

Abstract
Background:
It is unclear whether continuing anti-fibrotic therapy until the time of lung transplant increases the risk of complications in patients with idiopathic pulmonary fibrosis.
Objectives:
To investigate whether the time between discontinuation of anti-fibrotic therapy and lung transplant in patients with idiopathic pulmonary fibrosis affects the risk of complications.
Methods:
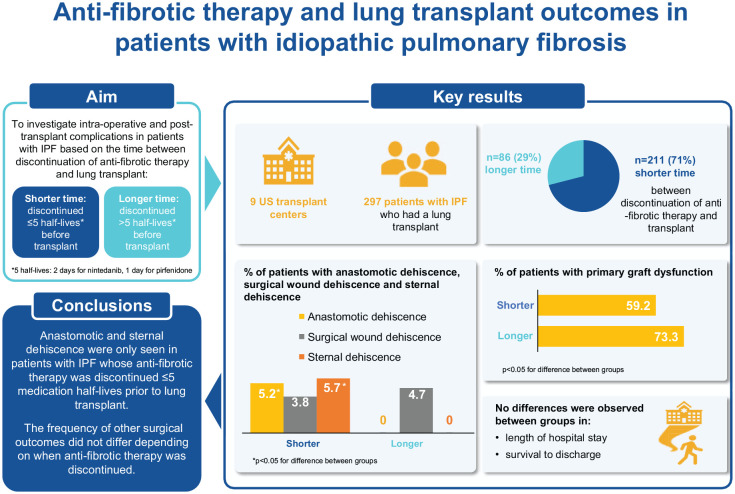
We assessed intra-operative and post-transplant complications among patients with idiopathic pulmonary fibrosis who underwent lung transplant and had been treated with nintedanib or pirfenidone continuously for ⩾ 90 days at listing. Patients were grouped according to whether they had a shorter (⩽ 5 medication half-lives) or longer (> 5 medication half-lives) time between discontinuation of anti-fibrotic medication and transplant. Five half-lives corresponded to 2 days for nintedanib and 1 day for pirfenidone.
Results:
Among patients taking nintedanib (n = 107) or pirfenidone (n = 190), 211 (71.0%) had discontinued anti-fibrotic therapy ⩽ 5 medication half-lives before transplant. Anastomotic and sternal dehiscence occurred only in this group (anastomotic: 11 patients [5.2%], p = 0.031 vs patients with longer time between discontinuation of anti-fibrotic medication and transplant; sternal: 12 patients [5.7%], p = 0.024). No differences were observed in surgical wound dehiscence, length of hospital stay, or survival to discharge between groups with a shorter versus longer time between discontinuation of anti-fibrotic therapy and transplant.
Conclusion:
Anastomotic and sternal dehiscence only occurred in patients with idiopathic pulmonary fibrosis who discontinued anti-fibrotic therapy < 5 medication half-lives before transplant. The frequency of other intra-operative and post-transplant complications did not appear to differ depending on when anti-fibrotic therapy was discontinued.
Registration:
clinicaltrials.gov NCT04316780: https://clinicaltrials.gov/ct2/show/NCT04316780
Keywords: interstitial lung disease, pulmonary fibrosis, surgery, tyrosine kinase, wound healing
Graphical abstract
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease (ILD) characterized by decline in lung function and high mortality. 1 Nintedanib and pirfenidone are anti-fibrotic medications approved for the treatment for IPF, which slow the rate of decline in forced vital capacity (FVC).2,3 Real-world data suggest that these medications improve life expectancy in patients with IPF,4–8 but the mortality associated with the disease remains high. Lung transplantation prolongs survival in select patients with IPF and should be discussed with appropriate patients.9,10
Nintedanib is an intracellular inhibitor of tyrosine kinases, including the platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF) receptors, and inhibits the migration and proliferation of fibroblasts, their differentiation into myofibroblasts, and the deposition of extracellular matrix components.11–13 The mechanism of action of pirfenidone has not been established, but it has been shown to inhibit processes involved in fibrosis, including fibroblast activation. 14 Based on these effects, concern has been raised that anti-fibrotic medications may increase the risk of complications such as intra-operative bleeding or delayed wound healing in patients undergoing lung transplant. However, no guidelines have been issued regarding when anti-fibrotic therapy should be discontinued prior to lung transplant. In this study, we investigated intra-operative and post-transplant complications in patients with IPF based on the time between discontinuation of anti-fibrotic therapy and lung transplant.
Methods
This was a retrospective study (clinicaltrials.gov NCT04316780) conducted at nine US transplant centers. The study protocol was approved by an independent ethics committee or institutional review board at each participating center. The study included all patients with IPF listed for lung transplant between 1 July 2015 and 30 June 2019 who had been treated with nintedanib or pirfenidone continuously for ⩾ 90 days at the time of listing for transplant. Patients who underwent additional interventions (e.g. coronary artery bypass grafting, valve replacement) at the time of their lung transplant were excluded.
Data were collected from medical records to assess complications during transplant and in the 6 months after transplant in three groups of patients based on the time between discontinuation of anti-fibrotic medication and transplant: (1) shortest: patients discontinued anti-fibrotic medication ⩽ 5 medication half-lives before transplant; (2) intermediate; patients discontinued anti-fibrotic medication between 5 medication half-lives and 28 days before transplant; and (3) longest: patients discontinued anti-fibrotic medication > 28 days before transplant (Supplementary Figure S1). For nintedanib, 5 half-lives correspond to 2 days 15 and for pirfenidone, 5 half-lives correspond to 1 day. 16 For the analyses of intra-operative and post-transplant complications, the intermediate and longest groups were combined such that patients were grouped according to whether they had a shorter (⩽ 5 medication half-lives) or longer (> 5 medication half-lives) time between discontinuation of anti-fibrotic medication and transplant (Supplementary Figure S1).
In each group, we investigated the percentages of patients who received intra-operative and post-operative extracorporeal membrane oxygenation (ECMO) or cardiopulmonary bypass and packed red blood cells (PRBCs); the units of PRBCs received; the percentages of patients with primary graft dysfunction, anastomotic dehiscence, surgical wound dehiscence and sternal dehiscence (irrespective of whether intervention was needed); the percentage who returned to the operating room; the percentage who died in hospital; time between transplant and extubation and between transplant and final chest tube removal; length of stay in intensive care unit (ICU); and length of hospital stay. We also investigated the percentages of patients alive 30 days, 90 days, and 6 months after transplant; with acute cellular rejection during 6 months after transplant; and with bronchiolitis obliterans syndrome or restrictive allograft syndrome during 6 months after transplant. The investigators were not provided with specific definitions for all the complications to be reported, but primary graft dysfunction was defined according to ISHLT consensus guidelines 17 and acute cellular rejection was graded according to histologic grading guidelines. 18 A web-based Research Electronic Data Capture (REDCap) database 19 was used for data entry and storage.
Analyses were intended to be descriptive and the study was not powered for a specific outcome; a sample size of 320 patients was estimated to be feasible and to allow for sufficient representation across the groups. Characteristics and outcomes of the two groups were compared using chi-square tests for categorical variables, and t-tests or Wilcoxon rank sum tests for continuous variables, depending on skewness. All tests were two-sided with alpha = 0.05. Data were analyzed at Tufts Medical Center, Boston, MA, using Stata v16 (StataCorp LLP, College Station, TX).
Results
Patients
A total of 320 patients with IPF taking nintedanib or pirfenidone were listed for transplant (Supplementary Table S1). Nineteen patients died awaiting transplant (Supplementary Table S2). At the time of the analysis, 4 patients were awaiting transplant. Thus, our analyses were based on 297 patients who underwent transplant, of whom 107 were taking nintedanib and 190 were taking pirfenidone at the time of listing. Among these patients, 211 (71.0%) had a shorter time between discontinuation of anti-fibrotic medication and transplant (⩽ 5 medication half-lives) and 86 (29.0%) had a longer time between discontinuation of anti-fibrotic medication and transplant (> 5 medication half-lives).
Patient characteristics at the time of transplant in groups by shorter and longer time between discontinuation of anti-fibrotic medication and transplant are shown in Table 1. The groups did not differ except that at transplant, the group with the shorter time between discontinuation of anti-fibrotic medication and transplant had a lower lung allocation score (LAS). 20 Patient characteristics in groups by type of anti-fibrotic medication and by shortest, intermediate and longest time between discontinuation of anti-fibrotic medication and transplant are shown in Supplementary Table S3. Right heart catheterization results before transplant in groups by shorter and longer time between discontinuation of anti-fibrotic medication and transplant are shown in Supplementary Table S4.
Table 1.
Patient characteristics at transplant by time between discontinuation of anti-fibrotic medication and transplant (N = 297).
| Characteristic | Time between discontinuation of anti-fibrotic medication and transplant | p-value | |
|---|---|---|---|
| Shorter (n = 211) | Longer (n = 86) | ||
| Age (years), mean (SD) | 65.9 (6.8) | 67.5 (5.4) | 0.052 |
| Time on transplant list (days) | 37.0 (10.0, 89.0) | 23.0 (11.0, 61.0) | 0.19 |
| Lung allocation score, mean (SD) | 44.9 (15.2) | 49.0 (14.8) | 0.033 |
| Taking prednisone a | 71 (33.8) | 21 (24.4) | 0.11 |
| Prednisone dose | 10.0 (10.0, 20.0) | 15.0 (7.5, 20.0) | 0.83 |
| Bilateral transplant | 126 (59.7) | 61 (70.9) | 0.069 |
| ECMO prior to transplant | 3 (1.4) | 1 (1.2) | 0.86 |
ECMO, extracorporeal membrane oxygenation; SD, standard deviation.
Data are n (%) or median (Q1, Q3) unless otherwise indicated. Shorter: discontinued anti-fibrotic medication ⩽ 5 medication half-lives before transplant; longer: discontinued anti-fibrotic medication > 5 medication half-lives before transplant.
Information not available for one patient.
Surgical outcomes
No differences were noted across the groups by time between discontinuation of anti-fibrotic medication and transplant in the percentages of patients who received intra-operative ECMO or cardiopulmonary bypass or post-operative ECMO (Table 2). The percentages of patients who received intra- or post-operative red blood cell transfusions were higher in the group with the longer time between discontinuation of anti-fibrotic medication and transplant (Table 2). No differences were observed between patients taking nintedanib and pirfenidone at the time of listing in the percentage who received intra-operative red blood cell transfusions (50.5% and 47.9%, respectively; p = 0.67) or post-operative red blood cell transfusions (36.4% and 35.3%, respectively; p = 0.84).
Table 2.
Surgical outcomes by time between discontinuation of anti-fibrotic medication and transplant (N = 297).
| Outcome | Time between discontinuation of anti-fibrotic medication and transplant | p-value | |
|---|---|---|---|
| Shorter (n = 211) | Longer (n = 86) | ||
| Received intra-operative ECMO/CPB | 100 (47.4) | 45 (52.3) | 0.44 |
| Received post-operative ECMO | 21 (10.0) | 13 (15.1) | 0.20 |
| Received intra-operative PRBCs | 94 (44.5) | 51 (59.3) | 0.021 |
| Units of intra-operative PRBCs a | 2.0 (2.0, 4.0) | 2.0 (1.0, 4.0) | 0.23 |
| Received post-operative PRBCs | 59 (28.0) | 47 (54.7) | <0.001 |
| Units of post-operative PRBCs b | 2.0 (1.0, 4.0) | 2.0 (1.0, 3.0) | 0.24 |
| Primary graft dysfunction | 125 (59.2) | 63 (73.3) | 0.023 |
| Anastomotic dehiscence | 11 (5.2) | 0 (0.0) | 0.031 |
| Surgical wound dehiscence | 8 (3.8) | 4 (4.7) | 0.73 |
| Sternal dehiscence | 12 (5.7) | 0 (0.0) | 0.024 |
| Transplant to extubation (days) | 1.0 (1.0, 2.0) | 1.0 (1.0, 3.0) | 0.65 |
| Time from transplant to final chest tube removal | 11.0 (8.0, 16.0) | 12.0 (9.0, 18.0) | 0.066 |
| Returned to operating room | 55 (26.1) | 25 (29.1) | 0.60 |
| Length of stay in ICU (days) c | 5.0 (3.0, 10.0) | 5.0 (3.0, 11.0) | 0.69 |
| Length of hospital stay (days) c | 16.0 (11.5, 31.0) | 18.0 (13.0, 32.0) | 0.12 |
| Died in hospital | 11 (5.2) | 3 (3.5) | 0.52 |
CPB, cardiopulmonary bypass; ECMO, extracorporeal membrane oxygenation; ICU, intensive care unit; PRBCs, packed red blood cells.
Data are n (%) or median (Q1, Q3). Shorter: discontinued anti-fibrotic medication ⩽ 5 medication half-lives before transplant; longer: discontinued anti-fibrotic medication > 5 medication half-lives before transplant.
Among patients who received intra-operative PRBCs.
Among patients who received post-operative PRBCs.
Among 283 patients who survived to discharge.
The proportion of patients with any grade primary graft dysfunction at any time-point was lower in the group with the shorter time between discontinuation of anti-fibrotic medication and transplant (59.2% vs 73.3%; p = 0.023) (Table 2). Anastomotic dehiscence was reported only in patients with the shorter time between discontinuation of anti-fibrotic medication and transplant (11 patients [5.2%]; p = 0.031 vs patients with longer time between discontinuation of anti-fibrotic medication and transplant) (Table 2; Figure 1). Surgical wound dehiscence was reported at a similar rate across the two groups (Table 2; Figure 1). Sternal dehiscence was reported only in patients with the shorter time between discontinuation of anti-fibrotic medication and transplant (12 patients [5.7%]; p = 0.024 vs patients with longer time between discontinuation of anti-fibrotic medication and transplant) (Table 2; Figure 1). Of the nine participating centers, five primarily continued anti-fibrotic medication until the time of transplant. The anastomotic dehiscence events occurred at three of these five centers, while the sternal dehiscence events occurred at two of these five centers.
Figure 1.

Percentages of patients with anastomotic dehiscence, surgical wound dehiscence and sternal dehiscence by time between discontinuation of anti-fibrotic medication and transplant.
Shorter: discontinued anti-fibrotic medication ⩽ 5 medication half-lives before transplant; longer: discontinued anti-fibrotic medication > 5 medication half-lives before transplant.
Other surgical outcomes did not differ between the groups, including time to extubation, time to final chest tube removal, and need for post-operative return to the operating room (Table 2). There were no significant differences between the groups in risk of in-hospital death (Table 2). Among the 283 patients discharged, the median length of stay in the ICU and the median length of hospital stay did not differ between the groups (Table 2). Surgical outcomes in groups by type of anti-fibrotic medication and by shortest, intermediate and longest time between discontinuation of anti-fibrotic medication and transplant are shown in Supplementary Table S5.
Post-discharge outcomes
Among 297 patients who underwent transplant, 283 were alive 30 days after transplant, 278 were alive 90 days after transplant, and 270 were alive 6 months after transplant. The proportions of patients who were alive at these timepoints did not differ between the groups (Table 3), but a higher proportion of patients were discharged home in the group with the longer time between discontinuation of anti-fibrotic medication and transplant (Table 3). The proportion of patients with acute cellular rejection in the 6 months after transplant did not differ between the groups (Table 3). One patient (in the group with the shorter time between discontinuation of anti-fibrotic medication and transplant) had chronic lung allograft dysfunction (CLAD) in the 6 months after transplant. No patients resumed anti-fibrotic medication after discharge. Post-discharge outcomes by type of anti-fibrotic medication and by shortest, intermediate and longest time between discontinuation of anti-fibrotic medication and transplant are shown in Supplementary Table S6.
Table 3.
Discharge and post-discharge outcomes by time between discontinuation of anti-fibrotic medication and transplant (N = 297).
| Time between discontinuation of anti-fibrotic medication and transplant | p-value | ||
|---|---|---|---|
| Outcome | Shorter (n = 211) | Longer (n = 86) | |
| Discharged home a | 161 (76.3) | 75 (87.2) | 0.035 |
| Alive 30 days after transplant | 201 (95.7) b | 82 (95.3) | 0.89 |
| Alive 90 days after transplant | 196 (93.8) c | 82 (95.3) | 0.60 |
| Alive 6 months after transplant | 190 (90.9) c | 80 (93.0) | 0.55 |
| Acute rejection during 6 months after transplant | 87 (42.0) d | 39 (45.3) | 0.60 |
| Bronchiolitis obliterans syndrome or restrictive allograft syndrome during 6 months after transplant | 1 (0.5) e | 0 (0) | 0.52 |
| Resumed anti-fibrotic medication | 0 (0) d | 0 (0) | |
Data are n (%). Shorter: discontinued anti-fibrotic medication ⩽ 5 medication half-lives before transplant; longer: discontinued anti-fibrotic medication > 5 medication half-lives before transplant.
Patient went home following discharge as opposed to being transferred to a skilled nursing, acute rehabilitation, or long-term acute care facility.
N = 210.
N = 209.
N = 207.
N = 204.
Discussion
Our study, based on chart review of 320 patients with IPF, indicates that there is variability in the timing of discontinuation of anti-fibrotic therapy prior to transplant, but most patients (71%) discontinued anti-fibrotic therapy ⩽ 5 medication half-lives before transplant. Our analyses did not suggest differences in the proportions of patients with surgical wound dehiscence across groups based on when anti-fibrotic therapy was discontinued. Anastomotic and sternal dehiscence, however, were only seen in patients whose anti-fibrotic therapy was discontinued ⩽ 5 medication half-lives prior to transplant. The time between discontinuation of anti-fibrotic therapy and transplant did not appear to affect the length of ICU stay or hospital stay or the proportion of patients who survived to discharge, or to 30 days, 60 days, or 6 months after transplant.
Previous studies have suggested that the use of anti-fibrotic medications does not have an adverse impact on the incidence of post-transplant complications in patients with IPF.21–27 A single-center study of 62 patients found that surgical complications were similar between patients who continued taking anti-fibrotic therapy in the 4 weeks before transplant and those who were naïve to anti-fibrotic therapy or discontinued it > 4 weeks before transplant. 22 An analysis of 132 patients with ILD, of whom 78 had IPF, found no differences in post-operative outcomes between patients who had versus had not taken anti-fibrotic therapy in the 4 weeks before transplant. 23 However, unlike the current study, these two studies did not precisely define when anti-fibrotic therapy was stopped relative to transplant. In a multi-center Australian study, there was a trend toward an increased rate of anastomotic dehiscence (7.5% vs 2.2%; p = 0.08) among patients with IPF who were taking anti-fibrotic therapy up to the day of lung transplant (n = 40) compared to those who were not (n = 186), findings similar to our observations. 24 In a multi-center Spanish study of 164 patients with IPF, there were no differences in the incidence of post-transplant complications between patients who were versus were not taking anti-fibrotic therapy at the time of transplant, although chest wall dehiscences appeared earlier in patients who had used anti-fibrotic drugs. 27
In general, the reported incidence of anastomotic dehiscence after lung transplant ranges between 1% and 10%,28,29 but few data are available on the rate of dehiscence after lung transplant in patients with IPF or other ILDs. In a recent analysis of data from the US Scientific Registry of Transplant Recipients (SRTR) database, among 4359 patients with ILD who underwent transplant between 2012 and 2019, the proportion of patients with dehiscence was 1.4%. 30
While this work and others have focused on the potential risks of taking anti-fibrotic therapy prior to transplant, there is also interest in the possibility that anti-fibrotic therapy may lessen certain post-transplant complications. In our study, the risk of primary graft dysfunction was lower in patients with a shorter time between discontinuation of anti-fibrotic medication and lung transplant. In a retrospective analysis of 62 patients with IPF at a single center, compared with patients not taking anti-fibrotic therapy, those taking anti-fibrotic therapy at transplant had a significant improvement in primary graft dysfunction grade in the 72 hours after transplant. No patients in this study had anastomotic dehiscence, but the proportion with airway stenosis requiring intervention was significantly lower in the patients taking anti-fibrotic therapy at transplant (2.6% vs 26.1%; p = 0.009). 26 Further research is needed into whether antifibrotic therapy may partly protect against primary graft dysfunction.
As an inhibitor of the VEGF receptor, nintedanib may increase the risk of bleeding. 15 An analysis of global pharmacovigilance data, based on a cumulative 60,107 patient-years of exposure, showed that bleeding was reported in patients with IPF treated with nintedanib at a rate of 36.8 events per 1000 patient-years, lower than the rate reported in Phase III trials. 31 Data from the open-label extension of the Phase III INPULSIS trials of nintedanib suggested that the risk of bleeding did not increase with duration of use. 32 In our study, the percentage of patients who received intra- or post-operative red blood cell transfusion was lower in the group that discontinued anti-fibrotic medication ⩽ 5 medication half-lives prior to transplant. However, it should be noted that there are differences among centers in protocols for blood transfusion and that our methodology may not have captured all bleeding events.
We were unable to analyze the effect of discontinuation of anti-fibrotic therapy on the progression of IPF prior to lung transplant, which, given the progressive nature of IPF and the risk of mortality associated with its progression,33,34 is an important consideration in decisions on when to discontinue anti-fibrotic therapy. A transplant center’s typical wait time may influence decisions on when antifibrotic therapy is stopped, as the risk of disease progression prior to transplant will be higher at centers with a longer wait time. This is also a consideration for patients with progressive fibrosing ILDs other than IPF, who may also be given anti-fibrotic therapy to slow ILD progression and be eligible for lung transplantation. 1
Strengths of our study include that it was multi-center and collected data for 6 months following lung transplant. We precisely defined the interval between anti-fibrotic therapy discontinuation and transplant. Although this is the largest published study to date that has investigated potential associations between anti-fibrotic medication use prior to lung transplant and surgical complications, our analyses of potential differences in complications among groups based on the timing of discontinuation of anti-fibrotic therapy prior to transplant were limited by the small number of patients in some of the groups. A larger study would be required to detect possible differences in rare complications. The nature of chart review means that our analysis of anastomotic dehiscence was limited by how each center monitored for this complication using bronchoscopy; no guidelines were provided to the investigators for defining or grading the severity of anastomotic dehiscence. The small number of dehiscence events were concentrated at three centers, all of which primarily continued anti-fibrotic medications until transplant. The potential association between the timing of discontinuation of anti-fibrotic medication and the risk of dehiscence should be explored in future studies that take into account possible center-level effects.
Based on our findings, the decision to continue anti-fibrotic agents following listing for transplant should include an individualized conversation about the risks and benefits for each candidate. It may be reasonable to stop these medications for a patient with a high LAS who is anticipated to have a short waitlist time, particularly if there are other risk factors for airway complications. In contrast, for a patient with a low LAS and risk factors for prolonged waitlist time, it may be reasonable to continue anti-fibrotic medication to prevent more rapid disease progression.
In conclusion, in our study anastomotic and sternal dehiscence were only seen in patients with IPF whose anti-fibrotic therapy was discontinued ⩽ 5 medication half-lives prior to lung transplant. The frequency of other surgical outcomes was similar across groups based on when anti-fibrotic therapy was discontinued relative to lung transplant. Further study will be needed to determine the optimal time to discontinue anti-fibrotic drugs prior to lung transplant.
Supplemental Material
Supplemental material, sj-docx-1-tar-10.1177_17534666231165912 for Anti-fibrotic therapy and lung transplant outcomes in patients with idiopathic pulmonary fibrosis by Todd L. Astor, Hilary J. Goldberg, Laurie D. Snyder, Andrew Courtwright, Ramsey Hachem, Tahuanty Pena, Lorenzo Zaffiri, Gerard J. Criner, Marie M. Budev, Tany Thaniyavarn, Thomas B. Leonard, Shaun Bender, Aliaa Barakat, Janis L. Breeze and Peter LaCamera in Therapeutic Advances in Respiratory Disease
Acknowledgments
The authors thank the patients who participated in this study. The authors thank Arthur Dea of St. Elizabeth’s Medical Center, Boston, MA, USA, for his assistance for data co-ordination. This study was funded by Boehringer Ingelheim Pharmaceuticals, Inc (BIPI). The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors did not receive payment for development of this article. Writing support was provided by Elizabeth Ng and Wendy Morris of FleishmanHillard, London, UK, which was contracted and funded by BIPI. The authors were fully responsible for all content and editorial decisions, were involved at all stages of development and provided their approval on the final version. BI was given the opportunity to review this article for medical and scientific accuracy as well as intellectual property considerations.
Footnotes
Supplemental material: Supplemental material for this article is available online.
ORCID iD: Peter LaCamera  https://orcid.org/0000-0002-4317-3708
https://orcid.org/0000-0002-4317-3708
Contributor Information
Todd L. Astor, Division of Pulmonary and Critical Care Medicine, Massachusetts General Hospital, Boston, MA, USA
Hilary J. Goldberg, Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Boston, MA, USA
Laurie D. Snyder, Division of Pulmonary, Allergy and Critical Care Medicine, Duke University, Durham, NC, USA
Andrew Courtwright, Division of Pulmonary, Allergy, and Critical Care, University of Pennsylvania, Philadelphia, PA, USA.
Ramsey Hachem, Division of Pulmonary and Critical Care Medicine, Washington University, St. Louis, MO, USA.
Tahuanty Pena, Division of Pulmonary, Critical Care and Occupational Medicine, University of Iowa, Iowa City, IA, USA.
Lorenzo Zaffiri, Division of Pulmonary, Allergy and Critical Care Medicine, Duke University, Durham, NC, USA.
Gerard J. Criner, Department of Thoracic Medicine and Surgery, Lewis Katz School of Medicine at Temple University, Philadelphia, PA, USA
Marie M. Budev, Respiratory Institute, Cleveland Clinic, Cleveland, OH, USA
Tany Thaniyavarn, Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Boston, MA, USA.
Thomas B. Leonard, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA
Shaun Bender, Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA.
Aliaa Barakat, ILD Collaborative, Boston, MA, USA.
Janis L. Breeze, Tufts Clinical and Translational Science Institute, Tufts University, Boston, MA, USA; Institute for Clinical Research and Health Policy Studies, Tufts Medical Center, Boston, MA, USA
Peter LaCamera, Division of Pulmonary, Critical Care and Sleep Medicine, St. Elizabeth’s Medical Center, 736 Cambridge Street, Boston, MA 02135, USA.
Declarations
Ethics approval and consent to participate: The study protocol was approved by an independent ethics committee or institutional review board at each participating center. The requirement for informed consent to participate was waived by the independent ethics committee/institutional review board at each participating center.
Consent for publication: Not applicable.
Author contributions: Todd L. Astor: Conceptualization; Data curation; Formal analysis; Writing – review & editing.
Hilary J. Goldberg: Data curation; Formal analysis; Writing – review & editing.
Laurie D. Snyder: Data curation; Formal analysis; Writing – review & editing.
Andrew Courtwright: Data curation; Formal analysis; Writing – review & editing.
Ramsey Hachem: Data curation; Formal analysis; Writing – review & editing.
Tahuanty Pena: Data curation; Formal analysis; Writing – review & editing.
Lorenzo Zaffiri: Data curation; Formal analysis; Writing – review & editing.
Gerard J. Criner: Data curation; Formal analysis; Writing – review & editing.
Marie M. Budev: Data curation; Formal analysis; Writing – review & editing.
Tany Thaniyavarn: Data curation; Formal analysis; Writing – review & editing.
Thomas B. Leonard: Conceptualization; Formal analysis; Writing – review & editing.
Shaun Bender: Conceptualization; Formal analysis; Writing – review & editing.
Aliaa Barakat: Conceptualization; Formal analysis; Writing – review & editing.
Janis L. Breeze: Conceptualization; Formal analysis; Writing – review & editing.
Peter LaCamera: Conceptualization; Data curation; Formal analysis; Writing – review & editing.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Boehringer Ingelheim Pharmaceuticals, Inc (BIPI).
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: T.L.A., H.J.G., A.C., T.P., M.M.B., T.T., A.B., J.L.B. and P.L. have nothing to disclose other than receiving research funding from BIPI for this project. L.D.S. and L.Z. are faculty members in the Duke Clinical Research Institute, which receives funding support from BIPI to co-ordinate the IPF-PRO/ILD-PRO Registry. R.H. reports grants from Bristol Myers Squibb and Therakos, royalties or licenses from UpToDate, consulting fees from Transmedics, and payment for presentations from CareDx. G.J.C. reports grants and consulting fees from AstraZeneca, BI, Broncus, Chiesi, GlaxoSmithKline, Lungpacer, Mereo BioPharma, Nuvaira, Olympus, Patara Pharma, PneumRx, Pulmonx, ResMed and Respironics; grants from Alung, Fisher & Paykel and Galapagos; and consulting fees from BTG, EOLO, NGM and Verona Pharma. T.B.L. is an employee of BIPI. S.B. was an employee of BIPI at the time that this study was conducted.
Availability of data and materials: The data collected in this study will not be shared beyond the investigators.
References
- 1.Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022; 205: e18–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. [DOI] [PubMed] [Google Scholar]
- 3.King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. [DOI] [PubMed] [Google Scholar]
- 4.Guenther A, Krauss E, Tello S, et al. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res 2018; 19: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dempsey TM, Sangaralingham LR, Yao X, et al. Clinical effectiveness of antifibrotic medications for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2019; 200: 168–174. [DOI] [PubMed] [Google Scholar]
- 6.Behr J, Prasse A, Wirtz H, et al. Survival and course of lung function in the presence or absence of antifibrotic treatment in patients with idiopathic pulmonary fibrosis: long-term results of the INSIGHTS-IPF registry. Eur Respir J 2020; 56: 1902279. [DOI] [PubMed] [Google Scholar]
- 7.Kang J, Han M, Song JW.Antifibrotic treatment improves clinical outcomes in patients with idiopathic pulmonary fibrosis: a propensity score matching analysis. Sci Rep 2020; 10: 15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moon SW, Kim SY, Chung MP, et al. Longitudinal changes in clinical features, management, and outcomes of idiopathic pulmonary fibrosis: a nationwide cohort study. Ann Am Thorac Soc 2021; 18: 780–787. [DOI] [PubMed] [Google Scholar]
- 9.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leard LE, Holm AM, Valapour M, et al. Consensus document for the selection of lung transplant candidates: an update from the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2021; 40: 1349–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 2008; 68: 4774–4782. [DOI] [PubMed] [Google Scholar]
- 12.Wollin L, Maillet I, Quesniaux V, et al. Anti-fibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 2014; 349: 209–220. [DOI] [PubMed] [Google Scholar]
- 13.Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 2015; 45: 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conte E, Gili E, Fagone E, et al. Effect of pirfenidone on proliferation, TGF-beta-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci 2014; 58: 13–19. [DOI] [PubMed] [Google Scholar]
- 15.Boehringer Ingelheim Pharmaceuticals, Inc. OFEV® (nintedanib) prescribing information, 2022, https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Ofev/ofev.pdf (accessed 8 September 2022).
- 16.Genentech USA, Inc. ESBRIET® (pirfenidone) prescribing information. 2019, https://www.gene.com/download/pdf/esbriet_prescribing.pdf (accessed 16 November 2020).
- 17.Snell GI, Yusen RD, Weill D, et al. Report of the ISHLT working group on primary lung graft dysfunction, part I: definition and grading-A 2016 consensus group statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2017; 36: 1097–1103. [DOI] [PubMed] [Google Scholar]
- 18.Stewart S, Fishbein MC, Snell GI, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant 2007; 26: 1229–1242. [DOI] [PubMed] [Google Scholar]
- 19.Harris PA, Taylor R, Thielke R, et al. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009; 42: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egan TM, Murray S, Bustami RT, et al. Development of the new lung allocation system in the United States. Am J Transplant 2006; 6: 1212–1227. [DOI] [PubMed] [Google Scholar]
- 21.Delanote I, Wuyts WA, Yserbyt J, et al. Safety and efficacy of bridging to lung transplantation with antifibrotic drugs in idiopathic pulmonary fibrosis: a case series. BMC Pulm Med 2016; 16: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leuschner G, Stocker F, Veit T, et al. Outcome of lung transplantation in idiopathic pulmonary fibrosis with previous anti-fibrotic therapy. J Heart Lung Transplant 2018; 37: P268–P274. [DOI] [PubMed] [Google Scholar]
- 23.Lambers C, Boehm P, Lee S, et al. Effect of antifibrotics on short-term outcome after bilateral lung transplantation: a multicentre analysis. Eur Respir J 2018; 51: 1800503. [DOI] [PubMed] [Google Scholar]
- 24.Mackintosh JA, Munsif M, Ranzenbacher L, et al. Risk of anastomotic dehiscence in patients with pulmonary fibrosis transplanted while receiving anti-fibrotics: experience of the Australian lung transplant collaborative. J Heart Lung Transplant 2019; 38: 553–559. [DOI] [PubMed] [Google Scholar]
- 25.Veit T, Leuschner G, Sisic A, et al. Pirfenidone exerts beneficial effects in patients with IPF undergoing single lung transplantation. Am J Transplant 2019; 19: 2358–2365. [DOI] [PubMed] [Google Scholar]
- 26.Combs MP, Fitzgerald LJ, Wakeam E, et al. Pre-transplant antifibrotic therapy is associated with resolution of primary graft dysfunction. Ann Am Thorac Soc 2022; 19: 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mora Cuesta VM, Iturbe Fernández D, Aguado Ibáñez S, et al. Antifibrotics and lung transplantation: a Spanish multicentre case-controlled study. Respirology 2022; 27: 1054–1063. [DOI] [PubMed] [Google Scholar]
- 28.Mahajan AK, Folch E, Khandhar SJ, et al. The diagnosis and management of airway complications following lung transplantation. Chest 2017; 152: 627–638. [DOI] [PubMed] [Google Scholar]
- 29.Crespo MM, McCarthy DP, Hopkins PM, et al. ISHLT consensus statement on adult and pediatric airway complications after lung transplantation: definitions, grading system, and therapeutics. J Heart Lung Transplant 2018; 37: 548–563. [DOI] [PubMed] [Google Scholar]
- 30.Choi B, Messika J, Courtwright A, et al. Airway complications in lung transplant recipients with telomere-related interstitial lung disease [abstract]. J Heart Lung Transplant 2021; 40: S156–S157. [DOI] [PubMed] [Google Scholar]
- 31.Lasky JA, Criner GJ, Lazarus HM, et al. Safety of nintedanib in patients with idiopathic pulmonary fibrosis: global pharmacovigilance data. Adv Ther 2020; 37: 4209–4219. [DOI] [PubMed] [Google Scholar]
- 32.Crestani B, Huggins JT, Kaye M, et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study, INPULSIS-ON. Lancet Respir Med 2019; 7: 60–68. [DOI] [PubMed] [Google Scholar]
- 33.Doubková M, Švancara J, Svoboda M, et al. EMPIRE registry, Czech part: impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J 2018; 12: 1526–1535. [DOI] [PubMed] [Google Scholar]
- 34.Brown KK, Martinez FJ, Walsh SLF, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J 2020; 55: 2000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-tar-10.1177_17534666231165912 for Anti-fibrotic therapy and lung transplant outcomes in patients with idiopathic pulmonary fibrosis by Todd L. Astor, Hilary J. Goldberg, Laurie D. Snyder, Andrew Courtwright, Ramsey Hachem, Tahuanty Pena, Lorenzo Zaffiri, Gerard J. Criner, Marie M. Budev, Tany Thaniyavarn, Thomas B. Leonard, Shaun Bender, Aliaa Barakat, Janis L. Breeze and Peter LaCamera in Therapeutic Advances in Respiratory Disease