

Abstract
This review outlines the neuropathogenesis of HIV, from initial HIV entry into the central nervous system (CNS) to chronic infection, focusing on key advancements in the last five years. Discoveries regarding acute HIV infection reveal timing and mechanisms of early HIV entry and replication in the CNS, early inflammatory responses, and establishment of genetically distinct viral reservoirs in the brain. Recent studies additionally explore how chronic HIV infection is maintained in the CNS, examining how the virus remains in a latent “hidden” state in diverse cells in the brain, and how this leads to sustained pathological inflammatory responses. Despite viral suppression with antiretroviral therapy, HIV can persist and even replicate in the CNS, and associate with ongoing neuropathology including CD8+ T-lymphocyte mediated encephalitis. Crucial investigation to advance our understanding of the immune mechanisms that both control viral infection and lead to pathological consequences in the brain is necessary to develop treatments to optimize long-term neurologic health in people living with HIV.
Introduction
There are approximately 1.2 million people living with HIV (PLWH) in the United States, and 37.7 million people worldwide (1, 2). Of these individuals, 18–50% will develop neurological and cognitive changes, which can include memory loss, behavioral changes, decline in mental acuity, concentration difficulties, and motor dysfunction — a collection of symptoms currently termed HIV-associated neurocognitive disorder (HAND) (3, 4). While combination antiretroviral therapy (ART) has dramatically reduced the prevalence of the most severe form of HAND (HIV-associated dementia, or HAD), milder forms of HAND remain a significant challenge in the clinical care of PLWH.
Advances in our understanding of the immune-mediated mechanisms of HIV neuropathogenesis have led to promising developments in the management and treatment of individuals with HAND. New developments in the use of biomarkers identify signs of neuroinflammation and neuronal injury that correlate with neurocognitive impairment. Improved ability to identify predictive factors of HAND may allow for earlier, more individualized interventions in the neurological care of PLWH. Finally, nanotechnology-based ART delivery mechanisms, CRISPR/Cas gene editing, and immunomodulatory therapies are novel approaches that show promise in ameliorating neuronal injury and neurocognitive impairment in PLWH.
1. Acute HIV
1.1. Characterizing acute HIV
Acute HIV infection typically occurs during the first four weeks following viral transmission, when the virus replicates and spreads from the site of entry through the blood to tissues, and the immune response rapidly evolves, culminating in development of anti-HIV antibodies. This period can be accompanied by symptoms of acute retroviral syndrome including fever, nausea, myalgia, and headache. Acute HIV is divided into five stages of infection, termed Fiebig stages (5). Fiebig 1 is defined as HIV-RNA ≥ 100 copies/mL with negative p24 antigen and negative HIV IgM. In Fiebig II, the p24 antigen becomes detectable. In Fiebig III, HIV IgM becomes positive. Fiebig IV is defined by an indeterminate p31 western blot (WB) and Fiebig V is defined by a positive WB. Much of our understanding of acute HIV infection has been informed by the RV254 study in Thailand, which has enrolled more than 675 antibody-negative acute HIV participants, at a median of 19 days post infection, who initiate ART during acute infection (6). In this cohort, HIV RNA has been detected in the CSF as early as 8 days post infection (7). In a study from this cohort of 117 participants in different stages of acute infection (median 18 days post-infection), quantifiable HIV RNA presented in the CSF most often at Fiebig stage III or later and peaked at Fiebig IV, compared to plasma HIV RNA which peaked earlier at Fiebig III (8). The presence of HIV RNA in the CSF in acute infection was associated with decreased CD4+ T cell count, higher CD8+ T cell count, higher plasma HIV RNA, and an increased frequency of acute retroviral syndrome (8). CSF immune system activation markers sCD14, IL16, CXCL10, CCL2, and sCD163 increased with CSF HIV RNA, with the greatest increase in activation occurring in a patient who had higher levels of HIV RNA in the CSF compared to plasma (8). These findings demonstrate that inflammation in the CNS occurs early in acute infection.
1.2. Entry of HIV into the CNS and resident cells
HIV putatively enters the CNS through infected lymphocytes and monocytes that cross the blood brain barrier (BBB) (Fig. 1). HIV viral proteins and inflammatory responses disrupt the tight junctions of the BBB, promoting the entry of virus-carrying immune cells into the CNS (9, 10). Pro-inflammatory cytokines and chemokines increase trafficking of immune cells to the brain, further increasing HIV viral load and activating immune responses from resident macrophages and microglia. HIV virus may also directly cross the BBB via transcytosis, mediated by gp120 (11, 12). However, studies in humans and non-human primates have shown that leukocytes are critical in the infection of the CNS. A study in macaques used the anti-alpha 4 integrin antibody to block trafficking of leukocytes across the BBB during acute infection, preventing integration of Simian Immunodeficiency Virus (SIV) DNA in the brain (13). In a study of CSF samples from 72 ART-naive human participants, those with CSF leukocytosis had significantly higher levels of HIV RNA in the CSF compared to those without leukocytosis (14). In another macaque model, CD4+ T cells with replication-competent SIV DNA were shown to infiltrate the neuroparenchyma and traffic to the site of multinucleated giant cells (15). More recently, in a small study of CSF cellular pellets from human study participants with acute HIV Fiebig stage III/IV, active viral transcription in CSF CD4+ T cells was detected in all six participants, underscoring the likely role of CD4+ T cells in trafficking HIV to the CNS in the early stages of infection (16).
Figure 1. Entry of HIV across the blood brain barrier (BBB) into the CNS.

It is proposed that HIV is trafficked into the blood brain barrier through CD4+ T lymphocytes and C14+ CD16+ monocytes. Dopamine can increase the trafficking of CD14+ CD16+ monocytes across the BBB into the CNS. The HIV protein Tat (trans-activator of transcription) disrupts endothelial tight junctions in the BBB, further promoting the influx of monocytes into the CNS. Infected microglia, astrocytes, and macrophages release inflammatory cytokines and chemokines, contributing to an inflammatory environment that potentiates the recruitment of lymphocytes. Created with BioRender.com.
Once HIV enters the CNS, it infects resident cells including macrophages, microglia, and astrocytes (Fig. 1). HIV typically requires the CD4 receptor and either the coreceptor CXCR4 or CCR5 to enter and subsequently integrate its viral genome into the host cell. In the presence of inflammatory stimuli, astrocytes can endocytose macrophages containing HIV, resulting in infection despite the lack of CD4 or CCR5 (11, 17–19). Though astrocytes were originally thought to be unable to productively replicate viral DNA, a recent in-vitro study of human astrocytes has demonstrated a CD4-independent, CXCR4-dependent mechanism of viral entry in astrocytes that results in production of infectious viral particles (20). Infected astrocytes can deregulate cytochrome C processes, disrupt glialneuronal signaling, induce apoptosis in neighboring cells, and increase migration of CD4+ T cells across the BBB via chemoattractant production (3, 21–23).
Monocytes may also have a role in trafficking HIV virus into the CNS. Of particular interest are HIV+ CD14+ CD16+ monocytes, which have been shown to preferentially cross the BBB in response to the chemokine CCL2 in an in vitro human BBB model (Fig. 1) (4, 24). In this study, the junctional proteins JAM-A and ALCAM and the CCL2 receptor CCR2 were increased on HIV+ CD14+ CD16+ monocytes compared to monocytes of the same type that were exposed to, but uninfected by, HIV (24). Furthermore, the CCR2/CCR5 antagonist cenicriviroc and antibodies against JAM-A and ALCAM decreased trafficking across the BBB (24). Interestingly, dopamine has been shown to enhance this migration of CD14+ CD16+ monocytes via a D1-like receptor in a human BBB model, pointing to the importance of investigating the relationship between substance use and HIV infection (Fig. 1) (25–27).
1.3. HIV proteins mediate viral entry
HIV proteins have a key role in the initial infection of the CNS. Tat (transactivator of transcription) is one of the first viral proteins to be expressed and is associated with increased neurovirulence, neuronal apoptosis, and oxidative stress (28, 29). Tat binds to the transactivation response element (TAR), a 59 bp RNA loop at the 5’ end of HIV transcripts, to promote viral transcription (30). Tat also plays a key role in inciting BBB disruption and inflammation, including the activation of the complement system in the CNS and recruitment of monocytes (28, 31). Tat increases the permeability of the BBB by disrupting endothelial tight junctions (9, 10, 32), and induces the production of MCP-1 by astrocytes, which increases monocyte migration across the BBB (Fig. 1) (33). Tat is also thought to trigger microglial activation, activating NLRP3 inflammasomes which increase TNFα and IL6 via caspase1 and IL-1β, contributing to the pro-inflammatory environment (11, 34, 35). Tat also has a direct neurotoxic effect via dysregulation of the kynurenin pathway, dysfunction of the glutamatergic system and overexcitation of NMDAR receptors, disruption of calcium regulating and mitophagy systems in the mitochondria, and mitochondrial damage (Fig. 2) (30, 35–39).
Figure 2. Causes of Neurological Damage in CNS HIV Infection.
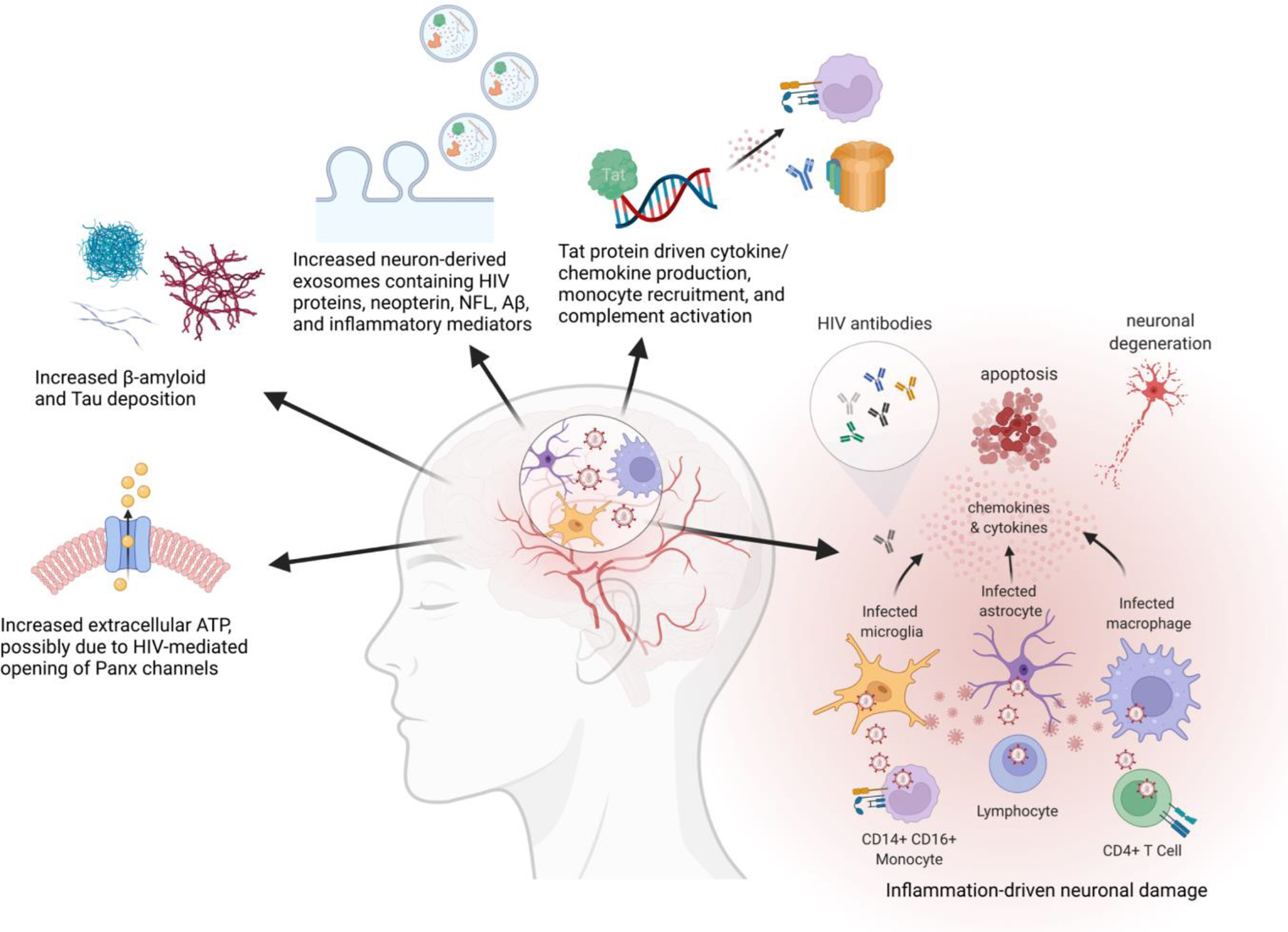
HIV in the CNS can cause neurological damage through numerous mechanisms including inflammation and Tat protein activity. Biomarkers of neuronal damage include neuron-derived exosomes, increased β-amyloid and Tau protein deposition, and increased extracellular ATP. Key: Panx = pannexin, NFL = neurofilament light chain, Aβ= amyloid beta. Created with BioRender.com.
1.4. Early Inflammatory Response in the CNS
An inflammatory environment in the CNS is established early in acute HIV infection, with activated CD4+ and CD8+ lymphocytes and monocytes migrating across the BBB and activating local inflammatory cells (40). As early as the first Fiebig stage, there is a rise in inflammatory markers in the CSF, including the lymphocyte chemokine CXCL10, monocyte chemokine CCL2, and macrophage activation biomarker neopterin (6, 7, 41). By acute HIV infection (AHI) stage 3 (comparable to Fiebig III), there is an expansion of activated CD8+ T cells and CD4+ T cells in the CSF (40). CD8+ T cells comprise the majority of white blood cells in the CSF in acute infection; elevations in frequency of these cells are observed as early as Fiebig I-II (42, 43). In a study of participants in the first 20 days after infection with HIV, the frequency of activated CD8+ T cells (defined as CD38+ CD127− CD8+ T cells) in CSF was increased at 43% in those in AHI stage 3 compared to 4.6% in uninfected controls and 6.7% in those in AHI stage 1–2 (40). In 268 participants from the same cohort at an average of 19 days post-infection and across all Fiebig stages, the frequency of activated CD8+ T cells in blood and monocyte activation marker sCD14 in plasma was increased compared to uninfected controls (44, 45). Similar to that seen with the CD8+ T cells, there was no significant increase in frequency of activated CD4+ T cells amongst participants in Fiebig I, only in the later Fiebig stages (44). The blood CD4/CD8 ratio was shown to decrease in later Fiebig stages compared to Fiebig I (44). In CSF, activated CD8+ T cells in acute infection were found to be HIV-specific and positively correlated with CSF levels of neopterin, sCD163, and IP10, markers of myeloid cell activation (40, 46). An upregulation of genes associated with CD8+ T cell cytolytic effector function and TCR signaling were observed as early as AHI stage 1, increasing in later stages (40). Interestingly, CD8+ T cells in the CSF from participants in AHI stage 3 displayed a unique TCR V-beta repertoire compared to those in the periphery, with 40% unique V-beta usage. This suggests that there is a local expansion of CD8+ T cells in the CNS that may reflect early viral compartmentalization. HIV-specific CD8+ T cells have been associated with reduced viral replication and decreased reservoir seeding (47). As discussed later in this review, it is unclear to what extent the infiltration of HIV-targeting cytotoxic T cells protects the CNS versus instigates pathogenic and persistent inflammatory changes, such HIV CD8+ encephalitis at its extreme (Fig. 2) (22). Potential deleterious effects of this early immune response can be seen in magnetic resonance spectroscopy studies in the Thailand RV254 cohort, which showed an increase in inflammatory brain metabolites in early acute HIV compared with uninfected controls, despite early initiation of ART (48). Elevated choline/creatine was observed in the basal ganglia and occipital grey matter of the middle posterior cingulate gyrus in participants at a median of 14.5 days post-infection, indicative of early inflammatory changes (7). In a different study of ART-naïve individuals, neuroimaging showed a progressive increase in inflammatory markers in multiple brain regions within the first year of HIV infection (34).
1.5. Early establishment of viral reservoirs and compartmentalization
The immune environment established in the acute phase of HIV infection creates a unique selection pressure on HIV virus in the CNS, leading to the evolution of CNS viral strains distinct from those found in the blood — a process known as compartmentalization (49). Though the literature on neuropathogenesis of HIV has largely focused on monocyte-derived macrophages and microglia as the main reservoir of infection, more recent work suggests that HIV viral strains in the CNS begin as T cell tropic, infecting T cells, then later evolve to a macrophage-tropic lineage capable of infecting cells with low CD4 expression (Fig. 3) (14, 34, 50, 51). Compartmentalized, macrophage-tropic viruses are most often observed in the CNS of individuals with HIV-associated dementia (HAD), though HIV likely persists in both T cell and monocyte-derived cell populations (51, 52).
Figure 3. CNS HIV Persistence and Mechanisms of Latency.

There are many mechanisms by which HIV remains in a latent state in the CNS, including epigenetic modifications of HIV DNA, degradation of the HIV protein Tat, and transcriptional repressors of HIV. It is thought that HIV virus is largely T-cell tropic in the earlier stages of infection and resides in T lymphocytes. The inflammatory environment and specific selection pressures in the CNS can lead to a macrophage-tropic HIV virus later in infection that resides in the macrophages and microglia of the CNS. Created with BioRender.com.
The transmission of T-cell tropic virus has been demonstrated in a study that included 72 ART naïve participants enrolled in the first year of HIV infection (14). Compartmentalized viral populations were detected in 11% of samples in participants within the first 12 months of infection, with compartmentalization occurring at the earliest at 120 days. Compartmentalization was defined as meeting significance (p <0.05) for three tests: tree-based Slatkin-Maddison, Wright’s measure of population subdivision, and the nearest-neighbor statistic. Using the time to most recent common ancestor (TMRCA), it was noted that CSF populations were established much earlier than compartmentalization, in the first 2 months for some participants. Using viruses pseudotyped with env proteins from a sample of 24 participants, it was determined that all transmitted variants were R5 T-cell tropic, requiring CCR5 and CD4 for entry. Interestingly, env proteins from samples with compartmentalized viruses (4 months or more post-infection) were significantly better at infecting cells with low CD4 than those from equilibrated samples (p << 0.0001), though they did not meet the criteria for macrophage-tropic lineage (14). This suggests that, while transmitted virus remains R5 T-cell tropic in the CNS for at least the first two years, is it possible that macrophage-tropic lineages may begin to evolve, establishing a distinct reservoir in long-lived macrophages and microglia (Fig. 3).
CNS compartmentalization may be initiated during the earliest stages of HIV transmission to the CNS. In a recent study, transmitted/founder (T/F) HIV sequences from blood were compared to concurrent HIV sequences in CSF examining pol and env sequences in 18 Fiebig II-V ART-naïve AHI participants (median 22 days post-transmission) who had been infected with either a single or multiple T/F viruses (49). Single genome analysis and NGS showed that there was little difference in viral sequences between the plasma and CSF in individuals infected with a single T/F virus, likely reflecting the trafficking of virus and immune cell from systemic circulation into the CNS. In those infected with multiple T/F viruses, however, NGS showed differences in proportions of major and minor HIV variants between compartments. This enrichment of unique variants in the CNS within days of infection supports a model in which a “bottleneck” event, such as specific neurotropic cell entry requirements or enhanced ability to replicate in CNS cells, establishes early variant speciation (53). Of note, fully unique CSF sequences did not appear until later in infection, suggesting that compartmentalization may involve longitudinal replication/ evolution in addition to early bottleneck events. These studies suggest two mechanisms of the development of CNS compartmentalization. Ongoing CNS replication in untreated individuals likely leads to the evolution of new CNS HIV variants over time in the first year of infection. Additionally, while all initial founder HIV variants seeding the CNS are also present in the periphery, enrichment for specific variants based on immune selection or localized replication may be established during the acute phases of infection, establishing populations prone to divergence and compartmentalized evolution.
2. Chronic infection: maintenance of HIV in the CNS
2.1. Diverse HIV reservoirs in the CNS
HIV has been shown to reside in diverse cell populations in the CNS, including CD4+ T cells, macrophages, microglia, and potentially astrocytes and oligodendrocytes, persisting despite ART viral suppression (Fig. 3) (4, 19, 22, 32, 54). There is increasing evidence for the importance of CD4+ T cells in establishing and preserving HIV infection in the CNS. Normal immune surveillance involves the migration of T cells across the BBB, which can be exploited by HIV to enter the brain (55–57). Recently, use of a humanized T-cell only mouse model demonstrated that T cells can establish and maintain HIV infection in the brain in the absence of myeloid cells (55). Advances in sequencing technologies have illuminated other potential HIV reservoir populations in the CNS. Single-cell RNA sequencing methods have revealed a newly described rare population of myeloid cells found predominantly in the CSF, with a gene expression profile similar to that seen in microglia associated with neurodegenerative disease, exhibiting upregulation of APOE, AXL, and TREM2 (58). These and other methods such as single nucleus RNA sequencing of brain tissue may reveal previously unrecognized HIV reservoir cell types in the CNS compartment.
2.2. Latency and reactivation
HIV has evolved unique transcriptional regulatory mechanisms to maintain latency in the CNS (Fig. 3). Distinct polymorphisms at the Sp transcription factor motifs in the LTR decrease HIV transcriptional activity, creating a dormant proviral state that can more easily evade immune cell detection and ART (59). Attenuation of Tat has also been posited as a mechanism of latency induction (32). The lncRNA NRON induces Tat proteasomal degradation, decreasing transcription (60). Other mechanisms of latency induction include microRNAs, transcriptional suppressors including TRIM22, TRIM19, and COMMD1, and epigenetic modifications (Fig. 3) (32, 61–63). Interactions between different CNS cell types have also been shown to regulate HIV latency. For example, HIV emerges from latency in microglia in response to co-culture with damaged, but not healthy, neurons (64).
Of particular concern is the question of whether HIV reservoirs established in the CNS can reactivate after periods of latency, and “re-seed” and/ or provoke inflammatory responses in the systemic circulation (34). The discovery in 2015 that lymphatic vessels drain from the brain dura to deep cervical lymph nodes provides a plausible pathway through which this re-infection could occur (32, 65).
To address this question, post-mortem samples from six donors with HIV were examined for compartmentalization and re-population of HIV across different anatomical sites (66). The participants included four individuals who were virally suppressed on ART, and two with rebound viremia after having stopped ART for approximately two months at the end of life. X4 T-cell tropic HIV virus dominated the viral population in the majority of PBMCs and postmortem tissue sampled. In both of the two patients who stopped ART, there was a re-emergence of HIV DNA that was T-cell tropic with low sequence diversity. In one participant, viral migration was observed from the frontal lobe to the PBMCs, and from lymph nodes to the frontal lobe. In the other participant, bidirectional migration was observed between the occipital lobe and frontal lobe. Other participants who did not stop ART exhibited viral transitions from PBMCs to frontal lobes. This study suggests that HIV is able to establish and maintain compartmentalized reservoirs in the brain in the presence of ART, and that it has the potential to re-emerge to infect peripheral blood cells when ART is discontinued for as few as 53 days. Further studies of anatomical compartments across the stages of HIV are needed to ascertain whether CNS HIV persistence can lead to clinically-important systemic virologic resurgence in the setting of ART interruption and HIV remission and cure interventions.
3. Effect of ART on HIV replication, inflammation, and establishment of viral reservoirs
3.1. ART mitigates, but does not eliminate, neuropathogenesis of HIV
In the pre-ART era, 20–30% of people living with uncontrolled HIV developed HIV-associated dementia (HAD) (3). While cART has decreased the incidence of HAD dramatically, published studies suggest that 18–50% of PLWH on ART will develop HAND, though some caution that this is an overestimate due to the cognitive tests used to determine HAND and to confounding comorbidities in PLWH (3, 67–70). It is established that individuals on ART have significantly decreased virus in their CNS compared to those who are untreated, and individuals who start ART treatment early have decreased HIV DNA in their PBMCs compared to those who start treatment during chronic infection (6). Early initiation of ART can prevent the CD4 count from dropping to low levels, which is associated with an increased risk of neurocognitive impairment (22). However, ART has failed to completely eliminate virus from the CNS (3, 32, 71). Despite significant viral suppression, persistence of low-level viral replication, integrated virus, or viral proteins are posited to provoke inflammation in the CNS and cause lasting neurocognitive deficits (Fig. 2) (32).
Many studies have asked whether early treatment with ART can prevent the establishment and persistence of viral reservoirs in the CNS. There is particular interest in understanding how ART modulates the chronic inflammatory and neurotoxic milieu associated with HIV infection. Despite significantly reduced viral loads in the CNS of individuals on ART, inflammation remains. The presence of HIV-specific CD8+ T cells and CD4+ T cells has been shown up to 2 years and 96 weeks, respectively, after infection despite beginning ART within weeks of transmission (6, 43). Some studies have reported that neopterin and NFL levels remains elevated compared to uninfected controls despite ART (22, 34, 72). Markers of BBB impairment (CSF protein, CSF: blood albumin ratio) have also been shown to remain elevated despite 6 months on ART (6). Other inflammatory mediators such as Tat remain active during ART, promoting inflammation, activation of complement pathways, and chemotaxis of immune cells (28).
Other studies have demonstrated a decline in inflammatory markers with ART, particularly with early initiation of ART. Early ART has been associated with lower levels of microglial activation, some inflammation markers, and neuronal damage markers in the CSF (43, 44, 73). Using sTREM2 as a marker of microglial activation, one study revealed that the highest levels of activation were in untreated HIV+ individuals with low CD4+ T cell counts, whereas those on ART had CSF sTREM2 levels comparable to that seen in uninfected controls (74). Other studies have reported a normalization of CSF immune markers including neopterin, CSCL10, CCL2, and IL6 with early ART, though plasma immune markers remained elevated in this study (75). Interestingly, early ART treatment has been associated with decreased HIV antibody production in the CSF (76). ART has also been shown to decrease frequency of autoantibodies in HIV infection (77). Future work is needed to better elucidate the role of elevated autoantibodies and HIV antibodies in neuropathogenesis of HIV.
Early initiation of ART (<4 months post-infection) is associated with lower molecular diversity of HIV DNA in the CSF, and lower IL-6 in the CSF compared to late initiation of ART (>14 months post-infection) (78). Importantly, CNS compartmentalization was found in participants with both early and late initiation of ART, suggesting that the lower molecular diversity seen after early ART treatment does not prevent CNS compartmentalization. This finding is supported by additional studies that have also shown the persistence of HIV in compartmentalized reservoirs despite ART (50).
4. CSF escape: mechanisms and insights into cellular origins of HIV
4.1. CSF HIV escape: neurologically symptomatic vs. asymptomatic
CSF escape involves the replication of HIV in the CNS despite systemic viral suppression with ART, defined by detection of CSF HIV RNA with undetectable plasma levels or (when plasma RNA is detected) CSF RNA greater than plasma RNA (79). CSF escape may be neurologically asymptomatic or symptomatic, which differ in frequency, and may reflect distinct pathophysiological states. One study of 69 neurologically asymptomatic participants on long-term treatment with undetectable plasma viral load found a CSF escape prevalence of 10%, where CSF escape was defined as CSF HIV RNA > 50 copies/mL with undetectable plasma HIV RNA (80). Participants with CSF escape had significantly higher levels of CSF neopterin compared to those without escape, but there was no difference in CSF pleocytosis. In a different study of 204 participants in Thailand (RV254) started on ART during acute HIV infection, only one individual had CSF escape and did not have any neurological dysfunction (81). The significance of asymptomatic CSF escape merits further research – in particular, it is unclear how asymptomatic CSF escape may contribute to HIV neuropathogenesis.
Neurosymptomatic CSF escape can present with neurocognitive impairment, tremors, gait ataxia, sensory impairment, headaches, and has rarely been associated with CD8+ T cell and autoantibody-mediated encephalitis (82–85). Symptomatic CSF HIV escape is associated with an increase in inflammation, evidenced by CSF pleocytosis, elevated CSF protein, increased neopterin, and abnormal brain imaging (white matter hyperintensities and perivascular enhancement) (34, 82, 83, 85, 86). The brain imaging likely reflects the perivascular polyclonal lymphocytic infiltration, microglial nodules, and inflammation that have been observed on brain biopsies of individuals with symptomatic CSF escape (83, 85, 86). Risk factors for CSF HIV escape include chronic HIV infection (>10 years), low CD4 nadir, low level viremia, long exposure to ART, and drug resistance mutations (34, 85, 87, 88). Perivascular CD8+ T cell infiltration and predominately CD8+ T cell populations have been observed in individuals with symptomatic CSF HIV escape, pointing to CD8+ T cells as a key mediator of this inflammatory process (83).
4.2. Distinct mechanisms of CSF HIV escape
Studies in participants who exhibit CSF HIV escape, both symptomatic and asymptomatic, have helped elucidate the mechanisms by which HIV establishes reservoirs in the CNS, maintains latency, and reactivates (50). It is proposed that CSF HIV escape could result from either reactivation of latent HIV in the CNS or infection of the CNS from infected cells in the periphery. Measurement of HIV RNA in the CSF and plasma of 101 asymptomatic participants on stable ART revealed that 6% had CSF escape (defined as a concentration of HIV RNA in the CSF > 0.5 log copies/mL above that in plasma). Sequencing of viral envelope proteins identified two distinct mechanisms of CSF escape in this population: viral replication in long-lived macrophage cells in the CNS, and infiltration of the CNS with clonally-expanded T cells from other compartments. CSF escape viruses were analyzed from three patients: two displaying the T-cell tropic virus, and one displaying the macrophage-tropic virus. Sequence analysis showed that the macrophage-tropic virus displayed drug resistance mutations and rose from a rare macrophage-tropic lineage that split from the major R5 T cell tropic lineage before ART treatment. In contrast, the T-cell tropic variants displayed little sequence diversity and no DRMs, suggesting that it originated from either latent cells in the CNS or trafficked T cells from the periphery, rather than ongoing viral replication in the CNS. These studies support a model in which CSF escape can arise from either ongoing replication in long-lived macrophages in the CNS, or via T cells that are transiently reactivated from a latent state or infiltrate from the periphery.
Many studies have asked whether CSF HIV escape arises from the inability of ART to fully penetrate the BBB and reach infected cells within the CNS. Efavirenz is reduced 200-fold in the CSF relative to blood, emtricitabine is reduced 2-fold, and tenofovir is reduced 20-fold (52). In a study of 41 individuals with CSF HIV escape (71% symptomatic), over 80% of participants had HIV infection for greater than 10 years, with a long exposure to ART (87). The common NRTI M184V/I mutation was found in 74% of CSF samples, suggesting that drug resistant mutations may provoke CSF escape (87). Additional studies have also noted a relationship between CSF escape and antiretroviral resistance mutations (88). For many patients, changing their medication to an ART with better CNS penetration can resolve symptomatic CSF escape (83). In a study of 156 neurosymptomatic HIV+ participants on ART, CSF escape was detected in 28% of participants and CD26 levels were consistent with a T-cell origin virus, rather than a monocyte derived macrophage (MDM) origin virus (52). This suggests that the source of HIV virus in CSF escape is at least partially originating in T cells. Surprisingly, there was no significant difference in CSF ART concentrations in participants with CSF escape compared to those without escape, nor was there an association between drug ART concentration and the presence of drug resistance mutations (DRM) (52). This shows that DRMs are not necessary for CSF escape. Possible mechanisms to explain this T-cell tropic viral replication in the absence of DRMs include ongoing infection between T cells and CNS resident cells, trafficking of infected T cells into the CSF from other compartments, or reactivation of latently-infected T cells.
5. Chronic inflammation: impact on neurocognitive function and associated biomarkers
5.1. Inflammation is associated with neurocognitive impairment
It is thought that immune activation and neuroinflammation is the basis of neurological injury and neurocognitive impairment in both treated and untreated HIV infection (Fig. 2) (58, 89–91). HIV-associated neurocognitive disorder is divided into three categories: asymptomatic neurocognitive impairment, mild neurocognitive disorder, and HIV-associated dementia (HAD) (68).
As described in Section 2, the activation of immune cells in the CNS leads to a persistent neuroinflammatory state. The activation of macrophages and microglial cells leads to the release of a number of cytokines and chemokines, which is enhanced by the pro-inflammatory and neurotoxic effects of HIV proteins Tat and gp120 (Fig. 2) (4, 9, 11, 12). Increased neopterin and sCD163 (both biomarkers of monocyte activation) are increased in in the CSF of individuals with chronic HIV suppressed on ART (58). The chemokine CCL2 remains elevated in the brain of PLWH despite ART, and attracts CD14+ CD16+ monocytes to cross the BBB and enter the CNS (4, 92). Interestingly, dopamine has been shown to further increase the production of pro-inflammatory molecules, including IL-1β, IL-6, IL-18, CCL2, and CXCL8–10, despite treatment with ART (25, 93). Hence there is concern that in PLWH who are using substances, the deleterious effects of inflammation may be heightened.
Inflammation has been associated with neurocognitive decline. Levels of the macrophage and microglial activation marker neopterin have been shown to be associated with HAD in untreated infection (34, 94, 95). Elevated CSF neopterin has also been shown to associate with mild forms of HAND in individuals on ART (96). In a study of ART-treated, virally suppressed PLWH, even very low level HIV RNA in the CSF, detected below the limit of quantification on a standard clinical assay (20 copies/mL), was associated with decreased BBB integrity and decreased executive function compared to those without any CSF HIV RNA detected (97). Loss of executive function is a classic finding in HAND, often associated with volume loss in the prefrontal cortex and thalamus (97–99).
Tat has been shown to regulate gene expression of a number of genes involved in inflammation and damage, including C5, APBA1, BDNF, and CRLF2 (Fig. 2) (28). Tat-1B is associated with neuronal apoptosis, neuroinflammation, oxidative stress, and beta amyloid production, and has been shown to induce hippocampus neural degermation and mitochondrial membrane damage in mouse models (24, 35). More work is needed to understand how other HIV proteins in addition to Tat (such as gp120, Nef, and Vpr) may contribute to chronic neuroinflammation and damage.
Chronic activation of immune cells can lead to neurotoxicity. The detoxifying enzyme heme oxygenase-1 is degraded by the immunoproteasome in astrocytes in a IFN-γ dependent manner (100). Reduced heme oxygenase-1 leads to increased glutamate release and neurotoxicity, contributing to neuropathogenesis. A 3D human brain organoid model created to better understand mechanisms of HIV neuropathogenesis revealed that infection of microglia with HIV increased the pro-inflammatory markers TNFα and IL-1β and led to neuronal damage, as indicated by the loss of neuronal marker βIII-Tubulin (101).
HIV CD8 encephalitis (CD8E) is an example of immune-mediated neurological injury at its extreme. CD8E is a severe inflammatory disorder, with infiltration of the brain by CD8+ T cells resulting in cerebral swelling and diffuse white matter changes (102), presenting as an acute or subacute decline in cerebral function that can result in coma and death unless treated promptly with corticosteroids (102). It is most commonly seen in the setting of ART interruption, CSF HIV escape, co-infection with other pathogens, or immune reconstitution inflammatory syndrome (IRIS) after starting ART. CD8E and IRIS are similar mechanistically in that both involve pathologic infiltration of CD8+ T cells into the brain. However, IRIS refers specifically to the excessive inflammatory response that occurs as CD4+ T cells recover following initiation of ART therapy (usually within the past ~3 months), whereas CD8E does not necessarily follow this clinical timeline (103). They also differ in that the majority of IRIS cases are self-limiting(104). In an analysis of 23 cases of HIV CD8E, 74% of individuals were receiving ART, 17% had stopped taking ART in the past 1–5 months, and 68% had active CSF HIV escape (102). Histopathology showed microglial activation, astrocyte activation, perivascular T cell infiltration, and low levels of CD4+ T cells. CD8E underscores the damage that inflammatory reactions can wreak on the CNS. NMDA receptor antibody-mediated encephalitis has also been reported in the setting of CSF HIV escape (84). It is theorized that the virus-induced neuronal damage exposes neuronal antigens, thus initiating autoimmunity. It is unknown whether mild forms of HIV-associated neurologic impairment in individuals on stable ART associates with mild to moderate histopathology on a spectrum with CD8E.
5.2. Identifying biomarkers of inflammation, neurotoxicity, and HAND
Identifying biomarkers of inflammatory changes and neuronal damage in HIV would allow clinicians to better anticipate the development of HAND, and to better understand the progression of neurological damage in individual patients (105). Biomarkers may also provide a more accurate way to define and characterize HIV-associated neurological damage. The diagnosis of HAND, a diagnosis based on neurocognitive testing, has been recently questioned, particularly for those diagnosed with asymptomatic neurological impairment (ANI) (69). Concerns have been raised that performance on cognitive tests is confounded by comorbid conditions, socioeconomic status, inequities, and culture, among other factors — and that PLWH with low scores on cognitive tests will be falsely labeled as having a cognitive disorder. One proposal suggests focusing on a clinical history of cognitive decline, coupled with neuroimaging findings and novel biomarkers to more accurately define HIV neurological pathology in individuals.
Biomarkers in the systemic circulation are especially attractive, as opposed to CSF markers that require more invasive lumbar punctures to procure. Numerous soluble and cellular biomarkers noted above have been previously identified as associated with HAND. Novel markers more recently identified with neuronal injury and neuropathogenesis include extracellular vesicles (EVs), nanoscale vesicles that carry proteins and lipids and have been shown to transfer viral proteins and pro-inflammatory factors to target cells (Fig. 2) (106, 107). Increased numbers of EVs have been observed in the CNS of PLWH on ART, compared to uninfected controls (106). Using immunoblotting, higher levels of CSF NFL, neopterin, and the monocyte activation marker S100B have been demonstrated on EVs from PLWH, providing additional evidence for the lasting neuroinflammatory changes seen with HIV (106). EVs obtained from the blood may serve as a more effective biomarker in clinical practice due to their increased accessibility (107). Neuron-derived exosomes (NDE) from plasma can be isolated via antibodies against the neuronal cell adhesion molecule L1CAM. In 23 PLWH and 12 uninfected controls, NDEs were increased in those with neuropsychological impairment compared to those without impairment, though no difference was noted between PLWH compared to negative controls (108). Increased levels of NFL, amyloid beta (Aβ), and the alarmin HMGB1 were identified in NDEs in PLWH (108, 109). The demonstration that NDEs are found in plasma additionally raises the question of whether NDEs can assist in the reactivation and pathogenesis of HIV in the periphery by transferring viral proteins and inflammatory molecules outside of the CNS.
Recent work has also identified shed PrP(c) (non-pathogenic human prion protein) as a potential biomarker of neuronal damage (110). Shed PrP(c) is increased in the CSF of PLWH with neurocognitive deficits compared to those without impairment, and is thought to play a role in monocyte recruitment to the brain (110, 111). CCL2 and TNFα, inflammatory mediators increased in the CNS of people with HIV, promote the metalloproteinase ADAM10 to adopt its active conformation, enabling it to cleave PrPc (110, 112). Shed PrPc then increases secretion of CCL2, CXCL12, and IL8 from astrocytes, further compounding the immune activation and neurological damage (110).
Other markers of injury include amyloid beta protein deposits, shed APP, and tau (Fig. 2) (105). HIV infection can disrupt normal amyloid metabolism through the actions of Tat, which leads to accumulation of Aβ deposits. The oligomer Aβ–42 as well as Total tau (T-tau) have been shown to be decreased in CSF of PLWH, reflecting the increased tissue deposition in the brain (113). Tissue deposition of beta amyloid protein has been associated with memory deficits due to toxicity and disruption of synaptic plasticity (108). Increased Aβ–42 deposition and increased T-tau deposition are also associated with oxidative stress, measured with the DNA damage marker 8-oxo-dG (114). More recently, ATP has been proposed as a biomarker of HIV-associated cognitive impairment, able to detect neuropathogenesis at an earlier stage than other markers such as NFL, which is a late marker of brain damage and is not specific for HIV (Fig. 2) (115, 116). It is proposed that HIV may open pannexin (Panx) channels, mediating ATP release. Circulating levels of ATP are positively correlated with cognitive impairment in PLWH, and are associated with BBB compromise and infiltration of leukocytes (115).
Recent developments in imaging technologies also provide a promising strategy for the identification of inflammatory-mediated neurological damage. A novel approach to identifying synaptic injury with PET using the SVA2 ligand has demonstrated decreased synaptic density in the frontostriatalthalamic circuit and other cortical areas of PLWH on ART, compared to HIV uninfected controls (117). Further studies are needed to examine the association between synaptic injury and neuroinflammation and HIV persistence.
6. Treatments for HIV-associated neurocognitive disorder: recent advances and remaining challenges
6.1. Increasing efficacy of ART in the CNS
ART is the mainstay of HIV treatment, and has significantly decreased the morbidity and mortality of HIV infection, including CNS complications. However, challenges remain surrounding delivery of ART into the brain. Many modifications to ART and the delivery of ART have been proposed, including the use of prodrugs, intracerebral injections or implants, and liposome delivery. Particularly promising are the recent developments in nanoparticle-based delivery of ART into the brain (32). These include a novel integrase inhibitor (elvitegravir) nanoformulation which showed improved penetrance in an in vitro BBB model and increased HIV suppression in HIV human monocyte-derived macrophages (118), and nanodiamonds to deliver the NNRTI efavirenz across a BBB barrier, demonstrating increased and longer-lasting bioavailability in the CNS (119). Questions remain, however, about the potential neurotoxicity of ART medications that can more easily cross the BBB.
6.2. Gene editing approaches to eliminating viral reservoirs
Gene editing therapy using CRISPR/Cas technology provides a potential strategy to eliminate latent HIV from its reservoirs. Studies have asked whether CRISPR/Cas can be used to excise integrated HIV proviral DNA, perhaps to be used as an adjuvant to ART. Ongoing discovery of smaller Cas proteins, such as Cas12 or CasΦ, may prove useful in this context (120, 121). CRISPR/Cas9 has been employed to eliminate latent proviruses in infected astrocytes. Astrocytes were transduced with a novel astrocyte-targeting AAV vector containing a guide RNA for the HIV proviral LTR sequence (122). The vector successfully targeted astrocytes and inactivated LTR, preventing HIV reactivation (122). More recently, the combination of CRISPR/Cas9 gene editing and long-acting slow-effective release antiviral therapy (LASER ART) was shown to completely eliminate HIV in a mouse model, an exciting development in the search to find a cure for HIV (123).
6.3. Immunomodulatory strategies
A mainstay of approaches to treatment of CNS HIV is modulation of the immune system to mitigate the damaging effects of inflammation. The chronic neuroinflammation seen in HIV raises the question of whether anti-inflammatory medications may be neuroprotective. In a mouse model of HAND, the JAK1/2 inhibitor and rheumatoid arthritis drug baricitinib reversed glial activation and cognitive deficits (124). This suggests that modulation of the pathogenic immune response in HIV may help to prevent and treat neurocognitive impairment. Preliminary studies have identified other potentially useful immunomodulatory medications, including the MS drug dimethyl fumarate, the RRMS drug teriflunomide, and the antibiotic minocycline which is used in rheumatoid arthritis (125–128).
Other immunomodulatory approaches have sought to strengthen and refine the immune system’s response to HIV in the CNS. This had led to the development of CAR-based immunotherapies targeting HIV, and latency reversal agents (LRAs) targeting HIV reservoirs (129). Barber-Axthelm et al. genetically modified HSPCs to express a CAR containing a CD4 domain fused to a CD3ζ signaling domain, creating lymphocytes that target and lyse HIV-infected cells (129). They showed that the CD4CAR+ cells successfully migrate to the CNS, engraft, and persist in multiple locations, providing a promising strategy for targeting hard-to-reach reservoirs of HIV. Additional work is needed to determine the ability of these CAR cells to eliminate infection in HIV reservoirs and mitigate the neurocognitive impairments seen in chronic HIV infection. Lastly, latency reversal agents, or “shock and kill” approaches, involve provoking HIV from its latent state to an active state that makes it more easily targeted by the immune system (19). There is concern, however, that this approach may instigate damaging neuroinflammatory responses, which will be important to address in further studies (32, 130).
Conclusions
The past five years have seen significant strides in understanding the neuropathogenesis of HIV, though many questions still remain. Many individuals living with HIV struggle with lasting neurological and cognitive impairment, despite viral suppression with ART. The treatment of HIV associated neurocognitive disorder and CSF HIV escape continue to pose a significant challenge for clinicians.
The research advances described here have illuminated the profoundly immune-mediated mechanisms of HIV neuropathogenesis. It is critical that we develop a better understanding of neuroinflammation in HIV, from the initial entry of HIV into the CNS during acute infection to the persistence of latent HIV in reservoirs and reactivation during CSF HIV escape. Somewhat paradoxically, the very cells designed to rid the brain of HIV are ultimately those implicated in the neuronal damage wrought by this virus. The key to developing more effective treatments for HAND may rest on understanding this balance between neuroprotective and pathological immune activation in the CNS of individuals with HIV. Identification of HIV-specific biomarkers of CNS inflammation, neuronal injury, and neurocognitive decline will surely aid in these efforts.
Acknowledgements
We express our gratitude to the many individuals who have volunteered to partake in the studies reviewed here. Without their generosity, this work would not be possible.
Funding
LK is supported by NIH training grant 5T32GM136651–02. SS is supported by grants from the NIH including R01MH125737, R01MH125396, R01MH106466, and UM1DA051410.
Footnotes
Declaration of Conflicting Interests
LK and SS report no conflicting interests.
References
- 1.2020. HIV Surveillance Report: Supplemental Report, Centers for Disease Control and Prevention [Google Scholar]
- 2.2020. Estimated number of people (all ages) living with HIV, World Health Organization [Google Scholar]
- 3.Zayyad Z, Spudich S. 2015. Neuropathogenesis of HIV: from initial neuroinvasion to HIV-associated neurocognitive disorder (HAND). Curr HIV/AIDS Rep 12: 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veenstra M, Leon-Rivera R, Li M, Gama L, Clements JE, Berman JW. 2017. Mechanisms of CNS Viral Seeding by HIV(+) CD14(+) CD16(+) Monocytes: Establishment and Reseeding of Viral Reservoirs Contributing to HIV-Associated Neurocognitive Disorders. mBio 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiebig EW, Wright DJ, Rawal BD, Garrett PE, Schumacher RT, Peddada L, Heldebrant C, Smith R, Conrad A, Kleinman SH, Busch MP. 2003. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS 17: 1871–9 [DOI] [PubMed] [Google Scholar]
- 6.Spudich S, Peterson J, Fuchs D, Price RW, Gisslen M. 2019. Potential for early antiretroviral therapy to reduce central nervous system HIV-1 persistence. AIDS 33 Suppl 2: S135–S44 [DOI] [PubMed] [Google Scholar]
- 7.Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, Suwanwela NC, Jagodzinski L, Michael N, Spudich S, van Griensven F, de Souza M, Kim J, Ananworanich J, Group RSS. 2012. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis 206: 275–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan P, Patel P, Hellmuth J, Colby DJ, Kroon E, Sacdalan C, Pinyakorn S, Jagodzinski L, Krebs S, Ananworanich J, Valcour V, Spudich S, Team RSS. 2018. Distribution of Human Immunodeficiency Virus (HIV) Ribonucleic Acid in Cerebrospinal Fluid and Blood Is Linked to CD4/CD8 Ratio During Acute HIV. J Infect Dis 218: 937–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. 2003. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res 74: 255–65 [DOI] [PubMed] [Google Scholar]
- 10.Xu R, Feng X, Xie X, Zhang J, Wu D, Xu L. 2012. HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res 1436: 13–9 [DOI] [PubMed] [Google Scholar]
- 11.Rojas-Celis V, Valiente-Echeverria F, Soto-Rifo R, Toro-Ascuy D. 2019. New Challenges of HIV-1 Infection: How HIV-1 Attacks and Resides in the Central Nervous System. Cells 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banks WA, Kastin AJ, Akerstrom V. 1997. HIV-1 protein gp120 crosses the blood-brain barrier: role of adsorptive endocytosis. Life Sci 61: PL119–25 [DOI] [PubMed] [Google Scholar]
- 13.Campbell JH, Ratai EM, Autissier P, Nolan DJ, Tse S, Miller AD, Gonzalez RG, Salemi M, Burdo TH, Williams KC. 2014. Anti-alpha4 antibody treatment blocks virus traffic to the brain and gut early, and stabilizes CNS injury late in infection. PLoS Pathog 10: e1004533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sturdevant CB, Joseph SB, Schnell G, Price RW, Swanstrom R, Spudich S. 2015. Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog 11: e1004720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CA, Beasley E, Sundar K, Smelkinson M, Vinton C, Deleage C, Matsuda K, Wu F, Estes JD, Lafont BAP, Brenchley JM, Hirsch VM. 2020. Simian Immunodeficiency Virus-Infected Memory CD4(+) T Cells Infiltrate to the Site of Infected Macrophages in the Neuroparenchyma of a Chronic Macaque Model of Neurological Complications of AIDS. mBio 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma V, Creegan M, Tokarev A, Hsu D, Slike BM, Sacdalan C, Chan P, Spudich S, Ananworanich J, Eller MA, Krebs SJ, Vasan S, Bolton DL, Rv254/Search, Teams RSS. 2021. Cerebrospinal fluid CD4+ T cell infection in humans and macaques during acute HIV-1 and SHIV infection. PLoS Pathog 17: e1010105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell RA, Chojnacki J, Jones DM, Johnson E, Do T, Eggeling C, Padilla-Parra S, Sattentau QJ. 2017. Astrocytes Resist HIV-1 Fusion but Engulf Infected Macrophage Material. Cell Rep 18: 1473–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll-Anzinger D, Al-Harthi L. 2006. Gamma interferon primes productive human immunodeficiency virus infection in astrocytes. J Virol 80: 541–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ash MK, Al-Harthi L, Schneider JR. 2021. HIV in the Brain: Identifying Viral Reservoirs and Addressing the Challenges of an HIV Cure. Vaccines (Basel) 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li GH, Maric D, Major EO, Nath A. 2020. Productive HIV infection in astrocytes can be established via a nonclassical mechanism. AIDS 34: 963–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertin J, Jalaguier P, Barat C, Roy MA, Tremblay MJ. 2014. Exposure of human astrocytes to leukotriene C4 promotes a CX3CL1/fractalkine-mediated transmigration of HIV-1-infected CD4(+) T cells across an in vitro blood-brain barrier model. Virology 454–455: 128–38 [DOI] [PubMed] [Google Scholar]
- 22.Subra C, Trautmann L. 2019. Role of T Lymphocytes in HIV Neuropathogenesis. Curr HIV/AIDS Rep 16: 236–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee KM, Chiu KB, Renner NA, Sansing HA, Didier PJ, MacLean AG. 2014. Form follows function: astrocyte morphology and immune dysfunction in SIV neuroAIDS. J Neurovirol 20: 474–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams DW, Calderon TM, Lopez L, Carvallo-Torres L, Gaskill PJ, Eugenin EA, Morgello S, Berman JW. 2013. Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS One 8: e69270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nickoloff-Bybel EA, Calderon TM, Gaskill PJ, Berman JW. 2020. HIV Neuropathogenesis in the Presence of a Disrupted Dopamine System. J Neuroimmune Pharmacol 15: 729–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calderon TM, Williams DW, Lopez L, Eugenin EA, Cheney L, Gaskill PJ, Veenstra M, Anastos K, Morgello S, Berman JW. 2017. Dopamine Increases CD14(+)CD16(+) Monocyte Transmigration across the Blood Brain Barrier: Implications for Substance Abuse and HIV Neuropathogenesis. J Neuroimmune Pharmacol 12: 353–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coley JS, Calderon TM, Gaskill PJ, Eugenin EA, Berman JW. 2015. Dopamine increases CD14+CD16+ monocyte migration and adhesion in the context of substance abuse and HIV neuropathogenesis. PLoS One 10: e0117450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvallo L, Lopez L, Fajardo JE, Jaureguiberry-Bravo M, Fiser A, Berman JW. 2017. HIV-Tat regulates macrophage gene expression in the context of neuroAIDS. PLoS One 12: e0179882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McRae M 2016. HIV and viral protein effects on the blood brain barrier. Tissue Barriers 4: e1143543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams ME, Zulu SS, Stein DJ, Joska JA, Naude PJW. 2020. Signatures of HIV-1 subtype B and C Tat proteins and their effects in the neuropathogenesis of HIV-associated neurocognitive impairments. Neurobiol Dis 136: 104701. [DOI] [PubMed] [Google Scholar]
- 31.Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, Ranga U, Tyor WR, Prasad VR. 2008. HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci 28: 10010–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonti S, Sharma AL, Tyagi M. 2021. HIV-1 persistence in the CNS: Mechanisms of latency, pathogenesis and an update on eradication strategies. Virus Res 303: 198523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bethel-Brown C, Yao H, Hu G, Buch S. 2012. Platelet-derived growth factor (PDGF)-BB-mediated induction of monocyte chemoattractant protein 1 in human astrocytes: implications for HIV-associated neuroinflammation. J Neuroinflammation 9: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spudich SS. 2016. Immune activation in the central nervous system throughout the course of HIV infection. Curr Opin HIV AIDS 11: 226–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thangaraj A, Periyasamy P, Liao K, Bendi VS, Callen S, Pendyala G, Buch S. 2018. HIV-1 TAT-mediated microglial activation: role of mitochondrial dysfunction and defective mitophagy. Autophagy 14: 1596–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samikkannu T, Atluri VS, Arias AY, Rao KV, Mulet CT, Jayant RD, Nair MP. 2014. HIV-1 subtypes B and C Tat differentially impact synaptic plasticity expression and implicates HIV-associated neurocognitive disorders. Curr HIV Res 12: 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. 2001. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78: 457–67 [DOI] [PubMed] [Google Scholar]
- 38.Hu XT. 2016. HIV-1 Tat-Mediated Calcium Dysregulation and Neuronal Dysfunction in Vulnerable Brain Regions. Curr Drug Targets 17: 4–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Var SR, Day TR, Vitomirov A, Smith DM, Soontornniyomkij V, Moore DJ, Achim CL, Mehta SR, Perez-Santiago J. 2016. Mitochondrial injury and cognitive function in HIV infection and methamphetamine use. AIDS 30: 839–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kessing CF, Spudich S, Valcour V, Cartwright P, Chalermchai T, Fletcher JL, Takata H, Nichols C, Josey BJ, Slike B, Krebs SJ, Sailsuta N, Lerdlum S, Jagodzinski L, Tipsuk S, Suttichom D, Rattanamanee S, Zetterberg H, Hellmuth J, Phanuphak N, Robb ML, Michael NL, Ananworanich J, Trautmann L. 2017. High Number of Activated CD8+ T Cells Targeting HIV Antigens Are Present in Cerebrospinal Fluid in Acute HIV Infection. J Acquir Immune Defic Syndr 75: 108–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valcour VG, Spudich SS, Sailasuta N, Phanuphak N, Lerdlum S, Fletcher JL, Kroon ED, Jagodzinski LL, Allen IE, Adams CL, Prueksakaew P, Slike BM, Hellmuth JM, Kim JH, Ananworanich J, Group SRS. 2015. Neurological Response to cART vs. cART plus Integrase Inhibitor and CCR5 Antagonist Initiated during Acute HIV. PLoS One 10: e0142600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ho EL, Ronquillo R, Altmeppen H, Spudich SS, Price RW, Sinclair E. 2013. Cellular Composition of Cerebrospinal Fluid in HIV-1 Infected and Uninfected Subjects. PLoS One 8: e66188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Subra FF C, Buranapraditkun S, Chan P, Sacdalan C, Tangnaree K, Rolland M, Krebs S, Sailasuta N, Tovanabutra S, Paul R, Michael NL, Robb M, Ananworanich J, Spudich S, Trautmann L 2019. CSF HIV Specific T Cells Persist During ART and Associate with Lower CNS Inflammation. Presented at CROI, Seattle, WA [Google Scholar]
- 44.Ananworanich J, Sacdalan CP, Pinyakorn S, Chomont N, de Souza M, Luekasemsuk T, Schuetz A, Krebs SJ, Dewar R, Jagodzinski L, Ubolyam S, Trichavaroj R, Tovanabutra S, Spudich S, Valcour V, Sereti I, Michael N, Robb M, Phanuphak P, Kim JH, Phanuphak N. 2016. Virological and immunological characteristics of HIV-infected individuals at the earliest stage of infection. J Virus Erad 2: 43–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shive CL, Jiang W, Anthony DD, Lederman MM. 2015. Soluble CD14 is a nonspecific marker of monocyte activation. AIDS 29: 1263–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burdo TH, Lentz MR, Autissier P, Krishnan A, Halpern E, Letendre S, Rosenberg ES, Ellis RJ, Williams KC. 2011. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. J Infect Dis 204: 154–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takata H, Buranapraditkun S, Kessing C, Fletcher JL, Muir R, Tardif V, Cartwright P, Vandergeeten C, Bakeman W, Nichols CN, Pinyakorn S, Hansasuta P, Kroon E, Chalermchai T, O’Connell R, Kim J, Phanuphak N, Robb ML, Michael NL, Chomont N, Haddad EK, Ananworanich J, Trautmann L, Rv254/Search, the RVSSG. 2017. Delayed differentiation of potent effector CD8(+) T cells reducing viremia and reservoir seeding in acute HIV infection. Sci Transl Med 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sailasuta N, Ross W, Ananworanich J, Chalermchai T, DeGruttola V, Lerdlum S, Pothisri M, Busovaca E, Ratto-Kim S, Jagodzinski L, Spudich S, Michael N, Kim JH, Valcour V, teams RSp. 2012. Change in brain magnetic resonance spectroscopy after treatment during acute HIV infection. PLoS One 7: e49272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tovanabutra S, Sirijatuphat R, Pham PT, Bonar L, Harbolick EA, Bose M, Song H, Chang D, Oropeza C, O’Sullivan AM, Balinang J, Kroon E, Colby DJ, Sacdalan C, Hellmuth J, Chan P, Prueksakaew P, Pinyakorn S, Jagodzinski LL, Sutthichom D, Pattamaswin S, de Souza M, Gramzinski RA, Kim JH, Michael NL, Robb ML, Phanuphak N, Ananworanich J, Valcour V, Kijak GH, Sanders-Buell E, Spudich S, Core MVS, Team RSS. 2019. Deep Sequencing Reveals Central Nervous System Compartmentalization in Multiple Transmitted/Founder Virus Acute HIV-1 Infection. Cells 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joseph SB, Kincer LP, Bowman NM, Evans C, Vinikoor MJ, Lippincott CK, Gisslen M, Spudich S, Menezes P, Robertson K, Archin N, Kashuba A, Eron JJ, Price RW, Swanstrom R. 2019. Human Immunodeficiency Virus Type 1 RNA Detected in the Central Nervous System (CNS) After Years of Suppressive Antiretroviral Therapy Can Originate from a Replicating CNS Reservoir or Clonally Expanded Cells. Clin Infect Dis 69: 1345–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R. 2011. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog 7: e1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lustig G, Cele S, Karim F, Derache A, Ngoepe A, Khan K, Gosnell BI, Moosa MS, Ntshuba N, Marais S, Jeena PM, Govender K, Adamson J, Kloverpris H, Gupta RK, Harrichandparsad R, Patel VB, Sigal A. 2021. T cell derived HIV-1 is present in the CSF in the face of suppressive antiretroviral therapy. PLoS Pathog 17: e1009871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balinang JC D; Jobe O; Sanders-Buell E; Chenine A; Mann B; Merbah M; Alvarez-Carbonell D; Bose M; Barrows B; et al. October 21–25 2018. Differential Infection of Cultured Peripheral and CNS Cells by Distinct Transmitted/Founder HIV-1 Infectious Molecular Clones. Presented at Proceedings of the HIV Research for Prevention (HIVR4P 2018), Madrid, Spain [Google Scholar]
- 54.Spudich S, Clements JE. 2019. HIV persistence in the central nervous system during antiretroviral therapy: evidence and implications. AIDS 33 Suppl 2: S103–S6 [DOI] [PubMed] [Google Scholar]
- 55.Honeycutt JB, Liao B, Nixon CC, Cleary RA, Thayer WO, Birath SL, Swanson MD, Sheridan P, Zakharova O, Prince F, Kuruc J, Gay CL, Evans C, Eron JJ, Wahl A, Garcia JV. 2018. T cells establish and maintain CNS viral infection in HIV-infected humanized mice. J Clin Invest 128: 2862–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smolders J, Remmerswaal EB, Schuurman KG, Melief J, van Eden CG, van Lier RA, Huitinga I, Hamann J. 2013. Characteristics of differentiated CD8(+) and CD4 (+) T cells present in the human brain. Acta Neuropathol 126: 525–35 [DOI] [PubMed] [Google Scholar]
- 57.Strazielle N, Creidy R, Malcus C, Boucraut J, Ghersi-Egea JF. 2016. T-Lymphocytes Traffic into the Brain across the Blood-CSF Barrier: Evidence Using a Reconstituted Choroid Plexus Epithelium. PLoS One 11: e0150945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farhadian SF, Mehta SS, Zografou C, Robertson K, Price RW, Pappalardo J, Chiarella J, Hafler DA, Spudich SS. 2018. Single-cell RNA sequencing reveals microglia-like cells in cerebrospinal fluid during virologically suppressed HIV. JCI Insight 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gray LR, Cowley D, Welsh C, Lu HK, Brew BJ, Lewin SR, Wesselingh SL, Gorry PR, Churchill MJ. 2016. CNS-specific regulatory elements in brain-derived HIV-1 strains affect responses to latency-reversing agents with implications for cure strategies. Mol Psychiatry 21: 574–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li J, Chen C, Ma X, Geng G, Liu B, Zhang Y, Zhang S, Zhong F, Liu C, Yin Y, Cai W, Zhang H. 2016. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat Commun 7: 11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turrini F, Marelli S, Kajaste-Rudnitski A, Lusic M, Van Lint C, Das AT, Harwig A, Berkhout B, Vicenzi E. 2015. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology 12: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marcello A, Ferrari A, Pellegrini V, Pegoraro G, Lusic M, Beltram F, Giacca M. 2003. Recruitment of human cyclin T1 to nuclear bodies through direct interaction with the PML protein. EMBO J 22: 2156–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maine GN, Mao X, Komarck CM, Burstein E. 2007. COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J 26: 436–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alvarez-Carbonell D, Ye F, Ramanath N, Garcia-Mesa Y, Knapp PE, Hauser KF, Karn J. 2019. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog 15: e1008249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H, Alitalo K. 2015. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 212: 991–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chaillon A, Gianella S, Dellicour S, Rawlings SA, Schlub TE, De Oliveira MF, Ignacio C, Porrachia M, Vrancken B, Smith DM. 2020. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J Clin Invest 130: 1699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei J, Hou J, Su B, Jiang T, Guo C, Wang W, Zhang Y, Chang B, Wu H, Zhang T. 2020. The Prevalence of Frascati-Criteria-Based HIV-Associated Neurocognitive Disorder (HAND) in HIV-Infected Adults: A Systematic Review and Meta-Analysis. Front Neurol 11: 581346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. 2007. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69: 1789–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nightingale S, Dreyer AJ, Saylor D, Gisslen M, Winston A, Joska JA. 2021. Moving on From HAND: Why We Need New Criteria for Cognitive Impairment in Persons Living With Human Immunodeficiency Virus and a Proposed Way Forward. Clin Infect Dis 73: 1113–8 [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, Liu M, Lu Q, Farrell M, Lappin JM, Shi J, Lu L, Bao Y. 2020. Global prevalence and burden of HIV-associated neurocognitive disorder: A meta-analysis. Neurology 95: e2610–e21 [DOI] [PubMed] [Google Scholar]
- 71.Langford D, Marquie-Beck J, de Almeida S, Lazzaretto D, Letendre S, Grant I, McCutchan JA, Masliah E, Ellis RJ. 2006. Relationship of antiretroviral treatment to postmortem brain tissue viral load in human immunodeficiency virus-infected patients. J Neurovirol 12: 100–7 [DOI] [PubMed] [Google Scholar]
- 72.Jessen Krut J, Mellberg T, Price RW, Hagberg L, Fuchs D, Rosengren L, Nilsson S, Zetterberg H, Gisslen M. 2014. Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS One 9: e88591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.D’Antoni ML, Byron MM, Chan P, Sailasuta N, Sacdalan C, Sithinamsuwan P, Tipsuk S, Pinyakorn S, Kroon E, Slike BM, Krebs SJ, Khadka VS, Chalermchai T, Kallianpur KJ, Robb M, Spudich S, Valcour V, Ananworanich J, Ndhlovu LC, Rv254/Search S, Groups RSS. 2018. Normalization of Soluble CD163 Levels After Institution of Antiretroviral Therapy During Acute HIV Infection Tracks with Fewer Neurological Abnormalities. J Infect Dis 218: 1453–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gisslen M, Heslegrave A, Veleva E, Yilmaz A, Andersson LM, Hagberg L, Spudich S, Fuchs D, Price RW, Zetterberg H. 2019. CSF concentrations of soluble TREM2 as a marker of microglial activation in HIV-1 infection. Neurol Neuroimmunol Neuroinflamm 6: e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hellmuth J, Slike BM, Sacdalan C, Best J, Kroon E, Phanuphak N, Fletcher JLK, Prueksakaew P, Jagodzinski LL, Valcour V, Robb M, Ananworanich J, Allen IE, Krebs SJ, Spudich S. 2019. Very Early Initiation of Antiretroviral Therapy During Acute HIV Infection Is Associated With Normalized Levels of Immune Activation Markers in Cerebrospinal Fluid but Not in Plasma. J Infect Dis 220: 1885–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burbelo PD, Price RW, Hagberg L, Hatano H, Spudich S, Deeks SG, Gisslen M. 2018. Anti-Human Immunodeficiency Virus Antibodies in the Cerebrospinal Fluid: Evidence of Early Treatment Impact on Central Nervous System Reservoir? J Infect Dis 217: 1024–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bundell C, Brunt SJ, Cysique LA, Brusch A, Brew BJ, Price P. 2018. The high frequency of autoantibodies in HIV patients declines on antiretroviral therapy. Pathology 50: 313–6 [DOI] [PubMed] [Google Scholar]
- 78.Oliveira MF, Chaillon A, Nakazawa M, Vargas M, Letendre SL, Strain MC, Ellis RJ, Morris S, Little SJ, Smith DM, Gianella S. 2017. Early Antiretroviral Therapy Is Associated with Lower HIV DNA Molecular Diversity and Lower Inflammation in Cerebrospinal Fluid but Does Not Prevent the Establishment of Compartmentalized HIV DNA Populations. PLoS Pathog 13: e1006112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Winston A, Antinori A, Cinque P, Fox HS, Gisslen M, Henrich TJ, Letendre S, Persaud D, Price RW, Spudich S. 2019. Defining cerebrospinal fluid HIV RNA escape: editorial review AIDS. AIDS 33 Suppl 2: S107–S11 [DOI] [PubMed] [Google Scholar]
- 80.Eden A, Fuchs D, Hagberg L, Nilsson S, Spudich S, Svennerholm B, Price RW, Gisslen M. 2010. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis 202: 1819–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Handoko R, Chan P, Jagodzinski L, Pinyakorn S, Ubolyam S, Phanuphak N, Sacdalan C, Kroon E, Dumrongpisutikul N, Paul R, Valcour V, Ananworanich J, Vasan S, Spudich S, Team SRS. 2021. Minimal detection of cerebrospinal fluid escape after initiation of antiretroviral therapy in acute HIV-1 infection. AIDS 35: 777–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Almeida SM, Rotta I, de Pereira AP, Tang B, Umlauf A, Ribeiro CEL, Letendre S, Ellis RJ, Group HIVNRC. 2020. Cerebrospinal fluid pleocytosis as a predictive factor for CSF and plasma HIV RNA discordance and escape. J Neurovirol 26: 241–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manesh A, Barnabas R, Mani S, Karthik R, Abraham OC, Chacko G, Kannangai R, Varghese GM. 2019. Symptomatic HIV CNS viral escape among patients on effective cART. Int J Infect Dis 84: 39–43 [DOI] [PubMed] [Google Scholar]
- 84.Moloney PB, Hutchinson S, Heskin J, Mulcahy F, Langan Y, Conlon NP, Linas BP, Takahashi C, Cervantes-Arslanian AM. 2020. Possible N-methyl-D-aspartate receptor antibody-mediated encephalitis in the setting of HIV cerebrospinal fluid escape. J Neurol 267: 1348–52 [DOI] [PubMed] [Google Scholar]
- 85.Peluso MJ, Ferretti F, Peterson J, Lee E, Fuchs D, Boschini A, Gisslen M, Angoff N, Price RW, Cinque P, Spudich S. 2012. Cerebrospinal fluid HIV escape associated with progressive neurologic dysfunction in patients on antiretroviral therapy with well controlled plasma viral load. AIDS 26: 1765–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Narvid J, Callen A, Talbott J, Uzelac A, Dupont SM, Chow F, Price RW, Rehani B. 2018. Brain MRI Features of CSF Human Immunodeficiency Virus Escape. J Neuroimaging 28: 601–7 [DOI] [PubMed] [Google Scholar]
- 87.Mukerji SS, Misra V, Lorenz D, Cervantes-Arslanian AM, Lyons J, Chalkias S, Wurcel A, Burke D, Venna N, Morgello S, Koralnik IJ, Gabuzda D. 2017. Temporal Patterns and Drug Resistance in CSF Viral Escape Among ART-Experienced HIV-1 Infected Adults. J Acquir Immune Defic Syndr 75: 246–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nightingale S, Geretti AM, Beloukas A, Fisher M, Winston A, Else L, Nelson M, Taylor S, Ustianowski A, Ainsworth J, Gilson R, Haddow L, Ong E, Watson V, Leen C, Minton J, Post F, Pirmohamed M, Solomon T, Khoo S. 2016. Discordant CSF/plasma HIV-1 RNA in patients with unexplained low-level viraemia. J Neurovirol 22: 852–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Swanta N, Aryal S, Nejtek V, Shenoy S, Ghorpade A, Borgmann K. 2020. Blood-based inflammation biomarkers of neurocognitive impairment in people living with HIV. J Neurovirol 26: 358–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hong S, Banks WA. 2015. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun 45: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vera JH, Guo Q, Cole JH, Boasso A, Greathead L, Kelleher P, Rabiner EA, Kalk N, Bishop C, Gunn RN, Matthews PM, Winston A. 2016. Neuroinflammation in treated HIV-positive individuals: A TSPO PET study. Neurology 86: 1425–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kamat A, Lyons JL, Misra V, Uno H, Morgello S, Singer EJ, Gabuzda D. 2012. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J Acquir Immune Defic Syndr 60: 234–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nolan RA, Muir R, Runner K, Haddad EK, Gaskill PJ. 2019. Role of Macrophage Dopamine Receptors in Mediating Cytokine Production: Implications for Neuroinflammation in the Context of HIV-Associated Neurocognitive Disorders. J Neuroimmune Pharmacol 14: 134–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yilmaz A, Yiannoutsos CT, Fuchs D, Price RW, Crozier K, Hagberg L, Spudich S, Gisslen M. 2013. Cerebrospinal fluid neopterin decay characteristics after initiation of antiretroviral therapy. J Neuroinflammation 10: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brew BJ, Dunbar N, Pemberton L, Kaldor J. 1996. Predictive markers of AIDS dementia complex: CD4 cell count and cerebrospinal fluid concentrations of beta 2-microglobulin and neopterin. J Infect Dis 174: 294–8 [DOI] [PubMed] [Google Scholar]
- 96.Eden A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslen M. 2007. Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J Infect Dis 196: 1779–83 [DOI] [PubMed] [Google Scholar]
- 97.Farhadian SF, Mistry H, Kirchwey T, Chiarella J, Calvi R, Chintanaphol M, Patel P, Landry ML, Robertson K, Spudich SS. 2019. Markers of CNS Injury in Adults Living With HIV With CSF HIV Not Detected vs Detected <20 Copies/mL. Open Forum Infect Dis 6: ofz528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pfefferbaum A, Rogosa DA, Rosenbloom MJ, Chu W, Sassoon SA, Kemper CA, Deresinski S, Rohlfing T, Zahr NM, Sullivan EV. 2014. Accelerated aging of selective brain structures in human immunodeficiency virus infection: a controlled, longitudinal magnetic resonance imaging study. Neurobiol Aging 35: 1755–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jernigan TL, Archibald SL, Fennema-Notestine C, Taylor MJ, Theilmann RJ, Julaton MD, Notestine RJ, Wolfson T, Letendre SL, Ellis RJ, Heaton RK, Gamst AC, Franklin DR Jr., Clifford DB, Collier AC, Gelman BB, Marra C, McArthur JC, McCutchan JA, Morgello S, Simpson DM, Grant I, Group C. 2011. Clinical factors related to brain structure in HIV: the CHARTER study. J Neurovirol 17: 248–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kovacsics CE, Gill AJ, Ambegaokar SS, Gelman BB, Kolson DL. 2017. Degradation of heme oxygenase-1 by the immunoproteasome in astrocytes: A potential interferon-gamma-dependent mechanism contributing to HIV neuropathogenesis. Glia 65: 1264–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dos Reis RS, Sant S, Keeney H, Wagner MCE, Ayyavoo V. 2020. Modeling HIV-1 neuropathogenesis using three-dimensional human brain organoids (hBORGs) with HIV-1 infected microglia. Sci Rep 10: 15209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lucas SB, Wong KT, Nightingale S, Miller RF. 2021. HIV-Associated CD8 Encephalitis: A UK Case Series and Review of Histopathologically Confirmed Cases. Front Neurol 12: 628296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sharma SK, Soneja M. 2011. HIV & immune reconstitution inflammatory syndrome (IRIS). Indian J Med Res 134: 866–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huis in ‘t Veld D, Sun HY, Hung CC, Colebunders R. 2012. The immune reconstitution inflammatory syndrome related to HIV co-infections: a review. Eur J Clin Microbiol Infect Dis 31: 919–27 [DOI] [PubMed] [Google Scholar]
- 105.Gendelman HE. 2020. Predictive biomarkers for cognitive decline during progressive HIV infection. EBioMedicine 51: 102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guha D, Mukerji SS, Chettimada S, Misra V, Lorenz DR, Morgello S, Gabuzda D. 2019. Cerebrospinal fluid extracellular vesicles and neurofilament light protein as biomarkers of central nervous system injury in HIV-infected patients on antiretroviral therapy. AIDS 33: 615–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pulliam L, Sun B, Mustapic M, Chawla S, Kapogiannis D. 2019. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J Neurovirol 25: 702–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sun B, Dalvi P, Abadjian L, Tang N, Pulliam L. 2017. Blood neuron-derived exosomes as biomarkers of cognitive impairment in HIV. AIDS 31: F9–F17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Paudel YN, Shaikh MF, Chakraborti A, Kumari Y, Aledo-Serrano A, Aleksovska K, Alvim MKM, Othman I. 2018. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front Neurosci 12: 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Megra BW, Eugenin EA, Berman JW. 2017. The Role of Shed PrP(c) in the Neuropathogenesis of HIV Infection. J Immunol 199: 224–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roberts TK, Eugenin EA, Morgello S, Clements JE, Zink MC, Berman JW. 2010. PrPC, the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammation. Am J Pathol 177: 1848–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, Kruse AC, Blacklow SC. 2017. Structural Basis for Regulated Proteolysis by the alpha-Secretase ADAM10. Cell 171: 1638–48 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.de Almeida SM, Ribeiro CE, Rotta I, Piovesan M, Tang B, Vaida F, Raboni SM, Letendre S, Potter M, Batistela Fernandes MS, Ellis RJ, Group HIVNRC. 2018. Biomarkers of neuronal injury and amyloid metabolism in the cerebrospinal fluid of patients infected with HIV-1 subtypes B and C. J Neurovirol 24: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ellis RJ, Moore DJ, Sundermann EE, Heaton RK, Mehta S, Hulgan T, Samuels D, Fields JA, Letendre SL. 2020. Nucleic acid oxidation is associated with biomarkers of neurodegeneration in CSF in people with HIV. Neurol Neuroimmunol Neuroinflamm 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Velasquez S, Prevedel L, Valdebenito S, Gorska AM, Golovko M, Khan N, Geiger J, Eugenin EA. 2020. Circulating levels of ATP is a biomarker of HIV cognitive impairment. EBioMedicine 51: 102503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bandera A, Taramasso L, Bozzi G, Muscatello A, Robinson JA, Burdo TH, Gori A. 2019. HIV-Associated Neurocognitive Impairment in the Modern ART Era: Are We Close to Discovering Reliable Biomarkers in the Setting of Virological Suppression? Front Aging Neurosci 11: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Weiss JJ, Calvi R, Naganawa M, Toyonaga T, Farhadian SF, Chintanaphol M, Chiarella J, Zheng MQ, Ropchan J, Huang Y, Pietrzak RH, Carson RE, Spudich S. 2021. Preliminary In Vivo Evidence of Reduced Synaptic Density in Human Immunodeficiency Virus (HIV) Despite Antiretroviral Therapy. Clin Infect Dis 73: 1404–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gong Y, Chowdhury P, Nagesh PKB, Rahman MA, Zhi K, Yallapu MM, Kumar S. 2020. Novel elvitegravir nanoformulation for drug delivery across the blood-brain barrier to achieve HIV-1 suppression in the CNS macrophages. Sci Rep 10: 3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Roy U, Drozd V, Durygin A, Rodriguez J, Barber P, Atluri V, Liu X, Voss TG, Saxena S, Nair M. 2018. Characterization of Nanodiamond-based anti-HIV drug Delivery to the Brain. Sci Rep 8: 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pausch P, Al-Shayeb B, Bisom-Rapp E, Tsuchida CA, Li Z, Cress BF, Knott GJ, Jacobsen SE, Banfield JF, Doudna JA. 2020. CRISPR-CasPhi from huge phages is a hypercompact genome editor. Science 369: 333–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yan WX, Hunnewell P, Alfonse LE, Carte JM, Keston-Smith E, Sothiselvam S, Garrity AJ, Chong S, Makarova KS, Koonin EV, Cheng DR, Scott DA. 2019. Functionally diverse type V CRISPR-Cas systems. Science 363: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kunze C, Borner K, Kienle E, Orschmann T, Rusha E, Schneider M, Radivojkov-Blagojevic M, Drukker M, Desbordes S, Grimm D, Brack-Werner R. 2018. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia 66: 413–27 [DOI] [PubMed] [Google Scholar]
- 123.Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, Chen C, Mancuso P, Sariyer R, Ferrante P, Donadoni M, Robinson JA, Sillman B, Lin Z, Hilaire JR, Banoub M, Elango M, Gautam N, Mosley RL, Poluektova LY, McMillan J, Bade AN, Gorantla S, Sariyer IK, Burdo TH, Young WB, Amini S, Gordon J, Jacobson JM, Edagwa B, Khalili K, Gendelman HE. 2019. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat Commun 10: 2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gavegnano C, Haile WB, Hurwitz S, Tao S, Jiang Y, Schinazi RF, Tyor WR. 2019. Baricitinib reverses HIV-associated neurocognitive disorders in a SCID mouse model and reservoir seeding in vitro. J Neuroinflammation 16: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ambrosius B, Gold R, Chan A, Faissner S. 2019. Antineuroinflammatory drugs in HIV-associated neurocognitive disorders as potential therapy. Neurol Neuroimmunol Neuroinflamm 6: e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cross SA, Cook DR, Chi AW, Vance PJ, Kolson LL, Wong BJ, Jordan-Sciutto KL, Kolson DL. 2011. Dimethyl fumarate, an immune modulator and inducer of the antioxidant response, suppresses HIV replication and macrophage-mediated neurotoxicity: a novel candidate for HIV neuroprotection. J Immunol 187: 5015–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Read SW, DeGrezia M, Ciccone EJ, DerSimonian R, Higgins J, Adelsberger JW, Starling JM, Rehm C, Sereti I. 2010. The effect of leflunomide on cycling and activation of T-cells in HIV-1-infected participants. PLoS One 5: e11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sacktor N, Miyahara S, Evans S, Schifitto G, Cohen B, Haughey N, Drewes JL, Graham D, Zink MC, Anderson C, Nath A, Pardo CA, McCarthy S, Hosey L, Clifford D, team AA. 2014. Impact of minocycline on cerebrospinal fluid markers of oxidative stress, neuronal injury, and inflammation in HIV-seropositive individuals with cognitive impairment. J Neurovirol 20: 620–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barber-Axthelm IM, Barber-Axthelm V, Sze KY, Zhen A, Suryawanshi GW, Chen IS, Zack JA, Kitchen SG, Kiem HP, Peterson CW. 2021. Stem cell-derived CAR T cells traffic to HIV reservoirs in macaques. JCI Insight 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ait-Ammar A, Kula A, Darcis G, Verdikt R, De Wit S, Gautier V, Mallon PWG, Marcello A, Rohr O, Van Lint C. 2019. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front Microbiol 10: 3060. [DOI] [PMC free article] [PubMed] [Google Scholar]