

Abstract
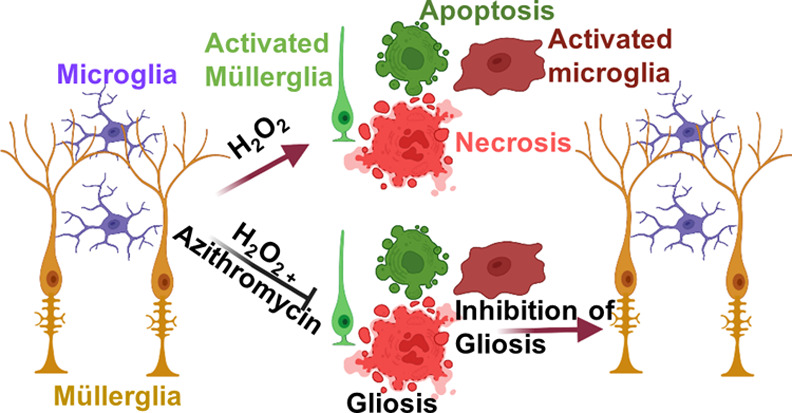
The reactivity of retinal glia in response to oxidative stress has a significant effect on retinal pathobiology. The reactive glia change their morphology and secret cytokines and neurotoxic factors in response to oxidative stress associated with retinal neurovascular degeneration. Therefore, pharmacological intervention to protect glial health against oxidative stress is crucial for maintaining homeostasis and the normal function of the retina. In this study, we explored the effect of azithromycin, a macrolide antibiotic with antioxidant, immunomodulatory, anti-inflammatory, and neuroprotective properties against oxidative stress-induced morphological changes, inflammation, and cell death in retinal microglia and Müller glia. Oxidative stress was induced by H2O2, and the intracellular oxidative stress was measured by DCFDA and DHE staining. The change in morphological characteristics such as the surface area, perimeter, and circularity was calculated using ImageJ software. Inflammation was measured by enzyme-linked immunosorbent assays for TNF-α, IL-1β, and IL-6. Reactive gliosis was characterized by anti-GFAP immunostaining. Cell death was measured by MTT assay, acridine orange/propidium iodide, and trypan blue staining. Pretreatment of azithromycin inhibits H2O2-induced oxidative stress in microglial (BV-2) and Müller glial (MIO-M1) cells. We observed that azithromycin inhibits oxidative stress-induced morphological changes, including the cell surface area, circularity, and perimeter in BV-2 and MIO-M1 cells. It also inhibits inflammation and cell death in both the glial cells. Azithromycin could be used as a pharmacological intervention on maintaining retinal glial health during oxidative stress.
Keywords: microglia, Müller glia, oxidative stress, cell death, inflammation, azithromycin
Introduction
Müller glia, microglia, and astrocytes are the three different glial cells present in the mammalian retina. Müller glia are involved in structural stabilization, maintaining homeostasis, neurotransmitter recycling, and neuronal survival in the retina, whereas microglia are distributed throughout the inner retina and involved in phagocyting apoptotic cells and cellular debris, cutting back dysfunctional synapse, providing trophic factors, regulating synaptic function, and remodeling the extracellular matrix.1−3 Glia become active and exhibit morphological and functional changes after homeostasis has been disturbed by external stress. Their activation state is determined by the nature and severity of the stress.3−5 The activated microglia change their shape from ramified morphology with a small cellular body to amoeboid shape with a large cellular body, increase in cell proliferation, and increase in IBA1 expression.6,7 IBA-1 plays important role in the actin-cross-linking involved in membrane ruffling of microglia. Since membrane ruffling is essential for the morphological changes from ramified microglia to activated amoeboid microglia, microglial activation is associated with increased IBA-1 expression.8 However, the reactive Müller glia are characterized by an increase in intermediate filament proteins including glial fibrillar acidic protein (GFAP), nestin, and vimentin and downregulation of proteins involved in its homeostatic functions.4 The reactive astrocytes are characterized by an increase in GFAP expression, proliferation, and cell adhesion with an increase in the production of inflammatory markers and oxidative stress molecules.5
Oxidative stress plays an important role in retinal pathogenesis. It stimulates inflammation and cell death in retinal neuronal and glial cells.9−12 Increases in oxidative stress result in the oxidation of lipid, DNA, and protein and induction of mitochondrial injury, leading to neuronal and glial degeneration.13 Microglia are the resident macrophages that play an important role in the innate immune system of the retina. Under stress, microglia secret proinflammatory cytokines and reactive oxygen and nitric oxide species with the activation of nicotinamide adenine dinucleotide phosphate oxidase (NOX-2 and NOX-4).14 Under stress, microglia secret proinflammatory cytokines and reactive oxygen species (ROS) with the activation of nicotinamide adenine dinucleotide phosphate oxidase.14 Microglia are considered to be the main source of oxidative stress in the eye as they have high concentrations of antioxidants, which makes them more oxidative stress-resistant.13 However, oxidative stress induces changes in microglia and Müller glia from the noninflammatory to the proinflammatory state and causes glial cell death.15−18 The reactive glia secret inflammatory cytokines and neurotoxic factors contributing significantly to disease pathology in various retinal diseases such as retinopathy of prematurity, diabetic retinopathy, ischemic retinopathy, glaucoma, age-related macular degeneration, and retinitis pigmentosa.1,2,18 Apoptosis of Müller glia leads to apoptotic cell death in photoreceptors, and glial degeneration has implication toward the neurodegeneration with loss in trophic factors.3,19,20 Glial cells are, therefore, a potential cell target for several therapeutic approaches to treat retinal diseases.21,22
Many studies focused on the role of glia on neurovascular abnormality; however, pharmacological intervention on protecting glial health against oxidative stress has received little attention. By controlling the stress factors and protecting glial health, many of the retinal diseases associated with neurovascular abnormalities may be controlled. Therefore, in this study, we explored the effect of azithromycin (AZM), an FDA-approved macrolide antibiotic known for its antioxidant, immunomodulatory, anti-inflammatory, and neuroprotective properties, used for infectious ocular diseases such as bacterial conjunctivitis, trachoma, blepharitis, and endophthalmitis experimented in different disease models.23−29 It has an interesting pharmacokinetics profile, has a high accumulation rate in cells, specifically phagocytic cells and tissues, and has a plasma half-life of >40 h.30 AZM also can act on macrophages and alter the inflammatory M1 to anti-inflammatory M2 phase.27 Here, we have focused on studying the effect of AZM on oxidative stress-induced changes in morphology, inflammation, and cell death for microglia and Müller glia as both cells show phagocytosis properties which may lead to high intracellular accumulation of AZM. It has the potential to open a new avenue for the application of AZM in inflammatory retinal diseases where microglial activation and Müller glial activation play an important role in disease progression, including neuronal and vascular degeneration.
Results
Pretreatment of AZM Inhibits Oxidative Stress in Microglial and Müller Glial Cells
The inhibition of oxidative stress by AZM was evaluated in retinal glial cells such as microglial (BV-2) and Müller glial (MIO-M1) cells. For this purpose, oxidative stress was induced by H2O2 with the presence and absence of AZM, and the intracellular ROS was determined by dichlorofluorescein (DCF) and dihydroethidium (DHE) levels. The fluorogenic substrate 2′,7-dichlorofluorescein diacetate is a cell-permeable dye, which, after entering into the cell, can be hydrolyzed into nonfluorescent 2′,7-dichlorofluorescein by intracellular esterase and then oxidized to fluorescent 2′,7-dichlorofluorescein by ROS within the cytosol; the resulting change in fluorescence intensity could be used to measure the intracellular generation of ROS.31 DHE is a cell-permeable compound that oxidized on reaction with superoxide to 2-hydroxyethidium, which binds to DNA in the nucleus and fluoresces red.32 AZM did not show any change in ROS in control BV-2 or MIO-M1 cells (Figures 1 and S1).
Figure 1.

AZM inhibits H2O2-induced intracellular ROS production in BV-2 microglial and MIO-M1 Müller glial cells in vitro. (A) Representative images of DCFDA staining for BV2 cells, (B) spectrofluorometric measurement of DCFDA biochemical assay for BV2 cells, (C) representative images of DCFDA staining for MIO-M1 cells, (D) spectrofluorometric measurement of DCFDA biochemical assay for MIO-M1 cells. Red bars indicate fluorescence quantified in control cells, faint red bars indicate AZM-treated cells, green bars indicate H2O2-treated cells, and faint green bars indicate AZM-pretreated H2O2-treated cells. DMEM was taken as 100% and it was compared with the rest of the groups. All the results were presented as mean ± SD, n = 6, and one-way ANOVA was performed with Tukey’s multiple comparisons, ****p < 0.0001, black stars are differences as compared to DMEM, and blue stars are differences as compared to the H2O2.
There was a significant increase in fluorescence intensity, indicating that with the H2O2 treatment, the ROS was increased in both BV-2 and MIO-M1 cells (Figures 1 and S1). The results also showed that the AZM inhibits the intracellular ROS production at all concentrations which is comparable to the control without any AZM or H2O2 treatment (7.5, 15, and 30 μM), and we did not see any dose dependence in the neutralization of intracellular ROS by AZM (Figure 1). It shows the complete neutralization of ROS at these concentrations as all are comparable to the control; however, at lower concentrations of AZM (3.35 to 7.5 μM), it shows partial neutralization of ROS. A similar but more pronounced effect was also observed at the 24 h time points with no major change in fluorescence intensity (Figure S1). In contrast, DHE staining shows dose dependence at 7.5 to 15 μM concentration (Figure S1), which might be due to their detection method; DHE detects superoxide, while DCFH-DA detects hydrogen peroxide (in the presence of peroxidases), hydroxyl radical, and peroxynitrite.
AZM Inhibits the Oxidative Stress-Induced Change in Cellular Morphology in Retinal Glia
To evaluate the change in morphology, BV-2 microglial and MIO-M1 Müller glial cells were induced with H2O2 in the presence and absence of AZM. Under a phase-contrast microscope after 30 min (Figure 2), it was observed that the morphology of BV2-cells did not change after AZM treatment as compared to the control; however, under H2O2 treatment, the morphology of the BV-2 cells changed to amoeboid shape with a large cell body as compared to controls which confirmed that the AZM inhibited the glial cells to form proinflammatory or amoeboid morphology under oxidative stress (Figure 2A). With the AZM treatment, the surface area, circularity, and perimeter did not change in microglia as compared to the control, which indicated that AZM does not cause any change in resting microglia.
Figure 2.

Morphological changes in control and H2O2-treated BV-2 microglial and MIO-M1 Müller glial cells in vitro in the presence and absence of AZM evaluated by phase-contrast microscopy. (A) Phase-contrast image of BV-2 cells, (B) surface area of the BV-2 cells, (C) circularity of the BV-2 cells, (D) perimeter of the BV-2 cells, (E) phase contrast image of MIO-M1 cells, (F) surface area of the MIO-M1 cells, (G) circularity of the MIO-M1 cells, and (H) perimeter of the MIO-M1 cells. All the results were presented as mean ± SD, n = 6, and one-way ANOVA was performed with Tukey’s multiple comparison, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns represents p > 0.05, black stars as compared to DMEM, and blue stars are as compared to the H2O2.
The quantification data indicated an increase in the cell surface area (Figure 2B) and circularity (Figure 2C). However, there was no change in perimeter (Figure 2D), which would be consistent with activation of the microglia under oxidative stress. The surface area, circularity, and perimeter of the AZM and H2O2-treated groups do not vary significantly as compared to the control without AZM and H2O2. The results suggested that the pretreatment of AZM impeded the microglia from entering into the activated proinflammatory state, which was confirmed by a decrease in surface area and circularity with constant perimeters (Figure 2A–C).
With the AZM treatment, the surface area, circularity, and perimeter did not change in Müller glia as compared to the control, which indicated that AZM does not cause any gliosis. Under oxidative stress, Müller glia underwent drastic morphological changes. The cellular process/lamellipodial structures were reduced and became smaller. With higher concentrations of H2O2, the cells turned to more circular morphology and detached completely from the surface of the well plate (Figure 2E). The quantification data indicated that the surface area and perimeter are significantly higher than the H2O2-treated group and lower than the control without H2O2 and AZM treatment (Figure 2F,H), whereas the circularity is significantly lower than the H2O2-treated group and higher than the control without H2O2 and AZM treatment (Figure 2G). The data indicated the partial protection of Müller gliosis by AZM pretreatment.
AZM Inhibits Oxidative Stress-Induced Proinflammatory Cytokine Secretion by Microglia in Microglia and Reduces GFAP Expression in Müller Glia
Proinflammatory cytokines are produced predominantly by activated macrophages, microglia, and Müller glia and are involved in the upregulation of inflammatory reactions. There is abundant evidence that certain proinflammatory cytokines such as IL-1β, IL-6, and TNF-α are involved in the process of pathological changes in retinal diseases. The data indicated that BV-2 cells secreted more proinflammatory cytokines such as TNF-α and IL-1β but not IL-6 by H2O2, and pretreatment of AZM inhibited their secretion which is comparable to the control without AZM and H2O2 treatment (Figure 3A–C). However, H2O2 treatment did not increase cytokines such as TNF-α, IL-1β, and IL-6 production in Müller glia (data not shown) in all the treatment groups, although there was reduced expression of GFAP observed (Figure 3D,E).
Figure 3.

Confirmation of the anti-inflammatory effect of AZM on BV-2 microglial cells by ELISA. (A) TNF-α, (B) IL-1β, and (C) IL-6 secreted by BV-2 cells. Red bars indicate fluorescence quantification in control cells, faint red bars indicate AZM-treated cells, green bars indicate H2O2-treated cells, and faint green bars indicate AZM-pretreated H2O2-treated cells. (D) Representative images of the anti-GFAP antibody staining in Müller glia. (E) Quantification of fluorescence intensity from GFAP immunostaining images. All the results were presented as mean ± SD, n = 6, and one-way ANOVA was performed with Tukey’s multiple comparison, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns represents p > 0.05, black stars are as compared to DMEM and blue stars are as compared to the H2O2. AZM inhibits oxidative stress-induced cell death in retinal glia.
The cytoprotective effect of AZM on H2O2-induced cell death was evaluated by AO/PI live–dead staining (green, live cells; red, necrotic cells; and yellow, apoptotic cells), trypan blue live–dead assay, and MTT assay (Figures 4, 5, and S2).
Figure 4.

Visualization of live and dead cells by acridine orange/propidium iodide (AO/PI) staining. AO gives green fluorescence for live cells, and PI gives red fluorescence for necrotic cells and yellow for apoptotic cells. (A) Representative images of AO/PI staining for BV-2 cells, (B) quantification of the percentage PI-positive cells compared to the total number of microglial cells (n = 8), (C) representative images of AO/PI staining for MIO-M1 cells, (D) quantification of the percentage of PI-positive cells compared to the total number of MIO-M1 cells (n = 8). All the results were presented as mean ± SD, and one-way ANOVA was performed with Tukey’s multiple comparisons, **p < 0.01, ***p < 0.001, ****p < 0.0001, black stars as compared to DMEM, and blue stars are as compared to the H2O2.
Figure 5.

MTT assay measurement of the cell viability. (A) MTT assay for BV-2 microglial cells and (B) MTT assay for MIO-M1 cells. All the results were presented as mean ± SD, n = 6, and one-way ANOVA was performed with Tukey’s multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns represents p > 0.05; black stars as compared to DMEM, and blue stars are as compared to the H2O2.
For this, BV-2 microglial and MIO-M1 Müller glial cells were induced with H2O2 in the presence and absence of AZM. To standardize the effect of concentration of H2O2 and incubation time, AO/PI staining and MTT assay were performed at 30 min and 24 h. It was observed that at 30 min time points, the cell death (75 and 150 μM) did not have a significant change as compared to the control; however, 300 μM in 50 and 95% cell death at 30 min and 24 h respectively, 600 μM H2O2 treatment showed 100% cell death in both time points in BV-2 cells (Figures 5 and S3). The MTT assay indicated that 300 μM H2O2 induces 50% cell death in MIO-M1 cells at 30 min time point (Figures 4 and 5). AZM could rescue 100 and 70% of cells from dying in BV-2 and MIO-M1 cells, respectively (Figure 5). Furthermore, 12 and 20% cells were PI and trypan blue-positive cells in BV2 and MIO-M1, respectively, indicating that besides necrosis, other modes of cell death may be involved for rest 20% cell death. AZM could inhibits H2O2-induced cell death quantified by a significant reduction in the number of PI-positive cells (Figures 4 and S4) and trypan blue-positive cells (Figure S4) in both microglia and Müller glia. However, at higher concentrations (>30 μM), AZM causes cell death, suggesting that lower concentrations are necessary for therapeutic application (Figure 5A,B).
Discussion
The microglia and Müller glia play significant roles in maintaining homeostasis and normal function in the retina. Microglia phagocytosed the apoptotic cells and cellular debris and invaded microorganisms, whereas Müller glia provide neuroprotection and maintain structural and functional integrity to the neuronal and vascular system.1−4 Although primary microglia and Müller glia are preferable options for drug evaluation, BV2 and MIO-M1 are suitable alternatives as they have similar transcriptome and proteome profiles as compared to their respective primary cells.33,34 Nonetheless, this raises a very important point regarding this work. The work presented her suggests that AZM may be an important target for addressing oxidative stress associated with retinal degeneration, but it is very much a first step in understanding how AZM might impact retinal disease. Longer term in vitro and in vivo work is necessary to further explore the role and mechanism. This work provides the impetus for that work.
Oxidative stress induces inflammatory and neurodegenerative retinal diseases. Microglia, being early responders, undergo structural and functional changes under oxidative stress. The ramified resting microglia change into amoeboid proinflammatory microglia during many retinal diseases such as diabetic retinopathy, age-related macular degeneration, and glaucoma, which are associated with oxidative stress.6,9,20,35−37 Inflammation in response to oxidative stress is a defensive mechanism. However, when oxidative stress overpowers the redox balance, it leads to the proinflammatory environment, which plays an important role in neurovascular degeneration in retinal diseases.16,38,39 Because of their defensive function, microglia have a very strong antioxidant system. Still, the oxidative stress becomes detrimental and leads to cell death in microglia.15,17,40,41 Interestingly, AZM could prevent the intracellular oxidative stress formation in glia which is corroborative with earlier findings, in which AZM exhibited antioxidant activity in the high glucose-induced podocyte.42 These antioxidant properties of AZM could be ensued by nuclear translocation of Nrf2 and hence increasing the expression of downstream antioxidant genes.43,44 By virtue of the inhibition of oxidative stress, the AZM pretreatment could prevent the microglia from entering into the proinflammatory phase and inhibits the secretion of inflammatory cytokines such as IL-1β and TNF-α. This is not quite surprising as AZM is known to have immunomodulatory properties by altering the proinflammatory (M1) microphase to anti-inflammatory (M2) microphase where it not only inhibits the secretion of IL-1β, and TNF-α but also enhances IL-10.27,45,46 AZM could inhibit oxidative stress-induced microglial cell death and hence can prevent neurodegeneration as evident from the neuroprotective properties of AZM in various ischemia-reperfusion injury models.28,47 Another important finding is that after intraperitoneal injection, the neuronal protection of AZM in the ischemia-reperfusion injury retina model indicates that it can cross the blood-retinal barrier after systemic administration.28 Microglia are known to secrete neuroprotective factors by releasing trophic factors and antioxidants; scavenging of toxic compounds and of cell debris released by the dying neurons occurs; hence, protection of microglia by AZM indirectly prevents neurodegeneration in the retina.3,48
Oxidative stress causes gliosis in the retina, which is believed to be an attempt by the Müller glia to maintain retinal homeostasis, involving morphological and functional changes.3,4 The change in morphology and increase in GFAP expression are well evident in our results under oxidative stress corroborating with the previous reports.49,50 Although the mechanism of changes in morphology is unknown, there might be an increase in cell adhesion molecules and remodeling of cytoskeletal protein.50,51 Müller glia respond against oxidative stress and protect the photoreceptors and neuronal cells by secreting neurotrophic factors and antioxidants.3,52 The chronic gliosis not only increases GFAP expression, which leads to neuronal cell death with an increase in stiffness, but also increases vascular breakdown.3 However, oxidative stress itself is detrimental for the glia, leading to an increase in cell death.15,17,41 The glial dysfunction or death indirectly causes neuronal and vascular injury with loss of supportive functions. Interestingly, AZM pretreatment significantly inhibits the oxidative stress in Müller glia, oxidative stress-induced GFAP expression, and glial cell death.
Ultimately, this work is a starting point for looking at the impact of AZM administration to address diseases with oxidative stress. One of the most important questions that need to be answered beyond further unpacking the mechanism is understanding what the window of opportunity is regarding the administration of the drug. The majority of retinal diseases are diagnosed relatively late. Whether AZM administration can be helpful at later timepoints will be important to determine moving forward in this work.
Conclusions
In conclusion, the inhibition of oxidative stress, inflammation, and cell death by inhibiting oxidative stress in glia by AZM can potentially benefit the neurovascular entity in retinal diseases. However, more mechanistic insights are needed to understand the phenomena. Furthermore, the AZM-loaded nanoparticle-based sustained drug delivery system may have potential and should be investigated for chronic retinal diseases where oxidative stress is a major component of the disease etiology.
Experimental Methods
Cell Culture
BV-2 microglial and MIO-M1 Müller glial cells were maintained in DMEM high glucose medium and low glucose medium, respectively, with 10% FBS and 1% antibiotics in a humidified 5% CO2 incubator at 37 °C as reported earlier.53,54
DCF and DHE Staining
The staining for DCF/DHE was performed as reported earlier.55 For staining of DCF/DHE, 104 cells were seeded on 24-well plates for imaging and allowed to grow for 48 h to reach 70% confluency. Cells were then incubated with DMEM (without FBS and antibiotics) and 7.5 and 15 μM AZM for 30 min followed by treatment with 300 μM H2O2 for 30 or 15 min for BV-2 and MIO-M1 cells, respectively. The medium was removed, and BV-2/MIO-M1 cells were treated with CM-H2DCFDA/DHE (10/5 μmol/L) in HBSS/serum-free medium and incubated for 30 min. The remaining was removed, and cells were incubated with DMEM without FBS. Images were captured in a fluorescence microscope (Cytation-5 (Biotek, Agilent Technologies, USA)) at constant exposure for all groups.
DCF and DHE Biochemical Assay
ROS production was quantified by the DCF and DHE assay. BV-2 and MIO-M1 cells were seeded in 96-well plates for 48 h to reach 70% confluency. Cells were then incubated with DMEM without FBS and 1% antibiotics with or without AZM for 30 min followed by treatment with 300 μM H2O2 for 30 min. The medium was removed, and BV-2/MIO-M1 cells were treated with CM-H2DCFDA/DHE (10/5 μM/L) in HBSS/serum-free medium and incubated for 30 min. The remaining HBSS with CM-H2DCFDA and DMEM with DHE was removed by serum-free DMEM. The DCF/DHE fluorescence was measured with a spectrofluorometer (Spectramax 2, Molecular Devices, SanJose, CA).
Morphological Evaluation of Microglia and Müller Glia
For morphological evaluation, 104 cells were seeded on 24-well plates for imaging and allowed to grow for 48 h to reach 70% confluency. Cells were then incubated with DMEM (without FBS and antibiotics) and 7.5 and 15 μM AZM for 30 min followed by treatment with 300 μM H2O2 for 30 or 15 min for BV-2 and MIO-M1 cells, respectively. The phase-contrast image was captured at 10× (Cytation 5 (Biotek, Agilent Technologies, USA)). Computer-based analysis of the morphology of individual cells was performed with NIH ImageJ (Bethesda, MD, Version 1.441) using the measurement function. Briefly, the image was opened in ImageJ, the scale was set, the outline of the cell was defined by the polygonal selection, and cellular area and perimeter were measured. The circularity was calculated using the following formula: (defined as 4π (area of the cell)/(cell perimeter2). About 40 cells were measured from each group.56
Enzyme-Linked Immunosorbent Assay
To assess anti-inflammatory activity, 104 cells were seeded on 24-well plates for imaging and allowed to grow for 48 h to reach 70% confluency. Cells were then incubated with DMEM (without FBS and antibiotics) and 7.5 and 15 μM AZM for 30 min followed by treatment with 300 μM H2O2 for 30 or 15 min for BV-2 and MIO-M1 cells, respectively. Cells without any treatment were used as the negative control. The medium was aspirated, and ELISA was performed for TNF-α, IL-1β, and IL6, as described by the kit R & D system (DY-410, DY-401, and Dy406).
Immunocytochemistry
For immunolabeling with GFAP, the cultures were fixed in 4% paraformaldehyde for 15 min. After washing with PBS, the cells were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, USA) in PBS for 5 min and for 10 min. It was followed by blocking with 4% bovine serum albumin + 1% normal goat serum in 0.25% Triton X-100 (Sigma-Aldrich, USA) for 1 h at room temperature and, subsequently, in a mixture of the primary anti-GFAP rabbit antibody (G9269 Sigma-Aldrich, USA) overnight at 4 °C. After washing in PBST 0.25% Triton X-100, goat anti-rabbit conjugated with Texas red secondary antibodies applied for 2 h at room temperature followed by DAPI staining for 5 min followed by three washing with 0.1% Triton X-100.
Images were captured using a citation 5 (Biotek, Agilent Technoloies, USA) fluorescence microscope, and quantification was performed as reported earlier.57,58
AO/PI Staining
To access cell death, AO and PI staining was performed. Cells were culture in a tissue culture flask. Cells were trypsinized and 106 cells/mL in 100 μL of DMEM or 0, 7.5, and 15 μM AZM and incubated for 30 min followed by treatment with H2O2 (300 μM) for 30/15 min for BV-2 and MIO-M1 cells, respectively, serum-free medium. Cells were then centrifuged at 1200 RPM for 5 min and redispersed in 10 μL of medium and then stained with a 1:1 ratio of cell and the mixture of 1 μg/mL AO and Pi each, dropped on a glass slide and mounted with a coverslip. Immediately, the image was captured using a fluorescence microscope, and cells were counted using image software with the plugin function (ImageJ plugin cell counter).
Cell Proliferation Assay
The cell proliferation assay was determined after H2O2 and AZM treatment by MTT assay in BV2 and MIO-M1 cells to see the cytoprotective effect of AZM. In this process, 104 cells were seeded on 96-well plates. After 48 h of cell seeding, the medium was removed and a new medium with different concentrations of AZM (0, 7.5, 15, and 30 μM) in DMEM was treated after 30 min of incubation with AZM, and the cells were further treated with H2O2 (300 μM) for 1 h for BV2 and 15 min for MIO-M1 in serum-free medium. The medium containing the treatment was removed with fresh 100 μL of medium containing 25 μL of MTT reagent from a kit (ab211091, Abcam, USA) was added and incubated at 37 °C for 3 h. After 3 h the medium absorbance was measured at 570 nm. The percentage of cell viability was calculated as compared with control cells (100% cell viability) without any treatment.59,60
Statistical Analysis
Experimental data in this study were analyzed using Graph Pad Prism 9 software. All quantitative variables are shown as means ± SD from at least 6 samples and the experiments have been repeated at least 2 times. Differences between three or more groups were compared using a one-way analysis of variance (ANOVA). There are two sets of comparison done one is control with all other groups and H2O2 with H2O2 and AZM treated groups. The probability value, p < 0.05 was considered statistically significant.
Acknowledgments
We thank Dr. Astrid Limb for providing the MIO-M1 cells, Dr. R. Kannan Wilmer Eye Institute for providing BV-2 microglial cells, and UMBC core facility for providing the Biospa live cell analysis system for imaging. BioRender was used to draw the TOC.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsbiomedchemau.2c00013.
Oxidative stress at 30 min determined by DHE, oxidative stress at 24 h by DCFDA, effect of H2O2 concentration on BV-2 microglial cell death at 30 min and the protection by AZM at 24 h determined by AO/PI, and effect of H2O2 concentration on BV-2 microglial cell death at 30 min determined by trypan blue assay (Figure S4) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wang M.; Wong W. T. Microglia-Müller cell interactions in the retina. Adv. Exp. Med. Biol. 2014, 801, 333–338. 10.1007/978-1-4614-3209-8_42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch K.; Jadhav A. P.; Trimarchi J. M.; Stadler M. B.; Roska B.; Sun B. B.; Cepko C. L. The transcriptome of retinal Müller glial cells. J. Comp. Neurol. 2008, 509, 225–238. 10.1002/cne.21730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coorey N. J.; Shen W.; Chung S. H.; Zhu L.; Gillies M. C. The role of glia in retinal vascular disease. Clin. Exp. Optom. 2012, 95, 266–281. 10.1111/j.1444-0938.2012.00741.x. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Pandey R. K.; Miller L. J.; Singh P. K.; Kanwar M. Muller glia in retinal innate immunity: a perspective on their roles in endophthalmitis. Crit. Rev. Immunol. 2013, 33, 119–135. 10.1615/critrevimmunol.2013006618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin E. S.; Huang Q.; Gurel Z.; Sorenson C. M.; Sheibani N. High glucose alters retinal astrocytes phenotype through increased production of inflammatory cytokines and oxidative stress. PLoS One 2014, 9, e103148 10.1371/journal.pone.0103148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Arjona M. d. M.; Grondona J. M.; Granados-Durán P.; Fernández-Llebrez P.; López-Ávalos M. D. Microglia Morphological Categorization in a Rat Model of Neuroinflammation by Hierarchical Cluster and Principal Components Analysis. Front. Cell. Neurosci. 2017, 11, 235. 10.3389/fncel.2017.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A.; Steele M. R.; Vetter M. L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. 10.1002/cne.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovens I.; Nyakas C.; Schoemaker R. A novel method for evaluating microglial activation using ionized calcium-binding adaptor protein-1 staining: cell body to cell size ratio. Neuroimmunol. Neuroinflammation 2014, 1, 82–88. 10.4103/2347-8659.139719. [DOI] [Google Scholar]
- Kowluru R. A.; Chan P.-S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. 10.1155/2007/43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.-Y.; Fu Z.-J.; Ma H.; Jang W.-C.; So K.-F.; Wong D.; Lo A. C. Y. Effect of lutein on retinal neurons and oxidative stress in a model of acute retinal ischemia/reperfusion. Invest. Ophthalmol. Visual Sci. 2009, 50, 836–843. 10.1167/iovs.08-2310. [DOI] [PubMed] [Google Scholar]
- Izzotti A.; Bagnis A.; Sacca S. The role of oxidative stress in glaucoma. Mutat. Res. 2006, 612, 105–114. 10.1016/j.mrrev.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Beatty S.; Koh H.-H.; Phil M.; Henson D.; Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- Lassmann H.; van Horssen J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta, Mol. Basis Dis. 2016, 1862, 506–510. 10.1016/j.bbadis.2015.09.018. [DOI] [PubMed] [Google Scholar]
- Wilkinson B. L.; Landreth G. E. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J. Neuroinflammation 2006, 3, 30. 10.1186/1742-2094-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou R. C.-W.; Wu C.-C.; Huang J.-R.; Chen Y.-S.; Jeng K.-C. G. Oxidative toxicity in BV-2 microglia cells: sesamolin neuroprotection of H2O2 injury involving activation of p38 mitogen-activated protein kinase. Ann. N. Y. Acad. Sci. 2005, 1042, 279–285. 10.1196/annals.1338.050. [DOI] [PubMed] [Google Scholar]
- Solleiro-Villavicencio H.; Rivas-Arancibia S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. 10.3389/fncel.2018.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft-Kehler A. K.; Gurubaran I. S.; Desler C.; Rasmussen L. J.; Skytt D. M.; Kolko M. Oxidative Stress-Induced Dysfunction of Müller Cells During Starvation. Invest. Ophthalmol. Visual Sci. 2016, 57, 2721–2728. 10.1167/iovs.16-19275. [DOI] [PubMed] [Google Scholar]
- Eastlake K.; Luis J.; Limb G. A. Potential of Müller Glia for Retina Neuroprotection. Curr. Eye Res. 2020, 45, 339–348. 10.1080/02713683.2019.1648831. [DOI] [PubMed] [Google Scholar]
- Dubois-Dauphin M.; Poitry-Yamate C.; de Bilbao F.; Julliard A. K.; Jourdan F.; Donati G. Early postnatal Müller cell death leads to retinal but not optic nerve degeneration in NSE-Hu-Bcl-2 transgenic mice. Neuroscience 2000, 95, 9–21. 10.1016/s0306-4522(99)00313-9. [DOI] [PubMed] [Google Scholar]
- Zalfa C.; Verpelli C.; D’Avanzo F.; Tomanin R.; Vicidomini C.; Cajola L.; Manara R.; Sala C.; Scarpa M.; Vescovi A. L.; De Filippis L. Glial degeneration with oxidative damage drives neuronal demise in MPSII disease. Cell Death Dis. 2016, 7, e2331 10.1038/cddis.2016.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid K.; Akhtar-Schaefer I.; Langmann T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10, 1975. 10.3389/fimmu.2019.01975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devoldere J.; Peynshaert K.; De Smedt S. C.; Remaut K. Müller cells as a target for retinal therapy. Drug Discovery Today 2019, 24, 1483–1498. 10.1016/j.drudis.2019.01.023. [DOI] [PubMed] [Google Scholar]
- Cucoranu I.; Clempus R.; Dikalova A.; Phelan P. J.; Ariyan S.; Dikalov S.; Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. 10.1161/01.res.0000187457.24338.3d. [DOI] [PubMed] [Google Scholar]
- West S. K. Azithromycin for control of trachoma. Community Eye Health 1999, 12, 55–56. [PMC free article] [PubMed] [Google Scholar]
- Luchs J. Efficacy of topical azithromycin ophthalmic solution 1% in the treatment of posterior blepharitis. Adv. Ther. 2008, 25, 858–870. 10.1007/s12325-008-0096-9. [DOI] [PubMed] [Google Scholar]
- Romero-Aroca P.; Sararols L.; Arias L.; Casaroli-Marano R. P.; Bassaganyas F. Topical azithromycin or ofloxacin for endophthalmitis prophylaxis after intravitreal injection. Clin. Ophthalmol. 2012, 6, 1595–1599. 10.2147/OPTH.S35795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B. S.; Sundareshan V.; Cory T. J.; Hayes D. Jr.; Anstead M. I.; Feola D. J. Azithromycin alters macrophage phenotype. J. Antimicrob. Chemother. 2008, 61, 554–560. 10.1093/jac/dkn007. [DOI] [PubMed] [Google Scholar]
- Varano G. P.; Parisi V.; Adornetto A.; Cavaliere F.; Amantea D.; Nucci C.; Corasaniti M. T.; Morrone L. A.; Bagetta G.; Russo R. Post-ischemic treatment with azithromycin protects ganglion cells against retinal ischemia/reperfusion injury in the rat. Mol. Vision 2017, 23, 911–921. [PMC free article] [PubMed] [Google Scholar]
- Geudens N.; Timmermans L.; Vanhooren H.; Vanaudenaerde B. M.; Vos R.; Van De Wauwer C.; Verleden G. M.; Verbeken E.; Lerut T.; Van Raemdonck D. E. M. Azithromycin reduces airway inflammation in a murine model of lung ischaemia reperfusion injury. Transplant Int. 2008, 21, 688–695. 10.1111/j.1432-2277.2008.00670.x. [DOI] [PubMed] [Google Scholar]
- Lebel M. Pharmacokinetic properties of clarithromycin: A comparison with erythromycin and azithromycin. Can. J. Infect. Dis. Med. Microbiol. 1993, 4, 148–152. 10.1155/1993/168061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubacher J. L.; Bols N. C. Chemically de-acetylated 2’,7’-dichlorodihydrofluorescein diacetate as a probe of respiratory burst activity in mononuclear phagocytes. J. Immunol. Methods 2001, 251, 81–91. 10.1016/s0022-1759(01)00308-8. [DOI] [PubMed] [Google Scholar]
- Bucana C.; Saiki I.; Nayar R. Uptake and accumulation of the vital dye hydroethidine in neoplastic cells. J. Histochem. Cytochem. 1986, 34, 1109–1115. 10.1177/34.9.2426339. [DOI] [PubMed] [Google Scholar]
- Henn A.; Lund S.; Hedtjärn M.; Schrattenholz A.; Pörzgen P.; Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009, 26, 83–94. 10.14573/altex.2009.2.83. [DOI] [PubMed] [Google Scholar]
- Lawrence J. M.; Singhal S.; Bhatia B.; Keegan D. J.; Reh T. A.; Luthert P. J.; Khaw P. T.; Limb G. A. MIO-M1 cells and similar muller glial cell lines derived from adult human retina exhibit neural stem cell characteristics. Stem Cells 2007, 25, 2033–2043. 10.1634/stemcells.2006-0724. [DOI] [PubMed] [Google Scholar]
- Tezel G. The immune response in glaucoma: A perspective on the roles of oxidative stress. Exp. Eye Res. 2011, 93, 178–186. 10.1016/j.exer.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G. l. n.; Yang X.; Luo C.; Peng Y.; Sun S. L.; Sun D. Mechanisms of Immune System Activation in Glaucoma: Oxidative Stress-Stimulated Antigen Presentation by the Retina and Optic Nerve Head Glia. Invest. Ophthalmol. Visual Sci. 2007, 48, 705–714. 10.1167/iovs.06-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi T. S.; Lee I.; Hüttemann M.; Kumar A.; Nantwi K. D.; Singh L. P. TXNIP Links Innate Host Defense Mechanisms to Oxidative Stress and Inflammation in Retinal Muller Glia under Chronic Hyperglycemia: Implications for Diabetic Retinopathy. Exp. Diabetes Res. 2012, 2012, 438238. 10.1155/2012/438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera J. C.; Dabouz R.; Noueihed B.; Omri S.; Tahiri H.; Chemtob S. Ischemic Retinopathies: Oxidative Stress and Inflammation. Oxid. Med. Cell. Longevity 2017, 2017, 3940241. 10.1155/2017/3940241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda T.; Shimazawa M.; Hara H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid. Med. Cell. Longevity 2017, 2017, 9208489. 10.1155/2017/9208489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhardt F.; Haslund-Vinding J.; Jaquet V.; McBean G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. 10.1111/bph.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-U.; Hwang C. N.; Sun H.-N.; Jin M.-H.; Han Y.-H.; Lee H.; Kim J.-M.; Kim S.-K.; Yu D.-Y.; Lee D.-S.; Lee S. H. Peroxiredoxin I is an indicator of microglia activation and protects against hydrogen peroxide-mediated microglial death. Biol. Pharm. Bull. 2008, 31, 820–825. 10.1248/bpb.31.820. [DOI] [PubMed] [Google Scholar]
- Xing Y. W.; Liu K. Z. Azithromycin inhibited oxidative stress and apoptosis of high glucose-induced podocytes by inhibiting STAT1 pathway. Drug Dev. Res. 2021, 82, 990–998. 10.1002/ddr.21801. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Cuevas S.; Armando I.; Jose P. Azithromycin induces sestrin2 expression through Nrf2 signaling pathway in lung epithelial cells stimulated with cigarette smoke extract (869.11). FASEB J. 2014, 28, 869.11. 10.1096/fasebj.28.1_supplement.869.11. [DOI] [Google Scholar]
- Cuevas S.; Yang Y.; Armando I.; Jose P. A. Mechanisms involved in the antioxidant properties of azithromycin in lung epithelial cells stimulated with cigarette smoke extract. FASEB J. 2016, 30, 982.2. 10.1096/fasebj.30.1_supplement.982.2. [DOI] [Google Scholar]
- Zhang B.; Bailey W. M.; Kopper T. J.; Orr M. B.; Feola D. J.; Gensel J. C. Azithromycin drives alternative macrophage activation and improves recovery and tissue sparing in contusion spinal cord injury. J. Neuroinflammation 2015, 12, 218. 10.1186/s12974-015-0440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrančić M.; Banjanac M.; Nujić K.; Bosnar M.; Murati T.; Munić V.; Stupin Polančec D.; Belamarić D.; Parnham M. J.; Eraković Haber V. Azithromycin distinctively modulates classical activation of human monocytes in vitro. Br. J. Pharmacol. 2012, 165, 1348–1360. 10.1111/j.1476-5381.2011.01576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantea D.; Certo M.; Petrelli F.; Bagetta G. Neuroprotective Properties of a Macrolide Antibiotic in a Mouse Model of Middle Cerebral Artery Occlusion: Characterization of the Immunomodulatory Effects and Validation of the Efficacy of Intravenous Administration. Assay Drug Dev. Technol. 2016, 14, 298–307. 10.1089/adt.2016.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polazzi E.; Monti B. Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog. Neurobiol. 2010, 92, 293–315. 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Mahaling B.; Baruah N.; Ahamad N.; Maisha N.; Lavik E.; Katti D. S. A non-invasive nanoparticle-based sustained dual-drug delivery system as an eyedrop for endophthalmitis. Int. J. Pharm. 2021, 606, 120900. 10.1016/j.ijpharm.2021.120900. [DOI] [PubMed] [Google Scholar]
- Bringmann A.; Wiedemann P. Müller Glial Cells in Retinal Disease. Ophthalmologica 2012, 227, 1–19. 10.1159/000328979. [DOI] [PubMed] [Google Scholar]
- Martinez-De Luna R. I.; Ku R. Y.; Aruck A. M.; Santiago F.; Viczian A. S.; San Mauro D.; Zuber M. E. Müller glia reactivity follows retinal injury despite the absence of the glial fibrillary acidic protein gene in Xenopus. Dev. Biol. 2017, 426, 219–235. 10.1016/j.ydbio.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García M.; Vecino E. Role of Müller glia in neuroprotection and regeneration in the retina. Histol. Histopathol. 2003, 18, 1205–1218. [DOI] [PubMed] [Google Scholar]
- Wang B.; Navath R. S.; Romero R.; Kannan S.; Kannan R. Anti-inflammatory and anti-oxidant activity of anionic dendrimer-N-acetyl cysteine conjugates in activated microglial cells. Int. J. Pharm. 2009, 377, 159–168. 10.1016/j.ijpharm.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlin K. J.; Lim L.; Grice E. A.; Campochiaro P. A.; Zack D. J.; Adler R. A method for analysis of gene expression in isolated mouse photoreceptor and Müller cells. Mol. Vision 2004, 10, 366–375. [PubMed] [Google Scholar]
- Datta S.; Choudhury D.; Das A.; Das Mukherjee D.; Das N.; Roy S. S.; Chakrabarti G. Paclitaxel resistance development is associated with biphasic changes in reactive oxygen species, mitochondrial membrane potential and autophagy with elevated energy production capacity in lung cancer cells: A chronological study. Tumour Biol. 2017, 39, 1010428317694314. 10.1177/1010428317694314. [DOI] [PubMed] [Google Scholar]
- Wang M.; Ma W.; Zhao L.; Fariss R. N.; Wong W. T. Adaptive Müller cell responses to microglial activation mediate neuroprotection and coordinate inflammation in the retina. J. Neuroinflammation 2011, 8, 173. 10.1186/1742-2094-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaling B.; Katti D. S. Understanding the influence of surface properties of nanoparticles and penetration enhancers for improving bioavailability in eye tissues in vivo. Int. J. Pharm. 2016, 501, 1–9. 10.1016/j.ijpharm.2016.01.053. [DOI] [PubMed] [Google Scholar]
- Mahaling B.; Katti D. S. Physicochemical properties of core–shell type nanoparticles govern their spatiotemporal biodistribution in the eye. Nanomedicine 2016, 12, 2149–2160. 10.1016/j.nano.2016.05.017. [DOI] [PubMed] [Google Scholar]
- Mahaling B.; Srinivasarao D. A.; Raghu G.; Kasam R. K.; Bhanuprakash Reddy G.; Katti D. S. A non-invasive nanoparticle mediated delivery of triamcinolone acetonide ameliorates diabetic retinopathy in rats. Nanoscale 2018, 10, 16485–16498. 10.1039/c8nr00058a. [DOI] [PubMed] [Google Scholar]
- Chatterjee N.; Mahaling B.; Sivakumar S.; Bharadwaj P. K. A highly selective and sensitive “Turn-On” fluorescence chemosensor for the Cu2+ ion in aqueous ethanolic medium and its application in live cell imaging. J. Photochem. Photobiol., A 2016, 330, 110–116. 10.1016/j.jphotochem.2016.07.011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.