N4R and hTau0N3R-expressing animals and MetBlu-mediated aggregate inhibition. Representative AFM images of aggregates from insoluble Tau fractions in head lysates of hTau0N4R and hTau0N3R-expressing flies. The images were taken at random points from the mica carrying the indicated samples with a scan rate of 1 Hz–2 Hz. Scale bar, 200 nm. A, Insoluble Tau fraction from adult pan-neuronally expressing hTau0N4R flies at the ON condition (1) or transgene repression conditions (2) and after treatment of repressed animals with 250 μm MetBlu (3). Insoluble hTau under transgene repression was significantly elevated in number and size (2) compared with lysates from the ON condition (1). Treatment with 250 μm MetBlu for 10 d at the OFF condition reduced the size and the number of aggregates, although short filaments appeared (3). Range of the filaments, <40 (thin arrow), 50–140 nm (small arrowheads) to 240–390 nm (thick arrow) and short filaments (asterisk). B, Insoluble Tau fraction from adult pan-neuronally expressing hTau0N3R flies at the ON condition (1), the ON condition with simultaneous treatment with 50 μm MetBlu (2), or 100 μm MetBlu (3). Treatment with 50 μm MetBlu for 12 d at 30°C reduced the size of filaments, whereas treatment with 100 μm MetBlu did not affect the size of the aggregates much but yielded short filaments. Range of the aggregates, from 40 to 60 nm (thin arrow), 80–150 nm (small arrowhead), >175 nm (thick arrow), and short filaments (asterisk).
In agreement with transgene expression (Fig. 2B) and biochemical assessment (Fig. 3C,D), induction of the hTau0N4R isoforms yielded few and rather small (<40 nm) apparent aggregates (Fig. 6Α1). Significantly, maintaining the flies under conditions restrictive to transgene expression, following initial induction resulted in accumulation of very large aggregates (>300 nm wide) in the CNS of these animals (Fig. 6A2, large arrowhead). However, these aggregates appeared highly reduced both in size and abundance if hTau0N4R-expressing flies were maintained on 250 μm MetBlu-laced food during this period (Fig. 6A3), and some appeared filamentous in shape (Fig. 6A3, star). Therefore, these results agree with the biochemically detectable aggregate accumulation on transgene silencing (Fig. 2E), the effect of maintaining the animals on 250 μm MetBlu on aggregates (Fig. 3D), and the emergence of PSD-M deficits in the latter animals (Fig. 4C).
Conversely, medium aggregates (50–80 nm) were apparent in CNS lysates of hTau0N3R-expressing flies (Fig. 6B1). These were highly reduced in abundance and size if these transgene-expressing animals were simultaneously maintained on 50 μm MetBlu-laced food (Fig. 6B2). Interestingly, maintaining these animals on 100 μm MetBlu, which did not affect their PSD-M performance (Fig. 5I) or the level of biochemically detected aggregates (Fig. 5B), did not appear to affect the abundance or size of the aggregates and appeared conducive to formation of filamentous forms of the protein (Fig. 6B3, star).
Collectively, these results provide additional support for the conclusion that large hTau aggregates do not impair processes requisite for PSD-M, but the smaller, likely soluble, aggregates do. Therefore, the presence of aggregates correlates with comparatively normal PSD-M formation.
Aggregates in adult mushroom body neurons do not impair PSD-M
The mushroom body neurons (MBs) are implicated in PSD-M formation and recall (Davis, 2005; Cognigni et al., 2018), and relatively low chronic Tau expression therein has been reported to precipitate learning and short-term memory deficits while leaving these neurons structurally intact in the short term (Mershin et al., 2004). However, whether adult-specific hTau expression within these neurons results in consolidated memory deficits in a manner analogous to those observed in human sporadic Tauopathy patients has not been examined systematically.
To determine whether PSD-M is compromised by adult-specific hTau0N4R accumulation within these neurons, this hTau isoform was specifically expressed in adults under the strong pan-MB neuron driver LeoGal4 (Messaritou et al., 2009) for 12 d posteclosion. Surprisingly, expression levels of hTau0N4R under LeoGal4 were highly elevated compared with those expressing pan-neuronally under ElavGal4 (Fig. 7A; ANOVA, F(1,11) = 37.416, p = 0.0001), and this was verified independently with the hTau0N4R2a double transgenic strain (Fig. 7B; ANOVA, F(1,11) = 34.926, p = 0.0001). This is surprising, considering the small number of neurons expressing hTau under LeoGal4 (Messaritou et al., 2009) compared with pan-neuronal expression under ElavGal4 (Robinow and White, 1988), and indicates that a large excess of hTau accumulates within ∼4000 of these MB neurons (Aso et al., 2009) during the 12 d of transgene expression. However, despite this vast hTau accumulation, PSD-M was unaffected both in hTau0N4R (Fig. 7C; ANOVA, F(2,39) = 1.527, p = 0.2306) and hTau0N4R2a-expressing (Fig. 7D; ANOVA, F(2,39) = 2.706, p = 0.0799) animals. These data support the notion that expression levels alone do not correlate with and predict neuronal dysfunction.
Figure 7.

Insoluble aggregates in adult-specific hTau0N4R-expressing animals within mushroom bodies are permissive to PSD-M. A–B, Representative Western blots from head lysates of flies accumulating hTau0N4R pan-neuronally (ElavGal4; TubGal80ts) for 12 d at 30°C compared with flies expressing hTau0N4R only in mushroom body neurons (LeoGal4; TubGal80ts), probed with the 5A6 anti-Tau antibody. The level of Syx in the lysates was used as control for quantifications. For the quantification, Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM values relative to respective levels in flies accumulating pan-neuronally the 0N4R isoform for 12 d, which was set to one. The stars indicate significant differences from the control genotype; n ≥ 5 per genotype in A and B. C, D, Bars represent the mean PIs and ± SEM for the number of indicated experimental replicates (n). The genotypes of all animals are indicated below each bar. C, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies kept for 12 d in the ON condition; n ≥ 13 per genotype. Statistical details are found in Table 4. D, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies from the hTau0N4R2a double transgenics kept for 12 d in the ON condition; n ≥ 13 per genotype. Ε, Representative Western blot of soluble (left) and insoluble (right) fractions generated from adult heads of flies accumulating hTau0N4R pan-neuronally or limited to mushroom body neurons for 12 d at 30°C, probed with the 5A6 anti-Tau antibody. Syx levels were used as control for quantifications. For the quantification, Tau levels were normalized using the Syx loading control and are shown as a ratio of their mean ± SEM values relative to respective levels in flies accumulating hTau0N4R pan-neuronally, which was set to one. The mean ± SEM is shown for each group. The star indicates significant differences from the control genotype; n ≥ 5 for both genotypes. F–H, Bars represent the mean PIs and ± SEM for the number of indicated experimental replicates (n). Stars indicate significant differences. F, Immediate Performance after Extended Conditioning (5X) of hTau0N4R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 10 μm MetBlu; n ≥ 11 per genotype. G, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies kept for 12 d in the ON condition in the absence (0 μm) or presence of 10 μm MetBlu; n ≥ 11 per genotype. H, Twenty-four-hour Spaced Conditioning memory (PSD-M) performance of hTau0N4R-expressing flies from the hTau0N4R2a double transgenics, kept for 12 d in the ON condition in the absence (0 μm) or presence of 10 μm MetBlu; n ≥ 11 per genotype.
As for the soluble 0N4R, insoluble species were highly abundant in head lysates of hTau0N4R-expressing flies under LeoGal4, compared with those under ElavGal4 (Fig. 7E; ANOVA for Soluble, F(1,11) = 291.294.499, p = 1.0 × 10−8 and ANOVA for Insoluble, F(1,11) = 49.499, p = 3.6 × 10−5), suggesting that in accord with the results and the hypothesis above, their accumulation could suppress hTau-dependent dysfunction of MB neurons. Given the dependence of MetBlu toxicity on temperature and hence fly metabolism and the rather limited number of neurons targeted, we opted to inhibit aggregate formation with 10 μm MetBlu, a concentration without significant effects on the viability of control (Fig. 3A) or flies expressing hTau0N4R pan-neuronally (Fig. 3B). Hence, MetBlu at 10 μm was fed to adult flies during the 12 d of transgene expression to inhibit aggregate formation and address the hypothesis-borne prediction that this treatment will result in PSD-M deficits. Indeed, MetBlu treatment did not affect learning/immediate memory after Extended Conditioning (Fig. 7F; ANOVA, F(1,23) = 0.719, p = 0.4797), indicating the expected lack of toxicity of 10 μm MetBlu. In contrast, PSD-M was significantly impaired in animals expressing hTau0N4R (Fig. 7G; ANOVA, F(1,23) = 6.768, p = 0.0163) or hTau0N4R2a (Fig. 7H; ANOVA, F(1,23) = 6.192, p = 0.0209) in their MBs compared with untreated controls, which presented memory levels in the expected normal range. Therefore, insoluble hTau aggregate accumulation within the MBs, even at the excessive levels under LeoGal4, do not precipitate neuronal dysfunction manifested as PSD-M deficits in contrast to soluble species that ostensibly do.
Discussion
Reversal of adult onset hTau-driven neuroplasticity deficits
Time-dependent memory deficits, likely reflective of disease progression, characterize most sporadic Tauopathies involving nonmutated Tau, such as AD (Lee et al., 2001; Delacourte, 2005). This time dependence of associative learning and PSD-M attenuation is clearly emulated in our adult onset hTau transgene expression model (Fig. 1; Sealey et al., 2017). However, it has been unclear whether these cognitive deficits are the consequence of irreversibly dysfunctional or degenerating neurons. Evidence from regulatable expression transgenic mouse models expressing the FTDP-linked mutations P301L and ΔK280 in the 0N4R isoform indicated that switching off hTau expression improved the associated memory impairment, without a reduction in large aggregates (Santacruz et al., 2005; Sydow et al., 2011; Van der Jeugd et al., 2012).
To our knowledge, this report is the first to demonstrate reversal of memory deficits on attenuation of wild-type hTau expression. Together with the mouse data, these results support the hypothesis that learning and PSD-M deficits are not consequent of irreversibly damaged neurons expressing the wild type or mutant hTau isoforms. Rather, cognitive deficits result from dysfunctional, but otherwise apparently healthy, neurons and therefore may be pharmacologically reversible in patients as well, at least before later degenerative stages of the disease (Braak and Braak, 1996; Lee et al., 2001; Papanikolopoulou and Skoulakis, 2020).
Significantly, we also demonstrate that excess 0N3R or 0N4R hTau in the fly CNS specifically compromises the apparent rate of learning and PSD-M, but learning per se and PSI-M remain intact. These results add further credence to the interpretation that excess hTau alone does not result in generally dysfunctional fly CNS, but rather it compromises specific processes and mechanisms essential for protein synthesis-dependent consolidated memory. Because recall of PSD-M requires neurotransmission from the MBs in Drosophila (McGuire et al., 2001), it appears likely that the compromised memory when soluble hTau expression is limited to the MBs (Fig. 7G,H) reflects deficits in synaptic function as previously proposed (Wang and Mandelkow, 2016).
Why does accumulation of ostensibly small soluble hTau aggregates impair PSD-M? Direct evidence of a physiological function of Tau as a negative regulator of translation was uncovered for the homologous Drosophila protein. Knock-out mutants of Drosophila Tau (dTau) present elevated translation and enhanced PSD-M, whereas overexpression of the protein impairs both processes (Papanikolopoulou et al., 2019). Therefore, elevation of small insoluble hTau aggregates likely impairs translation and precipitates the specific PSD-M deficits but spares the translation independent memory (PSI-M). PSI-M has also been reported to depend at least in part on regulated filamentous actin (F-actin) stability (Kotoula et al., 2017). Excess hTau in the fly CNS has been reported to stabilize F-actin (Fulga et al., 2007), providing a plausible explanation why PSI-M remains intact under these conditions.
Adult CNS-specific hTau aggregation correlates with suppression of neuroplasticity deficits
In agreement with the FTDP mouse models (Santacruz et al., 2005; Sydow et al., 2011; Van der Jeugd et al., 2012), aggregates not only persist in Drosophila for at least 10 d after transgene silencing but apparently make up a significant fraction of the hTau0N4R isoform in the fly CNS (Fig. 2E). Conversely, insoluble species make up a significant fraction of the hTau0N3R isoform in the fly CNS even when this transgene is fully transcriptionally active for 12 d (Fig. 5A). The greater aggregation propensity of hTau0N3R may reflect its elevated phosphorylation state relative to its hTau0N4R counterpart in the Drosophila CNS (Sealey et al., 2017) and/or its reduced affinity for microtubules. Either of these scenarios likely renders a significant number of hTau0N3R proteins more prone to aggregation (Goode et al., 2000). A large increase in insoluble hTau0N4R without silencing the transgene was observed when expression of this isoform was confined to ∼4500 of the MB neurons with the very strong LeoGal4 driver, relative to the levels attained under similar conditions with the pan-neuronally expressed (∼1 × 105 neurons) ElavGal4 (Fig. 7E). This clearly demonstrates that aggregation is favored by excessive local hTau accumulation as within the confines of particular neurons in agreement with in vitro experiments (Montejo de Garcini et al., 1986; von Bergen et al., 2005).
Insoluble hTau aggregates have been linked to neurodegenerative Tauopathies (Delacourte and Buee, 2000; Geschwind, 2003; Trojanowski and Lee, 2005), and larger ones such as NFTs may in fact contribute to toxicity in later stages of the disease. However, evidence supporting a cardinal role for soluble Tau oligomers in neuronal dysfunction and toxicity has been increasing (Cowan and Mudher, 2013; Cowan et al., 2015), whereas direct evidence for the role of large insoluble aggregates in these processes remains scant (Cowan et al., 2015; Arendt et al., 2016; Wang and Mandelkow, 2016). In addition, aggregates are relatively abundant in patients with primary age-related tauopathy, but these individuals seldom present cognitive deficits (Jellinger et al., 2015; Jellinger, 2019). Neurons may in fact degenerate when they are devoid of NFTs (Wittmann et al., 2001; Papanikolopoulou and Skoulakis, 2011; Wang and Mandelkow, 2016), and this is a hypothesis currently under investigation in our fly model.
The data herein provide three experimental scenarios strongly supporting the view that insoluble aggregates, whose exact conformation(s) is unclear at the moment, are protective or permissive of neuronal activities that underlie associative PSD-M. Our data extend the findings from regulatable mouse FTDP models that aggregates remain, whereas cognition improves (Santacruz et al., 2005; Sydow et al., 2011; Van der Jeugd et al., 2012), to posit that insoluble aggregates are in fact permissive if not protective of neuroplasticity. The results from the three experimental scenarios supporting this notion are discussed below.
Silencing the transgene in hTau0N4R-expressing animals results in reversal of their PSD-M deficit, and in fact correlates well with an increase in large insoluble aggregates (Figs. 2F,G, 6A2). In contrast, the PSD-M deficit was sustained by MetBlu-mediated inhibition of aggregation after transgene silencing (Figs. 4C, 6A3). In accord with this, MetBlu concentrations that do not affect hTau0N4R aggregation were not deleterious to PSD-M (Fig. 4F). Moreover, inhibition of aggregation while the hTau0N4R transgene was expressed exaggerated the PSD-M deficit of these flies (Fig. 5J). In the MB-limited expression setting of hTau0N4R, the excessive aggregates within these neurons (Fig. 7E) are the likely reason that PSD-M remained normal after 12 d of transgene expression (Fig. 7C,D) but was compromised after MetBlu mediated inhibition of aggregation (Fig. 7G,H). Finally, in the case of hTau0N3R adult-specific expression reported to spare learning and PSD-M (Sealey et al., 2017), we now provide evidence that this correlates nicely with the relative abundance of aggregates (Figs. 5A,B, 6B1) as MetBlu-mediated inhibition of their formation precipitated robust PSD-M deficits (Figs. 5E, 6B2).
Collectively, therefore, insoluble hTau aggregates in the fly CNS, at least, do not impair neuronal processes requisite for efficient learning and PSD-M such as regulated translation. This agrees with the suggestion that aggregate formation may reflect protective cellular response(s) to excess hyperphosphorylated Tau (Wang and Mandelkow, 2016). Conversely, our data support the idea that small soluble oligomers or monomeric to trimeric hTau species impede essential for PSD-M neuroplasticity. Oligomeric hTau may also be the neurotoxicity culprit as inhibiting hTau aggregation at relatively low MetBlu concentrations (50–250 μm), with minimal effect on control flies (Fig. 3A) under increased metabolic conditions (30°C), yielded a highly significant reduction in the life span of flies expressing hTau isoforms (Fig. 3B, Table 2). Therefore, inhibition of aggregation with the resultant excess of oligomeric species, or both, are also toxic to the CNS, precipitating premature lethality.
Preventive (Hochgräfe et al., 2015) or therapeutic (Santacruz et al., 2005; Sydow et al., 2011; Van der Jeugd et al., 2012) treatment with MetBlu in mouse models of FTDP recovers cognition. However, the effects of the drug on bona fide mouse AD models have not been assessed to our knowledge. In contrast, MetBlu has been tried on patients, even in Phase III trials as an antiaggregation therapeutic for AD with very poor results (Gauthier et al., 2016), most likely because it does not inhibit soluble Tau species including small oligomers (Soeda et al., 2015), which apparently accumulate to high levels. Furthermore, recent results from a mouse FTDP model indicate that soluble hTau oligomers carrying the P301L mutation appear solely responsible for Tauopathy progression (Shin et al., 2020). Collectively then, and in light of our own results, disaggregation-promoting pharmaceuticals (Dominguez-Meijide et al., 2020) should be considered with caution as they can easily lead to dispersal of larger protective Tau species to increase the availability of the toxic smaller soluble oligomers. Conversely pharmaceutical agents that may encourage the sequestration of toxic smaller oligomers into innocuous larger aggregates should be explored.
Footnotes
This work was supported in part by Phenotypos Grant MIS: 5002135, General Secretariat for Research and Technology Grant 2018ΣΕ01300001, Greece and the European Union–European Regional Development Fund, and the Flagship Initiative for Neurodegenerative Diseases Research on the Basis of Precision Medicine Project Infrastructures for National Research Networks for Precision Medicine and Climate Change. We thank the Bloomington Drosophila Stock Center for stocks; Dr. Martin Chow, University of Kentucky, for the 0N4R cDNA; Dr. Katerina Papanikolopoulou, Biomedical Sciences Research Centre (BSRC) Alexander Fleming, for help with construction of the double transgenic line; Maro Loizou, BSRC Alexander Fleming, for technical help; and Dr. Iris Nandhakumar, University of Southampton, for help with the Atomic Force Microscopy experiments.
The authors declare no competing financial interests.
References
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K (2001) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 98:6923–6928. 10.1073/pnas.121119298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25:(5446–5454. 10.1523/JNEUROSCI.4637-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis A, Broderick JA, Kosik KS (1995) Relative exon affinities and suboptimal splice site signals lead to non-equivalence of two cassette exons. Nucleic Acids Res 23:3585–3593. 10.1093/nar/23.17.3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Stieler JT, Holzer M (2016) Tau and tauopathies. Brain Res Bull 126:238–292. 10.1016/j.brainresbull.2016.08.018 [DOI] [PubMed] [Google Scholar]
- Aso Y, Grübel K, Busch S, Friedrich AB, Siwanowicz I, Tanimoto H (2009) The mushroom body of adult Drosophila characterized by GAL4 drivers. J Neurogenet 23:156–172. 10.1080/01677060802471718 [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104:3312–3317. 10.1073/pnas.0611511104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E (1996) Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol 92:197–201. 10.1007/s004010050508 [DOI] [PubMed] [Google Scholar]
- Cognigni P, Felsenberg J, Waddell S (2018) Do the right thing: neural network mechanisms of memory formation, expression and update in Drosophila. Curr Opin Neurobiol 49:51–58. 10.1016/j.conb.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CM, Mudher A (2013) Are tau aggregates toxic or protective in tauopathies? Front Neurol 4:114. 10.3389/fneur.2013.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CM, Quraishe S, Hands S, Sealey M, Mahajan S, Allan DW, Mudher A (2015) Rescue from tau-induced neuronal dysfunction produces insoluble tau oligomers. Sci Rep 5:17191. 10.1038/srep17191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL (2005) Olfactory memory formation in Drosophila: from molecular to systems neuroscience. Annu Rev Neurosci 28:275–302. 10.1146/annurev.neuro.28.061604.135651 [DOI] [PubMed] [Google Scholar]
- Delacourte A (2005) Tauopathies: recent insights into old diseases. Folia Neuropathol 43:244–257. [PubMed] [Google Scholar]
- Delacourte A, Buee L (2000) Tau pathology: a marker of neurodegenerative disorders. Curr Opin Neurol 13:371–376. [DOI] [PubMed] [Google Scholar]
- Dominguez-Meijide A, Vasili E, Outeiro T (2020) Pharmacological modulators of tau aggregation and spreading. Brain Sci 10:858. 10.3390/brainsci10110858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB (2007) Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol 9:139–148. 10.1038/ncb1528 [DOI] [PubMed] [Google Scholar]
- Gauthier S, Feldman H, Schneider L, Wilcock G, Frisoni G, Hardlund J, Moebius H, Bentham P, Kook K, Wischik D, Schelter B, Davis C, Staff R, Bracoud L, Shamsi K, Storey J, Harrington C, Wischik C (2016) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer's disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388:2873–2884. 10.1016/S0140-6736(16)31275-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH (2003) Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron 40:457–460. 10.1016/s0896-6273(03)00681-0 [DOI] [PubMed] [Google Scholar]
- Gillman P (2011) CNS toxicity involving methylene blue: the exemplar for understanding and predicting drug interactions that precipitate serotonin toxicity. J Psychopharmacol 25:429–436. 10.1177/0269881109359098 [DOI] [PubMed] [Google Scholar]
- Giong H, Subramanian M, Yu K, Lee J (2021) Non-rodent genetic animal models for studying tauopathy: review of Drosophila, zebrafish, and C. elegans models. IJMS 22:8465. 10.3390/ijms22168465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode B, Chau M, Denis P, Feinstein S (2000) Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. J Biol Chem 275:38182–38189. 10.1074/jbc.M007489200 [DOI] [PubMed] [Google Scholar]
- Gouzi JY, Bouraimi M, Roussou IG, Moressis A, Skoulakis EMC (2018) The Drosophila receptor tyrosine kinase alk constrains long-term memory formation. J Neurosci 38:7701–7712. 10.1523/JNEUROSCI.0784-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgräfe K, Sydow A, Matenia D, Cadinu D, Könen S, Petrova O, Pickhardt M, Goll P, Morellini F, Mandelkow E, Mandelkow E (2015) Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol Commun 3:25. 10.1186/s40478-015-0204-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa M, Arai T, Masuda-Suzukake M, Nonaka T, Yamashita M, Akiyama H, Hasegawa M (2012) Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS One 7:e52389. 10.1371/journal.pone.0052389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger K (2019) Primary age-related tauopathy (PART) and Alzheimer's disease (AD). Alzheimers Dement 15:720. 10.1016/j.jalz.2019.01.005 [DOI] [PubMed] [Google Scholar]
- Jellinger K, et al. (2015) PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 129:757–762. 10.1007/s00401-015-1407-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniyappan S, Chandupatla R, Mandelkow E, Mandelkow E (2017) Extracellular low-n oligomers of tau cause selective synaptotoxicity without affecting cell viability. Alzheimers Dement 13:1270–1291. 10.1016/j.jalz.2017.04.002 [DOI] [PubMed] [Google Scholar]
- Keramidis I, Vourkou E, Papanikolopoulou K, Skoulakis E (2020) Functional interactions of Tau phosphorylation sites that mediate toxicity and deficient learning in Drosophila melanogaster. Front Mol Neurosc 13:569520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmidis S, Grammenoudi S, Papanikolopoulou K, Skoulakis EMC (2010) Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J Neurosci 30:464–477. 10.1523/JNEUROSCI.1490-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotoula V, Moressis A, Semelidou O, Skoulakis EMC (2017) Drk-mediated signaling to Rho kinase is required for anesthesia-resistant memory in Drosophila. Proc Natl Acad Sci U S A 114:10984–10989. 10.1073/pnas.1704835114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. 10.1146/annurev.neuro.24.1.1121 [DOI] [PubMed] [Google Scholar]
- McGuire S, Le P, Davis RL (2001) The role of Drosophila mushroom body signaling in olfactory memory. Science 293:1330–1333. 10.1126/science.1062622 [DOI] [PubMed] [Google Scholar]
- McGuire SE, Mao Z, Davis RL (2004a) Spatiotemporal gene expression targeting with the TARGET and gene-switch systems in Drosophila. Sci STKE 2004:pl6. [DOI] [PubMed] [Google Scholar]
- McGuire SE, Roman G, Davis RL (2004b) Gene expression systems in Drosophila: a synthesis of time and space. Trends Genet 20:384–391. 10.1016/j.tig.2004.06.012 [DOI] [PubMed] [Google Scholar]
- Mershin A, Pavlopoulos E, Fitch O, Braden BC, Nanopoulos DV, Skoulakis EM (2004) Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem 11:277–287. 10.1101/lm.70804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaritou G, Leptourgidou F, Franco M, Skoulakis EM (2009) A third functional isoform enriched in mushroom body neurons is encoded by the Drosophila 14-3-3zeta gene. FEBS Lett 583:2934–2938. 10.1016/j.febslet.2009.08.003 [DOI] [PubMed] [Google Scholar]
- Moressis A, Friedrich AR, Pavlopoulos E, Davis RL, Skoulakis EMC (2009) A dual role for the adaptor protein DRK in Drosophila olfactory learning and memory. J Neurosci 29:2611–2625. 10.1523/JNEUROSCI.3670-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montejo de Garcini E, Serrano L, Avila J (1986) Self assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem Biophys Res Commun 141:790–796. 10.1016/s0006-291x(86)80242-x [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, Roussou IG, Gouzi JY, Samiotaki M, Panayotou G, Turin L, Skoulakis EMC (2019) Drosophila tau negatively regulates translation and olfactory long-term memory, but facilitates footshock habituation and cytoskeletal homeostasis. J Neurosci 39:8315–8329. 10.1523/JNEUROSCI.0391-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanikolopoulou K, Skoulakis EM (2011) The power and richness of modelling tauopathies in Drosophila. Mol Neurobiol 44:122–133. 10.1007/s12035-011-8193-1 [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, Skoulakis EM (2015) Temporally distinct phosphorylations differentiate Tau-dependent learning deficits and premature mortality in Drosophila. Hum Mol Genet 24:2065–2077. 10.1093/hmg/ddu726 [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, Skoulakis EMC (2020) Altered proteostasis in neurodegenerative tauopathies. Adv Exp Med Biol 1233:177–194. [DOI] [PubMed] [Google Scholar]
- Patterson K, Remmers C, Fu Y, Brooker S, Kanaan N, Vana L, Ward S, Reyes J, Philibert K, Glucksman M, Binder L (2011) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286:23063–23076. 10.1074/jbc.M111.237974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prifti E, Tsakiri E, Vourkou E, Stamatakis G, Samiotaki M, Papanikolopoulou K (2021) The two cysteines of tau protein are functionally distinct and contribute differentially to its pathogenicity in vivo. J Neurosci 41:797–810. 10.1523/JNEUROSCI.1920-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow S, White K (1988) The locus elav of Drosophila melanogaster is expressed in neurons at all developmental stages. Dev Biol 126:294–303. 10.1016/0012-1606(88)90139-x [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci 25:3020–3029. 10.1111/j.1460-9568.2007.05555.x [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Takashima A (2008) Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration. Curr Alzheimer Res 5:591–598. 10.2174/156720508786898442 [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. 10.1126/science.1113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer R, Adler H, Pickhardt M, Mandelkow E (2011) Lest we forget you—methylene blue…” Neurobiol Aging 32:2325.e7-16. [DOI] [PubMed] [Google Scholar]
- Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Grammenoudi S, Skoulakis EMC, Mudher A (2017) Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol Dis 105:74–83. 10.1016/j.nbd.2017.05.003 [DOI] [PubMed] [Google Scholar]
- Shi Y, et al. (2021) Structure-based classification of tauopathies. Nature 598:359–363. 10.1038/s41586-021-03911-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiells H, et al. (2020) Concentration-dependent activity of hydromethylthionine on clinical decline and brain atrophy in a randomized controlled trial in behavioral variant frontotemporal dementia. J Alzheimers Dis 75:501–519. 10.3233/JAD-191173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Kim D, Song J, Jeong H, Hyeon S, Kowall N, Ryu H, Pae A, Lim S, Kim Y (2020) Visualization of soluble tau oligomers in TauP301L-BiFC transgenic mice demonstrates the progression of tauopathy. Prog Neurobiol. Advance online publication. Retreived March 4, 2023. 10.1016/j.pneurobio.2020.101782 [DOI] [PubMed] [Google Scholar]
- Soeda Y, Yoshikawa M, Almeida OF, Sumioka A, Maeda S, Osada H, Kondoh Y, Saito A, Miyasaka T, Kimura T, Suzuki M, Koyama H, Yoshiike Y, Sugimoto H, Ihara Y, Takashima A (2015) Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat Commun 6:10216. 10.1038/ncomms10216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos I, Galas MC, Silva JM, Skoulakis E, Wegmann S, Maina MB, Blum D, Sayas CL, Mandelkow EM, Mandelkow E, Spillantini MG, Sousa N, Avila J, Medina M, Mudher A, Buee L (2017) Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol Commun 5:91. 10.1186/s40478-017-0489-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M (1998) Tau protein pathology in neurodegenerative diseases. Trends Neurosci 21:428–433. 10.1016/s0166-2236(98)01337-x [DOI] [PubMed] [Google Scholar]
- Spires-Jones T, Kopeikina K, Koffie R, de Calignon A, Hyman B (2011) Are tangles as toxic as they look? J Mol Neurosci 45:438–444. 10.1007/s12031-011-9566-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones T, Stoothoff W, de Calignon A, Jones P, Hyman B (2009) Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci 32:150–159. 10.1016/j.tins.2008.11.007 [DOI] [PubMed] [Google Scholar]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM (2011) Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci 31:2511–2525. 10.1523/JNEUROSCI.5245-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM (2005) Pathological tau: a loss of normal function or a gain in toxicity? Nat Neurosci 8:1136–1137. 10.1038/nn0905-1136 [DOI] [PubMed] [Google Scholar]
- Tully T, Preat T, Boynton SC, Del Vecchio M (1994) Genetic dissection of consolidated memory in Drosophila. Cell 79:35–47. 10.1016/0092-8674(94)90398-0 [DOI] [PubMed] [Google Scholar]
- Van der Jeugd A, Hochgräfe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D'Hooge R, Mandelkow EM (2012) Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol 123:787–805. 10.1007/s00401-012-0987-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E (2005) Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta 1739:158–166. 10.1016/j.bbadis.2004.09.010 [DOI] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:5–21. 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- Wischik C, Edwards P, Lai R, Roth M, Harrington C (1996) Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A 93:11213–11218. 10.1073/pnas.93.20.11213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik C, Staff R, Wischik D, Bentham P, Murray A, Storey J, Kook K, Harrington C (2015) Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J Alzheimers Dis 44:705–720. 10.3233/JAD-142874 [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. 10.1126/science.1062382 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wu KM, Yang L, Dong Q, Yu JT (2022) Tauopathies: new perspectives and challenges. Mol Neurodegener 17:28. 10.1186/s13024-022-00533-z [DOI] [PMC free article] [PubMed] [Google Scholar]