


Keywords: AKI, TIGIT, immune checkpoint therapy, T cells, inflammation
Abstract
Significance Statement
T cells mediate pathogenic and reparative processes during AKI, but the exact mechanisms regulating kidney T cell functions are unclear. This study identified upregulation of the novel immune checkpoint molecule, TIGIT, on mouse and human kidney T cells after AKI. TIGIT-expressing kidney T cells produced proinflammatory cytokines and had effector (EM) and central memory (CM) phenotypes. TIGIT-deficient mice had protection from both ischemic and nephrotoxic AKI. Single-cell RNA sequencing led to the discovery of possible downstream targets of TIGIT. TIGIT mediates AKI pathophysiology, is a promising novel target for AKI therapy, and is being increasingly studied in human cancer therapy trials.
Background
T cells play pathogenic and reparative roles during AKI. However, mechanisms regulating T cell responses are relatively unknown. We investigated the roles of the novel immune checkpoint molecule T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT) in kidney T cells and AKI outcomes.
Methods
TIGIT expression and functional effects were evaluated in mouse kidney T cells using RNA sequencing (RNA-Seq) and flow cytometry. TIGIT effect on AKI outcomes was studied with TIGIT knockout (TIGIT-KO) mice in ischemia reperfusion (IR) and cisplatin AKI models. Human kidney T cells from nephrectomy samples and single cell RNA sequencing (scRNA-Seq) data from the Kidney Precision Medicine Project were used to assess TIGIT's role in humans.
Results
RNA-Seq and flow cytometry analysis of mouse kidney CD4+ T cells revealed increased expression of TIGIT after IR injury. Ischemic injury also increased TIGIT expression in human kidney T cells, and TIGIT expression was restricted to T/natural killer cell subsets in patients with AKI. TIGIT-expressing kidney T cells in wild type (WT) mice had an effector/central memory phenotype and proinflammatory profile at baseline and post-IR. Kidney regulatory T cells were predominantly TIGIT+ and significantly reduced post-IR. TIGIT-KO mice had significantly reduced kidney injury after IR and nephrotoxic injury compared with WT mice. scRNA-Seq analysis showed enrichment of genes related to oxidative phosphorylation and mTORC1 signaling in Th17 cells from TIGIT-KO mice.
Conclusions
TIGIT expression increases in mouse and human kidney T cells during AKI, worsens AKI outcomes, and is a novel therapeutic target for AKI.
Introduction
AKI occurs in approximately 5% of hospitalized patients and up to 50% of patients in intensive care units.1–3 Ischemia reperfusion (IR) and nephrotoxins are common causes of AKI and involve both immune and nonimmune mechanisms. Prior studies demonstrated that T cells mediate both the early injury phase and the late recovery/repair phase of AKI. However, mechanisms by which T cells mediate AKI are incompletely understood.
This study investigated the role of a novel immune checkpoint molecule, T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (TIGIT) in AKI. We unexpectedly identified TIGIT upregulation on CD4+ T cells in the early phase of experimental AKI during discovery RNA-Seq analysis. TIGIT is a transmembrane glycoprotein that is significantly upregulated on memory and activated T cells and natural killer (NK) cells.4–6 TIGIT interacts with its ligand CD155, which is expressed by multiple cell types including kidney epithelium, to initiate coinhibitory signaling that dampens Th1 and Th17 responses and enhances Th2 response and Treg suppressive activity.6,7 In addition, simultaneous checkpoint blockade of TIGIT and PD1 was found to enhance antitumor immunity and survival in murine glioblastoma.8,9 Many clinical trials are underway for developing TIGIT-based immunotherapy for cancers and autoimmune diseases (clinicaltrials.gov). Current cancer immunotherapies targeting established coinhibitory receptors such as cytotoxic T-lymphocyte associated-4 and PD1 can be effective but also lead to increased incidence of AKI.10,11 Therefore, understanding TIGIT's role in AKI will be important for exploring novel therapies as well as mitigating immune checkpoint inhibitor-induced AKI.
Our data demonstrated increased TIGIT expression in post-IR kidney T cells of wild type (WT) mice that correlated with EM/CM phenotypes and a proinflammatory profile. In human kidney T cells, TIGIT expression increased after ischemia in nephrectomies from patients with renal cell carcinoma (RCC) as well as in patients with AKI. TIGIT knockout (TIGIT-KO) mice were protected from IR- and cisplatin-induced AKI and had metabolically distinct kidney T cells. These results provide important context for TIGIT-based immunotherapy for AKI.
Methods
Mice and Genotyping
Male C57BL/6J (WT, Stock no. 000664) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed under specific pathogen-free conditions at the Johns Hopkins University animal facility. TIGIT-KO mice on C57BL/6J background were a kind gift from Dr. Vijay Kuchroo (Harvard University) and were bred and maintained at the Johns Hopkins University animal facility. TIGIT-KO mice were generated by crossing homozygous TIGIT-floxed mice with cytomegalovirus (CMV) Cre mice, resulting in TIGIT deletion in all tissues in the progeny. TIGIT-KO status was confirmed before assigning mice for present studies by positive detection of CMV Cre gene (650bp PCR band) in tail tissue (Supplemental Figure 1) using the primer set and PCR program provided in Supplemental Tables 1 and 2, respectively. An internal positive control was used to confirm successful PCR conditions (324bp PCR band) during genotyping. Because our TIGIT-KO mouse colony is germline, i.e., homozygous for Cre and flox sequences, we used syngenic and age-matched WT mice (Jackson Laboratory) as controls. Eight to 10-week-old male mice were used in accordance with the Johns Hopkins University Institutional Animal Care and Use Committee approved protocols. Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines were followed to report data presented in this study.12
Induction of AKI
AKI was induced by either IR surgery or cisplatin injection as per established protocols.13,14 In brief, for IR surgery, mice were anesthetized with an intraperitoneal (i.p.) injection of pentobarbital (75 mg/kg) and renal pedicles dissected after a medial abdominal incision. A microvascular clamp (straight 1×8 mm, Roboz Surgical Instrument Co.) was placed on each renal pedicle for 25 minutes. Animals were kept hydrated with 1 ml of warm saline and at a constant body temperature (37°C). After appropriate ischemia time, the clamps were removed, and the kidneys were inspected for the restoration of blood flow. The incision was sutured, and the animals were allowed to recover with access to food and water ad libitum. Sham surgeries were performed similar to the IR group without clamping the renal pedicles. For nephrotoxic AKI model, cisplatin (cis-diammineplatinum II dichloride; Sigma-Aldrich) was dissolved in normal saline (1 mg/ml) on the day of injection. A single i.p. injection of cisplatin (25 mg/kg body weight) was given to induce AKI.
Assessment of Kidney Function
Blood samples were collected at 0, 24, 48, and 72 hours after IR or cisplatin injection, and serum creatinine (SCr) was measured to assess kidney function by Cobas Mira Plus automated analyzer system (Roche) using creatinine measurement reagents (Pointe Scientific Inc.).
Isolation of Kidney Mononuclear Cells and Splenocytes from Mice
Kidney mononuclear cells (KMNCs) and splenocytes were isolated according to an established protocol.15 Mice were anesthetized with i.p. ketamine hydrochloride (140 mg/kg) and xylazine (10 mg/kg) and exsanguinated to reduce the number of circulating immune cells. Both kidneys were removed, decapsulated, finely minced, and incubated in collagenase D (2 mg/ml; Sigma-Aldrich) solution for 30 minutes at 37°C. A single-cell suspension of the kidney digestion was achieved by mechanical disruption of the tissue using a 70-μm strainer (BD Bioscience), followed by centrifugation at room temperature using isotonic Percoll density gradient (GE Healthcare) (3000 revolutions per minute for 30 minute in brake off mode) to collect mononuclear cell population as per the manufacturer's instructions. Spleens were filtered through a 40-μm strainer (BD Bioscience) to prepare single-cell suspensions and incubated with ACK lysis buffer (Quality Biological) for 3 minutes to remove red blood cells. Collected cells from each organ were washed, and each pellet was resuspended in Roswell Park Memorial Institute media containing 5% FBS and counted.
T cell Activation and Intracellular Cytokine Analysis
To assess the effects of TIGIT on kidney T cell activation and cytokine production, freshly isolated KMNCs (approximately 1×106) were stained with the following antibodies. All antibodies were purchased from BioLegend unless specified otherwise: anti-mouse CD45-Spark Blue 550 (clone: 30.F11), TCRβ-Alexa Fluor 488 (clone: H57-597), CD8α-PerCP (clone: 53-6.7), TIGIT-BV421 (clone: 1G9), CD25-PE-Cy7 (clone: PC61.5), CD69-Alexa Fluor 700 (clone: H1.2F3), CD62L-BV785 (clone: MEL-14), CD44-BV570 (clone: 1M7), PD1-APC/Fire750 (clone: 29F.1412) or CD226-APC/Fire750 (clone: 10E5) and CD4-BV480 (clone: RM4-5, BD Biosciences). To measure intracellular cytokines, KMNCs were stimulated with leukocyte activation cocktail (BD Pharmigen) containing PMA, ionomycin, and brefeldin A before staining with IFNγ-BV650 (clone: XMG1.2), TNFα-BV711 (clone: MP6-XT22), IL17A-BV605 (clone: TC11-18H10.1), IL10-PE/Dazzle594 (clone: JES5-16E3), IL4-PerCP eFluor710 (clone: 11B11, Invitrogen), and FOXP3-Alexa Fluor647 (clone: MF-14) using Foxp3/Transcription Factor Staining Buffer Set (eBioscience). Dead cells were labeled with Zombie NIR (BioLegend) and excluded from the analyses. Labeled samples were analyzed with spectral cytometer, Aurora (Cytek). Live unmixing was performed onboard before running experimental samples using Spectroflo (Cytek) and unmixed .fcs files were analyzed with FlowJo software (BD Biosciences).
Assessment of Metabolic Markers in Kidney T Cells
Cell suspensions were prepared as described above for T cell activation and intracellular cytokine analysis and stained with the following antibodies for 30 minutes at 4°C. All antibodies were purchased from BioLegend unless specified otherwise: anti-mouse CD45-BV570 (clone: 30-F11), TCRβ-BV785 (clone: H57-597), CD8-BV510 (clone: 53-6.7), and CD4-APC-Fire810 (clone: GK1.5). Subsequently, cells were fixed and permeabilized for intracellular staining for metabolic markers using Foxp3/Transcription Factor Staining kit (ThermoFisher Scientific) as per instructions provided in the kit. Intracellular staining cocktail was prepared in 50 μL of permeabilization/wash buffer with the following antibodies: anti-mouse CPT1a-Alexa Fluor 488 (clone: 8F6AE9, Abcam), voltage-dependent anion channel 1-Alexa Fluor 532 (clone: 20B12AF2, Abcam), GLUT1-PE (clone: EPR3915, Abcam), pS6-Alexa Fluor 594 (clone: D68F8, Cell Signaling), hexokinase II-PE-Cy5 (clone: EPR20839, Abcam), H3K27me3-PE-Cy7 (clone: C36B11, Cell Signaling), and Tomm20-Alexa Fluor 647 (clone: EPR15581-54, Abcam). Cells were stained for 45 minutes at room temperature, washed with permeabilization/wash buffer, and resuspended in cell staining buffer. Dead cell staining, sample acquisition, and analysis were similar to T cell activation and intracellular cytokine analysis section.
Kidney CD4+ T Cell and CD45+ Immune Cell Flow Sorting
For FACS, KMNCs were preincubated with anti-CD16/CD32 Fc block (clone: 2.4G2, BD Biosciences) for 15 minutes on ice. WT, kidney CD4+ T cells were flow sorted by staining KMNCs with monoclonal Ab anti-TCRβ-BV421 (clone: H57-597, BioLegend) and anti-CD4-PerCP/Cy5.5 (clone: RM4-5, BioLegend) for 30 minutes at 4°C. WT and TIGIT-KO kidney CD45+ cells were flow sorted by staining KMNCs with anti-CD45-APC/Cy7 (clone: 30-F11, BioLegend) and live/dead aqua (ThermoFisher). Cells were sorted with a FACSAria II Cell Sorter (BD Biosciences). After sorting, purified CD4+ T cells from WT mice were used for bulk RNA-Seq analysis and CD45+ cells from WT and TIGIT-KO were used for single-cell RNA-Seq analysis (scRNA-Seq).
Bulk RNA-Seq Analysis of Kidney CD4+ T Cells
CD4+ T cells were flow sorted for bulk RNA-Seq studies from WT control and 24 hours post-IR mice.16 RNA was isolated from flow-sorted renal CD4+ T cells (approximately 3×104) using RNeasy mini kit (QIAGEN). Pooled cells from five to six mice were used for each flow sort experiment, and there were n=3 experiments/group for RNA-Seq analysis. Total RNA was fragmented and converted to Illumina NextSeq 500 compatible cDNA libraries. cDNA libraries were prepared from 1 ng of total RNA using SMARTer Stranded Total RNA-Seq Kit v2—Pico Input Mammalian kit (Takara). Libraries were multiplexed and sequenced with 60 million 36 base pair paired-end reads per sample using two flowcell lanes. Using Tophat version 2.1.0. and Cufflinks version 2.2.1 with default parameters, raw data were aligned to the RGCm38 mouse reference genome, and Fragments Per Kilobase Of Exon Per Million values were obtained. The gene expression of IR-injured renal CD4+ T cells was compared with normal control using a cutoff of P<0.05 and the minimum Fragments Per Kilobase Of Exon Per Million values of >1.0 in log2 notation. Complete bulk RNA-Seq data from this analysis has been deposited at National Center for Biotechnology Information's data repository Gene Expression Omnibus17 and is freely accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139171.
scRNA-Seq Analysis of Kidney Immune Cells
KMNCs were stained for anti-mouse CD45 antibody and Live dead aqua, and CD45+ immune cells from WT and TIGIT-KO mice at baseline and 24 hours after inducing kidney IR-AKI were isolated using FACS sorting. Purified cells were processed for high-throughput droplet encapsulation to capture and barcode individual cells using 10× chromium technology. In brief, sorted cells were processed through the GemCode Single Cell Platform (10× Genomics) as per the manufacturer's protocol. Single cells (approximately 10,000) were partitioned into gel beads and libraries prepared using the 10× Chromium system. Libraries were sequenced to a target depth of 100K reads/cell on an Illumina NovaSeq 6000. scRNA-Seq sequence reads were aligned to the mm10 reference genome with CellRanger (version 6.0.2). Subsequent quality control filtering, normalization, clustering, and differential gene expression analysis was performed using SCANPY (version 1.8.2).18 Specifically, potential doublets and negatives were removed by including only cells with gene counts >500, total counts <22,500, and percent mitochondrial gene counts <20%. Cell type classification was performed using SingleCellNet,19 and contaminating nonhematopoietic cells were excluded from further analysis. Genes were excluded if they were detected in fewer than five cells. The data were then normalized and log transformed before highly variable genes were identified. The data were scaled, and principal component analysis was performed. Leiden clustering was performed and visualized on a Uniform Manifold Approximation and Projection (UMAP) embedding. Differential gene expression analysis was performed with the rank_genes_groups function in SCANPY, and gene set enrichment analysis was performed with the Gseapy Enrichr function20 testing for enrichment of the MSigDB Hallmark signatures.21 scRNA-Seq data from this study are freely accessible at https://doi.org/10.5281/zenodo.7314510.
Histologic Evaluation of Kidney
The kidneys were harvested 72 hours after induction of AKI, and a transverse piece from each kidney was fixed with 10% buffered formalin phosphate and embedded with paraffin for histologic evaluation. Tissue sections (2–3 µm) were stained with hematoxylin and eosin (H&E). A renal pathologist (L.J.A), blinded to the experimental groups, scored the percentage of necrotic tubules out of total tubules in at least ten high-power fields, and the average percentage of tubular necrosis in all fields was presented as the renal tubular injury score of each mouse.
TIGIT Assessment in Human Kidney T Cells
Human kidney samples were collected, after informed consent, from grossly normal kidney sections from patients with RCC undergoing nephrectomy surgery either before or after the clamping of renal pedicles. Kidney tissue was digested according to an established protocol for isolation of KMNCs.22,23 Single-cell suspensions were preincubated with human Fc Block (clone: Fc1.3216, BD Biosciences) for 15 minutes and stained with anti-human fluorochrome-conjugated monoclonal antibodies CD45-BUV395 (clone: HI30, BD Biosciences), TCRα/β-BV421 (clone: IP26, BioLegend), CD4-PerCP/Cy5.5 (clone: OKT4, BioLegend), CD8α-BV605 (clone: RPA-T8, BioLegend), and TIGIT-PE (clone: A15153G, BioLegend) for 30 minutes at 4°C. CD45+TCRα/β+ cells were analyzed for TIGIT expression on CD4+, CD8+, and double negative (DN) T cells using LSRII flow cytometer, and data were analyzed using FlowJo software. Kidney precision medicine project database was explored using the online explorer tool to examine TIGIT expression in kidney T cells from healthy control and patients with AKI. This study was conducted in accordance with the Declaration of Helsinki and approved by the Johns Hopkins Medicine Institutional Review Board.
Statistical Analyses
Data were collected from at least three independent experiments and expressed as median and interquartile range (IQR=Q3−Q1). Statistical differences were analyzed using the unpaired nonparametric Mann-Whitney U test between two groups and Kruskal-Wallis one-way analysis of variance followed by the Dunn multiple comparison test for more than two groups using Prism 9 (GraphPad Software). Group size was calculated with a free online power calculation tool (stat.ubc.ca/∼rollin/stats/ssize/n2) to achieve a minimum of 80% power. Statistical significance was accepted at P<0.05.
Results
IR Injury Increased TIGIT Expression on Murine Kidney T Cells
Because CD4+ T cells play important modulatory roles during AKI, we first analyzed our previously published16 bulk RNA-Seq data from flow-sorted kidney CD4+ T cells to identify novel molecular players (Figure 1A). Among the cosignaling molecules identified in our RNA-Seq data, Tigit mRNA expression was significantly upregulated (adjusted P-value = 0.002) in CD4+ T cells from post-IR kidneys (52.91[88.09–48.07]) compared with control kidneys (20.54[26.85–17.95], Figure 1, B and C, Supplemental Table 3). Concurrently, TIGIT costimulatory partners Cd226 and Cd96 decreased significantly. In addition to Tigit, we also observed a significant increase in Ctla4 expression. However, checkpoint molecules Pd1, Lag3, and Tim3 and costimulatory molecules Cd28 and Icos were not affected. The 24-hour time point selected for this RNA-Seq study and the validation and mechanistic studies described below corresponds with the maximum increase in SCr levels in our IR model.
Figure 1.

Kidney IR injury increases TIGIT mRNA and protein expression in mouse kidney CD4+ T cells. (A) Schematic of steps involved during bulk RNA-Seq analysis of flow-sorted CD4+ T cells isolated from control and 24 hours post-IR kidneys of WT mice. Kidney mononuclear cells (KMNCs) were isolated and stained with anti T cell receptor (TCR) and CD4 antibodies to flow sort pure CD4+ T cells for bulk RNA-Seq analysis. (B) Volcano plot depicting all known cosignaling molecules identified after bulk RNA-Seq analysis. (C) Graph showing median and IQR values of Tigit mRNA in control and post-IR kidney CD4+ T cells from bulk RNA-Seq data with adjusted P value. n=3 per group with five to six mice per n. (D and E) Flow dot plots and corresponding graph showing TIGIT expression at protein level in CD4+, CD8+, and DN T cells from control (n=7), sham (n=5), and 24 hours post-IR (n=8) kidneys. (F) Absolute number of TIGIT-expressing CD4+, CD8+, and DN T cells from control, sham, and, 24 hours post-IR kidneys. Data in (E) and (F) were analyzed with Kruskal-Wallis one-way analysis of variance followed by Dunn multiple comparison post hoc analysis. Graphs represent median (IQR) values. *P≤0.05, **P≤0.01, ***P≤0.001, and ****P≤0.0001. IR, ischemia reperfusion; KMNC, kidney mononuclear cell; TIGIT, T-cell immunoreceptor with Ig and ITIM domains.
Validation of RNA-Seq data at the protein level, using the multiparameter spectral flow cytometry and gating strategy described in Supplemental Figure 2A, showed significant TIGIT expression in post-IR kidney CD4+ (IR=7.58[10.65–6.44]% versus control=1.52[2.08–1.26]%; P=0.0002), CD8+ (IR=10.30[11.25–7.89]% versus sham=3.74[7.12–2.13]%; P=0.02 and control=2.50[4.32–1.66]%; P=0.0008) and DN T cells (IR=7.42[10.28–4.37]% versus control=0.49[0.97–0.36]%; P<0.0001) compared with control mice (Figure 1, D and E). Quantification of absolute cell numbers showed significant increases in both TIGIT+CD4+ T cells in post-IR kidneys (171240[268884–86019]) compared with sham (52037[55702–34009]; P = 0.009) and control (51594[87334–30688]; P = 0.02) kidneys and TIGIT+DN T cells in the post-IR kidneys (57631[111788–34055]) compared with sham (11677[38666–7364]; P = 0.05) and control (15277[25220–6019]; P = 0.007) kidneys (Figure 1F). Splenic T cells from mice that underwent kidney IR injury showed no significant increase in TIGIT expression (Supplemental Figure 2B), further suggesting that renal IR injury increased TIGIT expression specifically in kidney T cells.
Ischemic Injury Increased TIGIT Expression in Human Kidney T Cells
To assess the relevance of this novel immune checkpoint molecule to human kidney injury, we evaluated TIGIT expression in T cells from human kidneys. Kidney tissue was collected from patients undergoing partial nephrectomy because of RCC. We collected kidney tissue either pre (nonischemic) or post (ischemic) renal pedicle clamping and carefully dissected the nonmalignant normal adjacent tissue. Expression of TIGIT was relatively low in T cells isolated from normal compared with ischemic tissue. We found significantly higher absolute numbers of TIGIT+CD8+ (63.0[121.9–39.75] versus 14.88[23.57–4.62], P = 0.03) and TIGIT+DN T cells (17.25[27.94–9.19] versus 0.68[0.68–0.00], P = 0.03) in ischemic kidney compared with normal kidney tissue (Figure 2A). We next explored the scRNA-Seq data of the National Institutes of Health (NIH) kidney precision medicine project, using the online explorer tool (https://atlas.kpmp.org/explorer/dataviz) to assess the status of TIGIT in kidney cells from well-characterized healthy controls (n=20) and patients with AKI (n=12). We observed significant TIGIT expression in patients with AKI compared with healthy controls that was restricted to Tregs, cytotoxic T cells, and NK cell populations (Figure 2, B–D and Supplemental Table 4).
Figure 2.

TIGIT expression increases in human kidney T cells after ischemic injury and in patients with AKI. (A) Flow dot plot and graphs showing percentage (left graph) of TIGIT+ CD4, CD8, and DN T cells in preclamp and postclamp human kidney. Corresponding bar graphs showing absolute numbers of TIGIT+ cells in the same preclamp and postclamp kidney tissue. Data were analyzed using the nonparametric Mann-Whitney test to compare preclamp and postclamp groups separately for CD4, CD8, and DN T cells. Graphs in Figure 2A represent median (IQR) values. (B) Reference UMAP of the NIH kidney precision medicine project (KPMP) scRNA-Seq data showing different kidney cell clusters identified for scRNA-Seq data set. (C and D) UMAPs showing TIGIT expression in T/NK cells cluster of healthy controls (n=20) and patients with AKI (n=12). Arrow showing position of T/NK cell cluster. *P≤0.05. KPMP, kidney precision medicine project; TIGIT, T-cell immunoreceptor with Ig and ITIM domains.
TIGIT-Expressing Kidney T Cells had a Proinflammatory and Memory Phenotype
To understand functional effects of TIGIT in kidney T cells, we assessed intracellular levels of proinflammatory and anti-inflammatory cytokines in TIGIT+ and TIGIT− subsets in WT mice, both at baseline and 24 hours post-IR. Overall, TIGIT+ T cells had a greater percentage and mean fluorescent intensity (MFI; per cell expression) of IFNγ and TNFα compared with TIGIT− T cells from post-IR kidneys. More specifically, the percentage of CD4+TIGIT+IFNγ+ cells were significantly higher compared with CD4+TIGIT−IFNγ+ subset (52.90[57.15–42.35]% versus 17.00[27.60–14.70]%, P = 0.008) after IR injury (Figure 3A). Correspondingly, CD4+TIGIT+ T cells had significantly higher per cell expression of IFNγ compared with CD4+TIGIT− T cells at baseline (10408[12556–6250] versus 5102[5415–4327], P≤0.008) and post-IR (9095[10494–7948] versus 3223[4568–2975], P≤0.008, Figure 3B). Similarly, the percentage of CD4+TIGIT+TNFα+ cells was significantly higher compared with CD4+TIGIT−TNFα+ cells after IR injury (53.60[67.15–45.20]% versus 34.10[45.15–26.35]%, P = 0.03, Figure 3C). At the per cell level, TNFα showed significantly higher MFI in CD4+TIGIT+ T cells compared with CD4+TIGIT− T cells post-IR (8609[11526–7441] versus 5309[7216–4248], P = 0.03, Figure 3D). In CD8 T cells, IFNγ or TNFα expression did not differ between TIGIT+ and TIGIT− subsets either at baseline or post-IR. However, we observed a significant decline in IFNγ-expressing TIGIT−CD8 T cells post-IR compared with baseline levels (58.20[66.85–55.70]% versus 30.90[55.70–21.45]%; P = 0.03) (Supplemental Figure 3A). Furthermore, TIGIT+DN T cells expressed increased levels of IFNγ compared with TIGIT−DN T cells after IR injury (40.00[53.35–27.75]% versus 17.20[24.30–15.65]%; P = 0.02). Similar to TIGIT−CD8+ cells, the percentage and MFI of TIGIT−DN T cells expressing IFNγ decreased after IR injury compared with basal levels (P≤0.01; Supplemental Figure 3, E and F).
Figure 3.

TIGIT+ kidney T cells have a proinflammatory profile and memory phenotype, and TIGIT+ Tregs are reduced after IR injury. Representative histograms and corresponding graphs showing percentage and MFI of proinflammatory cytokines IFNγ (A and B) and TNFα (C and D) in TIGIT+ and TIGIT− CD4+ T cells in control (baseline) and 24 hours after IR injury. TIGIT+ CD4+ T cells had significantly higher levels of IFNγ and TNFα compared with TIGIT− subsets after IR injury. (E and F) Percentage of naive, EM, and CM cells among TIGIT+ and TIGIT− CD4+ T cells at baseline and after IR injury. (G) Representative flow images showing gating strategy for assessing TIGIT+ and TIGIT− subsets of kidney CD4+ CD25+ Foxp3+ Tregs. (H and I) Percentage and absolute number of TIGIT+ and TIGIT− subsets of Tregs among CD4+ cells at baseline in WT kidneys. (J and K) Percentage and absolute number of TIGIT+ Treg cells in control and 24 hours post-IR kidneys of WT mice. Data between groups were analyzed using the unpaired nonparametric Mann-Whitney test. Graphs represent median (IQR) values. *P≤0.05 and **P≤0.01. CM, central memory; EM, effector memory; IR, ischemia reperfusion; TIGIT, T-cell immunoreceptor with Ig and ITIM domains.
Since TIGIT expression correlated with a proinflammatory phenotype, we hypothesized that TIGIT affected differentiation of kidney T cells to promote EM and CM phenotype. We measured the commonly used T cell memory phenotype markers, CD62L and CD44,24,25 to describe naive (CD62LhiCD44lo), EM (CD62LloCD44hi) and CM (CD62LhiCD44hi) phenotypes (Supplemental Figure 4A) of TIGIT+ and TIGIT− kidney T cells at baseline (Figure 3E) and 24 hours after IR injury (Figure 3F). Analysis of kidney CD4+ T cells showed predominantly EM phenotype in both TIGIT+ and TIGIT− subsets, which was comparable between the subsets at baseline (79.75[92.23–73.4]% versus 73.45[77.33–72.55]%, P = 0.2) and after IR injury (77.5[91.65–69.35]% versus 72.7[75.95–55.9]%, P = 0.07). The percentage of CD4+TIGIT+ T cells with CM phenotype was significantly greater than CD4+TIGIT− T cells both at baseline (12.75[18.35–4.81]% versus 0.78[2.2925–0.53]%, P = 0.0006) and after IR injury (10.3[16–3.17]% versus 0.99[1.12–0.49]%, P = 0.01). This finding was accompanied by significantly reduced percentage of naive cells in TIGIT+ subset compared with TIGIT− subset both at baseline (2.7[6.85–1.01]% versus 20.95[22.13–17.35]%, P≤0.001) and after IR injury (5.92[7.86–2.55]% versus 18.8[27.25–16.55]%, P≤0.001). Consistent with these observations, CD4+TIGIT+ T cells had an increased per cell expression (MFI) of CD44 and reduced expression of CD62L at baseline (P = 0.004 and P = 0.006, respectively, Supplemental Figure 4B) and after IR injury (P = 0.002 and P = 0.0002, respectively, Supplemental Figure 4C). TIGIT+, CD8+, and DN T cells were predominantly of EM phenotype at baseline and after IR injury (Supplemental Figure 4, D–G). MFI of CD44 was significantly higher on TIGIT+, CD8+, and DN T cells compared with TIGIT− cells (Supplemental Figure 4, H–K).
Kidney Tregs Were Predominantly TIGIT-Positive and Reduced after IR Injury
Because the role for TIGIT in kidney Tregs is unclear, we first assessed the relationship between TIGIT and kidney Tregs and then assessed the effect of IR on these cells. To study Treg-specific TIGIT expression, we used a gating strategy shown in Figure 3G. At baseline, the percentage of TIGIT+ Tregs was significantly higher than TIGIT− Tregs (62.30[72.25–60.55]% versus 33.30[38.85–27.20]%; P≤0.008, Figure 3H). This was supported by an increasing trend of absolute numbers of TIGIT+ Tregs compared with TIGIT− Tregs (1944[2245–722.6] versus 843.0[1012–447.2], Figure 3I). We also assessed the frequency of Treg cells in TIGIT+ and TIGIT− subsets of splenic CD4+ T cells at baseline, which showed significantly higher TIGIT− Tregs compared with TIGIT+ Tregs (65.4[69.1–57.6]% versus 33.8[41.5–29.9]%; P≤0.008, Supplemental Figure 5A). We further assessed the effect of IR injury on TIGIT+ Treg population in kidney and spleen. We found a significant reduction in the absolute number of TIGIT+ Tregs in post-IR kidneys (1944[2245–722.6] versus 283.2[486.6–167.2]; P = 0.02, Figure 3J), with a decreasing trend in the percentage of TIGIT+ Tregs (62.30[72.25–60.55]% versus 58.30[69.65–39.75]%; P = 0.2, Figure 3K) compared with control kidneys at baseline. There was no change in splenic TIGIT+ Treg percentage after kidney IR injury (Supplemental Figure 5B).
TIGIT-KO Mice had Reduced Kidney Injury after Experimental AKI
We next sought to assess the role of TIGIT in AKI pathophysiology by subjecting TIGIT-sufficient (WT) and TIGIT-deficient (TIGIT-KO) mice to a 25-minute bilateral IR surgery. TIGIT-KO (n=6) mice had significantly reduced SCr compared with WT mice (n=5) at 24 hours (1.05[1.67–0.72] mg/dl versus 2.64[2.89–2.46] mg/dl; P≤0.01), 48 hours (0.67[1.11–0.53] mg/dl versus 2.45[3.17–2.26] mg/dl; P≤0.01), and 72 hours (0.74[0.94–0.60] mg/dl versus 1.90[2.60–1.41] mg/dl; P≤0.01) time points (Figure 4A). Mice were euthanized at 72 hours post-IR for histologic evaluations of kidneys, which showed significantly reduced percentage of necrotic tubules in the outer medullary region of TIGIT-KO mice (50.4[57.0–44.0]% versus 71.7[76.5–70.0]%; P = 0.004) compared with WT mice (Figure 4B).
Figure 4.
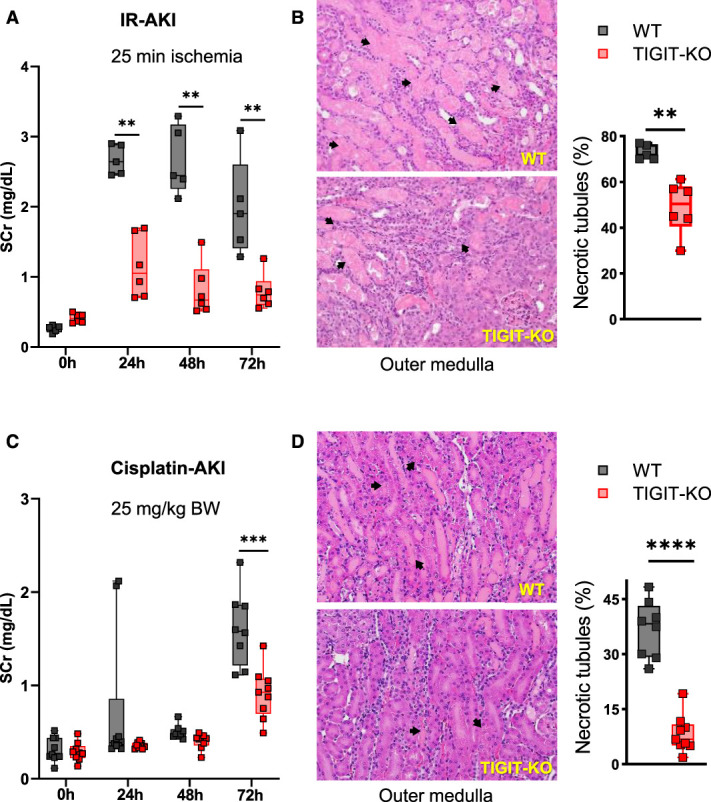
TIGIT-KO mice have reduced kidney injury after IR and cisplatin-induced AKI. (A) SCr levels in WT (n=5) and TIGIT-KO (n=6) mice at baseline (0 hour) and 24, 48, and 72 hours after IR injury. (B) H&E-stained kidney sections from WT and TIGIT-KO mice 72 hours after IR injury with arrows showing necrotic tubules. Corresponding graph on right showing percentage of necrotic tubules in the outer medullary region, 72 hours after IR injury. (C) SCr levels in WT (n=8) and TIGIT-KO (n=9) mice after cisplatin (25 mg/kg) injection. (D) H&E-stained kidney sections from WT and TIGIT-KO mice, 72 hours after cisplatin injection. Arrows showing necrotic tubules. Corresponding graphs showing percentage of necrotic tubules in the outer medullary region. SCr data in (A) and (C) were analyzed using multiple Mann-Whitney tests. Histology data in (B) and (D) were analyzed using the unpaired nonparametric Mann-Whitney test. Graphs represent median (IQR) values. **P≤0.01, ***P≤0.001, and ****P≤0.0001. IR, ischemia reperfusion; SCr, serum creatinine; TIGIT-KO, TIGIT knockout.
To further assess the role of TIGIT in nephrotoxic AKI, we treated WT and TIGIT-KO mice with 25 mg/kg cisplatin. TIGIT-KO mice (n=9) had significantly reduced SCr (0.93[1.07–0.69] mg/dl versus 1.58[1.86–1.22] mg/dl; P≤0.0003) compared with WT mice (n=8) 72 hours after cisplatin treatment (Figure 4C). Similar to the IR model, cisplatin-treated TIGIT-KO mice had significantly reduced percentage of necrotic tubules in the outer medullary region (6.80[10.85–5.1]% versus 38.25[42.1–29.5]%; P≤0.0001) compared with WT mice 72 hours after cisplatin treatment (Figure 4D). These results indicate that TIGIT contributes both IR and nephrotoxic AKI and plays a potential pathogenic role.
We then explored possible mechanisms through which TIGIT may be contributing towards AKI. We first immunophenotyped TIGIT-KO kidneys at baseline and found significantly reduced percentage of CD4+ T cells (48.3[48.6–44.3]% versus 51.8[55.9–47.9]%; P = 0.04) and increased CD8+ T cells (40.4[43.2–38.1]% versus 34.5[39.2–26.1]%; P = 0.01) compared with WT mice kidneys (Figure 5, A and B), although the absolute numbers were not different between groups (Figure 5C). In addition, kidneys of TIGIT-KO mice had increased percentage of Tregs (2.53[3.09–2.33]% versus 1.49[2.46–1.44]%; P = 0.02) compared with WT mice kidneys (Figure 5, D and E). However, absolute numbers of Tregs were comparable between WT and TIGIT-KO kidneys (Figure 5F).
Figure 5.

Immunophenotypic and functional assessment of kidney T cells from WT and TIGIT-KO kidneys at baseline and after IR injury. (A) Representative flow dot plot showing CD4, CD8, and DN T cells in WT and TIGIT-KO kidneys at baseline. (B) Baseline assessment of WT and TIGIT-KO mouse kidneys showing decreased percentage of CD4+ and increased percentage of CD8+ T cells in TIGIT-KO kidneys. (C) Absolute number of CD4+, CD8+, and DN T cells in WT and TIGIT-KO kidneys at baseline. (D) Representative flow dot plots showing Tregs in WT and TIGIT-KO kidneys at baseline. (E and F) Percentage and absolute number of Tregs among TCR+CD4+ cells in WT and TIGIT-KO mouse kidneys at baseline. (G) Percentage of PD1-expressing CD4+, CD8+, and DN T cells 24 hours after IR injury in WT and TIGIT-KO kidneys. (H and I) Percentage of naive, EM, and CM cells among TIGIT+ and TIGIT− CD4+ and CD8+ T cells from 24 hours post-IR WT and TIGIT-KO kidneys. (J and K) Percentage of IFNγ- and TNFα-expressing CD4+ and (L and M) CD8+ T cells from WT and TIGIT-KO kidneys, 24 hours after IR injury. Data between groups were analyzed using the unpaired nonparametric Mann-Whitney test. Graphs represent median (IQR) values. *P≤0.05 and **P≤0.01. CM, central memory; EM, effector memory; TIGIT-KO, TIGIT knockout.
Because PD1 expression on kidney T cells has been found to be important in AKI pathophysiology26 and PD1 and TIGIT coexpression affect T cell functions,27,28 we studied the effect of TIGIT on kidney T cell PD1 after AKI in WT and TIGIT-KO mice. We found no difference in PD1 expression on CD4+, CD8+, and DN T cells from WT and TIGIT-KO kidneys, 24 hours after IR injury (Figure 5G).
Assessment of CD62L and CD44 expression in 24 hours post-IR kidney T cells revealed no difference in the percentages of naive, EM, or CM CD4+ or CD8+ T cells in TIGIT-KO kidneys compared with WT kidneys (Figure 5, H and I). Furthermore, proinflammatory cytokines IFNγ and TNFα were comparable between WT and TIGIT-KO CD4+ T cells at baseline (Supplemental Figure 6, A and B) and 24 hours after IR injury (Figure 5, J and K). By contrast, CD8+ T cells isolated from TIGIT-KO mice produced increased levels of IFNγ at baseline (P = 0.001, Supplemental Figure 6C) and 24 hours post-IR (P = 0.03, Figure 5L). CD8+ T cells from post-IR kidneys of TIGIT-KO mice had increased expression of TNFα compared with WT kidneys (P = 0.03, Figure 5M), whereas its baseline expression was comparable between the groups (Supplemental Figure 6D).
TIGIT Expression Correlated with a Distinct Inflammatory and Metabolic Transcriptional Profile of Kidney T Cells
To further explore possible molecular mechanisms through which TIGIT mediates AKI, we performed scRNA-Seq analysis of flow-sorted CD45+ cells from WT and TIGIT-KO kidneys at baseline and 24 hours post-IR. scRNA-Seq data analysis showed all major immune cell types in both WT and TIGIT-KO kidneys on the basis of marker genes expressed by different clusters (Figure 6, A and B). Tigit expression was primarily restricted to T cell (Cd3d, Cd3e, and Cd3g expressing cells) and NK cell (Ncr1, Klrb1c, Klrk1, and Klrd1 expressing cells) populations among CD45+ cells in the WT mice (Figure 6C). As expected, Tigit expression was not detected in immune cells from TIGIT-KO mouse kidneys (Supplemental Figure 7A). Further annotation of T cell clusters showed the presence of effector and naive-like CD4+, CD8+, and DN T cell populations (Figure 6D). We used SingleCellNet19 and previously published reference data sets29–33 to annotate different subsets of kidney T cells as: Th1 (Cd4, Cd40lg), Th2 (Cd4, Ly6c2, Il4, Gata3), Th17 (Cd4, Il17r, Il17a, Rora), CD8 (Ccl5, Gzmk), and DN (Tcf12) T cells. In addition, naive-like cells were identified on the basis of the expression of Sell, Tcf7, and Il7r that either expressed Cd4 (naive-like CD4), Cd8 (naive-like CD8), and Cd4 and Cd8 (naive-like double-positive) or did not express Cd4 and Cd8 (naive-like DN). scRNA-Seq data further showed increased Tigit expression in WT T cells after IR injury (Supplemental Figure 7B) which was mostly restricted to Th17 cells (Figure 6E) compared with baseline, where most T cell subsets expressed Tigit, although to a lower extent (Supplemental Figure 7C). Further analysis of Th17 cluster identified several differentially expressed genes between WT and TIGIT-KO cells at baseline and after IR injury (Figure 6, F and G). Gene set enrichment analysis of differentially expressed genes between WT and TIGIT-KO mice identified a predominance of proinflammatory genes related to allograft rejection and TNFα signaling in Th17 cells (Figure 7A) from WT mice at baseline. Th17 cells from TIGIT-KO mice showed enrichment of oxidative phosphorylation (OXPHOS), mTORC1, DNA repair, and apoptosis-related genes at baseline (Figure 7B). Th17 cells from post-IR WT kidneys showed enrichment of genes important for mitotic spindle assembly and TNFα signaling response (Figure 7C), whereas Th17 cells from TIGIT-KO kidneys had an enrichment of metabolic genes related to Myc targets V1, OXPHOS, and mTORC1 as well as IFNα and IFNγ responses (Figure 7D).
Figure 6.

scRNA-Seq analysis of flow-sorted kidney CD45+ cells showing distinct inflammatory and metabolic differences between WT and TIGIT-KO kidney T cells at baseline and 24 hours post-IR injury. (A) Dot plot showing expression of marker genes used for annotating different immune cell clusters. (B) UMAP showing major immune cell populations identified in WT control kidneys. (C) UMAP showing Tigit expression primarily restricted to T/NK cell cluster among all kidney immune cells in WT control mice. (D) UMAP showing annotated T cell populations in WT and TIGIT-KO mice. (E) Violin plot representing Tigit expression in different T cell subsets with Th17 cells as major expressers, 24 hours post-IR in WT mice. (F and G) Volcano plots showing top differentially expressed genes (DEGs) in Th17 cells at baseline and 24 hours after IR injury between WT and TIGIT-KO mice. TIGIT-KO, TIGIT knockout.
Figure 7.
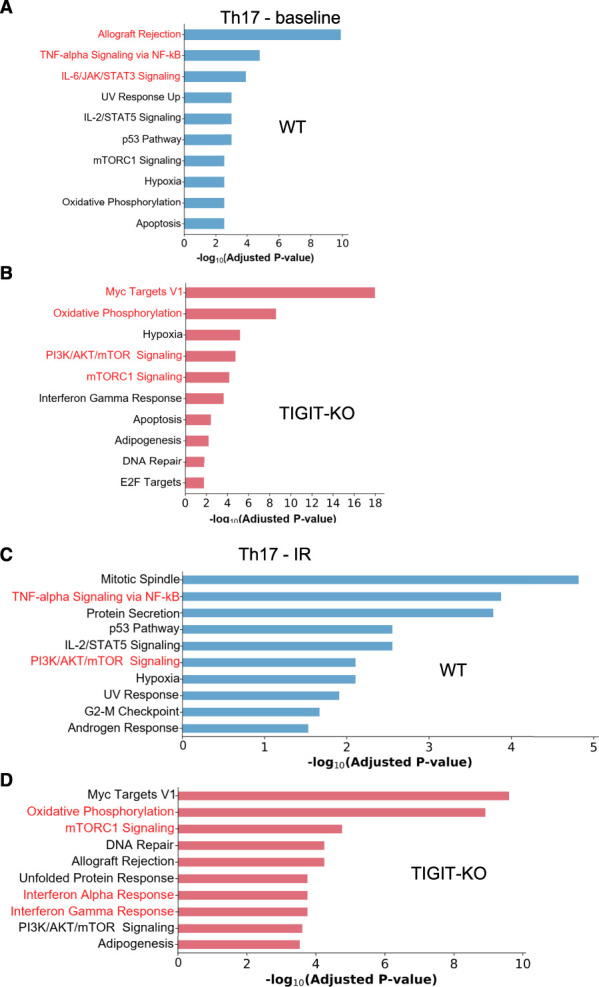
Gene set enrichment analysis (GSEA) of differentially expressed genes (DEGs) in Th17 cells from WT and TIGIT-KO kidneys at baseline and 24 hours post-IR injury. (A and B) GSEA plots representing cellular responses/pathways in WT (blue) and TIGIT-KO (red) Th17 cells at baseline. (C and D) GSEA plots of WT (blue) and TIGIT-KO (red) Th17 cells 24 hours after IR injury. mTOR, mammalian target of rapamycin.
In-vivo metabolite quantification in kidney T cells is challenging because of slow metabolite quenching and long T cell isolation protocol. We indirectly assessed metabolic changes between WT and TIGIT-KO kidney T cells at baseline and 24 hours post-IR by flow cytometry. We quantified GLUT1 and hexokinase II (HKII) expression to assess glycolysis, carnitine palmitoyltransferase 1a (CPT1a) to assess fatty acid oxidation, S6 ribosomal protein phosphorylation (pS6) for mammalian target of rapamycin (mTOR) signaling activity, voltage-dependent anion channel 1 and Tomm20 for mitochondrial OXPHOS activity, and H3K27Me3 for histone methylation. Although we observed no differences in these metabolic markers at baseline, post-IR CD4+ T cells from TIGIT-KO kidneys had increased GLUT1 and H3K27Me3 expression compared with WT (Figure 8, A and B).
Figure 8.
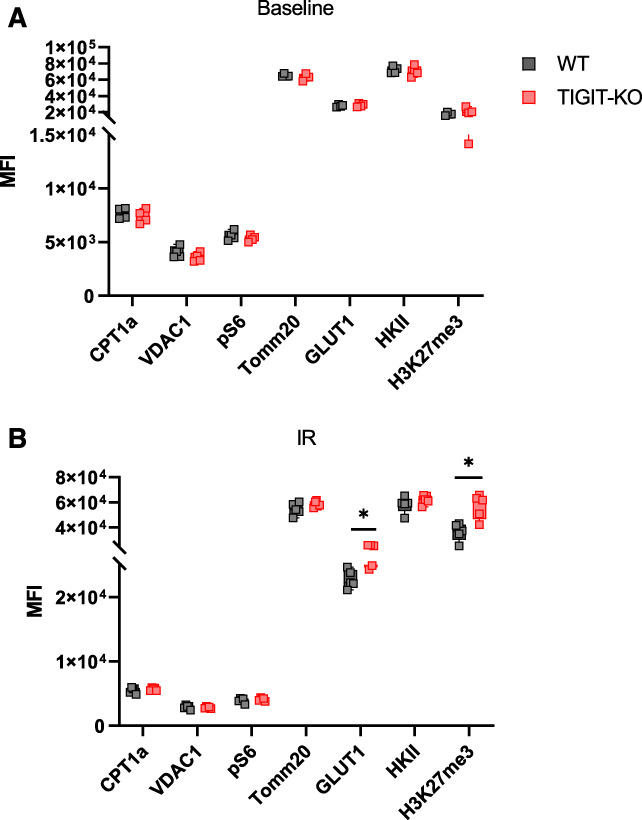
Metabolic assessment of kidney T cells in WT and TIGIT-KO mice at baseline and 24 hours post-IR injury. (A) Graph showing MFI values of the metabolism-related proteins CPT1a, VDAC1, pS6, TOMM20, GLUT1, HKII, and H3K27me3 in CD4+ T cells from WT and TIGIT-KO kidneys at baseline and (B) 24 hours after IR injury. Data between groups were analyzed using the unpaired nonparametric Mann-Whitney test. Graphs represent median (IQR) values. *P≤0.05. TIGIT-KO, TIGIT knockout; VDAC1, voltage-dependent anion channel 1.
Discussion
This study demonstrated that TIGIT expression increases in kidney T cells during murine and human AKI. In addition, these data demonstrated a pathogenic role for TIGIT in both experimental ischemic and nephrotoxic AKI. Several observations in this study could collectively explain the pathogenic mechanism of TIGIT during AKI. TIGIT+ CD4+ cells produced significantly higher proinflammatory cytokines than TIGIT− CD4+ cells in WT mice, despite a decreasing trend in both subsets after IR injury. It is important to note that our CD4+ T cell cytokine analysis included Tregs, which may have affected cytokine levels. TIGIT+ T cells also had a predominantly EM/CM phenotype. In this context, TIGIT-expressing donor-reactive CD4+ T cells had a highly polyfunctional cytokine profile and EM/CM phenotype in kidney transplant recipients.34 Importantly, scRNA-Seq analysis of WT kidney T cells demonstrated significant Tigit expression by Th17 cells, which have been implicated in promoting AKI.35 In addition, reduced CD4+ T cells and increased Tregs frequencies in TIGIT-KO kidneys could partly explain the protection seen in these mice, given a pathogenic role for CD4+ T cells36 and a reparative role for Tregs in AKI.37 scRNA-Seq data also showed enrichment of OXPHOS-, mTORC1-, and Myc-related genes in Th17 cells from TIGIT-KO mice. These metabolic genes play protective roles in ischemic injury by augmenting mitochondrial biogenesis38 and enhancing T cell activation and survival.39 This study's identification of metabolic gene signatures and increased GLUT1 and H3K27Me3 expression in TIGIT-KO kidney T cells provides new leads to understand TIGIT's role in T cell functions. Future studies should quantify actual metabolites and metabolic pathways affected by TIGIT to explore their mechanistic role in AKI.
Consistent with previous studies in other organs, kidney Tregs predominantly belong to the TIGIT+ T cell compartment.6,40,41 However, our observation that TIGIT+ kidney Tregs are significantly reduced after IR suggests that these cells are modified during ischemic AKI. TIGIT expression in prior studies was associated with both an increased immunosuppressive potential of Tregs and an activated and proinflammatory transcriptional profile of TIGIT-expressing Tregs.6,42
Our data demonstrating a pathogenic role for TIGIT in experimental AKI is supported by other studies that found a correlation between TIGIT expression and non-AKI disease severity. Elevated TIGIT on T cells positively correlated with markers of autoimmunity and inflammation in patients with rheumatoid arthritis and systemic lupus erythematosus disease activity index.43,44 Conversely, reduction in TIGIT-expressing polyfunctional CD4+ T cells after kidney transplant enhanced tolerance and improved graft survival.34 Our observation showing increased TIGIT expression after ischemic injury further implicates TIGIT in human AKI pathogenesis.
Our results demonstrating pathogenic TIGIT effects in AKI diverge from those demonstrating immunosuppressive TIGIT effects. This discrepancy could be due to differences in activating stimuli and the duration of T cell activation in different disease models. Furthermore, proportions of EM cells in TIGIT-KO and WT kidneys were different compared with TIGIT+ and TIGIT− T cell subsets in WT mice. Use of different gating strategies could account for this observation. Furthermore, constitutive TIGIT deletion in TIGIT-KO mice could modulate functions of CD226 and CD96 by promoting CD112- and CD113-mediated signaling. Moreover, TIGIT expression in NK cells could be an important mechanism in AKI in addition to TIGIT-independent effects of NK T cells, macrophages, dendritic cells, and neutrophils.
In conclusion, TIGIT expression increases in kidney T cells during AKI and mediates experimental ischemic and nephrotoxic AKI. TIGIT modulates kidney T cell composition, cytokine production, memory phenotype, and inflammatory and metabolic genes. Given the recent interest in TIGIT modulation for cancer and known kidney adverse effects of traditional immune checkpoint therapy, these results have relevance to both patients with AKI and patients with cancer.
Supplementary Material
ACKNOWLEDGMENTS
The authors are thankful to human kidney donors for providing valuable kidney samples for this work. The authors would also like to acknowledge the JHU Ross Flow Cytometry Core facility (S10OD026859) and The Single Cell & Transcriptomics Core for their help in this study. Kyungho Lee was supported by grants from Korea Health Industry Development Institute (HI19C1337), National Research Foundation of Korea (NRF-2021R1A6A3A03039863), and National Kidney Foundation Severing Maryland and Delaware. Johanna T Kurzhagen was supported by the Dr. Werner Jackstädt-Foundation award (S 134–10.117). Patrick Cahan was supported by grants from NIGMS and NIDDK (R35GM124725 and R01DK104662).
Footnotes
See related editorial, “The Importance of Immune Checkpoint Molecule TIGIT in AKI,” on pages 725–727.
Disclosures
L. Arend reports Advisory or Leadership Role: Vice President of Renal Pathology Society (no compensation). V. Kuchroo reports Consultancy: Alpine Immune Science, Biolegend, Celsius, Elpiscience, Equillium, GSK, ImmuneOncia, Incyte Corp, iTeos, Janssen, Kintai/SENDA (as of October 28, 2020), Larkspur, Novartis EHA-SWG Immunotherapy symposium, Novartis Sabatolimab, Pfizer, Reistone Biopharma, Rubius, Sanofi/Genzyme, Surface Oncology, Tizona, Vividion, Werewolf Tx; Ownership Interest: Bicara, Biocon Biologic Ltd, Biolegend, Celsius, Compass, Elpiscience, Equillium, Larskpur, Syngene Intl., Tizona, Trishula; Research Funding: Novartis; Honoraria: Reistone Biopharma (Recorded talk), Sanofi (Speaker Agreement), Surface Oncology (Speaker Agreement); Patents or Royalties: Becton Dickinson, Biolegend, Novartis; Advisory or Leadership Role: Board of Directors: Bicara, Biocon Pharmaceuticals, Syngene; Scientific Advisory Boards: Elpiscience, GSK, Kintai/SENDA (as of October 28, 2020), Larkspur, Novartis Sabatolmab, Reistone Biopharma, Repertoir, Rubius, Tizona, Vividion, Werewolf Tx; and Speakers Bureau: Reistone Biopharma (Recorded talk), Sanofi (Speaker Agreement), and Surface Oncology (Speaker Agreement). J. Kurzhagen reports Honoraria: Dr. Werner Jackstädt Foundation. P. Pierorazio reports Patents or Royalties: UpToDate: Chapter on Small Renal Masses. H. Rabb reports scientific advisory boards of Rapafusyn and Renibus Therapeutics. All remaining authors have nothing to disclose.
Funding
S. Noel was supported by Carl W. Gottschalk Research Scholar Grant from American Society of Nephrology (134535) and the Edward S. Kraus award from the Johns Hopkins School of Medicine, Division of Nephrology. H. Rabb and S. Noel were supported by grants from the NIDDK (R01DK123342 and R01DK104662) and philanthropic gifts to H. Rabb
Author Contributions
H. Rabb and S. Noel conceptualized the study and were responsible for funding, acquisition, investigation, and project administration; P. Cahan and S. Noel were responsible for data curation; L. Arend, P. Cahan, H. Rabb, and S. Noel were responsible for formal analysis; L. Arend, P. Cahan, S. Gharaie, J. Kurzhagen, K. Lee, S. Noel, and P. Pierorazio were responsible for methodology; V.K. Kuchroo, S. Noel, P. Pierorazio, and H. Rabb were responsible for resources; P. Cahan was responsible for software; S. Noel provided supervision; L. Arend and H. Rabb were responsible for visualization; H. Rabb and S. Noel wrote the original draft; and L. Arend, P. Cahan, S. Gharaie, V.K. Kuchroo, J. Kurzhagen, K. Lee, S. Noel, P. Pierorazio, and H. Rabb reviewed and edited the manuscript.
Data Sharing Statement
Bulk RNA-Seq data presented in this manuscript have been submitted to the National Center for Biotechnology Information Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139171) under accession number GSE139171. scRNA-Seq data from this study has been deposited at Zenodo, a data repository of CERN, and can be accessed at https://doi.org/10.5281/zenodo.7314510. scRNA-Seq data from Kidney precision medicine project is available at https://atlas.kpmp.org/explorer/dataviz.
Supplemental Material
This article contains the following supplemental material online at http://links.lww.com/JSN/D625
Supplemental Figure 1. Genotyping of TIGIT-KO mice.
Supplemental Figure 2. Gating strategy to assess TIGIT expression in Kidney and spleen.
Supplemental Figure 3. Intracellular cytokine analysis of TIGIT+ and TIGIT− subsets of CD8 and DN T cells.
Supplemental Figure 4. Analysis of memory phenotype in TIGIT+ and TIGIT− subsets of CD4+, CD8+, and DN kidney T cells.
Supplemental Figure 5. TIGIT assessment in splenic Tregs at baseline and post-IR.
Supplemental Figure 6. Intracellular cytokine analysis of CD4 and CD8 T cells from WT and TIGIT-KO kidneys at baseline.
Supplemental Figure 7. scRNA-Seq analysis of flow sorted kidney CD45+ cells from WT and TIGIT-KO kidney at baseline and 24 hours post-IR injury.
Supplemental Table 1. Primer sequences for CMV Cre and internal positive control used for genotyping TIGIT-KO mice.
Supplemental Table 2. PCR conditions for CMV Cre detection.
Supplemental Table 3. List of differentially expressed genes identified in bulk RNA-Seq analysis of flow-sorted CD4+ T cells from control and 24 hours post-IR kidneys of WT mice (Excel File).
Supplemental Table 4. KPMP scRNA-Seq data output result showing TIGIT expression by human kidney T cell subsets.
References
- 1.Hoste EAJ, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–1423. doi: 10.1007/s00134-015-3934-7 [DOI] [PubMed] [Google Scholar]
- 2.Kurzhagen JT, Dellepiane S, Cantaluppi V, Rabb H. AKI: an increasingly recognized risk factor for CKD development and progression. J Nephrol. 2020;33(6):1171–1187. doi: 10.1007/s40620-020-00793-2 [DOI] [PubMed] [Google Scholar]
- 3.Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ. Acute kidney injury. Nat Rev Dis Primers. 2021;7(1):52. doi: 10.1038/s41572-021-00284-z [DOI] [PubMed] [Google Scholar]
- 4.Joller N, Hafler JP, Brynedal B, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. 2011;186(3):1338–1342. doi: 10.4049/jimmunol.1003081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu X, Harden K, Gonzalez LC, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48–57. doi: 10.1038/ni.1674 [DOI] [PubMed] [Google Scholar]
- 6.Joller N, Lozano E, Burkett PR, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569–581. doi: 10.1016/j.immuni.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraus AK, Chen J, Edenhofer I, et al. The role of T cell costimulation via DNAM-1 in kidney transplantation. PLoS One. 2016;11(2):e0147951. doi: 10.1371/journal.pone.0147951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung AL, Maxwell R, Theodros D, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7(8):e1466769. doi: 10.1080/2162402x.2018.1466769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raphael I, Kumar R, McCarl LH, et al. TIGIT and PD-1 immune checkpoint pathways are associated with patient outcome and anti-tumor immunity in glioblastoma. Front Immunol. 2021;12:637146. doi: 10.3389/fimmu.2021.637146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertrand A, Kostine M, Barnetche T, Truchetet ME, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 2015;13(1):211. doi: 10.1186/s12916-015-0455-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seethapathy H, Zhao S, Chute DF, et al. The incidence, causes, and risk factors of acute kidney injury in patients receiving immune checkpoint inhibitors. Clin J Am Soc Nephrol. 2019;14(12):1692–1700. doi: 10.2215/CJN.00990119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Percie du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. doi: 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabb H, Ramirez G, Saba SR, et al. Renal ischemic-reperfusion injury in L-selectin-deficient mice. Am J Physiol. 1996;271(2):F408–F413. doi: 10.1152/ajprenal.1996.271.2.f408 [DOI] [PubMed] [Google Scholar]
- 14.Liu M, Grigoryev DN, Crow MT, et al. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009;76(3):277–285. doi: 10.1038/ki.2009.157 [DOI] [PubMed] [Google Scholar]
- 15.Ascon DB, Lopez-Briones S, Liu M, et al. Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury. J Immunol. 2006;177(5):3380–3387. doi: 10.4049/jimmunol.177.5.3380 [DOI] [PubMed] [Google Scholar]
- 16.Lee SA, Noel S, Kurzhagen JT, et al. CD4(+) T cell-derived NGAL modifies the outcome of ischemic acute kidney injury. J Immunol. 2020;204(3):586–595. doi: 10.4049/jimmunol.1900677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):15. doi: 10.1186/s13059-017-1382-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan Y, Cahan P. SingleCellNet: a computational tool to classify single cell RNA-Seq data across platforms and across species. Cell Syst. 2019;9(2):207.e2–213.e2. doi: 10.1016/j.cels.2019.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14(1):128. doi: 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database Hallmark gene set collection. Cell Syst. 2015;1(6):417–425. doi: 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martina MN, Noel S, Saxena A, et al. Double-Negative αβ T cells are early responders to AKI and are found in human kidney. J Am Soc Nephrol. 2016;27(4):1113–1123. doi: 10.1681/ASN.2014121214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noel S, Martina MN, Bandapalle S, et al. T lymphocyte-specific activation of Nrf2 protects from AKI. J Am Soc Nephrol. 2015;26(12):2989–3000. doi: 10.1681/ASN.2014100978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Zotto G, Principi E, Antonini F, et al. Comprehensive phenotyping of peripheral blood T lymphocytes in healthy mice. Cytometry A. 2021;99(3):243–250. doi: 10.1002/cyto.a.24246 [DOI] [PubMed] [Google Scholar]
- 25.Ascon DB, Ascon M, Satpute S, et al. Normal mouse kidneys contain activated and CD3+CD4- CD8- double-negative T lymphocytes with a distinct TCR repertoire. J Leukoc Biol. 2008;84(6):1400–1409. doi: 10.1189/jlb.0907651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaworska K, Ratajczak J, Huang L, et al. Both PD-1 ligands protect the kidney from ischemia reperfusion injury. J Immunol. 2015;194(1):325–333. doi: 10.4049/jimmunol.1400497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon S, Voillet V, Vignard V, et al. PD-1 and TIGIT coexpression identifies a circulating CD8 T cell subset predictive of response to anti-PD-1 therapy. J Immunother Cancer. 2020;8(2):e001631. doi: 10.1136/jitc-2020-001631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauvin JM, Pagliano O, Fourcade J, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. 2015;125(5):2046–2058. doi: 10.1172/jci80445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreatta M, Corria-Osorio J, Muller S, Cubas R, Coukos G, Carmona SJ. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat Commun. 2021;12(1):2965. doi: 10.1038/s41467-021-23324-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tibbitt CA, Stark JM, Martens L, et al. Single-cell RNA sequencing of the T helper cell response to house dust mites defines a distinct gene expression signature in airway Th2 cells. Immunity. 2019;51(1):169.e5–184.e5. doi: 10.1016/j.immuni.2019.05.014 [DOI] [PubMed] [Google Scholar]
- 31.Ding J, Smith SL, Orozco G, Barton A, Eyre S, Martin P. Characterisation of CD4+ T cell subtypes using single cell RNA sequencing and the impact of cell number and sequencing depth. Sci Rep. 2020;10(1):19825. doi: 10.1038/s41598-020-76972-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Zhu Y, Tian D, et al. Transcriptome landscape of double negative T cells by single-cell RNA sequencing. J Autoimmun. 2021;121:102653. doi: 10.1016/j.jaut.2021.102653 [DOI] [PubMed] [Google Scholar]
- 33.Szabo PA, Levitin HM, Miron M, et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat Commun. 2019;10(1):4706. doi: 10.1038/s41467-019-12464-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der List ACJ, Litjens NHR, Klepper M, Betjes MGH. Expression of senescence marker TIGIT identifies polyfunctional donor-reactive CD4+ T cells preferentially lost after kidney transplantation. Front Immunol. 2021;12:656846. doi: 10.3389/fimmu.2021.656846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basile DP, Ullah MM, Collet JA, Mehrotra P. T helper 17 cells in the pathophysiology of acute and chronic kidney disease. Kidney Res Clin Pract. 2021;40(1):12–28. doi: 10.23876/j.krcp.20.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burne MJ, Daniels F, El Ghandour A, et al. Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure. J Clin Invest. 2001;108(9):1283–1290. doi: 10.1172/jci200112080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gandolfo MT, Jang HR, Bagnasco SM, et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76(7):717–729. doi: 10.1038/ki.2009.259 [DOI] [PubMed] [Google Scholar]
- 38.de la Cruz Lopez KG, Toledo Guzman ME, Sanchez EO, Garcia Carranca A. mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Front Oncol. 2019;9:1373. doi: 10.3389/fonc.2019.01373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobs SR, Herman CE, Maciver NJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476–4486. doi: 10.4049/jimmunol.180.7.4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuhrman CA, Yeh WI, Seay HR, et al. Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol. 2015;195(1):145–155. doi: 10.4049/jimmunol.1402381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurtulus S, Sakuishi K, Ngiow SF, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125(11):4053–4062. doi: 10.1172/jci81187 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.de Lima MHF, Hiroki CH, de Fatima Borges V, et al. Sepsis-induced immunosuppression is marked by an expansion of a highly suppressive repertoire of FOXP3 +T regulatory cells-expressing TIGIT. J Infect Dis. 2022;225(3):531–541. doi: 10.1093/infdis/jiab405 [DOI] [PubMed] [Google Scholar]
- 43.Luo Q, Deng Z, Xu C, et al. Elevated expression of immunoreceptor tyrosine-based inhibitory motif (TIGIT) on T lymphocytes is correlated with disease activity in rheumatoid arthritis. Med Sci Monit. 2017;23:1232–1241. doi: 10.12659/msm.902454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mao L, Hou H, Wu S, et al. TIGIT signalling pathway negatively regulates CD4(+) T cell responses in systemic lupus erythematosus. Immunology. 2017;151(3):280–290. doi: 10.1111/imm.12715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Bulk RNA-Seq data presented in this manuscript have been submitted to the National Center for Biotechnology Information Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE139171) under accession number GSE139171. scRNA-Seq data from this study has been deposited at Zenodo, a data repository of CERN, and can be accessed at https://doi.org/10.5281/zenodo.7314510. scRNA-Seq data from Kidney precision medicine project is available at https://atlas.kpmp.org/explorer/dataviz.