

Summary
The glycerol-3-phosphate shuttle (G3PS) is a major NADH shuttle that regenerates reducing equivalents in the cytosol and produces energy in the mitochondria. Here, we demonstrate that G3PS is uncoupled in kidney cancer cells where the cytosolic reaction is ~4.5 times faster than the mitochondrial reaction. The high flux through cytosolic glycerol-3-phosphate dehydrogenase (GPD) is required to maintain redox balance and support lipid synthesis. Interestingly, inhibition of G3PS by knocking down mitochondrial GPD (GPD2) has no effect on mitochondrial respiration. Instead, loss of GPD2 upregulates cytosolic GPD on a transcriptional level and promotes cancer cell proliferation by increasing glycerol-3-phosphate supply. The proliferative advantage of GPD2 knockdown tumor can be abolished by pharmacologic inhibition of lipid synthesis. Taken together, our results suggest that G3PS is not required to run as an intact NADH shuttle but is instead truncated to support complex lipid synthesis in kidney cancer.
Graphical Abstract
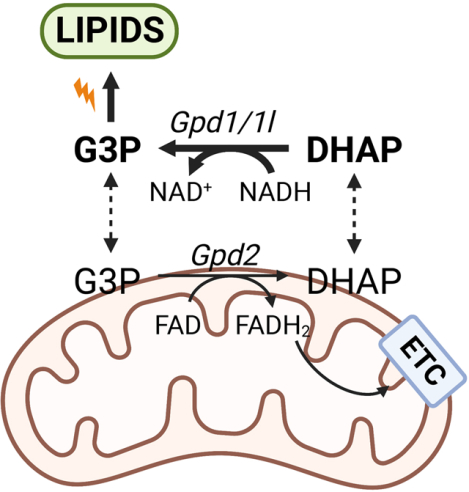
In Brief
Yao et al. demonstrate that the glycerol-3-phosphate shuttle is uncoupled in kidney cancers where the cytosolic reaction is faster than the mitochondrial reaction. High flux through cytosolic Gpd is important for redox homeostasis and lipid synthesis. Loss of mitochondrial Gpd promotes tumor growth and sensitizes tumors to lipid synthesis inhibitors
Introduction
Rapidly dividing cells upregulate glycolysis to support cellular proliferation1. To sustain high rates of glycolysis, NADH produced by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) needs to be promptly oxidized to regenerate NAD+ 2. Lactate fermentation has been implicated as a way to recycle reducing equivalents3,4. Interestingly, a recent study suggests that lactate dehydrogenase (LDH) operates as secondarily when mitochondrial NAD+ regeneration pathways are saturated5. Indeed, oxidizing NADH inside the mitochondria provides energy without the expense of secreting carbons as lactate. Because the inner mitochondrial membrane is impermeable to NAD+ and NADH, shuttles are required for the exchange of reducing equivalents between mitochondria and cytosol6.
The glycerol-3-phosphate shuttle (G3PS) is one of the two major NADH shuttles, along with the malate-aspartate shuttle (MAS). In G3PS, NAD+ is regenerated by cytosolic glycerol-3-phosphate dehydrogenase (GPD) by converting dihydroxyacetone phosphate (DHAP) into glycerol-3-phosphate (G3P). G3P freely diffuses into the intermembrane space of mitochondria and is oxidized by mitochondrial GPD2. GPD2 transfers electrons to the coenzyme Q, acting as an alternative route for the electron transport chain (ETC). In addition, G3P is the backbone for glycerolipids. Therefore, G3PS is a metabolic hub connecting glycolysis, lipid synthesis and oxidative phosphorylation.
The importance of G3PS in cancer is less understood compared to MAS7–9. Studies have reported confounding data on the expression of G3PS components in cancer. Cytosolic GPD1 is downregulated in breast cancer10,11 but enriched in brain tumor stem cells12. By contrast, mitochondrial GPD2 expression is increased in prostate cancer13,14. The differential expression of the cytosolic and mitochondrial GPDs suggests that G3PS may be dysregulated in cancer.
Here we analyzed the expression of cytosolic and mitochondrial GPDs using human TCGA datasets and found that the G3PS is uncoupled in a subset of cancers, including kidney cancer. Metabolic flux analysis determined that the rate of cytosolic reaction was ~4.5 times higher than that of the mitochondrial reaction in kidney cancer cells. This high flux through cytosolic GPD1/1L supported the synthesis of G3P and complex lipids. Inhibition of mitochondrial GPD2 promoted cell proliferation through upregulation of cytosolic GPD1/1L. The proliferative advantage of low GPD2 expressing tumor was abrogated by inhibition of lipid synthesis. These results reveal that G3PS may not be required to function as an intact NADH shuttle system in cancer, but rather functions as a critical orchestrator of lipid synthesis.
Results
Expression of cytosolic GPD1 is negatively correlated with mitochondrial GPD in cancer
In G3PS (Figure 1A), the conversion of DHAP into G3P is catalyzed by cytosolic enzymes GPD1 and GPD1L which are ~70% identical in protein sequence and redundant in function15. G3P is converted back to DHAP by mitochondrial GPD2. MAS involves more enzymes, including malate dehydrogenase 1 (MDH1) and glutamic-oxaloacetic transaminase 1 (GOT1) in the cytosol and malate dehydrogenase 2 (MDH2) and glutamic-oxaloacetic transaminase 1 (GOT2) in mitochondria (Figure S1A). Both shuttle systems contribute to the regeneration of reducing equivalents in the cytosol and energy production in the mitochondria.
Figure 1. Correlation analysis and metabolic flux analysis suggest that G3PS is uncoupled in kidney cancer.
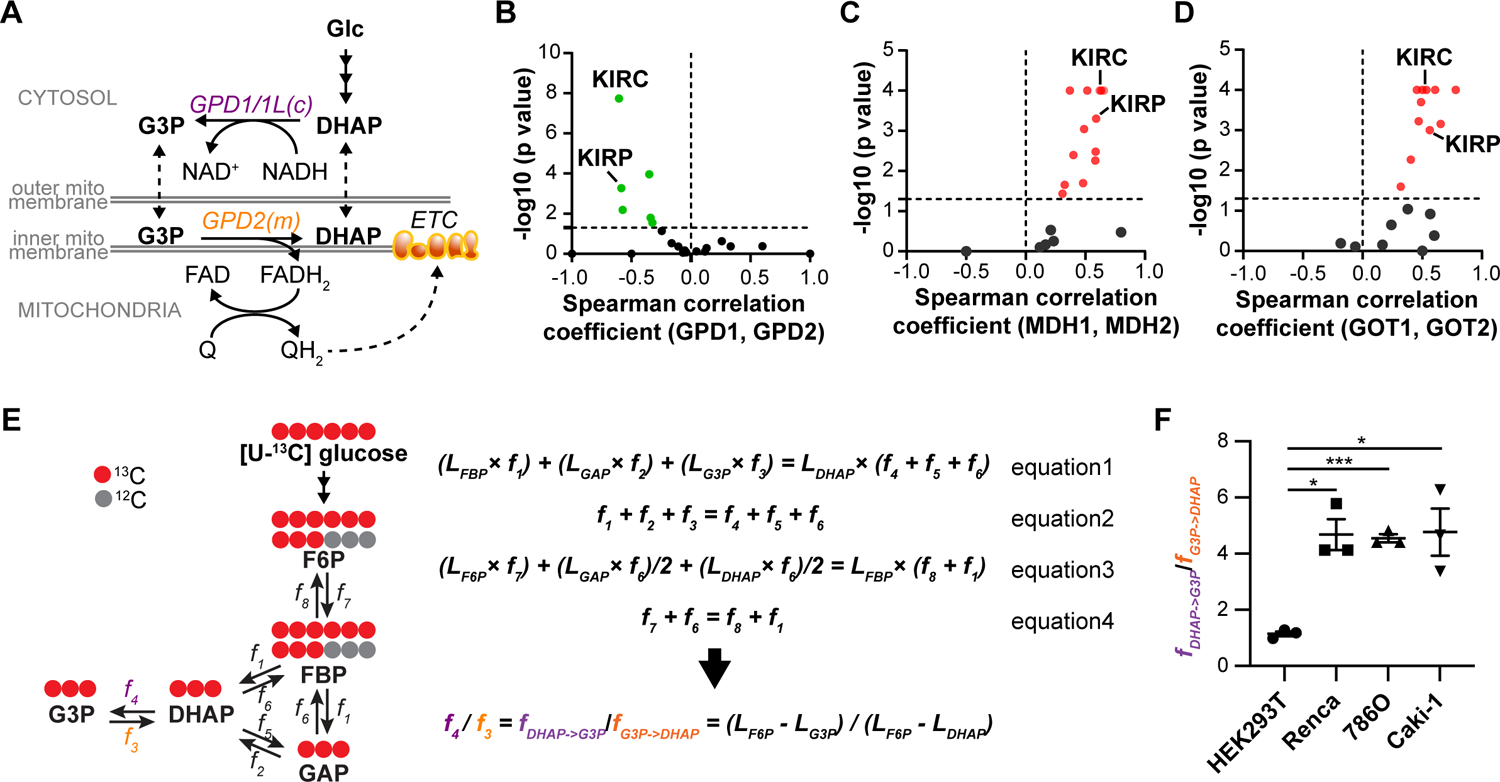
(A) Diagram of G3PS.
(B) Spearman correlation analysis of GPD1 and GPD2 expression in tumors (normalized to paired normal tissue). Green dots represent negative coefficients with p value <0.05.
(C) Spearman correlation analysis of MDH1 and MDH2 expression in tumors (normalized to paired normal tissue). Red dots represent positive coefficients with p value < 0.05.
(D) Spearman correlation analysis of GOT1 and GOT2 expression in tumors (normalized to paired normal tissue). Red dots represent positive coefficients with p value < 0.05.
(E) Metabolic flux analysis of G3PS.
(F) The ratio of cytosolic reaction rate over mitochondrial reaction rate of G3PS (fDHAP->G3P/fG3P->DHAP) in kidney cell lines.
Data are presented as mean ± SEM. *p<0.05, ***p<0.001. Experiments were performed with n=3 per condition at least two independent times.
To maintain a coupled shuttle system, we reasoned the rate of the cytosolic reaction would be balanced with the mitochondrial reaction to avoid the depletion or accumulation of shuttle intermediates. We hypothesized that cancer cells may coregulate the expression of GPD1 and GPD2 in the same direction to modulate G3PS activity. To test this idea, we performed a Spearman correlation analysis on gene expression from cancer patients by using the Cancer Genome Atlas (TCGA) database as well as from normal tissue with the Genotype-Tissue Expression (GTEx) dataset. As a proof of concept, we examined GPDs expression in energy-demanding normal tissues, such as skeletal muscle, heart and brain, that have been shown to rely on NADH shuttles to produce energy7,16. GPD1 and GPD2 had a strong positive correlation in these tissues, suggestive of a coupled G3PS system (Figure S1B).
Surprisingly, we observed a negative correlation between GPD1 and GPD2 in tumor (relative to matched normal tissue from the same patient) of many cancers, including kidney renal clear cell carcinoma (KIRC) and kidney renal papillary cell carcinoma (KIRP) (Figure 1B). For those cancers, patients with lower expression of GPD1 tended to have a higher expression of GPD2 and vice versa. By contrast, cytosolic and mitochondrial (MDH1/MDH2, GOT1/GOT2) components of MAS had strong positive correlation in both cancer and normal tissue (Figure 1C–D, S1C–D), indicating a coupled MAS. Taken together, these results imply that, unlike MAS, G3PS may be uncoupled in certain cancers.
Metabolic flux analysis reveals uncoupled G3PS in kidney cancer cells
To probe if the G3PS is uncoupled in cancer, we focused on KIRC, the strongest negatively correlated cancer type (Figure 1B). In KIRC patients, the expression of GPD1 is comparable between normal tissue and tumor whereas GPD2 expression is reduced in tumor (Figure S1E–F). To quantitatively assess the coupling of G3PS, we applied isotope-based metabolic flux analysis to directly measure the relative rate of the cytosolic reaction to the mitochondrial reaction. We performed studies in Renca mouse renal carcinoma cells, 786-O and Caki-1 human renal carcinoma cell lines, as well as in a non-cancerous human kidney cell line-HEK293T. We labeled cells with uniformly 13C-labeled glucose ([U-13C] glucose) until the labeling of metabolites reached steady state. At steady state, the labeling percentage of DHAP remained constant. Therefore, the influx of 13C from all three routes (FBP->DHAP, GAP->DHAP, G3P->DHAP) would equal the efflux of 13C (DHAP->G3P, DHAP->GAP, DHAP->FBP) (equation 1) (Figure 1E). Additionally, since the pool of DHAP remained constant at metabolic steady state, the total carbon influx to DHAP was balanced with the efflux (equation 2). The mass balance of labeled and total FBP are presented as equation 3 and 4 respectively. Here, L represents labeling percentage and f represents flux.
| equation 1 |
| equation 2 |
| equation 3 |
| equation 4 |
Among these reactions, the flux from DHAP to G3P (f4) represents the activity of cytosolic reaction of G3PS while the flux from G3P to DHAP (f3) represents the mitochondrial reaction. To solve the linear equations with 8 unknowns (f1–8), we made two assumptions. First, we assumed that the equilibrium between GAP and DHAP through triosephosphate isomerase is fast based on our observation that the M+1 isotopologue of DHAP was approximately half of the percentage of labeled FBP (when cells were labeled with 1-13C glucose at isotopic steady state) (Figure S1G)17. Thus, f2 ≈ f5 and LGAP ≈ LDHAP. Secondly, we assumed that f8<<f7 based on the fact that the labeling of F6P was significantly less compared to FBP when cells were labeled with either U-13C glycerol, U-13C glutamine or U-13C lactate (Table S1). This observation is consistent with previous studies reporting the negative ΔG of phosphofructokinase17,18. Taken together, we calculated the ratio of cytosolic reaction to mitochondrial reaction of G3PS as f4/f3=(LF6P-LG3P)/(LF6P-LDHAP). Note that we observed that GAP had different labeling kinetics compared to other glycolytic intermediates suggesting a separate pool away from glycolysis (Figure S1H–I). Thus, we used the labeling of DHAP to infer the labeling of GAP.
When G3PS is coupled, f4/f3 would approach 1. Indeed, in non-cancerous HEK293T cells, this ratio was 1.1 (Figure 1F). In comparison, this ratio was significantly higher in mouse and human kidney cancer cells (Figure 1F). In Renca cells, the rate of the cytosolic reaction was 4.5-fold higher than that of the mitochondrial reaction, demonstrating an unbalanced G3PS. Thus, this increased cytosolic flux may function beyond G3PS system to pose a distinct metabolic advantage for kidney cancers.
Knockdown of cytosolic and mitochondria GPD has opposite effects on cancer cell proliferation
To interrogate the role of G3PS, we used shRNA to knockdown Gpd1/Gpd1l or Gpd2 in Renca cells (Figure 2A). Since Gpd1 and Gpd1l are redundant in function and both isoforms are expressed in Renca cells, we created double knockdown cell lines of both genes (Gpd1/1l DKD #1 and #2) to abolish the cytosolic reaction. We validated the knockdown with metabolic flux analysis (Figure 2B). Interestingly, we found that Gpd1/1l DKD significantly attenuated cell growth in vitro, whereas Gpd2 KD promoted cell proliferation (Figure 2C). To test if the same trend held true in vivo, we subcutaneously implanted the empty vector control and the KD cell lines (Gpd1/1l DKD#1 or Gpd2 KD#1) into wildtype Balb/c mice. Consistent with the in vitro data, Gpd1/1l DKD tumors grew slower than control tumors (Figure 2D) whereas Gpd2 KD tumors grew significantly faster. To control for the off-target effects of Gpd1/1l DKD #1 and Gpd2 KD #1, we overexpressed shRNA-resistant Gpd1 and Gpd2 in KD cells respectively and restored their proliferative phenotypes (Figure S2A–D).
Figure 2. Knockdown of cytosolic GPD impairs kidney cancer cell proliferation and mitochondrial respiration whereas GPD2 knockdown promotes proliferation.
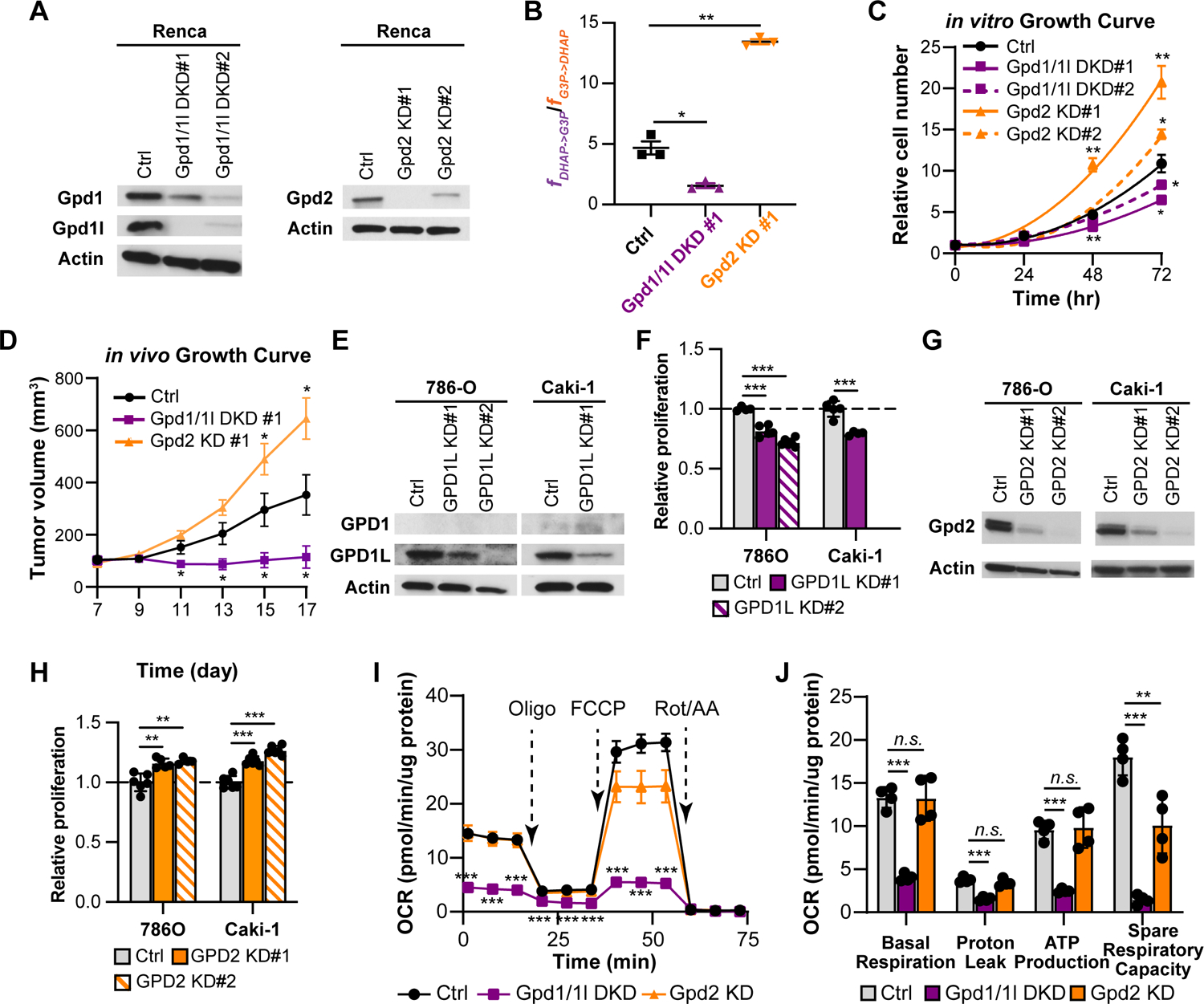
(A) Western blot analysis of Gpd1, Gpd1l, and Gpd2 in Renca knockdown cell lines. β-actin was used as a loading control.
(B) The ratio of cytosolic reaction rate over mitochondrial reaction rate of G3PS (fDHAP->G3P/fG3P->DHAP) in control, Gpd1/1l DKD, Gpd2 KD Renca cells.
(C) in vitro cell growth curves of control (ctrl) and knockdown (KD) Renca cell lines.
(D) in vivo tumor growth curves of ctrl and KD Renca cell lines.
(E) Western blot analysis of GPD1 and GPD1L in GPD1L knockdown (KD) cell lines. β-actin was used as a loading control.
(F) Relative cell numbers of ctrl and GPD1L KD cells after 3-day proliferation.
(G) Western blot analysis of GPD2 in GPD2 KD cell lines. β-actin was used as a loading control.
(H) Relative cell numbers of ctrl and GPD2 KD cells after 3-day proliferation.
(I) Oxygen consumption rate (OCR) of ctrl and KD Renca cells under basal and stress conditions. Oligo represents oligomycin. Rot/AA represents rotenone and antimycin A.
(J) Calculated parameters of mitochondrial respiration in ctrl and KD Renca cells (using results from Figure 2I).
Data are presented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, n.s. not statistically significant. Experiments were performed at least two independent times with an n=4–10 per condition.
Next, we expanded our results to human kidney cancer cells. Similar to previous findings, we observed low expression of GPD1 in these cell lines (Figure 2E, Figure S2E)19,20. Reducing the level of GPD1L alone was sufficient to reduce the proliferation (Figure 2F). Note that shRNA #2 targeting GPD1L did not yield any viable Caki-1 cells. For GPD2 KD, both shRNA constructs increased cellular proliferation (Figure 2G–H). Collectively, we show that blocking the cytosolic reaction of G3PS impairs the proliferation of kidney cancer cells, whereas blocking the mitochondrial reaction promotes proliferation. These results are consistent with the idea that G3PS is uncoupled and contributes to proliferation through a mechanism that goes beyond an intact G3PS circuit.
Gpd2 is dispensable for basal mitochondrial respiration whereas blocking Gpd1/1l inhibits respiration
G3PS oxidizes cytosolic reducing equivalents in the mitochondria to produce energy, and GPD2 transfers electrons to FAD and coenzyme Q contributing to mitochondrial respiration, which may be used as one readout of G3PS activity. We tested if the proliferative phenotypes were mediated by a change in oxidative phosphorylation. Intriguingly, Gpd2 KD had minimal effect on the oxygen consumption rate in Renca cells at basal conditions (Figure 2I). Only when cells were stressed with the proton uncoupler FCCP did we observe a significant decrease in the maximum respiration of Gpd2 KD cells (Figure 2I–J). This deficiency reflects a 43% decrease in spare respiratory capacity due to the loss of G3PS (Figure 2J). Other stress conditions, such as glucose starvation and LDH inhibition increased cellular reliance on G3PS (Figure S2F–H). In contrast, knocking down Gpd1/1l resulted in >70% decrease in the basal respiration rate compared to control cells along with decreased proton leak, ATP production and spare respiratory capacity, suggesting overall impaired mitochondrial activity (Figure 2J). These data suggest nonredundant functions for Gpd1/1l and Gpd2 on mitochondrial bioenergetics.
Loss of cytosolic Gpd impairs NAD+ regeneration and promotes aerobic glycolysis
To study how cytosolic Gpd contributes to mitochondrial respiration and cellular proliferation, we next investigated the metabolic adaptations in Gpd1/1l DKD Renca cells. Consistent with the loss of Gpd1/1l activity, we observed an elevated DHAP/G3P ratio in Gpd1/1l DKD cells (Figure 3A). Since cytosolic Gpd has been suggested to play an essential role in regenerating NAD+ 20, we hypothesized that cells may compensate for the loss of Gpd1/1l by upregulating another mechanism to regenerate reducing equivalents. Indeed, we observed a 2.5-fold increase in the glucose consumption and lactate secretion in Gpd1/1l DKD cells suggesting significantly elevated aerobic glycolysis (Figure 3B). In addition, Gpd1/1l DKD cells had increased labeling percentages in glycolytic intermediates from U-13C glucose (Figure 3C). By contrast, TCA cycle intermediates showed significantly reduced labeling percentages in Gpd1/1l DKD cells (Figure 3C). Additionally, when pyruvate dehydrogenase was inhibited with CPI-613, we observed a 70% decrease in oxygen consumption rate in control cells but minimal change in Gpd1/1l DKD cells suggesting pyruvate oxidation might be low in Gpd1/1l DKD cells (Figure S3A). Moreover, Gpd1/1l DKD cells had a 2.5-fold increase in alanine excretion (Figure S3B). This diversion of glucose carbon away from TCA cycle is consistent with our observation of impaired mitochondrial respiration in Gpd1/1l DKD cells.
Figure 3. Knockdown of Gpd1/1l leads to altered redox balance and impaired lipid synthesis in Renca cells.
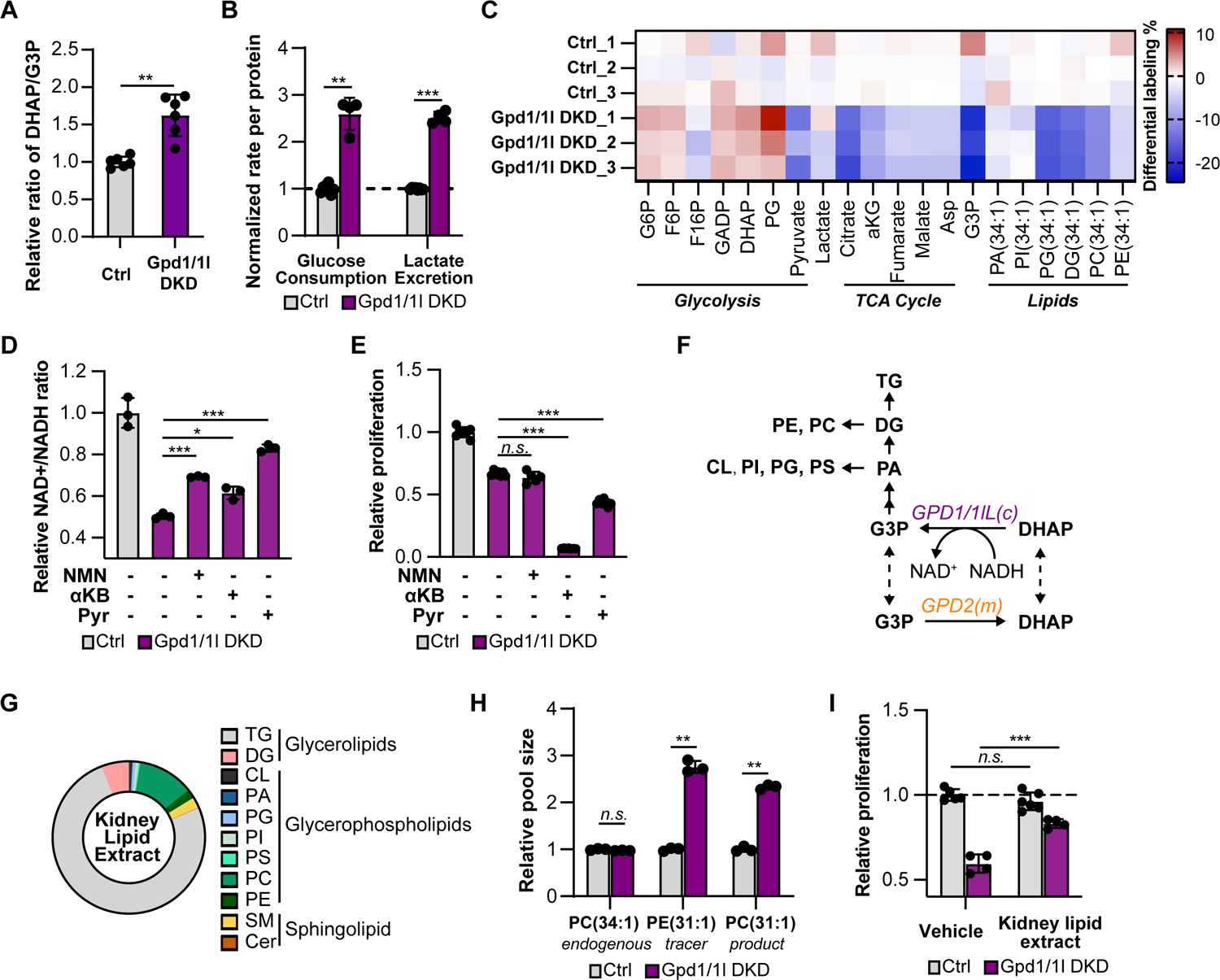
(A) The pool size ratio of DHAP to G3P in ctrl and Gpd1/1l DKD cells.
(B) Glucose consumption rate and lactate secretion rate in ctrl and Gpd1/1l DKD cells.
(C) Heat map showing the differential labeling percentages of glycolytic intermediates, TCA cycle intermediates and glycerolipid species in ctrl and Gpd1/1l DKD cells after 4hr-culture with U-13C glucose. Note that the labeling percentages in glycerolipids were the sum of odd-chain isotopologues starting from M+3.
(D) Relative NAD+/NADH ratios in ctrl and Gpd1/1l DKD cells treated with vehicle (water), 1 mM nicotinamide mononucleotide (NMN), 10 mM alpha-ketobutyrate (αKB) or 10 mM pyruvate (pyr) for 24 hours.
(E) Relative proliferation of ctrl and Gpd1/1l DKD cells treated with vehicle, 1 mM NMN, 10 mM αKB or 10 mM pyr after 3 days.
(F) Diagram of G3PS connected to complex lipid synthesis.
(G) Lipid composition in mouse kidney lipid extract.
(H) Intracellular pool sizes of endogenous PC(34:1), tracer PE(31:1) and its product PC(31:1) in ctrl and Gpd1/1l DKD cells.
(I) Relative proliferation of ctrl and Gpd1/1l DKD cells supplemented with vehicle (ethanol) or kidney lipid extract. The doubling time (DT) for ctrl+vehicle is 23.7±0.4hr. DT(ctrl+lipids) = 24.2±0.6hr. DT(Gpd1/1l DKD+vehicle) = 42±3hr. DT(Gpd1/1l DKD+lipids) = 32.8±0.6hr.
Data are presented as mean ±SEM. *p<0.05, **p<0.01, ***p<0.001, n.s. not statistically significant. Experiments were performed at least two independent times with an n=3–6 per condition.
Despite upregulated lactate production, we still observed a significant decrease in the NAD+/NADH ratio in Gpd1/1l DKD cells (Figure 3D). To test if the altered redox balance accounted for the impaired proliferation, we tried to restore the NAD+/NADH ratio with nicotinamide mononucleotide (NMN), alpha-ketobutyrate (αKB) or pyruvate (pyr). NMN is the precursor for NAD+ and can directly increase NAD+ pool21. αKB and pyruvate are electron acceptors that regenerate NAD+ by converting into αHB (alpha–hydroxybutyrate) and lactate respectively (Figure S3C)22. All three treatments significantly increased the NAD+/NADH ratio in Gpd1/1l DKD cells (Figure 3D). However, none of these treatments restored the proliferation of Gpd1/1l cells (Figure 3E) or rescued the mitochondrial respiration (Figure S3D). Taken together, these data suggest that cytosolic Gpd is important for maintaining redox balance, but restoring the redox balance alone is not sufficient to rescue Gpd1/1l DKD cells.
Impaired lipids synthesis contributes to the proliferative defect in Gpd1/1l DKD cells
Inhibition of cytosolic GPD not only blocks the regeneration of reducing equivalents but also the production of G3P. G3P provides the backbone of all glycerolipids, including structural lipids (e.g. phosphatidylethanolamine-PE, phosphatidylcholine-PC, phosphatidylinositol-PI, phosphatidylserine-PS, phosphatidylglycerol-PG, cardiolipin-CL), and signaling lipids (e.g. phosphatidic acid-PA, diacylglycerol-DG), and storage lipids (e.g. triacylglycerol-TG) (Figure 3F)23. Thus, we assessed if loss of Gpd1/1l limited the supply of G3P leading to impaired lipid synthesis. Although cells were cultured in media supplemented with physiologic levels of glycerol (300 mM), the majority of intracellular of G3P was synthesized from glucose (Table S4). Indeed, the labeling of G3P from U-13C glucose was significantly reduced in Gpd1/1l DKD cells (Figure 3C). In addition, many lipid species had decreased labeling percentages in odd-number isotopologues starting from M+3, corresponding to glycerol backbone in Gpd1/1l DKD cells (Figure 3C, Table S2). These data demonstrate that cytosolic Gpd is critical for G3P production and complex lipid synthesis.
We hypothesized that the shortage in lipid synthesis accounted for the proliferative defect in Gpd1/1l DKD cells. We failed to resuce the growth defect by providing exogenouse glycerol due to its poor uptake (Figure S3E–F). Thus, we probed if the downstream products of G3P, a mixture of complex lipids, could rescue proliferation. To mimic the lipid composition in Renca cells, we supplemented cell culture media with a lipid extract from mouse kidneys. Quantitative lipidomics showed that ~30% of the kidney lipid extract was polar lipids while the remainder was triacylglycerols (Figure 3G, Table S3). Since the mechanism of complex lipid transport into cells remains unclear24, we first confirmed that exogenous lipids could be imported and utilized by culturing cells in the presence of a lipid tracer PE(17:1/14:0) (i.e. PE(31:1)), which is composed of an odd chain fatty acid (17:1) and thus is not naturally present in mammalian cells25. We found that Gpd1/1l DKD cells had a 3-fold higher level of intracellular PE(31:1) and a 2.5-fold higher level of its downstream metabolite, PC (31:1), compared to control cells (Figure 3H). As a negative control, endogenous structural lipid PC(34:1) was present at similar level in both cells (Figure 3H). These results suggest that Gpd1/1l DKD cells not only scavenge more lipids from the media, but also readily metabolize them into complex lipids intracellularly.
After demonstrating that exogenous complex lipids could be taken up and metabolized, we supplemented cells with the kidney lipid extract which significantly rescued the proliferative defect in Gpd1/1l DKD cells (Figure 3I). Importantly, the proliferation of Gpd1/1l DKD cells was not restored by mouse heart lipid extract which was composed of predominantly neutral lipids (triacylglycerols) (Figure S3G–H, Table S3). Collectively, these data suggest that cytosolic Gpd is essential for cellular proliferation by providing G3P as the building blocks for certain polar lipids.
Gpd2 Knockdown leads to G3P accumulation and promotes complex lipid synthesis
In contrast to the defective proliferation in Gpd1/1l DKD cells, the loss of Gpd2 led to faster cell growth (Figure 2C–D). Interestingly, in both Gpd2 KD cell lines, we observed higher expression of Gpd1 on the mRNA and protein levels (Figure 4A–B). In comparison, Gpd1l expression remained similar in line with the absence of correlation between GPD1L and GPD2 (Figure 4A–B and Figure S4A). Higher Gpd1 expression increased G3P production in Gpd2 KD cells evidenced by a 30% higher labeling percentage in G3P from U-13C glucose whereas DHAP labeling percentage remained similar to controls (Figure 4C). Moreover, the pool size of G3P was 8 times higher in Gpd2 KD cells compared to control cells, whereas the DHAP pool remained similar (Figure 4D).
Figure 4. Gpd2 KD Renca cells have increased expression of Gpd1 contributing to elevated lipid synthesis and increased tumor growth.
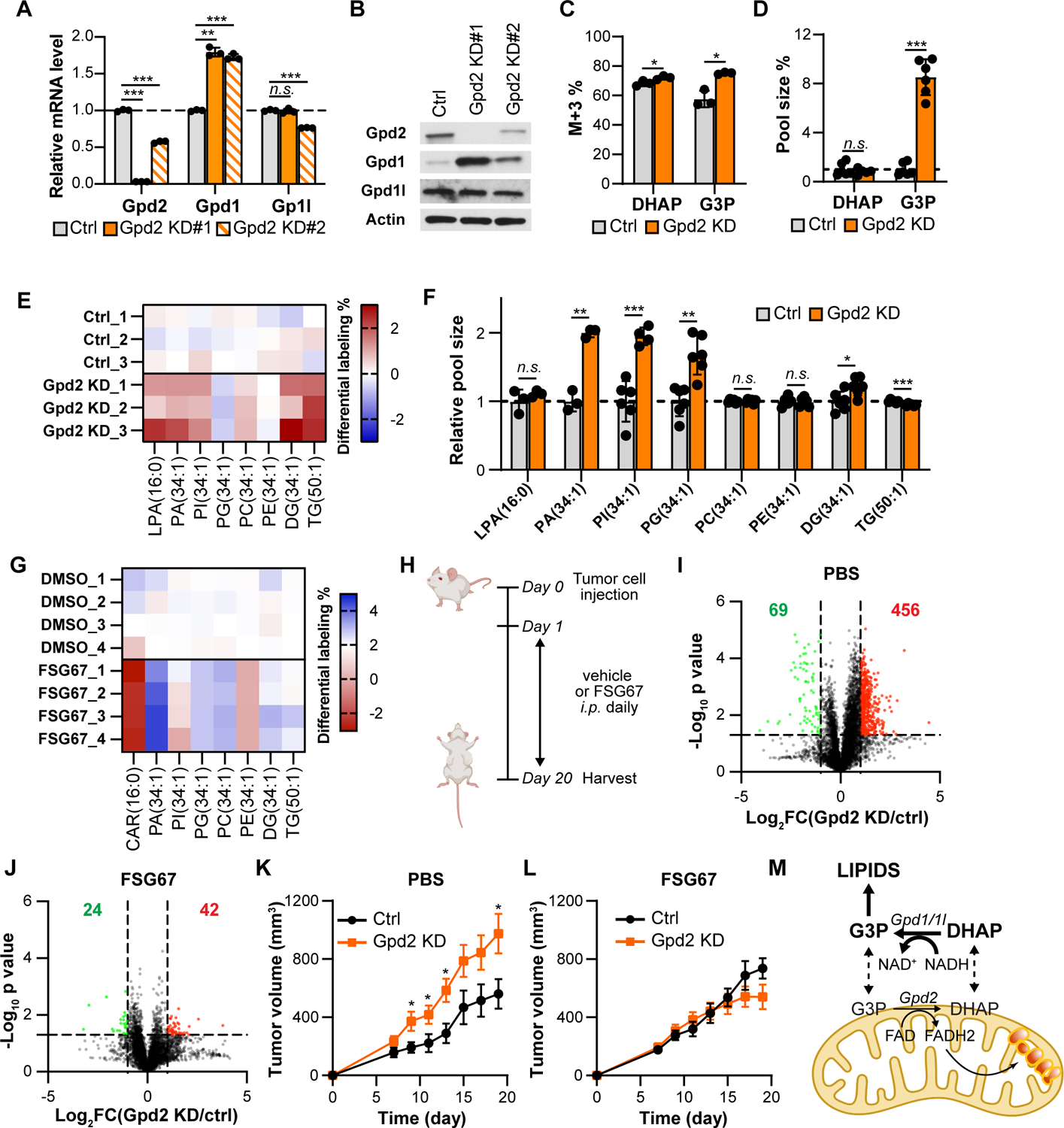
(A) Relative mRNA levels of Gpd1, Gpd1l and Gpd2 in ctrl and Gpd2 KD cells.
(B) Western blot analysis of Gpd1, Gpd1l and Gpd2 in ctrl and Gpd2 KD cells. β-actin was used as a loading control.
(C) Percentage differences of M+3 isotopologue in DHAP and G3P in ctrl and Gpd2 KD cells labeled with U-13C glucose for 4hr.
(D) Relative pool sizes of DHAP and G3P in ctrl and Gpd2 KD cells.
(E) Heat map showing the differential labeling percentage of glycerolipid species in ctrl and Gpd2 KD cells labeled with U-13C glucose for 4hr.
(F) Pool sizes of glycerolipid species in ctrl and Gpd2 KD cells.
(G) Percentages differences of 13C-labeled lipids in Gpd2 KD cells treated with vehicle control (DMSO) or FSG67 after culturing in U-13C glucose for 24hr.
(H) Experimental setup for FSG67 treatment. Ctrl and Gpd2 KD cells were implanted s.c. in the opposite flanks of the same Balb/c mouse on day 0. Vehicle ctrl (PBS) or FSG67 was injected daily i.p. from day 1.
(I-J) Volcano plot showing the log2 fold change and −log10 p value of the lipidome between control and Gpd2 KD tumors treated with PBS control (I) and FSG67 (J). Red dots represent lipid features with a fold change >2 and p value <0.05. Green dots represent lipid features with a fold change <0.5 and p value <0.05. The Red number on the right and the green number on the left represents the number of significantly upregulated and downregulated lipid features respectively.
(K) Tumor growth curves of ctrl and Gpd2 KD cells in BALB/c mice treated with PBS.
(L) Tumor growth curves of ctrl and Gpd2 KD cells in BALB/c mice treated with FSG67.
(M) Diagram summarizing the uncoupled glycerol phosphate shuttle in kidney cancers.
Data are presented as mean ±SEM. *p<0.05, **p<0.01, ***p<0.001, n.s. not statistically significant. Experiments were performed at least two independent times with an n=3–10 per condition.
We posited that the increased supply of G3P promoted lipid synthesis in Gpd2 KD cells. Indeed, Gpd2 KD cells had increased labeling percentages of numerous lipid species when labeled with U-13C glucose (Figure 4E). In addition, the pool sizes of many polar lipids (such as PA, PI, PG, and DG) were significantly larger in Gpd2 KD cells (Figure 4F). Taken together, these results suggest that the loss of Gpd2 leads to elevated expression of Gpd1 and increased supply of G3P, promoting lipid synthesis.
Inhibiting lipid synthesis is sufficient to block the proliferative advantage of GPD2 KD tumor
The majority of renal cell carcinomas have a clear cell phenotype characterized by abnormal lipid accumulation26. Even though GPD2 is mildly downregulated in KIRC tumors, lower GPD2 expression is significantly correlated with worse overall patient survival (Figure S4B, Figure S1F). The increased lipid synthesis in GPD2 KD tumors may contribute to their proliferative advantage. To test this idea, we used FSG67 to pharmacologically inhibit glycerol-3-phosphate acyltransferase (Gpat), the first step in glycerolipid synthesis (Figure S4C)27. The loss of GPD2 did not lead to a consistent change in the expression of GPAT 1–4 isoforms in cell lines tested (Figure S4D). When treated with FSG67, Renca cells had decreased labeling percentages in various lipid species from U-13C glucose (Figure 4G). This decrease was specifically attributed to the block of G3P incorporation instead of acyl chain, as the labeling of acylcarnitine which is independent of Gpat was not decreased (Figure 4G). Interestingly, cells with a lower expression of GPD2 were more sensitive to FSG67 treatment (Figure S4H–K). A fatty acid synthesis inhibitor GSK2194069 also inhibited the proliferation of GPD2 KD cells but in a less specific manner (Figure S4L–M).
We next tested if FSG67 affects the growth of Gpd2 KD tumors in vivo. Intraperitoneal injection of FSG67 led to the universal depletion of lipids in the plasma suggesting that lipid synthesis was blocked (Figure S4E)28. We treated Balb/c mice bearing a control tumor and Gpd2 KD tumor with PBS or FSG67 daily for ~3 weeks (Figure 4H). In PBS treated animals, Gpd2 KD tumors contained higher levels of lipids compared to control tumors (Figure 4I). Upon FSG67 treatment, these differences were diminished and tumors from both groups demonstrated similar lipid profiles, suggesting increased lipid synthesis was blocked in Gpd2 KD tumors (Figure 4J). We used a low concentration of FSG67 so the growth of control tumors was not significantly changed upon drug treatment (Figure 4K–L). Consistent with our hypothesis, FSG67 significantly reduced the growth of Gpd2 KD tumors down to the level of control tumors (Figure 4K–L). Similar to Renca tumors in Balb/c mice, the loss of GPD2 in 786-O tumors led to faster tumor growth and sensitized tumors to FSG67 treatment in SCID mice (Figure S4F–G). Taken together, these data demonstrate that increased tumor growth due to loss of GPD2 requires complex lipid synthesis rendering them more sensitive to GPAT inhibition.
Discussion
Compared to MAS, G3PS functions not only as a NADH shuttle system to transport cytosolic reducing equivalents into mitochondria, but also a metabolic hub connecting glycolysis, lipid synthesis and oxidative phosphorylation. Here, we present the first evidence to demonstrate that G3PS is uncoupled in a subset of human cancers (Figure 4L). Our results indicate that G3PS does not run as a complete circle in kidney cancer but rather is truncated to divert carbons to synthesize complex lipids.
Unlike LDH, cytosolic GPD regenerates 2 net NAD+ molecules per glucose by operating prior to GAPDH, the glycolytic enzyme that consumes NAD+. In comparison, LDH functions after GAPDH and thus does not contribute to net NAD+ regeneration29. Therefore, G3P synthesis presents a complementary and more efficient way to generate reducing equivalents, compared to lactate fermentation. Consistent with this notion, we found that Gpd1/1l DKD cells had 3-fold increase in lactate production yet a significantly reduced NAD+/NADH ratio suggesting that cytosolic GPD plays an essential role in maintaining redox homeostasis (Figure 3B, D).
In comparison to tumors, our correlation analysis suggests G3PS may be coupled in normal tissues, especially in energy-demanding tissues (Figure S1B). Interestingly, lipogenic normal tissues, such as liver and adipose show a negative correlation between the expression of cytosolic and mitochondrial components, implying that the coupling of G3PS may be regulated to balance cellular fuel resources into lipid synthesis or energy production.
The observed metabolic alterations might be relevant to clear cell renal cell carcinoma where majority of tumors harbor a VHL mutation leading to the constitutive stabilization of HIF and altered lipid metabolism30,31. Interestingly, when we classified TCGA data by VHL wildtype and mutant, we observed significant negative correlation in both groups, suggesting that the uncoupling of G3PS is independent of VHL status (Figure S4M–N).
Limitations of the study
The study presented here has several considerations. First, we focus on KIRC, whereas it would be important to investigate the coupling of G3PS in other cancer types and understand the relevant biological context. Next, we have not identified the specific lipid specie(s) essential and sufficient to rescue cellular proliferation of Gpd1/1l DKD cells. Moreover, a direct measurement of pryruvate oxidation is required to provide mechnismic sights on reduced respiration in Gpd1/1l cells. Finally, we calculate the flux of G3PS based on several assumptions that are detailed in the manuscript. We also observe a discrepancy between the labeling patterns of GAP and DHAP especially in kidney cancer cell lines, suggesting a separate GAP pool that equilibrates with lactate, which is yet to be validated.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Additional information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marcia Haigis. (marcia_haigis@hms.harvard.edu).
Materials availability
DNA constructs and other research reagents generated in this study will be distributed upon request to other research investigators under a Material Transfer Agreement.
Data and code availability
All datasets generated or analyzed during this study are included in the published article. Raw metabolomic datasets supporting the current study are available from the Lead Contact upon request. Western blot images have been deposited at Mendeley and are publicly available as of the date of publication through Mendeley Data. (https://data.mendeley.com/datasets/zynypyx9fj/draft?a=2d9866d8-956d-47a3-98a5-a954895bcea2).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Renca (mouse), 786-O (human), Caki-1 (human), A498 (human), HK2 (human), HepG2 (human) and HCT116 (human) cells were purchased from ATCC. Cells were cultured in DMEM without pyruvate supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. Cells were cultured at 37°C in a humidified 5% CO2 incubator.
Mice
5-week old BALB/c and NOD/SCID (NOD.Cg-Prkdcscid/J) female mice were purchased from Jackson Laboratories. All mouse colonies and experimental animals were maintained in the same animal facility at Harvard Medical School and housed in specific pathogen-free conditions. All animals were used in accordance with animal care guidelines from the Harvard Medical School Standing Committee on Animals and the National Institutes of Health. All mouse protocols were approved by the Harvard Medical Area Standing Committee on Animals.
METHOD DETAILS
Gene expression analysis with TCGA and GTEx dataset
Gene expression data in tumor and paired normal tissues were downloaded from the Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) by using Bioconductor 3.1. Raw counts (HTSeq-Counts) were normalized with DESeq2’s median of ratios. Primary tumor samples (samples encoded as “01” in TCGA data) were compared to their matched normal tissue (samples encoded as “11” in TCGA data) from the same patient. Tumor samples without matched normal tissue were excluded from further analysis. Fold change was calculated as the ratio of tumor to paired normal tissue in the same patient. Spearman correlation analysis of different genes in normal tissues was performed with GEPIA online tool with GTEx dataset.
Knockdown and overexpression of Gpd1, Gpd1l and Gpd2
shRNA constructs targeting mouse Gpd1l, mouse Gpd2 and human Gpd1l were cloned into the pMKO-GFP vector. pMKO-GFP empty vector was used as a control. shRNA constructs targeting mouse Gpd1, human Gpd1 and human Gpd2 cloned into the pLKO.1 vector were purchased from Horizon. pLKO.1 empty vector was used as a control. To create stable knockdown in mouse cells, shRNA constructs were transfected into Platinum-E (Cell Biolabs) with TurboFect (Thermo Fisher) and retroviruses were collected to infect Renca cells. To create stable knockdown in human cells, shRNA constructs were co-transfected into HEK293T cells with pRSV-Rev, pMD2-VSVG, and pMDLg/pRRE plasmids. Lentiviruses were collected to infect 786-O and Caki-1 cells. Successfully transduced cells were selected by sorting for GFP positive cells and/or with puromycin (2μg/mL). To rescue the knockdowns with overexpression of Gpd1 and Gpd2, the codon was optimized to be resistant to the shRNA used. The shRNA-resistant cDNA (GenScript) was cloned into a pcDNA3.1+ vector. pcDNA3.1+ empty vector was used as a control. Knockdown cells were transduced with overexpression plasmids using lipofectamine 3000 and assayed after 3–4 days post transduction.
Immunoblotting
Cells were trypsinized and collected by centrifugation. The cell pellets were resuspended in RIPA lysis buffer supplemented with EDTA-free protease inhibitor (Sigma), phosphatase inhibitor cocktail 2 (Sigma), and phosphatase inhibitor cocktail 3 (Sigma). Protein concentrations were determined by BCA assay (Thermo Fisher). Equal amounts of protein were loaded on a TGX 4%–20% gel (Biorad) and separated by SDS-PAGE under reducing conditions. Proteins were then transferred onto a nitrocellulose membrane and analyzed by immunoblotting. Beta-actin was used as a loading control.
Drug treatment, growth curve and proliferation assay
For growth curves, cells were seeded at low density and were counted by hemocytometer or flow cytometry every day up to 5 days. For proliferation assays, cells were plated a day prior to treatment and counted after 3–4 days of proliferation. The kidney lipid extract was prepared by extracting the kidney from an 8-week-old female BALB/c mouse with water/methanol/chloroform. The chloroform phase containing lipids was dried down and reconstituted in ethanol prior adding to cells. NMN (Sigma), aKB (Sigma) and glycerol (Sigma) were dissolved in water with pH adjusted to 7. PE(31:1) (Avanti) and heart lipid extract (Avanti) was dissolved in ethanol. FSG67 (Focus Biomolecules) was dissolved in DMSO. CPI-613 (APExBIO), GSK2837808A and GSK2194069 (Cayman) was dissolved in DMSO.
Mouse tumor models and drug treatment
6-week-old female Balb/c mice were anesthetized with 2.5% Avertin diluted in DPBS. 2.5×105 Renca cells (or indicated cells) were subcutaneously implanted in the flanks. 7 days post injection, tumors were measured with a caliper every 2–3 days. Tumor volumes were calculated using the following formula for ellipsoid volume: 0.5 × D × d2, where D is the long and d is the short diameter. For FSG67 treatment, 2.5×105 control cells and Gpd2 KD cells were subcutaneously implanted on each side of the abdominal flank of the same mouse. 1mg/mL FSG67 solution was made by dissolving FSG67 in PBS and sterile filtered through a 0.2μm filter. 100μL of FSG67 solution was administrated daily by intraperitoneal injection beginning one day after tumor injection.
Relative metabolic flux analysis
Cells were incubated with 5 mM uniformly 13C (U-13C) labeled glucose for 4 hr to reach isotopic steady state. The isotopologues of relevant metabolites were measured by liquid chromatography coupled mass spectrometry (LC-MS). The labeling percentages were corrected for natural abundance and isotope impurity. We only considered M+3 isotopologue for LG3P and LDHAP. We considered M+3 and M+6 isotopologues for LF6P. To assess gluconeogenesis, we labeled the cells with 0.5mM U-13C glycerol for 4hr, 5mM U-13C lactate for 12hr or 2mM U-13C glutamine for 24hr respectively. Since DHAP and G3P are freely diffused through outer mitochondrial membrane, the labeling percentages measured represent the one combined pool of each compound. See Table S4 for the isotopologue distribution for G3P, DHAP and F6P, which was used for metabolic flux analysis.
Oxygen consumption assays
The oxygen consumption rate (OCR) of cells was determined by using the Seahorse XF Extracellular Flux Analyzer (Seahorse Bioscience) and a XF Cell Mito Stress Test Kit. Cells were trypsinized and plated on a miniplate with the same seeding density 24 hours prior to the Seahorse assay. The assay medium consisted of 5 mM glucose, 500 μM glutamine, 250 μm pyruvate and 500 μM glycerol in Seahorse base medium. The OCR was monitored upon serial injections of oligomycin (oligo, 2 μM), FCCP (1 μM, optimized), and a rotenone/antimycin A mixture (rot/AA, 1 μM). OCR was normalized to the final protein amount in each well as determined by BCA assay. Data presented were corrected for non-mitochondrial respiration.
Metabolomic and lipidomic analysis
Cells, plasma, and tumor samples were quenched and extracted with a mixture of water, methanol and chloroform. The water-methanol phase containing polar metabolites was dried down and reconstituted with water/acetonitrile (1:1) with the volume normalized to cell number, volume, or tumor wet weight. The chloroform phase containing lipids was dried down and reconstituted with methanol/chloroform (9:1). The polar extract was separated on an iHILIC column (5 μm, 150 mm × 2.1 mm I.D., Nest Group). The lipid extract was separated on a Kinetex evo C18 column (2.6 um, 150 mm × 2.0 mm I.D., Phenomenex)32,33. Both columns were coupled to an Agilent 6546 LC/Q-TOF with an ESI source operated in negative and/or positive mode. The identity of each metabolite was confirmed by matching retention time and MS/MS fragmentation data to standard compounds and/or a database. For isotope tracing experiments, labeling percentages were corrected for natural abundance and isotope impurities from the tracer.
Media uptake and excretion analysis
After incubating cells in fresh media for 24 hours, the spent media was collected and analyzed. Known concentrations of U-13C internal standards (glucose, lactate, glutamine, glutamate, and palmitate; Cambridge Isotopes) were spiked into media samples before extraction34. Extractions were performed in glass to avoid plastic contamination as previously reported35. Samples were measured by LC/MS analysis, with the method described above. For each compound, the absolute concentrations were determined by calculating the ratio between the fully unlabeled peak from samples and the fully labeled peak from standards. The consumption rates (x) were normalized by cell growth over the experimental time period by using the equation below. N0 represents the starting cell number, t represents incubation time, DT represents doubling time, and Y represents nutrient utilization:
NAD+ and NADH assay
Cells were treated with drugs 24 hr prior to the assay. NAD+/NADH measurements were performed by using an NAD/NADH assay kit (Abcam) following manufacturer’s instructions. Briefly, cells were washed with PBS and extracted with provided extraction buffer by freeze/thaw cycles. After centrifugation, supernatants were equally divided into two parts. One part was assayed for total NAD+ and NADH amount. The other part was subjected to 60°C heating to degrade NAD+. All samples were incubated with NAD cycling enzyme and developer. The absolute quantitation was achieved by making a standard curve.
Real-time PCR analysis
RNA was extracted with RNeasy Mini Kit (Qiagen). cDNA was synthesized with an iScript cDNA synthesis kit (Bio-rad). Amplifications were run with RT-PCR by using premade primers (IDT). The results were normalized to a housekeeping gene, beta-actin.
Quantification and Statistical Analysis
Statistical analysis was performed using GraphPad Prism (v10.0.7) for Mac (GraphPad Software), R statistical programming language (v4.0.3, R-project.org) or Excel. Statistical values including the exact n, statistical test, and significance are reported in the Figure Legends. Statistical significance was defined as *p < 0.05 and unless indicated otherwise determined by 2-tailed Student’s t test or one-way ANOVA.
Supplementary Material
Table S2 (Related to Figure 3) Isotopologue distribution pattern of lipids when cells were labeled with U-13C glucose
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GPD1 | Proteintech | 27943-1 |
| GPD1L | Proteintech | 17263-1 |
| GPD2 | Proteintech | 17219-1 |
| Beta-ACTIN | Cell Signaling | 13E5 |
| Rabbit IgG HRP Linked Whole Ab | GE Healthcare | NA934V |
| Chemicals, peptides, and recombinant proteins | ||
| U-13C Glucose | Cambridge Isotope Laboratories | CLM-1396-2 |
| U-13C Glutamine | Cambridge Isotope Laboratories | CLM-1822-H |
| U-13C Glycerol | Cambridge Isotope Laboratories | CLM-1510-1 |
| U-13C Lactate | Cambridge Isotope Laboratories | CLM1579-0.5 |
| 1–13C Glucose | Cambridge Isotope Laboratories | CLM-420-0.5 |
| β-Nicotinamide mononucleotide | Sigma | N3501 |
| Sodium pyruvate | Thermo Fisher Scientific | 11360070 |
| 2-Ketobutyric acid | Sigma | K401 |
| PE(17:0-14:1) | Avanti | LM1104 |
| FSG67 | Focus biomolecules | 10-4577 |
| GSK2837808A | Cayman Chemical | 20626 |
| CPI-613 | APExBIO | A4333 |
| Glycerol | Acros | 327255000 |
| Puromycin | Invivogen | ant-pr-1 |
| Hear lipid extract | Avanti | 171201P |
| GSK2194069 | Cayman Chemical | 20022 |
| DMEM (high glucose, glutamine, no pyruvate) | Thermo Fisher Scientific | 11965118 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | 15140122 |
| iScript cDNA Synthesis Kit | BioRad | 1708891 |
| PerfeCTa SYBR® Green FastMix | Quantabio | 101414-270 |
| Buffer RLT | QIAGEN | 79216 |
| TurboFect™ Transfection Reagent | Thermo Scientific | R0533 |
| Fetal Bovine Serum (FBS) | Corning | 35-010-CV |
| Seahorse XF DMEM Medium | Agilent | 103575-100 |
| XF Mito Stress Test | Agilent | 103015-100 |
| Critical commercial assays | ||
| Western Lightning ECL | Perkin Elmer | NEL120001EA |
| BCA Protein Assay Kit | Thermo Fisher | 23227 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| NAD/NADH Cell-Based Assay Kit | Cayman Chemical | 600480 |
| Experimental models: Cell lines | ||
| Mouse: Renca | ATCC | CRL-2947 |
| Human: 786-O | ATCC | CRL-1932 |
| Human: Caki-1 | ATCC | HTB-46 |
| Human: A498 | ATCC | HTB-44 |
| Human: RPTEC | ATCC | PCS-400-010 |
| Human: HK2 | ATCC | CRL-2190 |
| Human: HepG2 | ATCC | HB-8065 |
| Human: HCT116 | ATCC | CCL247 |
| Human: HEK293T | ATCC | CRL-3216 |
| Human: Platinum-E | Cell Biolabs | RV-101 |
| Experimental models: Organisms/strains | ||
| BALB/c | Jackson Laboratory | 000651 |
| NOD/SCID | Jackson Laboratory | 001303 |
| Oligonucleotides | ||
| Gpd1l qPCR primers (mouse): Fwd- AACGTGAAATATCTCCCAGGAC Rev- TCTTGTGGATGAACTGGTGAG |
IDT | This manuscript |
| Gpd1 qPCR primers (mouse): Fwd- ACCCAACTTTCGCATCACT Rev- TGTCACCGAAGCCAAGC |
IDT | This manuscript |
| Gpd2 qPCR primers (mouse): Fwd- CATGAACTTGGATGTTGAGCAG Rev- AGTGGAAGCATGATAGGCAAC |
IDT | This manuscript |
| Actb qPCR primers (mouse): Fwd- GATTACTGCTCTGGCTCCTAG Rev- GACTCATCGTACTCCTGCTTG |
IDT | This manuscript |
| Gpam (Gpat1) qPCR primers (mouse): Fwd- TTCTTATCTGCCCAGTTCATCG Rev- GCTCTCCTTCCATTTCAGTGT |
IDT | This manuscript |
| Gpat2 qPCR primers (mouse): Fwd- GTGAAGAAAGAGGTACAGCGT Rev- CCATCTTCATCTGTCCCTTGT |
IDT | This manuscript |
| Agpat9 (Gpat3) qPCR primers (mouse): Fwd- ACCATAACAAGCAGTACAGACC Rev- GCTCTCTGAATGATCCCCATC |
IDT | This manuscript |
| Agpat6 (Gpat4) qPCR primers (mouse): Fwd- GAAATTGGAGCCACTGTTTACC Rev- GTCACCATGCCATACTTGCT |
IDT | This manuscript |
| GPAM (GPAT1) qPCR primers (human): Fwd- GCCATAACATCAAAGCACCATAC Rev- AGAACATCTTTCCGTCCATCTG |
IDT | This manuscript |
| GPAT2 qPCR primers (human): Fwd- CTTCCCTTGAGCAGTCCAC Rev- CACTCTCCTTCTCCCCACA |
IDT | This manuscript |
| AGPAT9 (GPAT3) qPCR primers (human): Fwd- GCTATTGTGTCCTACTGCCT Rev- CCAGTTCACTCAGCCAGTT |
IDT | This manuscript |
| AGPAT6 (GPAT4) qPCR primers (human): Fwd- GTACCTGCCTCCCATGACTA Rev- TCCTTCACCTTCTCCCTCTTC |
IDT | This manuscript |
| Gpd1l shRNA #1 (mouse): Fwd- CCGGCCACAAGATCTGCGATGAGATCTCGA GATCTCATCGCAGATCTTGTGGTTTTTG Rev- AATTCAAAAACCACAAGATCTGCGATGAGAT CTCGAGATCTCATCGCAGATCTTGTGG |
IDT | This manuscript |
| Gpd1l shRNA #2 (mouse): Fwd- CCGGGCTGACAGACATAATCAACAACTCGA GTTGTTGATTATGTCTGTCAGCTTTTTG Rev- AATTCAAAAAGCTGACAGACATAATCAACAA CTCGAGTTGTTGATTATGTCTGTCAGC |
IDT | This manuscript |
| Gpd1 shRNA #1 (mouse): Fwd- CCGGGCCAGCTTGATATTAAACTAACTCGAG TTAGTTTAATATCAAGCTGGCTTTTTG Rev- AATTCAAAAAGCCAGCTTGATATTAAACTAAC TCGAGTTAGTTTAATATCAAGCTGGC |
Horizon | This manuscript |
| Gpd2 shRNA #1 (mouse): Fwd- CCGGCGTGAGAGCCAAATGCGTTATCTCGA GATAACGCATTTGGCTCTCACGTTTTTG Rev- AATTCAAAAACGTGAGAGCCAAATGCGTTAT CTCGAGATAACGCATTTGGCTCTCACG |
IDT | This manuscript |
| Gpd2 shRNA #2 (mouse): Fwd- CCGGGCCACGAGATTTCTGTACTATCTCGA GATAGTACAGAAATCTCGTGGCTTTTTG Rev- AATTCAAAAAGCCACGAGATTTCTGTACTAT CTCGAGATAGTACAGAAATCTCGTGGC |
IDT | This manuscript |
| GPD1L shRNA #1 (human): Fwd- CCGGCGACATCATCCGAGAGAAGATCTCGA GATCTTCTCTCGGATGATGTCGTTTTTG Rev- AATTCAAAAACGACATCATCCGAGAGAAGAT CTCGAGATCTTCTCTCGGATGATGTCG |
IDT | This manuscript |
| GPD1L shRNA #2 (human): Fwd- CCGGGAAGCTAATCTCCGACATCATCTCGA GATGATGTCGGAGATTAGCTTCTTTTTG Rev- AATTCAAAAAGAAGCTAATCTCCGACATCAT CTCGAGATGATGTCGGAGATTAGCTTC |
IDT | This manuscript |
| Recombinant DNA | ||
| Mouse Gpd1 ORF | Genescript | Clone ID:OMu18825C |
| Mouse shRNA resistant Gpd2 ORF gcttccgcggacgcgtGCCACCATGGCGTTTCAAA AGGCAGTGAAGGGGACTATTCTTGTGGGTG GAGGAGCTCTGGCCACTGTTTTGGGACTCT CTCCGTTTGCTCATTACAGAAGGAAGCAAGT GAGCCTGGCATATGTGGAAGCAGCAGGATA CCTCACGGAGCCTGTGAACAGGGAACCTCC CTCCAGAGAAGCTCAGCTCATGACTTTGAAG AACACACCCGAATTTGACATCCTTGTTATCG GAGGCGGAGCCACAGGGTGTGGCTGTGCA CTAGATGCCGTCACCAGAGGACTGAAAACA GCCCTTGTAGAGAGAGATGACTTCTCATCG GGGACTAGCAGTAGAAGCACTAAATTGATCC ACGGTGGTGTGCGATACCTCCAGAAGGCTA TCATGAACTTGGATGTTGAGCAGTATAGGAT GGTGAAAGAAGCCCTTCACGAACGTGCCAA CTTACTAGAAATCGCTCCTCATTTATCAGCT CCGTTGCCTATCATGCTTCCACTTTACAAGT GGTGGCAGTTACCTTATTACTGGGTGGGAAT CAAGATGTATGACCTGGTTGCAGGGAGTCA ATGCCTGAAGAGCAGTTACGTCCTCAGCAAA TCCCGAGCCCTGGAGCATTTTCCCATGCTC CAGAAGGACAAGCTGGTAGGCGCCATTGTC TACTATGACGGACAACACAACGATGCACGG ATGAACCTCGCCATCGCCCTCACTGCTGCC AGGTACGGGGCTGCCACGGCCAATTACATG GAGGTGGTGAGCTTGCTCAAGAAGACAGAC CCTGAAACCGGCAAAGAGCGAGTGAGCGGT GCGCGGTGCAAGGATGTGCTCACAGGGCA GGAATTTGATGTCCGGGCTAAGTGTGTGATC AATGCCTCCGGCCCTTTCACAGACTCCGTG CGCAAAATGGATGATAAAAACGTTGTTCCCA TCTGCCAGCCCAGTGCCGGGGTCCATATTG TGATGCCCGGATACTACAGCCCTGAGAACA TGGGACTTCTTGATCCTGCAACCAGTGATGG CAGAGTGATTTTCTTCTTGCCTTGGGAGAAG ATGACAATTGCTGGCACCACTGATACGCCAA CGGACGTCACGCACCATCCTATTCCTTCAGA AGAAGACATTAACTTCATCCTGAATGAAGTG CGGAACTACCTGAGTTCTGACGTTGAAGTGA GAAGAGGGGATGTCTTGGCAGCCTGGAGTG GTATCCGTCCCCTTGTTACCGATCCCAAGTC TGCAGACACTCAGTCCATCTCTCGAAATCAT GTTGTGGACATCAGTGACAGCGGACTCATC ACAATAGCAGGTGGGAAGTGGACCACCTAC CGCTCCATGGCAGAAGATACCGTGGATGCA GCTGTCAAGTTTCACAACTTGAATGCGGGAC CGAGTAGGACTGTTGGGCTGTTCCTTCAAG GAGGCAAAGACTGGAGCCCCACACTCTACA TCAGGCTTGTCCAGGATTATGGGCTTGAGA GCGAGGTTGCACAACATCTGGCCAAAACCT ATGGTGACAAGGCTTTCGAGGTGGCCAAAA TGGCAAGTGTGACTGGAAAGCGGTGGCCTG TTGTTGGAGTGCGTCTTGTGTCAGAATTTCC ATACATTGAAGCAGAGGTGAAATACGGGATT AAGGAGTATGCCTGCACTGCAGTTGACATG ATCTCACGGCGCACCCGCCTGGCCTTTCTC AATGTTCAGGCTGCAGAGGAAGCCCTGCCT AGGATTGTTGAACTAATGGGAAGAGAGTTGA ACTGGAGTGAATTGAGGAAACAGGAAGAAC TTGAAACAGCTACCCGGTTCTTATATTACGA AATGGGCTACAAGTCTCGAACAGAACAACTT ACAGATAGCACTGAAATCAGCCTGCTGCCTT CAGACATCGATAGGTACAAGAAGAGATTTCA CAAGTTTGATGAAGATGAAAAAGGCTTCATT ACCATTGTTGATGTTCAGCGTGTCCTAGAGA GTATCAATGTACAAATGGACGAAAACACACT GCATGAAATTCTCTGCGAAGTAGATTTGAAC AAAAATGGACAGGTTGAGCTGCACGAGTTTC TGCAGCTGATGAGCGCAGTTCAGAAAGGAA GGGTCTCTGGAAGCCGACTTGCCATCCTGA TGAAAACTGCCGAGGAGAACTTGGACCGCA GAGTTCCAATCCCCGTGGACCGTAGTTGTG GAGGATTGTGAggatcccaattgatgc | IDT | This manuscript |
| Plasmid: pMKO.1 GFP | Addgene | 10676 |
| Plasmid: pLKO.1 | Addgene | 10878 |
| Plasmid: pcDNA3.1 | Invitrogen | V79020 |
| Plasmid: pLKO.1(shGPD2#1) | Horizon | RHS3979-201758926 |
| Plasmid: pLKO.1(shGPD2#2) | Horizon | RHS3979-201758902 |
| Software and algorithms | ||
| Prism 5 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| GEPIA (Gene Expression Profiling Interactive Analysis) | Laboratory of Z. Zhang, Peking University, Beijing, China | http://gepia.cancer-pku.cn/ |
| Agilent profinder | Agilent | https://www.agilent.com/en-us/support/software-informatics/masshunter-workstation-software/masshunter-profinder-b0600sp1 |
Highlights.
Glycerol-3-phosphate shuttle is uncoupled in some cancers such as kidney cancers
High flux through cytosolic Gpd supports lipid synthesis and cancer proliferation
Loss of mitochondrial Gpd upregulates the expression of cytosolic Gpd
Low-GPD2 expressing tumors are more sensitive to lipid synthesis inhibition
ACKNOWLEDGMENTS
C.-H. Y. was supported by AACR Anna D. Barker Basic Cancer Research Fellowship (20-40-01-YAO). This study was supported by the Ludwig Center at Harvard (M.C.H. and A.H.S.) and NIH grants U54-CA224088 (M.C.H. and A.H.S.). M.C.H received funding from NIH grants R01CA213062 and R01CA273461 and Massachusetts Life Sciences Center.
DECLARATION OF INTERESTS
M.C.H. and A.H.S. received unrelated research funding from Roche Pharmaceuticals. M.C.H. received funding from Agilent Technologies. M.C.H. and A.H.S. are advisors to Alixia Therapeutics. A.H.S is on advisory boards for Surface Oncology, SQZ Biotechnologies, Elpiscience, Selecta, Bicara and Monopteros, Bicara, and Fibrogen. She also is on scientific advisory boards for the Massachusetts General Cancer Center, Program in Cellular and Molecular Medicine at Boston Children’s Hospital, the Human Oncology and Pathogenesis Program at Memorial Sloan Kettering Cancer Center, Glaxo Smith Kline and Janssen. She is an academic editor for the Journal of Experimental Medicine. A.H.S has received unrelated funding from Novartis, Merck, AbbVie, Moderna, Vertex and Erasca. M.C.H is a Cell Metabolism and Molecular Cell advisory board member.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, Kim D, Le A, Yellen G, Albeck JG, and Locasale JW (2014). Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife 3. 10.7554/eLife.03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson DM, Goodfriend TL, and Kaplan NO (1964). LACTIC DEHYDROGENASES: FUNCTIONS OF THE TWO TYPES RATES OF SYNTHESIS OF THE TWO MAJOR FORMS CAN BE CORRELATED WITH METABOLIC DIFFERENTIATION. Science 143, 929–933. 10.1126/science.143.3609.929. [DOI] [PubMed] [Google Scholar]
- 4.Doherty JR, and Cleveland JL (2013). Targeting lactate metabolism for cancer therapeutics. J Clin Invest 123, 3685–3692. 10.1172/jci69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Stancliffe E, Fowle-Grider R, Wang R, Wang C, Schwaiger-Haber M, Shriver LP, and Patti GJ (2022). Saturation of the mitochondrial NADH shuttles drives aerobic glycolysis in proliferating cells. Molecular Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stein LR, and Imai S (2012). The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab 23, 420–428. 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mráček T, Drahota Z, and Houštěk J (2013). The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim Biophys Acta 1827, 401–410. 10.1016/j.bbabio.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Allen EL, Ulanet DB, Pirman D, Mahoney CE, Coco J, Si Y, Chen Y, Huang L, Ren J, Choe S, et al. (2016). Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep 17, 876–890. 10.1016/j.celrep.2016.09.052. [DOI] [PubMed] [Google Scholar]
- 9.Hosios AM, and Vander Heiden MG (2018). The redox requirements of proliferating mammalian cells. J Biol Chem 293, 7490–7498. 10.1074/jbc.TM117.000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoneten KK, Kasap M, Akpinar G, Gunes A, Gurel B, and Utkan NZ (2019). Comparative Proteome Analysis of Breast Cancer Tissues Highlights the Importance of Glycerol-3-phosphate Dehydrogenase 1 and Monoacylglycerol Lipase in Breast Cancer Metabolism. Cancer Genomics Proteomics 16, 377–397. 10.21873/cgp.20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou C, Yu J, Wang M, Yang J, Xiong H, Huang H, Wu D, Hu S, Wang Y, Chen XZ, and Tang J (2017). Identification of glycerol-3-phosphate dehydrogenase 1 as a tumour suppressor in human breast cancer. Oncotarget 8, 101309–101324. 10.18632/oncotarget.21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rusu P, Shao C, Neuerburg A, Acikgöz AA, Wu Y, Zou P, Phapale P, Shankar TS, Döring K, Dettling S, et al. (2019). GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem Cell 25, 241–257.e248. 10.1016/j.stem.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Pecinová A, Alán L, Brázdová A, Vrbacký M, Pecina P, Drahota Z, Houštěk J, and Mráček T (2020). Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer. Cells 9. 10.3390/cells9081764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowdhury SK, Raha S, Tarnopolsky MA, and Singh G (2007). Increased expression of mitochondrial glycerophosphate dehydrogenase and antioxidant enzymes in prostate cancer cell lines/cancer. Free Radic Res 41, 1116–1124. 10.1080/10715760701579314. [DOI] [PubMed] [Google Scholar]
- 15.Valdivia CR, Ueda K, Ackerman MJ, and Makielski JC (2009). GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol 297, H1446–1452. 10.1152/ajpheart.00513.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broeks MH, van Karnebeek CDM, Wanders RJA, Jans JJM, and Verhoeven-Duif NM (2021). Inborn disorders of the malate aspartate shuttle. J Inherit Metab Dis 44, 792–808. 10.1002/jimd.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park JO, Rubin SA, Xu YF, Amador-Noguez D, Fan J, Shlomi T, and Rabinowitz JD (2016). Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol 12, 482–489. 10.1038/nchembio.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanner LB, Goglia AG, Wei MH, Sehgal T, Parsons LR, Park JO, White E, Toettcher JE, and Rabinowitz JD (2018). Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst 7, 49–62.e48. 10.1016/j.cels.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balabanov S, Zimmermann U, Protzel C, Scharf C, Klebingat KJ, and Walther R (2001). Tumour-related enzyme alterations in the clear cell type of human renal cell carcinoma identified by two-dimensional gel electrophoresis. Eur J Biochem 268, 5977–5980. 10.1046/j.0014-2956.2001.02546.x. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Fu S, Wang G, Cao Y, Li L, Li X, Yang J, Li N, Shan Y, Cao Y, et al. (2021). Glycerol-3-phosphate biosynthesis regenerates cytosolic NAD(+) to alleviate mitochondrial disease. Cell Metab 33, 1974–1987.e1979. 10.1016/j.cmet.2021.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Berger F, Lau C, Dahlmann M, and Ziegler M (2005). Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280, 36334–36341. 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Meer G, and de Kroon AI (2011). Lipid map of the mammalian cell. J Cell Sci 124, 5–8. 10.1242/jcs.071233. [DOI] [PubMed] [Google Scholar]
- 24.Engelmann B, and Wiedmann MK (2010). Cellular phospholipid uptake: flexible paths to coregulate the functions of intracellular lipids. Biochim Biophys Acta 1801, 609–616. 10.1016/j.bbalip.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins B, West JA, and Koulman A (2015). A review of odd-chain fatty acid metabolism and the role of pentadecanoic Acid (c15:0) and heptadecanoic Acid (c17:0) in health and disease. Molecules 20, 2425–2444. 10.3390/molecules20022425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez DJ, and Simon MC (2018). Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim Biophys Acta Rev Cancer 1870, 23–31. 10.1016/j.bbcan.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wydysh EA, Medghalchi SM, Vadlamudi A, and Townsend CA (2009). Design and synthesis of small molecule glycerol 3-phosphate acyltransferase inhibitors. J Med Chem 52, 3317–3327. 10.1021/jm900251a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuhajda FP, Aja S, Tu Y, Han WF, Medghalchi SM, El Meskini R, Landree LE, Peterson JM, Daniels K, Wong K, et al. (2011). Pharmacological glycerol-3-phosphate acyltransferase inhibition decreases food intake and adiposity and increases insulin sensitivity in diet-induced obesity. Am J Physiol Regul Integr Comp Physiol 301, R116–130. 10.1152/ajpregu.00147.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, Ferreira R, Naamati A, Ali A, Lewis CA, et al. (2021). Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell 81, 691–707.e696. 10.1016/j.molcel.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, and Ratcliffe PJ (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275. 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 31.Qi X, Li Q, Che X, Wang Q, and Wu G (2021). The Uniqueness of Clear Cell Renal Cell Carcinoma: Summary of the Process and Abnormality of Glucose Metabolism and Lipid Metabolism in ccRCC. Front Oncol 11, 727778. 10.3389/fonc.2021.727778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao CH, Fowle-Grider R, Mahieu NG, Liu GY, Chen YJ, Wang R, Singh M, Potter GS, Gross RW, Schaefer J, et al. (2016). Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem Biol 23, 483–493. 10.1016/j.chembiol.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao CH, Wang L, Stancliffe E, Sindelar M, Cho K, Yin W, Wang Y, and Patti GJ (2020). Dose-Response Metabolomics To Understand Biochemical Mechanisms and Off-Target Drug Effects with the TOXcms Software. Anal Chem 92, 1856–1864. 10.1021/acs.analchem.9b03811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao CH, Wang R, Wang Y, Kung CP, Weber JD, and Patti GJ (2019). Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 8. 10.7554/eLife.41351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao CH, Liu GY, Yang K, Gross RW, and Patti GJ (2016). Inaccurate quantitation of palmitate in metabolomics and isotope tracer studies due to plastics. Metabolomics 12. 10.1007/s11306-016-1081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2 (Related to Figure 3) Isotopologue distribution pattern of lipids when cells were labeled with U-13C glucose
Data Availability Statement
All datasets generated or analyzed during this study are included in the published article. Raw metabolomic datasets supporting the current study are available from the Lead Contact upon request. Western blot images have been deposited at Mendeley and are publicly available as of the date of publication through Mendeley Data. (https://data.mendeley.com/datasets/zynypyx9fj/draft?a=2d9866d8-956d-47a3-98a5-a954895bcea2).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.