

Introduction and importance:
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is classified as a part of the spectrum of the white dot syndromes affecting the inner choroid and the outer retina. It is usually bilateral and affects young patients between the second and fourth decades. The authors report an unusual case of unilateral APMPPE mimicking Vogt–Koyanagi–Harada (VKH) disease where the fundus fluorescein angiography was instrumental in confirming the diagnosis.
Case presentation:
A 35-year-old male presented with decreased visual acuity in the right eye for 3 days. Fundus examination revealed minimal vitritis, disc edema, and multifocal yellowish placoid lesions. Optical coherence tomography (OCT) showed the accumulation of subretinal fluid with subretinal septations closely mimicking VKH. Fundus fluorescein angiography depicted features of early hypofluorescence and late staining of the placoid lesions, suggesting APMPPE. Subretinal fluid partly resolved within a week, and visual acuity improved to 6/9(20/30) in the affected eye after the use of oral NSAIDS. Complete resolution of subretinal fluid was seen after 6 weeks.
Clinical discussion:
The most distinguishing feature in this case is the unilateral presentation and macular serous retinal detachment with subretinal septa on OCT imaging, which are not the typical features in APMPPE but quite similar to the characteristic features in acute VKH disease.
Conclusion:
APMPPE and acute VKH disease may share some overlapping clinical manifestations and imaging findings on OCT. APMPPE is a self-resolving disease, unlike VKH, and early diagnosis can avoid unnecessary administration of steroids and related side effects.
Keywords: acute posterior multifocal placoid pigment epitheliopathy, case report, Harada disease, inflammatory choroidopathy
Introduction
HIGHLIGHTS
APMPPE and acute VKH can present with substantial overlap in clinical picture and imaging characteristics.
Spectral domain OCT and fluorescein angiogram can play a key role in illustrating both distinct and common features in these two conditions
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) was first described by Gass in 1968 as an inflammatory choroidopathy. APMPPE is classified as a part of the spectrum of the white dot syndromes affecting the inner choroid and the outer retina. It is usually bilateral and affects young patients between the second and fourth decades1. Reported cases have a male : female ratio of 1.2 : 1. Atypical features include age greater than 60 years, unilateral involvement, an interval of at least 6 months before involvement of the other eye, recurrence of the disease, and leakage from choroidal veins2.
Multimodal imaging features in APMPPE suggest that it primarily affects the choriocapillaris and inner choroid, resulting in secondary photoreceptor disruption and changes in the retinal pigment epithelium (RPE)3. Although its etiology is unknown, an immune driven nature has been hypothesized based on the association with viral prodrome, recent vaccination, and preexisting autoimmune diseases like sarcoidosis, psoriasis, diabetes mellitus. Also, numerous cases of APMPPE have been reported in patients with a positive tuberculosis immunological test without prior BCG vaccination4,5.
It is characterized by multiple whitish-yellow lesions at the outer retina, RPE, and choroid.
Fluorescein angiography shows typically early hypofluorescent dots, followed by hyperfluorescence during the late phase. Optical coherence tomography (OCT) shows a disruption of the inner/outer segment (IS/OS) due to placoid lesions and hyper-reflectivity of the outer retina. The prognosis of APMPPE is favorable with a good visual outcome. Spontaneous resolution typically occurs after few weeks of evolution. Eyes with atypical presentation, initial foveal involvement may be associated with poor visual recovery and thus may necessitate initial treatment. However, there is no clear consensus regarding the use of oral corticosteroids or any other treatment modalities6.
Cases describing the use of steroids advocate it is possible that it can modify the natural course of lesions, however, the choroidal reperfusion may also occur spontaneously in the course of the disease, without steroids6.
We report an unusual case of unilateral APMPPE mimicking Harada’s disease where the fundus fluorescein angiography was instrumental in confirming the diagnosis. This case report has been reported in line with the SCARE Criteria7.
Case report
A 35-year-old male presented with sudden painless diminution of vision in right eye (RE) for 4 days with feeling of distorted image for near. No history of preceding flu, recent vaccination, headache, and hearing loss was present. The decreased in vision was not associated with redness, flashes/floaters, or preceding trauma/surgery. A complete ophthalmological examination was done. Best corrected distant visual acuity was 6/36 (20/120) in the RE and 6/6 (20/20) in the left eye and near acuity was N18 and N6, respectively. Anterior segment examination and pupillary reflex was normal in both eyes. Intraocular pressure was normal. Examination of the fundus showed 1+ vitreous cells and haze in RE. The optic disc was hyperemic, edematous with blurred margins. There were multiple, deep, white-yellowish lesions with blurred borders in the posterior pole, associated with periretinal edema (Fig. 1). Left eye fundus was unremarkable (Fig. 2).
Figure 1.
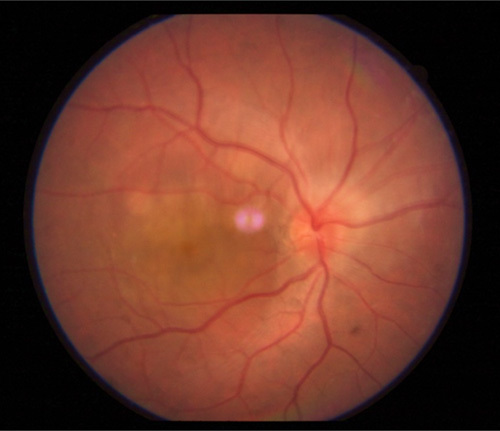
Multiple yellowish placoid lesions in the posterior pole with disc edema.
Figure 2.
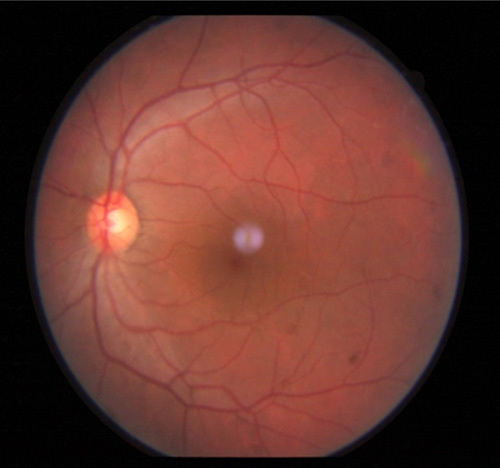
Normal fundus of left eye.
Routine laboratory investigations for uveitis were sent. Complete blood count was normal. Serology for syphilis, HIV were normal (Fig. 3). Mantoux test reported 8 mm of redness. Chest radiography showed minimal patchy changes in bilateral upper zone with no evidence of hilar/paratracheal lymphadenopathy. Serum ACE, serum calcium and QuantiFERON-TB Gold test were also normal. OCT macula of RE revealed subretinal fluid with bridging septas with interruption of the IS/OS junction (Figs 4–8). RE fundus fluorescein angiography showed early hypofluorescence followed by hyperfluorescence of the lesions during the late phase (Figs 9, 10).
Figure 3.

Resolved placoid lesions with pigmentary changes.
Figure 4.
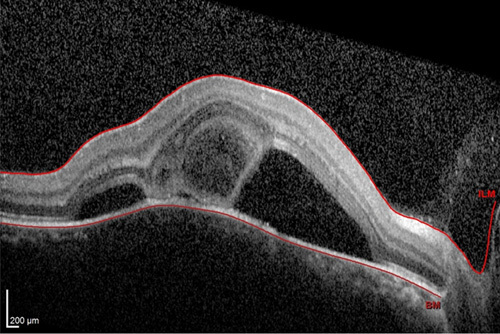
Spectral domain ocular coherence tomography of the right eye at initial presentation demonstrates subretinal fluid with bridging septa and disruption of inner/outer segment junction.
Figure 8.
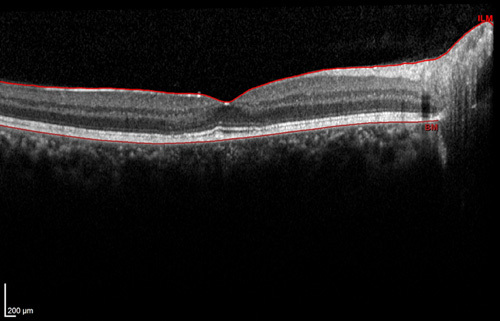
Optical coherence tomography right eye showing complete resolution of the subretinal fluid, with restoration of ellipsoid zone.
Figure 9.
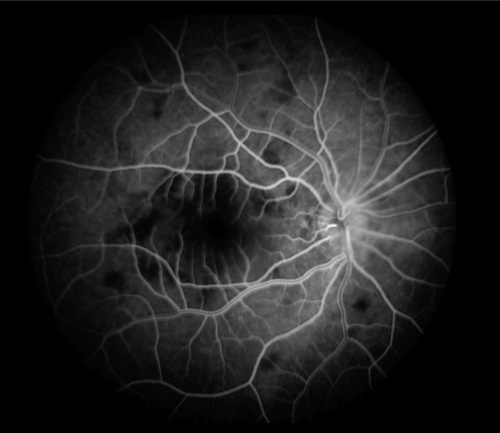
Laminar phase of fluorescein angiogram demonstrating early hypofluorescence of the placoid lesions.
Figure 10.
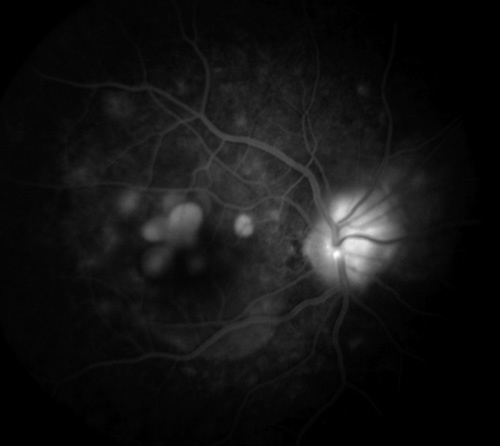
Late phase of fluorescein angiogram demonstrating late staining of the placoid lesions.
Figure 5.

Normal optical coherence tomography findings of left eye.
Figure 7.
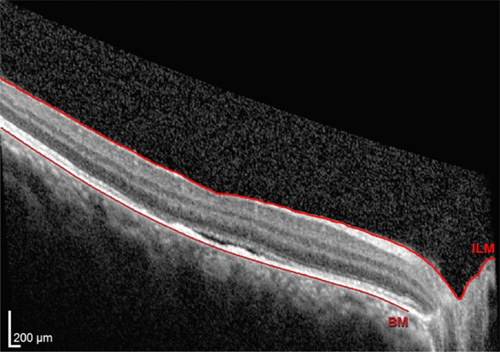
Optical coherence tomography right eye after 2 weeks with resolving edema and reduced disruption of the ellipsoid layer and photoreceptor segments.
Though an earlier provisional diagnosis was suspected to be unilateral Harada disease, fluorescein angiographic findings supported the diagnosis of APMPPE. Patient was started on oral NSAIDs and asked to follow-up after 1 week. After 1 week, repeat OCT showed a regression of serous fluid (Fig. 6) and improvement of visual acuity to 6/9 (20/30) and near vision to N12.
Figure 6.

Spectral domain ocular coherence tomography of the right eye after 1 week with significant resorption of subretinal fluid and irregularities of the retinal pigment epithelium layer.
After 6 weeks, there was complete resolution of subretinal fluid (Fig. 8) with VA of 6/6 (20/20) and near vision of N6. Disc edema and serous retinal detachment with bridging septa closely resembled unilateral Harada but absence of general symptoms, angiographic features were not in favor of Vogt–Koyanagi–Harada (VKH).
Despite the atypical unilateral presentation, the placoid lesions in the posterior pole and angiographic evidence of early hypofluorescence and late hyperfluorescence supported the diagnosis of APMPPE.
This case has been presented owing to its rarity and under-reporting.
Discussion
APMPPE and acute VKH disease may share some overlapping clinical manifestations and imaging findings on OCT. OCT findings of multilobular serous detachment with septations and increased choroidal thickening is considered to be a valuable diagnostic finding in acute VKH disease8. Characteristic features associated with the serous detachments in acute VKH include multiple lobulated septae which is hypothesized to be a portion of OS layer of the photoreceptors attached with inflammatory products such as fibrin9. The walls of the cystoid spaces are made of inflammatory products, such as fibrin and are located in the subretinal space. Fundus fluorescein angiography in the early phase shows multiple discrete hyperfluorescent dots at the RPE level that enlarge over time and in the late phase, if there is a serous detachment, the fluorescein pools beneath the subretinal space10.
Other typical OCT changes of the RPE and choroid layers for VKH disease include undulations of the RPE surface9.
Reported OCT findings in acute APMPPE lesions include increased reflectance of the outer retinal layers suggesting inflammatory cells with edema in the outer retinal layers and the loss of IS/OS junction, attenuation of photoreceptor layer and external limiting membrane and nonspecific changes in the choriocapillaris9. Fluorescein angiogram (FA) in early phase shows hypofluorescence secondary to either blocking of background choroidal fluorescence or choroidal hypoperfusion. Mid-phase and late-phase angiograms demonstrate staining of the acute lesions6.
The most distinguishing feature in our case is the unilateral presentation and macular serous retinal detachment with subretinal septa on OCT imaging which are not the typical features in APMPPE but quite similar to characteristic features in acute VKH disease. Studies have reported cases of unilateral APMPPE. In a study by Kutluturk and colleagues about 40% of the unaffected eye had signs of inflammation including vitreous-cells, vascular-sheathing, and ONH swelling. Thirty-five percent developed chorioretinal lesions during follow-up after 19.5±16.9 months11. However, our case did not show any signs of inflammation in the fellow eye at the initialpresentation and follow-up period of 3 months.
To our knowledge, five studies have recently reported similar characteristic features between APMPPE and VKH disease in acute phase9,12,13,14. A report by Li and colleagues presented a unilateral case with overlapping features of APMPPE and VKH similar to the present case but differed in clinical presentation. Anterior segment inflammation, OCT findings were in favor of unilateral VKH but fundus findings and FA were suggestive of APMPPE. Bilateral presentation was reported in four out of five studies.
Studies have shown that inflammatory etiology is involved in the pathogenesis of choroidal circulatory disturbance in APMPPE thus supporting the role of anti-inflammatory agents such as systemic corticosteroids, intravitreal injection of triamcinolone acetonide as a part of treatment. Oral NSAIDs has been used in a report by Kitamura et al.8 with good response. Because of the atypical presentation making the diagnosis difficult and absence of systemic neurological symptoms, we did not use corticosteroids. Instead, oral NSAID was used and showed a remarkable visual improvement within a few days.
Conclusion
This case highlights the importance of detecting characteristic FA findings to distinguish between the two entities. As APMPPE is a self-resolving disease unlike VKH, early diagnosis can avoid unnecessary administration of steroids and related side effects.
Ethical approval
None declared.
Consent
Written informed consent was obtained from the patient for the publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Sources of funding
None.
Author contribution
S.S.: study concept/design, preparation of manuscript. R.K.: study concept/design, manuscript revision. S.P., P.K.: review of manuscript. S.N.J.: design, revision of manuscript.
Conflicts of interest disclosure
The authors declare that they have no conflicts of interest.
Guarantor
Dr Sanket Parajuli.
Data availability
Data is available in the text.
Provenance and peer review
Not commissioned, externally peer reviewed.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Published online ■ ■
Contributor Information
Sadhana Sharma, Email: ssadnaa@gmail.com.
Ranju Kharel, Email: helloranju50@gmail.com.
Sanket Parajuli, Email: sanketparajuli@gmail.com.
Pratap Karki, Email: karkipratap@gmail.com.
Sagun N. Joshi, Email: Sagun.j3@gmail.com.
References
- 1. Gass JDM. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol 1968;80:177–185. [DOI] [PubMed] [Google Scholar]
- 2. Pagliarini S, Piguet B, Ffytche TJ, et al. Foveal involvement and lack of visual recovery in APMPPE associated with uncommon features. Eye (Lond) 1995;9(Pt 1):42–47. [DOI] [PubMed] [Google Scholar]
- 3. Park D, Schatz H, McDONALD HR, et al. Acute multifocal posterior placoid pigment epitheliopathy: a theory of pathogenesis. Retina 1995;15:351–352. [DOI] [PubMed] [Google Scholar]
- 4. Anderson K, Patel K, Webb L, et al. Acute posterior multifocal placoid pigment epitheliopathy associated with pulmonary tuberculosis. Br J Ophthalmol 1996;80:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jones NP. Acute posterior multifocal placoid pigment epitheliopathy. Br J Ophthalmol 1995;79:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fiore T, Iaccheri B, Androudi S, et al. Acute posterior multifocal placoid pigment epitheliopathy: outcome and visual prognosis. Retina 2009;29:994–1001. [DOI] [PubMed] [Google Scholar]
- 7. Agha RA, Franchi T, Sohrabi C, et al. for the SCARE Group. The SCARE 2020 Guideline: updating Consensus Surgical CAse REport (SCARE) Guidelines. Int J Surg 2020;84:226–230. [DOI] [PubMed] [Google Scholar]
- 8. Kitamura Y, Oshitari T, Kitahashi M, et al. Acute posterior multifocal placoid pigment epitheliopathy sharing characteristic OCT findings of Vogt-Koyanagi-Harada disease. Case Rep Ophthalmol Med 2019;2019:9217656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li B, Bentham RJ, Gonder JR. A case of unilateral and spontaneously resolving posterior uveitis with overlapping features of Vogt–Koyanagi–Harada disease and acute posterior multifocal placoid pigment epitheliopathy. Springerplus 2016;5:1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Betancourt RBS, Soler G, Mantilla RD, et al. Enfermedad de Vogt Koyanagi Harada. Reporte de un caso y revisión de la literatura. Rev Colomb Reumatol 2020;27:50–60. [Google Scholar]
- 11. Kutluturk I, Agarwal A, Shulman S, et al. The clinical characteristics of unilateral placoid pigment epitheliopathies. Ocul Immunol Inflamm 2021;29:1072–1079. [DOI] [PubMed] [Google Scholar]
- 12. Tanigawa M, Tsukahara Y, Yamanaka H. A case of acute posterior multifocal placoid pigment epitheliopathy demonstrating vogt-koyanagi-harada disease-like optical coherence tomography findings in the acute stage. Case Rep Ophthalmol 2013;4:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee GE, Lee BW, Rao NA, et al. Spectral domain optical coherence tomography and autofluorescence in a case of acute posterior multifocal placoid pigment epitheliopathy mimicking Vogt-Koyanagi-Harada disease: case report and review of literature. Ocul Immunol Inflamm 2011;19:42–47. [DOI] [PubMed] [Google Scholar]
- 14. Abu-Yaghi NE, Shokry AN. Acute posterior multifocal placoid pigment epitheliopathy versus Vogt-Koyanagi-Harada disease: tomographic characteristics. Jordan Med J 2015;171:1–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data is available in the text.