

Abstract
To date, more than 37 amyloidogenic proteins have been found to form toxic aggregates that are implicated in the progression of numerous debilitating protein misfolding diseases including Alzheimer's disease (AD). Extensive literature highlights the role of β‐amyloid (Aβ) aggregates in causing excessive neuronal cell loss in the brains of AD patients. In fact, major advances in our understanding of Aβ aggregation process, including kinetics, toxicity, and structures of fibrillar aggregates have been revealed by examining in vitro preparations of synthetic Aβ peptides. However, ongoing research shows that brain‐derived Aβ aggregates have specific characteristics that distinguish them from in vitro prepared species. Notably, the molecular structures of amyloid fibrils grown in the human brain were found to be markedly different than synthetic Aβ fibrils. In addition, recent findings report the existence of heterogeneous Aβ proteoforms in AD brain tissue in contrast to synthetically produced full‐length aggregates. Despite their high relevance to AD progression, brain‐derived Aβ species are less well‐characterized compared with synthetic aggregates. The aim of this review is to provide an overview of the literature on brain‐derived Aβ aggregates with particular focus on recent studies that report their structures as well as pathological roles in AD progression. The main motivation of this review is to highlight the importance of utilizing brain‐derived amyloids for characterizing the structural and toxic effects of amyloid species. With this knowledge, brain‐derived aggregates can be adopted to identify more relevant drug targets and validate potent aggregation inhibitors toward designing highly effective therapeutic strategies against AD.
Keywords: cortical brain extracts, Cryo‐EM, in vitro aggregation, natural amyloids, neurotoxicity, polymorphism, soluble oligomers, structural biology
1. INTRODUCTION
The misfolding and aggregation of amyloidogenic proteins characterize the progression of numerous protein misfolding diseases including the highly prevalent Alzheimer's disease (AD) (Chiti & Dobson, 2017; Dobson, 2004; Knowles et al., 2014; Lee et al., 2020). AD is an irreversible and progressive neurodegenerative disorder that accounts for the majority of dementia cases (60%–70%) (DeTure & Dickson, 2019; Prince et al., 2016). AD is clinically described by the progressive deterioration of cognitive functions and the gradual loss of memory which eventually interfere with many bodily functions (DeTure & Dickson, 2019; Jack et al., 2011). Neuropathologically, AD is characterized by neuronal cell loss due to two cellular hallmarks that were identified in the brains of AD patients, including the extracellular accumulation of β‐Amyloid (Aβ) peptide in the form of senile plaques, and the intracellular accumulation of hyper‐phosphorylated tau protein in the form of neurofibrillary tangles (Anand et al., 2014; Jack et al., 2018; Serrano‐Pozo et al., 2011). Studies have shown that Aβ monomers are produced in healthy brains at low concentrations where they hold potential physiological functions in maintaining neuronal activity and synaptic plasticity (Abramov et al., 2009; Giuffrida et al., 2009; Puzzo et al., 2015). However, excessive Aβ levels detected in AD patients induce the conversion of Aβ monomers into cytotoxic oligomers and fibrils (Puzzo et al., 2015; Resende et al., 2008). In fact, Aβ oligomers have been shown to play central roles in inducing neurotoxicity and synaptic loss in AD pathology (Carrillo‐Mora et al., 2014; Huang & Liu, 2020; Kayed & Lasagna‐Reeves, 2013).
In vitro aggregation of synthetic proteins has been extensively examined in literature with the aim of elucidating the pathways of protein aggregation (Chiti & Dobson, 2017). Such in vitro systems provide controlled conditions for monitoring and characterizing the formation of aggregated species (Villar‐Piqué et al., 2018). In fact, using in vitro aggregation models enabled substantial advances in understanding key aspects of the synthetic Aβ aggregation mechanism including kinetics of self‐assembly pathways (S. I. A. Cohen et al., 2013; Knowles et al., 2009), structure of synthetic Aβ aggregates (Ciudad et al., 2020; Colvin et al., 2016; Gremer et al., 2017; Hu et al., 2019; Lührs et al., 2005; Paravastu et al., 2008; Qiang et al., 2012; Schmidt et al., 2015; Schütz et al., 2015; Wälti et al., 2016; Xiao et al., 2015) and the cytotoxicity of oligomeric and fibrillar species (Bemporad & Chiti, 2012; Campioni et al., 2010; Cecchi & Stefani, 2013; De et al., 2019; Gonzalez‐Velasquez et al., 2008; Hoshi et al., 2003; Lorenzo & Yankner, 1996; Mannini et al., 2014; Ono et al., 2009; Sakono & Zako, 2010). Although in vitro parameters such as temperature, ionic strength, and pH can be tuned to resemble the in vivo environment, it is highly challenging to accurately mimic the complexity of the cellular microenvironment that, in turn, has profound effects on the actual in vivo aggregation process (Brody et al., 2017; Brody & Gross, 2014; Li & Stern, 2022).
In vitro synthetic fibrils have been shown by several studies to have polymorphic structures that are dependent on growth conditions rather than specific amino acid sequences (Diaz‐Espinoza, 2021; Fändrich et al., 2009; Petkova et al., 2005; Riek, 2017; Wu et al., 2010). These findings suggest that the molecular structure of amyloid species grown in the human brain cannot be determined by in vitro synthetic fibrils or fibrils extracted from the brains of transgenic animals (Brody & Gross, 2014). Previous studies reported that brain‐derived fibrils extracted from AD patients are morphologically distinct than purely synthetic fibrils (Lu et al., 2013; Paravastu et al., 2009). Particularly, the handedness of the fibrils' twist, key residue distances, residue contacts, and protofilament arrangements are reported to vary between synthetic and brain‐derived fibrils (Kollmer et al., 2019; Lu et al., 2013; Paravastu et al., 2008; Paravastu et al., 2009).
Ongoing research has shown that brain‐derived amyloids have specific characteristics that distinguish them from in vitro‐prepared fibrils. Aβ aggregates derived from brain‐extracts have higher resistance to proteinase K as compared to in vitro synthetic Aβ aggregates (Langer et al., 2011). Furthermore, recent findings suggest that brain‐derived Aβ oligomers have higher neurotoxicity than synthetic oligomers (Brody & Gross, 2014; Li & Stern, 2022). Importantly, the analysis of Aβ proteoforms present in AD brain tissues reveals the diversity of Aβ post‐translational modifications, particularly N‐terminal and C‐terminal truncations that are absent from synthetic full‐length Aβ peptides (Arai et al., 1999; Cabrera et al., 2018; Güntert et al., 2006; Kummer & Heneka, 2014; Milton, 2001; Rijal Upadhaya et al., 2014; Rostagno et al., 2018; Sergeant et al., 2003; Wildburger et al., 2017).
Despite their high relevance to AD progression, brain‐derived Aβ species are less characterized as compared to synthetic aggregates. In this work, we aim to review the literature on brain‐derived Aβ aggregates that have been purified from cerebral cortical tissues or meningeal tissues of deceased clinicopathologically confirmed AD patients. Next, we discuss the polymorphic features of synthetic and brain‐derived fibrils. Given the well‐known pathological roles of Aβ oligomers, we focus on literature findings that describe the neurotoxic roles of natural brain‐derived oligomers which have been extracted at autopsy from brain tissues of confirmed AD patients. Particularly, we highlight studies that utilized in vitro systems for examining the pathological roles of AD brain‐derived Aβ oligomers and dimers.
2. PROTEIN AGGREGATION IN HUMAN DISEASES
The high complexity of the cellular environment, which is crowded with a high concentration (300–400 mg/mL) of various proteins and other macromolecules, necessitates key assistive factors to maintain proteome homeostasis (proteostasis) (Balchin et al., 2016; Dobson, 2004; Vendruscolo, 2022). Molecular chaperones and folding catalysts assist globular proteins to achieve their native states (Balchin et al., 2016; Dobson, 2004; Knowles et al., 2014; Lee, 2005). In addition, tight regulations of the synthesis, stability, diversity, and degradation of intrinsically disordered proteins (IDPs) is essential for controlling their self‐ and non‐self‐interactions and preventing their potential pathological effects (Gsponer et al., 2008; Kundra et al., 2017; Theillet et al., 2014). Proteostasis is considered quite robust as it tightly regulates protein synthesis, protein turnover, the clearance of misfolded proteins, and the stress‐induced cellular responses (Balchin et al., 2016). Unsurprisingly, the failure of a protein to fold correctly and/or to be cleared if it gets misfolded result in cellular malfunction and eventually lead to disease progression (Dobson, 2004).
The exact physiological causes of protein aggregation and the cellular events that lead to their accumulation are not yet fully understood. However, aging and stress conditions, both genetic and environmental, are key factors that challenge the cellular capacity to maintain proteostasis. As a result, misfolded proteins may escape the cellular quality control checks and transform into intracellular or extracellular intractable aggregates giving rise to a wide range of protein misfolding diseases (Balchin et al., 2016; Dobson, 2004; Hipp et al., 2014; Vendruscolo, 2012). Particularly, aging is associated with decreased proteasomal and autophagy functions, which is considered a key risk factor for the development of numerous protein misfolding diseases (Balchin et al., 2016; Yacoubian TA Neurodegenerative Disorders, 2017). Aging and/or stress conditions could lead to different in vivo events that promote the aggregation of proteins and peptides. These in vivo events include denaturation of a natively folded protein, overexpression of a certain protein that overwhelms proteostasis, peptide fragmentation, and overproduction of a natively misfolded peptide (Eisenberg & Jucker, 2012). In fact, Aβ aggregation results from the fragmentation of its large amyloid precursor protein (APP) (Dobson, 2004). The resulting peptide fragments may not be able to fold correctly in the absence of their precursor proteins which makes them vulnerable to self‐interaction and aggregation (Dobson, 2004).
3. EXTRACTION METHODS AND PROPERTIES OF BRAIN‐DERIVED Aβ AGGREGATES
Amyloid formation in the brains of individuals developing sporadic AD is believed to accumulate for more than a decade before the clinical diagnosis of AD symptoms (DeTure & Dickson, 2019; Dubois et al., 2016). The senile plaques, containing extracellular Aβ deposits, are found to populate different regions of AD brain patients but their presence with high density is typically observed in the temporal lobe, particularly hippocampus and entorhinal cortex (DeTure & Dickson, 2019; Sideris et al., 2021; Smith, 2002). In addition to brain parenchyma deposition, Aβ aggregates are observed to accumulate in the cortical blood vessels with an estimated 85%–95% of AD cases having cerebral amyloid deposition (Attems et al., 2010; DeTure & Dickson, 2019).
Aβ deposits in brain tissues constitute both oligomeric and fibrillar species that can be experimentally extracted using appropriate extraction protocols. The soluble and insoluble brain extracts can be subjected to specific antibody treatment that differentiate different aggregate species depending on their specific conformations. It has been shown that Aβ oligomers acting as kinetic intermediates during the fibrillation process are recognized by A11 antibody (Kayed et al., 2003) whereas small fibrillar fragments that may act as seeds can be recognized by fibril‐specific OC antibody (Fritschi, Langer, et al., 2014).
The isolation of insoluble Aβ fibrils from senile plaques of cortical brain tissues, including the temporal, frontal, occipital or parietal regions was described by several papers (Fritschi, Langer, et al., 2014; Lu et al., 2013; Noguchi et al., 2009; Paravastu et al., 2009; Pedrero‐Prieto et al., 2019; Qiang et al., 2017; Roher & Kuo, 1999; Rostagno et al., 2018; Shankar et al., 2008). Although protocols have slight variations, the overall process is comparable as schematically summarized in Figure 1. Briefly, freshly frozen brain samples of the cerebral cortical tissues are minced and homogenized in an extraction buffer containing protease inhibitors. Next, centrifugation is performed to remove insoluble myelin, cellular debris, and an intermediate soluble supernatant; thus, leaving a pellet which contains the insoluble Aβ aggregates. The pellet can then be enzymatically hydrolyzed with collagenase and DNase I to remove contaminating collagen and DNA. Next, pellet is treated with detergent containing solutions to yield an amyloid‐enriched material that is further subjected in some studies to formic acid treatment to produce solubilized amyloid material (Fritschi, Langer, et al., 2014; Lu et al., 2013; Noguchi et al., 2009; Paravastu et al., 2009; Pedrero‐Prieto et al., 2019; Qiang et al., 2017; Roher & Kuo, 1999; Rostagno et al., 2018; Shankar et al., 2008).
FIGURE 1.
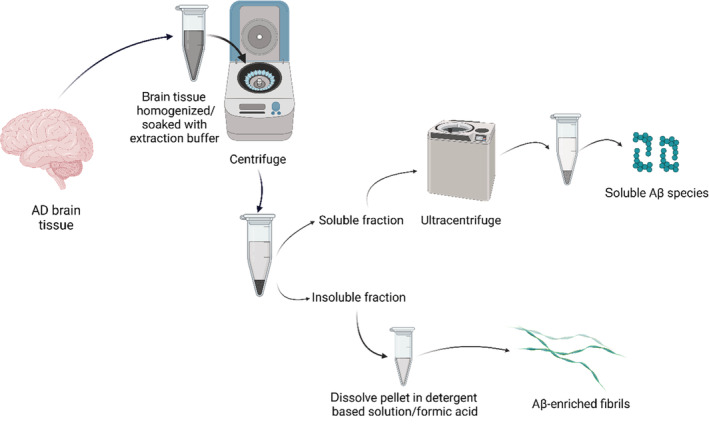
Schematic illustration of the overall extraction process of Aβ aggregates from brain tissues of deceased AD patients. Freshly frozen cortical brain samples are homogenized/soaked with extraction buffer and then centrifuged to obtain a soluble brain fraction supernatant and a mixture of insoluble brain fraction pellet. The insoluble pellet (1) is subjected to detergent‐containing buffer and/or formic acid to obtain solubilized amyloid‐enriched fibrils. On the other hand, the soluble brain fraction supernatant (2) is further subjected to ultracentrifugation and/or immunoprecipitation to selectively purify soluble Aβ oligomers and dimers.
The extraction of Aβ fibrils from vascular amyloid deposits of meningeal tissues was described by one paper that followed a fibril‐based water‐extraction protocol (Kollmer et al., 2019). Briefly, preliminary treatment and washing of tissue material with Tris calcium buffer is performed to remove soluble proteins. Next, an overnight collagenase treatment is applied and the resulting pellet is treated with 10 homogenization steps with Tris‐EDTA buffer where in each step, the supernatant is removed carefully and the resulting pellet is resuspended in Tris‐EDTA buffer. Next, 10 water‐based homogenization steps are performed to obtain the supernatant containing soluble amyloids in distilled water (Kollmer et al., 2019; Schubert et al., 1968).
In addition to the purification of insoluble amyloid fibrils, several studies described the purification of soluble AD brain extracts consisting of Aβ dimers (Brinkmalm et al., 2019; Brody & Gross, 2014; Jin et al., 2011; Lesné et al., 2013; Müller‐Schiffmann et al., 2016; Shankar et al., 2008; T. Yang et al., 2017) and small Aβ oligomers (Esparza et al., 2016; Fritschi, Langer, et al., 2014; Hong et al., 2018; Lesné et al., 2013; Li et al., 2011; Li & Stern, 2022; Noguchi et al., 2009; Sherman & Lesné, 2010; Sideris et al., 2021). The studies above performed the extraction of soluble brain fractions from supernatants retrieved by ultracentrifugation of AD brain tissues that were homogenized or soaked with an extraction buffer containing protease inhibitors.
Literature findings suggest the high stability and resistance properties of brain‐derived Aβ aggregates. Particularly, formaldehyde treatment of brain‐derived Aβ aggregates did not lead to their inactivation but slightly reduced their seeding activity in vivo (Fritschi, Cintron, et al., 2014). Moreover, brain‐derived Aβ aggregates have high resistance to proteinase K as compared to in vitro synthetic aggregates. A study showed that ultracentrifugation of brain‐extracts resulted in less than 0.05% of soluble Aβ species that had high in vivo seeding activity and Aβ deposition upon their intracerebral injection in APP transgenic mice (Langer et al., 2011). Another distinctive property of brain‐derived Aβ oligomers is their high binding affinity to calcium which enabled their purification using a specific calcium‐sensitive human monoclonal antibody (Stern et al., 2022). Interestingly, the reported Ca2+‐specific monoclonal antibody enabled the purification of amyloid fibrils in addition to soluble Aβ oligomers from AD cortical brain tissues (Stern et al., 2022).
The analysis of Aβ proteoforms present in AD brain tissues reveals the diversity of Aβ post‐translational modifications, particularly N‐terminal and C‐terminal truncations (Arai et al., 1999; Cabrera et al., 2018; Güntert et al., 2006; Kummer & Heneka, 2014; Milton, 2001; Rijal Upadhaya et al., 2014; Rostagno et al., 2018; Sergeant et al., 2003; Wildburger et al., 2017). Such posttranslational modifications of Aβ were found to impact the spreading, seeding, and neurotoxicity of amyloid deposits (Güntert et al., 2006; Kumar et al., 2011; Kumar et al., 2012; Kummer & Heneka, 2014; Rezaei‐Ghaleh et al., 2016; Rijal Upadhaya et al., 2014; Sergeant et al., 2003). N‐terminal truncations were found to be highly prevalent in AD brain extracts with an increase of 20% between Braak stages IV–VI (Güntert et al., 2006; Zampar et al., 2020). Several studies have used immunoprecipitation and mass spectrometry experiments to compare the prevalence of N‐terminal and C‐terminal truncated Aβ species between soluble and insoluble AD brain extracts. N‐terminal truncations were found to constitute 73% of the total identified Aβ proteoforms as opposed to C‐terminal truncations that had a lower prevalence ratio of 30% (Wildburger et al., 2017). In addition, N‐terminal truncations showed high aggregation propensities and were mainly purified from insoluble brain fractions unlike C‐terminal truncations that were mainly purified from soluble brain fractions (Cabrera et al., 2018; Rostagno et al., 2018; Wildburger et al., 2017; Zampar et al., 2020).
4. STRUCTURAL POLYMORPHISM OF SYNTHETIC AND BRAIN‐DERIVED Aβ FIBRILS
Fibrils formed by the self‐assembly of misfolded polypeptides share a common molecular subunit known as the cross‐β structure which consists of a repetitive two‐layered intermolecular β‐sheet motif (Diaz‐Espinoza, 2021). The pleated β‐sheets in the fibril core form by intermolecular hydrogen bonds that align parallel to the fibril axis and connect juxtaposed β‐strands of the misfolded polypeptide monomers (Diaz‐Espinoza, 2021; Fändrich et al., 2009; Pedersen et al., 2010; Riek, 2017; Wu et al., 2010). The side chains of the self‐assembled polypeptides extend perpendicular to the fibril axis and adopt various intra‐ and inter‐molecular interactions within the fibril (Fändrich et al., 2009). Different polypeptide sequences give rise to variations in the side‐chain spacing which defines the plane of the fibril cross‐section.
In the process of native protein folding, a given protein adopts a unique functional 3D conformation that consists of the same inter‐residue interactions. However, the process of fibril formation is different as the same protein can adopt different inter‐residue interactions or conformations during its self‐assembly giving rise to polymorphic fibrillar structures (Fändrich et al., 2009). In addition, polymorphism in fibrils can be described by other factors including fibril diameter, mass‐per‐length data, amount of fibrillar twisting, and protofilament number. Particularly, the protofilaments that form the substructure of mature amyloid fibrils can have different helical twisting which give rise to polymorphic fibrillar morphologies across different proteins. Electron microscopy (EM) can measure the fibril width and cross‐over distances which vary by the different arrangements of protofilaments (Fändrich et al., 2009; Petkova et al., 2005).
In fact, it has been experimentally shown in the literature that Aβ‐fibril polymorphism has a variable effect on neurodegenerative progression in AD (Condello et al., 2018; Tycko, 2015). An early study showed that different Aβ fibril morphologies are dependent on subtle variations in the fibril growth conditions. The study reported different toxic effect of polymorphic Aβ fibril morphologies on primary rat embryonic hippocampal neurons with Aβ fibrils grown under quiescent conditions having higher toxicity than those grown under agitated conditions (Petkova et al., 2005). At the in vivo level, studies showed differential neuropathological effects in transgenic mouse models injected with distinct polymorphic Aβ fibrils, including Aβ fibrils extracted from familial and sporadic AD cases, as well as synthetically produced Aβ fibrils with distinct C‐terminal variants (Ruiz‐Riquelme et al., 2021; Stöhr et al., 2014; Watts et al., 2014; Zhu et al., 2022). Synthetic Aβ fibrils injected into mice have been shown to be less potent in inducing Aβ deposition compared with brain‐derived fibrils (Stöhr et al., 2012). The different pathologies of AD cases might be correlated with structurally distinct polymorphic Aβ aggregates, as unique oligomeric particles have been detected in the cortical brain tissues of rapidly progressive AD cases (M. L. Cohen et al., 2015; M. Cohen et al., 2016). The findings above demonstrate that structural diversity of Aβ fibrils may play a central role in the progression of AD and that synthetic Aβ fibrils may not mimic and structure and toxic effect of brain‐derived Aβ aggregates.
Extensive research efforts have been devoted to elucidate the molecular structure of amyloid fibrils. Studies resolving the structures of in vitro fibrils grown from synthetic Aβ1–40 and Aβ1–42 reveal their polymorphic features as will be detailed in this section.
Most synthetic Aβ1–42 fibrils were reported to have twofold symmetry as two Aβ1–42 molecules assemble in a single fibril layer (see Figure 2, PDBs: 2NAO, 5KK3, and 5OQV). Nonetheless, the resolved Aβ1–42 fibrillar structures have different Aβ1–42 monomeric conformations within each fibril subunit. A study by Xiao et al. reported the structural model of synthetic Aβ1–42 fibrils having one Aβ1–42 molecule in each fibril layer which forms a triple parallel β‐motif (Figure 2a, PDB 2MXU) (Xiao et al., 2015). A high‐resolution structure of a disease‐relevant Aβ1–42 amyloid polymorph was determined using solid‐state nuclear magnetic resonance (ssNMR), and electron microscopy (Figure 2b, PDB: 2NAO). The fibrils had twofold symmetry where each Aβ1–42 molecule formed cross β‐sheets in a double horseshoe‐like morphology with residues 15–42 forming the fibril core while residues 1–14 appearing to be partially ordered as shown in Figure 2b (Wälti et al., 2016). The twofold symmetry of Aβ1–42 fibrils observed in previous work was also reported by Colvin et al. that used magic angle spinning NMR to determine an atomic resolution structure of Aβ1–42 monomorphic fibrils (Figure 2c, PDB: 5KK3) (Colvin et al., 2016). Each Aβ1–42 monomer had four β‐strands that assembled in an S‐shaped fold with residues 15–42 forming the fibril core. This Aβ1–42 fibrillar structure (PDB 5KK3) revealed the exposure of two hydrophobic residues (V40, A42) on the fibril surface which could explain the higher secondary nucleation rates observed for Aβ1–42 in comparison to Aβ1–40. A recent high‐resolution structure of Aβ1–42 fibrils was resolved using Cryo‐EM complemented with ssNMR and X‐ray diffraction experiments. In line with previous papers, the fibrillar Aβ1–42 structure (Figure 2d, PDB 5OQV) had a twofold symmetry consisting of a dimer spanning the fibril subunit. Unlike previous structures, the N‐terminus of each Aβ1–42 monomer in PDB 5OQV was involved in the fibril cross‐β core resulting in a LS‐shaped fibrillar subunit. Also, the dimer interface is formed by the C‐termini interaction from the two Aβ1–42 monomers unlike in previous ssNMR structures that elucidated a solvent‐exposed C‐termini (Gremer et al., 2017).
FIGURE 2.
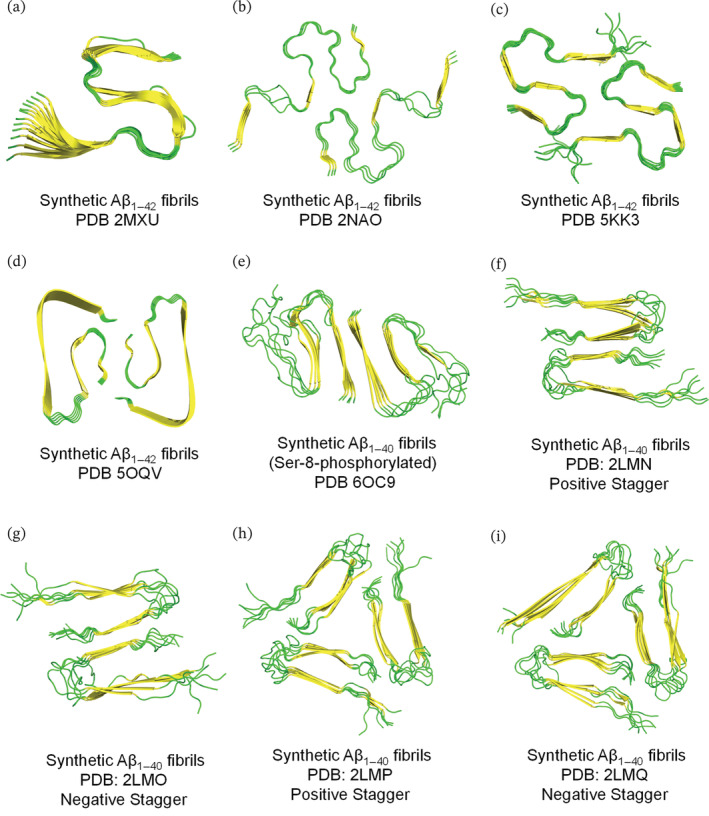
Resolved ssNMR/Cryo‐EM structures of synthetically prepared Aβ1–42/Aβ1–40 fibrils. Top view representation was used to demonstrate the fibril symmetry (i.e., conformation and number of molecules per fibril layer). (a–i) Fibrillar models are generated on PyMol using the protein data bank entries and are colored based on secondary structure (yellow for β‐sheets and green for loops). The terms, positive and negative stagger, in panels f–i describe the conformation of Aβ1–40 which spans two different z‐planes implying that each fibril layer is not occupied by a single Aβ1–40 molecule.
While for synthetic Aβ1–40 fibrils, a study revealed their structural polymorphism using solid state NMR coupled with electron microscopy experiments (Paravastu et al., 2008). In this study, synthetic Aβ1–40 fibrils with untwisted (striated ribbon) and periodically twisted morphologies had twofold and threefold symmetries about their fibril axes, respectively. In fact, the two fibrillar morphologies were produced using the same in vitro conditions except that striated ribbon fibrils were produced via agitation conditions while the twisted fibrils were produced via quiescent conditions (Paravastu et al., 2008; Petkova et al., 2006). The protofilaments of striated and twisted Aβ1–40 fibrils showed differences in the mass‐per‐length and solid‐state NMR data resulting in twofold and threefold symmetric models, respectively. However, both models had in‐register parallel β‐sheets that consist of similar β‐strands and non‐β strands segments but were mainly different in the symmetry, conformation of the non‐β strands segments and the overall quaternary structures (Paravastu et al., 2008; Petkova et al., 2006). As seen in Figure 2f–i, four structural models (PDBs 2LMN, 2LMO, 2LMP, and 2LMQ) were identified from synthetic Aβ1–40 fibrils in this work (Paravastu et al., 2008). The notions, positive and negative stagger describe the conformation of Aβ1–40 which spans two different z‐planes implying that each fibril layer is not occupied by a single Aβ1–40 molecule (Chen et al., 2018).
A study by Hu et al. elucidated the structure of synthetic fibrils produced from Ser‐8‐phosphorylated Aβ1–40 (Hu et al., 2019) (Figure 2e). The resolved structure, having a striated ribbon morphology with a twofold symmetry, is comparable to that of wild‐type Aβ1–40 fibrils produced using similar in vitro conditions (Hu et al., 2019; Paravastu et al., 2008; Petkova et al., 2006), but with variations in the fibrillar width (6 nm for wildtype vs. 8.5 nm for Ser‐8‐phosphorylated type). The N‐terminal residues of Ser‐8‐phosphorylated Aβ1–40 fibrils were involved in strong intra‐strand interactions with the amyloid core unlike the wildtype Aβ1–40 fibrils that have a highly dynamic N‐terminus that is not involved in the fibrillar core. In addition, the phosphorylated fibrils are shown to possess higher cross‐seeding capacity than non‐phosphorylated Aβ fibrils.
As established earlier, studies reporting the molecular structure of synthetic fibrils, produced by the in vitro aggregation of Aβ1–40, have shown that these structures are dependent on the growth conditions rather than the specific amino acid sequence. Hence, the molecular structure of amyloid fibrils that grow in AD patients cannot be primarily predicted by the in vitro derived synthetic amyloids.
Given that seeded growth of fibrils was shown to preserve molecular structures, a number of studies have used brain‐derived fibrils as seeds to grow in vitro labeled Aβ fibrils that preserve the molecular details of amyloids derived from human cortical tissues (Lu et al., 2013; Paravastu et al., 2009; Petkova et al., 2005; Qiang et al., 2017). In these studies, solid‐state NMR and electron microscopy experiments were employed to characterize fibrillar structures of Aβ as will be discussed next.
In the study by Paravastu et al. (2009), brain‐derived amyloid fibrils from two deceased AD patients were used to seed the in vitro growth of synthetic Aβ1–40 fibrils. Solid‐state NMR was used to determine the structures of isotopically labeled brain‐seeded Aβ fibrils. The structures were nearly identical between the two AD patients as they both had two co‐existing molecular structures with one structure, corresponding to 6.5 ± 1.0 nm single filament, being more predominant. On the other hand, the structures of the purely synthetic unseeded Aβ fibrils showed higher heterogeneity and were different from the brain‐derived fibrils.
Another study resolved the structures of brain‐derived Aβ fibrils extracted from occipital, parietal and temporal cortical brain tissues of two AD patients having different clinical AD histories, including mild and severe cortical atrophy as well as Lewy body dementia (Lu et al., 2013). For both patients, the resolved Aβ1–40 fibrillar structure had a threefold symmetry about the fibril axis. However, fibrillar structures of patient I and patient II were different as evident from NMR chemical shifts and TEM fibrillar morphologies that showed a periodic twist in the fibrils of patient II but not in those of patient I. The study also utilized structural calculations to accurately determine the full molecular structure of Aβ1–40 fibrils from patient I (Figure 3a, PDB 2M4J). Although threefold symmetry was identified for both brain‐derived (PDB 2M4J) and synthetic (PDB 2LMP) Aβ1–40 fibrils, variations in key residue distances are observed (Lu et al., 2013; Paravastu et al., 2008).
FIGURE 3.
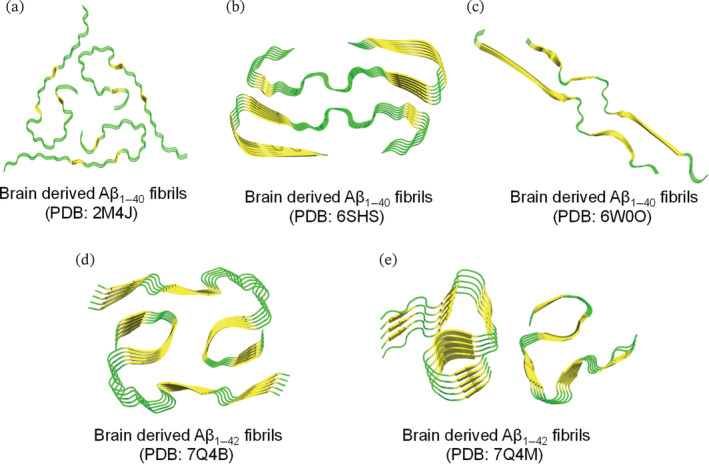
Resolved ssNMR/Cryo‐EM structures of natural brain‐derived Aβ1–42/Aβ1–40 fibrils extracted from deceased AD cases. Top view representation was used to demonstrate the fibril symmetry (i.e., conformation and number of molecules per fibril layer). (a–e) Fibrillar models of brain‐derived fibrils are generated on PyMol using the protein data bank entries and are colored based on secondary structure (yellow for β‐sheets and green for loops). (a) Threefold morphology of brain derived Aβ1–40 fibrils, PDB: 2M4J, from occipital, parietal and temporal cortical AD brain tissues. Structural calculations were performed to accurately determine the fibril structure of Aβ1–40 (PDB: 2M4J) that is fully consistent with the experimental data. (b) Twofold morphology of brain‐derived Aβ1–40 fibrils, PDB: 6SHS, from the meninges of severe AD and CAA (cerebral amyloid angiopathy). (c) Twofold morphology of brain‐derived Aβ1–40 fibrils, PDB: 6W0O, from cortical tissues of AD patients with slightly left‐handed twist as revealed by cryo‐EM. (d and e) Twofold morphologies of brain‐derived Aβ1–42 fibrils, extracted from cortical brain tissues of sporadic (type I in d) and familial (type II in e) AD cases.
A recent paper demonstrated the structural variations in Aβ1–40 and Aβ1–42 fibrils that were seeded with brain extracts of patients with different clinical AD subtypes. Aβ fibrils have been extracted from cortical brain tissues of patients having distinct AD phenotypes including typical AD, rapidly progressive AD and the posterior cortical atrophy variant of AD (a visual variant of an atypical AD) (Qiang et al., 2017). The findings of this study indicate that different AD clinical subtypes correspond to different amyloid fibrillar structures in brain tissues. Posterior cortical atrophy and typical variants shared a common predominant Aβ1–40 fibril structure. On the other hand, the rapidly progressive AD was characterized with increased polymorphism in Aβ1–40 fibrils that had additional fibrillar structures. Unlike Aβ1–40 fibrils, Aβ1–42 fibrils showed increased heterogeneity in all three AD clinical subtypes including the typical phenotype (Qiang et al., 2017). A high‐resolution Cryo‐EM structure was recently determined for the most predominant Aβ1–40 fibrils described by Qiang et al. (2017). The molecular structure (Figure 3c, PDB: 6W0O) has a twofold symmetry with slight left‐handed twist unlike the right‐handed twist identified for the meningeal Aβ fibril described below (Kollmer et al., 2019).
Recently, Kollmer et al. used Cryo‐EM to identify the structure of brain‐derived fibrils extracted at autopsy from the meninges of three severe AD patients (Kollmer et al., 2019). Although Cryo‐EM showed three major distinct morphologies of brain‐derived Aβ fibrils, a predominant structure was found to occupy brain amyloid extracts of all three AD patients (Kollmer et al., 2019). The predominant Aβ structure (Figure 3b, PDB: 6SHS) had a width of 7.4 nm, relatively short cross‐over distances and consisted of two peptide stacks (i.e., twofold symmetry with two Aβ molecules in each cross‐sectional fibril layer). Two other morphologies identified in the brain samples had larger width and cross‐over distances when compared to the main fibrillar structure described above. In fact, Cryo‐EM reconstruction of the other two morphologies revealed their similarity to the predominant morphology as they all share a common protofilament structure. However, the reported polymorphism in the three brain‐derived fibrillar structures is due to the number of the highly similar protofilaments where fibril morphology I contained one protofilament whereas fibril morphologies II and III contained two and three protofilaments, respectively. Interestingly, brain‐derived fibrils were found to be twisted with a right‐handed orientation unlike the left‐handed twisting observed in vitro synthetic fibrils. Also, brain‐derived fibrils were shown to be resistant to proteinase K (Serine protease) unlike the non‐resistant in vitro fibrils (Kollmer et al., 2019).
As for brain‐derived Aβ1–42 amyloid, in a recent study, the structures of Aβ1–42 filaments extracted from cortical brain tissues of 10 patients were resolved using cryo‐EM. Of the 10 patients, five had AD (two familial and three sporadic cases), while the remaining five had other, non‐AD age‐related diseases. Aβ1–42 fibrils extracted from brain tissues of patients with sporadic and familial AD were characterized by a twisted morphology (Y. Yang et al., 2022). However, in the sporadic AD cases, a dominant type of AD Aβ1–42 fibrils was found, termed type I, with a twofold symmetry consisting of two s‐shaped protofilaments packed together to form a predominantly hydrophobic interface. On the other hand, the familial AD cases and the other non‐AD conditions had a distinct Aβ1–42 fibril morphology, referred to as type II filament. Type II Aβ1–42 fibrils resemble those of type I in terms of their twofold s‐shaped protofilament arrangement, but the protofilament interface in type II fibrils is smaller, has more exposed hydrophobic residues, and is mainly stabilized by electrostatic interactions between K28 of one Aβ1–42 unit with the carboxyl group of A42 of the other protofilament (Y. Yang et al., 2022). The cryo‐EM structure of Aβ1–42, both type I and II (Figure 3d,e, PDBs: 7Q4B and 7Q4M), exhibits a left‐handed orientation, similar to the left‐handed Aβ1–40 cortical fibrils (PDB: 6W0O), but different from the right‐handed Aβ1–40 meningeal fibrils (PDB: 6SHS). In particular, cortical Aβ1–42 fibrils (PDBs: 7Q4B and 7Q4M) and cortical Aβ1–40 fibrils (PDB: 6W0O) share a common sub‐structure within the s‐shaped fold that is formed by the residues G25–G37 (Ghosh, Thurber, et al., 2021; Y. Yang et al., 2022). When comparing brain‐derived and in vitro‐produced Aβ1–42 fibrils, a common s‐shaped fold is observed, but the side chain interactions and orientations as well as the inter‐fibril arrangements differ between the brain‐derived and synthetic fibrils (Y. Yang et al., 2022).
Another study by Wickramasinghe et al. reported the structure of Aβ1–42 fibrils extracted from AD patients, using high sensitivity ssNMR (Wickramasinghe et al., 2021). Interestingly, this study reveals a new polymorphic structure of brain‐derived Aβ1–42 fibrils grown from bacterially expressed Aβ1–42 peptide (Wickramasinghe et al., 2021). The fibril structure consists of three β‐strands spanning residues, Tyr10–Asp23, Asn27–Vla36, and Val39–Ile41, which is different from the location of β‐strands of previously identified synthetic Aβ1–42 fibrils (Colvin et al., 2016; Gremer et al., 2017; Wälti et al., 2016; Xiao et al., 2015). Although Aβ1–42 has a substantial pathogenic effect in AD patients, only two studies have reported the structural details of brain‐derived Aβ1–42 fibrils. Future studies highlighting the structural details of Aβ1–42 fibrils in different clinical subtypes of AD cases would be necessary to investigate whether the polymorphic nature affects disease progression and type, and will be helpful to develop potential therapeutics against AD.
An important aspect of characterizing the structure of brain‐derived Aβ aggregates is to compare them with the amyloid plaques that have been detected in the brain tissues of some non‐demented elderly individuals. Characterizing the structural differences between amyloid deposits from non‐demented and AD individuals would shed more light on the relationship between amyloid deposits and neurodegenerative processes in AD. A study by Ghosh and co‐workers examined the structural details of brain‐derived Aβ1–40 and Aβ1–42 fibrils extracted from non‐demented as well as AD individuals (Ghosh, Yau, et al., 2021). The study reported similar ssNMR spectra of isotopically labeled Aβ fibrils seeded by Aβ extracts from cortical tissues of non‐demented and AD subjects, suggesting that similar polymorphs develop in both cases, with Aβ1–42 fibrils showing stronger variation between the two cases. However, statistical significances of polymorphic populations were observed in the cortical brain tissues of AD and non‐demented individuals indicating higher amounts of seed‐competent Aβ1–40 and Aβ1–42 aggregates. Importantly, the observed structural differences in Aβ1–42 fibrils between AD and non‐demented subjects indicate that fibrillar Aβ1–42 polymorphs are more predictive of cognitive impairment than Aβ1–40 polymorphs (Ghosh, Yau, et al., 2021).
Although brain‐derived Aβ fibrils have been shown to have different structures compared with synthetic fibrils, the polymorphic nature of brain‐derived fibrils themselves presents a challenge for screening effective drugs against AD. Structure‐based design of effective inhibitors against AD would require proper identification of ligand‐binding sites on polymorphic brain‐derived Aβ fibrils (Fändrich et al., 2018). It has been found that different amyloid tracers used for in vivo imaging of Aβ can bind to different surface sites on Aβ fibrils extracted from brain homogenates of patients with sporadic and autosomal dominant AD (Ni et al., 2013; Ni et al., 2017). The presence of multiple binding sites on polymorphic brain‐derived fibrils may also accommodate different ligand structures. However, future studies should further investigate the nature of binding sites on the surface of fibrils to facilitate the discovery of effective therapeutics against AD.
5. CHARACTERIZATION OF SOLUBLE BRAIN‐DERIVED Aβ OLIGOMERS
A growing body of evidence demonstrates that soluble oligomers of Aβ and other amyloidogenic proteins are the principal toxic entities (Li & Selkoe, 2020; Sherman & Lesné, 2010; Sideris et al., 2021; Walsh et al., 2002; T. Yang et al., 2017). As extracellular species, soluble Aβ‐derived diffusible ligands (ADDLs), have been shown to exert their toxic effects on neurons, astrocytes, microglia, and vasculature through binding to several cell receptors and inducing downstream signaling events that eventually cause neurodegeneration (Carrillo‐Mora et al., 2014; Catalano et al., 2006; Fani et al., 2022; Gong et al., 2003; Huang & Liu, 2020; Kayed & Lasagna‐Reeves, 2013; Sengupta et al., 2016; Wen et al., 2018). Of interest, Aβ species purified from soluble AD brain extracts were shown to significantly correlate with cognitive impairments than insoluble‐derived Aβ aggregates; a finding that indicates the pathological role of early soluble Aβ aggregates (Gong et al., 2003; Koss et al., 2016; Koss et al., 2018). Importantly, impaired synaptic function and inhibited hippocampal long‐term potentiation were observed in mice injected with cell media containing Aβ oligomers but not fibrils (Walsh et al., 2002). In addition, post‐mortem examination of brain tissues of AD and non‐demented elderly individuals indicates the presence of Aβ plaques but not the soluble fibrillar oligomers that are correlated with cognitive dysfunction (Tomic et al., 2009). In non‐demented subjects with Aβ plaques, toxic Aβ oligomers were not localized in postsynaptic densities and therefore did not induce synaptic loss (Zolochevska et al., 2018). In a recent review investigating possible mechanisms preventing non‐demented individuals with Aβ plaques from cognitive impairment, it was found that Aβ oligomers do not bind to synapses and therefore do not induce synaptotoxicity and inflammation through glial activation (Kok et al., 2022).
As demonstrated earlier, the resolved polymorphic structures of brain‐derived Aβ fibrils are markedly different than synthetic in vitro Aβ fibrils. Such findings stress on the need to characterize natural brain‐derived oligomers which could probably differ from their synthetic counterparts. In this section, we focus on literature findings that describe the neurotoxic roles of natural brain‐derived oligomers that have been extracted at autopsy from brain tissues of confirmed AD patients. Particularly, we highlight studies that utilized in vitro systems for examining the pathological roles of AD brain‐derived Aβ oligomers and dimers.
The presence of different soluble brain‐derived Aβ oligomers, including Aβ dimers, Aβ trimers, and Aβ*56 (i.e., 56 kDa Aβ oligomers), has been examined in brain extracts and CSF of cognitively intact and AD probable subjects (Lesné et al., 2013). The levels of Aβ oligomers were found to differ at different ages of cognitively intact individuals. Specifically, children and young subjects had negligible and undetected levels of Aβ*56 and Aβ dimers, but their levels were found to increase significantly in subjects in their 40s and 60s, respectively. On the other hand, Aβ trimers were detected in children and young subjects and their levels were found to gradually increase in subjects in their 70s. Importantly, AD probable subjects had the highest levels of Aβ dimers but not Aβ trimers or Aβ*56 (Lesné et al., 2013).
The synaptotoxicity of Aβ oligomers extracted from AD cortical tissues have been described by several in vivo models (Fritschi, Langer, et al., 2014; Li et al., 2011; Müller‐Schiffmann et al., 2016; Rush et al., 2018; Shankar et al., 2008). In these studies, brain‐derived Aβ oligomers have been particularly implicated in inhibiting hippocampal long‐term potentiation and long‐term memory in mouse models. It was shown that toxic Aβ dimers, stabilized with an intermolecular disulfide bridge, can be generated by expressing an amyloid precursor protein construct having Aβ‐S8C mutation in a transgenic mouse model. However, these soluble Aβ dimers were unable to generate insoluble Aβ plaques unless a crossbreeding with an Aβ‐plaque generating mouse model was performed (Müller‐Schiffmann et al., 2016). A thorough literature review on the pathological roles of soluble brain‐derived Aβ oligomers, particularly synaptic toxicity and hippocampal synaptic dysfunction, has been very recently published by Li and Stern (2022). In addition to using in vivo models, few studies employed in vitro toxicity assays and reported key pathological roles of natural Aβ oligomers from AD brain extracts we detail next.
An early study by Noguchi et al. showed that soluble Aβ spherical assemblies (10–15 nm, >100 kDa) derived from brain tissues of AD patients induced neurodegeneration of mature human neuronal cells and mature rat hippocampal neurons (Noguchi et al., 2009). Later in 2014, a study by Fritschi et al. showed that soluble Aβ seeds extracted from AD neocortical brain tissues had higher in vitro and in vivo seeding potencies as compared to Aβ seeds extracted from cerebrospinal fluid of the same AD patients (Fritschi, Langer, et al., 2014). For in vivo experiments, soluble Aβ extracts were injected into APP transgenic mice and after 8‐month follow‐up, a high deposition of Aβ was detected in the hippocampus of mice injected with soluble brain extracts but not for those injected with cerebrospinal fluid. Similarly, in vitro experiments of synthetic Aβ aggregation seeded with soluble brain‐derived Aβ extracts resulted in more elongated Aβ fibrils as compared to those formed by seeding with CSF‐derived Aβ oligomers (Fritschi, Langer, et al., 2014). Another recent study characterized Aβ amyloids that were derived from brain extracts of post‐mortem AD brains (olfactory cortex, amygdala, and hippocampus). Such brain‐derived amyloids were shown to internalize in in vitro endothelial cells as well in vivo mice brains that were inoculated with Aβ brain extracts intracerebrally. In this study, Aβ enriched brain‐extracts were characterized using proteomics based‐liquid chromatography–tandem mass spectrometry approach that revealed the exclusive presence of specific proteins, that is, Ring finger protein 213, in brain‐derived extracts of AD patients (Pedrero‐Prieto et al., 2019).
Using neuronal cells derived from human induced pluripotent stem cells and an in vivo mouse model, Hong et al. compared the toxicity of soluble brain‐derived Aβ oligomers obtained by the typical vigorous tissue homogenization method with those obtained by a more gentle buffer‐soaked extraction method (Hong et al., 2018; Jin et al., 2018). Despite retrieving smaller amounts of Aβ oligomers from the gentle extraction method, the obtained small diffusible Aβ fractions had comparable neurotoxic and LTP impairment effects to those exerted by Aβ homogenized extracts. The paper suggests that majority of soluble Aβ extracts are relatively non‐toxic and that only a small pool of diffusible Aβ oligomers are the main toxic entities (Hong et al., 2018).
A more thorough examination of the distribution of brain‐derived Aβ oligomers in different brain regions at an early stage of AD, that is, Braak stage III, was reported recently by Sideris et al. (2021). In their study, soluble Aβ oligomers, extracted by soaking tissues of eight different brain regions, were analyzed for their size, structure, morphology, and pathological roles including inflammation, lipid membrane penetration, and toxicity. The findings reveal the presence of Aβ oligomers in all examined brain regions that were all capable of inducing inflammation, toxicity, and membrane lipid permeabilization, to varying levels. Particularly, Aβ oligomers of hippocampal extracts, with <100 nm in length and 2 nm diameter, were the most potent neurotoxic and inflammatory species (Sideris et al., 2021).
In addition to the aforementioned studies, which examined brain‐derived high molecular weight Aβ oligomers, other studies have shown that low molecular weight oligomers (T. Yang et al., 2017), particularly dimers, are more potent species that can potentially induce synaptotoxicity (Brinkmalm et al., 2019; Brody & Gross, 2014; Jin et al., 2011; Shankar et al., 2008). Using an in vitro model of hippocampal neurons, brain‐derived soluble Aβ dimers from cortical tissues of AD patients were shown to induce tau hyperphosphorylation and lead to notable disruption of cellular cytoskeleton at sub‐nanomolar levels, whereas at least two order of magnitudes of synthetic Aβ dimers were required to get the same neurotoxic effects (Brody & Gross, 2014; Jin et al., 2011).
6. CONCLUSIONS AND FUTURE PERSPECTIVES
The field of protein aggregation has largely relied on the use of synthetic proteins as the main species in in vitro aggregation systems. However, the reported polymorphic features of synthetic Aβ aggregates and their dependence on growth conditions suggest that amyloid species grown in the human brain cannot be predicted by fibrils produced in vitro. Indeed, recent work shows important differences between brain‐derived and synthetic aggregates, which are summarized in Table 1. Knowledge of the unique structural features of brain‐derived fibrils is extremely important for making new discoveries in the field of disease‐modifying therapies for AD and other neurodegenerative diseases. However, the polymorphic nature of brain‐derived Aβfibrils presents a challenge in the development of therapeutics for AD. Current knowledge of the polymorphic fibril structures of Aβ1–40 and Aβ1–42 in the brain may provide the basis for identifying potential binding sites that could be used in future studies to develop effective structure‐based drugs that either prevent formation of or disaggregate Aβ fibrils. Interestingly, in a recent paper, Seidler et al. used brain‐derived tau fibrils to perform a structure‐based discovery of small molecules that can disaggregate brain‐derived fibrils in vitro (Seidler et al., 2022). This work clearly demonstrates the importance of using amyloids from the brain to advance the discovery of potential inhibitory molecules against amyloid diseases. As the most relevant species, brain‐derived amyloids can be used as a starting material for growing disease‐relevant aggregates, which would enhance future in vitro studies of aggregation inhibition.
TABLE 1.
Brain‐derived versus synthetic Aβ fibrils.
| Brain‐derived Aβ fibrils | In vitro synthetic Aβ fibrils | |
|---|---|---|
| Fibrillar structural polymorphism |
|
|
| Proteinase K treatment | More resistance to proteinase K (Kollmer et al., 2019) | Less resistance to proteinase K (Kollmer et al., 2019) |
| Aβ post‐translational modifications | High diversity of post‐translational modifications especially in N‐terminal and C‐terminal truncations. (Arai et al., 1999; Cabrera et al., 2018; Güntert et al., 2006; Kummer & Heneka, 2014; Milton, 2001; Rijal Upadhaya et al., 2014; Rostagno et al., 2018; Sergeant et al., 2003; Wildburger et al., 2017) | No diversity in N‐terminal and C‐terminal truncations |
| Toxicity of Aβ oligomers | Higher neurotoxicity (Brody & Gross, 2014; Hong et al., 2018; Jin et al., 2011; Noguchi et al., 2009; Sideris et al., 2021) | Lower neurotoxicity (Brody & Gross, 2014; Jin et al., 2011) |
Future studies of brain‐derived Aβ amyloids should consider the extraction of Aβ aggregates, both oligomers and fibrils, from the parenchyma and cortical blood vessels of deceased AD patients with various AD clinical subtypes. The extracted amyloid material, both soluble and insoluble, can be characterized by high‐resolution mass spectrometry to identify the diversity of Aβ proteoforms, including post‐translational modifications. In addition, determining the percentage of Aβ aggregates in the total isolated protein content is useful to characterize other abundant proteins that may co‐localize with Aβ and influence its aggregation pathways. Next, the structure of the different Aβ proteoforms extracted from AD brain tissues would need to be elucidated and their toxicity assessed both in vitro and in vivo to correlate the toxicity of the aggregates with the different Aβ species, which will be important for the development of effective therapeutics against AD.
AUTHOR CONTRIBUTIONS
Kenana Al Adem: Conceptualization (equal); investigation (lead); methodology (lead); validation (equal); visualization (equal); writing – original draft (lead); writing – review and editing (equal). Sungmun Lee: Conceptualization (equal); funding acquisition (lead); resources (equal); supervision (lead); writing – review and editing (equal).
Al Adem K, Lee S. Structural polymorphism and cytotoxicity of brain‐derived β‐amyloid extracts. Protein Science. 2023;32(5):e4639. 10.1002/pro.4639
Review Editor: Jean Baum
REFERENCES
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid‐β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–76. [DOI] [PubMed] [Google Scholar]
- Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer's disease: past, present and future. Neuropharmacology. 2014;76:27–50. [DOI] [PubMed] [Google Scholar]
- Arai T, Akiyama H, Ikeda K, Kondo H, Mori H. Immunohistochemical localization of amyloid β‐protein with amino‐terminal aspartate in the cerebral cortex of patients with Alzheimer's disease. Brain Res. 1999;823:202–6. [DOI] [PubMed] [Google Scholar]
- Attems J, Yamaguchi H, Saido TC, Thal DR. Capillary CAA and perivascular Aβ‐deposition: two distinct features of Alzheimer's disease pathology. J Neurol Sci. 2010;299:155–62. [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer‐Hartl M, Hartl FU. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. [DOI] [PubMed] [Google Scholar]
- Bemporad F, Chiti F. Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure‐toxicity relationships. Chem Biol. 2012;19:315–27. [DOI] [PubMed] [Google Scholar]
- Brinkmalm G, Hong W, Wang Z, Liu W, O'Malley TT, Sun X, et al. Identification of neurotoxic cross‐linked amyloid‐β dimers in the Alzheimer's brain. Brain. 2019;142:1441–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Gross ML. The remarkable properties of amyloid‐β derived from human Alzheimer's disease brain: swinging the streetlight. Brain. 2014;137:2874–5. [DOI] [PubMed] [Google Scholar]
- Brody DL, Jiang H, Wildburger N, Esparza TJ. Non‐canonical soluble amyloid‐beta aggregates and plaque buffering: controversies and future directions for target discovery in Alzheimer's disease. Alzheimers Res Ther. 2017;9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera E, Mathews P, Mezhericher E, Beach TG, Deng J, Neubert TA, et al. Aβ truncated species: implications for brain clearance mechanisms and amyloid plaque deposition. Biochim Biophys Acta BBA Mol Basis Dis. 2018;1864:208–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. 2010;6:140–7. [DOI] [PubMed] [Google Scholar]
- Carrillo‐Mora P, Luna R, Colín‐Barenque L. Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev. 2014;2014:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano S, Dodson E, Henze D, Joyce J, Krafft G, Kinney G. The role of amyloid‐Beta derived diffusible ligands (ADDLs) in Alzheimers disease. Curr Top Med Chem. 2006;6:597–608. [DOI] [PubMed] [Google Scholar]
- Cecchi C, Stefani M. The amyloid‐cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys Chem. 2013;182:30–43. [DOI] [PubMed] [Google Scholar]
- Chen M, Schafer NP, Wolynes PG. Surveying the energy landscapes of Aβ fibril polymorphism. J Phys Chem B. 2018;122:11414–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68. [DOI] [PubMed] [Google Scholar]
- Ciudad S, Puig E, Botzanowski T, Meigooni M, Arango AS, Do J, et al. Aβ(1‐42) tetramer and octamer structures reveal edge conductivity pores as a mechanism for membrane damage. Nat Commun. 2020;11:3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Appleby B, Safar JG. Distinct prion‐like strains of amyloid beta implicated in phenotypic diversity of Alzheimer's disease. Prion. 2016;10:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, et al. Rapidly progressive Alzheimer's disease features distinct structures of amyloid‐β. Brain. 2015;138:1009–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, et al. Proliferation of amyloid‐β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci. 2013;110:9758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, et al. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J Am Chem Soc. 2016;138:9663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C, Lemmin T, Stöhr J, Nick M, Wu Y, Maxwell AM, et al. Structural heterogeneity and intersubject variability of Aβ in familial and sporadic Alzheimer's disease. Proc Natl Acad Sci. 2018;115:E782–91. 10.1073/pnas.1714966115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S, Wirthensohn DC, Flagmeier P, Hughes C, Aprile FA, Ruggeri FS, et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat Commun. 2019;10:1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz‐Espinoza R. Recent high‐resolution structures of amyloids involved in neurodegenerative diseases. Front Aging Neurosci. 2021;13:782617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004;15:3–16. [DOI] [PubMed] [Google Scholar]
- Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, et al. Preclinical Alzheimer's disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esparza TJ, Wildburger NC, Jiang H, Gangolli M, Cairns NJ, Bateman RJ, et al. Soluble amyloid‐beta aggregates from human Alzheimer's disease brains. Sci Rep. 2016;6:38187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fändrich M, Meinhardt J, Grigorieff N. Structural polymorphism of Alzheimer Aβ and other amyloid fibrils. Prion. 2009;3:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fändrich M, Nyström S, Nilsson KPR, Böckmann A, LeVine H, Hammarström P. Amyloid fibril polymorphism: a challenge for molecular imaging and therapy. J Intern Med. 2018;283:218–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fani G, La Torre CE, Cascella R, Cecchi C, Vendruscolo M, Chiti F. Misfolded protein oligomers induce an increase of intracellular Ca2+ causing an escalation of reactive oxidative species. Cell Mol Life Sci. 2022;79:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschi SK, Cintron A, Ye L, Mahler J, Bühler A, Baumann F, et al. Aβ seeds resist inactivation by formaldehyde. Acta Neuropathol (Berl). 2014;128:477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschi SK, Langer F, Kaeser SA, Maia LF, Portelius E, Pinotsi D, et al. Highly potent soluble amyloid‐β seeds in human Alzheimer brain but not cerebrospinal fluid. Brain. 2014;137:2909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh U, Thurber KR, Yau W‐M, Tycko R. Molecular structure of a prevalent amyloid‐β fibril polymorph from Alzheimer's disease brain tissue. Proc Natl Acad Sci. 2021;118:e2023089118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh U, Yau W‐M, Collinge J, Tycko R. Structural differences in amyloid‐β fibrils from brains of nondemented elderly individuals and Alzheimer's disease patients. Proc Natl Acad Sci. 2021;118:e2111863118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, et al. Amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer's disease‐affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci. 2003;100:10417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Velasquez FJ, Kotarek JA, Moss MA. Soluble aggregates of the amyloid‐β protein selectively stimulate permeability in human brain microvascular endothelial monolayers: soluble amyloid‐β protein aggregates induce blood‐brain barrier permeability. J Neurochem. 2008;107:466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, et al. Fibril structure of amyloid‐β(1–42) by cryo‐electron microscopy. Science. 2017;358:116–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322:1365–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güntert A, Döbeli H, Bohrmann B. High sensitivity analysis of amyloid‐beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience. 2006;143:461–75. [DOI] [PubMed] [Google Scholar]
- Hipp MS, Park S‐H, Hartl FU. Proteostasis impairment in protein‐misfolding and ‐aggregation diseases. Trends Cell Biol. 2014;24:506–14. [DOI] [PubMed] [Google Scholar]
- Hong W, Wang Z, Liu W, O'Malley TT, Jin M, Willem M, et al. Diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in Alzheimer's disease brain. Acta Neuropathol (Berl). 2018;136:19–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, et al. Spherical aggregates of β‐amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase‐3β. Proc Natl Acad Sci. 2003;100:6370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z‐W, Vugmeyster L, Au DF, Ostrovsky D, Sun Y, Qiang W. Molecular structure of an N‐terminal phosphorylated β‐amyloid fibril. Proc Natl Acad Sci. 2019;116:11253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Liu R. The toxicity and polymorphism of β‐amyloid oligomers. Int J Mol Sci. 2020;21:4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, O'Nuallain B, Hong W, Boyd J, Lagomarsino VN, O'Malley TT, et al. An in vitro paradigm to assess potential anti‐Aβ antibodies for Alzheimer's disease. Nat Commun. 2018;9:2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid β‐protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci. 2011;108:5819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. [DOI] [PubMed] [Google Scholar]
- Kayed R, Lasagna‐Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis JAD. 2013;33(Suppl 1):S67–78. [DOI] [PubMed] [Google Scholar]
- Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–96. [DOI] [PubMed] [Google Scholar]
- Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–7. [DOI] [PubMed] [Google Scholar]
- Kok FK, van Leerdam SL, de Lange ECM. Potential mechanisms underlying resistance to dementia in non‐demented individuals with Alzheimer's disease neuropathology. J Alzheimers Dis. 2022;87:51–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, et al. Cryo‐EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer's brain tissue. Nat Commun. 2019;10:4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss DJ, Dubini M, Buchanan H, Hull C, Platt B. Distinctive temporal profiles of detergent‐soluble and ‐insoluble tau and Aβ species in human Alzheimer's disease. Brain Res. 2018;1699:121–34. [DOI] [PubMed] [Google Scholar]
- Koss DJ, Jones G, Cranston A, Gardner H, Kanaan NM, Platt B. Soluble pre‐fibrillar tau and β‐amyloid species emerge in early human Alzheimer's disease and track disease progression and cognitive decline. Acta Neuropathol (Berl). 2016;132:875–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Rezaei‐Ghaleh N, Terwel D, Thal DR, Richard M, Hoch M, et al. Extracellular phosphorylation of the amyloid β‐peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer's disease: extracellular phosphorylation of the amyloid β‐peptide. EMBO J. 2011;30:2255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Singh S, Hinze D, Josten M, Sahl H‐G, Siepmann M, et al. Phosphorylation of amyloid‐β peptide at serine 8 attenuates its clearance via insulin‐degrading and angiotensin‐converting enzymes. J Biol Chem. 2012;287:8641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer MP, Heneka MT. Truncated and modified amyloid‐beta species. Alzheimers Res Ther. 2014;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundra R, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M. Protein homeostasis of a metastable subproteome associated with Alzheimer's disease. Proc Natl Acad Sci. 2017;114:E5703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Aβ seeds are potent inducers of cerebral β‐amyloid deposition. J Neurosci. 2011;31:14488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Hsp20, a novel α‐crystallin, prevents Aβ fibril formation and toxicity. Protein Sci. 2005;14:593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Choi MC, Al Adem K, Lukman S, Kim T‐Y. Aggregation and cellular toxicity of pathogenic or non‐pathogenic proteins. Sci Rep. [cited 20 March, 2020]. http://www.nature.com/articles/s41598-020-62062-3. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, et al. Brain amyloid‐β oligomers in ageing and Alzheimer's disease. Brain. 2013;136:1383–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long‐term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B‐containing NMDA receptors. J Neurosci. 2011;31:6627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Selkoe DJ. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer's brain. J Neurochem. 2020;154:583–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Stern AM. Bioactive human Alzheimer brain soluble Aβ: pathophysiology and therapeutic opportunities. Mol Psychiatry. [cited 28 April, 2022]. https://www.nature.com/articles/s41380-022-01589-5. 2022;27:3182–91. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA. Amyloid fibril toxicity in Alzheimer's disease and diabetes. Ann N Y Acad Sci. 1996;777:89–95. [DOI] [PubMed] [Google Scholar]
- Lu J‐X, Qiang W, Yau W‐M, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β‐amyloid fibrils in Alzheimer's disease brain tissue. Cell. 2013;154:1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lührs T, Ritter C, Adrian M, Riek‐Loher D, Bohrmann B, Döbeli H, et al. 3D structure of Alzheimer's amyloid‐β(1–42) fibrils. Proc Natl Acad Sci. 2005;102:17342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannini B, Mulvihill E, Sgromo C, Cascella R, Khodarahmi R, Ramazzotti M, et al. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem Biol. 2014;9:2309–17. [DOI] [PubMed] [Google Scholar]
- Miller Y, Ma B, Nussinov R. The unique Alzheimer's β‐amyloid triangular fibril has a cavity along the fibril axis under physiological conditions. J Am Chem Soc. 2011;133:2742–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton NGN. Phosphorylation of amyloid‐β at the serine 26 residue by human cdc2 kinase. Neuroreport. 2001;12:3839–44. [DOI] [PubMed] [Google Scholar]
- Müller‐Schiffmann A, Herring A, Abdel‐Hafiz L, Chepkova AN, Schäble S, Wedel D, et al. Amyloid‐β dimers in the absence of plaque pathology impair learning and synaptic plasticity. Brain. 2016;139:509–25. [DOI] [PubMed] [Google Scholar]
- Ni R, Gillberg P, Bogdanovic N, Viitanen M, Myllykangas L, Nennesmo I, et al. Amyloid tracers binding sites in autosomal dominant and sporadic Alzheimer's disease. Alzheimers Dement. 2017;13:419–30. [DOI] [PubMed] [Google Scholar]
- Ni R, Gillberg P‐G, Bergfors A, Marutle A, Nordberg A. Amyloid tracers detect multiple binding sites in Alzheimer's disease brain tissue. Brain. 2013;136:2217–27. [DOI] [PubMed] [Google Scholar]
- Noguchi A, Matsumura S, Dezawa M, Tada M, Yanazawa M, Ito A, et al. Isolation and characterization of patient‐derived, toxic, high mass amyloid β‐protein (Aβ) assembly from Alzheimer disease brains. J Biol Chem. 2009;284:32895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Condron MM, Teplow DB. Structure–neurotoxicity relationships of amyloid β‐protein oligomers. Proc Natl Acad Sci. 2009;106:14745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Leapman RD, Yau W‐M, Tycko R. Molecular structural basis for polymorphism in Alzheimer's β‐amyloid fibrils. Proc Natl Acad Sci. 2008;105:18349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R. Seeded growth of β‐amyloid fibrils from Alzheimer's brain‐derived fibrils produces a distinct fibril structure. Proc Natl Acad Sci. 2009;106:7443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JS, Andersen CB, Otzen DE. Amyloid structure—one but not the same: the many levels of fibrillar polymorphism: the structural ambiguity of glucagon amyloids. FEBS J. 2010;277:4591–601. [DOI] [PubMed] [Google Scholar]
- Pedrero‐Prieto CM, Flores‐Cuadrado A, Saiz‐Sánchez D, Úbeda‐Bañón I, Frontiñán‐Rubio J, Alcaín FJ, et al. Human amyloid‐β enriched extracts: evaluation of in vitro and in vivo internalization and molecular characterization. Alzheimers Res Ther. 2019;11:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Leapman RD, Guo Z, Yau W‐M, Mattson MP, Tycko R. Self‐propagating, molecular‐level polymorphism in Alzheimer's ß‐amyloid fibrils. Science. 2005;307:262–5. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Yau W‐M, Tycko R. Experimental constraints on quaternary structure in Alzheimer's β‐amyloid fibrils. Biochemistry. 2006;45:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince M, Comas‐Herrera A, Knapp M, Guerchet M, Karagiannidou M. World Alzheimer report 2016: improving healthcare for people living with dementia: coverage, quality and costs now and in the future. London, UK: Alzheimer's Disease International (ADI); 2016. [cited 15 September, 2016]. https://www.alz.co.uk/research/world-report-2016 [Google Scholar]
- Puzzo D, Gulisano W, Arancio O, Palmeri A. The keystone of Alzheimer pathogenesis might be sought in Aβ physiology. Neuroscience. 2015;307:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau W‐M, Lu J‐X, Collinge J, Tycko R. Structural variation in amyloid‐β fibrils from Alzheimer's disease clinical subtypes. Nature. 2017;541:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau W‐M, Luo Y, Mattson MP, Tycko R. Antiparallel β‐sheet architecture in Iowa‐mutant β‐amyloid fibrils. Proc Natl Acad Sci. 2012;109:4443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resende R, Ferreiro E, Pereira C, Resende de Oliveira C. Neurotoxic effect of oligomeric and fibrillar species of amyloid‐beta peptide 1–42: involvement of endoplasmic reticulum calcium release in oligomer‐induced cell death. Neuroscience. 2008;155:725–37. [DOI] [PubMed] [Google Scholar]
- Rezaei‐Ghaleh N, Amininasab M, Kumar S, Walter J, Zweckstetter M. Phosphorylation modifies the molecular stability of β‐amyloid deposits. Nat Commun. 2016;7:11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R. The three‐dimensional structures of amyloids. Cold Spring Harb Perspect Biol. 2017;9:a023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijal Upadhaya A, Kosterin I, Kumar S, von Arnim CAF, Yamaguchi H, Fändrich M, et al. Biochemical stages of amyloid‐β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer's disease. Brain. 2014;137:887–903. [DOI] [PubMed] [Google Scholar]
- Roher AE, Kuo Y‐M. Isolation of amyloid deposits from brain. Methods in enzymology. Volume 309. Amsterdam, The Netherlands: Elsevier; 1999. p. 58–67. [cited 7 January, 2004]. https://linkinghub.elsevier.com/retrieve/pii/S0076687999090060 [DOI] [PubMed] [Google Scholar]
- Rostagno A, Neubert TA, Ghiso J. Unveiling brain Aβ heterogeneity through targeted proteomic analysis. In: Sigurdsson EM, Calero M, Gasset M, editors. Amyloid proteins. Vol. 1779. Methods in molecular biology. New York, NY: Springer New York; 2018. p. 23–43. 10.1007/978-1-4939-7816-8_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Riquelme A, Mao A, Barghash MM, Lau HHC, Stuart E, Kovacs GG, et al. Aβ43 aggregates exhibit enhanced prion‐like seeding activity in mice. Acta Neuropathol Commun. 2021;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush T, Martinez‐Hernandez J, Dollmeyer M, Frandemiche ML, Borel E, Boisseau S, et al. Synaptotoxicity in Alzheimer's disease involved a dysregulation of Actin cytoskeleton dynamics through cofilin 1 phosphorylation. J Neurosci. 2018;38:10349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 2010;277:1348–58. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Rohou A, Lasker K, Yadav JK, Schiene‐Fischer C, Fändrich M, et al. Peptide dimer structure in an Aβ(1–42) fibril visualized with cryo‐EM. Proc Natl Acad Sci. 2015;112:11858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert MPM, Zucker‐Franklin D, Rimon A, Franklin EC. The characterization of soluble amyloid prepared in water. J Clin Invest. 1968;47:924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, et al. Atomic‐resolution three‐dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew Chem Int Ed. 2015;54:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler PM, Murray KA, Boyer DR, Ge P, Sawaya MR, Hu CJ, et al. Structure‐based discovery of small molecules that disaggregate Alzheimer's disease tissue derived tau fibrils in vitro. Nat Commun. 2022;13:5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta U, Nilson AN, Kayed R. The role of amyloid‐β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, et al. Truncated beta‐amyloid peptide species in pre‐clinical Alzheimer's disease as new targets for the vaccination approach: β‐amyloid peptides in early amyloid deposits. J Neurochem. 2003;85:1581–91. [DOI] [PubMed] [Google Scholar]
- Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia‐Munoz A, Shepardson NE, Smith I, et al. Amyloid‐β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MA, Lesné SE. Detecting Aβ*56 oligomers in brain tissues. In: Roberson ED, editor. Alzheimer's disease and frontotemporal dementia. Vol. 670. Methods in molecular biology. Totowa, NJ: Humana Press; 2010. p. 45–56. 10.1007/978-1-60761-744-0_4 [DOI] [PubMed] [Google Scholar]
- Sideris DI, Danial JSH, Emin D, Ruggeri FS, Xia Z, Zhang YP, et al. Soluble amyloid beta‐containing aggregates are present throughout the brain at early stages of Alzheimer's disease. Brain Commun. 2021;3:fcab147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD. Imaging the progression of Alzheimer pathology through the brain. Proc Natl Acad Sci. 2002;99:4135–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern AM, Liu L, Jin S, Liu W, Meunier AL, Ericsson M, et al. A calcium‐sensitive antibody isolates soluble amyloid‐β aggregates and fibrils from Alzheimer's disease brain. Brain. 2022;145:2528–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, et al. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci. 2014;111:10329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, et al. Purified and synthetic Alzheimer's amyloid beta (Aβ) prions. Proc Natl Acad Sci. 2012;109:11025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theillet F‐X, Binolfi A, Frembgen‐Kesner T, Hingorani K, Sarkar M, Kyne C, et al. Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs). Chem Rev. 2014;114:6661–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. Amyloid polymorphism: structural basis and neurobiological relevance. Neuron. 2015;86:632–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendruscolo M. Proteome folding and aggregation. Curr Opin Struct Biol. 2012;22:138–43. [DOI] [PubMed] [Google Scholar]
- Vendruscolo M. Lipid homeostasis and its links with protein misfolding diseases. Front Mol Neurosci. 2022;15:829291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar‐Piqué A, Schmitz M, Candelise N, Ventura S, Llorens F, Zerr I. Molecular and clinical aspects of protein aggregation assays in neurodegenerative diseases. Mol Neurobiol. 2018;55:7588–605. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long‐term potentiation in vivo. Nature. 2002;416:535–9. [DOI] [PubMed] [Google Scholar]
- Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, et al. Atomic‐resolution structure of a disease‐relevant Aβ(1–42) amyloid fibril. Proc Natl Acad Sci. 2016;113:E4976–84. 10.1073/pnas.1600749113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Condello C, Stöhr J, Oehler A, Lee J, DeArmond SJ, et al. Serial propagation of distinct strains of Aβ prions from Alzheimer's disease patients. Proc Natl Acad Sci. 2014;111:10323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Fang F, Guo S‐H, Zhang Y, Peng X‐L, Sun W‐M, et al. Amyloid β‐derived diffusible ligands (ADDLs) induce abnormal autophagy associated with Aβ aggregation degree. J Mol Neurosci. 2018;64:162–74. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe A, Xiao Y, Kobayashi N, Wang S, Scherpelz KP, Yamazaki T, et al. Sensitivity‐enhanced solid‐state NMR detection of structural differences and unique polymorphs in pico‐ to nanomolar amounts of brain‐derived and synthetic 42‐residue amyloid‐β fibrils. J Am Chem Soc. 2021;143:11462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildburger NC, Esparza TJ, LeDuc RD, Fellers RT, Thomas PM, Cairns NJ, et al. Diversity of amyloid‐beta proteoforms in the Alzheimer's disease brain. Sci Rep. 2017;7:9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Bowers MT, Shea J‐E. Molecular structures of quiescently grown and brain‐derived polymorphic fibrils of the Alzheimer amyloid Aβ9‐40 peptide: a comparison to agitated fibrils. PLoS Comput Biol. 2010;6:e1000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, et al. Aβ(1–42) fibril structure illuminates self‐recognition and replication of amyloid in Alzheimer's disease. Nat Struct Mol Biol. 2015;22:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian TA Neurodegenerative Disorders . Drug discovery approaches for the treatment of neurodegenerative disorders. Amsterdam, The Netherlands: Elsevier; 2017. p. 1–16. [cited 20 September, 2016]. https://linkinghub.elsevier.com/retrieve/pii/B9780128028100000015 [Google Scholar]
- Yang T, Li S, Xu H, Walsh DM, Selkoe DJ. Large soluble oligomers of amyloid β‐protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci. 2017;37:152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Arseni D, Zhang W, Huang M, Lövestam S, Schweighauser M, et al. Cryo‐EM structures of amyloid‐β 42 filaments from human brains. Science. 2022;375:167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampar S, Klafki HW, Sritharen K, Bayer TA, Wiltfang J, Rostagno A, et al. N‐terminal heterogeneity of parenchymal and vascular amyloid‐β deposits in Alzheimer's disease. Neuropathol Appl Neurobiol. 2020;46:673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Schrader JM, Irizarry BA, Smith SO, Van Nostrand WE. Impact of Aβ40 and Aβ42 fibrils on the transcriptome of primary astrocytes and microglia. Biomedicine. 2022;10:2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolochevska O, Bjorklund N, Woltjer R, Wiktorowicz JE, Taglialatela G. Postsynaptic proteome of non‐demented individuals with Alzheimer's disease neuropathology. J Alzheimers Dis. 2018;65:659–82. [DOI] [PMC free article] [PubMed] [Google Scholar]