

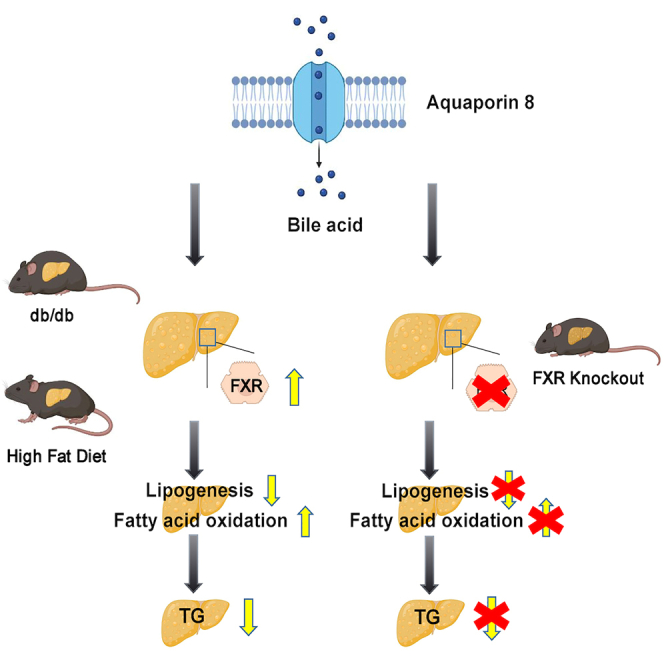
Summary
Aquaporin-8(AQP8), is a transmembrane channel protein that abounds in liver, which mainly promotes water transport, modulating bile acid formation. However, its role in hepatic lipid metabolism remains unclear. In this study, we found the expression of AQP8 was reduced in liver specimens of patients with NAFLD, high-fat diet (HFD)-induced mice and genetically obese db/db mice. Knockdown of AQP8 in hepatocytes exacerbated the intracellular lipid accumulation induced by free fatty acid (FFA) mixtures. In contrast, hepatic AQP8 overexpression activated farnesoid X receptor (FXR), inhibiting gene expression associated with lipogenesis, which further reduced intrahepatic triglyceride overload in obese mice. FXR knockout abrogated the ameliorating effect of AQP8 overexpression on NAFLD in mice. These findings indicate that AQP8 overexpression protects against fatty liver through activating the FXR pathway.
Subject areas: Molecular biology, Endocrinology, Cell biology
Graphical abstract
Highlights
-
•
Hepatic AQP8 expression is decreased in patients with NAFLD and obese mice
-
•
Knockdown of AQP8 in hepatocytes exacerbates lipid accumulation
-
•
Hepatic AQP8 overexpression improves hepatosteatosis in db/db and HFD mice
-
•
AQP8 regulates hepatic lipid metabolism in an FXR depedent manner
Molecular biology; Endocrinology; Cell biology
Introduction
Non-alcoholic fatty liver disease (NAFLD), which is characterized by an abnormal hepatic accumulation of triglycerides (TGs), has become a major threat to human health affecting 25% of general population worldwide.1 The clinical pathological spectrum of NAFLD ranges from simple hepatocellular fat deposits to non-alcoholic steatohepatitis and further progression toward cirrhosis.2 Ectopic fat accumulation in the liver is accompanied by insulin resistance, hypertension, and type 2 diabetes mellitus, and NAFLD is recognized as the hepatic component of a cluster of conditions that are associated with metabolic disorders.3,4 Currently, specific therapies for this disease are limited to lifestyle modifications, such as exercising and changing dietary habits to achieve weight loss.5 Consequently, further studies are needed to identify the key molecular regulators of NAFLD development.
Mammalian aquaporins (AQPs) are a family of 13 integral membrane proteins, functionally divided into three typical classes: classical AQPs (AQP0, AQP1, AQP2, AQP4, AQP5, AQP6, and AQP8), which function as water-selective transporters; aquaglyceroporins (AQP3, AQP7, AQP9, and AQP10), which transport water, glycerol as well as other small solutes; and super AQPs (AQP11 and AQP12).3,6 The expression of aquaglyceroporins has been documented in numerous tissue types, including adipose tissue, liver, and pancreas, in which they are reported to play important metabolic roles.6,7,8,9
AQP8, a member of the family of transmembrane channel proteins, appears to be the most abundant AQP in hepatocytes,10,11 in which it not only facilitates the passive transport of water across the plasma membrane but also shows high permeability to urea, ammonia, and hydrogen peroxide (H2O2).12,13 It is located in the canalicular membranes of hepatocytes and functions as a rate-limiting step for bile secretion by promoting water secretion through the canalicular membrane.7,14,15 Knockdown of AQP8 has been found to impair basal canalicular water movement, leading to approximately 70% decline in canalicular volume. AQP8 has also been shown to be localized in intracellular vesicles16,17 and exists as a non-glycosylated protein in the mitochondria.18 Glucagon can regulate AQP8 by transporting vesicles to the hepatocellular membrane via cAMP-protein kinase A (PKA), and this process is involved in phosphatidylinositol 3-kinase (PI3K) signaling pathways and microtubule-associated proteins, together with an upregulation of AQP8 expression, which in turn contribute to a significant increase in hepatocellular water permeability.19 Given its efficient transport of ammonia and H2O2, AQP8 plays an important role in mitochondrial ammonia detoxification via ureagenesis and H2O2-mediated cell signaling.20 In addition, in hepatocellular mitochondrial membranes, AQP8 interacts with stearoyl-CoA response element binding proteins (SREBPs), mediating the regulation of hepatocyte cholesterol biosynthesis.21 Nonetheless, it is yet to be determined whether AQP8 has beneficial effects on NAFLD.
In this study, we detected reduced levels of hepatic AQP8 expression in both human patients with NAFLD and obese mice. By using db/db obese mice and mice fed an high-fat diet (HFD), both of which have previously been shown to mimic liver steatosis seen in humans, as well as in vitro NAFLD models, we examined whether AQP8 can inhibit the pathogenesis of NAFLD both in vivo and in vitro. We also assessed whether AQP8 plays a role in regulating de novo lipogenesis to ameliorate hepatosteatosis via a farnesoid X receptor (FXR)-dependent pathway. All these findings highlight the role of AQP8 as a potential target for the treatment of NAFLD.
Results
AQP8 expression is downregulated in the livers of patients with NAFLD and obese mice
Compared to other tissues in mice, the level of AQP8 mRNA is dramatically elevated in the liver (Figure S1A). To characterize the involvement of AQP8 in the pathogenesis of NAFLD, we examined the levels of AQP8 expression in the livers of human subjects with NAFLD through AQP8 immunohistochemistry, gene and protein expression, which indicated reductions (Figures 1A and S1B). We subsequently examined the expression of AQP8 in the livers of db/db mice and C57BL/6J mice fed a long-term HFD for 8 weeks. Among the obese mice, we also detected the reduction in levels of AQP8 (Figures 1B, 1C, S1C, and S1D). We also fed mice a short-term HFD and treated HepG2 cells with 1 mM FFA mixtures at different times to mimic hepatic steatosis. Compared with the respective control groups, exposure to HFD for 7 days and treatment with FFA mixtures for 12 h were found to promote significant reductions in AQP8 mRNA (Figures 1D and 1E) and protein expression (Figures S2A andS2B). These findings indicate that in both humans and rodents, the expression of AQP8 is consistently attenuated under conditions of hepatic steatosis.
Figure 1.

Downregulation of AQP8 in the livers of NAFLD patients and obese mice and effects of hepatic AQP8 overexpression in vitro
Hepatic mRNA and protein levels of AQP8 decreased in patients and obese mouse model: (A) NAFLD patients (n = 8 per group), (B) db/db mice (n = 6 per group), (C) high fat diet (HFD) mice (n = 7 per group), (D) short-term HFD mice (n = 5 per group) (See also Figure S2A), (E) HepG2 cells without or with FFA(OA:PA = 2:1) treatment for the indicated time(See also Figure S2B). Primary mouse hepatocytes (PMHs) and HepG2 cells were transfected with either ad-GFP or ad-AQP8 with or without FFA treatment.
(F) AQP8 mRNA expression of PMHs.
(G) Analysis of AQP8 and lipogenic genes expression (See also Figure S2C).
(H) Oil red O staining (200 X) of HepG2 cells.
(I) TG measurement of PMHs. The data represent the mean ± SEM values. ∗p < 0.05 ∗∗p < 0.01 ∗∗∗p < 0.001.
To further clarify the function of AQP8 in an in vitro model of NAFLD, we infected primary mouse hepatocytes (PMHs) and HepG2 cells with either Ad-AQP8 or Ad-GFP, and these cells were also exposed to FFA mixtures. Subsequent RT–PCR analyses revealed that AQP8 gene expression was significantly increased in PMHs in response to overexpression of AQP8 (Figure 1F). In hepatocytes, although lipogenic genes were found to be enhanced by FFA treatment, these genes were inhibited by the overexpression of AQP8 (Figures 1G and S2C). Consistently, we observed an exacerbation of lipid accumulation in the HepG2 hepatocytes treated with FFA mixtures, whereas accumulation was attenuated in cells overexpressing hepatic AQP8 as revealed by Oil Red O staining (Figure 1H). Measurements of intracellular TG levels in PMHs also revealed that in response to FFA mixtures administration, there was a significant increase in TG content, whereas TG levels were reduced following the overexpression of AQP8 compared with that in the Ad-GFP group (Figure 1I). In contrast, lipid contents in hepatocytes co-treated with Ad-shAQP8 and FFA overload were exacerbated (Figure S3).
Hepatic overexpression of AQP8 attenuates liver steatosis in db/db mice
To further examine the effects of AQP8 overexpression on hepatic lipid metabolism in vivo, we employed db/db mice and WT mice infected with either Ad-AQP8 or vehicle Ad-GFP. Analysis of gene expression revealed a marked increase in the expression of AQP8 in the livers of db/db and WT mice with adenoviral AQP8 overexpression (Figure 2A). Hepatic AQP8 overexpression had little effect on body weight or food intake (Figures 2B and 2C). However, in Ad-AQP8 mice, we observed reductions in fasting blood glucose levels (Figure 2D). In addition, we detected reductions in the serum levels of TG (Figure 2E), TC (Figure 2F), LDL-c (Figure 2G), and total bile acids (Figure 2H), whereas serum levels of β-hydroxybutyrate (β-OHB) were elevated (Figure 2I).
Figure 2.

Effects of AQP8 on body weight and lipid metabolism in db/db mice
db/db mice were i.v. injected with either ad-GFP or ad-AQP8 and mice were sacrificed at day 10.
(A) Relative AQP8 mRNA levels in liver tissue samples.
(B) Body weight of the four groups were monitored for 10 days.
(C) Food intake of the four groups were monitored for 10 days.
(D–I) Serum levels of FBG, TG, TC, LDL-c, TBA and β-OHB. The data represent the mean ± SEM values, n = 8–11. ∗p<0.05 ∗∗p<0.01 ∗∗∗p<0.001.
Hematoxylin-eosin (H&E) staining revealed prominent hepatosteatosis in the livers of db/db mice with Ad-GFP, whereas in db/db mice with Ad-AQP8 there was a pronounced reduction in the numbers of lipid droplets (Figure 3A). In parallel, there were reductions in the ratio of liver weight to body weight (LW/BW) of db/db and WT mice with Ad-AQP8 and also reductions in TG contents in the livers of db/db mice (Figures 3B and 3C). De novo lipogenesis plays a vital role in the pathogenesis of NAFLD,22 and in the present study, we performed RT–PCR to examine the expression of genes related to lipogenesis, fatty acid oxidation, bile acid metabolism, and inflammation. The expression of genes and proteins associated lipogenesis, namely, FAS, SREBP-1c, and ChREBP, were significantly inhibited by hepatic overexpression of AQP8 in db/db and WT mice (Figures 3D, 3H, and 3I).
Figure 3.

Histological and molecular changes in the livers of db/db mice with AQP8 overexpression
(A) Histological analysis of hematoxylin-eosin-stained liver sections, magnification: 200×, scale bar, 100 μm.
(B) The analysis of liver weight in four groups. The count is liver weight per kilogram of body weight.
(C) Quantification of hepatic TG contents.
(D–G) Analysis of mRNA levels on hepatic lipogenesis, fatty acid oxidation, bile acid metabolism and inflammation-related genes; β-actin was used as an internal control.
(H and I) Western blot assays analysis of the accumulation of AQP8 and lipogenic genes in the liver tissue sample of wildtype littermates and db/db mice. The data represent the mean ± SEM values, n = 8–11. ∗p<0.05 ∗∗p<0.01 ∗∗∗p<0.001.
The steady-state balance of hepatic TGs is linked to the consumption of fatty acids via mitochondrial β-oxidation,23 and in db/db mice with hepatic overexpression of AQP8, we detected increases in the expression of key genes associated with fatty acid oxidation (Figure 3E). As shown in Figure 3F, the mRNA expression of hepatic FXR, SHP, and BSEP was significantly increased in livers overexpressing AQP8. To identify potential FXR agonists and antagonists, and we used ultra-high performance liquid chromatography–tandem mass spectrometry to quantify serum bile acids from db/db mice. As our result showed that, AQP8 overexpression altered the bile acid profile with elevated cholic acid (CA), chenodeoxycholic acid (CDCA) (Figure S4), which have been identified as effective ligands for FXR.24,25 In addition, we also observed deregulation of the bile acid synthesis gene Cyp7a1 which inhibited by FXR (Figure 3F), as well as reductions in the expression levels of genes associated with inflammation, namely, CCL7, CCL3, IL10, and TNF-α in mice with AQP8 overexpression (Figure 3G).
Hepatic overexpression of AQP8 improves HFD-induced liver steatosis
The HFD mouse model has been used extensively to mimic NAFLD,26 and in the present study, we evaluated the effects of hepatic overexpression of AQP8 on lipid changes attributable to consumption of HFD. To confirm the overexpression efficiency of mice, we performed RT–PCR and western blot analyses of AQP8 and accordingly detected marked increases in AQP8 gene expression levels in the livers of Ad-AQP8 group mice relative to those in Ad-GFP group mice (Figure 4A). Neither body weight nor food intake was affected by the hepatic overexpression of AQP8 (Figures 4B and 4C), whereas we detected reductions in the serum levels of TG, TC, LDL-c, and total bile acids (Figures 4D–4G) and an increase in β-OHB (Figure 4H) in the Ad-AQP8 groups.
Figure 4.

Effects of AQP8 on body weight and lipid metabolism in HFD mice
HFD mice were i.v. injected with either ad-GFP or ad-AQP8 and mice were sacrificed at day 10.
(A) Relative AQP8 mRNA levels in liver tissue samples.
(B) Body weight of the four groups were monitored for 10 days.
(C) Food intake of the four groups were monitored for 10 days.
(D–H) Serum levels of TG, TC, LDL-c, TBA and β-OHB. The data represent the mean ± SEM values, n = 8–10. ∗p<0.05 ∗∗p<0.01 ∗∗∗p<0.001.
HFD mice with hepatic overexpression of AQP8 were characterized by a more pronounced attenuation of hepatic steatosis (as revealed by H&E staining) (Figure 5A), with reductions in the LW/BW ratio (Figure 5B) and hepatic TG contents (Figure 5C). RT–PCR performed to examine the expression of genes associated with lipogenesis (FAS, SREBP-1c, and ChREBP) revealed that the expression of genes and proteins was downregulated in response to the hepatic overexpression of AQP8 (Figures 5D, 5H, and 5I). Hepatic AQP8 overexpression was also found to promote an increase in the oxidation of fatty acids in HFD and NCD mice, as indicated by increases in the expression levels of the fatty acid oxidation-related genes Acox1, FATP1, LCAD, and PPARα (Figure 5E). Overexpression of AQP8 also promoted the upregulated expression of genes associated with bile acid metabolism (FXR, SHP, and BSEP) and inhibited the expression of Cyp7a1 mRNA (Figure 5F). In line with expectations, we found that consumption of HFD contributed to a significant increase in the expression of inflammation-related genes in liver tissues, whereas the hepatic overexpression of AQP8 prevented increases in the expression of inflammatory genes such as CCL5, CCL8, IL1β, and TNF-α (Figure 5G).
Figure 5.

Histological and molecular changes in the livers of HFD mice with AQP8 overexpression
(A) Histological analysis of hematoxylin-eosin-stained liver sections, magnification: 200×, scale bar, 100 μm.
(B) The analysis of liver weight in four groups. The count is liver weight per kilogram of body weight.
(C) Quantification of hepatic TG contents.
(D–G), Analysis of mRNA levels on hepatic lipogenesis, fatty acid oxidation, bile acid metabolism and inflammation-related genes; β-actin was used as an internal control.
(H and I) Western blot assays analysis of the levels of AQP8 and lipogenic genes in the liver tissue sample of NCD and HFD mice. The data represent the mean ± SEM values, n = 8–10. ∗p<0.05 ∗∗p<0.01 ∗∗∗p<0.001.
Taken together, these findings indicate that hepatic overexpression of AQP8 contributes to the regulation of hepatic TG metabolism and mitigates NAFLD development by suppressing the liver lipogenic program.
FXR is required for overexpressed AQP8-mediated hepatic steatosis improvement
FXR/Nr1h4, a potent bile acid nuclear receptor, is highly expressed in the liver and exerts important effects on the regulation of bile acids, lipids, cholesterol, and carbohydrate metabolism.27,28,29,30 Activation of FXR by the specific agonist GW4064 was observed to promote the upregulated expression of AQP8 mRNA in vitro, whereas other specific agonists had no significant effects (Figure 6A). To obtain further experimental evidence indicating that AQP8 is upregulated by FXR activation at transcriptional level, we subsequently investigated the effects of the FXR-specific agonist GW4064 on AQP8 expression using dual-luciferase reporter assays. Activation of FXR by GW 4064 was found to induce high levels of AQP8 activity (Figure 6B). To confirm the role of FXR in the regulation of AQP8 expression, we performed FXRE mutagenesis31 and chromatin immunoprecipitation (ChIP) assay. Using the online software JARSPAR (https://jaspar.genereg.net/matrix/MA1110.1/), we predicted the putative FXR Response Elements (FXREs) in the promoter region of AQP8, on the basis of which, we cloned the human AQP8 gene promoter (containing the fragment −1500 to +100) in the pGL4.14-luciferase reporter plasmid (Figure S5A), which was used to transfect HepG2 cells with or without an FXR overexpression plasmid. Consistent with our finding regarding the FXR-specific agonist GW4064, we observed that AQP8 promoter activity was also enhanced by FXR overexpression (Figure 6C), thereby indicating that this region may harbor a regulatory element. Mutation of FXREs within the construct pGL4.14-AQP8 was found to abolish the luciferase activity induced by the FXR overexpression plasmid (Figures 6C and S5B), confirming that FXR enhances the transcriptional activity of AQP8 promoter. Moreover, the ChIP assay demonstrated that the anti-FXR antibody precipitated the DNA fragment containing the FXREs, indicating that FXR binds directly to the FXREs of the AQP8 promoter region in HepG2 cells (Figure 6D). In contrast, we found that knockdown of the FXR gene in mice resulted in a downregulation of hepatic AQP8 expression (Figure 6E). Accordingly, it is reasonable to assume that if the improvement of hepatic steatosis in response to the overexpression of AQP8 is dependent mainly on FXR, AQP8 overexpression would no longer be effective if FXR is depleted. To verify this assumption, we used FXR-KO mice to investigate whether blocking FXR could reverse the benefits of AQP8 overexpression. To this end, FXR-KO and FXR-WT mice were fed an HFD for 2 weeks and then intravenously injected with either Ad-GFP or Ad-AQP8 for 10 days. We accordingly detected increases in the mRNA level of AQP8 in FXR-KO mice with hepatic overexpression of AQP8 (Figure 6F), although there were no significant reductions in total bile acid levels in these mice (Figure 6G). Furthermore, overexpression of AQP8 in the livers of FXR-KO mice had no effect on serum levels of TG, TC, or LDL-c (Figures 6H–6J). Moreover, whereas a reduction in LW/BW values was observed in db/db and HFD mice with Ad-AQP8 (Figures 3B and 5B), these effects were reversed in FXR-KO mice (Figure 6K). Similarly, despite infection with Ad-AQP8, we detected increased hepatic TG contents and lipid droplets in FXR-KO mice (Figures 6L and 6M).
Figure 6.

FXR regulates AQP8 expression and FXR knockout inhibits the improvement of AQP8 in NAFLD
(A) PMHs were treated with DMSO, PPARα agonist Fenofibrate, PPARβ/δ agonist GW0742, PPARγ agonist Rosiglitazone, FXR agonist GW4064, LXR agonist T0901317 and RT–PCR analysis of AQP8 gene expression.
(B) Dual-luciferase reporter assay was conducted to determine the effect of FXR agonist GW4064 on AQP8 activity.
(C) Dual-luciferase reporter assay was conducted to determine the effect of FXR overexpression on AQP8 activity.
(D) The interaction of FXR with AQP8 promoter containing putative FXRE was confirmed by chromatin immunoprecipitation (ChIP) in HepG2 cells (n = 3 individual experiments) using an FXR antibody, IgG was used as a negative control.
(E) Relative AQP8 mRNA levels in the livers of FXR wild-type (WT) and knockout (KO) mice.
(F) FXR-WT and FXR-KO were fed an HFD for two weeks and then i.v injected with either ad-GFP or ad-AQP8 and relative AQP8 mRNA levels in the livers of three groups.
(G–J) Serum levels of TBA, TG, TC, LDL-c.
(K) The analysis of liver weight in three groups. The count is liver weight per kilogram of body weight.
(L) Quantification of hepatic TG contents.
(M) Histological analysis of hematoxylin-eosin-stained liver sections, magnification: 200×, scale bar, 100 μm. The data represent the mean ± SEM values. ∗p<0.05 ∗∗p<0.01 ∗∗∗p<0.001.
Analysis of the levels of lipogenic genes and proteins in liver sections obtained from FXR-KO mice with Ad-AQP8 revealed no significant downregulation (Figures 7A and 7E). In addition, we demonstrated that hepatic overexpression of AQP8 promoted increases in the expression of genes associated with fatty acid oxidation, but these changes were reversed by FXR knockout (Figure 7B). Furthermore, we found that overexpression of AQP8 in FXR knockout mice had no further effect on the expression of genes associated with bile acid metabolism such as SHP and BSEP (Figure 7C). Knockout of FXR was also found to promote increases in the expression of inflammation-related genes (Figure 7D).
Figure 7.

Molecular changes in the liver of FXR knockout mice with AQP8 overexpression
(A–D) Analysis of mRNA on hepatic lipogenesis, fatty acid oxidation, bile acid metabolism and inflammation-related genes; β-actin was used as an internal control.
(E) Western blot assays analysis of the accumulation of AQP8 and lipogenic genes in the liver tissue samples of the three groups. (p)SREBP-1c, precursors SREBP-1c; (n)SREBP-1c, nuclear SREBP-1c. The data represent the mean ± SEM values, n = 9–11.∗p<0.05 ∗∗p<0.01 ∗∗∗p<0.001.
Discussion
Given recent increases in the incidence and prevalence of NAFLD, this disease has become a global health issue of major concern.32 It is also becoming the leading cause of chronic liver disease, and despite considerable efforts to identify therapeutic targets, there is as yet no established therapy for this disease.33 In this study, we sought to characterize a member of the family of mammalian AQPs, AQP8, which facilitates the passage of water and other small solutes, such as hydrogen peroxide and ammonia, across cell membranes.9,34 To date, only a small number of classical AQPs have been established to play roles in the regulation of metabolic disease development and pathology6,8,35,36, However, whether AQP8 also plays a role in metabolic diseases has yet to be determined.
Our findings in this study revealed reduced levels of AQP8 in the livers of NAFLD patients and obese animal models, providing clinical and basic pathological insights into the association between AQP8 expression and hepatic steatosis. We therefore performed adenovirus-mediated overexpression or knockdown of AQP8 in hepatocytes and mouse models to examine whether AQP8 plays a role in NAFLD development. Under physiological conditions, the steady-state concentrations of intrahepatic TGs are low, and the liver does not serve as a storage depot for fat.22 The main cause of hepatic lipid perturbation during NAFLD is the excessive accumulation of TG,37 and in the present study, we observed increases in TG levels and an exacerbation of hepatic steatosis in mice fed an HFD and those with spontaneous genetic obesity. However, in both HFD and db/db mice, hepatic overexpression of AQP8 was found to contribute to a reduction of TG content and mitigation of hepatic steatosis.
Accumulating evidence indicates that hepatic lipid homeostasis is tightly controlled by de novo fatty acid synthesis, oxidation, lipoprotein uptake, and secretion,38,39 and it has been established that NAFLD development is driven by a dysfunction in lipogenesis or fatty acid oxidation.40 Consequently, alleviating hepatic lipid accumulation by effectively inhibiting lipogenesis and promoting fatty acid oxidation could represent promising approaches for the treatment of NAFLD.40 Moreover, lipogenesis is primarily controlled at the transcriptional level,41,42 and it has been established that downregulation of the expression of SREBP-1c, a vital transcriptional regulator of the enzymes involved in de novo lipogenesis, and ChREBP, which induces the expression of enzymes involved in de novo lipogenesis, can contribute to the treatment of NAFLD. In the present study, we found that hepatic overexpression of AQP8 promoted the downregulated expression of genes involved in lipogenesis, such as SREBP-1c, FAS, and ChREBP, and markedly enhanced the expression of fatty acid oxidation-associated genes in HFD and db/db mice. In addition, overexpression of AQP8 reduced abnormal lipid accumulation in mouse primary hepatocytes, despite FFA overload. In contrast, suppression of AQP8 triggered lipogenic gene expression and lipid deposition. The findings of both our in vivo and in vitro studies provide evidence in support of the hypothesis that AQP8 may improve hepatic steatosis by regulating lipogenesis.
With respect to hepatic steatosis intervention, bile acids, their receptors (such as FXR and TGR5), and transporters (such as ASBT, the ileal apical sodium-dependent bile acid transporter) have emerged as potential therapeutic targets.43 The bile acid/FXR-mediated pathways may induce SHP synthesis, which further suppresses the transcriptional function of SREBP-1c, leading to the suppression of lipogenic gene expression.44 In addition, the effect of activated FXR in enterocytes is mediated by fibroblast growth factor 15 (FGF15, mouse) or FGF19 (human), which in turn activates the hepatic FGF receptor to repress hepatic lipogenesis and stimulate fatty acid β-oxidation.44 As a water channel, AQP8 contributes to the secretion of water from hepatocytes into the bile canalicular lumen,12,14 leading to bile dilution, whereas a reduction in AQP8 expression may contribute to cholestasis.45 In the present study, although available data showed that adenovirus-mediated hepatic overexpression of AQP8 in db/db and HFD mice promoted a reduction in the levels of serum bile acids and mitigated hepatic steatosis, the evidence about bile acid levels in liver, gall bladder, and intestine which is not provided may further demonstrate the effect of AQP8 on BA metabolism and hepatic steatosis.
Recent findings have highlighted that intestinal lipid absorption is another important pathway mediating hepatic triglyceride metabolism.46,47,48 Activation of FXR has been established alter the composition of bile acid pools with or without affecting the overall pool size and reducing intestinal lipid absorption.46 AQP8 acts as an important water channel in hepatocyte bile formation49 and intestine,50 and consequently, we should not exclude the possibility that the overexpression of AQP8 potentially activates FXR and further reduces lipid absorption via an alteration in bile acid composition or pool size. At molecular levels, it is speculated that the overexpression of AQP8 contributes to increased expression of FXR and FXR target genes such as SHP, and in vitro, we also found that AQP8 can be activated by FXR. In contrast, the knockdown of FXR in mice was found to abrogate the beneficial effects of hepatic AQP8 overexpression on TG accumulation, providing compelling evidence to indicate that FXR is necessary for the AQP8-associated improvements in fatty liver disease and that similar to SHP, AQP8 might also be one of the target genes of FXR and provides positive feedback to FXR. Importantly, in this regard, we found that knockout of FXR abolished the beneficial effects of AQP8 overexpression on hepatic steatosis with respect to inhibiting fatty acid oxidation (Figure 7B), promoting inflammation (Figure 7D), increasing the serum levels of bile acid (Figure 6G) and probably increasing intestinal lipid absorption which exacerbated fatty liver development. However, the mechanisms underlying these processes remain to be elucidated.
Currently, FXR agonists are being investigated for their potential therapeutic application in NAFLD. However, the use of FXR agonists such as obeticholic acid has proved to be challenging, owing to notable adverse effects, including pruritus and increases in LDL-c.51 Further investigations are needed to evaluate the beneficial effects of other agonists on NAFLD, a multifactorial disease that involves a number of different contributory mechanisms. For example, it has been established that cholestasis is associated with NAFLD,52 and the findings of a previous study using an experimental cholestasis model have indicated that defective expression of AQP8 in the canalicular membrane contributes to the development of bile secretory dysfunction.49 In the present study, we observed a reduction in the expression of AQP8 in the liver specimens of patients with NAFLD, as well as in the livers of obese mice, whereas the overexpression of AQP8 contributed to a resolution of NAFLD, associated with an improvement on bile acid metabolism, in obese mice. These findings accordingly indicate that targeting AQP8 could represent an important therapeutic approach in the treatment of cholestatic associated with hepatic steatosis. In addition, in steatotic livers, lipid accumulation causes hepatocyte swelling and further impairs microcirculation, thereby curtailing vital supplies of oxygen and water45 and thus overexpression of AQP8 contributes to the improvement of impaired microcirculation and lipid disorders in patients with NAFLD. Consequently, targeting the expression of AQP8 may serve as a strategy for improving impaired microcirculation and cholestasis and further lipid disorders in patients with NAFLD.
In summary, our findings clearly indicate that hepatic AQP8 overexpression attenuates abnormal lipid deposition in experimental mice and cultured hepatocytes mainly via FXR activation, which reinforced the insight that AQP8 functioned as a potential FXR agonist will be helpful to provide therapeutic actions on NAFLD.
Limitations of the study
Despite our important observations, this study does have certain limitations. First, adenovirus overexpression typically results in artificially high levels of the gene/protein of interest and the potential for non-specific effects, including off-target effects. Second, AQP8 is also expressed in other tissues such as pancreas and intestine, and thus our study exclusively focusing changes on hepatic AQP8 expression may have overlooked the contribution of AQP8 in other tissues to the improvement of hepatic steatosis. Third, in addition to changes in gene expression, lipogenesis or fatty acid oxidation process assays should be performed to complement our limited observations, providing more compelling evidence.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AQP8 | Sigma | RRID:AB_1839763 |
| FAS | Cell Signaling Technology | Cat# cs-3180 |
| Hsp90 | Santa Cruz | RRID:AB_675659 |
| SREBP-1c | Thermo Fisher Scientific | RRID:AB_2194231 |
| ChREBP | Thermo Fisher Scientific | RRID:AB_11153460 |
| Bacterial and virus strains | ||
| Ad-GFP | Xitubio, Shanghai | N/A |
| Ad-AQP8 | Xitubio, Shanghai | N/A |
| Ad-shAQP8 | Xitubio, Shanghai | N/A |
| Biological samples | ||
| RIPA | BIOCOLOR | Cat# R0095 |
| Trizol Reagent | Life Technologies | Cat# 15596026 |
| PVDF membranes | Millipore | Cat# IPVH00010 |
| NEFA HR color reagent | Wako Life Sciences | Cat# 294-63601 |
| β-hydroxybutyrate Assay kit | Jian cheng, Nanjing, China | Cat# H69 |
| Triglyceride Quantification Kit | Biovision | K622–100 |
| Experimental models: Cell lines | ||
| HepG2 | ATCC | Cat#HB-8065 |
| Hepatocyte | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Vital River Laboratories | N/A |
| Mouse:C57BLKS/J-Leprdb/Leprdb | Model Animal Research Center of Nanjing University | N/A |
| Mouse: Nr1h4tm1Gonz/J | Jackson Laboratory | RRID: IMSR_JAX:004144 |
| Software and algorithms | ||
| GraphPad Prism 8 | Graphpad | https://www.graphpad.com/ |
| Other | ||
| High fat diet | Diet Research | D12492i |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhiguo Zhang (zhangzhiguo@outlook.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human livers samples
A total of 15 adults (age>18 years, 9 males, 6 females) who underwent liver biopsy for normal control liver specimens and suspected NAFLD were enrolled for analysis. Written informed consent was obtained from each participant for collection of samples. Ethical approval (Y2020-622) was obtained from the Zhongshan Hospital Research Ethics Committee.
Mouse models
The animal protocol was reviewed and approved by the Animal Care Committee of Shanghai Jiao Tong University School of Medicine. Obese C57BLKS/J-Leprdb/Leprdb (db/db) male mice and their wild-type littermates were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). C57BL/6J male mice were purchased from Vital River Laboratories (Beijing, China). Nr1h4tm1Gonz/J (also known as FXR-KO) and wildtype control mice were purchased from the Jackson Laboratory (RRID: IMSR_JAX:004144; Bar Harbor, ME, USA) and maintained on a C57BL/6J background. At 8 weeks of age, the mice were housed at controlled humidity and temperature conditions (55±5% and 22 ± 2°C, respectively) with a 12-hour light/dark cycle. HFD-induced obese mice had free access to high fat diet (D12492, Research Diets, New Brunswick, New Jersey, USA) or chow diet and water were given ad libitum for indicated time. For the experiments with db/db mice, db/db mice fed with a normal chow diet were utilized.
Mouse experiments
Each mouse was intravenously injected via tail vein with Ad-GFP/Ad-AQP8 at dose of 5 × 109 plaque-forming units. All of the mice were sacrificed on the tenth day after viral administration, and their livers and total blood samples were collected for analysis after a-12 hours-fast.
Method details
Cell cultures and adenovirus infection
Isolation of C57BL/6J mouse primary hepatocytes were previously reported.53 Mouse primary hepatocytes and HepG2 cells (AmericanType Culture Collection, China) were cultured in DMEM supplemented with 10% of fetal bovine serum, 1% of L-glutamine, and 1% of penicillin-streptomycin solution and maintained in a humidified atmosphere of 5% CO2 at 37°C. Mouse primary hepatocytes and HepG2 cells were infected with adenovirus encoding green fluorescent protein(Ad-GFP; Xitubio Shanghai) or adenovirus expressing AQP8 short hairpin RNA(Ad-shAQP8; Xitubio Shanghai) or adenovirus expressing AQP8(Ad-AQP8; Xitubio Shanghai) for 12-24 hours.
Free fatty acid treatment in vitro
HepG2 cells and mouse primary hepatocytes were incubated with 0.5% of BSA-conjugated FFAs (oleic acid and palmitic acid at the proportion of 2:1, Sigma, USA) at final concentration of 1 mM for indicated time after 12-24 hours of adenovirus transfection. Controls were kept with 0.5% of BSA but no FFAs. Then, the cells were harvested for subsequent analysis.
Free fatty acid measurement
Serum free fatty acid levels were measured with NEFA HR color reagent (Wako Life Sciences) according to the manufacturer’s instructions. All serum samples were from mice fasted for 12 hours.
Serum β-hydroxybutyrate measurement
Serum β-hydroxybutyrate(β-OHB) was measured with mouse β-hydroxybutyrate Assay kit (Jian cheng, Nanjing, China). Under 450 nm wavelength, optical density (OD) of each well was detected.
Hepatic and cellular TG measurement
Frozen livers were homogenized in 1 ml of 5% NP-40 buffer and triglyceride concentration was measured using a Triglyceride Quantification Kit (Biovision, K622–100) adhering to the manufacturer's instructions. Data were normalized to liver weight. Cells were resuspended in 5% NP-40 buffer, and TG content was detected as described above.
Serum analysis
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), total cholesterol and triglyceride levels were measured using detection kits(KHB, China). Specific steps adhered to the manufacturer's instructions.
Quantitative polymerase chain reaction
Total RNA from each sample was extracted using the Trizol reagent (Invitrogen) and purified. Quantitative real-time–PCR (RT–PCR) was performed using complementary DNA reverse-transcribed from total RNA, according to the manufacturer's instructions.
Oil Red O staining
Cells were washed twice with PBS and then fixed with 4% Paraformaldehyde for 30 minutes. After three washes in PBS, cells were stained in freshly diluted oil Red O working solution for 15 min to measure intracellular lipid accumulation.
Histochemistry
Livers were removed and fixed in 4% paraformaldehyde overnight. Embedded in paraffin, the livers were cut into parallel slices. After hematoxylin and eosin (H&E) staining staining, the slices were visualized and imaged under a light microscope.
Luciferase reporter assay
A dual-luciferase assay was applied for functional characterization by co-transfecting hFXR construct with the pGL4.14-hAQP8, pGL4.14-hMut-AQP8, and phRL-CMV plasmids (constitutively expressing Renilla luciferase) into HepG2 cells. The firefly luciferase activity was normalized by Renilla luciferase activity.
ChIP assay
Chromatin immunoprecipitation (ChIP) experiment was performed essentially as described by Upstate Biotechnology with modifications. Briefly, cells were cross-linked and harvested in SDS lysis buffer. Chromatins were then sonicated to an average length of 100 to 500 bp and immunoprecipitated with antibodies against FXR (Cell Signal Technology) and taking IgG as a control. DNA fragments were subjected to quantitative PCR (qPCR) using primers flanking FXRE on the AQP8 promoter(forwardprimer:5′- ATGTGAGGCTAGTGCTCTTCT− 3′, and reverse primer: 5′ − CCAAGATGAAGGAGGCAACTGA − 3′).
Quantification and statistical analysis
Results were performed with GraphPad Prism version 8.0 (GraphPad Software Inc., San Diego, CA, USA) and presented as mean ± SEM. When comparing two groups, the unpaired Student’s t-test was performed, except when variances were statistically different. For analysis of more than two groups, statistical analysis were compared by one-way ANOVA with post-hoc test. Two-tailed p-values less than 0.05 were considered statistically significant.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 82070828, 82270896, 81770863), partly supported by the Shanghai Medical and Health Development Foundation (grant number DMRFP_II_01 to Y.Z.), and the Program for Shanghai Outstanding Medical Academic Leader (grant number 2019LJ07 to Y.Z.).
Author contributions
Conceptualization, Z.Z.G., H.L.Y., W.H., and X.M.Q.; Methodology, X.M.Q., Q.X., H.L.Y., W.H., and Y.J.; Investigation, X.M.Q., Q.X., and Y.S.S.; Writing – Original Draft, X.M.Q. and Z.Z.G.; Writing – Review and Editing, X.M.Q. and Z.Z.G.; Funding Acquisition, Z.Z.G. and Z.Y.F.; Resources, Z.Z.G., Z.Y.F., W.J.Q., L.W.R., and G.Y.Y.; Supervision, Z.Z.G., Z.Y.F., and P.Y.
Declaration of interests
The authors declare no competing interests.
Published: April 1, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106561.
Contributor Information
Yifei Zhang, Email: feifei-a@163.com.
Ying Peng, Email: pennyppp@126.com.
Zhiguo Zhang, Email: zhangzhiguo@outlook.com.
Supplemental information
Data and code availability
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original codes.
-
•
Any additional information available from the lead contact upon request.
References
- 1.Cotter T.G., Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology. 2020;158:1851–1864. doi: 10.1053/j.gastro.2020.01.052. [DOI] [PubMed] [Google Scholar]
- 2.Anstee Q.M., Targher G., Day C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013;10:330–344. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 3.Aron-Wisnewsky J., Vigliotti C., Witjes J., Le P., Holleboom A.G., Verheij J., Nieuwdorp M., Clément K. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020;17:279–297. doi: 10.1038/s41575-020-0269-9. [DOI] [PubMed] [Google Scholar]
- 4.Powell E.E., Wong V.W.S., Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397:2212–2224. doi: 10.1016/S0140-6736(20)32511-3. [DOI] [PubMed] [Google Scholar]
- 5.Sheka A.C., Adeyi O., Thompson J., Hameed B., Crawford P.A., Ikramuddin S. Nonalcoholic steatohepatitis: a review. JAMA. 2020;323:1175–1183. doi: 10.1001/jama.2020.2298. [DOI] [PubMed] [Google Scholar]
- 6.Calamita G., Perret J., Delporte C. Aquaglyceroporins: drug targets for metabolic diseases? Front. Physiol. 2018;9:851. doi: 10.3389/fphys.2018.00851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernardino R.L., Marinelli R.A., Maggio A., Gena P., Cataldo I., Alves M.G., Svelto M., Oliveira P.F., Calamita G. Hepatocyte and Sertoli cell aquaporins, recent advances and Research trends. Int. J. Mol. Sci. 2016;17:1096. doi: 10.3390/ijms17071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.da Silva I.V., Díaz-Sáez F., Zorzano A., Gumà A., Camps M., Soveral G. Aquaglyceroporins are differentially expressed in beige and white adipocytes. Int. J. Mol. Sci. 2020;21:610. doi: 10.3390/ijms21020610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krüger C., Waldeck-Weiermair M., Kaynert J., Pokrant T., Komaragiri Y., Otto O., Michel T., Elsner M. AQP8 is a crucial H(2)O(2) transporter in insulin-producing RINm5F cells. Redox Biol. 2021;43:101962. doi: 10.1016/j.redox.2021.101962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huebert R.C., Splinter P.L., Garcia F., Marinelli R.A., LaRusso N.F. Expression and localization of aquaporin water channels in rat hepatocytes. Evidence for a role in canalicular bile secretion. J. Biol. Chem. 2002;277:22710–22717. doi: 10.1074/jbc.M202394200. [DOI] [PubMed] [Google Scholar]
- 11.Carreras F.I., Gradilone S.A., Mazzone A., García F., Huang B.Q., Ochoa J.E., Tietz P.S., Larusso N.F., Calamita G., Marinelli R.A. Rat hepatocyte aquaporin-8 water channels are down-regulated in extrahepatic cholestasis. Hepatology. 2003;37:1026–1033. doi: 10.1053/jhep.2003.50170. [DOI] [PubMed] [Google Scholar]
- 12.Ferri D., Mazzone A., Liquori G.E., Cassano G., Svelto M., Calamita G. Ontogeny, distribution, and possible functional implications of an unusual aquaporin, AQP8, in mouse liver. Hepatology. 2003;38:947–957. doi: 10.1053/jhep.2003.50397. [DOI] [PubMed] [Google Scholar]
- 13.Ma T., Yang B., Verkman A.S. Cloning of a novel water and urea-permeable aquaporin from mouse expressed strongly in colon, placenta, liver, and heart. Biochem.Biophys. Res. Commun. 1997;240:324–328. doi: 10.1006/bbrc.1997.7664. [DOI] [PubMed] [Google Scholar]
- 14.Larocca M.C., Soria L.R., Espelt M.V., Lehmann G.L., Marinelli R.A. Knockdown of hepatocyte aquaporin-8 by RNA interference induces defective bile canalicular water transport. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G93–G100. doi: 10.1152/ajpgi.90410.2008. [DOI] [PubMed] [Google Scholar]
- 15.Marinelli R.A., Tietz P.S., Caride A.J., Huang B.Q., LaRusso N.F. Water transporting properties of hepatocyte basolateral and canalicular plasma membrane domains. J. Biol. Chem. 2003;278:43157–43162. doi: 10.1074/jbc.M305899200. [DOI] [PubMed] [Google Scholar]
- 16.Elkjaer M.L., Nejsum L.N., Gresz V., Kwon T.H., Jensen U.B., Frøkiaer J., Nielsen S. Immunolocalization of aquaporin-8 in rat kidney, gastrointestinal tract, testis, and airways. Am. J. Physiol. Renal Physiol. 2001;281:F1047–F1057. doi: 10.1152/ajprenal.0158.2001. [DOI] [PubMed] [Google Scholar]
- 17.Soria L.R., Gradilone S.A., Larocca M.C., Marinelli R.A. Glucagon induces the gene expression of aquaporin-8 but not that of aquaporin-9 water channels in the rat hepatocyte. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;296:R1274–R1281. doi: 10.1152/ajpregu.90783.2008. [DOI] [PubMed] [Google Scholar]
- 18.Calamita G., Ferri D., Gena P., Liquori G.E., Cavalier A., Thomas D., Svelto M. The inner mitochondrial membrane has aquaporin-8 water channels and is highly permeable to water. J. Biol. Chem. 2005;280:17149–17153. doi: 10.1074/jbc.C400595200. [DOI] [PubMed] [Google Scholar]
- 19.Gradilone S.A., García F., Huebert R.C., Tietz P.S., Larocca M.C., Kierbel A., Carreras F.I., Larusso N.F., Marinelli R.A. Glucagon induces the plasma membrane insertion of functional aquaporin-8 water channels in isolated rat hepatocytes. Hepatology. 2003;37:1435–1441. doi: 10.1053/jhep.2003.50241. [DOI] [PubMed] [Google Scholar]
- 20.Soria L.R., Marrone J., Calamita G., Marinelli R.A. Ammonia detoxification via ureagenesis in rat hepatocytes involves mitochondrial aquaporin-8 channels. Hepatology. 2013;57:2061–2071. doi: 10.1002/hep.26236. [DOI] [PubMed] [Google Scholar]
- 21.Danielli M., Marrone J., Capiglioni A.M., Marinelli R.A. Mitochondrial aquaporin-8 is involved in SREBP-controlled hepatocyte cholesterol biosynthesis. Free Radic. Biol. Med. 2019;131:370–375. doi: 10.1016/j.freeradbiomed.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 22.Kawano Y., Cohen D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013;48:434–441. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adeva-Andany M.M., Carneiro-Freire N., Seco-Filgueira M., Fernández-Fernández C., Mouriño-Bayolo D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion. 2019;46:73–90. doi: 10.1016/j.mito.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 24.de Aguiar Vallim T.Q., Tarling E.J., Edwards P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ovadia C., Perdones-Montero A., Spagou K., Smith A., Sarafian M.H., Gomez-Romero M., Bellafante E., Clarke L.C.D., Sadiq F., Nikolova V., et al. Enhanced microbial bile acid deconjugation and impaired ileal uptake in pregnancy repress intestinal regulation of bile acid synthesis. Hepatology. 2019;70:276–293. doi: 10.1002/hep.30661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang C., Xie C., Li F., Zhang L., Nichols R.G., Krausz K.W., Cai J., Qi Y., Fang Z.Z., Takahashi S., et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest. 2015;125:386–402. doi: 10.1172/JCI76738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J., Li Y., Chen W.D., Xu Y., Yin L., Ge X., Jadhav K., Adorini L., Zhang Y. Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology. 2014;59:1761–1771. doi: 10.1002/hep.26714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makishima M. Nuclear receptors as targets for drug development: regulation of cholesterol and bile acid metabolism by nuclear receptors. J. Pharmacol. Sci. 2005;97:177–183. doi: 10.1254/jphs.fmj04008x4. [DOI] [PubMed] [Google Scholar]
- 29.Dong B., Young M., Liu X., Singh A.B., Liu J. Regulation of lipid metabolism by obeticholic acid in hyperlipidemic hamsters. J. Lipid Res. 2017;58:350–363. doi: 10.1194/jlr.M070888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan Z.Q., Li K.W. Role of farnesoid X receptor in cholestasis. J. Dig. Dis. 2016;17:501–509. doi: 10.1111/1751-2980.12378. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X., Huang S., Gao M., Liu J., Jia X., Han Q., Zheng S., Miao Y., Li S., Weng H., et al. Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proc. Natl. Acad. Sci. USA. 2014;111:2277–2282. doi: 10.1073/pnas.1323977111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang D.Q., El-Serag H.B., Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021;18:223–238. doi: 10.1038/s41575-020-00381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefan N., Häring H.U., Cusi K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019;7:313–324. doi: 10.1016/S2213-8587(18)30154-2. [DOI] [PubMed] [Google Scholar]
- 34.Galli M., Hameed A., Żbikowski A., Zabielski P. Aquaporins in insulin resistance and diabetes: more than channels! Redox Biol. 2021;44:102027. doi: 10.1016/j.redox.2021.102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castle N.A. Aquaporins as targets for drug discovery. Drug Discov. Today. 2005;10:485–493. doi: 10.1016/S1359-6446(05)03390-8. [DOI] [PubMed] [Google Scholar]
- 36.Verkman A.S., Anderson M.O., Papadopoulos M.C. Aquaporins: important but elusive drug targets. Nat. Rev. Drug Discov. 2014;13:259–277. doi: 10.1038/nrd4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donnelly K.L., Smith C.I., Schwarzenberg S.J., Jessurun J., Boldt M.D., Parks E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bălănescu A., Bălănescu P., Comănici V., Stan I., Acs B., Prisăcariu L., Brezan F., Ciomârtan T., Gherghina I. Lipid profile pattern in pediatric overweight population with or without NAFLD in relation to IDF criteria for metabolic syndrome: a preliminary study. Rom. J. Intern. Med. 2018;56:47–54. doi: 10.1515/rjim-2017-0040. [DOI] [PubMed] [Google Scholar]
- 39.Rotman Y., Sanyal A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut. 2017;66:180–190. doi: 10.1136/gutjnl-2016-312431. [DOI] [PubMed] [Google Scholar]
- 40.Sun N., Shen C., Zhang L., Wu X., Yu Y., Yang X., Yang C., Zhong C., Gao Z., Miao W., et al. Hepatic Krüppel-like factor 16 (KLF16) targets PPARα to improve steatohepatitis and insulin resistance. Gut. 2021;70:2183–2195. doi: 10.1136/gutjnl-2020-321774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferré P., Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010;12:83–92. doi: 10.1111/j.1463-1326.2010.01275.x. [DOI] [PubMed] [Google Scholar]
- 42.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duan S., Li X., Fan G., Liu R. Targeting bile acid signaling for the treatment of liver diseases: from bench to bed. Biomed. Pharmacother. 2022;152:113154. doi: 10.1016/j.biopha.2022.113154. [DOI] [PubMed] [Google Scholar]
- 44.Arab J.P., Karpen S.J., Dawson P.A., Arrese M., Trauner M. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology. 2017;65:350–362. doi: 10.1002/hep.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asai Y., Yamada T., Tsukita S., Takahashi K., Maekawa M., Honma M., Ikeda M., Murakami K., Munakata Y., Shirai Y., et al. Activation of the hypoxia inducible factor 1α Subunit pathway in steatotic liver contributes to formation of cholesterol gallstones. Gastroenterology. 2017;152:1521–1535.e8. doi: 10.1053/j.gastro.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 46.Clifford B.L., Sedgeman L.R., Williams K.J., Morand P., Cheng A., Jarrett K.E., Chan A.P., Brearley-Sholto M.C., Wahlström A., Ashby J.W., et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33:1671–1684.e4. doi: 10.1016/j.cmet.2021.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang S., Suh J.M., Reilly S.M., Yu E., Osborn O., Lackey D., Yoshihara E., Perino A., Jacinto S., Lukasheva Y., et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015;21:159–165. doi: 10.1038/nm.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sayin S.I., Wahlström A., Felin J., Jäntti S., Marschall H.U., Bamberg K., Angelin B., Hyötyläinen T., Orešič M., Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 49.Lehmann G.L., Larocca M.C., Soria L.R., Marinelli R.A. Aquaporins: their role in cholestatic liver disease. World J. Gastroenterol. 2008;14:7059–7067. doi: 10.3748/wjg.14.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou Y., Wang L., Yi D., Ding B., Yang Z., Li J., Chen X., Qiu Y., Wu G. N-acetylcysteine reduces inflammation in the small intestine by regulating redox, EGF and TLR4 signaling. Amino Acids. 2013;45:513–522. doi: 10.1007/s00726-012-1295-x. [DOI] [PubMed] [Google Scholar]
- 51.Younossi Z.M., Ratziu V., Loomba R., Rinella M., Anstee Q.M., Goodman Z., Bedossa P., Geier A., Beckebaum S., Newsome P.N., et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–2196. doi: 10.1016/S0140-6736(19)33041-7. [DOI] [PubMed] [Google Scholar]
- 52.Trauner M., Fuchs C.D. Novel therapeutic targets for cholestatic and fatty liver disease. Gut. 2022;71:194–209. doi: 10.1136/gutjnl-2021-324305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li W.C., Ralphs K.L., Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol. 2010;633:185–196. doi: 10.1007/978-1-59745-019-5_13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original codes.
-
•
Any additional information available from the lead contact upon request.