

ABSTRACT
Oral, gut, and tumor microbiota have been implicated as important regulators in the carcinogenesis and progression of gastrointestinal malignancies. However, few studies focused on the existence and association of resident microbes within different body regions. Herein, we aim to reveal the durability of the oral-gut-tumor microbiome and its diagnostic performance in hepatocellular carcinoma (HCC). Our study included two cohorts: a retrospective discovery cohort of 364 HBV-HCC patients and 160 controls with oral or fecal samples, a prospective validation cohort of 91 cases, and 124 controls for matching samples, as well as 48 HBV, and 39 HBV-cirrhosis patients for gut microbial patterns examined by 16S rRNA gene sequencing. With the random forest analysis, 10 oral and 9 gut genera that could distinguish HCC from controls in the retrospective cohort were validated among the prospective matching participants, with area under the curve (AUC) values of 0.7971 and 0.8084, respectively. When influential taxa were merged, the AUC of the consistent classifier increased to 0.9405. The performance continued to improve to 0.9811 when combined with serum levels of alpha-fetoprotein (AFP). Specifically, microbial biomarkers represented by Streptococcus displayed a constantly increasing trend during the disease transition. Furthermore, the presence of several dominant microbiota species was confirmed in hepatic tumor and non-tumor tissues with fluorescence in situ hybridization (FISH) and 5 R 16S rRNA gene sequencing. Overall, our findings based on the oral-gut-tumor microbiota provide a reliable approach for the early detection of HCC.
KEYWORDS: Microbiota, hepatocellular carcinoma, 16S rRNA gene, AFP, fluorescence in situ hybridization, early detection
Introduction
Hepatocellular carcinoma (HCC) remains one of the most fatal malignancies with a global increase in incidence and mortality, resulting in over 700,000 deaths annually1. Due to the lack of precisely predictive biomarkers and the asymptomatic nature of the disease, a considerable proportion of patients are detected at an advanced stage to miss the optimal timing for surgical eradication. To date, HCC has been diagnosed with serum AFP levels and imaging examination2. However, as the sole standard predictor for HCC, AFP levels can be elevated in other conditions, such as active hepatitis, gonad embryoma, secondary liver cancer, and pregnancy, restricting the disease specificity to some degree. Therefore, it is urgent to develop a brand-new indication with reasonably high accuracy to boost life expectancy.
Recently, there has been a surge of interest in the etiological role of gut microbiota in a variety of cancers. Referring to HCC, it has a complex etiology that comprises alcohol, hepatitis virus, and aflatoxin3, which are frequently accompanied by visible alterations in the microbiome. For instance, convergent evidence has confirmed that hepatitis virus infection can elicit structural changes in the intestinal barrier and increase microbial translocation via the gut-liver axis, resulting in impaired mucosal immunological function, and an excessive long-term immune response that contributes to the development of liver diseases, including fibrosis, cirrhosis, and even HCC4,5. Simultaneously, the oral microbiota has been widely linked to digestive system diseases, such as oral squamous cell carcinoma (OSCC)6, inflammatory bowel disease (IBD),7 and liver cirrhosis8, indicating microbial migration of the intestine from the oral cavity. In particular, significant changes occur in the oral microbiome of patients with liver carcinoma and cirrhosis, which are expected to distinguish HCC from healthy conditions as noninvasive biomarkers9. Moreover, altered gut microbiome profiles have been implicated in the diagnosis of HCC, and Ren et al. established a diagnostic model with successful cross-region validation10. However, previous studies merely focused on the link between the oral or fecal microbiome and HCC, which remains as yet unexplored in the translation of combined improvements into a novel microbiome signature for early detection as a signal of imminent HCC development.
Nowadays, distinct intratumor microbiota compositions have been verified in several cancers with low biomasses that are mostly localized to cancer and immune cells, and are probably generated from circulating bacteria11. In addition, it was identified that the dominant microbiome Proteobacteria in pancreatic cancer was similar to that in duodenal microbiome makeup, which reflected retrograde bacterial invasion from the gut to the pancreatic duct12. Similarly, the presence of tumor-associated microbiota has been characterized in HCC patients with hepatitis virus13, which may assist in illustrating the potential pathological contribution of intratumoral microbiota from either the portal vein or bile duct to HCC. For this reason, the so-called oral-gut-liver axis has been proposed, in which dysregulated oral bacteria infiltrate the gut, disseminate negative events to the resident intestinal microbiome, and result in liver diseases8. Here, we prospectively evaluated a cohort of 91 newly diagnosed patients with HCC and 124 controls. Both oral and fecal samples, along with suitable hepatic tumor and normal tissues, were collected for microbial composition examination with 16S rRNA gene amplicon sequencing, IHC and FISH assays. Based on a set of 19 integrated genera, we identified a distinct microbial signature with high prediction accuracy. More importantly, HCC-enriched species were investigated in tumorous tissues, with a possible relationship with oral and gut residents, disclosing a potential involvement in HCC pathogenesis.
Methods
Patient enrollment and sample collection
Briefly, our study comprised two independent cohorts: a retrospective cohort of 364 patients with newly diagnosed HBV-HCC and 160 control individuals (healthy volunteers and in patients with hepatic hemangioma) without HCC-related risk factors were recruited from the First Affiliated Hospital of Wenzhou Medical University from May 2019 to September 2020. The 16S rRNA gene amplicons of oral or gut microbiome and clinical features were obtained. In the prospective population (ClinicalTrials.gov identifiers NCT04637048), we collected oral (tongue surface) and matching fecal samples from 124 patients with HCC and 91 controls between March and October 2021. Meanwhile, 16S rRNA gene amplicons of the fecal microbiome for the patients with HBV-Cirrhosis (n = 39) and HBV-Noncirrhosis (n = 48) were obtained. Moreover, 46 HCC tissues, 42 paracancerous tissues, and 11 healthy tissues from the control group (hepatic hemangioma) were analyzed by 5 R 16S rRNA gene sequencing, following a strictly sterile procedure with a blank control to avoid cross-contamination11. Clinical data pertaining to age, gender, BMI, and follow-up information were acquired from the same hospital structured questionnaires. Serum AFP levels were examined with an electrochemiluminescence immunoassay according to the manufacturer’s instructions from the Medical Laboratory Center. Written informed consent and ethics approval were obtained from the study participants and the Ethics Committee in Clinical Research (ECCR) of the First Affiliated Hospital of Wenzhou Medical University. Prior to the tongue surface samples collection with the sterile cotton swab, the oral health condition was evaluated among the participants, excluding potential tongue-related diseases that might alter the innate oral microbiota, such as Sjogren’s syndrome, Meige’s syndrome, atrophic glossitis, oral leukoplakia, and tongue fungal infection, etc. In addition, participants were instructed to gargle before sampling during non-eating times. Following that, oral and fecal samples were collected in sterile tubes and temporarily moved to the −20°C freezer in each ward, and were permanently stored at −80°C for DNA extraction and taxonomic analysis. For a subset of individuals, tumor and paracancerous tissues were obtained during surgery, immediately flash-frozen in liquid nitrogen, and stored at −80°C. All samples were shipped with dry ice.
Inclusion criteria
Patients with HBV-related HCC (clinically and histologically diagnosed) and healthy participants aged over 18 were enrolled.
Exclusion criteria
(1) Alcoholic liver cancer, nonalcoholic fatty liver cancer, HCV-related liver cancer, etc. (2) intrahepatic cholangiocarcinoma and mixed liver cancer (3) history of other malignancies (4) antibiotics or probiotics usage within 1 month of sample collection (5) presence of hypertension, diabetes, and other metabolic diseases (6) Crohn’s disease, ulcerative colitis, and inflammatory bowel disease.
The inclusion and exclusion criteria were applied identically to each cohort.
Collection of sterile tissues
Tumor and normal hepatic tissues were dissected in the operating room using sterile scalpels that were changed before dissecting different samples to avoid cross-contamination. The cutting tissue was placed in a sterile and nucleus-free cryopreservation tube with a corresponding blank collection tube as a negative control for 5 R 16S rRNA gene sequencing, which was left in the collection area with the lid open for 30 s and immediately stored with the sample tubes.
DNA/RNA extraction, amplification, sequencing, and analysis
Genomic DNA of oral and fecal samples was extracted with the EZNA® Soil DNA kit in accordance with the manufacturer’s guidelines (D4015, Omega, Inc., USA). Genomic DNA from hepatic tumor and non-tumor samples was extracted with an EZNA® Tissue DNA kit (D3396, Omega, Inc., USA) in a standard protocol. The isolated DNA was immediately stored at −80°C for amplification. For oral and gut DNA, the 16S rRNA gene V3-V4 region was amplified with slightly modified microbiome primers (515F-805 R) in a two-step PCR protocol14. To characterize the intratumor microbiota, a modified 5 R 16S rRNA gene amplification was done after amplifying five regions on the multiplexed 16S rRNA genes11. The amplification products were purified with AMPure XT beads (Beckman Coulter, USA) and quantified with Qubit (Invitrogen, USA). Subsequently, the amplicon pools were applied for sequencing, and the quantity and size of the amplified libraries were evaluated on Agilent 2100 Bioanalyzer (Agilent, USA). Samples were sequenced on the Illumina NovaSeq platform from the manufacturer’s recommendations, which were supported by Lc-Bio Technologies Co., Ltd (Hangzhou, China). Afterward, raw reads were analyzed with QIIME2 software, and quality filtration was performed in fqtrim software (V.0.9.4) to acquire high-quality clean tags. DABA2 software was employed to construct the sequence and feature tables containing the amplicon sequence variants (ASVs). A taxonomic analysis for species annotation was conducted using BLAST with the database of SILVA and NT-16S. MAFFT software was used to explore the predominant species in different groups and diverse sequence alignments. The alpha diversity of specimens was characterized by the Chao1 and Shannon indices, which were determined with QIIME2. Beta diversity was described by principal component analysis (PCA) and principal coordinates analysis (PCoA) with R ade4 and vegan packages, and the P value was analyzed by the Wilcoxon test.
Shotgun metagenomic sequencing
A number of backup gut samples from the HCC (n = 42) and control (n = 34) groups in the prospective cohort were further analyzed by the shotgun metagenomic sequencing. Total genomic DNA was obtained from backup gut samples with the EZNA® Stool DNA kit. After evaluation of the purity and concentration, genomic DNA was pooled for sequencing on an Illumina Hiseq 2500 platform with NovaSeq Reagent Kits. Raw data were sorted with assigned barcodes and filtered out before analysis. The Partek Flow and MetAMOS pipeline software15 were applied for the annotation and assembly of sequences. Afterward, the non-redundant Unigenes sets were obtained through sequencing clustering for relevant functional profiles. The Diamond program was used to annotate the functional profiling of KEGG pathway information by comparing it with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database16. A one-way ANOVA analysis was conducted to assess the statistical difference in functional items with Statistical Analysis of Metagenomic Profiles (STAMP)17.
Microbial data statistical analyses
All steps of data analyses were performed in the R Statistical Computing platform. The continuous variables were represented as mean ± standard error of mean (SEM) or median with interquartile range, and categorical data were described with frequency. Differences in alpha diversity were verified with an analysis of Wilcoxon. The beta diversity between differences in sample community composition was presented as PCA and PCoA with a weighted Unifrac index. The relationship between community composition and microbiome extrinsic element was analyzed using the PERMANOVA and VEGAN R packages. The FDR method of Bonferroni correction was applied to avoid a type I error (false positives) for multiple testing of multivariate microbiota data. The Bonferroni-adjusted P values were considered to compensate for the significant level [α = 0.05, (n = 812 for retrospective oral test, P = 6.2 × 10−5; n = 989 for the retrospective fecal test, P = 5.1 × 10−5; n = 766 for prospective oral test, P = 6.5 × 10−5; n = 782 for the prospective fecal test, P = 6.4 × 10−5)]. Wilcoxon-rank-sum analysis was utilized to evaluate the significant difference in microbial abundance with the STAMP software, and the statistically enriched microorganisms were determined using a linear discriminant analysis effect size (LEfSe) algorithm with an LDA score of more than three (Bonferroni-adjusted P < 0.05). The predominant data flow that occurred during the disease progression at phylum and genus levels was visualized with a bubble plot, heatmap and Sankey plot using the R pheatmap, stats, ggplot2, and ggalluvia packages, respectively. The SparCC algorithm18 was performed to identify the microbial correlation of biomarkers between HCC and control groups (correlation p < 0.05 and |rho| > 0.1). Redundancy Analysis (RDA) was conducted to select several relevant clinical variables from the fecal microbiota with the R vegan package.
Establishment of multivariable statistical models and evaluation
Classification models were established with the random forest algorithm in the RandomForest R package. Following that, taxa with poor richness and prevalence were excluded from the multivariable statistical training models for HCC prediction. Certain taxa identified between HCC and the control group were integrated separately for the model construction of the oral and fecal microbiome. The area under the receiver operating curve (ROC) with validation scores was used to evaluate the performance in the prediction models. Alternatively, features with a ROC<0.5 were eliminated after the cross-validation, therefore sorting out features enriched in HCC. The selected influential taxa in oral and fecal microbiota were merged to culminate in a new classifier. Likewise, the ROC value of the combined classifier was calculated by the pROC R package as well. The oral and fecal samples were collected for taxonomic analysis immediately after the participants were enrolled in the validation cohort, and genera-based signatures were applied for the identification of HCC patients based on the imaging examination plus AFP or pathological test. All data processing, model training, predictions, and evaluation were conducted by the SIAMCAT R package.
Evaluation of intestinal permeability
Intestinal permeability was evaluated by quantification of serum lipopolysaccharide (LPS), zonulin-1 (ZO-1) and fecal calprotectin. Accordingly, a competitive enzyme-link immunosorbent assay (ELISA) was conducted for the quantitative determinant of ZO-1 (ZO-1, Immundiagnostik AG, Bensheim, Germany), and calprotectin (Elabscience, Houston, USA). The corresponding antibody was pre-coated in the microplate with 100 μl standards, serum, and fecal suspensions (feces were dissolved in PBS and vortexed for 30 min) were incubated at 37°C for 90 min. The biotinylated detection antibodies for ZO-1, calprotectin, and avidin-HRP conjugate were converted to each microplate. The concentration of ZO-1 and calprotectin was determined in a microplate reader with an absorbance of 450 nm, and bicinchoninic acid (BCA, Thermo Fisher Scientific, USA) was used for the quantification of fecal protein concentrations. The bacterial LPS was detected and measured by the chromogenic LAL endotoxin assay (Piscataway, NJ, USA), which was based on a colorimetric reaction in which the endotoxin could activate a proenzyme in the Limulus Amebocyte Lysate (LAL). 100 μl standard, serum sample, and LAL reagent were dispensed into endotoxin-free vials, then the mixture was incubated with 100 μl reconstituted chromogenic substrate solution for 6 min at 37°C. The reaction was stopped with the 500 μl reconstituted stop solution (hydrochloric acid with LAL reagent water), and the absorbance at 545 nm was measured in a microplate reader. The endotoxin concentration was calculated from a standard curve.
Immunohistochemistry (IHC) assays
IHC staining was conducted in a standard procedure, which included deparaffinization and rehydration stages. The antigens were retrieved by slightly boiling the citrate buffer in a microwave for 15 min at low-to-medium power. Endogenous catalase activity was quenched with 3% hydrogen peroxide for 10 min. After blockage with 5% bovine serum albumin for 1 hour, the samples were incubated with primary LPS (Lipopolysaccharide Core, HM6011) and LTA (Lipoteichoic acid, HM2048-200UG) antibodies (1:50) at 4°C overnight, followed by incubation with secondary antibody for an hour at room temperature in a humid chamber. The slices were stained with DAB (1:1000 dissolved in 30% hydrogen peroxide) and terminated with washing water. The sections were then incubated with hematoxylin for 3 min and sealed with neutral resin. The images were captured and analyzed using a Leica DM6000 microscope with CytoVision software.
Fluorescence in situ hybridization (FISH)
The FISH test was carried out using a probe that targeted the 16S rRNA gene sequence of a specific bacterial taxon. The probes were displayed in Supplemental Table S2. HCC tumor specimens were obtained during the surgical resection and immediately stored in liquid nitrogen under sterile circumstances. After that, the samples were preserved in a −80°C freezer for long-term storage. The samples were transferred to an appropriate cutting temperature mold and cut on a cryotome into several sections of 4 μm. All materials were sanitized using ethanol after sample processing. Sample slices of 4 μm thickness were mounted on the slides. Tissues were fixed in fresh 4% paraformaldehyde, and bacterial wall permeabilization was enhanced by 1 mg/ml lysozyme at room temperature (RT) for 10 min, followed by treatment with 20 μg/ml proteinase K for 10 min at RT. The probes were diluted to 1 μM in hybridization buffer with 10% dextran sulfate, 25% formamide, 1 mg/mL E. coli tRNA, 0.02% BSA, and 2× SSC buffer. Tissue sections were hybridized with specific probes overnight at 42°C after pre-hybridization in the hybridization buffer at 37°C for an hour. The unbounded probes were washed with wash buffer for 30 min at RT. The sections were stained with 1 ng/ml DAPI for 5 min at RT in the dark. The germ-free (GF) section denoted a clinical HCC tumor sample that was determined to be sterile and devoid of colonized bacteria using 5 R 16S rRNA gene sequencing, and it was selected as a control for the hybridization protocol. The FISH images were captured with a Nikon Eclipse Ci-L microscope. The images were analyzed and scored on the basis of the fluorescence signal.
Results
Clinicopathological features of participants in the study
All the participants were recruited from our hospital with the same criteria. The characteristics of the two cohorts were depicted in Figure 1a. Clinicopathological variables including age, gender, BMI, alcohol consumption, smoking history, Child-Pugh classification, serum AFP level, and liver function indexes et al. were presented in Figure 1b (for the prospective cohort) and Supplemental Table S1 (for the retrospective cohort). Several basic variables, such as age, gender, and the Child-Pugh score showed no significant difference among the groups, whether in the retrospective or prospective study. In addition, microbial-related factors like obesity, cigarette smoking, alcohol consumption, and non-antibiotic drugs usage, such as chemotherapy agents, proton-pump inhibitors (PPIs), nonsteroidal anti-inflammatory drugs (NSAIDs), atypical antipsychotics (AAPs), osmotic laxative and Chinese medicine, were generally matched. Patients with diabetes or hypertensive who had previously taken metformin, angiotensin II receptor blocker (ARB), angiotensin-converting enzyme inhibitors (ACEI), thiazide, or similar medication were also excluded, removing potential confounders to affect microbiome profiles and group discrimination.
Figure 1.
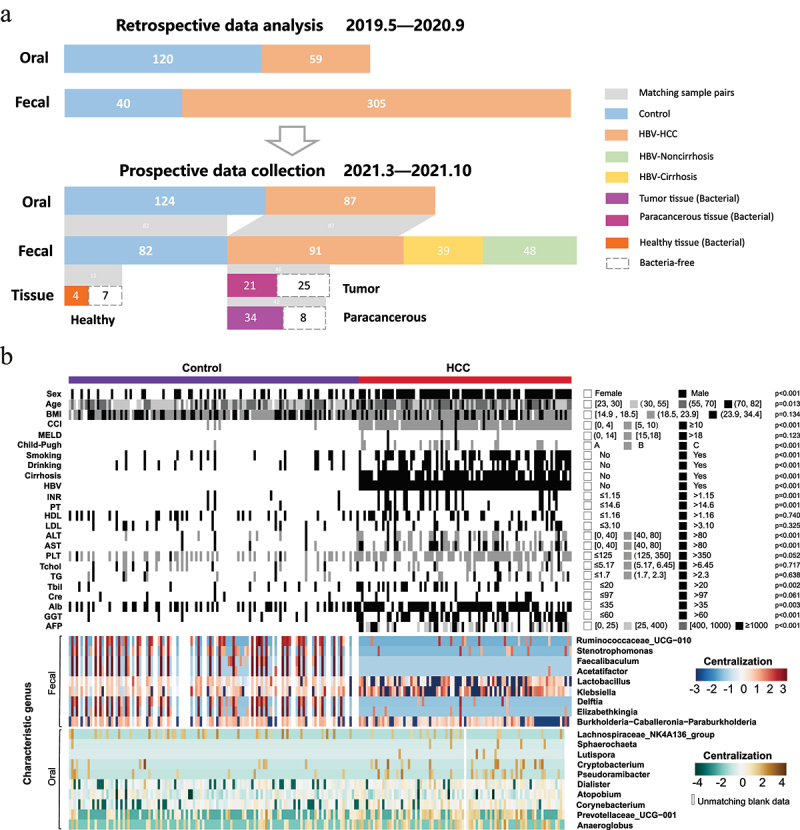
Community analysis of clinicopathological features in the longitudinal study. (a) Overview of the study population. Grey bands between bar plots represent samples of matching body regions within individuals. (b) Clinicopathological information and normalized abundance (log10 transformation and centralization) of influential genera (10 oral genera and 9 gut genera) shown as heatmap between HCC (n = 91) and the control groups (n = 124) in the prospective study with the statistical p values. The blank cell in the “oral” and “fecal” panels represented the unmatching data of corresponding individuals.
Abbreviations: BMI, body mass index; CCI, Charlson comorbidity index; MELD, model for end-stage liver disease; HBV, hepatitis B virus; INR, international normalized ratio; PT, prothrombin time; HDL, high-density lipoprotein; LDL, low-density lipoprotein; ALT, alanine transaminase; AST, aspartate transaminase; PLT, platelet; Tchol, total cholesterol; TG, Triglyceride; Tbil, Total bilirubin; Cre, creatinine; Alb, albumin; GGT, γ-Glutamyl transpeptidase; AFP, alpha-fetoprotein.
The microbial alterations discriminate HCC patients from controls with oral and fecal samples analyzed by the 16S rRNA gene sequencing
In the prospective study, we collected both oral and gut samples from a cohort between March and October 2021 for the 16S rRNA gene sequencing. As illustrated in Figure 2a–d, the HCC group showed separated clusters from the control group with statistical β-diversity (PCA and PCoA analyses, p < 0.05), whether in oral or gut samples. Besides, the statistical taxonomic analysis was conducted at the levels of phylum and genus to determine the prevalent microbiota composition. At the phylum level, Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, and Fusobacteria dominated the two groups in terms of relative abundance taxa. In oral samples, Firmicutes and Actinobacteria showed a significant increase in the HCC group compared with the control group (Firmicutes: 26.81% versus 32.91%; Actinobacteria: 9.74% versus 13.38%), whereas the relative abundance of Proteobacteria, and Fusobacteria were significantly enriched in the subjects of the control group (Proteobacteria: 28.45% versus 21.35%; Fusobacteria: 11.11% versus 9.09%) (Figure 2e). In gut samples, from the stacked bar plot of four predominant phyla, it was identified that the HCC group had a considerably greater average Proteobacteria composition (20.22%) than the control group (16.15%) (Figure 2g). The top 30 genera were plotted at the genus level, while the rest were merged as Others (Figure 2f–h). The box plots generated certain statistical general that comprised 10 and 21 abundant taxa (abundance>1%, Bonferroni-adjusted p < 0.05) in the oral and fecal samples correspondingly (Figure 2i,j). Furthermore, LEfSe analysis was used to identify the bacterial differences in the cladogram (Figure 2k,l) with the criteria of LDA score>3 (Bonferroni-adjusted p < 0.05). Particularly, Streptococcus was more abundant in HCC than that in the control group in both samples. Similarly, the microbiota diversity and taxonomic profiling of the retrospective cohort were analyzed and manifested in Supplemental Figure S1. In short, the microbiota profiles alter in HCC and differ from those in healthy conditions, proposing a microbial transformation of the disease.
Figure 2.
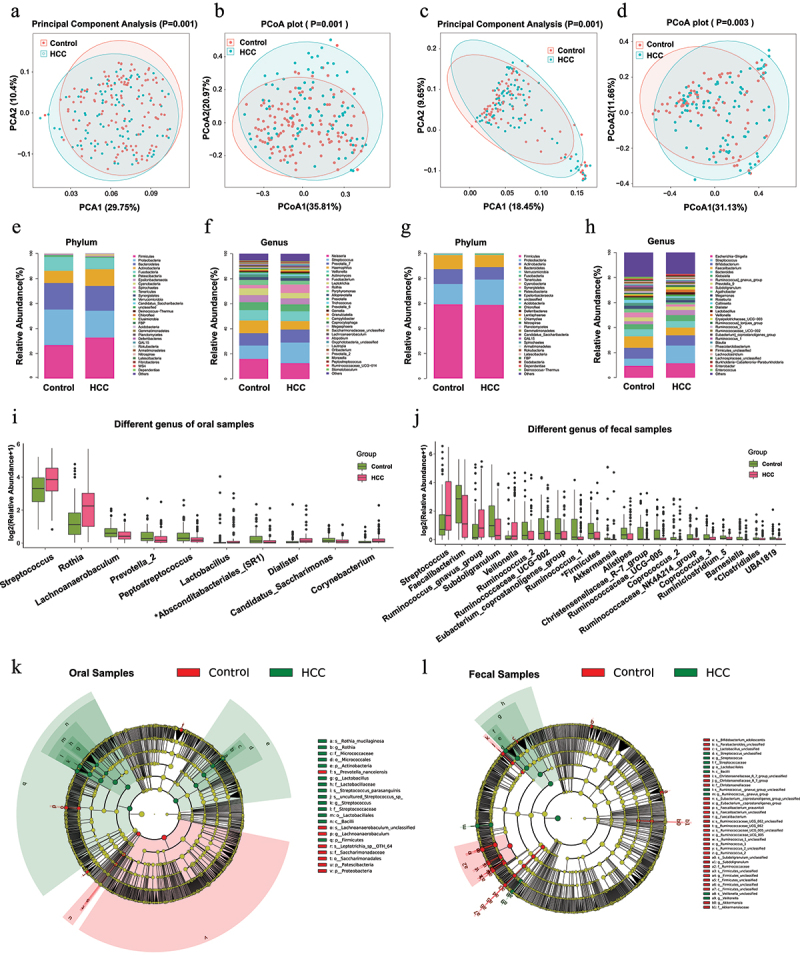
The microbial composition and difference between HCC and the control groups in the prospective study.
(a-d) The distributional difference of oral (a-b) and gut (c-d) microbiota was assessed using principal component analysis (PCA) and principal coordinates analysis (PCoA) with the weighted Unifrac index. P value was calculated by the analysis of similarities (ANOSIM). Red and blue plots represented the control and HCC samples.
(e-h) Stacked bar plot of mean proportions of oral (e-f) and gut (g-h) derived taxonomic composition between HCC and the control groups at phylum and genus levels.
(i-j) Statistically differential genera of oral and gut microbiota were evaluated with box plots (Bonferroni-adjusted P < 0.05, mean abundance>1%). The “*” before the taxa represented the unclassified genus.
(k-l) Predominant taxa distribution between groups in a phylogenetic tree with cladogram computed by linear discriminant analysis effect size (LEfSe) algorithm (LDA [log10 transformation] > 3, Bonferroni-adjusted P < 0.05). The circles radiating from inside to outside represented the classification level from Kingdom to Species. Each node on different levels represented the specific taxon, with the diameter corresponding to the relative abundance. Taxa with significant differences (LDA>3, Bonferroni-adjusted P < 0.05) were highlighted and labeled between HCC and the control groups with red and green nodes. Significant taxa were comprehensively described in another panel.
Gut microbial alterations during the transition from healthy status to cirrhosis and HCC
Well-known factors for HCC, HBV and cirrhosis play critical roles in the disruption of microbial composition. Therefore, HBV individuals with and without cirrhosis were enrolled in our prospective study to investigate the changes of intestinal microbiota in the progression from healthy condition to HCC with the taxonomic resolution of 16S rRNA gene amplicons. Among the control, non-cirrhosis, cirrhosis, and HCC groups, the bubble plot and heatmap revealed that the top 5 phyla and top 30 genera were shared across the four groups (Figure 3a,b). The Sankey plot, in particular, was constructed to exhibit the branch association between phylum and genus degree of taxa with different colors (Figure 3c). And the size of the bubble and the breadth of the branch represented the relative richness of each genus among the groups. The average composition of Streptococcus, Subdoligranulum, Prevotella_9, Agathobacter, Faecalibacterium, Bifidobacterium, Ruminococcus]_gnavus_group, Klebsiella, Escherichia-Shigella, and Bacteroides showed the predominant abundance at the genus level, with Firmicutes accounted for the majority at the phylum level. Statistically, the control individuals had the highest abundance of Faecalibacterium (9.12%), Bacteroides (5.65%), Prevotella_9 (3.16%), and Agathobacter (2.94%), and the lowest abundance of Streptococcus (5.67%) and Escherichia-Shigella (9.55%) among the groups. And the HCC group had the highest proportion of Ruminococcus]_gnavus_group (4.08%) and the lowest proportion of Subdoligranulum (1.73%). Of note, Streptococcus (14.14% in HCC, 14.95% in cirrhosis) and Escherichia-Shigella (11.5% in HCC, 13.21% in cirrhosis) were considerably more abundant in HCC and cirrhosis groups than those in the control group (Streptococcus 5.67%, Escherichia-Shigella 9.55%), accompanied by an ascending trend during disease progression. The average abundance of Agathobacter (1.58%) in HCC and Prevotella_9 (0.27%) in the cirrhosis group, on the other hand, was significantly decreased. Additionally, we attempted to characterize the microbiome phenotypes at the organism level with the BugBase tool that incorporated three potential characteristics, including anaerobic, contains mobile elements and facultatively anaerobic. Consequently, the scatter plot depicted an apparent decrease in anaerobic (healthy individuals: 87.23%, HBV-Noncirrhosis: 81.11%, HBV-cirrhosis: 71.51%, HBV-HCC: 73.65%; p = 0.0047), and an increase in mobile elements (healthy individuals: 2.36%, HBV-Noncirrhosis: 6.23%, HBV-cirrhosis: 16.40%, HBV-HCC: 16.28%; p = 0.0001), and facultatively anaerobic (healthy individuals: 5.74%, HBV-Noncirrhosis: 9.33%, HBV-cirrhosis: 18.44%, HBV-HCC: 20.54%; p = 1.425594e-05) throughout the disease evolution (Figure 3d–f), which might indirectly reflect the changes in the intestinal epithelial hypoxia as the disease progressed. Moreover, the altered intestinal permeability caused by epithelial hypoxia was evaluated between the control and HCC groups. As illuminated in Figure 3g–i, the HCC group showed enhanced intestinal permeability than the control group indicated with significantly elevated levels of serum LPS [(6.678 ± 0.78) EU/ml versus (4.025 ± 0.57) EU/ml, p = 0.0083], ZO-1 [(147.60 ± 26.26) pg/ml versus (38.67 ± 8.30) pg/ml, p = 0.0002] and fecal calprotectin [(1.84 ± 0.17) pg/mg versus (0.57 ± 0.05) pg/mg, p < 0.0001]. Taken together, these data showed the landscape of intestinal hypoxia and microbial alterations during hepatic carcinogenesis.
Figure 3.
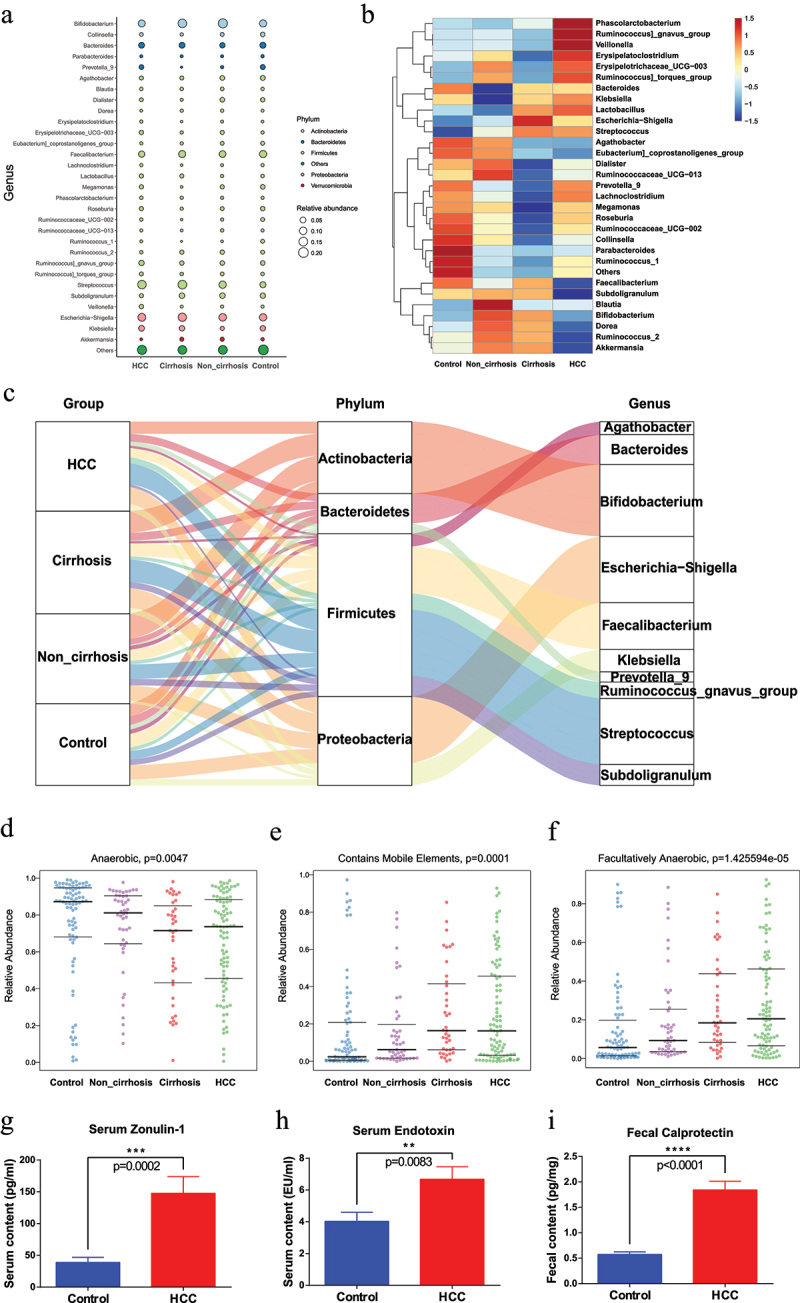
Dynamic microbial alteration in the HCC transition from healthy condition (fecal samples obtained from control group, non_cirrhosis group, cirrhosis group and HCC group) of the prospective cohort.
(a) Bubble plot of the gut genus affiliation to phylum with different colors and the abundance of genera with bubble size.
(b) Heatmap of different gut genera among the control, non-cirrhosis, cirrhosis, and HCC group. Color in the heatmap was utilized to describe specific general abundance within the four groups, with blue indicating lower abundance and red indicating higher abundance.
(c) Sankey plot of the taxonomic data changes with the breadth of the branch at genus (right side) and phylum (middle) levels during the disease progression (left side). The color and width of the branches represented the flow of specific genera within different phyla.
(d-f) Characteristic analysis of fecal microbiota phenotype among the four groups with Anaerobic (d), Contains mobile elements (e) and Facultatively anaerobic (f).
(g) Metagenomic sequencing validation of the mobile genetic element database between HCC (n = 42) and Control (n = 24) groups
(h-j) The levels of serum Zonulin-1 (g), LPS (h), and fecal calprotectin (i) between the control and HCC groups, with the data representing mean ± SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001; ns, no significance.
Microbial relevance with clinical features and functional profiling between the groups
After recognition of the difference between HCC and the control group, SparCC network analysis was used to clarify the interactive correlation of the prevalent genera with each other (Figure 4A–B). Relevantly, Streptococcus was identified in both oral and intestinal correlation networks, where it exhibited positive and negative connections with a total of 11 genera. According to the 16s rRNA gene amplicons, the underlying relationship between clinical features and fecal microbiome was detected with the RDA plot in which the distribution of microbiome was positively related with MELD, Child-Pugh, Age, CCI, AFP, AST, ALT, GGT, Cre, LDL, Tchol, PT and INR, and negatively related with ALB, BMI, HDL, TG, and PLT in HCC patients. Streptococcus and Veillonella showed a consistent tendency whereas Faecalibacterium, Akkermansia, Ruminococcaceae_UCG−002, Rumin-ococcaceae_UCG−005, Ruminococcus_2, and Subdoligranulum showed the contrary (Figure 4c–e). More importantly, for direct identification of the functional profiles, a number of backup gut samples from the HCC (n = 42) and control (n = 34) groups in the prospective cohort were further analyzed by the shotgun metagenomic sequencing. Methodologically, the Diamond algorithm was constructed to annotate and calculate the statistical difference in the KEGG pathways between the two groups (Figure 5, adjusted p < 0.05). In the majority of the top 20 statistical KEGG items, the HCC group showed more activities than the control group, including Arginine and proline metabolism; Alanine, aspartate, and glutamate metabolism; Cysteine and methionine metabolism; Arginine biosynthesis; Valine, leucine, and isoleucine biosynthesis, which underscored the significance of secondary metabolites, namely the amino acid synthesis and metabolism in the HCC microbiome.
Figure 4.
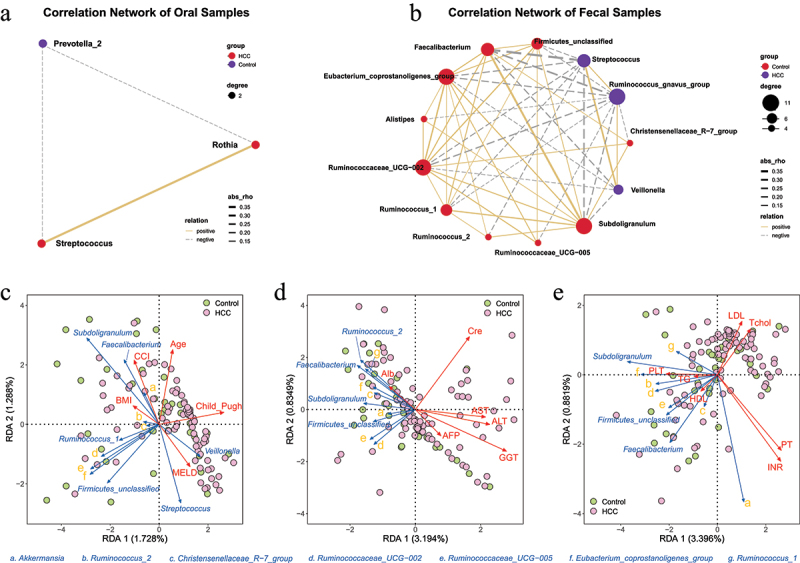
Microbial biomarkers (LDA>3, Bonferroni-adjusted P value<0.05) intercorrelation and relevance with clinical characteristics between HCC and the control groups in the prospective cohort.
(a-b) SparCC network of the abundance of genera with node size and the link between nodes with a correlation coefficient |rho| more than 0.1 by default in oral (a) and fecal (b) samples, with positive and negative correlations represented by solid and dashed lines, respectively (correlation p < 0.05).
(c-e) RDA analysis of clinical feature and fecal genus in HCC and control groups, with the acute and obtuse angle indicating the positive and negative association between specific genus and clinical indicator.
Figure 5.
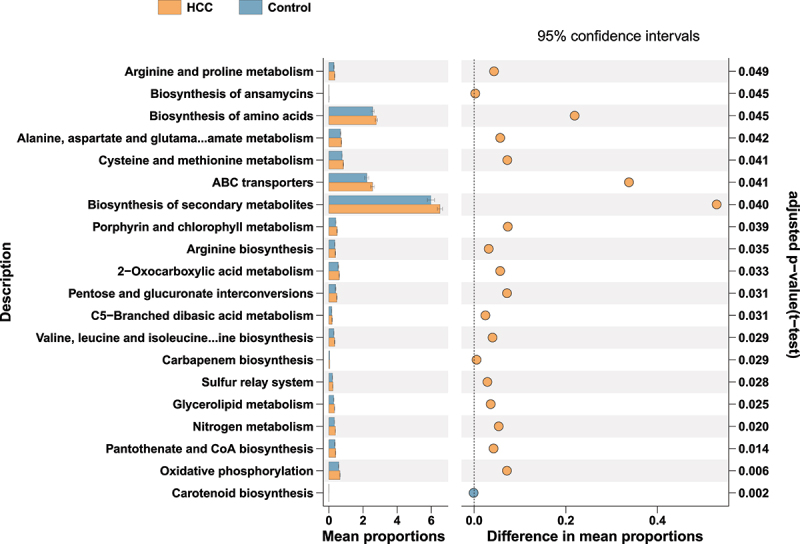
The major KEGG pathways between HCC and the control groups of the prospective cohort with the metagenomic sequencing data of fecal samples.
Differential shotgun metagenomic sequence-based KEGG pathways in gut microbiota between the two groups detected by Diamond software. The top 20 items are listed along with the appropriate 95% confidence intervals and adjusted-p values.
The performance of a combined oral and fecal microbiota-based model in the diagnosis of HCC
Having recognized the existence of a microbial signal for HCC at the coarse level of general community composition based on the 16S rRNA gene sequencing, the random forest method was employed to visualize specific taxa that contributed to the diagnostic potential between HCC and the control group. In the retrospective study, the top 20 influential taxa were identified in oral or fecal microbiota, as represented by the mean decrease accuracy of each taxon in Figure 6a–b. After qualification, 10 (Anderoglobus, Corynebacterium, Prevotellaceae_UCG-001, Pseudoramibacter, Atopobi-um, Lutispora, Cryptobacterium, Dialister, Lachnospiraceae_NK4A136_group, Sphaerochaeta) and 9 (Elizabethkingia, Burkholderia_Caballeron-ia_Paraburkholderia, Klebsiella, Delftia, Faecalib-aculum, Acetatifactor, Lactobacillus, Rumino-coccaceae_UCG-010, Stenotrophomonas) genera with disease-specific univariate (Figure 1b) were applied to establish oral and fecal models with favorable ROC values of 0.7971 and 0.8084 in the prospective cohort for validation, which was comparable to the accuracy of AFP signature (AUC = 0.8505, Figure 6c). Taken together, the oral and fecal influential taxa were merged to develop a combined prediction model. Afterward, the classification accuracy of such a prediction model between HCC and the controls was verified with an AUC of 0.9405, indicating its potent differential diagnosis capability in the prospective population. Combined with AFP, the performance of the diagnostic model further improved to 0.9811. Generally, our results explained the diagnostic potential of the microbiota and established related classifiers with favorable accuracy.
Figure 6.
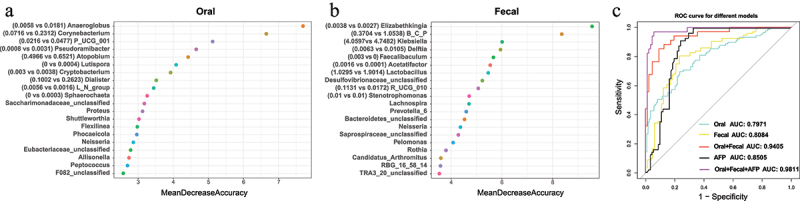
Establishment of classification models from the retrospective data and validation for distinguishing HCC from the control in the prospective cohort.
(a-b) The top 20 influential oral (a) and gut (b) genera for distinguishing HCC from the control were identified by random forest analysis from the retrospective data. Each genus is ranked with the mean decrease in accuracy. The mean relative abundance (%) of the predictive genus derived from the oral (n = 10) and fecal (n = 9) models was described as Control vs HCC between the two groups of the prospective data.
Abbreviations: B_C_P (Burkholderia_Caballeronia_Paraburkholderia), R_UCG_010 (Ruminococcaceae_UCG_010), L_N_group (Lachnospiraceae_NK4A136_group), P_UCG_001 (Prevotellaceae_UCG_001).
(c) External longitudinal-validation results of the AFP model, oral model, fecal model, oral-fecal model, and the combination model of AFP with microbial features in the prospective cohort were shown as ROC curves with corresponding colors.
Associations between the oral, intestinal, and hepatic microbiomes
The biliary duct anatomically interacts with the duodenum, which proposed an evident link for the microbiota to colonize the hepatic tissues and facilitate HCC development. To corroborate the presence of several influential taxa described in the prospective study, we taxonomically profiled the biopsies of HCC tumors (n = 46), normal hepatic tissues from paracancerous regions (n = 42), and hepatic hemangioma (n = 11) with 5 R 16S rRNA gene sequencing, following a strict procedure to eliminate putative bacterial contaminants. A relatively rich and diverse hepatic microbiome with the top 30 genera and corresponding 16 phyla were observed in over 50% of the samples (Figure 7a–b). Among these, a series of genera including Enterococcus, Escherichia-Shigella, Faecalibacterium, Streptococcus, Delftia, Bifidobacterium, Prevotella, Parabacteroides, and Stenotrophomonas were abundant in tumors (abundance>1%), along with their presence in the oral or gut samples of HCC patients. In particular, among the differential taxa in oral and fecal samples, the species Streptococcus parasanguinis was identified in tumors with a relatively low abundance. We then characterized the microbes using IHC assays with LPS (for gram-negative bacteria) and LTA (for gram-positive bacteria) staining, which showed the presence of microbiota as punctate dots (Figure 7c). FISH assays with specific primers were utilized to further verify the prevalence of Streptococcus parasanguinis, Streptococcus mitis, Streptococcus salivarius, Delftia acidovorans, Parabacteroides distasonis, and Stenotrophomonas maltophilia in tumor tissues. Owing to the decreased sensitivity of FISH, Stenotrophomonas maltophilia, Parabacteroides distasonis, Streptoco-ccus salivarius, and Streptococcus mitis were detectable in the tested samples (Figure 7d). In summary, the concordant amplicon, IHC, and FISH data suggested that distinct hepatic microbial residents may be derived from the oral and the gut.
Figure 7.
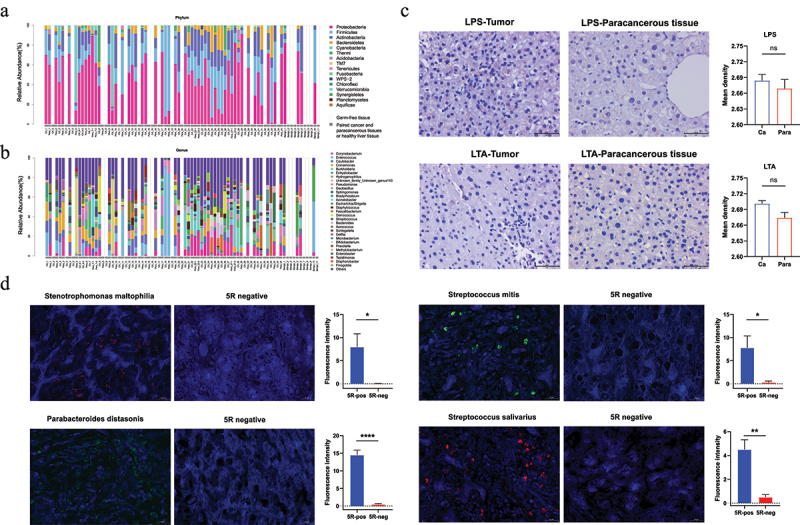
Presence of microbiota in hepatic tissues with different conditions of the prospective cohort.
(a-b) Microbial composition in hepatic tumor and healthy tissue samples at phylum (a) and genus (b) levels, with top 16 phyla and 30 genera as determined by 5 R 16S amplicon data, with different colors representing the corresponding taxa. The matched samples were put next to each other (cancerous and paracancerous tissues) within the gray bar, along with benign liver tissues from the hepatic hemangioma inpatients. Two random regions of each large tissue were selected for microbial analysis, e.g. CA_6-1, CA_6-2. The blank bars of Germ-free samples were marked as GF.
(c) IHC of LPS and LTA staining in hepatic cancer, paracancerous tissues, and control samples as indicated by punctate dots (magnification x400). The expression of LPS and LTA between tumor and the paracancerous group showed no significant difference, *P < 0.05, **P < 0.01, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, no significance.
(d) Representative microscope images for Parabacteroides distasonis, Streptococcus mitis, Streptococcus salivarius, and Stenotrophomonas maltophilia with Fam and Cy3 fluorescent dyes, and DAPI was used for nucleus staining in tumor and control samples (GF tissues) (magnification x400). The expression of fluorescence density between tumor and control samples showed significant difference, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, no significance.
Discussion
As an insidious malignancy, HCC remains a formidable challenge that requires extensive efforts for early detection to mitigate its adverse burden. Herein, this was the first time we determined that the oral microbiota of HBV-HCC patients differed significantly from that of healthy individuals, as well as a similar gut microbial trend that was confirmed in the literature. These alterations thus prompted several distinct microbiota-based classifiers to predict HCC incidence. Importantly, microbial data of oral and fecal counterparts in the matching prospective cohort confirmed the diagnostic superiority of a combined classifier with an impressive AUC value. Furthermore, we identified the presence of hepatic microbiota and disclosed a potential relationship across microbes in the cancer ecosystem, mouth, and intestine.
Previous studies have explored associations between digestive tract diseases and oral19 or gut20,21 microbiome with the taxonomic resolution of 16S rRNA gene amplicons, including early liver carcinoma with cirrhosis9. In particular, the gut microbiome has also been identified to effectively predict HCC incidence10, demonstrating its diagnostic potential for classification. In our retrospective study, we delineated the microbial composition of oral and fecal samples with 16S rRNA gene sequencing in HCC and healthy individuals. The taxa heterogeneity and distribution described with β diversity and the LEfSe algorithm (Supplemental Figure S1) significantly distinguished HCC from the control group. Subsequently, with the random forest algorithm, several influential oral (n = 10) and gut (n = 9) genera were recognized to improve the diagnostic efficiency of HCC patients from healthy individuals. For validation, both tongue and matching fecal samples were prospectively collected from 124 HCC patients and 91 control individuals, with taxonomic profiles showing consistent results. Respectively, we developed amplicon-derived classifiers that could accurately predict HCC based on characteristic oral and fecal microbial genera with AUC values of 0.7971 and 0.8084 (Figure 6c), which showed comparable performance to AFP (AUC = 0.8505), as the sole FDA-approved marker for HCC22. Moreover, the oral-gut-liver axis has been introduced to play a pivotal role in the maintenance of metabolism and the pathophysiology of hepatic diseases through blood circulation and enteral routine23,24, which was confirmed with the intrinsic connection among the microbes and their correlation to the clinical markers in SparCC and RDA analyses (Figure 4). Reportedly, a combination of oral and fecal microbiota has been proposed to enhance the sensitivity of predictive models for pancreatic ductal adenocarcinoma and colorectal cancer25,26. Therefore, the optimal 19 microbial biomarkers were merged to establish a distinct microbiota classification that considerably increased the accuracy to 0.9405 (Figure 6c). Importantly, when paired with the AFP level, the combined prediction model’s accuracy further improved to 0.9811 (Figure 6c), suggesting its complementary effect to the serum testable markers. These results indicated that oral and gut microbiota-targeted markers may become promising noninvasive tools for early diagnosis, demonstrating for the first time that an integrated analysis of the microbiome from subjects with HCC was conducted using oral and fecal samples.
To further explore when the alterations of microbiota occur in the participants, HBV and HBV-cirrhosis patients were enrolled and the relative abundance of potentially pathogenic bacteria was also evaluated to indirectly reflect the difference in microbial composition across HCC disease stages. Consequently, a couple of genera were identified to be crucial elements, some of which had been confirmed in the occurrence and progression of HCC, including Streptococcus 27, Prevotella_9 28, Faecalibacterium 29, and Bacteroides 30. Interestingly, these potential pathogens represented by Streptococcus showed a continuous accumulation from the healthy condition to HCC, which was speculated to be related to the successive decline in the gastrointestinal mucosal barrier. Further microbial phenotype analyses showed a decreasing trend in anaerobic bacteria, and an increasing trend in mobile elements and facultatively anaerobic bacteria (Figure 3d–f), indicating that disruption of epithelial hypoxia and accumulation of harmful bacteria were linked to gut dysbiosis and inflammatory reactions that increased intestinal permeability31 for intestinal bacterial translocation during carcinogenesis. In addition, improved intestinal permeability was also confirmed with elevated levels of serum LPS, ZO-1 and fecal calprotectin in HCC patients compared with the control individuals (Figure 3g–i). Consistently, as a typical facultative anaerobic genus, Streptococcus displayed a continuous accumulation from healthy conditions to HCC in the Sankey plot (abundance: 5.67% to 14.14%, Figure 3c), signifying its potential relationship with HCC. Notably, emerging lines of evidence have linked microbiota metabolites and dysfunctional gut barriers to the occurrence of HCC. A variety of metabolites, including choline, bile acids (BAs), short-chain fatty acids (SCFAs) and amino acids have been reported to serve as critical signaling factors and metabolic substrates that affect liver function32. Accordingly, genera Streptococcus and Lactobacillus were enriched in HCC, whereas the Akkermansia, Prevotella_2, Subdoligranulum and Faecalibacterium decreased, all of which were associated with BAs synthesis, especially with lithocholic acid (LCA) and deoxycholic acid (DCA)33–35. Intriguingly, Faecalibacterium was involved in the production of butyrate36 and acted as a SCFAs-related bacterium that modulated inflammatory reactions. Meanwhile, KEGG pathway analyses discovered several biological metabolic pathways in HCC with the metagenomic sequencing (Figure 5), particularly the biosynthesis and metabolism of Arginine37, proline38, alanine, aspartate39, glutamate40, cysteine and methionine41, which may provide a promising prospect for the regulatory role of gut microbes in the biosynthesis and metabolism of amino acids during HCC development with metagenomic sequencing. With regard to the function of the gut barrier, of those exactly defined genera that were differentially distributed in control and HCC participants, alterations in the relative abundance of Akkermansia, Bacteroides and Lactobacillus were related to intestinal permeability42,43 that contributed to the gut microbiota shifting for gut-liver reactions. Besides, dysbiosis of Bacteroides and Bifidobacterium in the tumor lesion microbiota was correlated with cirrhosis and HBV-related HCC11,44. It is known that the leaky gut and bacterial translocation could facilitate microbial metabolites to the liver, followed by the impairment of BAs metabolism and inflammation via Toll-like receptors (TLRs) signaling, eventually leading to the progression of tumorigenesis45. Additionally, the present study has also shown a positive relationship between the gut microbiome and clinical outcomes in patients with HBV-related HCC46. Therefore, all this evidence underscored the importance of certain signature-derived microbiota in the development of liver disease and early detection of HCC, which reminded us that the intervention of gut microbiota in the precancerous stage may slowdown the deterioration among the patients.
Clinically, cohort studies have revealed that features of tissue-derived bacteria are correlated with cancer risks47, treatment response,11 and cancer prognosis48 through various mechanisms, including increased mutation rate, modulation of oncogenic genes and pathways, and regulation of tumor immune microenvironment49–51. In particular, a landmark work reported by Cai and his colleagues52 innovatively identified the intracellular microbiota that could protect circulating tumor cells from mechanical stress by reorganizing the actin cytoskeleton in breast cancer. Alternatively, bacteria had also been postulated to contribute to tumorigenesis through indirect mechanisms such as metabolites, biofilms, inflammation, and immunosuppression53. In our study, after strictly aseptic collection and careful filtration of the 16S rRNA gene amplicon datasets with different controls to account for contamination during the procedures, several taxa could be traced from the gut and hepatic tissues with univariate enrichment in lesions, suggesting possible associations between HCC and intestinal microbiota. Based on specific antibodies and primers, IHC and FISH assays were used to describe the landscape of microbiome and certain abundant species in hepatic samples. Similarly, a significantly elevated level of oral-intestinal strain transmission was observed in patients with HCC, particularly of signature taxa, indicating that they originated intraindividually from the oral cavity. These findings illustrated that the oral, gut, and hepatic microbiomes may be intricately linked, which will be essential to disclose their respective roles in HCC etiology.
To our knowledge, it was the first longitudinal cohort study focusing on the diagnostic potential of the combined oral-gut microbiome in HBV-HCC patients, and the microbial relationship between liver tumors and the oral-gut axis had been systematically introduced through the prospectively matched population. A majority of HCC in our country was caused by HBV and developed into cirrhosis, which would influence the dynamic microbiome along with the predominant geographical variances54–56. Hence, with the relatively large size of HBV-HCC patients, our co-territorial study achieved a few convincing results with internal validation and less geographical bias. However, there were certain limitations to our research. On the basis of differential abundance analysis, more robust methods including ANCOM-BC57, LinDA,58 and LOCOM59 were required with a unique focus on the compositional nature of the original data and powerful performance in the FDR control. Sequential samples were obtained from a specific disease status without dynamic samples of the same patient over time, and the data were derived from the same race in a single center (Asia), therefore future study was required to verify the general application for the predictive signature in different races from other regions (e.g. North America, Africa). And it was innovative to demonstrate the visible presence and exact location of viable bacteria within tumor tissues under the corresponding medium, which may assist in clarifying its potential carcinogenic role in additional works. Nevertheless, the present study described a distinct microbiome signature that enabled robust performance for HCC detection with unique specificity, complementary to current biomarkers, and the possibility of effective HCC screening and monitoring. Beyond the use for early diagnosis, we believe that the panel of microbial species may be relevant to hepatic carcinogenesis, providing promising insights into proper HCC prevention and intervention.
Supplementary Material
Acknowledgments
The authors would like to thank Peng Wu, Qingqing Liang, Wei Wang, and Gaoyang He of Lc-Bio Technologies Co., Ltd (Hangzhou, China) for their technical support in this work. We thank the anonymous reviewers and the editors for their helpful remarks and useful feedback that improved this paper.
Funding Statement
The work was supported by the National Natural Science Foundation of China [81772628, 81703310, 82072685]; the Research Foundation of the National Health Commission of China–Major Medical and Health Technology Project for Zhejiang Province [WKJ-ZJ-1706]
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author, Gang Chen. The data are not publicly available due to their containing information that could compromise the privacy of research participants.
List of abbreviations
- HCC
hepatocellular carcinoma
- AUC
area under the curve
- AFP
alpha-fetoprotein
- FISH
fluorescence in situ hybridization
- OSCC
oral squamous cell carcinoma
- IBD
inflammatory bowel disease
- PCA
principal component analysis
- PCoA
principal coordinates analysis
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- STAMP
statistical analysis of metagenomic profiles
- SEM
standard error of mean
- LEfSe
linear discriminant analysis effect size
- FDR
false discovery rate
- RDA
redundancy analysis
- ROC
receiver operating curve
- LPS
lipopolysaccharide
- ZO-1
zonulin-1
- ELISA
enzyme-link immunosorbent assay
- LAL
limulus amebocyte lysate
- IHC
immunohistochemistry
- LTA
lipoteichoic acid
- RT
room temperature
- GF
Germ-free
- PPIs
proton-pump inhibitors
- NSAIDs
nonsteroidal anti-inflammatory drugs
- AAPs
atypical antipsychotics
- ARB
angiotensin II receptor blocker
- ACEI
angiotensin converting enzyme inhibitors
- ANOSIM
analysis of similarities
- BMI
body mass index
- CCI
charlson comorbidity index
- MELD
model for end-stage liver disease
- HBV
hepatitis B virus
- INR
international normalized ratio
- PT
prothrombin time
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- ALT
alanine transaminase
- AST
aspartate transaminase
- PLT
platelet
- Tchol
total cholesterol
- TG
triglyceride
- Tbil
total bilirubin
- Cre
creatinine
- Alb
albumin
- GGT
γ-glutamyl transpeptidase
- BAs
bile acids
- SCFAs
short-chain fatty acids
- LCA
lithocholic acid
- DCA
deoxycholic acid
- TLRs
toll-like receptors
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2201159
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A.. Global cancer statistics 2018: gLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–19. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, Roberts LR, Heimbach JK. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American association for the study of liver diseases. Hepatol. 2018;68(2):723–750. doi: 10.1002/hep.29913. [DOI] [PubMed] [Google Scholar]
- 3.Center MM, Jemal A. International trends in liver cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2011;20(11):2362–2368. doi: 10.1158/1055-9965.EPI-11-0643. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, Wang Y, Zhu B, Li L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatol. 2011;54(2):562–572. doi: 10.1002/hep.24423. [DOI] [PubMed] [Google Scholar]
- 5.Sandler NG, Koh C, Roque A, Eccleston JL, Siegel RB, Demino M, Kleiner DE, Deeks SG, Liang TJ, Heller T, et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterol. 2011;141(4):1220–1230. doi: 10.1053/j.gastro.2011.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang SF, Huang HD, Fan WL, Jong YJ, Chen MK, Huang CN, Chuang CY, Kuo YL, Chung WH, Su SC et al. Compositional and functional variations of oral microbiota associated with the mutational changes in oral cancer. Oral Oncol. 2018;77:1–8. doi: 10.1016/j.oraloncology.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, Kiguchi Y, Yasuma K, Watanabe E, Tanoue T, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Sci. 2017;358(6361):359–365. doi: 10.1126/science.aan4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513(7516):59–64. doi: 10.1038/nature13568. [DOI] [PubMed] [Google Scholar]
- 9.Lu H, Ren Z, Li A, Zhang H, Jiang J, Xu S, Luo Q, Zhou K, Sun X, Zheng S, Li L. Deep sequencing reveals microbiota dysbiosis of tongue coat in patients with liver carcinoma. Sci Rep. 2016;6(1):33142. doi: 10.1038/srep33142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, Xie H, Chen X, Shao L, Zhang R, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68(6):1014–1023. doi: 10.1136/gutjnl-2017-315084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E, et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Sci. 2020;368(6494):973–980. doi: 10.1126/science.aay9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Sci. 2017;357(6356):1156–1160. doi: 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komiyama S, Yamada T, Takemura N, Kokudo N, Hase K, Kawamura YI. Profiling of tumour-associated microbiota in human hepatocellular carcinoma. Sci Rep. 2021;11(1):10589. doi: 10.1038/s41598-021-89963-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Logue JB, Stedmon CA, Kellerman AM, Nielsen NJ, Andersson AF, Laudon H, Lindström ES, Kritzberg ES. Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. Isme J. 2016;10(3):533–545. doi: 10.1038/ismej.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Treangen TJ, Koren S, Sommer DD, Liu B, Astrovskaya I, Ondov B, Darling AE, Phillippy AM, Pop M. MetAMOS: a modular and open source metagenomic assembly and analysis pipeline. Genome Biol. 2013;14(1):R2. doi: 10.1186/gb-2013-14-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–62. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformat. 2014;30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8(9):e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao L, Xu T, Huang G, Jiang S, Gu Y, Chen F. Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell. 2018;9(5):488–500. doi: 10.1007/s13238-018-0548-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung JJY, Coker OO, Chu E, Szeto CH, Luk STY, Lau HCH, Yu J. Gastric microbes associated with gastric inflammation, atrophy and intestinal metaplasia 1 year after helicobacter pylori eradication. Gut. 2020;69(9):1572–1580. doi: 10.1136/gutjnl-2019-319826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Du L, Shi D, Kong C, Liu J, Liu G, Li X, Ma Y. Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat Commun. 2021;12(1):6757. doi: 10.1038/s41467-021-27112-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuzery AK, Levin J, Chan MM, Chan DW. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteom. 2013;10(1):13. doi: 10.1186/1559-0275-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acharya C, Sahingur SE, Bajaj JS. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight. 2017;2. doi: 10.1172/jci.insight.94416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bajaj JS, Betrapally NS, Hylemon PB, Heuman DM, Daita K, White MB, Unser A, Thacker LR, Sanyal AJ, Kang DJ, et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatol. 2015;62(4):1260–1271. doi: 10.1002/hep.27819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kartal E, Schmidt TSB, Molina-Montes E, Rodriguez-Perales S, Wirbel J, Maistrenko OM, Akanni WA, Alhamwe BA, Alves RJ, Carrato A, et al. A faecal microbiota signature with high specificity for pancreatic cancer. Gut. 2022;71(7):1359–1372. doi: 10.1136/gutjnl-2021-324755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flemer B, Warren RD, Barrett MP, Cisek K, Das A, Jeffery IB, Hurley E, O‘riordain M, Shanahan F, O‘toole PW. The oral microbiota in colorectal cancer is distinctive and predictive. Gut. 2018;67(8):1454–1463. doi: 10.1136/gutjnl-2017-314814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inoue T, Nakayama J, Moriya K, Kawaratani H, Momoda R, Ito K, Iio E, Nojiri S, Fujiwara K, Yoneda M, et al. Gut dysbiosis associated with hepatitis C virus infection. Clin Infect Dis. 2018;67(6):869–877. doi: 10.1093/cid/ciy205. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Wang D, Qiao S, Wu X, Cao S, Wang L, Su X, Li L. Metabolic and microbial signatures in rat hepatocellular carcinoma treated with caffeic acid and chlorogenic acid. Sci Rep. 2017;7(1):4508. doi: 10.1038/s41598-017-04888-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ponziani FR, De Luca A, Picca A, Marzetti E, Petito V, Del Chierico F, Reddel S, Paroni Sterbini F, Sanguinetti M, Putignani L, Gasbarrini A. Gut dysbiosis and fecal calprotectin predict response to immune checkpoint inhibitors in patients with hepatocellular carcinoma. Hepatol Commun. 2022;6(6):1492–1501. doi: 10.1002/hep4.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang YX, Chan AWH, Wei H, Yang X, Sung JJY, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. 2021;70(4):761–774. doi: 10.1136/gutjnl-2019-319664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byndloss MX. Microbial management. Science. 2020;369:153. doi: 10.1126/science.abc5619. [DOI] [PubMed] [Google Scholar]
- 32.Di Ciaula A, Baj J, Garruti G, Celano G, De Angelis M, Wang HH, Di Palo DM, Bonfrate L, Wang DQ, Portincasa P. Liver steatosis, gut-liver axis, microbiome and environmental factors. A never-ending bidirectional cross-talk. J Clin Med. 2020;9(8):2648. doi: 10.3390/jcm9082648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aymeric L, Donnadieu F, Mulet C, du Merle L, Nigro G, Saffarian A, Bérard M, Poyart C, Robine S, Regnault B, et al. Colorectal cancer specific conditions promote streptococcus gallolyticus gut colonization. Proceedings of the National Academy of Sciences of the United States of America; 2018; 115:E283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang N, Xu J, Wang X, Chen N, Su L, Liu Y. The spatial landscape of the bacterial community and bile acids in the digestive tract of patients with bile reflux. Front Microbiol. 2022;13:835310. doi: 10.3389/fmicb.2022.835310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan H, Wei W, Hu L, Zhang Y, Zhang H, Liu J. Reduced feeding frequency improves feed efficiency associated with altered fecal microbiota and bile acid composition in pigs. Front Microbiol. 2021;12:761210. doi: 10.3389/fmicb.2021.761210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA: acetate CoA-transferase gene. Environ Microbiol. 2010;12(2):304–314. doi: 10.1111/j.1462-2920.2009.02066.x. [DOI] [PubMed] [Google Scholar]
- 37.Missiaen R, Anderson NM, Kim LC, Nance B, Burrows M, Skuli N, Carens M, Riscal R, Steensels A, Li F, et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. 2022;34(8):1151–67 e7. doi: 10.1016/j.cmet.2022.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding Z, Ericksen RE, Escande-Beillard N, Lee QY, Loh A, Denil S, Steckel M, Haegebarth A, Wai Ho TS, Chow P, et al. Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J Hepatol. 2020;72(4):725–735. doi: 10.1016/j.jhep.2019.10.026. [DOI] [PubMed] [Google Scholar]
- 39.De Falco P, Lazzarino G, Felice F, Desideri E, Castelli S, Salvatori I, Ciccarone F, Ciriolo MR. Hindering NAT8L expression in hepatocellular carcinoma increases cytosolic aspartate delivery that fosters pentose phosphate pathway and purine biosynthesis promoting cell proliferation. Redox Biol. 2023;59:102585. doi: 10.1016/j.redox.2022.102585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu X, He Y, Han Z, Liu W, Liu D, Zhang X, Chen L, Qi L, Chen L, Luo Y, et al. PNO1 inhibits autophagy-mediated ferroptosis by GSH metabolic reprogramming in hepatocellular carcinoma. Cell Death Dis. 2022;13(11):1010. doi: 10.1038/s41419-022-05448-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li JT, Yang H, Lei MZ, Zhu WP, Su Y, Li KY, Zhu WY, Wang J, Zhang L, Qu J, et al. Dietary folate drives methionine metabolism to promote cancer development by stabilizing MAT IIA. Signal Transduct Target Ther. 2022;7:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao Y, Kuang Z, Li C, Guo S, Xu Y, Zhao D, Hu Y, Song B, Jiang Z, Ge Z, et al. Gut akkermansia muciniphila ameliorates metabolic dysfunction-associated fatty liver disease by regulating the metabolism of L-aspartate via gut-liver axis. Gut Microbes. 2021;13(1):1–19. doi: 10.1080/19490976.2021.1927633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ram AK, Vairappan B, Srinivas BH. Nimbolide attenuates gut dysbiosis and prevents bacterial translocation by improving intestinal barrier integrity and ameliorating inflammation in hepatocellular carcinoma. Phytother Res. 2022;36(5):2143–2160. doi: 10.1002/ptr.7434. [DOI] [PubMed] [Google Scholar]
- 44.Liu Q, Li F, Zhuang Y, Xu J, Wang J, Mao X, Zhang Y, Liu X.. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019;11(1):1. doi: 10.1186/s13099-018-0281-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plaza-Diaz J, Solis-Urra P, Rodriguez-Rodriguez F, Olivares-Arancibia J, Navarro-Oliveros M, Abadia-Molina F, Álvarez-Mercado AI. The gut barrier, intestinal microbiota, and liver disease: molecular mechanisms and strategies to manage. Int J Mol Sci. 2020;21(21):21. doi: 10.3390/ijms21218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang H, Ren Z, Gao X, Hu X, Zhou Y, Jiang J, Lu H, Yin S, Ji J, Zhou L, et al. Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 2020;12(1):102. doi: 10.1186/s13073-020-00796-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chng KR, Chan SH, Ng AHQ, Li C, Jusakul A, Bertrand D, Wilm A, Choo SP, Tan DMY, Lim KH, et al. Tissue microbiome profiling identifies an enrichment of specific enteric bacteria in opisthorchis viverrini associated cholangiocarcinoma. EBioMedicine. 2016;8:195–202. doi: 10.1016/j.ebiom.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178(4):795–806 e12. doi: 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garrett WS. Cancer and the microbiota. Science. 2015;348:80–86. doi: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, Smith CK, Trinchieri G. Microbes and cancer. Annu Rev Immunol. 2017;35(1):199–228. doi: 10.1146/annurev-immunol-051116-052133. [DOI] [PubMed] [Google Scholar]
- 51.Ramirez-Labrada AG, Isla D, Artal A, Arias M, Rezusta A, Pardo J, Gálvez EM. The influence of lung microbiota on lung carcinogenesis, immunity, and immunotherapy. Trends Cancer. 2020;6(2):86–97. doi: 10.1016/j.trecan.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Fu A, Yao B, Dong T, Chen Y, Yao J, Liu Y, Li H, Bai H, Liu X, Zhang Y, et al. Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell. 2022;185(8):1356–72 e26. doi: 10.1016/j.cell.2022.02.027. [DOI] [PubMed] [Google Scholar]
- 53.Knippel RJ, Drewes JL, Sears CL. The cancer microbiome: recent highlights and knowledge gaps. Cancer Discov. 2021;11(10):2378–2395. doi: 10.1158/2159-8290.CD-21-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jia W, Rajani C, Xu H, Zheng X. Gut microbiota alterations are distinct for primary colorectal cancer and hepatocellular carcinoma. Protein Cell. 2021;12(5):374–393. doi: 10.1007/s13238-020-00748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clooney AG, Eckenberger J, Laserna-Mendieta E, Sexton KA, Bernstein MT, Vagianos K, Sargent M, Ryan FJ, Moran C, Sheehan D, et al. Ranking microbiome variance in inflammatory bowel disease: a large longitudinal intercontinental study. Gut. 2021;70(3):499–510. doi: 10.1136/gutjnl-2020-321106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 2018;562(7728):583–588. doi: 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11:3514. doi: 10.1038/s41467-020-17041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou H, He K, Chen J, Zhang X. LinDA: linear models for differential abundance analysis of microbiome compositional data. Genome Biol. 2022;23(1):95. doi: 10.1186/s13059-022-02655-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu Y, Satten GA, Hu YJ. LOCOM: a logistic regression model for testing differential abundance in compositional microbiome data with false discovery rate control. Proceedings of the National Academy of Sciences of the United States of America. 2022; 119:e2122788119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author, Gang Chen. The data are not publicly available due to their containing information that could compromise the privacy of research participants.