

Abstract
Serotonin deficiency in major depressive disorder (MDD) has formed the basis of antidepressant drug development and was originally attributed to induction of the major tryptophan (Trp)-degrading enzyme, liver Trp 2,3-dioxygenase (TDO), by cortisol, leading to decreased Trp availability to the brain for serotonin synthesis. Subsequently, the serotonin deficiency was proposed to involve induction of the extrahepatic Trp-degrading enzyme indoleamine 2,3-dioxygenase (IDO) by proinflammatory cytokines, with inflammation being the underlying cause. Recent evidence, however, challenges this latter concept, as not all MDD patients are immune-activated and, when present, inflammation is mild and/or transient. A wide range of antidepressant drugs inhibit the activity of liver TDO and bind specifically to the enzyme, but not to IDO. IDO induction is not a major event in MDD, but, when it occurs, its metabolic consequences may be masked and overridden by upregulation of kynurenine monooxygenase (KMO), the gateway to production of modulators of immune and neuronal functions. KMO appears to be activated in MDD by certain proinflammatory cytokines and antidepressants with anti-inflammatory properties may block this activation. We demonstrate the ability of the antidepressant ketamine to dock (bind) to KMO. The pathophysiology of MDD may be underpinned by both the serotonin deficiency and glutamatergic activation mediated respectively by TDO induction and N-methyl-D-aspartate receptor activation. Inhibition of TDO and KMO should be the focus of MDD pharmacotherapy.
Keywords: Major depressive disorder; Indoleamine 2,3-dioxygenase; Kynurenine monooxygenase; Proinflammatory cytokines; Serotonin deficiency; Tryptophan 2,3-dioxygenase
Core Tip: Antidepressant drugs inhibit the activity of liver tryptophan 2,3-dioxygenase (TDO) and bind specifically to the enzyme, but not to indoleamine 2,3-dioxygenase (IDO). IDO induction is not a major event in the pathophysiology of major depressive disorder (MDD), but may be masked and overridden by upregulation of kynurenine monooxygenase (KMO), the gateway to production of modulators of immune and neuronal functions. The pathophysiology of MDD may be underpinned by both the serotonin deficiency and glutamatergic activation mediated respectively by TDO induction and N-methyl-D-aspartate receptor activation. Inhibition of TDO and KMO should be the focus of MDD pharmacotherapy.
INTRODUCTION
Serotonin deficiency in major depressive disorder
Antidepressant drug development has been based on the monoamine hypothesis of affective disorders that postulates a deficiency of one or more monoamines and the need to develop agents that can inhibit either their breakdown (monoamine oxidase inhibitors) or their reuptake and subsequent degradation [tricyclic antidepressants (TCAs), serotonin-specific and serotonin-noradrenaline reuptake inhibitors]. The serotonin deficiency was originally suggested from observations by Curzon and Bridges[1] that patients with depression exhibit raised cortisol levels that can induce the synthesis of liver tryptophan 2,3-dioxygenase (TDO; formerly Trp pyrrolase) resulting in accelerated peripheral Trp degradation and hence a decrease in its availability to the brain for 5-HT synthesis. Brain (Trp) is the major determinant of 5-HT synthesis, because Trp hydroxylase exists partially (≤ 50%) saturated with its Trp substrate[2]. Depriving the brain of Trp by the acute Trp depletion test reverses antidepressant-induced remission in depressed patients[3].
Inhibition of liver TDO by antidepressant drugs
Curzon[4] suggested that TDO is a biochemical factor in depressive illness and Samsonova and Lapin were the first to demonstrate TDO inhibition by some TCAs[5]. Subsequently, a wide range of antidepressant drugs of various structures and pharmacological profiles were shown to inhibit TDO activity in rat liver in vitro and after administration[6-13]. TDO inhibition occurs with doses as small as 0.5 mg/kg body weight[7] and involves prevention of conjugation of the inactive apoenzyme with its heme activator and cofactor. TDO inhibition by antidepressants therefore depends on extent of the heme saturation of the apoenzyme and also of its glucocorticoid induction by cortisol, with some antidepressants being better inhibitors of heme-activated TDO, whereas others are better inhibitors of cortisol-induced TDO[13]. Ability of antidepressants to lower raised cortisol levels is another determinant of TDO inhibition[13].
Further evidence of the targeting of TDO by antidepressant drugs was provided by our group, using molecular docking in silico: A technique for screening potential inhibitors of target proteins[14,15], but is also useful to confirm known inhibitors. We demonstrated high docking scores (strong binding) of many antidepressants, including amoxapine, citalopram, fluoxetine, fluvoxamine, moclobemide, paroxetine, seproxetine, sertraline[16], tianeptine and venlafaxine, but not the non-antidepressant drugs pargyline and mefenamic acid[17] to the crystal structure of TDO, but no docking to that of the extrahepatic IDO. Thus, antidepressants target TDO and this could explain their ability to restore serotonin homeostasis, at least in part, by reversing the defective serotonin synthesis. As TDO is the first and rate-limiting enzyme of the kynurenine (Kyn) pathway (KP), a description of this pathway and its intermediates may be useful at this point (Figure 1).
Figure 1.

Tryptophan metabolism to serotonin and kynurenine metabolites. AA: Anthranilic acid; ALAAD: Aromatic L-amino acid decarboxylase; ACMS: 2-amino-3-carboxymuconic acid-6-semialdehyde; FAMID: N-formylkynurenine formamidase; 3-HAA: 3-hydroxyanthranilic acid; 3HAAO: 3-hydroxyanthranilic acid 3,4-dioxygenase; 3-HK: 3-hydroxykynurenine; 5-HT: 5-hydroxytryptamine (serotonin); 5-HTP: 5-hydroxytryptophan; IDO: Indoleamine 2,3-dioxygenase; KA: Kynurenic acid; Kyn: Kynurenine; KAT: Kynurenine aminotransferase; KYNU: Kynureninase; KMO: Kynurenine monooxygenase (Kyn hydroxylase); NFK: N-formylkynurenine; QA: Quinolinic acid; Trp: Tryptophan; TDO: Tryptophan 2,3-dioxygenase; XA: Xanthurenic acid; TPH2: Tryptophan hydroxylase isoform 2.
The KP of Trp metabolism
The KP is the major pathway of Trp degradation, accounting for 95% of dietary Trp metabolism, with the hepatic pathway contributing 90% and the extrahepatic pathway the remaining 5%, though contribution of the latter pathway is significantly increased by immune activation of IDO by interferons and other proinflammatory cytokines[18]. The liver contains all the enzymes leading to NAD+ synthesis, whereas other tissues express fewer enzymes[18]. Production of Trp metabolites in extrahepatic tissues including immune cells will therefore depend on the enzymes present. In the absence of TDO or IDO induction, the Kyn produced in liver or IDO-containing tissues can be transported elsewhere for further metabolism. Activity of the KP controls plasma Trp availability for cerebral serotonin synthesis and results in production of a range of Kyn metabolites that influence immune and neuronal functions[18].
Control of plasma Trp availability
Control of plasma Trp availability to tissues including the brain is primarily the function of liver TDO. Thus, deletion of the tdo2 gene in mice increases plasma (Trp) by up to 12.7-fold[19,20] and brain (Trp) by 10.6-fold[21]. The increase in brain (Trp) is associated in increased 5-HT synthesis[21]. By contrast, deletion of the IDO1 or IDO2 gene does not alter Trp availability to the brain or 5-HT synthesis[21]. Trp availability for cerebral 5-HT synthesis is therefore determined by hepatic TDO activity in the first place, but also, secondarily, but more immediately, by 2 other determinants: albumin binding of plasma Trp and extent of competition for entry into the brain from a group of large neutral or competing amino acids (CAA), mainly Val Leu, Ile, Phe and Tyr. The best predictor of likely change in brain Trp and hence 5-HT synthesis is the ratio in plasma of Trp/CAA[13]. Plasma Trp exists largely (90%-95%) bound to albumin, with 5%-10% being freely available for tissue uptake. Binding is determined by levels of albumin and of the physiological displacers of bound Trp, nonesterified fatty acids (NEFAs). Free Trp can therefore be easily altered by dietary, hormonal and pharmacological factors that influence Trp binding[22]. Under certain conditions wherein displacement of bound Trp is strong and sustained, the rise in free Trp will be associated with a decrease in total Trp, due to the rapid equilibration between the free and total fractions. Investigators should be careful in interpreting the changes in Trp disposition under these conditions[22], and whereas most investigators measure the ratio of total Trp/CAA, the ratio of free Trp/CAA is a more accurate measure in situations other than after acute Trp depletion or loading, as these are associated with parallel changes in both free and total Trp.
Neuronal and immune properties of KP metabolites
Neuronal activity is influenced by the cytoprotective kynurenic acid (KA) and excitotoxic quinolinic acid (QA) as antagonist and agonist respectively of the N-methyl-D-aspartate (NMDA) receptors of the excitatory amino acid glutamate[23], with the balance between QA and KA determining the level of neuronal excitability. Immune function is influenced by several Kyn metabolites. 3-hydroxykynurenine (3-HK), 3-hydroxyanthranilic acid (3-HAA) and QA are the main proinflammatory metabolites that undermine T-cell function[18], whereas picolinic acid (PA) is anti-inflammatory. KA is dually acting[24]. KMO is the gateway for production of these metabolites and the pathway favours Kyn oxidation by KMO to 3-HK and hydrolysis of the latter to 3-HAA by KYNU, because of the relatively high affinity of both enzymes towards their respective substrates[18]. 3-HAA 3,4-dioxygenase is the most active of KP enzymes, hence the rapid conversion of 3-HAA to an unstable intermediate that cyclises non-enzymically to QA[18]. Affinity of Kyn aminotransferase (KAT) towards its Kyn and 3-HK substrates is much weaker, hence the relatively low production of KA and xanthurenic acid, formation of both of which requires increased levels of the KAT substrates[18]. KMO gene deletion in mice decreases plasma 3-HK and QA by about 86% and 92% respectively and increases concentrations of Kyn, KA and AA by 15-, 133- and 4-fold respectively[25]. Thus, KMO inhibition diverts Kyn metabolism towards the cytoprotective KA. By contrast, activation of KMO can enhance the conversion of Kyn to 3-HK leading to decreased Kyn levels: An effect that will neutralise the rise in Kyn expected with TDO and/or IDO induction. This may explain the frequent observation that the reported increase in the Kyn/Trp ratio after mild immune activation is mostly due to the decrease in Trp, rather than an increase in Kyn[26]. These aspects may help explain some of the KP changes in MDD and their modulation by antidepressants (see below). Investigators use the plasma Kyn/Trp ratio to express indirectly IDO activity. While this may apply correctly in in vitro culture or cell systems, this ratio is not specific for IDO in in vivo situations, as other factors also alter it, including TDO activity, flux of plasma free Trp down the KP and activities of KMO and KYNU[26].
Inflammation and glutamatergic activity in major depressive disorder
The involvement of inflammation in major depressive disorder (MDD) was first proposed following the observation[27] that hepatitis C patients treated with interferon-alpha (IFN-α) become depressed. As this cytokine is an IDO inducer, the concept that serotonin deficiency in MDD is underpinned by IDO induction secondarily to inflammation was born. Extrapolating from hepatitis C to MDD was however unwise, given that Trp metabolism is already compromised by this virus and the use of IFN-α can only potentiate the effect of the virus on Trp metabolism, in particular IDO induction[17]. Although many studies of the immune status in MDD followed, recent studies suggested that not all MDD patients are immune-activated and, when present, inflammation is mild and/or transient and its reversal does not reflect clinical outcome[28,29]. Elevation of proinflammatory cytokines in some patients is not associated with an inflamed subgroup, but is due to a right shift of the immune marker distribution[30]. In MDD, reports of changes in the Kyn/Trp ratio used to express IDO activity are contradictory, partly because of the non-specificity of this ratio[26]. Whereas considerable evidence exists for liver TDO inhibition leading to elevation of plasma and brain Trp and enhanced serotonin synthesis in rats by a broad range of antidepressant drugs[6-8], and emerging evidence for inhibition of the accelerated Trp degradation in MDD by escitalopram causing an increase in plasma Trp[31], little is known about potential IDO inhibition by antidepressants. The absence of docking of antidepressants to IDO[17] suggests that this extrahepatic enzyme does not play a role in MDD antidepressant therapy and this notion is supported by the observations that the Kyn/Trp ratio does not link inflammation with depressive symptoms[32], and that IDO expression in monocytes of MDD patients is not different from that in healthy controls[33], though this may reflect patient heterogeneity.
Antidepressants and KP enzymes
As stated above, antidepressant drugs inhibit and target liver TDO, but not extrahepatic IDO[17]. Even when IDO is induced in some MDD patients by proinflammatory cytokines, it is most likely that the effect of this induction (as well as that of TDO) will be masked, superseded or overridden by changes in subsequent KP enzymes, notably KMO. KMO activation in MDD is gaining ground and may be another important biochemical event in the pathophysiology of this disorder. KMO activation can result in decreased levels of the Kyn substrate and consequently in the Kyn transamination product KA, as has been reported in unmedicated MDD patients[34,35]. While many antidepressants have previously been proposed to act by reducing inflammation and hence inhibition of IDO induction, it is more likely that they act by inhibiting KMO activation by cytokines, notably, interleukin-1β and IFN-γ[36], as has been demonstrated with ketamine[29], escitalopram[31] and sertraline[37]. Additionally, whereas ketamine does not dock to either TDO or IDO and so is unlikely to inhibit either enzyme[17], we demonstrate here for the first time its strong docking to KMO (Figure 2), that is comparable with that of the reference ligand Kyn itself, and suggest that KMO inhibition is likely to be one mechanism of action of ketamine.
Figure 2.
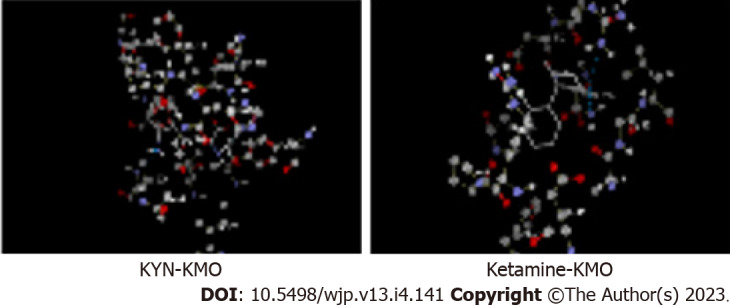
Molecular docking of kynurenine and ketamine to kynurenine monooxygenase. Docking was performed using the Molegro virtual Docker software as described previously[16,17], but with kynurenine monooxygenase. With kynurenine (top), the best scored docking solution of 5y66 with the reference ligand, amino acids in the active site are presented in ball and stick with element colour and ligand is presented in thick lines with element colour (where carbon is grey, oxygen is red, nitrogen is blue, sulphur is yellow and hydrogen is white). Blue lines represent the hydrogen bonds in between the ligand and the active site of 5y66. Docking parameters were as follows: Kynurenine: Molecular weight (207.206), docking score (-95.4662), re-rank score (-81.6652), root mean square deviation (RMSD) (2.19513), torsion (4), H-bond (-6.03152). Amino acid residues at the kynurenine monooxygenase (KMO) active site are: Gly321, IIe224, Phe319, Met373, His320, Ala56, Leu226, Phe319, ligand binding amino acid residues are: Tyr404, His320, and Leu226. With ketamine (bottom), molecular weight (237.725), docking score (-81.8059), re-rank score (-65.0284), RMSD (zero), torsion (2), H-bond (-0.0342033). Amino acid residues at the KMO active site are: Asp112, Arg39, Gly16, Ala15, Glu37, Phe131, Arg111, Arg100, ligand binding amino acid residues are: Asn 115, Arg111. KMO: Kynurenine monooxygenase; KYN: Kynurenine.
The role of anti-inflammatory drugs in MDD therapy
Current evidence suggests that anti-inflammatory strategies have not met with the anticipated success in MDD therapy and that only MDD patients with chronic inflammation may benefit from such therapy, while other patients with low-grade inflammation may experience harm[38]. Of 6 non-steroidal anti-inflammatory drugs tested, only salicylic acid (the active aspirin metabolite) docks to TDO[17] and aspirin appears to be an effective single or adjunctive therapy of MDD if given in relatively small doses and for short- to medium-term durations, but not for long-term use, which does not protect against depression and can actually induce it[18]. Mechanisms of these opposite effects of aspirin involve modulation by salicylate of Trp metabolism and disposition in opposite direction[17].
Serotonin and glutamate interactions
KMO activation leads to production of the excitotoxic Kyn metabolite and NMDA receptor agonist QA. Glutamatergic activity can therefore be enhanced in at least some MDD patients. Mutual interactions between serotonergic and glutamatergic activities are well established and may be important determinants of MDD pathophysiology. Thus, serotonin modulates glutamate neurotransmission in several brain regions, especially those involved in cognition, motor function and nociception, with raphe neurons being immune-positive for glutamate[39]. The cognitive and emotional disorders in MDD due to defective serotonin function can in part be due to disruption of the serotonin control over glutamate and gamma-aminobutyric acid neurotransmission[40,41]. On the other hand, NMDA receptors may regulate behavior by modulating serotonin and dopamine function[42,43]. Though complex in nature, this mutual interaction can be viewed simplistically as low serotonin losing control over glutamate neurotransmission and inhibition of the latter facilitating serotonin function (Figure 3).
Figure 3.
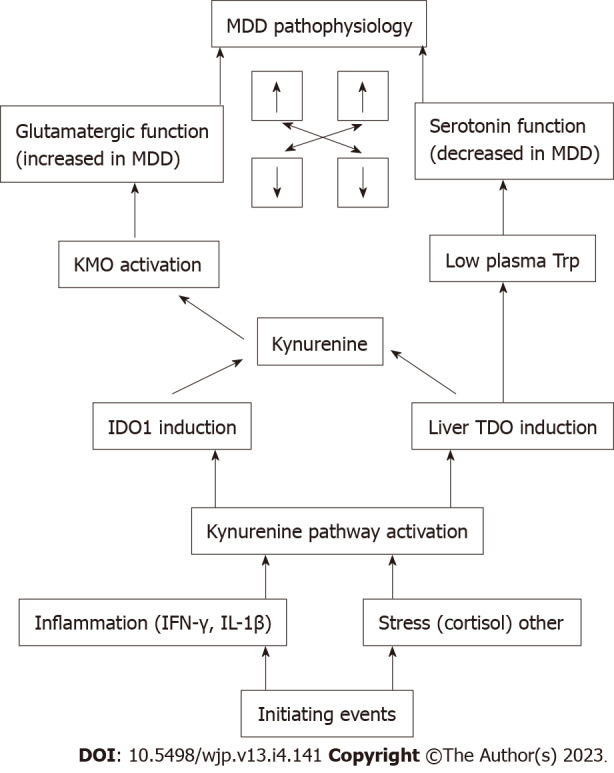
Schematic representation of the proposed role of the kynurenine pathway in the pathophysiology of major depressive disorder. Mutual interactions between serotonergic and glutamatergic functions are indicated by the bidirectional arrows for increased or decreased function. MDD: Major depressive disorder; KMO: Kynurenine monooxygenase; Trp: Tryptophan; IDO: Indoleamine 2,3-dioxygenase; TDO: Tryptophan 2,3-dioxygenase; IFN: Interferon; IL: Interleukin.
CONCLUSION
We hypothesized that the KP is at the center of MDD pathophysiology and a target of antidepressant therapy. Figure 3 outlines the potential role of the KP in MDD and emphasizes the importance of TDO and KMO, the inhibition of which is likely to become the focus of pharmacotherapy of MDD. Studies on the frequency of KMO enhancement and its mechanism(s) in MDD are needed. Effects of antidepressants other than ketamine and escitalopram on enzymes and metabolites of the KP should be investigated. KMO inhibition therapy could be explored in MDD. We suggest that lowering glutamatergic activity by KMO inhibition can restore serotonin function and should be recognized as a new mechanism of antidepressant action, perhaps exemplified by ketamine. The rapid and novel mode of action of ketamine should encourage the search for safer and longer-acting alternatives. The re-quirement of normal serotonin levels in the antidepressant-like efficacy of ketamine in rodent models[44] may explain in part the transient efficacy of the drug in humans. Furthermore, studies with the two ketamine enantiomers suggest that NMDA receptor antagonism is not the sole mechanism of the drug’s antidepressant action[45]. KMO inhibition and blockade of progress of the KP to QA formation is thus far a potential mechanism of ketamine action. Addressing the above issues may provide a way forward in the search for the ideal MDD pharmacotherapy.
Footnotes
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: November 25, 2022
First decision: December 12, 2022
Article in press: March 21, 2023
Specialty type: Psychiatry
Country/Territory of origin: United Kingdom
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Cheng J, China; Kaur M, United States S-Editor: Wang JJ L-Editor: A P-Editor: Wang JJ
Contributor Information
Abdulla A-B Badawy, Formerly School of Health Sciences, Cardiff Metropolitan University, Cardiff CF5 2YB, United Kingdom. badawyabdulla@yahoo.com.
Shazia Dawood, Pharmacy and Allied Health Sciences, Iqra University, Karachi 7580, Pakistan.
Samina Bano, Biochemistry, Karachi University, Karachi 75270, Pakistan.
References
- 1.Curzon G, Bridges PK. Tryptophan metabolism in depression. J Neurol Neurosurg Psychiatry. 1970;33:698–704. doi: 10.1136/jnnp.33.5.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlsson A, Lindqvist M. Dependence of 5-HT and catecholamine synthesis on concentrations of precursor amino-acids in rat brain. Naunyn Schmiedebergs Arch Pharmacol. 1978;303:157–164. doi: 10.1007/BF00508062. [DOI] [PubMed] [Google Scholar]
- 3.Delgado PL, Charney DS, Price LH, Aghajanian GK, Landis H, Heninger GR. Serotonin function and the mechanism of antidepressant action. Reversal of antidepressant-induced remission by rapid depletion of plasma tryptophan. Arch Gen Psychiatry. 1990;47:411–418. doi: 10.1001/archpsyc.1990.01810170011002. [DOI] [PubMed] [Google Scholar]
- 4.Curzon G. Tryptophan pyrrolase--a biochemical factor in depressive illness? Br J Psychiatry. 1970;116:571–572. doi: 10.1192/bjp.116.534.571. [DOI] [PubMed] [Google Scholar]
- 5.Samsonova ML, Lapin IP. Antidepressants and liver tryptophan pyrrolase activity. Biochem Pharmacol. 1973;22:1499–1507. doi: 10.1016/0006-2952(73)90327-4. [DOI] [PubMed] [Google Scholar]
- 6.Badawy AA, Evans M. Inhibition of rat liver tryptophan pyrrolase activity and elevation of brain tryptophan concentration by administration of antidepressants. Biochem Pharmacol. 1981;30:1211–1216. doi: 10.1016/0006-2952(81)90299-9. [DOI] [PubMed] [Google Scholar]
- 7.Badawy AA, Evans M. Inhibition of rat liver tryptophan pyrrolase activity and elevation of brain tryptophan concentration by acute administration of small doses of antidepressants. Br J Pharmacol. 1982;77:59–67. doi: 10.1111/j.1476-5381.1982.tb09269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badawy AA, Morgan CJ. Effects of acute paroxetine administration on tryptophan metabolism and disposition in the rat. Br J Pharmacol. 1991;102:429–433. doi: 10.1111/j.1476-5381.1991.tb12190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badawy AA, Morgan CJ, Dacey A, Stoppard T. The effects of lofepramine and desmethylimipramine on tryptophan metabolism and disposition in the rat. Biochem Pharmacol. 1991;42:921–929. doi: 10.1016/0006-2952(91)90054-9. [DOI] [PubMed] [Google Scholar]
- 10.Badawy AA-B, Morgan JC, Bano S, Buckland P, McGuffin P. Mechanism of enhancement of rat brain serotonin synthesis by acute fluoxetine administration. J Neurochem. 1996;66:436–437. [PubMed] [Google Scholar]
- 11.Bano S, Sherkheli MA. Inhibition of tryptophan - pyrrolase activity and elevation of brain tryptophan concentration by fluoxetine in rats. J Coll Physicians Surg Pak. 2003;13:5–10. [PubMed] [Google Scholar]
- 12.Bano S, Gitay M, Ara I, Badawy A. Acute effects of serotonergic antidepressants on tryptophan metabolism and corticosterone levels in rats. Pak J Pharm Sci. 2010;23:266–272. [PubMed] [Google Scholar]
- 13.Badawy AA. Tryptophan: the key to boosting brain serotonin synthesis in depressive illness. J Psychopharmacol. 2013;27:878–893. doi: 10.1177/0269881113499209. [DOI] [PubMed] [Google Scholar]
- 14.Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Molecules. 2015;20:13384–13421. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawood S, Zarina S, Bano S. Docking studies of antidepressants against single crystal structure of tryptophan 2, 3-dioxygenase using Molegro Virtual Docker software. Pak J Pharm Sci. 2014;27:1529–1539. [PubMed] [Google Scholar]
- 17.Dawood S, Bano S, Badawy AA. Inflammation and serotonin deficiency in major depressive disorder: molecular docking of antidepressant and anti-inflammatory drugs to tryptophan and indoleamine 2,3-dioxygenases. Biosci Rep. 2022;42 doi: 10.1042/BSR20220426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badawy AA. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int J Tryptophan Res. 2017;10:1178646917691938. doi: 10.1177/1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanai M, Funakoshi H, Takahashi H, Hayakawa T, Mizuno S, Matsumoto K, Nakamura T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol Brain. 2009;2:8. doi: 10.1186/1756-6606-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terakata M, Fukuwatari T, Kadota E, Sano M, Kanai M, Nakamura T, Funakoshi H, Shibata K. The niacin required for optimum growth can be synthesized from L-tryptophan in growing mice lacking tryptophan-2,3-dioxygenase. J Nutr. 2013;143:1046–1051. doi: 10.3945/jn.113.176875. [DOI] [PubMed] [Google Scholar]
- 21.Too LK, Li KM, Suarna C, Maghzal GJ, Stocker R, McGregor IS, Hunt NH. Deletion of TDO2, IDO-1 and IDO-2 differentially affects mouse behavior and cognitive function. Behav Brain Res. 2016;312:102–117. doi: 10.1016/j.bbr.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 22.Badawy AA. Plasma free tryptophan revisited: what you need to know and do before measuring it. J Psychopharmacol. 2010;24:809–815. doi: 10.1177/0269881108098965. [DOI] [PubMed] [Google Scholar]
- 23.Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993;45:309–379. [PubMed] [Google Scholar]
- 24.Wirthgen E, Hoeflich A, Rebl A, Günther J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front Immunol. 2017;8:1957. doi: 10.3389/fimmu.2017.01957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giorgini F, Huang SY, Sathyasaikumar KV, Notarangelo FM, Thomas MA, Tararina M, Wu HQ, Schwarcz R, Muchowski PJ. Targeted deletion of kynurenine 3-monooxygenase in mice: a new tool for studying kynurenine pathway metabolism in periphery and brain. J Biol Chem. 2013;288:36554–36566. doi: 10.1074/jbc.M113.503813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Badawy AA, Guillemin G. The Plasma [Kynurenine]/[Tryptophan] Ratio and Indoleamine 2,3-Dioxygenase: Time for Appraisal. Int J Tryptophan Res. 2019;12:1178646919868978. doi: 10.1177/1178646919868978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonaccorso S, Marino V, Biondi M, Grimaldi F, Ippoliti F, Maes M. Depression induced by treatment with interferon-alpha in patients affected by hepatitis C virus. J Affect Disord. 2002;72:237–241. doi: 10.1016/s0165-0327(02)00264-1. [DOI] [PubMed] [Google Scholar]
- 28.Beurel E, Toups M, Nemeroff CB. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron. 2020;107:234–256. doi: 10.1016/j.neuron.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kopra E, Mondelli V, Pariante C, Nikkheslat N. Ketamine's effect on inflammation and kynurenine pathway in depression: A systematic review. J Psychopharmacol. 2021;35:934–945. doi: 10.1177/02698811211026426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osimo EF, Pillinger T, Rodriguez IM, Khandaker GM, Pariante CM, Howes OD. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain Behav Immun. 2020;87:901–909. doi: 10.1016/j.bbi.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halaris A, Myint AM, Savant V, Meresh E, Lim E, Guillemin G, Hoppensteadt D, Fareed J, Sinacore J. Does escitalopram reduce neurotoxicity in major depression? J Psychiatr Res. 2015;66-67:118–126. doi: 10.1016/j.jpsychires.2015.04.026. [DOI] [PubMed] [Google Scholar]
- 32.Quak J, Doornbos B, Roest AM, Duivis HE, Vogelzangs N, Nolen WA, Penninx BW, Kema IP, de Jonge P. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology. 2014;45:202–210. doi: 10.1016/j.psyneuen.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Arteaga-Henriquez G, Burger B, Weidinger E, Grosse L, Moll N, Schuetze G, Schwarz M, Wijkhuijs A, Op de Beeck G, Berghmans R, Versnel MA, Arolt V, Müller N, Drexhage HA. Activation and deactivation steps in the tryptophan breakdown pathway in major depressive disorder: A link to the monocyte inflammatory state of patients. Prog Neuropsychopharmacol Biol Psychiatry. 2021;107:110226. doi: 10.1016/j.pnpbp.2020.110226. [DOI] [PubMed] [Google Scholar]
- 34.Ogyu K, Kubo K, Noda Y, Iwata Y, Tsugawa S, Omura Y, Wada M, Tarumi R, Plitman E, Moriguchi S, Miyazaki T, Uchida H, Graff-Guerrero A, Mimura M, Nakajima S. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci Biobehav Rev. 2018;90:16–25. doi: 10.1016/j.neubiorev.2018.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Kuwano N, Kato TA, Setoyama D, Sato-Kasai M, Shimokawa N, Hayakawa K, Ohgidani M, Sagata N, Kubo H, Kishimoto J, Kang D, Kanba S. Tryptophan-kynurenine and lipid related metabolites as blood biomarkers for first-episode drug-naïve patients with major depressive disorder: An exploratory pilot case-control study. J Affect Disord. 2018;231:74–82. doi: 10.1016/j.jad.2018.01.014. [DOI] [PubMed] [Google Scholar]
- 36.Liu JJ, Raynal S, Bailbé D, Gausseres B, Carbonne C, Autier V, Movassat J, Kergoat M, Portha B. Expression of the kynurenine pathway enzymes in the pancreatic islet cells. Activation by cytokines and glucolipotoxicity. Biochim Biophys Acta. 2015;1852:980–991. doi: 10.1016/j.bbadis.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Al-Hakeim HK, Twayej AJ, Al-Dujaili AH, Maes M. Plasma Indoleamine-2,3-Dioxygenase (IDO) is Increased in Drug-Naï ve Major Depressed Patients and Treatment with Sertraline and Ketoprofen Normalizes IDO in Association with Pro-Inflammatory and Immune- Regulatory Cytokines. CNS Neurol Disord Drug Targets. 2020;19:44–54. doi: 10.2174/1871527319666200102100307. [DOI] [PubMed] [Google Scholar]
- 38.Raison CL. Forward. In: Berk M, Leboyer M, Sommer IE. Immunopsychiatry: Facts and Prospects. Switzerland: Springer Nature, 2021. [Google Scholar]
- 39.Ciranna L. Serotonin as a modulator of glutamate- and GABA-mediated neurotransmission: implications in physiological functions and in pathology. Curr Neuropharmacol. 2006;4:101–114. doi: 10.2174/157015906776359540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aristieta A, Morera-Herreras T, Ruiz-Ortega JA, Miguelez C, Vidaurrazaga I, Arrue A, Zumarraga M, Ugedo L. Modulation of the subthalamic nucleus activity by serotonergic agents and fluoxetine administration. Psychopharmacology (Berl) 2014;231:1913–1924. doi: 10.1007/s00213-013-3333-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ugedo L, De Deurwaerdère P. Serotonergic control of the glutamatergic neurons of the subthalamic nucleus. Prog Brain Res. 2021;261:423–462. doi: 10.1016/bs.pbr.2020.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Miyamoto Y, Yamada K, Noda Y, Mori H, Mishina M, Nabeshima T. Hyperfunction of dopaminergic and serotonergic neuronal systems in mice lacking the NMDA receptor epsilon1 subunit. J Neurosci. 2001;21:750–757. doi: 10.1523/JNEUROSCI.21-02-00750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fullana N, Gasull-Camós J, Tarrés-Gatius M, Castañé A, Bortolozzi A, Artigas F. Astrocyte control of glutamatergic activity: Downstream effects on serotonergic function and emotional behavior. Neuropharmacology. 2020;166:107914. doi: 10.1016/j.neuropharm.2019.107914. [DOI] [PubMed] [Google Scholar]
- 44.Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, Pereira EFR, Albuquerque EX, Thomas CJ, Zarate CA Jr, Gould TD. Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol Rev. 2018;70:621–660. doi: 10.1124/pr.117.015198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zanos P, Gould TD. Intracellular Signaling Pathways Involved in (S)- and (R)-Ketamine Antidepressant Actions. Biol Psychiatry. 2018;83:2–4. doi: 10.1016/j.biopsych.2017.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]