

ABSTRACT
Streptococcus pyogenes, also known as group A Streptococcus, causes a wide variety of diseases ranging from mild noninvasive to severe invasive infections. To identify possible causes of colonization-to-invasive switches, we determined the genomic sequences of 10 isolates from five pairs each composed of an invasive strain and a carriage strain originating from five infectious clusters. Among them, one pair displayed a single-nucleotide difference in covS, encoding the sensor histidine kinase of the two-component CovRS system that controls the expression of 15% of the genome. In contrast to previously described cases where the invasive strains harbor nonfunctional CovS proteins, the carriage strain possessed the mutation covST115C, leading to the replacement of the tyrosine at position 39 by a histidine. The CovSY39H mutation affected the expression of the genes from the CovR regulon in a unique fashion. Genes usually overexpressed in covS mutant strains were underexpressed and vice versa. Furthermore, the covS mutant strain barely responded to the addition of the CovS-signaling compounds Mg2+ and LL-37. The variations in the accumulation of two virulence factors paralleled the transcription modifications. In addition, the covST115C mutant strain showed less survival than its wild-type counterpart in murine macrophages. Finally, in two murine models of infection, the covS mutant strain was less virulent than the wild-type strain. Our study suggests that the CovSY39H protein compromises CovS phosphatase activity and that this yields a noninvasive strain.
IMPORTANCE Streptococcus pyogenes, also known as group A Streptococcus, causes a wide variety of diseases, leading to 517,000 deaths yearly. The two-component CovRS system, which responds to MgCl2 and the antimicrobial peptide LL-37, controls the expression of 15% of the genome. Invasive strains may harbor nonfunctional CovS sensor proteins that lead to the derepression of most virulence genes. We isolated a colonization strain that harbors a novel covS mutation. This mutant strain harbored a transcriptome profile opposite that of other covS mutant strains, barely responded to environmental signals, and was less virulent than the wild-type strain. This supports the importance of the derepression of the expression of most virulence genes, via mutations that impact the phosphorylation of the regulator CovR, for favoring S. pyogenes invasive infections.
KEYWORDS: adaptation, covRS, environmental signal, group A Streptococcus, infection, two-component regulatory system
INTRODUCTION
Streptococcus pyogenes (group A Streptococcus [GAS]) is an important human pathogen responsible for a large variety of clinical manifestations ranging from mild superficial infections to more life-threatening invasive infections, including necrotizing fasciitis (NF) and streptococcal toxic shock syndrome (STSS) (1). Sequencing of the variable extremity of the emm gene is at the basis of epidemiological surveys of GAS infections, and more than 225 different emm genotypes have been described, with variable distributions worldwide; emm1 is predominant in high-income countries (2–4). The molecular mechanisms that enable GAS to cause a large range of diseases are unknown. How GAS can regulate the expression of a variety of gene products involved in GAS adaptation and survival in the human host influences the severity of infection (5).
The CovRS (control of virulence) system (initially termed CsrRS [6]) is the best-studied system among the 13 two-component signal transduction systems (TCSs) identified in the GAS genome. The CovRS system is responsible for regulating, directly or indirectly, approximately 10 to 15% of the GAS genome, mainly as a repressor (7–10). Comparisons with TCSs in other species, genetic evidence, and in vitro studies have indicated that CovS is a member of the bifunctional HisKA family of histidine kinases (11) and that CovR is a transcriptional regulator whose activity on promoters is controlled by its phosphorylation state (12–16). CovR is phosphorylated, most probably directly, by CovS and can be phosphorylated by the S. pyogenes serine/threonine kinase (SP-STK) and exogenous acetyl-phosphate (12–15, 17, 18). CovR can be dephosphorylated by CovS, which also has phosphatase activity, and by the S. pyogenes serine/threonine phosphatase (SP-STP), whose gene is cotranscribed with that of SP-STK (19–22). The CovS phosphorylation state is influenced by environmental signals, including extracellular Mg2+, LL-37, elevated temperatures, acidic pH, and high osmolarity (19, 23–26). Mg2+ and LL-37 have opposite effects on the expression of the CovRS-controlled genes: Mg2+ enhances the repression of CovRS-repressed genes, and LL-37 has the paradoxical effect of stimulating their expression (19, 23–26). A cluster of 3 amino acid residues from the CovS extracellular domain, D148-ED152, interacts with these environmental signals (26). Mechanistically, a high concentration of magnesium increases the level of phosphorylated CovR in a CovS-dependent manner (20); the effect of Mg2+ is mediated mainly by the impairment of CovS phosphatase activity and not by enhanced kinase activity (20). Conversely, LL-37 signaling increases CovS phosphatase activity (21). In addition, the loss of CovS phosphatase activity impairs the pathogenicity of S. pyogenes (21). An orphan regulator, RocA, binds to CovS in the membrane and enhances its autokinase activity or inhibits its phosphatase activity (27, 28). Finally, the in vivo selection of covRS mutants leads to strains with enhanced virulence both in humans and in animal models, highlighting the major impact of the CovRS system on GAS virulence (10, 23, 29–34). The mutations all correspond to a loss or a diminution of phosphorylated CovR, by either the absence of CovR or a mutation affecting the capacity of CovS to phosphorylate CovR.
Various groups among the genes controlled by CovRS have been identified (21, 35). A group of CovRS-repressed genes includes, among others, slo, which encodes streptolysin O (SLO), a pore-forming toxin that also facilitates the entry of the NAD-glycohydrolase into eukaryotic cells (36, 37); speA, which encodes a superantigen; and the has operon, encoding the biosynthetic enzymes of the antiphagocytic polysaccharide capsule (6). In both covS- and covR-deleted strains, these genes are overexpressed (8). Another group of genes, sometimes described as being activated by CovRS, includes speB, encoding the extracellular cysteine protease SpeB, a major virulence factor of S. pyogenes (38–45), and grab an α2-macroglobulin-binding protein (10, 35, 46–48). These genes are underexpressed in covS mutant strains and strains harboring point mutations in covR but are overexpressed in a covR-deleted strain (10, 35, 46–48). CovR represses their expression more efficiently than CovR phosphorylated on aspartate 53 (49). Also, speB expression is upregulated at high bacterial cell densities (50).
In France, the epidemiology of GAS invasive infections is analyzed by the French national reference center for streptococci (CNR-Strep [https://cnr-strep.fr/]), which collects and analyzes invasive GAS strains sent on a voluntary basis by laboratories located throughout the national territory. GAS strains isolated as part of the investigations conducted around clusters from contacts with no symptoms or noninvasive GAS infection are also sent to and appraised by the CNR-Strep.
In order to identify the factors responsible for the noninvasive-to-invasive switch, we sought to determine the genetic differences between invasion and colonization GAS strains. Ten isolates from five pairs of GAS strains, belonging to five independent clusters, each constituted by an invasive GAS strain and an asymptomatic-colonization GAS strain, have been sequenced. Here, we report the characterization of the strains from one emm1 pair in which, in contrast to the cases described until now, the carriage strain harbored a mutation in covS. The transcription profiles, responses to environmental signals, virulence factor production, cell interactions, and in vivo virulence of these strains have been assessed, and differences between the wild-type (WT) and the covS mutant strains were observed.
RESULTS
A carriage pharyngeal strain harbors a mutation in covS.
To identify the factors responsible for the shift from noninvasive to invasive strains, we selected five pairs of GAS strains. These were isolated during five independent clustered cases, with an invasive infection, yielding the “invasive strain,” and a swab from a family member with no or a noninvasive infection, yielding the “colonization strain” (Table 1). Sequencing of these strains identified two pairs (pairs 1 and 2) where the covS genes differed between the strains, two pairs (pairs 3 and 4) where mutations were in the carriage strains in sfbX or immediately upstream of the sof-sfbX operon, and one pair (pair 5) in which a com gene was lost. In pair 2, composed of emm11 strains, the invasive strain displayed a single mutation in the covS gene leading to a frameshift and a stop codon at the 26th position, whereas the colonization strain harbored a wild-type covS gene (Table 1). The presence of such mutations in invasive strains has previously been described for GAS strains of different emm types (10, 30, 31, 33, 34).
TABLE 1.
Selected pairs of clinical group A Streptococcus strains
| Pair | Strain | Sexc | Age (yrs) | Sample origin | Clinical symptom(s) | emm genotypea | PFGE patternb | Type of mutation |
|---|---|---|---|---|---|---|---|---|
| 1 | M1-CovS-WT | M | 36 | Blood culture | NF + STSS | emm1 | 1-A | Point mutation in covS |
| M1-CovS-Y39H | M | 7 | Throat swab | Pharyngeal carriage | emm1 | 1-A | ||
| 2 | M11CovS | F | 56 | Blood culture | Peritonitis + STSS | emm11 | 11-A3 | Frameshift in covS |
| M11WT | M | 54 | Throat swab | Pharyngeal carriage | emm11 | 11-A3 | ||
| 3 | 2003-223 | M | 72 | Pus | Ethmoiditis | emm77 | 77-A | Frameshift in sfbX |
| 2003-225 | F | 43 | Cutaneous | Soft skin infection | emm77 | 77-A | ||
| 4 | 2009-126 | M | 75 | Blood culture | Bacteremia | emm77 | Mutation upstream of sof | |
| 2009-125 | M | 80 | Cutaneous | Impetigo | emm77 | |||
| 5 | 2007-635 | M | 47 | Blood culture | Bacteremia | emm94 | Loss of the com gene | |
| 2007-949 | F | 85 | Cutaneous | Cellulitis | emm94 | |||
The sequences of the pair 1 strains that are of the emm1 genotype were compared to published complete genome sequences of emm1 strains and found to be very similar to the genome sequence of the epidemic strain MGAS5005 (GenBank accession number NC_007297.2) and strain A20 (GenBank accession number CP003901.1) isolated from a patient with necrotizing fasciitis, with 60 to 65 single-nucleotide polymorphisms (SNPs) and up to 14 indels of 1 to 21 nucleotides (nt) (51–53). A single-nucleotide difference was observed between the strains from pair 1; it is located in the covS gene, covST115C. The covS gene from one strain was identical to the wild-type covS gene (53); this strain was named M1-CovS-WT. This single-nucleotide difference in covS led to the replacement of the tyrosine residue at position 39 by a histidine residue (Y39H). This replacement did not appear in the sequences obtained when performing a BLAST analysis of the first 100 amino acid residues of CovS against the nonredundant protein sequences from S. pyogenes (NCBI database, January 2023). In fact, tyrosine 39, which is located at the end of the first membrane-spanning domain, is highly conserved in CovS sequences (Fig. 1) (54). Altogether, these results suggest that the Y39H modification is not a polymorphism but may be a mutation; we named the mutant strain M1-CovS-Y39H. In contrast to what has been described until now and was found for the strains from the emm11 strain pair, the mutant M1-CovS-Y39H strain is the carriage strain, while the invasive strain harbors a wild-type covS allele (Table 1). To our knowledge, this is the first description of this covS mutation and the first report of a carriage strain harboring a covS mutation (34, 55). We consequently decided to study this strain further.
FIG 1.
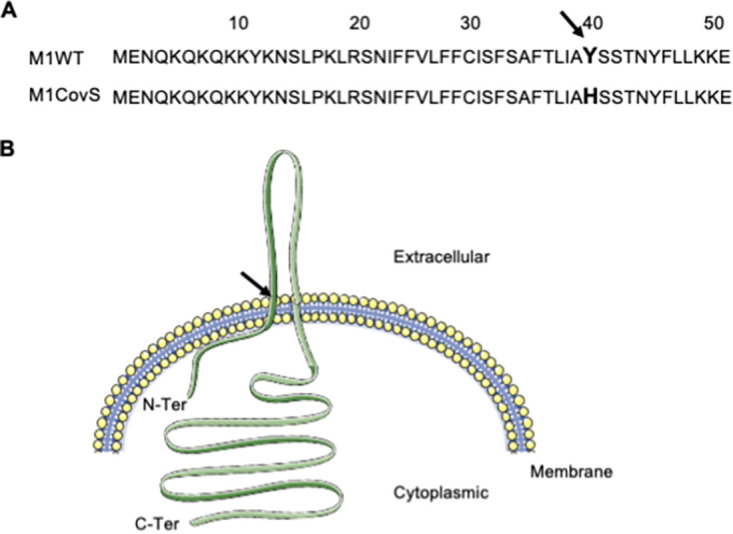
Localization of the Y39H mutation. (A) Amino acid sequences of the first 50 amino acid residues in the M1-CovS-WT and M1-CovS-Y39H strains. The 39th amino acid residue is in boldface type. (B) Localization of the mutation in a schematic representation of CovS. Black arrow, 39th residue. (Modified from reference 54.)
The CovSY39H mutation yields unique transcriptomic profiles.
The growth of the M1-CovSY39H strain was identical to that of the M1-CovS-WT strain in Todd-Hewitt (TH) broth supplemented with 0.2% yeast extract (THY broth), indicating that the Y39H mutation did not alter the growth of the M1-CovS-Y39H strain in vitro (data not shown). To characterize the consequences of the CovSY39H mutation, we compared the transcriptomic profiles, determined by transcriptome sequencing (RNA-seq), of the M1-CovS-WT and M1-CovS-Y39H strains grown in THY broth and harvested at entry into stationary phase (optical density at 600 nm [OD600] = 0.7) (Fig. 2A).
FIG 2.
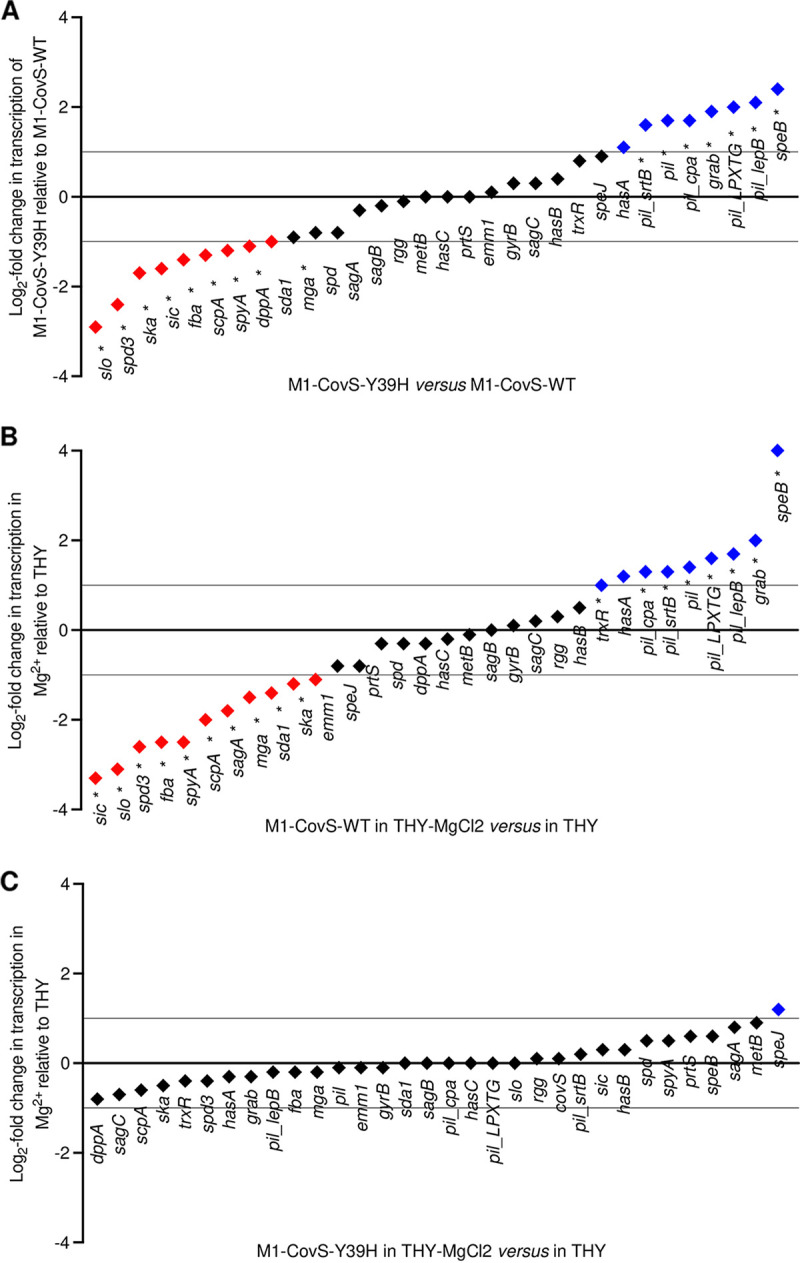
The M1-CovS-Y39H strain displays an atypical transcriptomic profile and responds poorly to an environmental cue. Shown are log2 fold transcript differences in selected virulence genes between the M1-CovS-Y39H and M1-CovS-WT strains grown in THY broth (A), M1-CovS-WT grown in THY broth supplemented with MgCl2 and THY broth (B), and M1-CovS-Y39H grown in THY broth supplemented with MgCl2 and THY broth (C), as determined by RNA-seq analysis (see Table S2 in the supplemental material). Genes are in black; those that are overexpressed and underexpressed are highlighted with a blue diamond above the zero bar and a red diamond below the zero bar, respectively. *, statistically significant after a multiple-testing-adjustment procedure.
Statistical analyses of the RNA-seq data showed that 81 genes were differentially expressed, with a statistically significant difference (see Table S2 in the supplemental material) and a minimum fold change of 2. In particular, the expression levels of the genes encoding streptolysin O (slo), streptokinase (ska), C5A-peptidase (scpA), secreted inhibitor of complement (sicC3-like ADP-ribosyltransferase [spyA]), and fibronectin-binding protein (fbaA), all known virulence factors (56, 57), were lower in the M1-CovS-Y39H strain than in the M1-CovS-WT strain (Fig. 2A). In contrast, the expression levels of the genes encoding exotoxin B (speB) and immunoglobulin G-binding protein G (grab) and genes from the pilus locus, or fibronectin-collagen-T antigen (FCT), were higher in the M1-CovS-Y39H strain than in the M1-CovS-WT strain. The has genes were expressed at very low levels, and their expression was not significantly different between the strains. In addition to variations in the expression of genes involved in virulence, the two strains also differed in the expression of metabolic genes, such as genes involved in pyrimidine metabolism (including the pyrR regulatory gene) and sugar metabolism. To confirm the RNA-seq results, we performed quantitative real-time PCR (qRT-PCR) analysis of several virulence genes from independent RNA samples (Fig. 3A). The speB gene was overexpressed and the ska and slo genes were underexpressed in the M1-CovS-Y39H strain compared to the M1-CovS-WT strain. The transcription of grab, speA, and has was not or was barely affected. Overall, these results confirmed the RNA-seq data, although the transcription differences were somewhat weaker in the qRT-PCR experiments. These data indicate that the variations in gene expression between M1-CovS-Y39H and M1-CovS-WT are opposite those classically described between covS and wild-type strains (23–26).
FIG 3.

The CovSY39H variant responds poorly to environmental cues. Log2 fold transcript differences in selected genes between the M1-CovS-Y39H and M1-CovS-WT strains grown in THY broth (A), the M1-CovS-WT and M1-CovS-Y39H strains grown in THY broth supplemented or not with MgCl2 (B), and the M1-CovS-WT and M1-CovS-Y39H strains grown in THY broth supplemented or not with LL-37 (C) were determined by qRT-PCR. Genes above or below the zero bar are those that are overexpressed or underexpressed, respectively. Mean values and standard deviations (SD) are indicated (n = 4 [A and C] and n = 3 [B]). P values were determined by an unequal-variance t test on ΔCT (*, P < 0.05; **, P < 0.01; ****, P < 0.001).
To further explore the phenotypes of both strains, we tested the influence of Mg2+ on their transcriptomic profiles (Fig. 2B and C, Fig. 3B, and Tables S1 and S2) (23, 24, 26). The addition of 15 mM Mg2+ to the growth medium did not impact the growth of these strains (data not shown). Important changes in gene expression were observed for the M1-CovS-WT strain in response to Mg2+, with at least 202 genes being differentially expressed (Table S2). Regarding virulence genes, speB, grab, genes of the pilus locus, and one has gene were overexpressed in the presence of Mg2+. In contrast, sci, slo, spyA, and spd3, among others, were underexpressed (Fig. 2B). qRT-PCR confirmed the RNA-seq results and showed that the addition of Mg2+ results in the overexpression of speB and grab and the underexpression of speA, ska, and slo (Fig. 3B). These results indicate that the addition of Mg2+ elicited the same effects, as those described previously, on the expression of GAS virulence genes in wild-type strains (23, 24, 26).
The addition of Mg2+ to the growth medium of the M1-CovS-Y39H strain led to slight transcription modifications. However, no statistically significant difference was observed by either RNA-seq or qRT-PCR (Fig. 2C, Fig. 3B, and Table S2). This suggests that the MgCl2 effect was strongly attenuated in the M1-CovS-Y39H strain compared to the M1-CovS-WT strain.
To further analyze the functionality of the CovSY39H protein, we tested the consequences on the expression of the chosen genes of the addition of the antimicrobial peptide LL-37, which shifts the equilibrium toward CovS phosphatase activity, to the growth medium (19, 23–26). The addition of 100 nM LL-37 to the growth medium did not impact the growth of these strains (data not shown). Only the expression of the speB gene was modified for the M1-CovS-WT strain in response to LL-37 (Fig. 3C). However, this change was lost with the M1-CovS-Y39H strain. This suggests that the effect of LL-37 was attenuated in the M1-CovS-Y39H strain compared to the M1-CovS-WT strain.
These results suggest that the CovSY39H variant decreases the capacity of the CovS sensor protein to transfer the environmental signals to the regulatory protein.
The CovSY39H variant limits the influence of Mg2+ and LL-37 on SpeB and SLO accumulation.
To determine the phenotypic consequences of the transcription variations, we assayed the levels of SpeB and SLO accumulation produced by the M1-CovS-WT and M1-CovS-Y39H strains cultured in THY broth and THY broth containing MgCl2 at the late (OD600 = 0.6 to 0.7) and early (OD600 = 0.3 to 0.4) exponential phases, respectively (Fig. 4). Proteins from the supernatants were precipitated, and immunoblot analysis was performed with specific anti-SpeB (Fig. 4A) or anti-SLO (Fig. 4B) antibodies.
FIG 4.
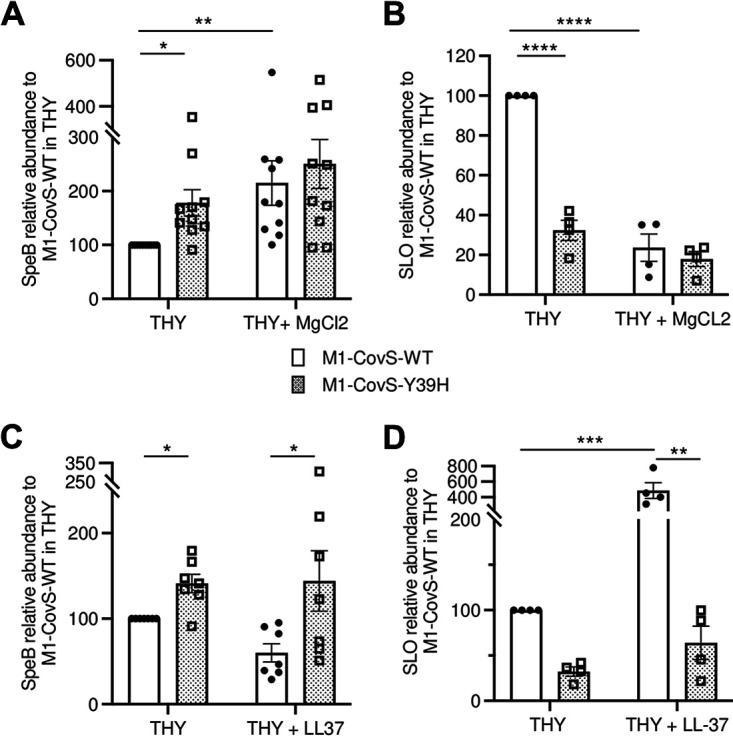
Effect of the CovSY39H variant on SpeB and SLO production. Shown are the relative abundances of the accumulated SpeB (A and C) or SLO (B and D) in the supernatants of M1-CovS-WT (closed circles) and M1-CovS-Y39H (open squares) grown in THY broth or THY broth with MgCl2 (A and B) or in THY broth or THY broth with LL-37 (C and D) compared to those in the supernatants of the wild-type strain grown in THY broth. Strains were grown to the late (OD600 = 0.6 to 0.7) and early (OD600 = 0.3 to 0.4) exponential phases for SpeB and SLO quantifications, respectively. The experiments were carried out for the SpeB and the SLO quantifications 10 and 4 times, respectively. Statistical analysis was performed by a Kruskal-Wallis test with a Dunn posttest and by two-way ANOVA with a Bonferroni posttest for SpeB and SLO analyses, respectively (Prism 9). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
SpeB was more abundant in the supernatant of the M1-CovS-Y39H strain than in the supernatant of the M1-CovS-WT strain when they were grown in THY broth (Fig. 4A and B). SpeB was more abundant in the M1-CovS-WT supernatant when bacteria were grown in the presence of added MgCl2 than in its absence (Fig. 4A). However, this MgCl2 effect was lost in the mutant strain, supporting the transcription results (Fig. 3B and Fig. 4A). Furthermore, no significant difference in the levels of SpeB was observed between the supernatants of M1-CovS-WT and M1-CovS-Y39H grown in the presence of MgCl2.
SLO was more abundant in the M1-CovS-WT supernatant than in the M1-CovS-Y39H supernatant grown in THY broth (Fig. 4B). Whereas SLO was more abundant in the supernatants of M1-CovS-WT grown in the absence than in the presence of added MgCl2, this was not the case for the M1-CovS-Y39H strain, where there was no difference (Fig. 4B). Finally in the presence of MgCl2, both strains accumulated the same levels of SLO.
This fully supports the transcription data and strengthens the conclusion that the CovSY39H protein has a reduced capacity to respond to the MgCl2 signal.
We also compared the levels of production of SpeB and SLO by both strains when they were grown in the absence or the presence of the antimicrobial peptide LL-37. The addition of LL-37 barely modified the production of SpeB, whose concentration was higher in the M1-CovS-H39Y strain in THY broth and THY broth supplemented with LL-37. The addition of LL-37 increased the relative abundance of SLO in the supernatant of M1-CovS-WT. In contrast, it had no effect on the relative abundance of SLO in the M1-CovS-Y39H supernatant. These results indicate that CovSY39H does not respond to LL-37.
Altogether, these results suggest that CovSY39H is impaired in its capacity to respond to environmental signals.
The M1-CovS-Y39H strain is impaired in its macrophage survival capacity.
The role of macrophages in the early steps of GAS infection remains unclear; they can kill GAS, or the bacteria can survive and even multiply in the macrophages (58–60). They may constitute a reservoir for GAS dissemination as macrophage intracellular persistence and multiplication have been described in an emm1 GAS strain (60). A phagocytosis assay was performed with murine RAW264.7 macrophages at a multiplicity of infection (MOI) of 10 bacteria per macrophage (Fig. 5). After a 45-min infection period, the M1-CovS-WT and M1-CovS-Y39H strains were phagocytosed at similar levels (not shown). The macrophage survival assay indicated that at 2 h postinfection, the M1-CovS-WT strain survived significantly better than the M1-CovS-Y39H strain (P = 0.0062). No significant differences were observed at other time points. This suggests that the CovSY39H variant weakens GAS survival in macrophages.
FIG 5.
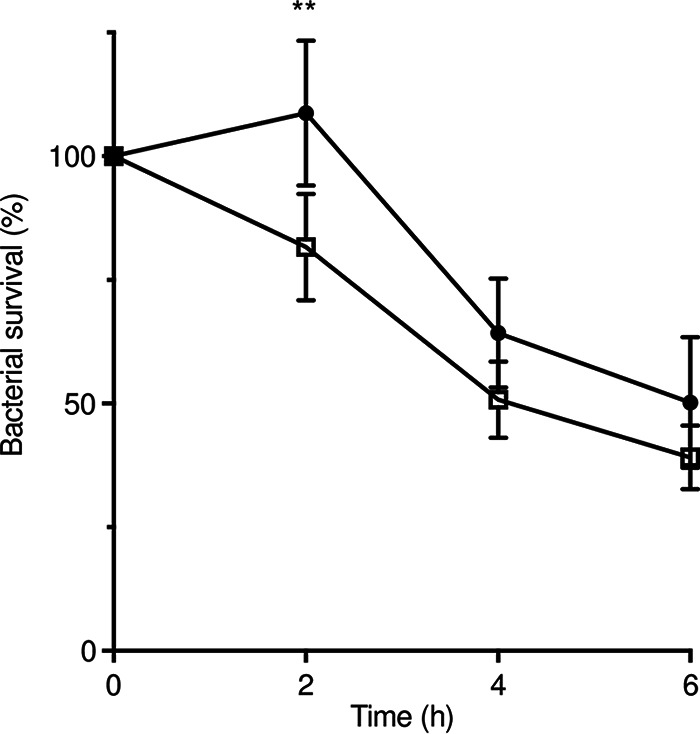
The M1-CovS-WT strain survives better than the M1-CovS-Y39H mutant strain inside macrophages. A macrophage survival assay was performed with mouse RAW264.7 macrophages infected with M1-CovS-WT (closed circles) and M1-CovS-Y39H (open squares) at an MOI of 10. GAS survival is expressed as a percentage of intracellular bacteria at T2, T4, and T6 relative to those at T0. The results are representative of data from 7 independent experiments performed in triplicate. Error bars represent the means and standard errors of the means (**, P < 0.01).
The M1-CovS-Y39H strain is less virulent than the M1-CovS-WT strain.
With the classical covS mutant strains being more virulent in models of invasive infections (29, 31, 54, 61) and the M1-CovS-Y39H strain displaying an atypical phenotype, we assessed whether the virulence of this strain would be modified. Mice were infected intravenously with the M1-CovS-WT and M1-CovS-Y39H strains, and survival was monitored (Fig. 6A). The M1-CovS-Y39H strain was less virulent (P = 0.0051). To mimic natural infection and correspond to the location where the M1-CovS-Y39H strain was swabbed, mice were also infected intranasally with the same strains (Fig. 6B). We confirmed on the following days that both bacterial strains were found intranasally and in the throat, and we monitored colonization (Fig. S1). Whereas the CFU counts in M1-CovS-WT-infected mice increased during the first days, leading to the death of three mice, and then dropped in the surviving ones, they decreased in the M1-CovS-Y39H-infected mice during the first days and then stabilized. However, since the M1-CovS-WT strain killed more mice than the M1-CovS-Y39H strain from day 5 postinfection onward, determining a colonization difference was impossible. Again, the M1-CovS-WT strain tended to be more virulent than the M1-CovS-Y39H mutant strain (P = 0.057). This indicates that with both infection models, the CovSY39H variant impairs GAS virulence.
FIG 6.

The M1-CovS-WT strain is more virulent than the M1-CovS-Y39H mutant strain. Mice were infected with the M1-CovS-WT and M1-CovS-Y39H strains intravenously (n = 10) at 4 × 107 and 5 × 107 CFU, respectively (A), and intranasally (n = 6) at 5 × 108 and 1.2 × 109 CFU, respectively (B). The survival of the animals was scored for a period of 14 days. Dashed lines, M1-CovS-WT; dotted lines, M1-CovS-Y39H. **, P < 0.001 (by log rank and Gehan-Breslow-Wilcoxon tests).
DISCUSSION
To identify the factors involved in the switch from colonization to invasive strains, we isolated pairs of colonization and invasive strains from infectious clusters and sequenced their genomes. Two pairs displayed mutations in covS. One followed a previously described schema where the covS mutant strain is the invasive strain (10, 29–34).
The other pair was atypical, with the colonization strain harboring a covS mutation, covST115C. We assessed whether the covS mutant strain (M1-CovS-Y39H) is less virulent than its WT counterpart (M1-CovS-WT) using two murine infection models. In the first one, bacteria were injected intravenously, enabling the evaluation of GAS survival in the blood. The second one, intranasal infection, takes into account that the oropharynx is regarded as the most common site of colonization by GAS and mimics a relevant natural portal of entry (62). In both models, the M1-CovS-Y39H strain was less virulent than the M1-CovS-WT strain. These results strongly support that the resultant variant of the CovS sensor protein yields a less virulent strain, in contrast to all spontaneous CovS variants isolated thus far (10, 29–34). The immune pressure selects in vivo covR or covS mutations that enable the switch from a colonization phenotype to an invasive phenotype, and among the immune system components, macrophages play an important role, either killing or being a reservoir for GAS (58–60). Our data indicate that whereas the M1-CovS-WT strain multiplied during the first hours after phagocytosis, the elimination of the M1-CovS-Y39H strain was already under way, suggesting a weaker survival capacity of this strain. These results account for this mutant clinical isolate being less invasive than the wild-type isolate and, consequently, the noninvasive strain of the pair.
The mutation replaces the tyrosine at position 39 with a histidine (CovSY39H). The amino acid residue at position 39 is the first residue, starting from the N terminus of the protein, of the extracellular domain (54). Most spontaneous mutations in covS lead to a stop codon and, hence, a deleted CovS protein or are located in the cytoplasmic domain of CovS that starts at position 215 (34). Two notable exceptions are a triple mutation in the first membrane-spanning α-helix and one in the small cytoplasmic domain preceding this helix; yet both strains, like the other characterized spontaneous mutant isolates, overexpress SLO (34).
A comparison of the gene expression profile of the strain harboring the CovSY39H variant to those of its wild-type counterparts indicated that this mutation yielded an effect opposite that induced by other natural CovS variants on the expression of genes belonging to the CovR regulon (10, 35). Genes that are usually overexpressed, such as slo or ska, in covS mutant strains were underexpressed in the M1-CovS-Y39H strain, and conversely, those that are usually underexpressed in covS mutant invasive strains, such as speB or grab, were overexpressed in the M1-CovS-Y39H strain. Furthermore, this was confirmed at the protein level. SpeB and SLO were more and less abundant, respectively, in the supernatants of the covST115C mutant than in the supernatants of the wild-type strain. Since speB and slo are repressed by unphosphorylated and phosphorylated CovR, respectively (49, 63), the CovSY39H protein could yield an excess of phosphorylated CovR in the absence of additional environmental signals. This could be due to excessive kinase activity. This would be in contrast to classical spontaneous mutations in covS recovered from GAS invasive isolates, which lead to CovS proteins that have lost the capacity to phosphorylate CovR. Alternatively, the increased CovR phosphorylation could result from impaired phosphatase activity, such as that described previously for constructed mutant strains (21).
Mg2+ and LL-37 are environmental signals that favor CovR-phosphorylated and nonphosphorylated states, respectively, by acting on CovS phosphatase activity (20). The effect of Mg2+ is mediated mainly by an impairment of CovS phosphatase activity, whereas LL-37 signaling increases it (20, 21). To assess whether CovSY39H is impaired in its phosphatase activity, we tested whether M1-CovS-Y39H responds to the addition of any of these signals. The transcriptomic profiles, or the expression levels of given genes, and the accumulation of SpeB and SLO were therefore compared after cultivating the M1-CovS-WT and the M1-CovS-Y39H strains in the absence or presence of MgCl2 or LL-37. The effects of both Mg2+ and LL-37 were weakened in the M1-CovS-Y39H strain. These data indicate that the CovSY39H protein has a defective capacity to respond to both of these environmental signals.
The strain in which a cluster of acidic residues (D148-ED152), thought to interact with these environmental signals, had been mutated displayed hyporesponsiveness to environmental signals (26). Constructed strains deficient in phosphatase activity also display high levels of phosphorylated CovR and similar hyporesponsive phenotypes (20). Hence, there is experimental evidence showing that a decrease in the relative concentration of phosphorylated CovR weakens the response to environmental signals. However, in contrast to previous observations made with genetically modified strains, the transcriptional variation due to the CovSY39H variant was not as strong as that due to the addition of Mg2+ (63). The localization of the CovSY39H mutation is original as the tyrosine at position 39 is the first residue of the extracellular domain. It is not located in the cluster where environmental signals are thought to interact, nor is it directly involved in the phosphatase activity of CovS, and this may cause it to be less stringent than the constructed ones. Altogether, these data indicate that Y39 plays an important role in CovS protein activity. Its mutation to a histidine may induce a conformational change that may constrain the CovS protein in a conformation in which the phosphatase activity is decreased.
The documentation of CovS sequence variants, identified either by analyzing in vivo-selected GAS strains or by comparing the in vivo properties of constructed covS mutants, indicates that all covS mutant strains have an invasive phenotype, with two exceptions. The strain with the triple mutation in the extracellular acidic residue cluster is more susceptible than the wild-type strain to opsonophagocytic killing (26). Also, the total loss of CovS phosphatase activity impairs GAS pathogenicity, including mouse oropharynx colonization (21). Mutations in other regulators have also been shown to decrease virulence or increase colonization. One example is mutations in the Rgg2 and Rgg3 transcriptional regulators that respond to short hydrophobic peptide signals. In contrast to the deletion of the transcriptional activator Rgg2, which leads to diminished colonization, the deletion of the transcriptional repressor Rgg3 increased colonization in the murine oropharynx (62). The mutations may also alter the expression of a regulator. In both the M1-CovS-Y39H strain and the constructed phosphatase-deficient strain, mga, which encodes an S. pyogenes master regulator, is less expressed than in their wild-type counterparts (21). Interestingly, carrier strains have a 12-bp deletion in the mga promoter region (64). This deletion yields decreased mga transcripts, and strains harboring it are less virulent than the wild-type strains, potentially contributing to the asymptomatic-carrier state. The CovSY39H variant could therefore, in addition to directly modifying the expression of numerous virulence factors, indirectly control those that are activated by Mga. Until now, no spontaneous covS mutant displaying a colonization phenotype has been isolated. The M1-CovS-Y39H strain was isolated from a healthy carrier during an epidemiological survey concerning an invasive case of GAS infection and displayed an in vivo virulence defect.
Previous studies of naturally occurring covS mutant invasive strains demonstrated that these isolates were outcompeted in noninvasive infections and that there is a fitness cost in the nasopharynx for these strains (35, 65). Those studies, together with ours, support the requirement for wild-type covRS in GAS to permit the full cycle leading to dissemination and invasion after colonization. Finally, further analysis of the CovSY39H protein, including the determination of its structure and its interaction with CovR and its membrane regulator RocA, would broaden our knowledge of the GAS CovRS TCS and possibly other TCSs.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
All clinical GAS strains listed in Table 1 were sent on a voluntary basis to the CNR-Strep by laboratories located throughout the national territory. Clinical characteristics were obtained from questionnaires sent with the isolates. Data collected included the sex and date of birth of the patient, the date and origin of the sample, geographical area, and clinical manifestations. Strains were stored in 2% glycerol Todd-Hewitt (TH) broth at −80°C. GAS strains were cultured in static TH broth supplemented with 0.2% yeast extract (THY broth) at 37°C without agitation. When necessary, 15 mM MgCl2 or 100 nM LL-37 was added to THY broth.
GAS strain sequencing and sequence analyses.
Complete genome sequencing was carried out on five pairs of GAS strains using Illumina technology, with a read length of 51 nt and >200-fold coverage. Libraries were constructed by using the Illumina TrueSeq kit according to the manufacturer’s instructions. Illumina short reads were assembled by using Velvet software (66). In each pair, one assembly was chosen as a reference sequence to align the reads of both strains by using the Burrows-Wheeler aligner (BWA) (67). SNP calling was performed using SAMtools MPILEUP and varFilter (68). All putative SNPs were visually verified by using Tablet (69). The alignment of the reference genome reads allowed the identification of errors in the Velvet assembly. In pairs where SNPs were identified, the search for the directionality of the mutation was carried out using NCBI BLASTP against the nonredundant protein sequence database and specifying Streptococcus pyogenes (taxonomy identifier [Taxid] 1314), as described previously by Almeida et al. (70). The two strains from pair 1 were found to be of the emm1 type and to differ by a unique SNP in the covS coding DNA sequence (CDS) at codon 39 (TAT versus CAT) leading to either a tyrosine or a histidine. The sequence of the WT strain (isolated from blood) was further completed as described previously (GenBank accession number LR031521) (51) and used as a reference sequence to confirm the SNP uniqueness in the strain isolated from the throat (GenBank accession number ERR9468758) by using breseq (71).
RNA isolation and Illumina RNA-seq.
The S. pyogenes M1-CovS-WT and M1-CovS-Y39H strains were cultured at 37°C in THY broth supplemented or not with MgCl2, and cells were harvested at the late exponential growth phase (OD600 = 0.7 to 0.8), as described previously (26). Total RNA was extracted, as described previously (72), from three independent cultures under each condition. Residual DNA was removed using DNase (Turbo DNA-free kit; Ambion). RNA integrity was analyzed using an Agilent Bioanalyzer (Agilent Biotechnologies). Strand-specific RNA-seq libraries ranging in size from 100 to 200 bp (insert size of 30 to 130 bp) were prepared as previously described (51). Multiplexed libraries (6 samples per lane) were sequenced on the Hi-Seq 2000 platform (Illumina), generating 6,300,000 to 40,000,000 50-bp-long reads per sample.
RNA-seq data analysis.
The M1-CovS-WT sequence (GenBank accession number LR031521) was used as a reference sequence to map sequencing reads. RNA-seq data were analyzed using bowtie-0.12.7, Rsamtools (version 1.26.2), Genomic-Alignments (version 1.10.1), and Genomic Features (version 1.26.2) in R 3.3.1 as previously described (51, 73). For differential expression analysis, normalization and statistical analyses were performed by using the SARTools package and DESeq2 (74, 75). P values were calculated and adjusted for multiple testing using the false discovery rate-controlling procedure (68).
First-strand cDNA synthesis and quantitative PCR.
A total of 0.5 μg of total RNA was used for first-strand cDNA synthesis using SuperScript II reverse transcriptase and random primers according to the manufacturer’s instructions (Invitrogen, Life Technologies). Quantitative PCR (qPCR) was carried out with SYBR green PCR kits (Applied Biosystems, Life Technologies) using six pairs of primers (see Table S1 in the supplemental material). Relative quantification of the expression of specific genes was performed with the 2−ΔΔCT method using gyrA as the housekeeping reference gene, and values were expressed as log2 fold changes. Each assay was performed on independent biological triplicates, and the statistical significance of the differences was inferred by using a two-tailed unequal-variance t test (Welch’s test) on the ΔCq values.
SpeB and SLO accumulation assays.
Cultures grown overnight in static THY medium, supplemented or not with MgCl2 or LL-37, were diluted 1:30 in the appropriate growth medium and incubated at 37°C, and the OD600 was monitored. Since SpeB, but not SLO, was assayed at a growth phase during which its accumulation becomes consequent, the medium was supplemented with the cysteine protease inhibitor E-64 (28 μM) for the SpeB assays. Supernatant proteins were precipitated in the presence of 0.6 N TCA (2,2,2-trichloroacetic acid) for 1 h at 0°C. After centrifugation, pellets were resuspended in 50 mM Tris (pH 8.0), and total proteins were quantified using the bicinchoninic acid (BCA) assay kit (Thermo-Fisher). Defined concentrations of total proteins were loaded onto a nitrocellulose membrane. The detection of SpeB or SLO was performed by incubation with specific anti-SpeB or anti-SLO rabbit antibodies (Abcam) overnight at 4°C. Horseradish peroxidase (HRP)-coupled goat anti-rabbit secondary antibody (Zymed) was added, and detection was performed with enhanced chemiluminescence (ECL reagent; GE Healthcare). The results of immunoblot assays were analyzed using ImageJ software. The Kruskal-Wallis test or one-way analysis of variance (ANOVA) was used (Prism 9).
Macrophage survival assay.
The RAW264.7 murine macrophage cell line was used to perform phagocytosis and survival assays. Twenty-four-well plates were seeded with 5 × 105 RAW264.7 cells per well in Dulbecco’s modified Eagle’s medium (DMEM) (high glucose) supplemented with 10% fetal calf serum at 37°C under a 5% CO2-enriched atmosphere. After 24 h, mid-logarithmic-phase bacterial cultures, resuspended in DMEM (high glucose) supplemented with 2 mM l-glutamine, were added at a multiplicity of infection (MOI) of 10. The survival assay was carried out as previously described (76). The plates were centrifuged at 6,600 rpm for 10 min. The infected cells were then incubated for 45 min at 37°C, washed three times with phosphate-buffered saline (PBS), and treated with penicillin-streptomycin for 30 min. At time zero (T0) (corresponding to 30 min after the addition of antibiotics) and at specific time points thereafter, intracellular GAS were recovered, and serial dilutions were plated. The results were expressed as the percentage of intracellular GAS relative to GAS at T0. For all experiments, 7 independent assays in triplicate were carried out. Two-way ANOVA with a Bonferroni posttest was used (Prism 9).
Animal experiments.
The procedures for animal experiments were performed according to the guidelines of the European Commission for the handling of laboratory animals, directive 86/609/EEC (http://ec.europa.eu/environment/chemicals/lab_animals/home_en.htm), and were approved by the Université Paris Descartes ethics committee. Female CD1 mice (6 weeks old; Charles River Laboratories, France) were either injected intravenously in the tail with 4 × 107 to 5 × 107 CFU of exponentially growing bacteria or infected intranasally with 5 × 108 to 1.2 × 109 CFU of exponentially growing bacteria in a volume of 5 μL per nostril. Animal survival was monitored for 10 or 15 days. The log rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests were used to analyze the data (Prism 9).
Data availability.
The genome sequence data for the five pairs of strains have been deposited at the DDBJ/EMBL/GenBank database (BioProject accession number PRJEB52031 and SRA accession number ERP136702).
ACKNOWLEDGMENTS
We thank C. Méhats for her helpful discussions.
This work was supported by the High Council for Scientific and Technological Cooperation between France-Israel Complexity in Biology program and by the INSERM, CNRS, Université de Paris, Santé Publique France, Centre National de Référence des Streptocoques, and Assistance Publique-Hôpitaux de Paris.
Footnotes
Supplemental material is available online only.
Contributor Information
Agnès Fouet, Email: agnes.fouet@inserm.fr.
Michael J. Federle, University of Illinois at Chicago
REFERENCES
- 1.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. 10.1128/CMR.13.3.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carapetis JR, Steer AC, Mulholland EK, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 3.Bisno AL, Brito MO, Collins CM. 2003. Molecular basis of group A streptococcal virulence. Lancet Infect Dis 3:191–200. 10.1016/s1473-3099(03)00576-0. [DOI] [PubMed] [Google Scholar]
- 4.Li Z, Sakota V, Jackson D, Franklin AR, Beall B. Active Bacterial Core Surveillance/Emerging Infections Program Network. 2003. Array of M protein gene subtypes in 1064 recent invasive group A streptococcus isolates recovered from the active bacterial core surveillance. J Infect Dis 188:1587–1592. 10.1086/379050. [DOI] [PubMed] [Google Scholar]
- 5.Kreikemeyer B, McIver KS, Podbielski A. 2003. Virulence factor regulation and regulatory networks in Streptococcus pyogenes and their impact on pathogen-host interactions. Trends Microbiol 11:224–232. 10.1016/s0966-842x(03)00098-2. [DOI] [PubMed] [Google Scholar]
- 6.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30:209–219. 10.1046/j.1365-2958.1998.01057.x. [DOI] [PubMed] [Google Scholar]
- 7.Graham MR, Smoot LM, Migliaccio CAL, Virtaneva K, Sturdevant DE, Porcella SF, Federle MJ, Adams GJ, Scott JR, Musser JM. 2002. Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc Natl Acad Sci USA 99:13855–13860. 10.1073/pnas.202353699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Churchward G. 2007. The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol Microbiol 64:34–41. 10.1111/j.1365-2958.2007.05649.x. [DOI] [PubMed] [Google Scholar]
- 9.Dalton TL, Collins JT, Barnett TC, Scott JR. 2006. RscA, a member of the MDR1 family of transporters, is repressed by CovR and required for growth of Streptococcus pyogenes under heat stress. J Bacteriol 188:77–85. 10.1128/JB.188.1.77-85.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willett JW, Kirby JR. 2012. Genetic and biochemical dissection of a HisKA domain identifies residues required exclusively for kinase and phosphatase activities. PLoS Genet 8:e1003084. 10.1371/journal.pgen.1003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Churchward G, Bates C, Gusa AA, Stringer V, Scott JR. 2009. Regulation of streptokinase expression by CovR/S in Streptococcus pyogenes: CovR acts through a single high-affinity binding site. Microbiology (Reading) 155:566–575. 10.1099/mic.0.024620-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Federle MJ, Scott JR. 2002. Identification of binding sites for the group A streptococcal global regulator CovR. Mol Microbiol 43:1161–1172. 10.1046/j.1365-2958.2002.02810.x. [DOI] [PubMed] [Google Scholar]
- 14.Gao J, Gusa AA, Scott JR, Churchward G. 2005. Binding of the global response regulator protein CovR to the sag promoter of Streptococcus pyogenes reveals a new mode of CovR-DNA interaction. J Biol Chem 280:38948–38956. 10.1074/jbc.M506121200. [DOI] [PubMed] [Google Scholar]
- 15.Gusa AA, Scott JR. 2005. The CovR response regulator of group A streptococcus (GAS) acts directly to repress its own promoter. Mol Microbiol 56:1195–1207. 10.1111/j.1365-2958.2005.04623.x. [DOI] [PubMed] [Google Scholar]
- 16.Horstmann N, Myers KS, Tran CN, Flores AR, Shelburne SA, III.. 2022. CovS inactivation reduces CovR promoter binding at diverse virulence factor encoding genes in group A Streptococcus. PLoS Pathog 18:e1010341. 10.1371/journal.ppat.1010341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gusa AA, Gao J, Stringer V, Churchward G, Scott JR. 2006. Phosphorylation of the group A streptococcal CovR response regulator causes dimerization and promoter-specific recruitment by RNA polymerase. J Bacteriol 188:4620–4626. 10.1128/JB.00198-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horstmann N, Saldana M, Sahasrabhojane P, Yao H, Su X, Thompson E, Koller A, Shelburne SA, III.. 2014. Dual-site phosphorylation of the control of virulence regulator impacts group A streptococcal global gene expression and pathogenesis. PLoS Pathog 10:e1004088. 10.1371/journal.ppat.1004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dalton TL, Scott JR. 2004. CovS inactivates CovR and is required for growth under conditions of general stress in Streptococcus pyogenes. J Bacteriol 186:3928–3937. 10.1128/JB.186.12.3928-3937.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horstmann N, Sahasrabhojane P, Saldana M, Ajami NJ, Flores AR, Sumby P, Liu C-G, Yao H, Su X, Thompson E, Shelburne SA. 2015. Characterization of the effect of the histidine kinase CovS on response regulator phosphorylation in group A Streptococcus. Infect Immun 83:1068–1077. 10.1128/IAI.02659-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horstmann N, Tran CN, Brumlow C, DebRoy S, Yao H, Nogueras Gonzalez G, Makthal N, Kumaraswami M, Shelburne SA. 2018. Phosphatase activity of the control of virulence sensor kinase CovS is critical for the pathogenesis of group A Streptococcus. PLoS Pathog 14:e1007354. 10.1371/journal.ppat.1007354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agarwal S, Agarwal S, Pancholi P, Pancholi V. 2011. Role of serine/threonine phosphatase (SP-STP) in Streptococcus pyogenes physiology and virulence. J Biol Chem 286:41368–41380. 10.1074/jbc.M111.286690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gryllos I, Grifantini R, Colaprico A, Jiang S, Deforce E, Hakansson A, Telford JL, Grandi G, Wessels MR. 2007. Mg2+ signalling defines the group A streptococcal CsrRS (CovRS) regulon. Mol Microbiol 65:671–683. 10.1111/j.1365-2958.2007.05818.x. [DOI] [PubMed] [Google Scholar]
- 24.Gryllos I, Levin JC, Wessels MR. 2003. The CsrR/CsrS two-component system of group A Streptococcus responds to environmental Mg2+. Proc Natl Acad Sci USA 100:4227–4232. 10.1073/pnas.0636231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gryllos I, Tran-Winkler HJ, Cheng M-F, Chung H, Bolcome R, III, Lu W, Lehrer RI, Wessels MR. 2008. Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc Natl Acad Sci USA 105:16755–16760. 10.1073/pnas.0803815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran-Winkler HJ, Love JF, Gryllos I, Wessels MR. 2011. Signal transduction through CsrRS confers an invasive phenotype in group A Streptococcus. PLoS Pathog 7:e1002361. 10.1371/journal.ppat.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biswas I, Scott JR. 2003. Identification of rocA, a positive regulator of covR expression in the group A streptococcus. J Bacteriol 185:3081–3090. 10.1128/JB.185.10.3081-3090.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lynskey NN, Velarde JJ, Finn MB, Dove SL, Wessels MR. 2019. RocA binds CsrS to modulate CsrRS-mediated gene regulation in group A Streptococcus. mBio 10:e01495-19. 10.1128/mBio.01495-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. 2001. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis 183:1043–1054. 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 30.Garcia AF, Abe LM, Erdem G, Cortez CL, Kurahara D, Yamaga K. 2010. An insert in the covS gene distinguishes a pharyngeal and a blood isolate of Streptococcus pyogenes found in the same individual. Microbiology (Reading) 156:3085–3095. 10.1099/mic.0.042614-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollands A, Pence MA, Timmer AM, Osvath SR, Turnbull L, Whitchurch CB, Walker MJ, Nizet V. 2010. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A streptococcus M1T1 clone. J Infect Dis 202:11–19. 10.1086/653124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horstmann N, Sahasrabhojane P, Suber B, Kumaraswami M, Olsen RJ, Flores A, Musser JM, Brennan RG, Shelburne SA, III.. 2011. Distinct single amino acid replacements in the control of virulence regulator protein differentially impact streptococcal pathogenesis. PLoS Pathog 7:e1002311. 10.1371/journal.ppat.1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shea PR, Beres SB, Flores AR, Ewbank AL, Gonzalez-Lugo JH, Martagon-Rosado AJ, Martinez-Gutierrez JC, Rehman HA, Serrano-Gonzalez M, Fittipaldi N, Ayers SD, Webb P, Willey BM, Low DE, Musser JM. 2011. Distinct signatures of diversifying selection revealed by genome analysis of respiratory tract and invasive bacterial populations. Proc Natl Acad Sci USA 108:5039–5044. 10.1073/pnas.1016282108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog 6:e1000832. 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trevino J, Perez N, Ramirez-Pena E, Liu Z, Shelburne SA, III, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun 77:3141–3149. 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madden JC, Ruiz N, Caparon M. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in Gram-positive bacteria. Cell 104:143–152. 10.1016/s0092-8674(01)00198-2. [DOI] [PubMed] [Google Scholar]
- 37.Zhu L, Olsen RJ, Lee JD, Porter AR, DeLeo FR, Musser JM. 2017. Contribution of secreted NADase and streptolysin O to the pathogenesis of epidemic serotype M1 Streptococcus pyogenes infections. Am J Pathol 187:605–613. 10.1016/j.ajpath.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berge A, Bjorck L. 1995. Streptococcal cysteine proteinase releases biologically active fragments of streptococcal surface proteins. J Biol Chem 270:9862–9867. 10.1074/jbc.270.17.9862. [DOI] [PubMed] [Google Scholar]
- 39.Collin M, Olsen A. 2001. Effect of SpeB and EndoS from Streptococcus pyogenes on human immunoglobulins. Infect Immun 69:7187–7189. 10.1128/IAI.69.11.7187-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eriksson A, Norgren M. 2003. Cleavage of antigen-bound immunoglobulin G by SpeB contributes to streptococcal persistence in opsonizing blood. Infect Immun 71:211–217. 10.1128/IAI.71.1.211-217.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo C-F, Lin Y-S, Chuang W-J, Wu J-J, Tsao N. 2008. Degradation of complement 3 by streptococcal pyrogenic exotoxin B inhibits complement activation and neutrophil opsonophagocytosis. Infect Immun 76:1163–1169. 10.1128/IAI.01116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nyberg P, Rasmussen M, Von Pawel-Rammingen U, Bjorck L. 2004. SpeB modulates fibronectin-dependent internalization of Streptococcus pyogenes by efficient proteolysis of cell-wall-anchored protein F1. Microbiology (Reading) 150:1559–1569. 10.1099/mic.0.27076-0. [DOI] [PubMed] [Google Scholar]
- 43.Terao Y, Mori Y, Yamaguchi M, Shimizu Y, Ooe K, Hamada S, Kawabata S. 2008. Group A streptococcal cysteine protease degrades C3 (C3b) and contributes to evasion of innate immunity. J Biol Chem 283:6253–6260. 10.1074/jbc.M704821200. [DOI] [PubMed] [Google Scholar]
- 44.Wei L, Pandiripally V, Gregory E, Clymer M, Cue D. 2005. Impact of the SpeB protease on binding of the complement regulatory proteins factor H and factor H-like protein 1 by Streptococcus pyogenes. Infect Immun 73:2040–2050. 10.1128/IAI.73.4.2040-2050.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weckel A, Guilbert T, Lambert C, Plainvert C, Goffinet F, Poyart C, Mehats C, Fouet A. 2021. Streptococcus pyogenes infects human endometrium by limiting the innate immune response. J Clin Invest 131:e130746. 10.1172/JCI130746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aziz RK, Pabst MJ, Jeng A, Kansal R, Low DE, Nizet V, Kotb M. 2004. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol 51:123–134. 10.1046/j.1365-2958.2003.03797.x. [DOI] [PubMed] [Google Scholar]
- 47.Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V. 2007. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13:981–985. 10.1038/nm1612. [DOI] [PubMed] [Google Scholar]
- 48.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun 67:5298–5305. 10.1128/IAI.67.10.5298-5305.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiang-Ni C, Kao C-Y, Hsu C-Y, Chiu C-H. 2019. Phosphorylation at the D53 but not the T65 residue of CovR determines the repression of rgg and speB transcription in emm1- and emm49-type group A streptococci. J Bacteriol 201:e00681-18. 10.1128/JB.00681-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Do H, Makthal N, VanderWal AR, Rettel M, Savitski MM, Peschek N, Papenfort K, Olsen RJ, Musser JM, Kumaraswami M. 2017. Leaderless secreted peptide signaling molecule alters global gene expression and increases virulence of a human bacterial pathogen. Proc Natl Acad Sci USA 114:E8498–E8507. 10.1073/pnas.1705972114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosinski-Chupin I, Sauvage E, Fouet A, Poyart C, Glaser P. 2019. Conserved and specific features of Streptococcus pyogenes and Streptococcus agalactiae transcriptional landscapes. BMC Genomics 20:236. 10.1186/s12864-019-5613-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis 192:771–782. 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 53.Zheng P-X, Chung K-T, Chiang-Ni C, Wang S-Y, Tsai P-J, Chuang W-J, Lin Y-S, Liu C-C, Wu J-J. 2013. Complete genome sequence of emm1 Streptococcus pyogenes A20, a strain with an intact two-component system, CovRS, isolated from a patient with necrotizing fasciitis. Genome Announc 1:e00149-12. 10.1128/genomeA.00149-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin J-N, Chang L-L, Lai C-H, Lin H-H, Chen Y-H. 2014. Association between polymorphisms in the csrRS two-component regulatory system and invasive group A streptococcal infection. Eur J Clin Microbiol Infect Dis 33:735–743. 10.1007/s10096-013-2005-7. [DOI] [PubMed] [Google Scholar]
- 55.Beres SB, Carroll RK, Shea PR, Sitkiewicz I, Martinez-Gutierrez JC, Low DE, McGeer A, Willey BM, Green K, Tyrrell GJ, Goldman TD, Feldgarden M, Birren BW, Fofanov Y, Boos J, Wheaton WD, Honisch C, Musser JM. 2010. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci USA 107:4371–4376. 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoff JS, DeWald M, Moseley SL, Collins CM, Voyich JM. 2011. SpyA, a C3-like ADP-ribosyltransferase, contributes to virulence in a mouse subcutaneous model of Streptococcus pyogenes infection. Infect Immun 79:2404–2411. 10.1128/IAI.01191-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Seaghdha M, Wessels MR. 2013. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from xenophagic killing. PLoS Pathog 9:e1003394. 10.1371/journal.ppat.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldmann O, Rohde M, Chhatwal GS, Medina E. 2004. Role of macrophages in host resistance to group A streptococci. Infect Immun 72:2956–2963. 10.1128/IAI.72.5.2956-2963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thulin P, Johansson L, Low DE, Gan BS, Kotb M, McGeer A, Norrby-Teglund A. 2006. Viable group A streptococci in macrophages during acute soft tissue infection. PLoS Med 3:e53. 10.1371/journal.pmed.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hertzen E, Johansson L, Wallin R, Schmidt H, Kroll M, Rehn AP, Kotb M, Morgelin M, Norrby-Teglund A. 2010. M1 protein-dependent intracellular trafficking promotes persistence and replication of Streptococcus pyogenes in macrophages. J Innate Immun 2:534–545. 10.1159/000317635. [DOI] [PubMed] [Google Scholar]
- 61.Tatsuno I, Okada R, Zhang Y, Isaka M, Hasegawa T. 2013. Partial loss of CovS function in Streptococcus pyogenes causes severe invasive disease. BMC Res Notes 6:126. 10.1186/1756-0500-6-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gogos A, Federle MJ. 2020. Colonization of the murine oropharynx by Streptococcus pyogenes is governed by the Rgg2/3 quorum sensing system. Infect Immun 88:e00464-20. 10.1128/IAI.00464-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Finn MB, Ramsey KM, Dove SL, Wessels MR. 2021. Identification of group A Streptococcus genes directly regulated by CsrRS and novel intermediate regulators. mBio 12:e01642-21. 10.1128/mBio.01642-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flores AR, Olsen RJ, Wunsche A, Kumaraswami M, Shelburne SA, III, Carroll RK, Musser JM. 2013. Natural variation in the promoter of the gene encoding the Mga regulator alters host-pathogen interactions in group A Streptococcus carrier strains. Infect Immun 81:4128–4138. 10.1128/IAI.00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alam FM, Turner CE, Smith K, Wiles S, Sriskandan S. 2013. Inactivation of the CovR/S virulence regulator impairs infection in an improved murine model of Streptococcus pyogenes naso-pharyngeal infection. PLoS One 8:e61655. 10.1371/journal.pone.0061655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289–300. [Google Scholar]
- 69.Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193–202. 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 70.Almeida A, Villain A, Joubrel C, Touak G, Sauvage E, Rosinski-Chupin I, Poyart C, Glaser P. 2015. Whole-genome comparison uncovers genomic mutations between group B streptococci sampled from infected newborns and their mothers. J Bacteriol 197:3354–3366. 10.1128/JB.00429-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151:165–188. 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lamy M-C, Zouine M, Fert J, Vergassola M, Couve E, Pellegrini E, Glaser P, Kunst F, Msadek T, Trieu-Cuot P, Poyart C. 2004. CovS/CovR of group B streptococcus: a two-component global regulatory system involved in virulence. Mol Microbiol 54:1250–1268. 10.1111/j.1365-2958.2004.04365.x. [DOI] [PubMed] [Google Scholar]
- 73.Langille MGI, Hsiao WWL, Brinkman FSL. 2010. Detecting genomic islands using bioinformatics approaches. Nat Rev Microbiol 8:373–382. 10.1038/nrmicro2350. [DOI] [PubMed] [Google Scholar]
- 74.Varet H, Brillet-Gueguen L, Coppee J-Y, Dillies MA. 2016. SARTools: a DESeq2- and EdgeR-based R pipeline for comprehensive differential analysis of RNA-Seq data. PLoS One 11:e0157022. 10.1371/journal.pone.0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. 2013. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8:1765–1786. 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 76.Dinis M, Plainvert C, Kovarik P, Longo M, Fouet A, Poyart C. 2014. The innate immune response elicited by group A streptococcus is highly variable among clinical isolates and correlates with the emm type. PLoS One 9:e101464. 10.1371/journal.pone.0101464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Plainvert C, Doloy A, Loubinoux J, Lepoutre A, Collobert G, Touak G, Trieu-Cuot P, Bouvet A, Poyart C. CNR-Strep Network. 2012. Invasive group A streptococcal infections in adults, France (2006-2010). Clin Microbiol Infect 18:702–710. 10.1111/j.1469-0691.2011.03624.x. [DOI] [PubMed] [Google Scholar]
- 78.Cady A, Plainvert C, Donnio P-Y, Loury P, Huguenet D, Briand A, Revest M, Kayal S, Bouvet A. 2011. Clonal spread of Streptococcus pyogenes emm44 among homeless persons, Rennes, France. Emerg Infect Dis 17:315–317. 10.3201/eid1702.101022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 and Tables S1 and S2. Download jb.00039-23-s0001.pdf, PDF file, 1.3 MB (1.3MB, pdf)
Data Availability Statement
The genome sequence data for the five pairs of strains have been deposited at the DDBJ/EMBL/GenBank database (BioProject accession number PRJEB52031 and SRA accession number ERP136702).