

Abstract
T cell receptor (TCR)–based immunotherapy has emerged as a promising therapeutic approach for the treatment of patients with solid cancers. Identifying peptide–human leukocyte antigen (pHLA) complexes highly presented on tumors and rarely expressed on healthy tissue in combination with high-affinity TCRs that when introduced into T cells can redirect T cells to eliminate tumor but not healthy tissue is a key requirement for safe and efficacious TCR-based therapies. To discover promising shared tumor antigens that could be targeted via TCR-based adoptive T cell therapy, we employed population-scale immunopeptidomics using quantitative mass spectrometry across ~1500 tumor and normal tissue samples. We identified an HLA-A*02:01-restricted pan-cancer epitope within the collagen type VI α-3 (COL6A3) gene that is highly presented on tumor stroma across multiple solid cancers due to a tumor-specific alternative splicing event that rarely occurs outside the tumor microenvironment. T cells expressing natural COL6A3-specific TCRs demonstrated only modest activity against cells presenting high copy numbers of COL6A3 pHLAs. One of these TCRs was affinity-enhanced, enabling transduced T cells to specifically eliminate tumors in vivo that expressed similar copy numbers of pHLAs as primary tumor specimens. The enhanced TCR variants exhibited a favorable safety profile with no detectable off-target reactivity, paving the way to initiate clinical trials using COL6A3-specific TCRs to target an array of solid tumors.
INTRODUCTION
Adoptive T cell therapy targeting CD19-expressing tumors has resulted in durable remissions for individuals with previously incurable disease (1), helping to establish cell and gene therapy as a new pillar of cancer medicine (2). However, attempts to broaden the types of tumors targeted by adoptive T cell therapy have proven challenging because of a range of hurdles, including finding targets that are tumor specific, the immunosuppressive nature of the tumor microenvironment (TME), and tumor immune escape mechanisms (3–5). To overcome some of these obstacles, much effort has been focused on identifying tumor-specific mutations and peptides derived thereof (neoepitopes), which are presented by human leukocyte antigen (HLA) (6). Although this would allow engineered T cells to solely target tumor tissue (7), these peptides are limited to indications with high mutational burden and are rarely shared across patients. This has triggered exploration into the potential of RNA-based neoepitopes, such as those derived from RNA editing (8) or from alternative splicing (9) that may be more universally present within various tumors. Whereas many studies consider the transcriptome and proteome for neoepitopes, the immunopeptidome is often only analyzed through HLA binding predictions (10, 11). Considering the complexity of the antigen processing pathway (12), mass spectrometry (MS) has proven useful in determining which peptides can be targeted (10). These efforts need to include sufficient normal samples to provide a valid baseline to assess tumor selectivity and to cover tissue-specific differences (13). Furthermore, these data allow the concomitant identification of appropriate similar peptides that are also presented on normal tissues, to assess off-target reactivity and allow early de-risking for clinical development during T cell receptor (TCR) discovery and development. In addition, targeted MS enables the absolute quantitation of HLA peptides, which is important because the strength of the T cell response is controlled by the number of peptide-HLA (pHLA) complexes located in a particular immuno-logical synapse (14). Thus, tumors, as well as normal tissues, that present low amounts of a targeted pHLA will likely not be sufficiently recognized by infused T cells.
The TME is a unique mixture of hematopoietic and mesenchymal cells that support tumor growth by providing nutrients via neovascularization, cytokines, and extracellular matrix, as well as recruiting fibroblasts and suppressive immune cells (15). Because the TME is essential for tumor growth, genetically stable, present in the vast majority of solid tumors, and unique to tumors (16), agents that specifically target or modify the TME are being developed (17). For example, fibroblast-associated protein (FAP) has emerged as a promising target because it is highly expressed in epithelial cancer with limited expression in normal tissues (18). Preclinical models of chimeric antigen receptor-T (CAR-T) cells targeting FAP demonstrate that this approach effectively eliminates tumors due to the dependence of cancer cells on FAP-expressing stroma cells. There was no off-tumor activity observed in one model (19), although other models suggested bone marrow toxicity, which may be unique to FAP-directed therapies (20). Thus, new agents may be required to take advantage of the therapeutic opportunity of targeting the TME.
Here, we performed a direct and unbiased analysis of HLA-presented peptides using population-scale immunopeptidomics data acquired by MS to identify pHLAs frequently and abundantly presented on tumor tissue and scarcely presented on normal tissue. We identified a prevalently shared pan-cancer HLA-A*02:01-restricted collagen type VI α-3 (COL6A3) epitope that is only expressed via an alternative splicing event within the TME. We identified several TCRs that could recognize this peptide in the context of HLA-A*02:01 while avoiding off-target recognition of the most similar peptides found in the acquired HLA peptidomes. One of these TCRs was affinity-enhanced so that both CD4 and CD8 T cells could target cells that express physiologically relevant copy numbers of this pHLA. Assessment of antitumor activity in in vitro and in vivo models demonstrated that engineered T cells expressing a subset of affinity-enhanced TCRs specifically and efficiently eliminated tumors but did not react against normal tissue. These data warrant clinical evaluation of T cells genetically modified with COL6A3-specific TCRs in patients with a broad range of different solid tumors.
RESULTS
Population-scale quantitative immunopeptidomics identifies a prevalent high-copy number pan-cancer epitope derived from a tumor-specific splice variant of COL6A3
The human immunopeptidome was analyzed by a target discovery platform that uses label-free liquid chromatography–MS (LC-MS) for identification and relative quantitation of HLA-restricted peptides combined with the expression analysis of corresponding mRNAs (fig. S1) (21). HLA-A*02-presented peptides were analyzed across 739 tumor and 673 normal tissue samples from HLA-A*02-positive donors after immunoprecipitation with an HLA-A*02-specific antibody, enabling a population-scale view on the HLA-A*02 peptidome. On the basis of these presentation profiles, peptides were subsequently filtered for epitopes rarely found on healthy tissue and abundantly found across many tumor types. This approach revealed a peptide (COL6A3-FLNV) from exon 6 of COL6A3 (COL6A3-E6, ENSE0000107376). The peptide sequence (FLLDGSANV, P12111 p642–650) was confirmed by coelution of the corresponding synthetic isotope-labeled peptide using LC-MS (fig. S2A) (22). Examination of the immunopeptidomics data revealed that COL6A3-FLNV was present on 28% (n = 210) of all tumors and on only 1% (n = 7) of all normal tissue samples (Fig. 1A). Of note, two of the most pronounced normal tissue hits were derived from placenta, a tissue usually not found in patients with cancer. Various types of cancer, including esophageal cancer, breast cancer, and small and non–small cell lung carcinoma, showed significant overpresentation of up to 23-fold (P < 0.001) higher average COL6A3-FLNV–presenting pHLAs on the cell surface relative to critical normal tissues (table S1). Subsequently, highly sensitive targeted MS was applied to a subset of samples using parallel reaction monitoring LC-MS with stable isotope-labeled synthetic peptides as internal standards as previously described (8) to experimentally determine the absolute copy number of peptides presented on the cell surface. A set of 233 tumor and normal tissue samples was selected on the basis of the availability of retention samples prioritizing prevalent cancer indications and normal tissues with higher risk of on-target toxicity. COL6A3-FLNV was presented on 55 of 134 tumor samples with an average of 228 copies per cell (CpC) (Fig. 1B) and up to 1928 CpC for an individual tumor sample. Among the 99 normal tissue samples, the peptide was detected and quantified on six tissues with an average of 28 CpC and a maximum of 49 CpC on one thyroid sample. The increased number of COL6A3-FLNV–positive normal tissues compared to the population-scale relative peptide quantitation data (Fig. 1A) underlines the increased sensitivity of the targeted MS approach and confirmed the low abundance on normal tissues. The measurement of the absolute number of peptide copies bound to HLAs confirms a promising therapeutic window to target this pHLA complex through TCR-based adoptive T cell therapy.
Fig. 1. Population-scale immunopeptidomics and transcriptomics identify a shared pan-cancer epitope from a tumor-specific splice variant of COL6A3.
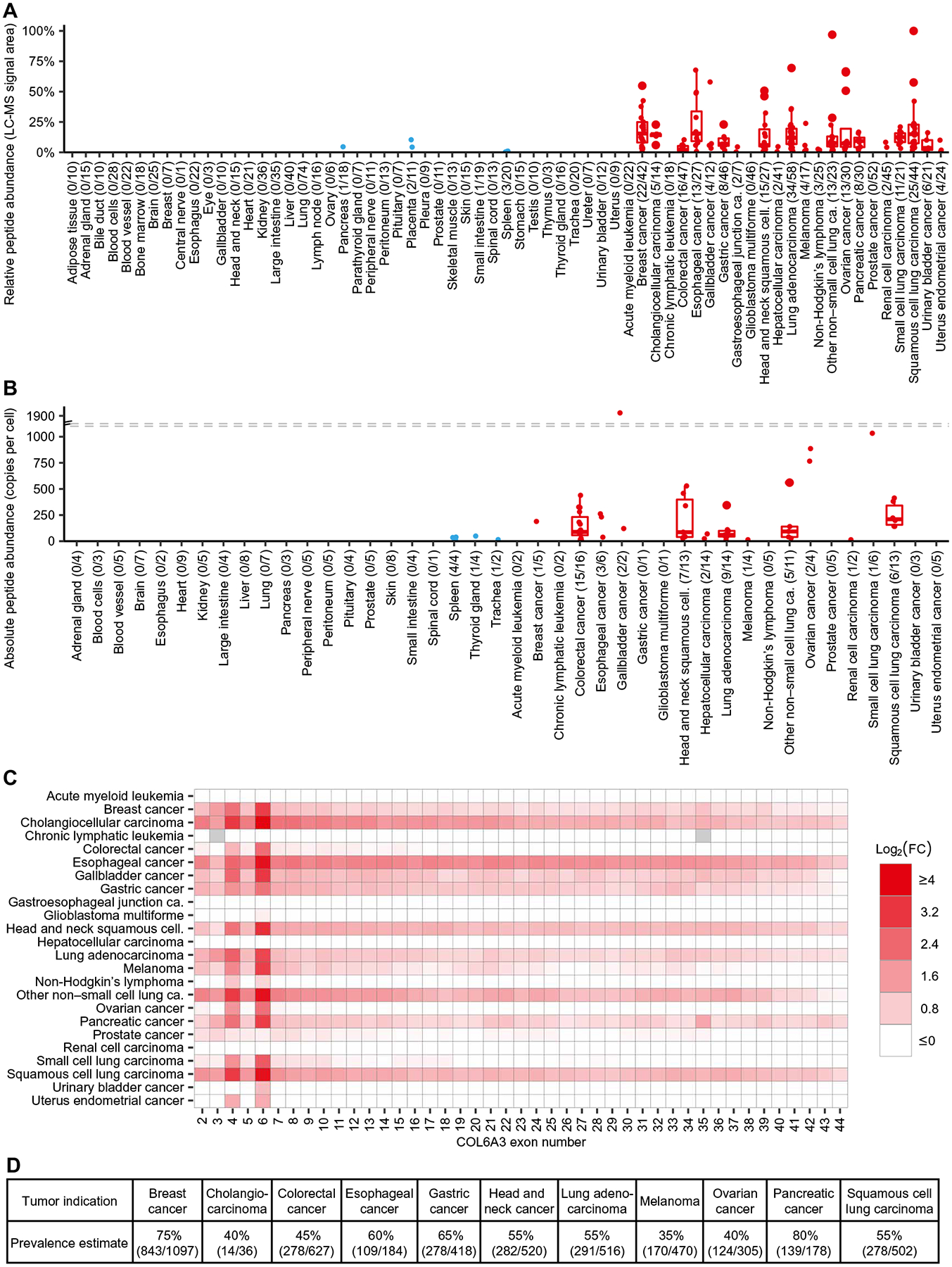
(A) Relative abundance of HLA-bound COL6A3-FLNV peptide isolated from tumor (red) and normal samples (blue). Each dot represents the median from five technical replicates of samples for which the peptide was detected and quantified. The number of donors with peptide detection, as well as the total number per group, is indicated in parentheses. (B) Absolute peptide copy numbers per cell were determined in a subset of samples. Each dot represents the mean across triplicates of each sample. (C) Heatmap of average log fold changes (FC) between tumor and average normal tissue samples for all protein-coding COL6A3 exons as determined by RNA sequencing. Missing log fold changes due to zero expression were indicated in gray. (D) Estimates for prevalence of patients positive for COL6A3-FLNV in selected tumor indications with exon 6 overexpression based on TCGA patient cohorts.
Previously, alternative splicing of COL6A3 has been described to be based on three cassette exons, exons 3, 4, and 6 (23). For exon 6, its association with various cancers has been demonstrated by reverse transcription polymerase chain reaction (PCR) (24). On the basis of the ENSEMBL version 77 annotation (http://www.ensembl.org), there are 44 exons in COL6A3, of which 43 are protein coding. We observed junction spanning reads for exon 6 in the very same sample used for peptide confirmation showing an exon-inclusion ratio (percent spliced-in index) of 51.5% (fig. S2B) (25). Quantification of mRNA for all 43 protein-coding exons by RNA sequencing further confirmed the up-regulation of the two cassette exons 4 and 6 in contrast to other exons of COL6A3 (Fig. 1C). Although for some tumor indications all COL6A3 exons were slightly overexpressed, most of the up-regulation was exon specific, indicating reduced exon skipping in tumors. In agreement with the peptide-level quantification, tumor selectivity was observed for multiple indications, showing overexpression for esophageal cancer, breast cancer, and non–small cell lung carcinoma, among others (table S1).
The paired transcriptome and immunopeptidome data were used to estimate an MS-based mRNA expression threshold for COL6A3-FLNV that can be used for patient inclusion and prevalence estimation as previously described (26). Target prevalence was estimated in indications showing overexpression. To base these estimates on a larger patient cohort, data from the Cancer Genome Atlas (TCGA; https://cancer.gov/tcga) were integrated with discovery data of this study (fig. S1) to estimate the target prevalence of COL6A3-FLNV across several tumor types (Fig. 1D). Together, these data indicate that the expression of exon 6 of COL6A3 is highly enriched within tumors, and this results in the presentation of a prevalent pan-cancer epitope.
COL6A3-E6 expression is enriched within the tumor stroma
Tumors isolated from patients are a heterogeneous mixture of tumor, stroma, vascular, and immune cells. The spatial localization of COL6A3-E6 expression within the bulk tumor mass was analyzed histologically by in situ hybridization (ISH) in different tumor types. Regions rich in stroma or tumor cells were either assessed by a hematoxylin and eosin (H&E) stain or by analyzing the expression of cell type–specific markers. Tumor cells were marked by a probe set cocktail targeting eight different epithelial markers [pancytokeratin (panCK)], whereas cancer-associated fibroblasts were identified by smooth muscle actin α2 (ACTA2), also known as α-smooth muscle actin (αSMA). Chromogenic costainings on serial sections revealed clear colocalization of COL6A3-E6 expression with ACTA2 expression (Fig. 2A). In contrast, panCK-positive tumor cells rarely displayed COL6A3-E6 expression. Overall, we analyzed 30 tumor tissues derived from eight different cancer types. All 30 tumor samples displayed clear stroma-associated expression of COL6A3-E6. In addition, we observed less intense staining within the tumor cells in 4 of the 30 samples, indicating that the inclusion of exon 6 into COL6A3 can occur predominantly but not exclusively in the tumor stroma (Fig. 2, B and C, and table S2).
Fig. 2. COL6A3-E6 expression is enriched within the tumor stroma.
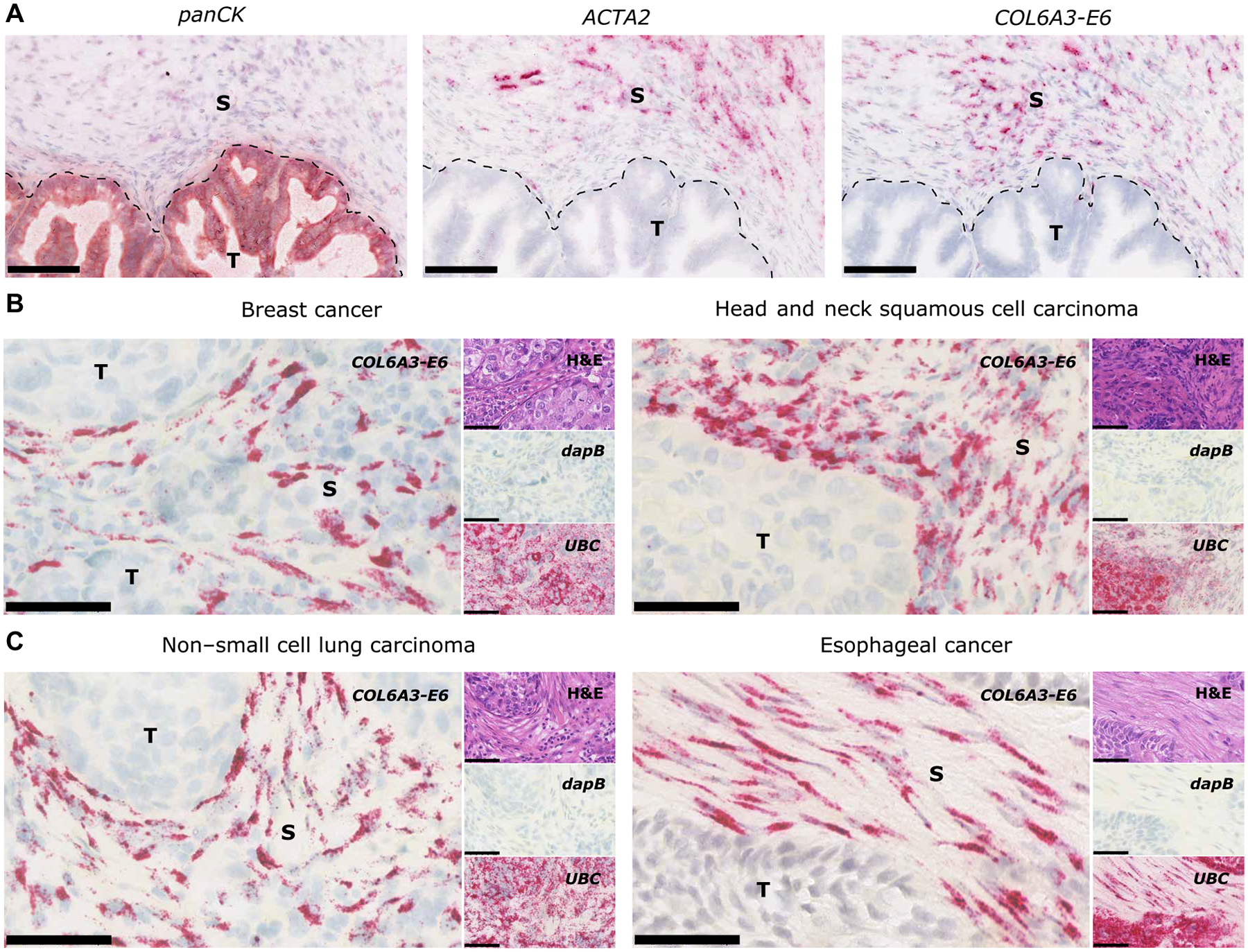
(A) Representative images of serial sections of a gastric cancer FFPE sample with a tumor (T) marker probe set [pancytokeratin (panCK)], a stromal (S) marker probe set [smooth muscle actin α2 (ACTA2)], and a COL6A3-E6 probe set. Scale bars, 100 μm. Representative images of COL6A3-E6 ISH in (B) breast cancer (left) and head and neck squamous cell carcinoma (right) and (C) non–small cell lung carcinoma (left) and esophageal cancer (right). Insets on the right side of each picture show the same region on consecutive sections stained with H&E or reference negative and positive control probe sets for the bacterial gene locus of dihydrodipicolinate reductase (dapB) or human ubiquitin C (UBC), respectively. Probe set hybridization is visualized as punctate red dots or clusters of red dots using Fast Red. Chromogenic color detection varies between red and russet depending on the target signal intensity and the used kits. Nuclei are stained in violet blue by means of hematoxylin. In case of the H&E section, eosin stains the extracellular matrix and cytoplasm pink. Scale bars, 60 μm.
Discovery and characterization of COL6A3-FLNV–specific TCRs
We next sought to obtain COL6A3-FLNV–specific TCRs so that the immunotherapeutic potential of this target could be ascertained (fig. S1). To do this, we employed a stepwise approach for using repeated stimulation of T cells from HLA-A*02-positive and HLA-A*02-negative blood donors with bead-coupled COL6A3-FLNV:HLA-A*02 complexes to enrich the antigen-specific T cells (fig. S3A). Among 16 different blood donors, we identified 91 unique TCR sequences. All TCRs were reexpressed in human CD8 T cells upon transfection with TCR encoding mRNA and characterized for their functional response to COL6A3-FLNV using peptide-loaded K562 cells stably expressing HLA-A*02 (K.A2). Using a COL6A3-FLNV dilution series, we identified those TCRs with the highest sensitivity in COL6A3-FLNV recognition and selected the two best-performing TCRs (D10 and F9) together with a low-sensitivity TCR (H3) for further in-depth analysis (table S3). H3 and D10 were obtained from T cells of an HLA-A*02–positive donors, whereas F9 was derived from an HLA-A*02–negative individual. We first produced soluble TCR variants of D10, F9, and H3 and used surface plasmon resonance (SPR) analysis to determine the binding affinity toward COL6A3-FLNV in complex with HLA-A*02:01. For the TCRs H3, F9, and D10, we measured binding equilibrium dissociation constants (KD’s) of ~100 μM, ~60 μM, and 16 μM, respectively (fig. S3B), which is in good agreement with TCR affinities observed among cancer-specific T cells (27).
To ascertain each TCR’s ability to specifically recognize COL6A3-FLNV, we generated lentiviral vectors that expressed each TCR pair by a T2A sequence and transduced primary human CD8 T cells (28). We observed strong COL6A3-FLNV–specific tetramer binding of CD8 T cells transduced with the F9 and D10 TCRs and less robust staining of T cells transduced with H3 TCR despite equivalent concentrations of lentiviral integration events [6.7 × 105 (H3), 7.3 × 105 (F9), and 1.5 × 106 (D10) per 106 cells], suggesting that the lower affinity of H3 TCR reduced its ability to stably bind tetramers (Fig. 3A).
Fig. 3. Natural COL6A3-FLNV–specific TCRs confer limited antitumor immunity in vivo.

(A) CD8 T cells were activated with anti-CD3/CD28 beads, transduced with the indicated TCR or left UTD, and stained with COL6A3-specific tetramer after 6 days of culture. Results are representative of three independent experiments with different donors performed in triplicate. (B) T cells from (A) after 11 days of culture were incubated with indicated artificial APCs (aAPCs) for 4 hours, and intracellular cytokine staining was performed. The average number of relevant pHLA copies per cell (pHLA-CpC) was determined by MS-based quantitation. Results are representative of three independent experiments with different donors performed in triplicate. Bioluminescence imaging of N6.COL6A3 (C) and N6.COL6A1 (D) tumors on the indicated days. Experimental schematic for in vivo study and survival curves of each on-target tumor group and off-target tumor groups can be found in fig. S5.
Efficient discovery of TCRs with superior safety profiles requires the early counterselection of TCRs based on potential off-targets. To select an initial set of the most likely off-target peptides displaying sequence similarity, the experimentally determined HLA-A*02 ligand space (fig. S1) was filtered for peptides presented on normal tissues sharing at least four consecutive amino acids with COL6A3-FLNV (table S4). We loaded these seven peptides, mainly derived from the collagen family and from the von Willebrand factor (VWF), onto K.A2 cells to determine whether COL6A3-specific T cells could recognize these COL6A3-FLNV–related sequences. Besides the cognate peptide COL6A3-FLNV that was recognized by all three TCRs, only one similar peptide from the α1 chain of type VI collagen (COL6A1) with the sequence ILLDGSASV (COL6A1-ILSV) was recognized by the D10 TCR and to a lesser extent by the F9 TCR, but not by H3 (fig. S4). To determine whether we could model physiologic copy numbers of pHLAs, we transduced K.A2 and NALM6 (N6) cells, which do not naturally express the exon 6 containing splice variant of COL6A3 or express the COL6A1 epitope, with a minigene containing 50 amino acids surrounding the COL6A3 or COL6A1 epitope (K.A2.COL6A3, K.A2.COL6A1, N6.COL6A3, or N6.COL6A1). Next, we performed absolute quantification by targeted MS to determine the average number of pHLA molecules on the cell surface of these cell lines. The N6 lines had high CpC for each pHLA (N6.COL6A1, 1695; N6.COL6A3, 6932) (Fig. 3B). In contrast, K.A2.COL6A3 had 244 CpC, which is within the range of what was observed for tissue from tumors (Fig. 1B), making this cell line a physiologic model. The K.A2.COL6A1 had high amounts of the expected pHLAs on its cell surface (2849). Knowing how many pHLAs were on each target cell surface provided an ideal way of studying the sensitivity, potency, and specificity of the different COL6A3-specific TCRs. T cells expressing the H3 TCR showed limited recognition of K.A2.COL6A3 or K.A2.COL6A1 and displayed a robust response against N6.COL6A3, but not N6.COL6A1, indicating that this TCR is specific to COL6A3-FLNV, but will only recognize tumors that highly express pHLAs. T cells expressing the F9 TCR could recognize K.A2.COL6A3 but had no recognition of K.A2.COL6A1, which has >10-fold more pHLAs on the cell surface. Likewise, we observed a robust response to N6.COL6A3 and only background responses to N6.COL6A1, indicating that higher-affinity F9 TCR is able to recognize the low pHLA-expressing K.A2.COL6A3, but not as robustly as it recognizes the cell line with more pHLAs on the surface (N6.COL6A3). D10 TCR, on the other hand, confers pronounced polyfunctional responses to all cell lines, indicating that it equally recognizes the COL6A1-ILSV and COL6A3-FLNV pHLAs, making it less suitable for use in adoptive T cell therapy (Fig. 3B).
To test whether COL6A3-specific TCRs had any ability to function in vivo, we used cell lines with defined COL6A3-FLNV and COL6A1-ILSV pHLA copy numbers because human tumor stroma does not engraft well in NSG models to permit strategies targeting the stroma to be robustly evaluated. Our initial studies used N6 lines described above because they expressed high, albeit nonphysiological, amounts of the relevant pHLAs, and we previously described an experimental model that allows differences in T cell potency to be evaluated (fig. S5A) (29). Eight days after the infusion of N6.COL6A3 or N6.COL6A1 cells, we infused 2.5 million untransduced (UTD) T cells or 2.5 million of each of the COL6A3-FLNV–specific TCR-transduced T cells and tracked tumor growth using bioluminescence imaging (Fig. 3, C and D). The antitumor activity of the H3 TCR-transduced T cells was limited. D10 TCR-transduced T cells durably controlled tumor growth and enabled long-term survival of the mice but did not show preference of COL6A3- over COL6A1-transduced tumors, confirming our in vitro findings. T cells transduced with the F9 TCR showed no control of COL6A1-expressing tumors but did delay tumor progression in all treated mice, and one mouse was cured of its COL6A3-expressing tumor (fig. S5, B and C). However, when compared to T cells expressing the D10 TCR, the COL6A3 tumor control mediated by the F9 TCR-expressing T cells was less, suggesting that further improvements to the in vivo activity of the F9 TCR could be made.
Affinity enhancement of a COL6A3-specific TCR imparts increased pHLA sensitivity and HLA class I recognition to CD4 T cells while maintaining specificity to the target pHLA
We postulated that an affinity-enhanced F9 TCR would augment in vivo activity of COL6A3-specific T cells. A yeast surface display approach was used for generating affinity-enhanced F9 TCR variants (fig. S1). As expected, the identified stable Vα/Vβ single-chain TCR (scTv) variants mainly harbored mutations in the framework regions of the TCR. However, a group of stable scTv variants encoded a shared mutation (G29R) in complementarity-determining region 1 (CDR1) of TCRα, whose enrichment in the yeast display process may have been caused by an improved binding to COL6A3- FLNV:HLA-A*02. In the second step, we generated yeast libraries with combinatorial mutations in each CDR of TCRα and TCRβ, respectively, and selected for improved binding of scTv variants. Analysis of the βCDR1 yeast library revealed scTv variants with increased COL6A3-FLNV tetramer binding but unchanged binding to a set of counterselection tetramers comprising each of the seven similar peptides (table S4), highlighting the selectivity in COL6A3-FLNV binding improvement. Among scTv variants with increased binding, we identified eight mutant βCDR1 sequences, which strongly deviated from the βCDR1 of the parental TCR. We next asked whether grafting of mutant CDR1 sequences onto the parental F9 TCR would improve COL6A3-FLNV recognition. We tested all mutant βCDR1 variants alone or in combination with the G29R mutation in αCDR1 and determined the reactivity of TCR mRNA-transfected CD8 T cells using T2 cells loaded with either COL6A3-FLNV or the off-target peptide COL6A1-ILSV. All mutant βCDR1 variants vastly improved TCR reactivity against COL6A3-FLNV, whereas COL6A1-ILSV reactivity remained unaltered in comparison to the F9 TCR (fig. S6, A and B). Together with the αG29R mutation, most of the βCDR1 TCR variants showed no further increase in COL6A3-FLNV reactivity but rather an increased reactivity to the COL6A1-ILSV off-target (fig. S6, C and D). However, for one βCDR1 TCR variant, the combination with αG29R (a1b4) resulted in an enhanced reactivity against COL6A3-FLNV yet still lacking recognition of the COL6A1-specific peptide, underlying the high specificity of this TCR variant (fig. S6E). We then produced soluble molecules of the highly reactive and specific mutant TCR variant (a1b4) and its mutant counterpart without αG29R mutation (awb4) and analyzed whether the increased COL6A3-FLNV reactivity was linked to an increased binding affinity of the TCRs. In agreement with previous SPR results (fig. S3B), we measured a binding affinity (KD) of 49 μM for the parental F9 TCR, whereas the affinity of the mutant TCR variants awb4 and a1b4 was 19 and 1.6 μM, respectively, and thus increased over the parental F9 TCR (fig. S6, F to H).
TCR affinities between 1 and 5 μM are within a physiologically relevant range and have been described as optimal for CD8 T cell activation (27, 30, 31) and potentially sufficient for CD4 T cell activation (32). We thus asked whether the affinity-enhanced F9 TCRs could impart COL6A3 exon 6 recognition in the context of HLA class I to CD4 T cells in a co-receptor–independent manner. For the analysis, we placed the F9 TCR and its affinity variants, awb4 and a1b4, respectively, into a lentiviral vector and transduced human T cells. In agreement with an increased affinity, the affinity-enhanced awb4 and a1b4 TCRs showed robust COL6A3-FLNV tetramer staining in both CD8 and CD4 T cells, whereas tetramer staining of the parental F9 TCR was restricted to CD8 T cells (Fig. 4A). We also assessed the functional response of T cells transduced with the mutant TCR variants using K.A2 target cells loaded with decreasing concentrations of either COL6A3-FLNV or the off-target peptide COL6A1-ILSV. When compared to the parental F9 TCR, both a1b4 and awb4 mutant TCRs triggered CD8 T cell activation at 10-fold to 100-fold lower COL6A3-FLNV concentrations (Fig. 4C), suggesting that TCR affinity enhancement enables COL6A3-FLNV targeting at lower peptide copy numbers. Furthermore, the mutant a1b4 TCR and, to a lesser extent, the mutant awb4 TCRs induced a COL6A3-FLNV–specific activation of CD4 T cells, whereas the F9 parental TCR was nonfunctional in CD4 T cells (Fig. 4B). We found that the low peptide copy numbers of COL6A3-FLNV required for a1b4 TCR-mediated CD4 T cell activation were comparable to those required for CD8 T cell activation (Fig. 4, B and C), indicating that this TCR is CD8 co-receptor independent. At very high peptide loading concentrations, both mutant TCR variants showed reduced (a1b4) or no (awb4) reactivity toward COL6A1-ILSV in comparison to the parental F9 TCR, confirming that TCR specificity is retained during affinity enhancement. In contrast, a1bw showed increased reactivity toward the COL6A1-ILSV epitope, indicating that some TCR combinations did not maintain COL6A3-FLNV specificity. Next, we assessed the ability of the parental F9 TCR and the engineered variants to recognize K.A2.COL6A3 and K.A2.COL6A1 cell lines (Fig. 4, F and G). T cells expressing the F9 TCR demonstrated a weak response against K.A2.COL6A3 and no response against K.A2.COL6A1 cell lines. In contrast, T cells expressing awb4 and, to a greater extent, a1b4 TCRs demonstrated enhanced recognition of K.A2.COL6A3 and no response against K.A2.COL6A1, which expressed ~10 fold more pHLAs than K.A2.COL6A3 (Fig. 3B). Moreover, examination of the ability to make multiple cytokines or mobilize CD107a in the same T cell revealed that T cells expressing a high-affinity TCR were more likely to be polyfunctional (Fig. 4, F and G, and fig. S7). Last, consistent with the peptide loading data, a1bw had enhanced recognition of both K.A2.COL6A3 and K.A2.COL6A1, making it not suitable for use in clinical adoptive T cell therapy studies.
Fig. 4. Affinity enhancement of a COL6A3-specific TCR imparts increased pHLA sensitivity and HLA class I recognition to CD4 T cells and maintained specificity to the target pHLA.
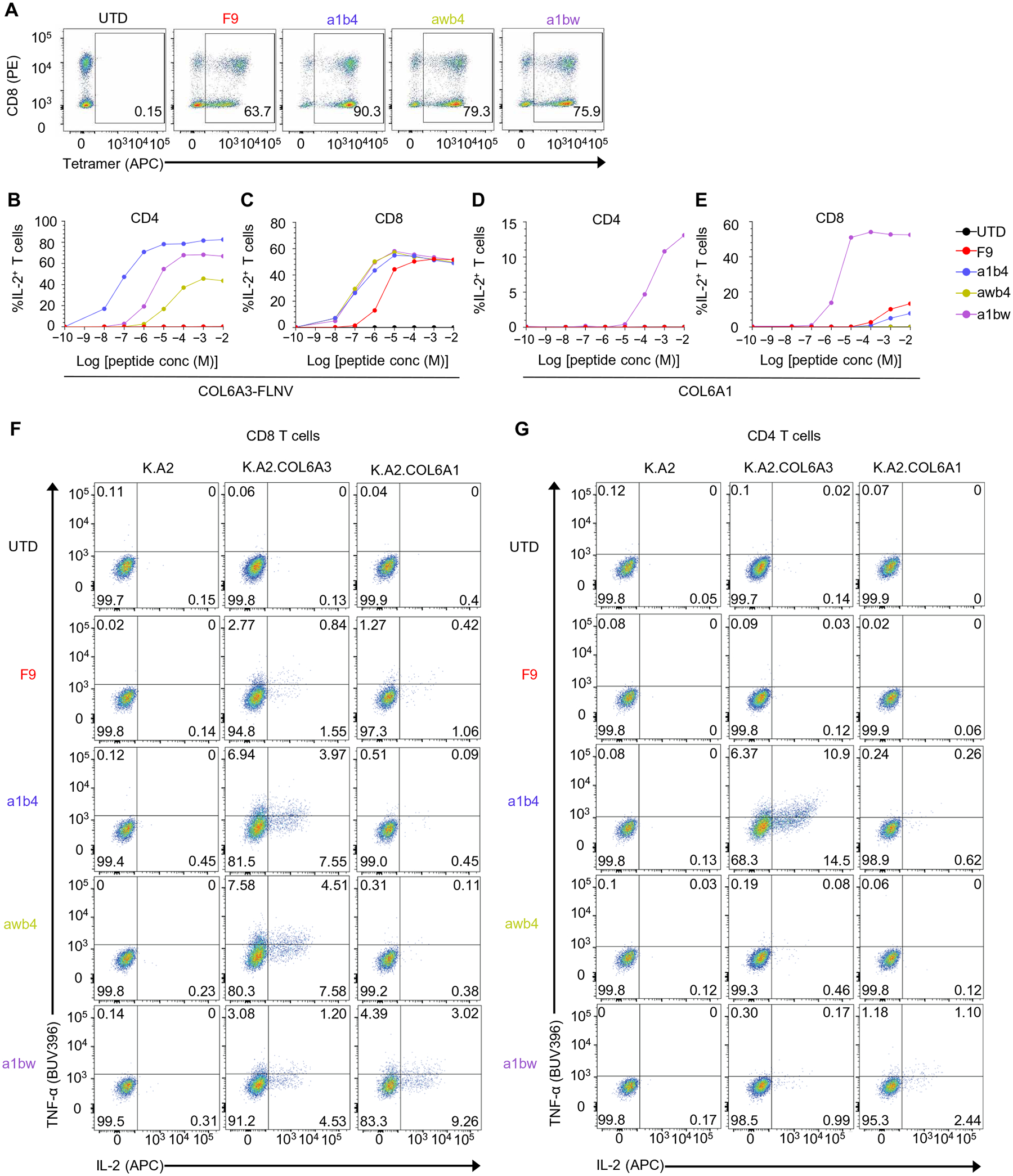
(A) A mixture of CD4 and CD8 T cells was activated with CD3/28-coated beads and left UTD or transduced with the indicated COL6A3-specific TCR. After 6 to 9 days of culture, a COL6A3-specific tetramer was used to stain the T cell populations. Results are representative of three independent experiments with different donors performed in triplicate. (B to E) K.A2 cells were loaded with indicated concentrations of (B and C) COL6A3-FLNV or (D and E) COL6A1-ILSV peptide and mixed with either (B and D) CD4 or (C and E) CD8 T cells expressing the indicated COL6A3-specific TCR. After 4 hours, intracellular IL-2 expression was measured by flow cytometry. (F and G) T cells from (A) were mixed with the indicated aAPCs for 4 hours, and TNF-α and IL-2 intracellular cytokine staining is shown for CD8 T cells (F) and for CD4 T cells (G). Results are representative of three independent experiments with different donors performed in triplicate.
T cells expressing affinity-enhanced COL6A3-specific TCRs recognize endogenously processed target peptide and show no signs of off-target reactivity
TCRs can recognize large numbers of closely related peptides (33), and this can have tragic outcomes in patients treated with T cells modified with affinity-enhanced TCRs (34). To further probe for potential off-target peptides that could be presented on normal tissues and recognized by COL6A3-specific TCRs, we performed a series of safety assays (fig. S1). First, we applied a mutagenesis method to define which residues within the COL6A3-FLNV sequence are necessary for TCR recognition (35). Only two amino acids (N8 and V9) were dispensable for F9 TCR recognition, whereas for recognition by awb4, amino acids D4 and V9 were dispensable. For a1b4, three amino acids (D4, A7, and V9) were fully dispensable, with additional two amino acids (F1 and L2) appearing less important, but still contributing to TCR recognition (Fig. 5, A to D). On the basis of these recognition motifs, we searched for similar peptides. Yet, instead of searching the human proteome restricted by HLA binding prediction algorithms, we searched within the experimentally determined HLA-A*02 immunopeptidomes obtained from normal tissues on population scale (fig. S1) to avoid false-positive and false-negative predictions. Peptides were required to contain a high number of residues that are either identical or similar to COL6A3-FLNV contact residues (Fig. 5D). Similarity was defined using the peptide-MHC (major histocompatibility complex) binding energy covariance matrix (36). T cells expressing affinity-enhanced TCRs (a1b4 and awb4) were tested against this panel of 30 motif-fitting target similar peptides that are naturally presented on normal tissues by HLA-A*02 and therefore could pose a potential safety risk. The abundance of peptide presentation on 673 normal tissues and the corresponding lack of recognition by T cells expressing a1b4 and awb4 TCRs is illustrated in Fig. 5 (E and F) for similar peptide 17 (SP17). Similarly, none of the other selected motif-fitting normal peptides were recognized (Fig. 5G and fig. S8), except for the target peptide, demonstrating the high specificity of affinity-enhanced a1b4 and awb4 TCRs.
Fig. 5. T cells expressing affinity-enhanced COL6A3-specific TCRs recognize endogenously processed target and show no signs of off-target reactivity.
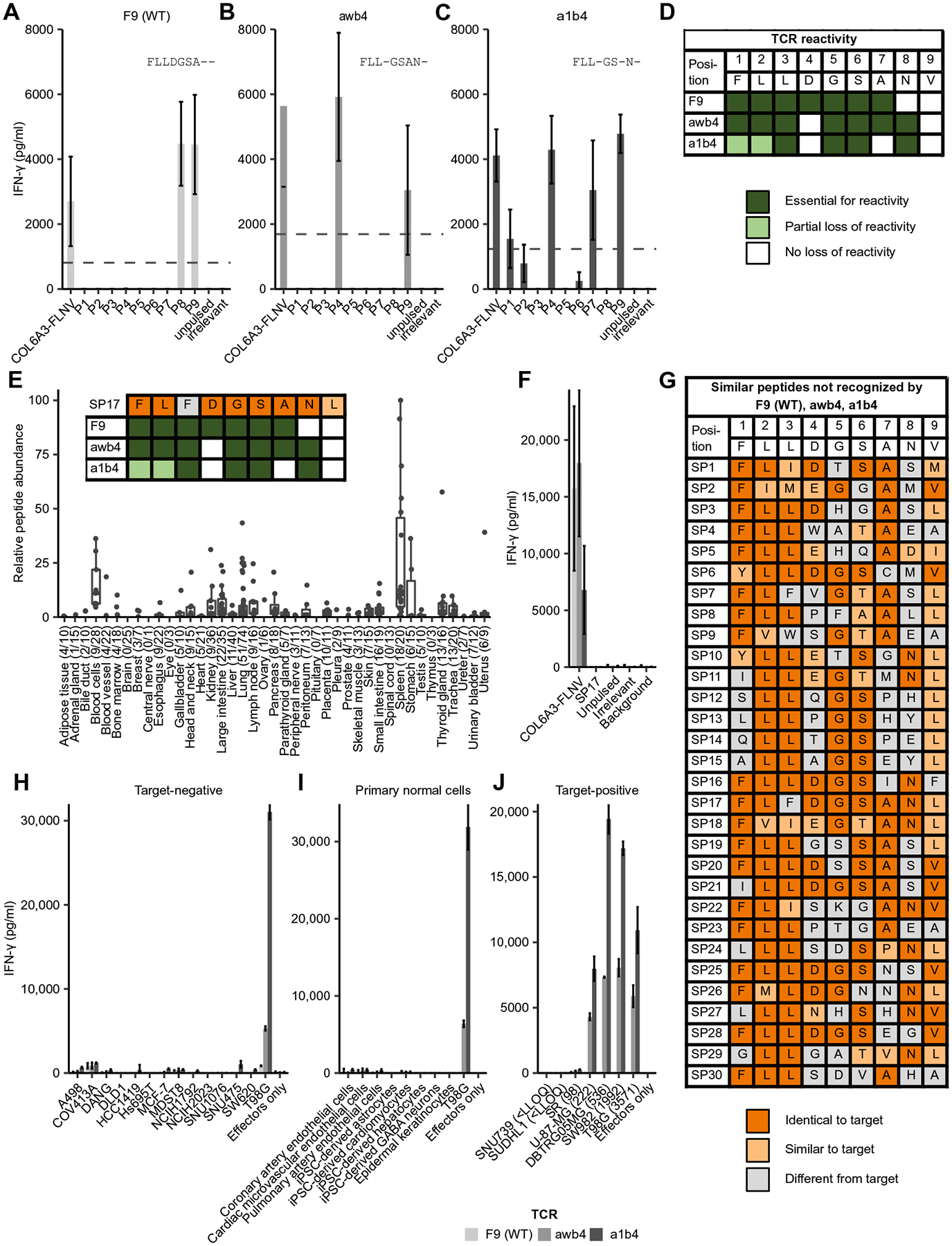
(A to C) Peptides containing a substitution at each position of the COL6A3-FLNV epitope were loaded on T2 cells and mixed with an equal number of T cells transduced with the parental COL6A3-FLNV–specific TCR F9 (A) or the COL6A3-FLNV–specific affinity-enhanced TCRs awb4 (B) or a1b4 (C). Bars show mean IFN-γ measured by enzyme-linked immunosorbent assay (ELISA), with error bars indicating the SD across four T cell donors. The dotted line indicates 30% of the COL6A3-FLNV IFN-γ secretion. Positions were defined as essential for T cell reactivity if responses for all donors were below this threshold. (D) Summary data highlighting TCR contact residues for each of the COL6A3-specific TCRs. Dark green indicates that the residue is essential for TCR recognition, light green indicates partial loss of activity due to mutation, and white indicates residues not important for TCR recognition. (E) Relative peptide abundance of HLA-A*02 peptide SP17 across 673 healthy tissue samples. The number of biological replicates is listed in parentheses for each group. Each dot represents the median of the quantified signals from five technical replicates. SP17 is identical to the target peptide COL6A3-FLNV in seven of nine residues and identical in six of seven residues that are essential for the target recognition of the awb4 TCR. Inset shows TCR recognition motif for each TCR overlaid with SP17. (F) SP17, COL6A3-FLNV, and control peptides were loaded on T2 cells, and the indicated TCR-transduced T cells were interrogated for IFN-γ secretion as above. (G) In total, 30 similar peptides based on the a1b4 and awb4 motifs were screened in IFN-γ secretion assays as described above, showing no recognition for any of the three COL6A3-specific TCRs. Identical and similar residues were indicated in dark and light orange shadings, respectively. Similarity was based on positive BLOSUM80 substitution matrix scores. (H to J) The indicated COL6A3-FLNV TCR-transduced primary human T cells were mixed with target-negative tumor cells (H), primary normal cells (I), or target-positive cells (J) at a 3:1 effector to target cell ratio for 48 hours. Bars show mean IFN-γ secretion measured by ELISA, and error bars indicate SD across three T cell donors. The number of COL6A3-FLNV CpC is listed in parenthesis. LLOQ, lower limit of quantitation; WT, wild type.
To further expand on specificity and safety of T cells expressing affinity-enhanced a1b4 and awb4 TCRs, we cocultured TCR-transduced T cells with a panel of target-negative cell lines of different tissue origins. Cell line selection was guided by RNA sequencing and high-sensitivity targeted MS. There was no or only minimal interferon-γ (IFN-γ) production by the TCR-transduced T cells when mixed with any of the 13 COL6A3-negative cell lines, but a strong response against T98G, the target-positive control cell line (Fig. 5H). Similarly, target-negative, primary normal cells of different organs or induced pluripotent stem cell (iPSC)–derived primary cells were not recognized by T cells expressing the parental TCR or the awb4 or a1b4 mutant TCR (Fig. 5I). Together, the lack of cross-recognition of target-similar peptides presented on normal tissues as well as the lack of recognition of target-negative cell lines and primary cells point toward a favorable off-target safety profile for T cells expressing the affinity-enhanced awb4 and a1b4 TCRs.
Next, we assessed the ability of T cells transduced with the engineered TCR variants awb4 and a1b4 to recognize endogenously processed target peptide COL6A3-FLNV in tumor cell lines (Fig. 5J). Seven different tumor cell lines were determined to be target-positive by RNA sequencing, with five cell lines covering a range from 98 to 3571 CpC based on targeted MS. T cells expressing COL6A3-specific TCRs secreted high amounts of IFN-γ when cultured with DBTRG05MG, SW982, and T98G cell lines that express several hundreds to thousands of target-HLA complexes on the cell surface. T cells expressing the a1b4 TCR produced more IFN-γ compared to those expressing the awb4 TCR, indicating that higher affinity results in improved T cell effector functions. U-87-MG with 222 CpC was recognized similarly to the K.A2.COL6A3 line that had a similar number of CpC (244), whereas SR with 98 CpC only induced minimal amounts of IFN-γ despite having twofold higher copy numbers than what was observed on the highest normal tissue, which was a thyroid sample with 49 CpC. These data highlight that affinity-enhanced COL6A3-specific TCRs maintain specificity and suggest that there is a therapeutic window by which tumors can be eliminated and healthy tissue can be ignored.
T cells expressing affinity-enhanced COL6A3-specific TCRs specifically control tumors in vivo expressing physiologic copy numbers of pHLAs on their cell surface
For T cells expressing COL6A3-specific TCRs to have therapeutic benefit, they will need to specifically recognize and kill tumor and tumor stroma that express pHLAs on their cell surface in a physiological range. Because K.A2.COL6A3 expressed pHLAs in that range and the corresponding control K.A2.COL6A1 expressed ~10-fold higher copies for the off-target peptide, we used these lines to test the in vivo efficacy of the COL6A3-specific, affinity-enhanced TCRs using the schema outlined in Fig. 6A. Tumor cells were engrafted in the subcutaneous flank and UTD T cells or T cells expressing COL6A3-specific TCRs were injected intravenously on day 8. Unlike our earlier studies using the N6 cell lines that displayed pHLAs in a high nonphysiologic range, we observed little ability of the F9-TCR–transduced T cells to control K.A2.COL6A3 tumor growth and improve survival relative to the infusion of UTD T cells (Fig. 6, B and C, and fig. S7, A and B). In contrast, both affinity-enhanced TCRs (a1b4 and awb4) were able to quickly and durably control K.A2.COL6A3 but not K.A2.COL6A1 tumor growth (Fig. 6, D, E, H, and I). After the last imaging on day 29, we isolated T cells from mice that cleared K.A2.COL6A3 and measured their response to K.A2.COL6A3 using an in vitro intracellular cytokine assay (Fig. 6, J and K, and data file S1). We observed that all T cells expressing COL6A3-specific TCRs maintained robust responses to cognate antigens after clearing tumors and residing in vivo for 21 days. However, we did note that T cells expressing a1b4 TCR maintained more robust and polyfunctional responses compared to those expressing awb4 TCR (fig. S7, C and D), suggesting that the improved responses conferred by a1b4 may be more durable. Together, these data suggest that T cells expressing either awb4 or a1b4 TCR may target and eliminate human tumor stroma.
Fig. 6. Adoptive transfer of T cells expressing affinity-enhanced COL6A3-specific TCRs are able to recognize and eliminate cells expressing pHLAs in a physiologic copy number range.
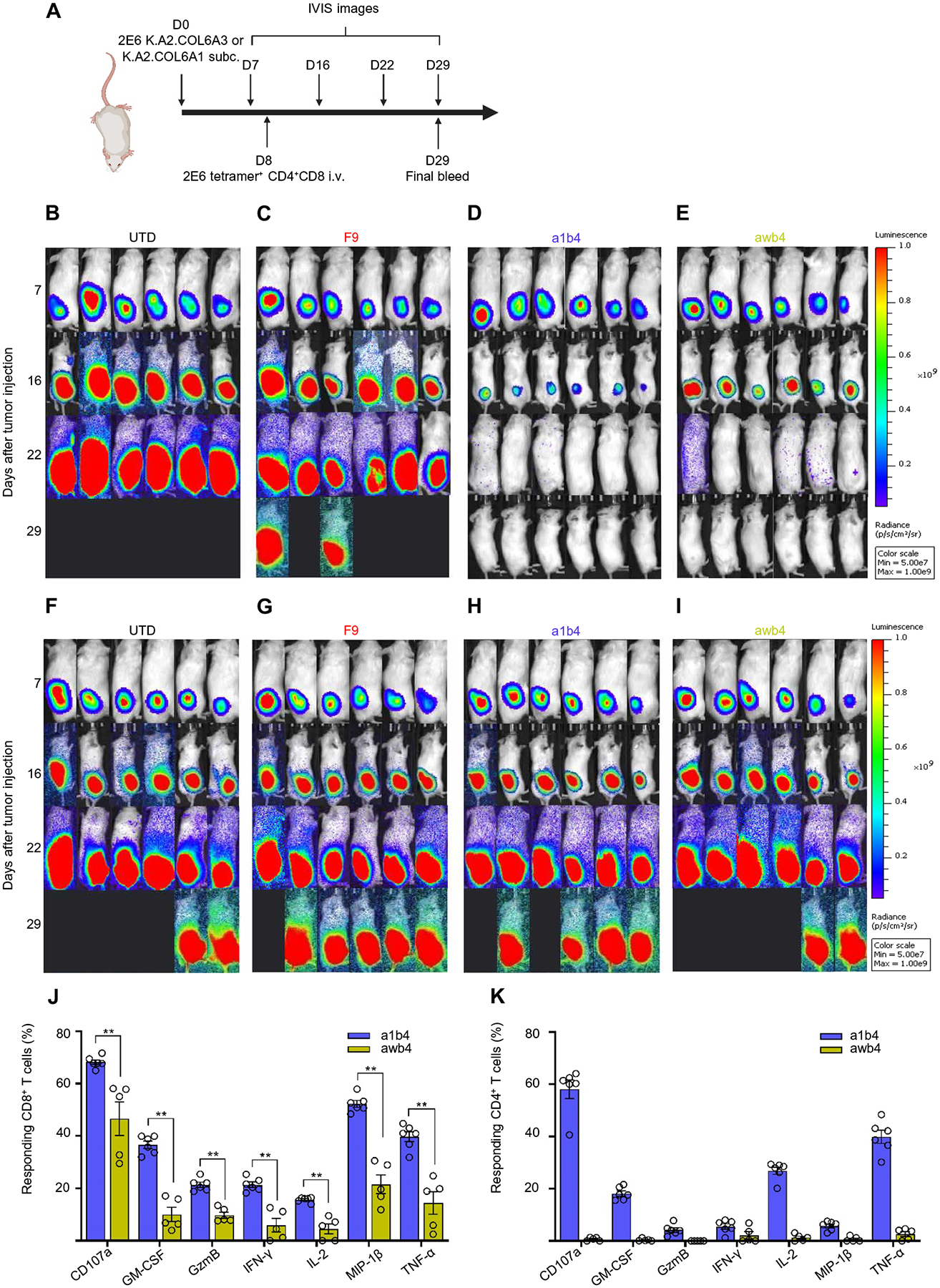
(A) Timeline and schematic of in vivo study. i.v., intravenous. (B to E) IVIS images of the K.A2.COL6A3-bearing NSG mice injected with UTD T cells (B), or T cells transduced with F9 TCR (C), a1b4 TCR (D), or awb4 TCR (E), respectively. (F to I) IVIS images of the K.A2.COL6A1-bearing NSG mice injected with UTD T cells (F), or T cells transduced with F9 TCR (G), a1b4 TCR (H), or awb4 TCR (I), respectively. Two million T cells expressing the indicated TCR were injected on day 8 after tumor injection. (J and K) At the study’s conclusion, splenocytes from non–tumor-bearing mice were isolated and incubated with K.A2.COL6A3 for 6 hours and percent of CD8 T cells (J) and percent of CD4 T cells (K) making the indicated cytokines are shown. Each dot represents T cells from an individual mouse. Wilcoxon rank sum test, two tailed was performed. *P < 0.05. Data are representative of three independent experiments. GM-CSF, granulocyte-macrophage colony-stimulating factor; GzmB, granzyme B.
DISCUSSION
Defining tumor-selective targets is key for the development of specific immunotherapies. RNA expression studies can highlight differentially expressed and spliced genes (9), yet this does not necessarily translate to the differential presentation of HLA-restricted peptides that are actual targets of TCR-based adoptive T cell therapies. Here, we used population-scale immunopeptidomics by quantitative LC-MS/MS to perform an unbiased search for such peptides differentially presented on tumors and normal tissues. Through this process, we identified the HLA-A2–restricted tumor stroma epitope COL6A3-FLNV within an alternatively spliced exon of COL6A3, which is presented in high copy numbers on the tumor stroma and shared by many patients across various tumor types.
As a critical component of the TME, tumor stroma has a profound effect on many hallmarks of cancer (37). Stroma has been shown to have crucial roles in tumor initiation, cancer progression, and metastasis, and its destruction has resulted in tumor clearance (38). Given its importance, several strategies have been put forth to target the TME. Among them, FAP has been a major focus of targeted therapy of stromal cells (39). In addition, CAR-T cells that recognize FAP-positive cells and target stromal cells specifically have been used in previous preclinical studies and demonstrated a reasonable safety profile (19). Although COL6A3 has been identified as a potential prognosis marker of colorectal carcinoma (40) and pancreatic cancer (41), there are so far no targeted therapies available. Because COL6A3 is secreted and becomes part of the extracellular matrix, approaches directed against the protein cannot target the stroma cells. On the other hand, COL6A3-FLNV allows direct targeting of stroma cells that actively transcribe and translate COL6A3, opening the intracellular target space for anticancer stroma therapy. Moreover, as COL6A3-specific T cells are destroying the tumor stroma, the COL6A3-FLNV peptide may be presented using a reverse cross-presentation process, and this may permit some direct tumor killing by the COL6A3-specific T cells. The first therapeutic approach to target COL6A3 exon 6 by an adoptive cell transfer (ACT) approach was undertaken as part of the IMA101 study (42) that used autologous nonengineered T cell products against multiple defined pHLA targets. Besides utilization in an ACT approach, the described TCRs can also be further developed into soluble bispecific TCRs (43), which provide an off-the-shelf opportunity with greater dosing flexibility.
The ability of TCRs to bind a wide array of similar pHLAs (33) makes the careful assessment of the potential off-target reactivity necessary to avoid major adverse events when using these TCRs in ACT. This is even more relevant if TCRs undergo engineering for affinity enhancement as seen in previous clinical trial failures like development of MAGE-A3 TCRs (44), which was stopped due to off-target toxicity related to a titin epitope (34). These findings highlight the need for and importance of reliable preclinical de-risking of TCR candidates, including additional preclinical screening assays aimed to enhance TCR specificity before clinical use, which have been thoroughly considered in our studies. Considering the homology between collagens and other proteins embodying VWF type A–like domains (45), the strength of our approach is that we screened off-targets that are naturally processed and presented on healthy tissue.
A number of studies have demonstrated the existence of optimal TCR affinity windows for key parameters of T cell function such as proliferation, antigen sensitivity, and polyfunctionality (46), above which signaling can render cells hyporesponsive and mediate undesired cross-reactivity to self-pHLAs. For example, T cells engineered with TCRs of very high affinities (KD < 1 nM) become cross-reactive to other pHLA complexes. TCRs with enhanced affinities risk increased reactivity with structurally related self-peptides and off-target toxicities. Many studies have found an affinity range of KD of ~1 to 10 μM to be optimal for signaling of engineered TCRs and avoiding cross-reactivity (34, 47–49). We have identified two TCRs that fit within this “optimal” window by measuring the number of pHLAs presented on the surface of tumor and healthy tissue. This provides key information regarding both the potential safety and efficacy of adoptively transferred T cells. The wild-type F9 TCR showed limited antitumor activity clearing some NALM6 tumors in mice that presented more than 1000 pHLAs on their cell surface. Given that most tumor samples we studied had around 300 copies of COL6A3-FLNV per cell, we would predict that the F9 would have little therapeutic benefit. Off-target toxicity was tested in an exaggerated scenario on engineered K562 cells (50) presenting the target peptide in a physiological range of primary tumor specimens and 10-fold higher copies for the most relevant off-target peptide. Last, we identified tumor cell lines with different pHLA target CpC and could demonstrate that cell lines with target copies in the physiological range of natural tumor stroma are recognized well by the affinity-enhanced COL6A3-specific TCRs, with responses declining with lower copy numbers suggesting that these TCRs will target tumors but ignore healthy tissues. We believe that stringent preclinical TCR development, including comprehensive in vitro and in vivo testing of TCRs combined with knowledge about pHLA CpC, is important to develop safe and effective immunotherapies.
Our study has some limitations; for instance, we had to use tumor lines rather than tumors with intact tumor stroma to study COL6A3-FLNV–specific T cells in vivo. Because of the inability of tumor stroma cells to survive for a long time in humanized mice, there are currently no in vivo models available to study immuno-therapies targeting human tumor stroma (51). This could in principle be studied in immunocompetent strains of HLA-A2 transgenic mice. However, the COL6A3 target is not conserved between humans and mice, making efficacy studies unreliable. Moreover, given all the sequence diversity between mouse and human, safety studies would be uninformative. We did, however, identify some tumor cell lines that also perform this alternative splicing event, although this was not generally observed in our ISH studies using primary patient material. Nonetheless, it does suggest that in some patient populations both the stroma and tumor could be targeted by these COL6A3-specific T cells, which may even enhance destruction of the tumor. Another limitation is that while the two affinity-enhanced TCRs showed no cross-reactivity in our studies, this does not guarantee lack of adverse events in patients when T cells expressing these TCRs are infused into cancer patients. The remaining risk of unexpected cross-reactivity in vivo should be addressed by clinical risk mitigation strategies like dose escalation.
We were able to characterize several TCRs that specifically recognized COL6A3-FLNV, and T cells expressing these TCRs were able to durably eradicate tumors, begging the obvious question of which one should be tested in the clinic. The wild-type F9 TCR has limited potency and thus would not be effective. In terms of TCR’s potential to become cross-reactive, awb4 appears to be slightly better than a1b4 TCR because seven of the nine amino acids within COL6A3-FLNV are required for awb4 TCR recognition, whereas six of the nine amino acids contribute to a1b4 recognition. Nevertheless, high specificity could be shown for both awb4 and a1b4 TCRs when we examined related peptides presented by HLA-A*02 in healthy tissue. Our results indicate that T cells expressing a1b4 TCR maintained more robust and potent responses compared to those expressing awb4 TCR, suggesting that the improved responses conferred by a1b4 may be more durable. In contrast to awb4, a1b4 shows full functionality in both CD8 and CD4 T cells without the need for CD8 cotransduction, a next-generation technology distinct from most TCRs being investigated in the TCR–T cell field. Activation of both T cell types has been reported as favorable for induction and maintenance CAR functional activity (52). In any case, further testing of COL6A3-specific T cells in a phase 1 clinical trial will be required to determine whether this could be an effective treatment to target the TME and induce durable tumor remissions.
MATERIALS AND METHODS
Study design
The main objective of this study was to characterize TCRs against a prevalent tumor-specific target to enable future clinical development of T cell therapies. For in vivo efficacy studies, six mice were randomized to each treatment group. Previously, this group size yielded statistically significant data for a similarly designed study (29). For all in vitro safety studies, three donors were used for T cell isolations to control for biological variability. Data analysis was performed in an unblinded manner.
Tissue acquisition and processing
Primary human tissue samples were extracted surgically or postmortem from 728 HLA-A*02–positive patients with cancer and 298 HLA-A*02–positive normal tissue donors, respectively. All specimens were procured by Immatics Biotechnologies GmbH with written informed consents and with appropriate ethics committee approvals. The resulting sample set of 673 normal and 739 cancer samples covered 38 different organs and 24 tumor types with a median group size of 18 donors. Tissue samples were snap-frozen in liquid nitrogen after excision and stored until isolation at −80°C for subsequent RNA and pHLA analyses. Tissue samples were fixed in formalin and embedded in paraffin (FFPE) for histological assessment.
HLA peptide isolation and relative quantification
After tissue homogenization and lysis, pHLA complexes were isolated by immunoprecipitation using BB7.2 (Department of Immunology, University of Tübingen, Germany) (53) coupled to cyanogen bromide–activated Sepharose resin (GE Healthcare Europe). Peptides were eluted from antibody-resin by acid treatment and purified by ultrafiltration. HLA peptidomics was performed using the XPRESIDENT platform as previously described (26). Briefly, peptidome samples were separated by reversed-phase ultraperformance LC (UPLC) (nanoAcquity Waters) using ACQUITY UPLC BEH C18 columns (75 μm × 250 mm, Waters) and a gradient ranging from 1 to 34.5% acetonitrile over the course of 70 or 190 min. MS was performed on online coupled Orbitrap mass spectrometers Fusion, Velos, and Linear trap quadrupole (Thermo Fisher Scientific) in data-dependent acquisition mode. Samples were analyzed in at least three replicate runs, acquiring MS/MS data in collision-induced dissociation (CID) and higher collisional energy dissociation mode, resulting in a total of 7271 runs. Data processing was performed using a proprietary pipeline, which combines database search, spectral clustering, feature detection, retention time alignment, and global normalization for the generation of population-scale, relative-quantitative peptide presentation profiles.
Absolute quantitation of peptides
pHLA copy numbers were determined using the AbsQuant method (US10545154B2) based on the number of cells within the investigated tissue, total amount of the isolated peptide, and target-specific pHLA isolation efficiency. Hereby, each of these three parameters was determined experimentally. The number of cells was determined on the basis of quantitation of DNA content in the investigated tissue sample. DNA was isolated using the QIAamp DNA Mini Kit (QIAGEN) from lysate aliquot, which was sampled during the isolation of HLA-restricted peptides from primary tissue. The DNA yield was quantified using the Qubit dsDNA High Sensitivity (HS) Assay Kit (Applied Biosystems/Thermo Fisher Scientific), and the number of cells was interpolated from DNA content using a standard curve derived from peripheral blood mononuclear cells. For absolute quantitation, a series of targeted nanoLC-MS/MS measurements was performed on an Orbitrap mass spectrometer (Thermo Fisher Scientific). Two differently isotopically labeled peptide equivalents were synthesized. One of the isotopically labeled equivalents was used as an absolute quantity reference and was spiked into retention vials of each human tissue sample, which was used for absolute quantitation. The other isotopically labeled equivalent was used to generate the peptide-specific standard curve. Thereby, one of the isotopically labeled equivalents was titrated, and the other one was used as mentioned before as an absolute quantity reference. The MS/MS spectra were acquired using parallel reaction monitoring restricting to labeled peptide masses for the analysis of standard curves and labeled and native peptide masses for the analysis of primary tissue samples. The MS/MS signals of selected fragment ions were extracted using Skyline 3.6.0 and translated into an absolute peptide amount using peptide-specific standard curves. The efficiency of pHLA isolation was established by spiking of refolded pHLA complex of the investigated peptide into the tissue lysate during the peptide isolation procedure and subsequent detection and quantitation of the peptide by nanoLC-MS/MS as described above for quantitation of natively presented peptide.
RNA isolation and sequencing
Immunopeptidome measurements were accompanied by paired transcriptome analysis for a subset of 715 samples by isolating total RNA using TRIzol (Invitrogen), followed by purification with an RNeasy mini kit (QIAGEN) according to the manufacturer’s protocol. RNA sequencing and expression quantification were performed by CeGaT. Briefly, 1 to 2 μg of total RNA were used as starting material for library preparation performed according to the Illumina protocol (TruSeq Stranded mRNA Library Prep Kit). Sequencing was performed on an Illumina HiSeq 2500 machine. For all experiments, a strand-specific protocol was used to generate single-end reads of a length of 50 nucleotides. The minimum number of reads was 43,700,000 per sample. The quality of the sequencing process was monitored using spike-ins derived from bacteriophage Phi-X. We determined normalization factors to allow intersample read count comparisons according to methods used for differential expression analysis of RNA-sequencing data.
HLA-A2 typing
DNA of donors was isolated from tissue or whole blood using the QIAamp DNA Mini Kit (QIAGEN) or the QIAamp DNA Blood Mini Kit (QIAGEN), respectively. In case of limited tissue amounts, the QIAamp Investigator Kit (QIAGEN) has been used. HLA genotyping for HLA-A*02 was performed by PCR and subsequent agarose gel electrophoresis using the Ambisolv Primer Mix PM002 (Life Technologies) and recombinant Taq polymerase (Life Technologies).
ISH by manual and automated RNAscope
ISH assays were performed on 5-μm-thick consecutive FFPE tissue sections according to manufacturer’s protocols [Advanced Cell Diagnostics (ACD)], document number 322150-USM for use with Leica Biosystems’ BOND RX or document number 330487-USM for manual RNAscope. At least two different pretreatment conditions were applied to all samples to achieve optimal staining results. Briefly, for the manual RNAscope assay (catalog nos. 320487 and 322360, ACD) sections were baked for 60 min at 60°C, deparaffinized in xylene, dehydrated in ethanol, and air-dried. All slides were incubated with hydrogen peroxide for 10 to 20 min at room temperature, target retrieval reagent for 15 to 30 min at 98°C, and protease plus for 15 to 30 min at 40°C. All probe sets were hybridized for 2 hours at 40°C, followed by RNAscope amplification and Fast Red chromogenic signal detection. Automated RNAscope 2.5 LS Reagent Kit–RED assays (catalog no. 322150, ACD) were performed on the Leica BOND RX platform (Leica Biosystems). Briefly, sections were baked for 90 min at 60°C before deparaffinization and dehydration on the instrument. Target retrieval was performed with Epitope Retrieval Buffer 2 (catalog no. AR9551, Leica) for 15 min at 88° or 95°C and subsequent protease treatment for 15 min at 40°C. Probe sets were hybridized for 2 hours at 42°C, followed by RNAscope amplification and Fast Red chromogenic signal detection applying the Bond Polymer Refine Red Detection (Leica, DS9390). The following RNAscope probe sets obtained from ACD were used in this study: dihydrodipicolinate reductase (dapB) (accession no. EF191515, manual catalog no. 310043; LS catalog no. 312038), ubiquitin C (UBC) (accession no. NM_021009, manual catalog no. 310041; LS catalog no. 312028), ACTA2 (accession no. NM_001613.2, manual catalog no. 311811), panCK (manual catalog no. 420421), and collagen α-3 (COL6A3-E6) (accession no. NM_004369.3, manual catalog no. 444601; LS catalog no. 839618). Specimens were scanned with ×40 magnification using the Hamamatsu NanoZoomer Digital Pathology System (Hamamatsu Photonics K.K.).
Microscopic evaluation
Tumor specimens mounted on glass slides and stained with RNAscope ISH were microscopically evaluated to assess staining quality. The optimal pretreatment conditions were determined on the basis of reference positive and negative probe sets to maximize the signal-to-noise ratio while maintaining tissue morphology. COL6A3-E6 target expression in stroma or tumor cells was evaluated by a trained pathologist using a semiquantitative scoring system. Staining intensity (SI) was measured in signal dots per cell, and the predominant phenotype was categorized in SI scores ranging from 0 to 4: SI0 = less than 1 dot in 10 cells, SI1 = 1 to 3 dots per cell, SI2 = 4 to 9 dots per cell, SI3 = 10 to 15 dots per cell, and SI4 = more than 15 dots or clustered dots. The frequency of stroma or tumor cells with the predominant SI score was measured in percent, and percentage points (PP) were assigned as follows: PP1 = 1 to 10%, PP2 = 11 to 50%, PP3 = 51 to 80%, and PP4 = 81 to 100%. Finally, a combined score—adapted from the immunoreactivity score (IRS)—was calculated for each sample as follows: IRS = SI × PP.
TCR identification and engineering
The α and β TCR chain sequences were isolated from T cells of HLA-A*02–positive and HLA-A*02–negative healthy donors. CD8 T cells were stimulated repeatedly with artificial antigen-presenting cells coated with target pHLA monomers and CD28 as previously described (54). Subsequently, single cells were sorted using COL6A3-FLNV-HLA-A*02 tetramers. The TCR nucleotide sequences were obtained using rapid amplification of complementary DNA ends (5′ RACE) and Sanger sequencing. TCR engineering was performed essentially as previously described (55). Briefly, the retrieved TCR variable domains were linked by a glycine-serine linker to form an scTv and subcloned into a yeast surface display vector. Stability engineering to accomplish correct folding of the scTv on the yeast surface was performed using random mutagenesis via error-prone PCR and subsequent selection of most functional clones using COL6A3-FLNV-HLA-A*02 tetramers. The CDR β1 of a stabilized clone was randomized in six positions using degenerate codons, creating a library of more than 6 × 107 scTv clones. This library was subjected to multiple rounds of flow cytometric selection with COL6A3-FLNV-HLA-A*02 tetramer or dimer, implying increasing stringency over the course. Simultaneously, counterselection with a high concentration of similar peptide tetramers was applied (table S4). Yeast clones were selected to single-cell level and investigated thoroughly for target and similar peptide binding. Outstanding mutated CDRs were then either grafted onto the parental TCR framework to see their functionality in cellular assays or transferred into a soluble molecule to determine affinity.
Affinity measurements
Binding affinities of the parental TCRs D10, F9, and H3 toward COL6A3-FLNV-HLA-A*02 were determined by SPR analysis in two independent runs using a Biacore 2000 instrument. Therefore, biotinylated HLA-A*02:01 in complex with COL6A3-FLNV was immobilized on streptavidin SPR chips, resulting in density of 247 response units (RU) (run 1) and 249 RU (run 2). Soluble TCR variants of D10, F9, and H3 diluted in 10 mM Hepes (pH 7.4), 150 nM NaCl, and 0.05% Tween 20 were applied at concentrations of 0, 1.6, 6.3, 25, and 100 μM, respectively, at a flow rate of 30 μl/min. Binding equilibrium dissociation constants (KD’s) were determined from both independent runs, and mean KD values for TCRs D10, F9, and H3 are indicated.
Binding affinities of mutant TCR versions were analyzed by bio-layer interferometry using an Octet Red384 instrument (forteBIO). Measurements were performed in phosphate-buffered saline (PBS) at 30°C. Streptavidin-coated biosensors were loaded with biotinylated HLA-A*02:01 complexed with COL6A3-FLNV or an unrelated control peptide. After 45 s of loading in an HLA solution (10 μg/ml), 1- to 1.5-nm adsorbed layer was measured. The biosensors were subsequently brought in contact with solutions of recombinant F9 wt, F9 awb4, and F9 a1b4 at concentrations of 0.1, 0.3, 1, 3.2, 10, 31.6, and 100 μM. Association of the TCRs to the biosensors and dissociation in PBS were recorded for 60 and 120 s, respectively. To correct for unspecific binding, signal observed with the control peptide was subtracted from the COL6A3-FLNV data. Data were analyzed, and the binding affinities were calculated using the Octet data analysis software (forteBIO).
Lentiviral vector construction and production
COL6A3-specific TCR sequences were synthesized (Integrated DNA Technologies, Coralville, IA) and placed into third-generation lentiviral transfer vector pTRPE (56). Minigene vectors encoding COL6A1 and COL6A3 epitopes were constructed by fusing mCherry or iRFP720 to COL6A1832–840 (P12109) or GFP or iRFP670 to COL6A3642–650 (NP_476508.2), respectively, within pELNS (57). Lentiviral vector supernatants were produced as previously described (52).
Cell lines and culture
To generate artificial APCs that presented COL6A1 or COL6A3 epitopes, K562 cells expressing HLA-A2 (50) or Nalm6 [American Type Culture Collection (ATCC)] cells were transduced with pELNSxv-GFP-COL6A1832–840 or mCherry-COL6A3642–650, respectively. Single-cell clones were generated via BD FACSJazz (BD Biosciences), and a clone that exhibited stable expression was selected. For bioluminescence imaging, K562-GFP-COL6A3 and K562-mCherry-COL6A1 cells were transduced with pTRPE-CBR-mCherry and pTRPE-CBG-GFP minigenes, respectively. In addition, NALM6-GFP-COL6A3 and NALM6-mCherry-COL6A1 cells were transduced with pTRPE-iRFP670 and pTRPE-iRFP720 minigenes, respectively. NALM6, K562, and NALM6 and K562–derived cell lines were maintained in RPMI 1960 (Gibco) supplemented with 10% fetal bovine serum (Gibco). The following cell lines were used: renal cell carcinoma cell line A498 from ATCC (HTB-44), ovarian epithelial–serous carcinoma cell line COV413A from Sigma-Aldrich (07071905), pancreas carcinoma cell line DANG from Deutsche Sammlung für Mikroorganismen und Zellkulturen (DSMZ) (ACC-249), glioblastoma cell line DBTRG05MG from ATCC (CRL-2020), colon adenocarcinoma cell line DLD1 from DSMZ (ACC-278), primary ductal carcinoma cell line HCC1419 from ATCC (CRL-2326), melanoma cell line Hs695T from ATCC (HTB-137), breast cancer cell line MCF7 from ATCC (HTB-22), colon carcinoma cell line MDST8 from Sigma-Aldrich (99011801), human lung adenocarcinoma cell line NCIH1792 from ATCC (CRL-5895), non–small cell lung cancer adenocarcinoma cell line NCIH2023 from ATCC (CRL-5912), laryngeal squamous cell carcinoma cell line SNU1076 from KCLB (01076), liver cancer cell line SNU475 from ATCC (CRL-2236), colon adenocarcinoma cell line SW620 from Sigma-Aldrich (87051203), hepatocellular carcinoma cell line SNU739 from KCLB (00739), anaplastic large cell lymphoma cell line SR from ATCC (CRL-2262), anaplastic large cell lymphoma cell line SUDHL1 from DSMZ (ACC-356), synovial sarcoma cell line SW982 from ATCC (HTB-93), glioblastoma cell line T98G from Sigma-Aldrich (92090213), glioma cell line U-87-MG from ATCC (HTB-14), and lymphoblast cell line T2 cells from ATCC (CRL-1992). The following primary cells were obtained from PromoCell: human coronary artery endothelial cells, human cardiac microvascular endothelial cells, human pulmonary artery endothelial cells, and normal human epidermal keratinocytes. iPSC-derived cells were from Fujifilm/Cellular Dynamics: iCell hepatocytes 2.0, iCell GABAneurons, iCell astrocytes, and iCell cardiomyocytes.
T cell transduction and expansion
Primary human CD4 and CD8 T cells from deidentified healthy volunteer donors were provided by the University of Pennsylvania Human Immunology Core (University of Pennsylvania Institutional Review Board no. 70596). All specimens were collected under a University Institutional Review Board–approved protocol, and written informed consent was obtained from each donor. T cells were maintained in Cell Therapy Systems (CTS) OpTmizer T Cell Expansion Serum Free Medium (Thermo Fisher Scientific) supplemented with CTS OpTmizer T cells Expansion Supplement (Thermo Fisher Scientific), GlutaMAX (Thermo Fisher Scientific), penicillin (100 U/ml; Gibco), and Hepes (10 mM; Gibco). T cells were stimulated with anti-CD3/CD28 Dynabeads (Invitrogen) at a bead to cell ratio of 3:1. Approximately 24 hours after activation, T cells were transduced with lentiviral vectors by adding the virus supernatant to T cell cultures. CD4 and CD8 T cells used for both in vitro and in vivo experiments were mixed at 1:1 ratio, activated, and transduced.
Flow cytometry
The following antibodies were used to stain primary human T cells: anti-CD3 BV605 (catalog no. 317322, BioLegend), CD4 BV421 (catalog no. 311507, BioLegend), CD8 phycoerythrin (PE) (BD Biosciences), tumor necrosis factor–α (TNF-α) mouse anti-human BUV395 (catalog no. 563996, BD Biosciences), IFN-γ mouse anti-human BUV737 (catalog no. 564620, BD Biosciences), interleukin-2 (IL-2) rat anti-human allophycocyamin (APC) (catalog no. 554567, BD Biosciences), and macrophage inflammatory protein-1β (MIP-1β) mouse anti-human PE (catalog no. 550078, BD Biosciences). HLA-A*02:01-peptide monomers were produced by protein refolding from HLA-A*02:01 and β2 microglobulin Escherichia coli inclusion bodies as previously described (58). Before tetramer straining, T cells were incubated with 500 nM dasatinib in the RPMI 1640 medium for 60 min at 37°C as previously described (59). Intracellular cytokine staining was performed as previously described (60). Flow cytometry was performed using a BD LSRFortessa cell analyzer and analyzed using FlowJo software v.10.
In vivo studies of engineered T cell efficacy
Six- to 12-week-old nonobese diabetic (NOD)/severe combined immunodeficient (SCID)/γ-chain−/− (NSG) mice were obtained from the Stem Cell and Xenograft Core of the Abramson Cancer Center, University of Pennsylvania under a University of Pennsylvania Institutional Animal Care and Use Committee–approved protocol. NSG mice were inoculated subcutaneously with K562-derived cells on the flank or intravenously with NALM6-dervived tumors. After 8 to 10 days, expanded human primary T cells (CD4 and CD8 T cells used were mixed at 1:1 ratio) were infused. Anesthetized mice were imaged using IVIS Spectrum (PerkinElmer) as previously described (29).
Statistical analysis
Statistics for Figs. 1 and 5 and table S2 were generated using R (version 3.6.1) and ggplot2 (version 3.2.1). We corrected P values reported in table S1 for multiple testing with the method described by Benjamini and Hochberg (61). All other statistical analyses were performed using GraphPad Prism version 7. For comparisons of two groups, two-tailed unpaired t tests were used. One-way analysis of variance (ANOVA) with Turkey post hoc test was used for comparison of three or more groups in a single condition. Statistical analysis for tumor volume was performed using a two-way repeated-measures ANOVA. Kaplan-Meier survival data were analyzed using a log-rank (Mantel-Cox) test. Data are expressed as means ± SEM of n experiments. P values less than 0.05 were considered significant.
Supplementary Material
Acknowledgments:
We thank C. June for playing a key role in establishing this collaboration between Immatics and Riley laboratory and the rest of the Center for Cellular Immunotherapies investigators for their helpful feedback. We also thank the sample management team at Immatics for tissue and cell line recruitment; the Target Research laboratory team for preparation of tissues, DNA, RNA, and peptides; the histology team for tissue staining; the MS team for peptide analysis and validation; the Chemistry, Manufacturing, and Controls team for peptide synthesis; the bioinformatics team for data analysis; and the Immunology team for TCR discovery, engineering, and characterization as well as HLA monomer refolding. We also thank S. Schecher for review of the manuscript. We thank Stem Cell and Xenograft Core at the University of Pennsylvania for assistance with the humanized mouse studies. We also thank the Human Immunology Core (P30-CA016520 and P30-AI045008) for providing purified human T cells.
Funding:
The research in this manuscript was funded by Immatics, Cancer Prevention Research Institute of Texas Foundation (Immatics, DP150029) to S.W., Brody Family Medical Trust Fund Fellowship from the Philadelphia Foundation to G.B.K., and an NIH grant (T32CA009140) to G.B.K. This study was supported in part by Tmunity Therapeutics to J.L.R.
Footnotes
SUPPLEMENTARY MATERIALS
www.science.org/doi/10.1126/scitranslmed.abo6135
Competing interests: J.F., S.B., A.M., F.U., H. Schuster, D.J.K., V.G., M.O., D.F., M.B., C. Song, G.S., C. Schraeder, F.H., M.R., C.-C.T., M. Hofmann, T.T., M. Hutt, L.A., M.J., A.A., S.M., O.S., H. Singh, C.R., S.W., C.W., D.M., and T.W. were employees of Immatics in the course of this work and may have securities from Immatics. J.L.R. is a cofounder and holds an equity interest in Tmunity Therapeutics. C.R., H. Singh, S.W., and T.W. are management board members of Immatics. All other authors have no competing interests. The following patent applications relate to subject matter in this paper: PCT/EP2011/053863 (entitled “Novel Immunotherapy against several tumors including gastrointestinal and gastric cancer”), disclosing subject matter coinvented by J.F., H. Singh, S.W., and T.W.; PCT/EP2017/066630 (entitled “T cell receptors and immune therapy using the same”), disclosing subject matter coinvented by S.B., S.W., D.M., and L.A.; PCT/EP2018/080176 (entitled “Novel engineered T cell receptors and immune therapy using the same”), disclosing subject matter coinvented by S.B., F.U., M. Hofmann, M. Hutt, C.W., D.M., and L.A.; PCT/EP2014/066755 (entitled “Novel immunotherapy against several tumors, such as lung cancer, including NSCLC”), disclosing subject matter coinvented by J.F., C. Song, H. Singh, S.W., and T.W.; PCT/EP2016/055817 (entitled “Novel peptides and combination of peptides for use in immunotherapy against pancreatic cancer and other cancers”), disclosing subject matter coinvented by J.F., A.M., M.O., H. Singh, O.S., C.W., and T.W.; PCT/EP2016/059053 (entitled “Novel peptides and combination of peptides for use in immunotherapy against lung cancer, including NSCLC and other cancers”), disclosing subject matter coinvented by J.F., A.M., C. Song, H. Singh, O.S., C.W., and T.W.; PCT/EP2016/060007 [entitled “Novel peptides and combination of peptides and scaffolds thereof for use in immunotherapy against colorectal carcinoma (CRC) and other cancers”], disclosing subject matter coinvented by J.F., A.M., H. Singh, O.S., and T.W.; and PCT/EP2016/079059 (entitled “Peptides and combination of peptides for use in immunotherapy against breast cancer and other cancers”), disclosing subject matter coinvented by J.F., A.M., H. Singh, O.S., and T.W.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. In response to reasonable requests, noncommercially available materials and experimental protocols that Immatics or J.L.R. has the right to provide will be made available to not-for-profit or academic requesters upon completion of a material transfer agreement. Requests may be made by contacting a corresponding author.
REFERENCES AND NOTES
- 1.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, Wasik M, Levine BL, Lacey SF, Melenhorst JJ, Porter DL, June CH, Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med 377, 2545–2554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guedan S, Ruella M, June CH, Emerging cellular therapies for cancer. Annu. Rev. Immunol 37, 145–171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majzner RG, Mackall CL, Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 8, 1219–1226 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Schietinger A, Philip M, Schreiber H, Specificity in cancer immunotherapy. Semin. Immunol 20, 276–285 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson KG, Stromnes IM, Greenberg PD, Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 31, 311–325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jhunjhunwala S, Hammer C, Delamarre L, Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 21, 298–312 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto TN, Kishton RJ, Restifo NP, Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med 25, 1488–1499 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Fritsche J, Roszik J, Williams LJ, Peng X, Chiu Y, Tsou CC, Hoffgaard F, Goldfinger V, Schoor O, Talukder A, Forget MA, Haymaker C, Bernatchez C, Han L, Tsang YH, Kong K, Xu X, Scott KL, Singh-Jasuja H, Lizee G, Liang H, Weinschenk T, Mills GB, Hwu P, RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun 9, 3919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahles A, Lehmann KV, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C; Cancer Genome Atlas Research Network, Rätsch G, Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 34, 211–224.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L, Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 11.The problem with neoantigen prediction. Nat. Biotechnol 35, 97 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V, MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst 105, 1172–1187 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Kubiniok P, Marcu A, Bichmann L, Kuchenbecker L, Schuster H, Hamelin DJ, Duquette JD, Kovalchik KA, Wessling L, Kohlbacher O, Rammensee HG, Neidert MC, Sirois I, Caron E, Understanding the constitutive presentation of MHC class I immunopeptidomes in primary tissues. iScience 25, 103768 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM, Direct observation of ligand recognition by T cells. Nature 419, 845–849 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Murciano-Goroff YR, Warner AB, Wolchok JD, The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 30, 507–519 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Junttila MR, de Sauvage FJ, Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L, Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol 27, 1482–1492 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Pure E, Blomberg R, Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 37, 4343–4357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, Solomides CC, June CH, Pure E, Albelda SM, Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res 2, 154–166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, Espeli M, Kraman M, McKenna B, Wells RJ, Zhao Q, Caballero OL, Larder R, Coll AP, O’Rahilly S, Brindle KM, Teichmann SA, Tuveson DA, Fearon DT, Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med 210, 1137–1151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinschenk T, Gouttefangeas C, Schirle M, Obermayr F, Walter S, Schoor O, Kurek R, Loeser W, Bichler KH, Wernet D, Stevanovic S, Rammensee HG, Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer Res. 62, 5818–5827 (2002). [PubMed] [Google Scholar]
- 22.Fritsche J, Kowalewski DJ, Backert L, Gwinner F, Dorner S, Priemer M, Tsou CC, Hoffgaard F, Romer M, Schuster H, Schoor O, Weinschenk T, Pitfalls in HLA ligandomics-how to catch a Li(e)gand. Mol. Cell. Proteomics 20, 100110 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Pan W, The biological role of the collagen alpha-3 (VI) chain and its cleaved C5 domain fragment endotrophin in cancer. Onco. Targets Ther 13, 5779–5793 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorsen K, Sorensen KD, Brems-Eskildsen AS, Modin C, Gaustadnes M, Hein AM, Kruhoffer M, Laurberg S, Borre M, Wang K, Brunak S, Krainer AR, Torring N, Dyrskjot L, Andersen CL, Orntoft TF, Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Mol. Cell. Proteomics 7, 1214–1224 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y, rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U.S.A 111, E5593–E5601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fritsche J, Rakitsch B, Hoffgaard F, Romer M, Schuster H, Kowalewski DJ, Priemer M, Stos-Zweifel V, Horzer H, Satelli A, Sonntag A, Goldfinger V, Song C, Mahr A, Ott M, Schoor O, Weinschenk T, Translating immunopeptidomics to immunotherapy-decision-making for patient and personalized target selection. Proteomics 18, e1700284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, Jakobsen BK, Different affinity windows for virus and cancer-specific T-cell receptors: Implications for therapeutic strategies. Eur. J. Immunol 42, 3174–3179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, Milicic A, Mahon T, Sutton DH, Laugel B, Moysey R, Cameron BJ, Vuidepot A, Purbhoo MA, Cole DK, Phillips RE, June CH, Jakobsen BK, Sewell AK, Riley JL, Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat. Med 14, 1390–1395 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medvec AR, Ecker C, Kong H, Winters EA, Glover J, Varela-Rohena A, Riley JL, Improved expansion and in vivo function of patient T cells by a serum-free medium. Mol. Ther. Methods Clin. Dev 8, 65–74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zoete V, Irving M, Ferber M, Cuendet MA, Michielin O, Structure-based, rational design of T cell receptors. Front. Immunol 4, 268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Presotto D, Erdes E, Duong MN, Allard M, Regamey PO, Quadroni M, Doucey MA, Rufer N, Hebeisen M, Fine-tuning of optimal TCR signaling in tumor-redirected CD8 T cells by distinct TCR affinity-mediated mechanisms. Front. Immunol 8, 1564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone JD, Kranz DM, Role of T cell receptor affinity in the efficacy and specificity of adoptive T cell therapies. Front. Immunol 4, 244 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, Dolton G, Clement M, Llewellyn-Lacey S, Price DA, Peakman M, Sewell AK, A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem 287, 1168–1177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, Bossi G, Vuidepot A, Powlesland AS, Legg A, Adams KJ, Bennett AD, Pumphrey NJ, Williams DD, Binder-Scholl G, Kulikovskaya I, Levine BL, Riley JL, Varela-Rohena A, Stadtmauer EA, Rapoport AP, Linette GP, June CH, Hassan NJ, Kalos M, Jakobsen BK, Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med 5, 197ra103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinnunen T, Buhot C, Narvanen A, Rytkonen-Nissinen M, Saarelainen S, Pouvelle-Moratille S, Rautiainen J, Taivainen A, Maillere B, Mantyjarvi R, Virtanen T, The immunodominant epitope of lipocalin allergen Bos d 2 is suboptimal for human T cells. Eur. J. Immunol 33, 1717–1726 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Kim Y, Sidney J, Pinilla C, Sette A, Peters B, Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics 10, 394 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanahan D, Weinberg RA, Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Engels B, Rowley DA, Schreiber H, Targeting stroma to treat cancers. Semin. Cancer Biol 22, 41–49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu R, Li H, Liu L, Yu J, Ren X, Fibroblast activation protein. Cancer Biol. Ther 13, 123–129 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Qiao J, Fang CY, Chen SX, Wang XQ, Cui SJ, Liu XH, Jiang YH, Wang J, Zhang Y, Yang PY, Liu F, Stroma derived COL6A3 is a potential prognosis marker of colorectal carcinoma revealed by quantitative proteomics. Oncotarget 6, 29929–29946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arafat H, Lazar M, Salem K, Chipitsyna G, Gong Q, Pan TC, Zhang RZ, Yeo CJ, Chu ML, Tumor-specific expression and alternative splicing of the COL6A3 gene in pancreatic cancer. Surgery 150, 306–315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsimberidou A, Guenther K, Alpert A, Andersson B, Coughlin Z, Fritsche J, Hilf N, Hwu P, Kalra M, Kuttruff-Coqui S, Maurer D, Mendrzyk R, Mohamed A, Norris B, Nowak A, Ort R, Reinhardt C, Richter F, Satelli A, Schoor O, Sieger K, Singh H, Vining D, Wagner C, Weinschenk T, Yee C, Walter S, 293 Results of the first-in-human clinical trial with personalized multi-target adoptive cell therapy (ACTolog IMA101). J. Immunother. Cancer 8, A179 (2020). [Google Scholar]
- 43.Dolgin E, First soluble TCR therapy opens “new universe” of cancer targets. Nat. Biotechnol 40, 441–444 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH, Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zanussi S, Doliana R, Segat D, Bonaldo P, Colombatti A, The human type VI collagen gene. mRNA and protein variants of the alpha 3 chain generated by alternative splicing of an additional 5-end exon. J. Biol. Chem 267, 24082–24089 (1992). [PubMed] [Google Scholar]
- 46.Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N, Identifying individual T cell receptors of optimal avidity for tumor antigens. Front. Immunol 6, 582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Border EC, Sanderson JP, Weissensteiner T, Gerry AB, Pumphrey NJ, Affinity-enhanced T-cell receptors for adoptive T-cell therapy targeting MAGE-A10: Strategy for selection of an optimal candidate. Onco. Targets Ther 8, e1532759 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, Pumphrey NJ, Jakobsen BK, Price DA, Ladell K, Sewell AK, T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin. Exp. Immunol 180, 255–270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, McGary K, Huang K, Boyer J, Corse E, Shao Y, Rosenberg SA, Restifo NP, Osman I, Krogsgaard M, T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc. Natl. Acad. Sci. U.S.A 110, 6973–6978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, Carroll RG, Riley JL, June CH, Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol. Ther 15, 981–988 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pu X, Zhang R, Wang L, Chen Y, Xu Y, Pataer A, Meraz IM, Zhang X, Wu S, Wu L, Su D, Mao W, Heymach JV, Roth JA, Swisher SG, Fang B, Patient-derived tumor immune microenvironments in patient-derived xenografts of lung cancer. J. Transl. Med 16, 328 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maldini CR, Gayout K, Leibman RS, Dopkin DL, Mills JP, Shan X, Glover JA, Riley JL, HIV-resistant and HIV-specific CAR-modified CD4+ T cells mitigate HIV disease progression and confer CD4+ T cell help in vivo. Mol. Ther 28, 1585–1599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG, Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351, 290–296 (1991). [DOI] [PubMed] [Google Scholar]
- 54.Walter S, Herrgen L, Schoor O, Jung G, Wernet D, Bühring H-J, Rammensee H-G, Stevanović S, Cutting edge: Predetermined avidity of human CD8 T cells expanded on calibrated MHC/anti-CD28-coated microspheres. J. Immunol 171, 4974–4978 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Stone JD, Chervin AS, Aggen DH, Kranz DM, T cell receptor engineering. Methods Enzymol. 503, 189–222 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Leibman RS, Richardson MW, Ellebrecht CT, Maldini CR, Glover JA, Secreto AJ, Kulikovskaya I, Lacey SF, Akkina SR, Yi Y, Shaheen F, Wang J, Dufendach KA, Holmes MC, Collman RG, Payne AS, Riley JL, Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLOS Pathog. 13, e1006613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richardson MW, Carroll RG, Stremlau M, Korokhov N, Humeau LM, Silvestri G, Sodroski J, Riley JL, Mode of transmission affects the sensitivity of human immunodeficiency virus type 1 to restriction by rhesus TRIM5α. J. Virol 82, 11117–11128 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garboczi DN, Hung DT, Wiley DC, HLA-A2-peptide complexes: Refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. U.S.A 89, 3429–3433 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lissina A, Ladell K, Skowera A, Clement M, Edwards E, Seggewiss R, van den Berg HA, Gostick E, Gallagher K, Jones E, Melenhorst JJ, Godkin AJ, Peakman M, Price DA, Sewell AK, Wooldridge L, Protein kinase inhibitors substantially improve the physical detection of T-cells with peptide-MHC tetramers. J. Immunol. Methods 340, 11–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ecker C, Guo L, Voicu S, Gil-de-Gomez L, Medvec A, Cortina L, Pajda J, Andolina M, Torres-Castillo M, Donato JL, Mansour S, Zynda ER, Lin PY, Varela-Rohena A, Blair IA, Riley JL, Differential reliance on lipid metabolism as a salvage pathway underlies functional differences of T cell subsets in poor nutrient environments. Cell Rep. 23, 741–755 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benjamini Y, Hochberg Y, Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. In response to reasonable requests, noncommercially available materials and experimental protocols that Immatics or J.L.R. has the right to provide will be made available to not-for-profit or academic requesters upon completion of a material transfer agreement. Requests may be made by contacting a corresponding author.