

SUMMARY
Interleukin-18 (IL-18) promotes natural killer (NK) and T cell production of interferon (IFN)-γ, a key factor in resistance to Toxoplasma gondii, but previous work has shown a limited role for endogenous IL-18 in control of this parasite. Although infection with T. gondii results in release of IL-18, the production of IFN-γ induces high levels of the IL-18 binding protein (IL-18BP). Antagonism of IL-18BP with a “decoy-to-the-decoy” (D2D) IL-18 construct that does not signal but rather binds IL-18BP results in enhanced innate lymphoid cell (ILC) and T cell responses and improved parasite control. In addition, the use of IL-18 resistant to IL-18BP (“decoy-resistant” IL-18 [DR-18]) is more effective than exogenous IL-18 at promoting innate resistance to infection. DR-18 enhances CD4+ T cell production of IFN-γ but results in CD4+ T cell-mediated pathology. Thus, endogenous IL-18BP restrains aberrant immune pathology, and this study highlights strategies that can be used to tune this regulatory pathway for optimal anti-pathogen responses.
In brief
Clark et al. use variants of the cytokine IL-18 to bypass the effects of the IL-18 binding protein to assess their impact on infection with Toxoplasma gondii. These approaches highlight that during infection, unopposed IL-18 enhances NK cell production of IFN-γ but can also promote CD4+ T cell-mediated pathology.
Graphical abstract
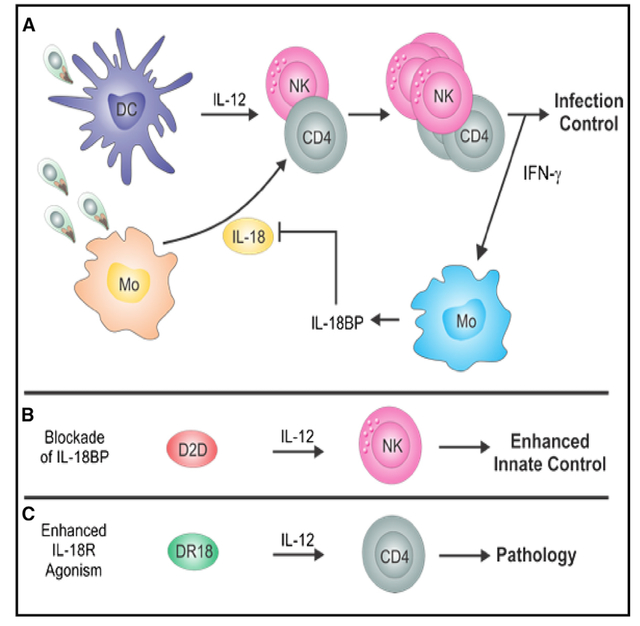
INTRODUCTION
Interleukin-18 (IL-18) is a member of the IL-1 family that was identified based on its ability to induce natural killer (NK) and T cell production of interferon (IFN)-γ and promote Th1 immunity.1,2 In current models, IL-18 acts synergistically with IL-12 to optimize production of IFN-γ and tumor necrosis factor (TNF)-α by innate lymphoid cells (ILCs) and T cells.3–6 Endogenous IL-18 contributes to optimal resistance to diverse intracellular pathogens that include Mycobacterium tuberculosis,7 Salmonella tymphimurium,8,9 Cryptosporidium parvum,10,11 and Leishmania major.12 However, sustained IL-18 activity is associated with autoinflammatory pathologies that include cryopyrinopathies, hereditary periodic fever syndromes, familial hemophagocytic lymphohistiocytosis,13 and macrophage activation syndromes, as well as viral and bacterial sepsis.14,15 These clinical examples illustrate the need for endogenous mechanisms to optimize host protective activity of IL-18 while limiting aberrant pathological effect of IL-18 signaling. Two distinct checkpoints exist to limit IL-18 production and activity. First, IL-18 is produced as a pro-cytokine without biological activity and requires caspase-mediated processing.16 Second, the ability of IL-18 to induce the production of IFN-γ results in the synthesis of a dedicated IL-18 binding protein (IL-18BP), which blocks the ability of IL-18 to signal.17
Innate immunity to the intracellular parasite T. gondii is dominated by the initial induction of IL-12, which promotes ILC production of IFN-γ and thereby limits parasite growth.18–21 However, parasite-specific CD4+ and CD8+ T cell responses are required for long-term control and host survival.22–24 Multiple IL-1 family members have been implicated in the development of protective innate and adaptive responses to Toxoplasma.25–29 Initial studies on the role of IL-18 in resistance to T. gondii demonstrated that infection resulted in increased production of IL-18, but it was not required for protective NK or T cell responses.30–33 However, exogenous IL-18 could be used to enhance innate resistance to toxoplasmosis.33 Therefore, it remains unknown why endogenous IL-18 does not have a more significant role in innate or adaptive resistance to T. gondii and to what extent host immunity is controlled by IL-18BP immune regulation during infection. Here, we set out to describe the events that lead to IL-18BP production during toxoplasmosis and pharmacologically dissect the contribution of IL-18 on host anti-parasitic response to T. gondii and the role of endogenous IL-18BP signaling using engineered IL-18 variants.
RESULTS
Infection-induced production of IL-18 and IL-18BP
Based on previous reports,28,33,34 studies were performed to determine whether the induction of IFN-γ caused by infection with T. gondii impacted the production of IL-18 and the IL-18BP. Thus, adaptive immune deficient Rag1−/− and wild-type (WT) mice were infected intraperitoneally and treated with an isotype control antibody or anti-IFN-γ and the levels of IL-18 and IL-18BP measured at the local site of infection. In naive mice, IFN-γ, IL-18, and IL-18BP levels were below the level of detection, but by 7 days post-infection (dpi), there was an increase in the levels of IFN-γ, IL-18, and IL-18BP. IL-18BP was the predominant signal, whose levels were approximately 10- to 100-fold greater than IL-18 in both Rag1−/− and WT mice (Figure 1A). Previous studies have shown that infection results in increased serum levels of IFN-γ and IL-18.33,34 IL-18BP levels were highest in WT mice compared with Rag1−/− mice, which correlated with greater levels of IFN-γ present in WT mice (Figure 1A). In mice treated with anti-IFN-γ, there was an approximate 10-fold increase in parasite burden (data not shown), and this was accompanied by reduced production of IL-18 and IL-18BP in both WT and Rag1−/− mice (Figure 1A). Previous studies have identified monocytes and macrophages as a major source of IL-18BP.35 Consistent with this, analysis of single-cell RNA sequencing (RNA-seq) of splenocytes from infected mice revealed that the predominant source of IL-18 and IL-18BP was monocytes. (Figure 1B). These results were confirmed via flow cytometry, where it was additionally noted that Sirpa+ class 2 conventional dendritic cells (cDC2s) produced IL-18BP (Figure 1C). Granulocytes produced high levels of Il18bp mRNA but little protein detectable by flow cytometry. (Figures 1B and 1C). Together, these data highlight that the infection-induced production of IL-18 is not simply a function of parasite burden and that IFN-γ signaling promotes IL-18BP production by monocytes and dendritic cells during toxoplasmosis.
Figure 1. IL-18 and IL-18BP are expressed during acute T. gondii infection.
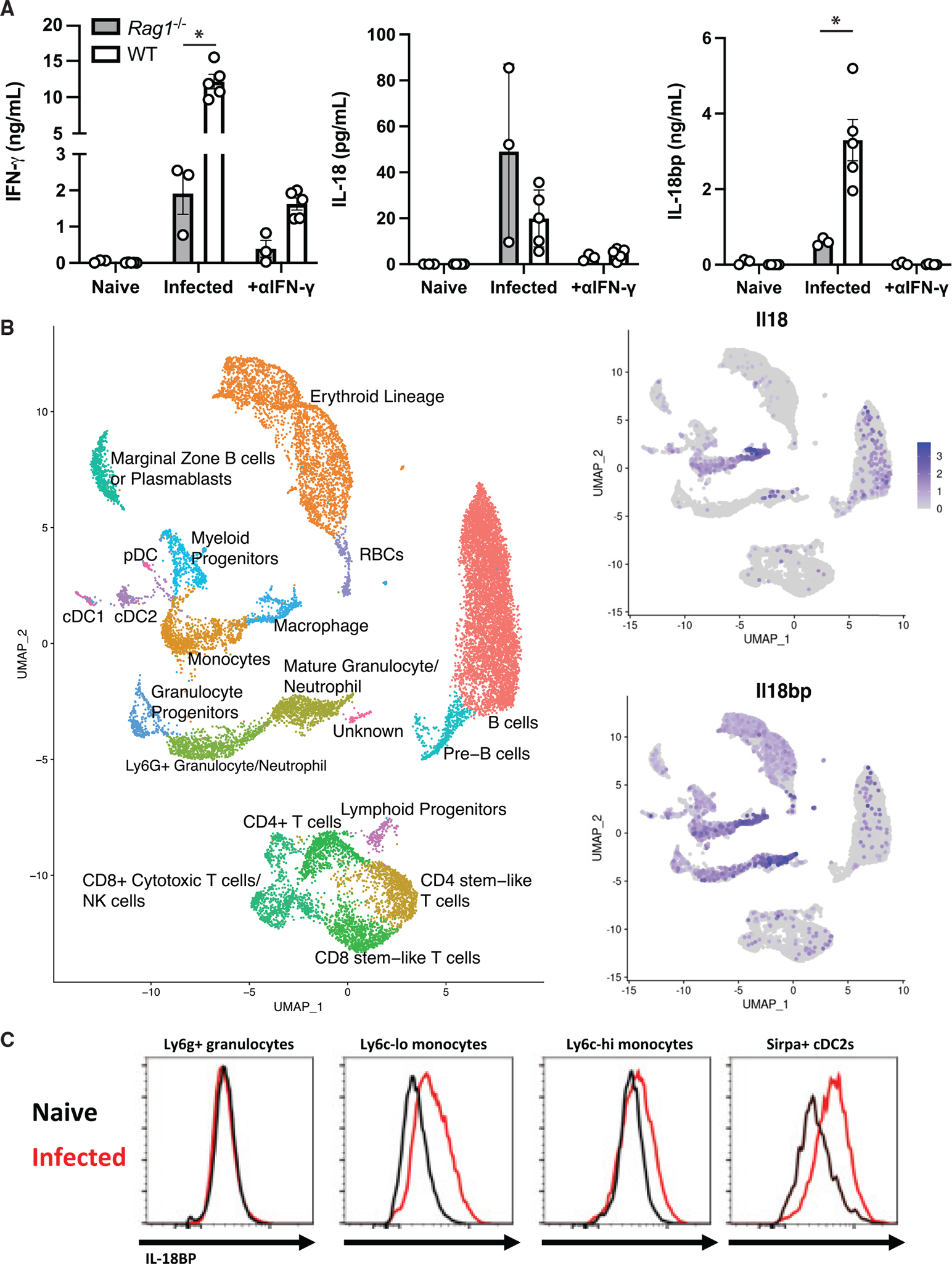
Mice were infected i.p. with T. gondii after 7 days.
(A) IFN-γ, IL-18, and IL-18BP in the peritoneal lavage were measured by ELISA directly ex vivo.
(B) Single-cell RNA-seq was performed on splenocytes from B6 mice infected with T. gondii at 14 dpi. UMAP analysis was used to generate clusters according to cellular lineage, and expression heatmaps for Il18 and Il18bp were overlaid.
Data are pooled from 2 independent experiments (A). RNA-seq data are representative of one biological replicate.
(C) Flow cytometry showing staining for IL-18BP in the populations described from naive (black) and infected (red) mouse spleens. *p < 0.05 (Student’s t test).
Data are displayed as mean ± standard error.
Expression of the IL-18 receptor during infection
To assess which immune populations would be sensitive to IL-18 and IL-18BP during infection, we examined the single-cell RNA-seq of splenocytes from infected mice to define which cell types express the IL-18 receptor. Expression of Il18r1 and Il18rap, which encode IL-18 receptor subunits IL-18Rα and IL18Rβ, respectively, were predominantly localized in the T cell and NK cell compartments, with expression also detected in the granulocyte fraction (Figure 2A). To visualize the distribution of the IL-18Rα at the protein level, we performed unbiased cluster determination by uniform manifold approximation and projection (UMAP) analysis of flow cytometry data from splenocytes of naive and day 10 infected WT mice. In naive mice, IL-18Rα was expressed on a population of NKp46+ group 1 ILCs cells that include NK cells and ILC1s and a small population of CD4+ T cells (Figures 2B and 2C). By 10 dpi, IL-18Rα expression was strongly induced on CD4+ and CD8+ T cells, as well as TCRb−NKp46+ NK/ILC1 populations. Analysis of the T cells from infected mice revealed that the IL-18 receptor was expressed only by CD11ahi (a marker of antigen experience) CD4+ and CD8+ T cells, as well as virtually all parasite-specific CD4+ and CD8+ T cells, identified by class I and class II tetramer staining (H2-Kb SVLAFRRL and I-Ab AVEIHRPVPGTAPPS)36,37 (Figure 2D). Together, these data indicate that during toxoplasmosis, effector lymphocytes are receptive to IL-18, but IL-18BP may limit anti-parasitic responses.
Figure 2. IL-18R is expressed during acute T. gondii infection.
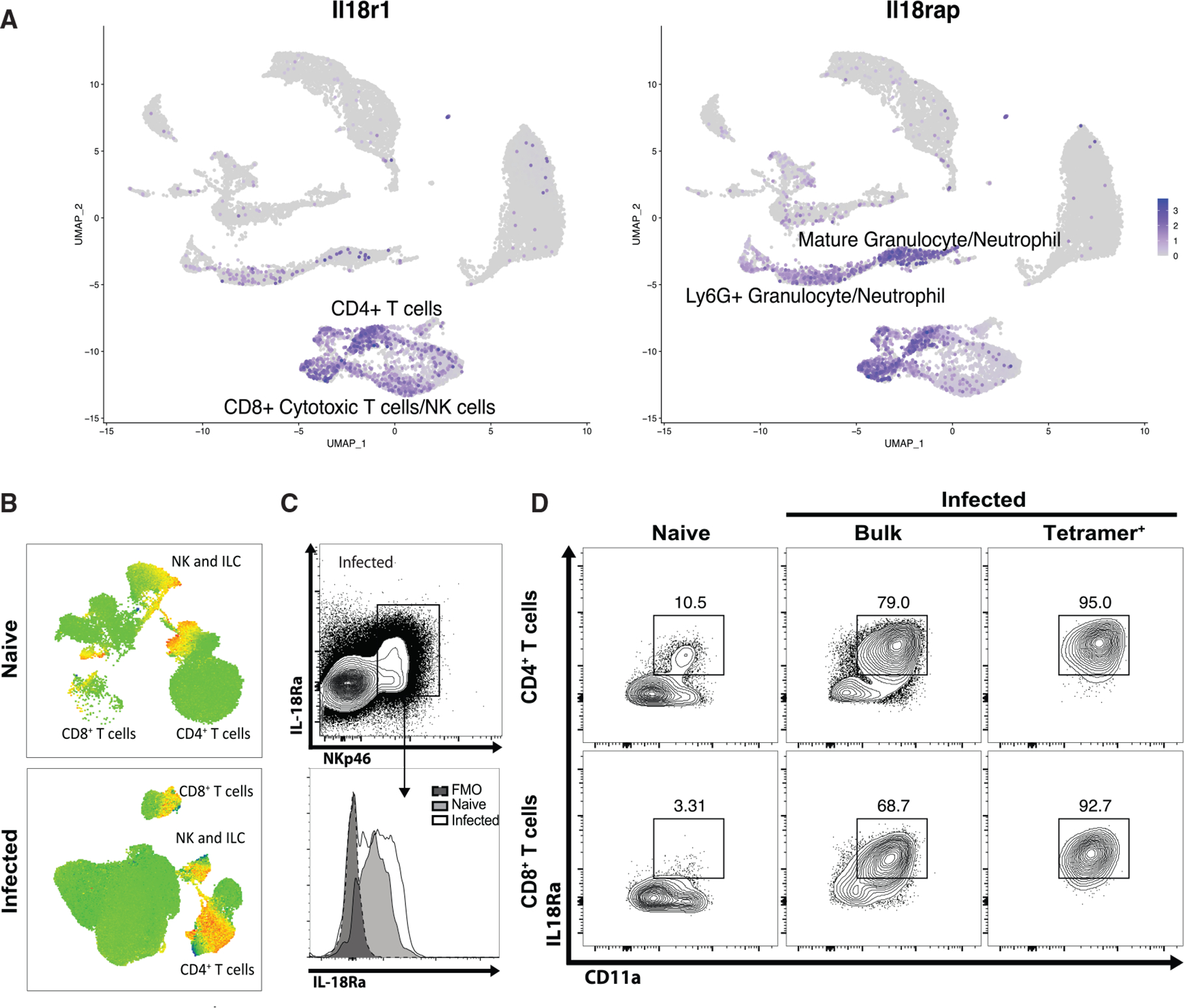
(A) As in Figure 1, single-cell RNA-seq was performed on splenocytes from B6 mice infected with T. gondii at 14 dpi. UMAP analysis was used to generate clusters according to cellular lineage, and expression heatmaps for Il18r1 and Il18rap were overlaid.
(B) Flow cytometry was performed on splenocytes from naive and infected B6 mice at 10 dpi. UMAP analysis was performed to generate clusters according to cellular lineage. Heat maps for IL-18Rα staining are shown.
(C) Rag1−/− mice were infected with T. gondii. At 7 dpi, flow cytometry was performed on peritoneal cells. Population show is pregated on live singlets.
(D) B6 mice were infected with T. gondii. At 10 dpi, splenocytes were analyzed by flow cytometry alongside naive controls. Populations shown are pregated on live CD4+ or CD8+ T cells.
Data are representative of three independent experiments (B–D).
Pharmacological inhibition of IL-18BP reveals endogenous IL-18 activity
To investigate the impact of the infection-induced IL-18BP on the ability of endogenous IL-18 to affect innate resistance to T. gondii, we sought to engineer a “decoy-to-the-decoy” (D2D) IL-18 variant that abrogated signaling capacity through the IL-18 receptor but selectively retained binding capacity to IL-18BP using directed evolution with yeast surface display (Figures 3A and 3B). This method was used to screen >300 million human IL-18 variants that were randomized at 16 contact positions for those that retained IL-18BP binding but lacked binding to IL-18 receptors. After three rounds of selection for mouse and viral IL-18BP orthologs and counter selection against human and mouse IL-18 receptors, we obtained a population that exclusively bound to IL-18BP with no detectable binding to either IL-18Rα ortholog (Figure 3C). Sequencing of the post-round 3 pool revealed 7 mutated residues that were used to identify a consensus sequence for D2D, which was confirmed to retain mIL-18BP binding with no binding to mIL-18Rα (Figures 3D and 3E). To confirm the functional properties of D2D to antagonize IL-18BP without agonizing the IL-18 receptor by itself, we stimulated in-vitro-derived NK cells with IL-18 or D2D in the presence of IL-12. While WT IL-18 was a potent inducer of IFN-γ, the D2D construct had no stimulatory activity (Figure 3F). Finally, to test the ability of D2D to antagonize IL-18BP, a reporter cell line engineered to detect IL-18 signaling was stimulated with a fixed ratio of IL-18 and IL-18BP, which produced minimal activity, and an escalating dose of D2D (Figure 3G). At increasing concentrations of D2D, IL-18 signaling was detected, indicating that D2D indeed neutralized the IL-18BP with an IC50 of 78.5 nM.
Figure 3. Generation and use of D2D to reveal endogenous IL-18 activity.
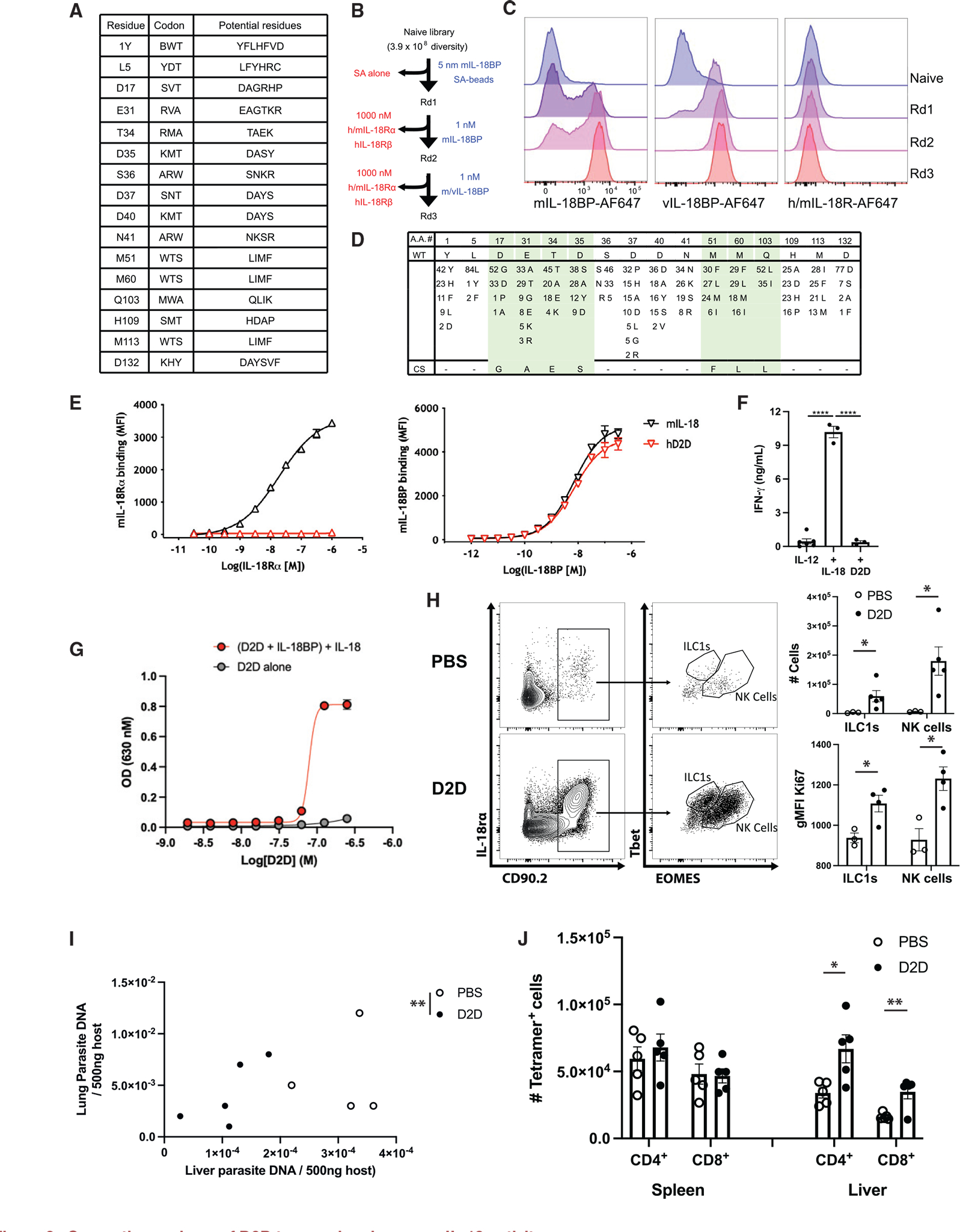
(A) Randomized positions of human (h) IL-18 to create D2D, with the corresponding degenerate codon and the potential amino acid at each position.
(B) Summary of the experimental design for directed evolution and yeast selection process to generate D2D. Yeast libraries were selected for mouse (m) and viral (v) IL-18BP binding and counter-selected with streptavidin (SA; round 1) and m/hIL-18Rα and hIL-18Rβ receptors (rounds 2 and 3) using magnetic-activated cell sorting (MACS; round 1) and subsequently fluorescence-activated cell sorting (FACS; rounds 2 and 3). Blue text (right) indicates positive selection reagent, and red text (left) shows the counter-selection reagent.
(C) Representative histogram assessing mIL-18BP (10 nM, left), vL-18BP (10 nM, middle), and IL-18 receptor (m/hIL-18Rα and hIL-18Rβ, 100 nM each, right) staining by flow cytometry of yeast display library after each round of selection.
(D) The sequences of 87 clones summarized for selected D2D variants, with differences for wild-type IL-18 indicated for each mutant at the given amino acid position (top). Number indicates number of clones that shared same residue change. Green shading highlights converging residues to form consensus sequence (bottom).
(E) Dose-response curves comparing binding of mIL-18 and human D2D displayed on yeast to mIL-18Rα and mIL-18BP.
(F) Quantification of INF-γ ELISA from supernatants of lymphokine activated killer cells stimulated with the cytokines listed.
(G) IL18B neutralization curve generated by addition of recombinant IL-18, IL-18BP, and D2D protein to IL-18 signaling reporter cell line.
(H) Rag1−/− mice were infected with T. gondii. At 7 dpi, flow cytometry of peritoneal cells was performed. Populations shown are pregated on live singlets. Quantification of cell numbers and intracellular staining shown on right.
(I) Quantification of parasite DNA isolated from host liver and lung tissue, Rag1−/− mouse, 7 dpi. Units are ng parasite DNA/500 ng host DNA. Statistical significance was determined by combined analysis of the parasite burden in lung and liver of individual mice.
(J) B6 mice were infected with T. gondii, and at 10 dpi, the number of tetramer+ CD4+ and CD8+ cells was analyzed by flow cytometry. Summary data are shown. NS, not significant (p > 0.05); *p < 0.05, **p < 0.01 (Student’s t test). Data are representative of 2 independent experiments (A–D). Data are displayed as mean ± standard error.
The ability of D2D to impact in vivo IL-18-IL-18BP interactions was next tested. When D2D was administered to infected Rag1−/− mice, there was a marked increase in numbers of ILC1s (17-fold) and NK cells (26-fold) associated with upregulation of the cell-cycle marker Ki67 (Figure 3H) at the site of infection. This antagonism of the infection-induced IL-18BP also resulted in lower parasite burden, as measured by qPCR for parasite DNA (Figure 3I). When WT mice were treated with D2D, there was no significant impact on the numbers of parasite-specific tetramer+ CD4+ and CD8+ T cells in the spleen, but increased numbers of tetramer+ CD4+ and CD8+ T cells were observed in the liver (Figure 3J). This was accompanied by a decrease in parasite burden in the liver as measured by qPCR (Figure S1). Thus, the induction of endogenous IL-18BP is an important determinant of the ability of endogenous IL-18 to direct the innate and adaptive immune response to T. gondii.
Full IL-18 pathway agonism elicits robust innate immune responses to T. gondii
Given the effect of enhancing endogenous IL-18 activity with D2D, we sought to determine the effect of full IL-18 pathway agonism by using “decoy-resistant” IL-18 (DR-18), an IL-18 variant that binds the IL-18 receptor with increased affinity but is not neutralized by IL-18BP.38 When compared with WT IL-18, DR-18 was a more potent stimulator of cell proliferation of in-vitro-derived NK cells based on expression of Ki67 and dilution of CellTrace Violet (Figure 4A, left) as well as IFN-γ production (Figure 4A, right). These cytokines were administered intraperitoneally (i.p.) to Rag1−/− mice at 1 dpi and every other day thereafter. At 7–10 dpi, parasites were readily detected in cytopsins of the peritoneal exudate cells (PECs) in mock-treated mice (Figure 4B–1, white arrows), and treatment with IL-18 or DR-18 resulted in an increased number of activated macrophages and a marked decrease in the frequency and number of infected cells (Figures 4B1 and 4B2). Analysis of parasite burdens in peripheral tissues revealed that relative to mock-treated mice, IL-18 treatment resulted in a 10-fold reduction of parasite DNA in the lungs and livers of infected mice (Figure 4C; data not shown). However, DR-18 treatment resulted in a 50- to 100-fold reduction in parasite levels compared with mock-treated mice. Compared with WT IL-18, DR-18-treated mice exhibited decreased parasitic burden, suggesting that IL-18 agonism (independent of IL-18BP) improved host anti-parasitic immunity.
Figure 4. DR18 treatment promotes systemic protective immunity in Rag1−/− mice.
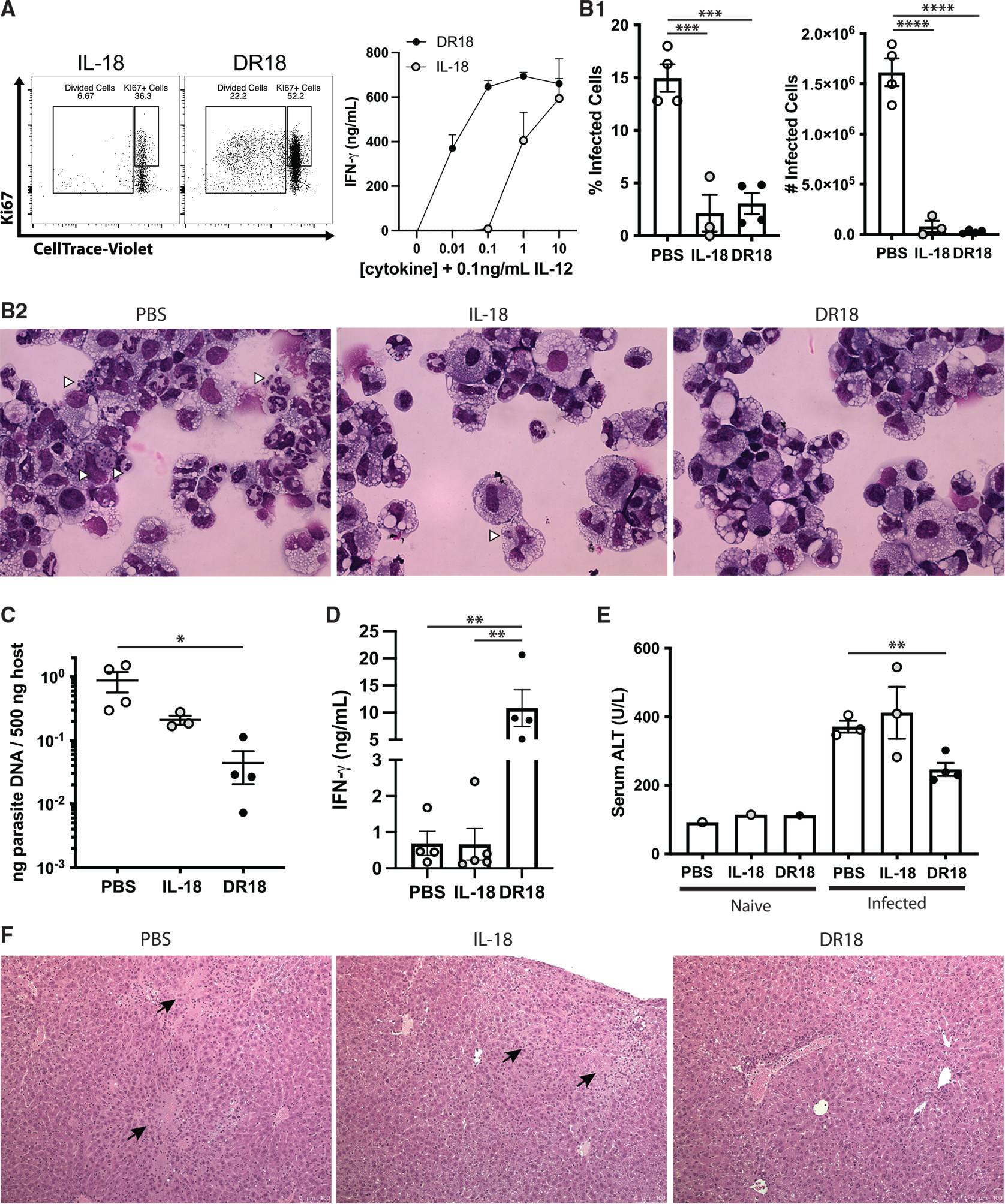
(A) Intracellular staining (left) and ELISA from supernatants (right) of lymphokine activated killer cells stimulated with the cytokines listed.
(B) Quantification of cytospins from peritoneal exudate cells of infected Rag1−/− mice at 7 dpi (top) and representative cytospins (bottom).
(C) Quantification of parasite DNA isolated from lung tissue.
(D) ELISA from serum of Rag1−/− mice at 7 dpi treated as described.
(E) Serum ALT values obtained from Rag1−/− mice at 7 dpi treated as described.
(F) Histology of livers from Rag1−/− mice at 7 dpi treated as described. Black arrows indicate necrotic lesions.
Data are representative of two (A) or 3 (B–F) independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (Student’s t test, A, ordinary one-way ANOVA with Tukey’s multiple comparisons test, B–D, and Welch’s ANOVA with Dunnett’s multiple comparisons test, E). Data are displayed as mean ± standard error.
In naive mice, IFN-γ is undetectable in the serum, and treatment with IL-18 or DR-18 did not result in elevated serum IFN-γ or altered numbers of innate lymphoid or myeloid cells (data not shown). In infected Rag1−/− mice at 7 dpi, mock-treated and IL-18-treated mice had similar IFN-γ levels, but serum IFN-γ was significantly elevated in DR18-treated mice (Figure 4D). Additionally, serum ALT, a marker of liver damage, revealed that infection in Rag1−/− mice was associated with increased levels of inflammation in the liver (Figures 4E and 4F). While this was unaffected by IL-18 treatment, mice treated with DR-18 showed lower ALT levels accompanied by reduced pathology (Figures 4E and 4F), likely associated with the reduced parasite burden in this tissue (Figure 4C).
Next, flow cytometry-based immunophenotyping was performed to determine the impact of IL-18 agonism on immune populations in infected Rag1−/− mice. Across all tissues assessed (peritoneum, spleen, and lung), IL-18 treatment had no effect on ILC1 or NK cell numbers. DR-18 treatment also did not impact ILC1 numbers but promoted higher NK cell numbers in all tissues examined (Figure 5A). Analysis of NK cells across multiple tissues using KLRG1 (a surface receptor indicative of NK cell maturation) combined with Ki67 revealed that in naive mice, treatment with either form of IL-18 resulted in a minimal increase in KLRG1 expression (Figures 5B and 5C). However, during infection of Rag1−/− mice, NK cell expression of KLRG1 was upregulated by IL-18 and further increased following DR-18 treatment (Figures 5B and 5C). Additionally, DR-18 treatment-induced mature NK cells expressed the highest levels of Ki67 (Figures 5B and 5C).
Figure 5. DR18 drives protective NK cell responses in Rag1−/− mice.
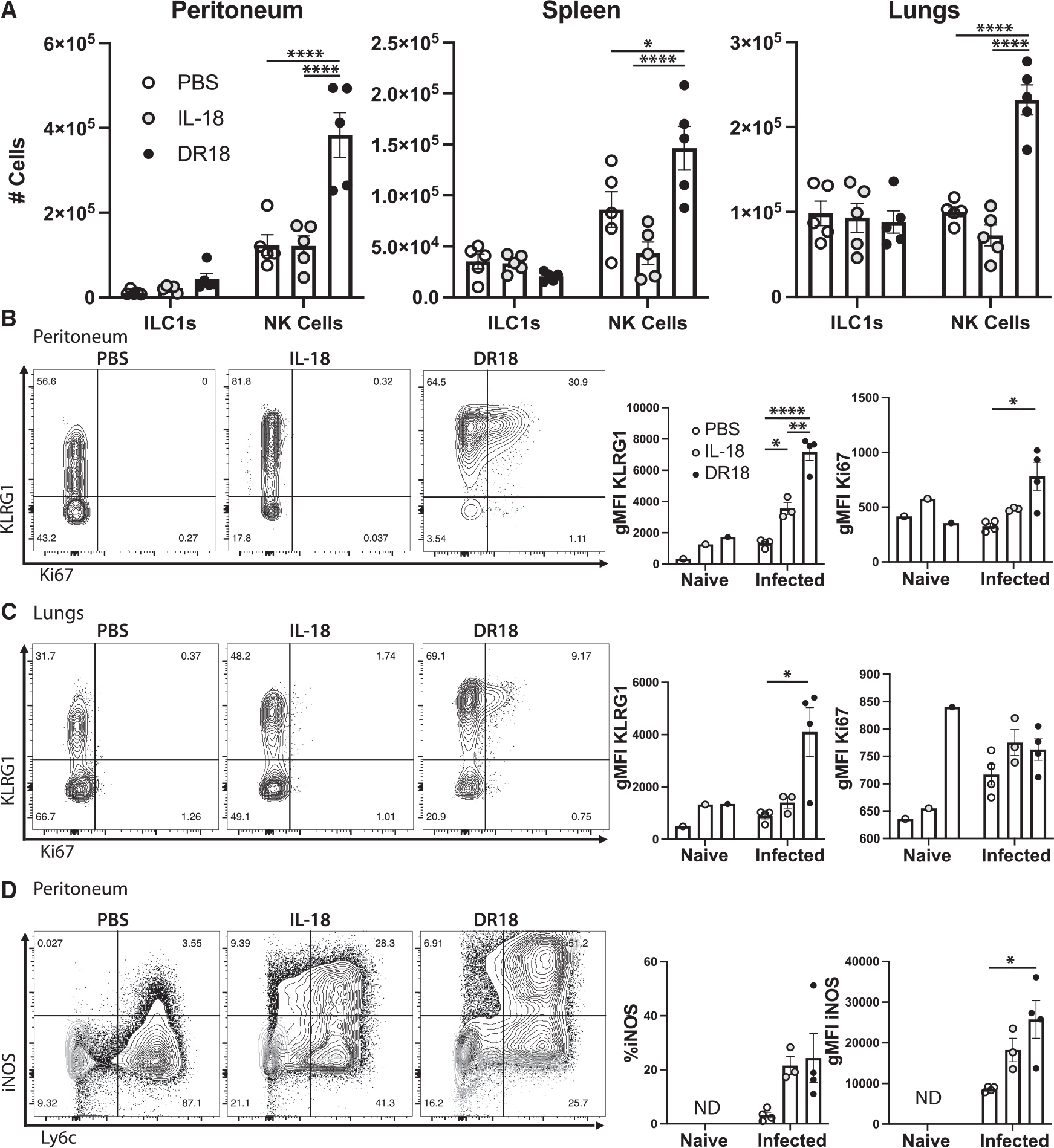
(A) Quantification of cells from Rag1−/− mice at 7 dpi from the tissues listed. ILC1s are defined as NK1.1+ Tbet+ EOMES− cells, and NK cells are NK1.1+ Tbet+/− EOMES+.
(B) Flow cytometry and quantification of NK cell phenotypes in the peritoneum and (C) in the lungs (D) of mice infected with T. gondii.
(D) Flow cytometric analysis and quantification of myeloid cells in the peritoneum. Populations shown are pregated on live CD11b+ Ly6g− singlets. ND, not detected. NS, not significant.
Data are representative of 3 (A–C) or 2 (D) independent experiments. *p < 0.05, **p < 0.01, ****p < 0.0001 (two-way ANOVA with Tukey’s multiple comparisons test, A–C, and one-way ANOVA with Tukey’s multiple comparisons test, D). Data are displayed as mean ± standard error.
The ability of IFN-γ to stimulate macrophage expression of the enzyme iNOS is required for the ability of these cells to produce reactive oxidative species and limit replication of T. gondii.39,40 In uninfected mice, the resting macrophage populations are typically iNOSlow, and IL-18 treatment had minimal impact on its expression (Figure 5D, light gray). As expected, in infected Rag1−/− mice, there was an increase in the population of Ly6Chi monocytes in the peritoneum, of which a small percentage co-expressed iNOS (Figure 5D). Treatment with either form of IL-18 led to an increase of iNOS+ Ly6Chi monocytes in the PECs but was most strongly induced by DR-18 treatment (Figure 5D). These data reveal that in Rag1−/− mice, exogenous DR-18 is more robust than IL-18 in its ability to promote the maturation and expansion of NK cells and production of IFN-γ associated with increased macrophage activation and parasite control.
Impact of DR18 on parasite-specific T cell responses
By 10 dpi, pathogen-specific T cells express the IL-18Rα, but the production of IFN-γ drives expression of high levels of IL-18BP (Figure 1A). To focus on the effects of IL-18 on the adaptive response, WT C57BL/6 mice were treated with IL-18 or DR-18 starting at 7 dpi, a time point when parasite-specific T cell responses can be reliably detected. In contrast to the results in Rag1−/− mice, sham- or IL-18-treated infected mice survived infection, but DR-18 treatment resulted in decreased survival (Figure 6A). This effect of DR-18 was associated with increased levels of pathology in the liver (Figure 6B), with a range of pathology apparent in histology (two examples are shown). Flow cytometric analysis revealed increased numbers of neutrophils (Figure 6C), and elevated serum IFN-γ levels were detected by ELISA (Figure 6D). This is in contrast to the results observed in D2D-treated WT mice, where no lethality was observed (data not shown). Analysis of parasite-specific CD4+ and CD8+ T cells showed that they were unaffected by treatment with WT IL-18 (Figure 6E), but DR-18 treatment resulted in a marked expansion of the parasite-specific CD4+ compartment in the spleen (Figure 6E). Previous studies have used the expression of KLRG1 and the chemokine receptors CXCR3 and CX3CR1 to distinguish different effector populations with distinct functional properties that develop during infection.41,42 One paradigm for T cell differentiation during Toxoplasma infection suggests that pathogen-specific T cells that are initially CXCR3+ CX3CR1− transition to an intermediate state characterized by co-expression of CXCR3, CX3CR1, and KLRG1, which finally lose CXCR3 but maintain KLRG1 and CX3CR1 as terminally differentiated effector T cells.42 Administration of DR-18 resulted in a higher frequency of the least-differentiated CXCR3+ CD4+ T cells among tetramer+ cells in the spleen and a corresponding decrease in KLRG1 single-positive cells (Figure 6F). DR-18 treatment also resulted in a diminution of the terminally differentiated KLRG1+ CX3CR1hi T cell population in the CD4+ T cell compartment and an increase in the double-negative population (Figure 6G). However, CD8+ T cell differentiation was not altered (Figure 6H). These data suggest that DR-18 treatment promotes a less-differentiated phenotype in antigen-specific CD4+ T cells during acute Toxoplasma infection. This agrees with the effects described in tumor studies, where treatment with DR18 was associated with expansion of terminal effectors and stem-like cells, although in that model, CD8+ T cells were preferentially affected.38
Figure 6. DR18 drives altered CD4+ T cell responses and immune pathology.
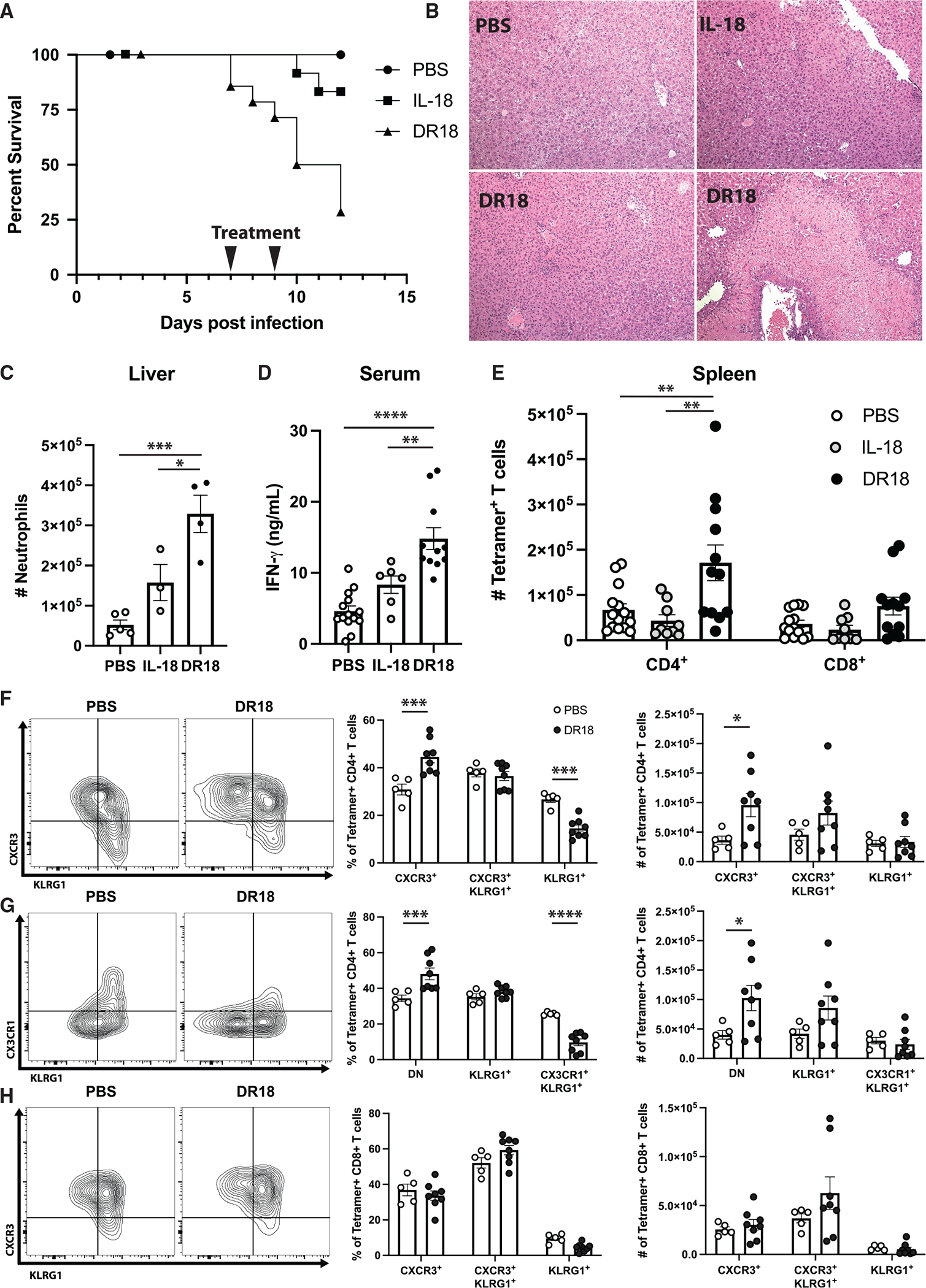
(A) Survival analysis of B6 mice infected with T. gondii and treated with PBS, IL-18, or DR18 at 7 and 9 dpi.
(B) Representative histology from experiment described in (A). Two images of DR18-treated mice are included to show variation in pathology observed.
(C) Flow cytometric analysis of livers from mice treated as described in (A), analyzed at 10 dpi. Neutrophils defined as CD11b+ Ly6g+ live singlets.
(D) ELISA analysis of serum from mice treated as described in (A).
(E) Quantification of tetramer+ CD4+ and CD8+ T cells in the spleens of mice at 10 dpi.
(F and G) Representative flow cytometry of tetramer+ CD4+ T cells (left) and quantification (right) from the spleens of B6 mice treated as described at 10 dpi.
(H) Analysis of tetramer+ CD8+ T cells as from (F) and (G).
Data are pooled from (A), (D), and (E) or are representative of (B, C, and F–H) 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (ordinary one-way ANOVA with Tukey’s multiple comparisons test, C and D, two-way ANOVA with Tukey’s multiple comparisons test, E, and two-way ANOVA with Šídák’s multiple comparisons test, F–H). Data are displayed as mean ± standard error.
To examine the impact of these treatments on IFN-γ production, a reporter mouse that expresses the surface protein Thy1.1 under the control of the IFN-γ promoter was used.43 In response to infection, CD4+ T cells were the major source of IFN-γ in the spleen (data not shown) and in the liver (Figure 7A). Treatment with DR-18 augmented IFN-γ in CD4+ T cells and, to a lesser extent, CD8+ T cells. Culture of splenocytes from infected and treated mice revealed that those treated with DR-18 produced high basal levels of IFN-γ (Figure 7B). Incubation of splenocytes from infected mice with soluble Toxoplasma antigen (STAg) stimulates parasite-specific T cells to produce IFN-γ. While IL-18 treatment enhanced this response, the highest levels were observed with splenocytes from DR-18-treated mice (Figure 7B). In addition, the inclusion of blocking antibody to the CD4 co-receptor, but not CD8, resulted in a marked decline in IFN-γ (Figure 7B). Likewise, STAg-specific restimulation of splenocytes resulted in elevated levels of granulocyte-macrophage colony-stimulating factor (GM-CSF) in infected mice treated with DR-18 compared with control mice (Figure 7C). Inclusion of α-CD4, but not α-CD8, abrogated GM-CSF production in the spleen and the liver (Figure 7C; data not shown). These data indicate that during toxoplasmosis, antigen-specific CD4+ T cells are the dominant source of IFN-γ and GM-CSF following DR-18 treatment.
Figure 7. CD4+ T cells produce enhanced cytokines in response to DR18.
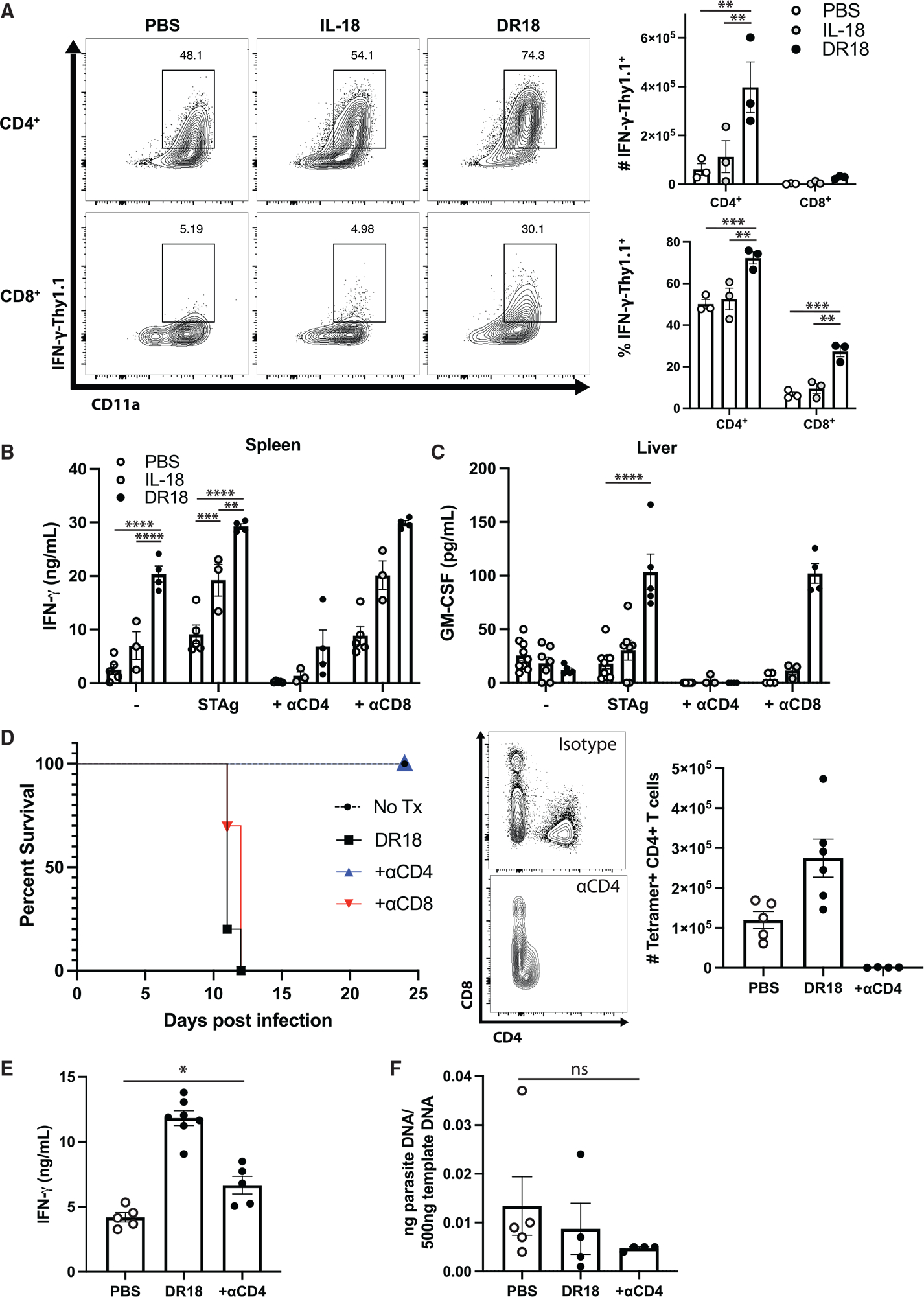
(A) Representative flow cytometry (left) and quantification (right) from the livers of IFN-γ-Thy1.1 reporter mice at 10 dpi. Population shown is pregated on live CD4+ or CD8+ T cells.
(B and C) ELISA analysis of ex vivo cultures from infected mice at 10 dpi stimulated as described. STAg, soluble Toxoplasma antigen.
(D) Survival analysis of B6 mice infected with T. gondii and treated with PBS, IL-18, or DR18 at 7 and 9 dpi (left) and validation of CD4+ T cell depletion by flow cytometry (right).
(E) ELISA analysis of serum IFN-γ from mice treated as described at 10 dpi.
(F) Quantification of parasite DNA isolated from livers of infected mice at 10 dpi.
Data are representative of 2 independent experiments. NS, not significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (ordinary one-way ANOVA with Tukey’s multiple comparisons test, A, two-way ANOVA with Tukey’s, B, or Šídák’s, C, multiple comparisons test, and one-way ANOVA with Tukey’s multiple comparisons test, E and F). Data are displayed as mean ± standard error.
Since DR-18 treatment resulted in an increase in the pathogen-specific T cell populations and their production of cytokines, we sought to determine if depletion of either of these populations would mitigate the lethal effects of DR-18. We thus administered CD4+ and CD8+ T cell-depleting antibodies 6 h prior to administration of DR-18 at 7 dpi. Depletion of CD8+ T cells (Figure S2) had no impact on survival, but the depletion of CD4+ T cells abrogated DR-18-induced mortality (Figure 7D). This was associated with a reduction of serum IFN-γ to levels comparable to infected, untreated mice (Figure 7E). Depletion of CD4+ T cells did not result in enhanced parasite burden in DR18-treated mice (Figure 7F). Together, these results establish that DR-18 treatment promotes the function and “stemness” of CD4+ T cells but results in immune pathology.
DISCUSSION
Previous studies have reported that infection with T. gondii leads to the release of IL-18.33,44 However, the studies presented here show that this is not simply a function of innate detection of parasite infiltration or damage associated with levels of parasite replication. Rather, the observation that IFN-γ is required for IL-18 secretion in inflammatory macrophages suggests a feedforward mechanism between IFN-γ and the molecular machinery that processes IL-18. The Nod-like receptor (NLR) family members are cytosolic molecules that can bind pathogen-associated molecular patterns (PAMPs) and nucleate the assembly of the inflammasome complex, which results in caspase-1-mediated processing of IL-18 to its bioactive form. In murine macrophages, NLRP1 and NLRP3 are implicated in the detection of T. gondii and production of IL-1 family members.44,45 However, the ability of IFN-γ-primed human macrophages to kill T. gondii leads to release of parasite DNA into the cytosol, where it can be sensed by the AIM2 inflammasome.46,47 Whether a similar process contributes to the parasite-induced, IFN-γ-dependent production of IL-18 observed here is unclear. In addition, as reported in other systems,48,49 the production of IL-18BP is proportional to the levels of circulating IFN-γ. This highlights the complex regulatory effects of IFN-γ on macrophages: it promotes activation to control T. gondii39,40,50,51 and enhances innate sensing of T. gondii and IL-18 secretion but also induces production of IL-18BP as a potent negative feedback mechanism that limits the impact of endogenous IL-18.
In order to dissect the role of IL-18BP and IL-18 signaling during toxoplasmosis, we engineered an IL-18 variant, D2D, that abrogated signaling capacity through the IL-18 receptor but selectively retained binding capacity to IL-18BP. In conjunction with our previously designed IL-18 variant, DR-18, an IL-18 agonist that is impervious to IL-18BP inhibition,38 we were able to dissect the impact of IL-18 signaling on innate and adaptive IL-12-mediated resistance to T. gondii. In the Rag1−/− model, the use of DR-18 emphasizes how unopposed IL-18 signals can elevate NK cell and ILC1 activities, while D2D treatment highlights that the increased level of IL-18BP characteristic of acute T. gondii infection is a major determinant of the contribution of endogenous IL-18 to parasite control. Thus, during toxoplasmosis, the rapid induction of high levels of IFN-γ promotes IL-18BP, which provides an explanation as to why endogenous IL-18 is not a major contributor to innate and adaptive mechanisms of resistance. Similarly, during infection with Leishmania sp., where IL-12 has a dominant role in the induction of IFN-γ required for resistance, several reports concluded that IL-18 is not critical for development of protective Th1 response and resolution of disease.52,53 Likewise, for Salmonella, IL-12-induced production of IFN-γ is important for bacterial control, but endogenous IL-18 is not required for this activity, although IL-18 does directly impact on enterocyte functions.9 For Mycobacterium tuberculosis, a system where the role of IL-12 and IFN-γ in resistance to infection is well established, the impact of IL-18 is less clear. One study reported that IL-18, but not the IL-18 receptor α chain, was required to survive challenge.54 However, others have noted comparably minor defects in immunity in Il18−/− mice, with elevated bacterial burdens and lower local IFN-γ in the lungs.7,55 It remains unclear in these other experimental systems what levels of IL-18BP are induced, and the availability of the D2D or other reagents that target the IL-18BP provides the opportunity to test the ability of endogenous IL-18 to impact responses to these pathogens.
In the models discussed above, where microbes drive high IL-12 and rapid IFN-γ, IL-18 does not appear to contribute to IFN-γ-mediated resistance. However, there are instances where the ability of IL-18 to promote IFN-γ is required for resistance to infection. For example, Cryptosporidium sp. are confined to enterocytes of the small intestine, do not require macrophage activation for parasite control, and are associated with low levels of IL-12, but enterocyte-derived IL-18 plays a co-dominant role to promote ILC production of IFN-γ and early innate resistance to this organism.10,11,56 In the case of vaccinia virus, IL-12 is not required for IFN-γ responses,57 but IL-18 is important for viral control.58 Interestingly, vaccinia virus encodes its own viral ortholog of IL-18BP that is produced early during infection as an immunoevasin. Using a murine model of vaccinia infection, we observed that D2D and DR-18 were able to reduce systemic viral burden in both prophylactic and therapeutic treatment regimens (O.-E.W. and A.M.R, unpublished data). This literature suggests that IL-18 is likely relevant in contexts where there are low levels of IL-12 and IFN-γ activity, in which IL-18 may provide a mechanism to enhance nascent responses immediately following pathogen sensing. Thus, in the contexts considered here, endogenous IL-18 may be more relevant to early and/or local responses within tissues than to the regulation of systemic responses. The discrepant greater effect of D2D compared with DR18 on the magnitude of tissue ILC responses lends further support for a local role of endogenous IL-18.
The properties of the engineered DR-18 construct suggest its potential as an anti-parasitic therapeutic beyond its originally intended use in cancer immunotherapy; in addition to its ability to evade the IL-18BP, DR-18 has a higher affinity for the IL18R than WT IL-1838 and is a more potent inducer of IFN-γ production. However, in the setting of T. gondii infection, this variant was associated with increased immune pathology, which was not seen in the tumor-treatment setting. Resistance to T. gondii requires a robust expansion of antigen-specific Th1-polarized CD4+ and CD8+ T cells to limit parasite replication and dissemination, but this response must be tempered to prevent the development of immune-mediated pathology. There are numerous examples where the CD4+ T cell responses associated with T. gondii result in damage to diverse tissues that include the gut, lungs, liver, and brain.59 The finding that treatment with the D2D augmented T cell responses in the liver (but not the spleen) supports the idea that IL-18 would be produced at sites of inflammation where it can influence the local response.
It has been suggested previously that IL-18 is inherently hepatotoxic in humans,60 but in our studies, IL-18-mediated hepatotoxicity was only observed in the presence of CD4+ T cells in infected mice. DR-18 treatment has previously not been found to elicit liver toxicity in tumor-bearing mice,38 and patients treated with very high doses of rIL-18 (up to 2 mg/kg/week or 1 mg/kg/day) did not exhibit significant hepatotoxicity.61,62 Consistent with these observations, we found that in Rag1−/− mice, the ability of DR-18 to limit parasite replication results in hepatoprotection, suggesting that IL-18-mediated damage to the liver is indirect and secondary to effects on CD4+ T cells. However, treatment of B6 mice with systemic DR-18 resulted in immune pathology mediated by antigen-experienced CD4+ T cell in the liver. This finding illustrates potential adverse consequence of sustained, unregulated IL-18 signaling in the context of infection and the important role that IL-18BP plays in limiting pathological T cell responses.
Previous studies have focused on the impact of IL-18 on CD8+ T cells, and the anti-tumor effect of DR-18 is dependent on tumor-infiltrating CD8+ T cells.38,63,64 In contrast, the studies presented here show that the DR-18-driven immune pathology is dependent on infection-induced CD4+ T cells. Multiple studies by our laboratory and others have concluded that CD4+ and CD8+ T cells represent broadly comparable sources of IFN-γ during toxoplasmosis—specifically following ex vivo PMA/ionomycin stimulation followed by intracellular staining of IFN-γ.27,65–67 Consequently, it was unanticipated that IFN-γ reporter mice would reveal that CD4+ T cells were the main source of IFN-γ and that this was augmented by DR-18. The reporter mouse, an artificial bacterial chromosome transgenic mouse that expresses the surface marker CD90.1/Thy1.1 under the IFN-γ promoter, may be a much more faithful representation of the endogenous IFN-γ production dominated by CD4+ T cells. Likewise, during STAg restimulation in the context of the CD4 and CD8 co-receptor blockade, CD4+ T cells were the main source of both IFN-γ and GM-CSF. The bias toward cytokine production by CD4+ T cells observed in these studies may explain their prominent role in immune pathology during toxoplasmosis.68–70 Notably, in genetic knockout models where CD4+ T cells mediate pathology during Toxoplasma infection, depletion of these cells does not increase acute susceptibility.
The duality of CD4+ T cells as mediators of both protective immunity as well as immune pathology highlights the importance of regulation of these cells. The cytokines IL-10 and IL-27 act in distinct fashion to limit these pathological CD4+ T cell responses: IL-10 inhibits accessory cell functions, while IL-27 directly limits the magnitude and duration of the T cell responses. The studies here highlight that IL-18BP acts on an orthogonal axis—by blocking the activity of IL-18 on T cells—providing an additional non-redundant regulatory mechanism that operates during infection and underscoring the critical importance of the pathway in mediating the balance between protective immunity and life-threatening immunopathology. A notable phenotypic similarity exists between the immune pathology observed in Il10−/− mice69 and mice treated with DR18 that suggests potential interplay or epistatic function of IL-10. However, previous studies have noted that IL-10 fails to inhibit IL-18 production and that Il10−/− mice infected with T. gondii produce normal levels of IL-18 and IL-18BP.34 These findings suggest that endogenous IL-18 serves as a powerful amplifier of incipient local responses to infection, which must be tightly controlled during systemic responses to limit pathology. However, in the service of medicine, pharmacological paradigms that can fine-tune these mechanisms, such as the D2D IL-18BP antagonist, present a potential avenue in host-directed immunotherapies for infectious disease.
Limitations of the study
There are two main limitations to this study. The first relates to the fact that the DR18 is not only a mutein with an inability to bind to the IL-18BP but that it also has a higher affinity for the IL-18R than IL-18. Thus, part of its biological activity may not solely be due to its ability to avoid the IL-18BP. The second is that while endogenous IL-18 is implicated in resistance to a subset of intracellular infections, there are other instances where IL-18 is not important. The studies presented here indicate that the IL-18BP is a major factor that limits the activity of IL-18 during toxoplasmosis, but additional studies using the D2D will be needed to understand whether this is a more universal phenomena relevant to other infections.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christopher Hunter (chunter@vet.upenn.edu)
Materials availability
The sequence of D2D and methods to produce it are described in the materials and methods.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication under GEO Accession # GSE207173. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not generate original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
B6 (C57BL/6NTac) (Taconic #B6-F), Rag1−/− (B6.129S7-Rag1tm1Mom/J) (Jackson #002216) mice were purchased from their respective vendors. IFN-γ-Thy1.1 (Ifngtm1(Thy1)Weav) reporter mice were generated by Casey Weaver.43 Mice were housed in a specific pathogen free environment at the University of Pennsylvania School of Veterinary Medicine and treated according to protocols approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania. Male and female (age 8–12 weeks at start of experiment) mice were used for all experiments.
Parasites, infection, and treatment
The ME49 strain of T. gondii was maintained by serial passage in Swiss Webster mice and used to generate banks of chronically infected CBA/ca mice, which were a source of tissue cysts for these experiments. For all experiments presented here, mice were infected intraperitoneally (i.p.) with 20 cysts ME49. Soluble toxoplasma antigen was prepared from tachyzoites of the RH strain as described previously.71 Treatment of infected mice was performed via IP injection beginning at 1 dpi, at 0.5 mg/kg (IL-18 and DR18) or 5 mg/kg (D2D). For antibody depletion experiments, 500 μg of anti-CD4 antibody clone GK1.5 or anti-CD8 antibody clone 2.43 were administered IP 6 h before cytokine treatment. For quantitative PCR (qPCR), DNA was isolated from tissues using the DNEasy DNA isolation kit (Qiagen) followed by qPCR measuring the abundance of the T. gondii gene B1 using the primers 5′-TCTTTAAAGCGTTCGTGGTC-3′ (forward) and 5′-GGAACTGCATCCGTTCATGAG-3′ (reverse).
METHOD DETAILS
Histology
For IHC detection of T. gondii and iNOS, tissues were fixed in 10% formalin solution and then paraffin embedded and sectioned. Sections were deparaffinized, rehydrated, Ag retrieved in 0.01 M sodium citrate buffer (pH 6.0), and endogenous peroxidase blocked by 0.3% H2O2 in PBS. After blocking with 2% normal goat serum, the sections were incubated either with rabbit anti-Toxoplasma Ab, anti-INOS Ab or isotype control. The sections were then incubated with biotinylated goat anti-rabbit IgG (Vector, Burlingame, CA), and ABC reagent was applied (Vectastain ABC Kit; Vector Labs). Then DAB substrate (Vector Labs) was used to visualize specific staining according to manufacturer’s instructions, and slides were counterstained with hematoxylin. To quantify parasite burden in the peritoneal exudate, 100,000 cells were used to prepare cytospins. Cells were methanol fixed and then stained with the Protocol Hema-3 Stain Set, and the ratio of infected cells to total cells in a field of view was calculated, with a minimum of 200 cells counted per sample.
Generation of lymphokine activated killer cells
Lymphokine Activated Killer cells (LAKs) were generated from Rag1−/− bone marrow as described previously.72,73 Briefly, whole bone marrow was plated at 1M cells/mL in cRPMI +400U/mL Proleukin human IL-2 (Peprotech). Fresh IL-2 was added every third day, and cells were used for experiments between days 7–10.
Antibody and cytokine reagents
IL-18 reagents: IL-18 ELISA (DY7625), IL-18BP ELISA (DY122), and GM-CSF ELISA (DY415) kits were purchased from R&D Biosystems. WT IL-18 and DR18 constructs were generated as described previously.38 D2D engineering and production is described below.
For flow cytometry the following antibodies were used: for analysis of NK cells and T cells: CD335 NKp46 (29A1.4, eBioscience), NK-1.1 (PK136, Biolegend), IFN-γ (XMG1.2, eBioscience), CD200R1 (OX110, eBioscience, Tbet (4B10, Biolegend), EOMES (Dan11-mag, eBioscience). IL-18Rα (P3TUNYA, eBiosciences), CD4 (GK1.5, eBiosciences or in context of GK1.5 mediated depletion, RM4–5, Biolegend), CD8α (53–6.7, Biolegend), CD8β (H35–17.2, eBiosciences), TCRβ (h57–597, eBiosciences), CD11a (2D7, BD), CXCR3 (CXCR3–173, Biolegend), KLRG1 (2F1, eBiosciences), CX3CR1 (SA011F11, Biolegend), CD90.1 (HIS51, eBiosciences). For analysis of myeloid cells: CD11b (M1/70, eBioscience), CD11c (N418, Biolegend), Ly6c (HK1.4, Biolegend), Ly6g (1A8, Biolegend), CCR2 CD192 (SA203G11, Biolegend), CD64 FcgRI (X54–5/7.1, Biolegend), MHC II I-A/I-E (m5/114.15.2, eBioscience), iNOS (CXNFT, eBioscience). Flow cytometry was performed on BD Fortessa, Symphony A3 Lite, and X-50 cytometers and data analysis was performed using Flowjo 9 and Flowjo 10 (Treestar), and Prism 9 (Graphpad). Uniform Manifold Approximation and Projection for Dimension Reduction (uMAP) analysis was performed using the uMAP plug-in (version: 1802.03426, 2018, ©2017, Leland McInness) for Flowjo (Version 10.53). The Euclidean distance function was utilized with a nearest neighbor score of 15, and a minimum distance rating of 0.5.
Directed evolution of D2D
Protein expression, purification, and biotinylation
The mature form of human (h) IL-18 (amino acids 37–193) and IL-18 D2D (amino acid substitutions D53G, E67A, T70E, D71S, M87F, M96L, and Q139L) were assembled as gene blocks (Integrated DNA Technologies, IDT) and cloned into a pSH vector for expression of N-terminal SUMO-tagged and C-terminal hexahistidine-tagged proteins in Escherichia coli BL21 (DE3) Rosetta strain (Fisher Scientific, 70,954–3). Protein expression was induced with 0.5 mM IPTG at 16°C for 20 h. The fusion protein was first purified using an Ni-NTA column (Fisher Scientific, P188223), followed by removal of the SUMO tag with the SUMO protease Ulp1. Next, the protein solution was buffer-exchanged to remove imidazole and re-applied to a second Ni-NTA column to remove the free SUMO tag. The eluted protein was concentrated and separated from aggregates by gel-filtration (Column SEC70, Bio-rad). Protein from the monodisperse peak was pooled and loaded on a final Ni-NTA column for endotoxin removal with 0.2% Triton X-114 at 4°C. Finally, the eluted protein was buffer exchanged into sterile, endotoxin free PBS using a PD-10 column (GE Healthcare) and flash-frozen in liquid nitrogen for storage at −80°C.
The mouse (m) IL-18Rα ectodomain, hIL-18Rα ectodomain, hIL-18Rβ ectodomain, mIL-18 binding protein (IL-18BP) and viral (v) IL-18BP (vaccinia and variola) were expressed by transient transfection of Expi293 cells (Thermo Fisher). Sequences were cloned into the pEZT_D_Lux vector with an N-terminal H7 signal peptide and a C-terminal AviTag and hexahistidine tag. Plasmids were transfected into Expi293 cells by ExpiFectamine 293 Transfection Kit (Thermo Fisher, A14524) per the manufacturer’s instructions. Cells were harvested 3–5 days after transfection. Proteins were captured from cell supernatant via Ni-NTA chelating resin and further purified by size exclusion chromatography (Column SEC70, Bio-rad) into a final buffer of HEPES buffered saline (HBS; 10 mM HEPES, pH 7.5, 150 mM NaCl).
For protein biotinylation, proteins were expressed with a C-terminal biotin acceptor tag (AviTag)-GLNDIFEAQKIEWHE. After Ni-NTA chromatography, protein biotinylation was carried out at room temperature for 2 h with soluble BirA ligase enzyme in 0.1 mM bicine (pH 8.3), 10 mM ATP, 10 mM magnesium acetate, and 0.5 mM biotin (Avidity, BIO500). Biotinylated proteins were then purified by gel-filtration as described above. Biotinylation efficiency was assessed using an SDS/PAGE streptavidin-shift assay.
Human D2D IL-18 library construction and selection
Sixteen residues in IL-18 which were in contact with both IL-18Rα and IL-18BP were identified by aligning the structure of hIL-18–hIL-18Rα–hIL-18Rβ complex (Protein DataBank (PDB) ID 3WO4) to the structure of hIL-18: ectromelia virus IL-18BP complex (PDB ID 3F62). A library randomizing these residues was constructed using assembly PCR with the degeneration primers. The PCR products were further amplified with primers containing homology to the pYAL vector and co-electroporated into EBY100 competent yeast together with linearized pYAL vector. The resulting library was later measured to contain 3.9 × 108 transformants.
Transformed yeast were recovered and expanded in SDCAA medium at 30 °C, induced by 1:10 dilution into SGCAA medium and cultured at 20 °C for 24–48 h. The appropriate numbers of induced yeast were used in each round to ensure at least tenfold coverage of the expected diversity, and not less than 108 cells. All selection steps were carried out at 4 °C using PBE buffer (PBS with 0.5% BSA and 2 mM EDTA). For round 1, the yeast library was counter-selected with anti-Cy5/Alexa Fluor 647 microbeads (Miltenyi, 130–091-395) with an LS MACS column (Miltenyi, 130–042-401) to remove non-specific binders. Positive selection was performed by labeling yeast with 5 nM biotinylated mIL-18BP, followed by magnetic selection with Alexa Fluor 647 microbeads and the LS MACS column. For subsequent rounds, the library was positively selected with 1 nM mouse IL-18BP (round 2) and 1 nM of both mouse and viral IL-18BP orthologs for (round 3) and counter selected with 1μM biotinylated h/mIL-18BRα and hIL-18Rβ. IL-18 display levels were determined by staining with AF488-conjugated anti-Myc (Cell Signaling Technologies). The library was selected by FACS sorting with a Sony SH800 cell sorter by excluding IL-18 receptor binders and gating the top 1% of display-normalized IL-18RBP binders. After each round of selection, recovered yeast were expanded in SDCAA medium at 30 °C overnight and later induced at 20 °C by a 1:10 dilution into SGCAA medium for 24–48 h.
IL-18 reporter assay
The IL-18 HEK-Blue assay (InvivoGen, hkb-hmil18) was performed according to the manufacturer’s instructions. Briefly, D2D was titrated onto 150 nM (final) of IL-18BP and incubated for 45 min before the mixture was transferred to IL-18 at 0.5 ng/mL (final). The three-component mixture or D2D alone was then used to stimulate 5 × 104 cells seeded into wells of a 96-well plate for 20 h at 37°C and 5% CO2. Twenty microliters of cell culture supernatant were then taken from each well and mixed with 180 μL of QUANTI-Blue solution in a separate 96-well plate, incubated for 15 min at 37°C, and then read in a microplate reader at 630 nm. The data were plotted and analyzed using Prism.
IC50 = 78.5 nM, which is ~1/2 of [IL18BP] used in the assay.
Single-cell RNA sequencing
Spleen tissue was manually disrupted with a razor blade in a 6cm dish and suspended in 1 mL plain PBS +5 mM CaCl2 + 10 mg/mL B. Licheniformis protease +125U/mL DNase/50 mg tissue, then manually disrupted by pipette every 2 min for 45 min at 4C. Digested tissue was then mashed through a 70uM filter and washed with 10% FBS RPMI. Cells were pelleted via centrifugation at 300 × g for 5 min @ 4C. The pellet was then ACK lysed at 4C for 5 min, followed by washing with 10% RPMI.
Library preparation was done according to manufacturer’s protocol. Single cell drop encapsulation, RT-PCR, amplification, and adapter ligation were performed with the 10X Chromium Controller platform with the Chromium Next GEM Single Cell 3′ GEM, Library and Gel Bead Kit v3.1, Chromium Next GEM Chip G Singe Cell Kit, and Single Index Kit T Set A (10X Genomics). Following encapsulation, samples were cleaned-up using Dynabeads (Invitrogen) according to manufacturer’s protocol. RNA and DNA quality was checked on a Tapestation (Agilent). Paired-end sequencing was performed on a NextSeq5000 (Illumina).
Raw sequence data was aligned and transcripts were counted with Cell Ranger (10X Genomics) using the mus musculus reference genome mm10–3.0.0. The R Seurat package was utilized to analyze single cell data and generate graphics, with cells with fewer than 1000 counts, greater than 25,000 counts, or greater than 20% mitochondrial RNA were excluded from analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as means ± standard error of the mean (SEM). For comparisons between two groups, the Student’s t-test was applied. For data with more than two datasets and one independent variable, one-way ANOVA coupled with Tukey’s multiple comparisons test was applied. When multiple datasets with two independent variables were analyzed, two-way ANOVA with Tukey’s multiple comparisons test was applied. Statistical details are indicated in figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rat anti-mouse CD4 clone GK1.5 | BioXCell | (Bio X Cell Cat# BE0003–1, RRID:AB_1107636) |
| Rat anti-mouse CD8 clone 2.43 | BioXcell | (BioXCell Cat# BE0061, RRID:AB_1125541) |
| CD335 (NKp46) Monoclonal Antibody (29A1.4), PE-eFluor™ 610, eBioscience | Thermo Fisher | (Thermo Fisher Scientific Cat# 61–3351-82, RRID:AB_2574606) |
| Brilliant Violet 711™ anti-mouse NK-1.1 Antibody | Biolegend | (BioLegend Cat# 108745, RRID:AB_2563286) |
| IFN gamma Monoclonal Antibody (XMG1.2), PerCP-Cyanine5.5, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 45–7311-82, RRID:AB_1107020) |
| CD200 Receptor Monoclonal Antibody (OX110), APC, eBioscience™ | Biolegend | (Thermo Fisher Scientific Cat# 17–5201-82, RRID:AB_10717289) |
| PE/Cyanine7 anti-T-bet Antibody | Biolegend | (BioLegend Cat# 644824, RRID:AB_2561761) |
| EOMES Monoclonal Antibody (Dan11mag), PE, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 12–4875-82, RRID:AB_1603275) |
| CD218a (IL-18Ra) Monoclonal Antibody (P3TUNYA), eFluor™ 450, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 48–5183-82, RRID:AB_2574069) |
| Alexa Fluor® 700 Rat Anti-Mouse CD4 | BD | (BD Biosciences Cat# 561025, RRID:AB_2034006) |
| Brilliant Violet 650™ anti-mouse CD8a Antibody | Biolegend | (BioLegend Cat# 100742, RRID:AB_2563056) |
| CD8b Monoclonal Antibody (eBioH35–17.2 (H35–17.2)), PE-Cyanine7, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 25–0083-82, RRID:AB_11218494) |
| CD8b Monoclonal Antibody (eBioH35–17.2 (H35–17.2)), FITC, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 11–0083-82, RRID:AB_657764) |
| TCR beta Monoclonal Antibody (H57–597), PE, eBioscience™ | Thermo Fisher | (BD Biosciences Cat# 562839, RRID:AB_2737830) |
| PerCP-Cy™5.5 Rat Anti-Mouse CD11a | BD | (BD Biosciences Cat# 562809, RRID:AB_2737809) |
| Brilliant Violet 650™ anti-mouse CD183 (CXCR3) Antibody | Biolegend | (BioLegend Cat# 126531, RRID:AB_2563160) |
| Brilliant Violet 711™ anti-mouse/human KLRG1 (MAFA) Antibody | Biolegend | (BioLegend Cat# 138427, RRID:AB_2629721) |
| Brilliant Violet 785™ anti-mouse CX3CR1 Antibody | Biolegend | (BioLegend Cat# 149029, RRID:AB_2565938) |
| CD90.1 (Thy-1.1) Monoclonal Antibody (HIS51), PE-Cyanine7, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 25–0900-82, RRID:AB_469640) |
| CD11b Monoclonal Antibody (M1/70), Brilliant Violet™ 650, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 416–0112-80 |
| APC/Cyanine7 anti-mouse CD11c Antibody | Biolegend | (BioLegend Cat# 117324, RRID:AB_830649) |
| Brilliant Violet 785™ anti-mouse Ly-6C Antibody | Biolegend | (BioLegend Cat# 128041, RRID:AB_2565852) |
| Brilliant Violet 711™ anti-mouse Ly-6G Antibody | Biolegend | (BioLegend Cat# 127643, RRID:AB_2565971) |
| iNOS Monoclonal Antibody (CXNFT), APC, eBioscience™ | Thermo Fisher | (Thermo Fisher Scientific Cat# 17–5920-82, RRID:AB_2573244) |
| Alexa Fluor® 700 anti-mouse Ki-67 Antibody | Biolegend | (BioLegend Cat# 652420, RRID:AB_2564285) |
| IFN gamma Monoclonal Antibody (AN-18), eBioscience | Biolegend | (Thermo Fisher Scientific Cat# 14–7313-85, RRID:AB_468472) |
| Biotin anti-mouse IFN-gamma | Biolegend | (BioLegend Cat# 505704, RRID:AB_315392) |
| Anti-Cy5/Alexa Flour 647 microbeads | Miltenyi | 130–091-395 RRID:AB_244369 |
| AF488 anti-Myc | Cell Signaling Technology | D84C12 RRID:AB_2798045 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Wild type mouse IL-18 | Aaron Ring | N/A |
| Decoy Resistant IL-18 (DR18) | Aaron Ring | N/A |
| Decoy to Decoy IL-18 (D2D) | Aaron Ring | N/A |
| rIL-18BP | Aaron Ring | N/A |
| Human IL-2 | Peprotech | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse IL-18 Duoset ELISA kit | R&D Systems | (R and D Systems Cat# DY7625–05, RRID:AB_2895548) |
| Mouse IL-18BP Duoset ELISA | R&D Systems | DY122 |
| Mouse GM-CSF Duoset ELISA | R&D Systems | DY415 |
| ExpiFectamine 293 Transfection Kit | Thermofisher | Cat#A14525 |
|
| ||
| Deposited data | ||
|
| ||
| Single cell sequencing of splenocytes following T. gondii infection | This paper | GEO Accession# GSE207173 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEKBlue IL-18 assay | Invivogen | (InvivoGen, hkb-hmil18) (RRID:CVCL_UF33) |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: B6 (C57BL/6NTac) (Taconic #B6-F) | Taconic | RRID:IMSR_TAC:b6 |
| Mouse: B6 Ragi(B6.129S7- Rag1tm1Mom/J) (Jackson #002216) | Jackson | RRID:MGI:3582299 |
| Mouse: B6 //hgtm1(Thy1)Weav | Jackson | RRID:MGI:3831299 |
| T. gondii ME49 Strain | Hunter lab | N/A |
| S. cerevisiae: Strain background: EBY100 | Dane Wittrup Lab | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer: Toxoplasma B1 Fwd: TCTTTAAAGCGTTCGTGGTC | Thermo Fisher | N/A |
| Primer: Toxoplasma B1 Rev: GGAACTGCATCCGTTCATGAG | Thermo Fisher | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSH vector | Aaron Ring Lab | N/A |
| pEZT_DLux | Aaron Ring Lab | N/A |
| pYAL Vector | Aaron Ring Lab | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Flowjo 10 | Treestar | FlowJo (RRID:SCR_008520) |
| Uniform Manifold Approximation and Projection for Dimension Reduction (uMAP) | McInnes, L ArXiv e-prints 1802.03426, 2018 | Umap (RRID:SCR_018217) |
| Cell Ranger | 10x Genomics | Cell Ranger (RRID:SCR_017344) |
| Seurat | OMICtools | OMICtools (RRID:SCR_002250) |
| Prism | Graphpad | GraphPad Prism (RRID:SCR_002798) |
Highlights.
Generation of reagents to dissect impact of IL-18 on resistance to infection
Antagonism of IL-18 binding protein enhances resistance to infection
IL-18 variant bypasses IL-18 binding protein and promotes innate resistance to infection
High-affinity IL-18 variant promotes CD4+ T cell-mediated immune pathology
ACKNOWLEDGMENTS
The authors would like the acknowledge the members of the Hunter and Ring labs for scientific and moral support. This work was supported by grants from the National Institute of Allergy and Infectious Diseases (5R01AI25563-05 and 5T32AI00753223 to C.A.H.), National Cancer Institute Immuno-Oncology Translation Network (U01CA233096 to A.M.R.), Gabrielle’s Angel Foundation (to A.M.R.), and an NIH Director’s Early Independence Award (DP5OD 023088 to A.M.R.).
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
A.M.R. and T.Z. are named inventors on a patent application that describes the DR-18 and D2D molecules. A.M.R. is the founder and a Director of Simcha Therapeutics, the commercial licensee of DR-18.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112147.
REFERENCES
- 1.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, and Akira S (1998). Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8, 383–390. 10.1016/S1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 2.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada Y, and Hattori K (1995). Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 378, 88–91. [DOI] [PubMed] [Google Scholar]
- 3.Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, and Kaneda K (1996). IFN-gamma-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J. Immunol. 157, 3967–3973. [PubMed] [Google Scholar]
- 4.Nakamura K, Okamura H, Wada M, Nagata K, and Tamura T (1989). Endotoxin-induced serum factor that stimulates gamma interferon production. Infect. Immun. 57, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Micallef MJ, Tanimoto T, Kohno K, Ikeda M, and Kurimoto M (1997). Interleukin 18 induces the sequential activation of natural killer cells and cytotoxic T lymphocytes to protect syngeneic mice from transplantation with Meth A sarcoma. Cancer Res. 57, 4557–4563. [PubMed] [Google Scholar]
- 6.Micallef MJ, Ohtsuki T, Kohno K, Tanabe F, Ushio S, Namba M, Tanimoto T, Torigoe K, Fujii M, Ikeda M, et al. (1996). Interferon-γ-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-γ production. Eur. J. Immunol. 26, 1647–1651. [DOI] [PubMed] [Google Scholar]
- 7.Kinjo Y, Kawakami K, Uezu K, Yara S, Miyagi K, Koguchi Y, Hoshino T, Okamoto M, Kawase Y, Yokota K, et al. (2002). Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis : a comparative study with IL-12p40. J. Immunol. 169, 323–329. 10.4049/jimmunol.169.1.323. [DOI] [PubMed] [Google Scholar]
- 8.Rauch I, Katherine AD, X Ji D, Jakob von M, Jeannette LT, Angus YL, Naomi HP, Janelle SA, Igor EB, Karsten G, and Russell EV (2017). NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and −8. Immunity 46, 649–659. 10.1016/j.immuni.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarret A, Jackson R, Duizer C, Healy ME, Zhao J, Rone JM, Bielecki P, Sefik E, Roulis M, Rice T, et al. (2020). Enteric nervous system-derived IL-18 orchestrates mucosal barrier immunity. Cell 180, 813–814. [DOI] [PubMed] [Google Scholar]
- 10.Sateriale A, Gullicksrud JA, Engiles JB, McLeod BI, Kugler EM, Henao-Mejia J, Zhou T, Ring AM, Brodsky IE, Hunter CA, and Striepen B (2021). The intestinal parasite Cryptosporidium is controlled by an enterocyte intrinsic inflammasome that depends on NLRP6. Proc. Natl. Acad. Sci. USA 118, e2007807118. 10.1073/pnas.2007807118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gullicksrud J, Sateriale A, Englies J, Gibson A, Shaw S, Hutchins Z, Martin L, Christian D, Taylor GA, Yamamoto M, et al. (2021). Cross-talk between enterocytes and innate lymphoid cell drives early IFN-γ-mediated control of Cryptosporidium. Preprint at bioRxiv. 10.1101/2021.03.13.435244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei XQ, Leung BP, Niedbala W, Piedrafita D, Feng GJ, Sweet M, Dobbie L, Smith AJ, and Liew FY (1999). Altered immune responses and susceptibility to Leishmania major and Staphylococcus aureus infection in IL-18-deficient mice. J. Immunol. 163, 2821–2828. [PubMed] [Google Scholar]
- 13.Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, Surrey LF, Russo P, Sleight A, Schiffrin E, et al. (2017). Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 139, 1698–1701. 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grobmyer SR, Lin E, Lowry SF, Rivadeneira DE, Potter S, Barie PS, and Nathan CF (2000). Elevation of IL-18 in human sepsis. J. Clin. Immunol. 20, 212–215. [DOI] [PubMed] [Google Scholar]
- 15.Pirhonen J, Sareneva T, Kurimoto M, Julkunen I, and Matikainen S (1999). Virus infection activates IL-1 and IL-18 production in human macrophages by a caspase-1-dependent pathway 1. J. Immunol. 162, 7322–7329. [PubMed] [Google Scholar]
- 16.Dinarello CA (1999). Interleukin-18. Methods 19, 121–132. [DOI] [PubMed] [Google Scholar]
- 17.Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, and Rubinstein M (1999). Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 10, 127–136. [DOI] [PubMed] [Google Scholar]
- 18.Hunter CA, and Sibley LD (2012). Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10, 766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gazzinelli RT, Hieny S, Wynn TA, Wolf S, and Sher A (1993). Interleukin 12 is required for the T-lymphocyte-independent induction of interferon gamma by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc. Natl. Acad. Sci. USA 90, 6115–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter CA, Subauste CS, Van Cleave VH, and Remington JS (1994). Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect. Immun. 62, 2818–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, and Sher A (1994). Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 153, 2533–2543. [PubMed] [Google Scholar]
- 22.Vollmer TL, Waldor MK, Steinman L, and Conley FK (1987). Depletion of T-4+ lymphocytes with monoclonal antibody reactivates toxoplasmosis in the central nervous system: a model of superinfection in AIDS. J. Immunol. 138, 3737–3741. [PubMed] [Google Scholar]
- 23.Gazzinelli R, Xu Y, Hieny S, Cheever A, and Sher A (1992). Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J. Immunol. 149, 175–180. [PubMed] [Google Scholar]
- 24.Lütjen S, Soltek S, Virna S, Deckert M, and Schlüter D (2006). Organ- and disease-stage-specific regulation of Toxoplasma gondii-specific CD8-T-cell responses by CD4 T cells. Infect. Immun. 74, 5790–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones LA, Roberts F, Nickdel MB, Brombacher F, McKenzie ANJ, Henriquez FL, Alexander J, and Roberts CW (2010). IL-33 receptor (T1/ST2) signalling is necessary to prevent the development of encephalitis in mice infected with Toxoplasma gondii. Eur. J. Immunol. 40, 426–436. [DOI] [PubMed] [Google Scholar]
- 26.Batista SJ, Still KM, Johanson D, Thompson JA, OʼBrien CA, Lukens JR, and Harris TH (2020). Gasdermin-D-dependent IL-1α release from microglia promotes protective immunity during chronic Toxoplasma gondii infection. Nat. Commun. 11, 3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Still KM, Batista SJ, O’Brien CA, Oyesola OO, Früh SP, Webb LM, Smirnov I, Kovacs MA, Cowan MN, Hayes NW, et al. (2020). Astrocytes promote a protective immune response to brain Toxoplasma gondii infection via IL-33-ST2 signaling. PLoS Pathog. 16, e1009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark JT, Christian DA, Gullicksrud JA, Perry JA, Park J, Jacquet M, Tarrant JC, Radaelli E, Silver J, and Hunter CA (2021). Il-33 promotes innate lymphoid cell-dependent IFN-γ production required for innate immunity to toxoplasma gondii. Elife 10, e65614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunter CA, Chizzonite R, and Remington JS (1995). IL-1 beta is required for IL-12 to induce production of IFN-gamma by NK cells. A role for IL-1 beta in the T cell-independent mechanism of resistance against intracellular pathogens. J. Immunol. 155, 4347–4354. [PubMed] [Google Scholar]
- 30.Yap GS, Ortmann R, Shevach E, and Sher A (2001). A heritable defect in IL-12 signaling in B10.Q/J mice. II. Effect on acute resistance to toxoplasma gondii and rescue by IL-18 treatment. J. Immunol. 166, 5720–5725. [DOI] [PubMed] [Google Scholar]
- 31.Vossenkämper A, Struck D, Alvarado-Esquivel C, Went T, Takeda K, Akira S, Pfeffer K, Alber G, Lochner M, Förster I, and Liesenfeld O (2004). Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur. J. Immunol. 34, 3197–3207. [DOI] [PubMed] [Google Scholar]
- 32.LaRosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, and Turka LA (2008). T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc. Natl. Acad. Sci. USA 105, 3855–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai G, Kastelein R, and Hunter CA (2000). Interleukin-18 (IL-18) enhances innate IL-12-mediated resistance to Toxoplasma gondii. Infect. Immun. 68, 6932–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zediak VP, and Hunter CA (2003). IL-10 fails to inhibit the production of IL-18 in response to inflammatory stimuli. Cytokine 21, 84–90. [DOI] [PubMed] [Google Scholar]
- 35.Harel M, Girard-Guyonvarc’h C, Rodriguez E, Palmer G, and Gabay C (2020). Production of IL-18 binding protein by radiosensitive and radio-resistant cells in CpG-induced macrophage activation syndrome. J. Immunol. 205, 1167–1175. [DOI] [PubMed] [Google Scholar]
- 36.Grover HS, Blanchard N, Gonzalez F, Chan S, Robey EA, and Shastri N (2012). The Toxoplasma gondii peptide AS15 elicits CD4 T cells that can control parasite burden. Infect Immun. 80, 3279–3288. 10.1128/IAI.00425-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson DC, Grotenbreg GM, Liu K, Zhao Y, Frickel EM, Gubbels M-J, Ploegh HL, and Yap GS (2010). Differential Regulation of Effector- and Central-Memory Responses to Toxoplasma gondii Infection by IL-12 Revealed by Tracking of Tgd057-Specific CD8+ T Cells. PLOS Pathogens 6, e1000815. 10.1371/journal.ppat.1000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, Fischer S, Jackson R, Flavell RA, Wang J, et al. (2020). IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scharton-Kersten TM, Yap G, Magram J, and Sher A (1997). Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J. Exp. Med. 185, 1261–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yap GS, and Sher A (1999). Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-γ- and tumor necrosis factor (TNF)-α- dependent host resistance to the intracellular pathogen, Toxoplasma gondii. J. Exp. Med. 189, 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu HH, Chan SW, Gosling JP, Blanchard N, Tsitsiklis A, Lythe G, Shastri N, Molina-París C, and Robey EA (2016). Continuous effector CD8 + T cell production in a controlled persistent infection is sustained by a proliferative intermediate population. Immunity 45, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, and von Andrian UH (2016). The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 45, 1270–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatton RD, Harrington LE, Luther RJ, Wakefield T, Janowski KM, Oliver JR, Lallone RL, Murphy KM, and Weaver CT (2006). A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity 25, 717–729. [DOI] [PubMed] [Google Scholar]
- 44.Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, Masters S, Sher A, Leppla SH, Moayeri M, et al. (2014). Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to toxoplasma gondii. mBio 5, e1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ewald SE, Chavarria-Smith J, and Boothroyd JC (2014). NLRP1 is an inflammasome sensor for toxoplasma gondii. Infect. Immun. 82, 460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fisch D, Bando H, Clough B, Hornung V, Yamamoto M, Shenoy AR, and Frickel EM (2019). Human GBP 1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 38, e100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fisch D, Clough B, Domart MC, Encheva V, Bando H, Snijders AP, Collinson LM, Yamamoto M, Shenoy AR, and Frickel EM (2020). Human GBP1 differentially targets Salmonella and toxoplasma to license recognition of microbial ligands and caspase-mediated death. Cell Rep. 32, 108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mühl H, Kämpfer H, Bosmann M, Frank S, Radeke H, and Pfeilschifter J (2000). Interferon-γ mediates gene expression of IL-18 binding protein in nonleukocytic cells. Biochem. Biophys. Res. Commun. 267, 960–963. [DOI] [PubMed] [Google Scholar]
- 49.Hurgin V, Novick D, and Rubinstein M (2002). The promoter of IL-18 binding protein: activation by an IFN-γ-induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein β. Proc. Natl. Acad. Sci. USA 99, 16957–16962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki Y, Orellana MA, Schreiber RD, and Remington JS (1988). Interferon-γ: the major mediator of resistance against toxoplasma gondii. Science 240, 516–518. [DOI] [PubMed] [Google Scholar]
- 51.Nathan CF, Murray HW, Wiebe ME, and Rubin BY (1983). Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J. Exp. Med. 158, 670–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sousa LMA, Carneiro MBH, Dos Santos LM, Natale CC, Resende ME, Mosser DM, and Vieira LQ (2015). IL-18 contributes to susceptibility to Leishmania amazonensis infection by macrophage-independent mechanisms. Cytokine 74, 327–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Monteforte GM, Takeda K, Rodriguez-Sosa M, Akira S, David JR, and Satoskar AR (2000). Genetically resistant mice lacking IL-18 gene develop Th1 response and control cutaneous Leishmania major infection. J. Immunol. 164, 5890–5893. [DOI] [PubMed] [Google Scholar]
- 54.Schneider BE, Korbel D, Hagens K, Koch M, Raupach B, Enders J, Kaufmann SHE, Mittrücker HW, and Schaible UE (2010). A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur. J. Immunol. 40, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugawara I, Yamada H, Kaneko H, Mizuno S, Takeda K, and Akira S (1999). Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 67, 2585–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bedi B, McNair NN, Förster I, and Mead JR (2015). IL-18 cytokine levels modulate innate immune responses and cryptosporidiosis in mice. J. Eukaryot. Microbiol. 62, 44–50. [DOI] [PubMed] [Google Scholar]
- 57.van Den Broek M, Bachmann MF, Köhler G, Barner M, Escher R, Zinkernagel R, and Kopf M (2000). IL-4 and IL-10 antagonize IL-12-mediated protection against acute vaccinia virus infection with a limited role of IFN-γ and nitric oxide synthetase 2. J. Immunol. 164, 371–378. [DOI] [PubMed] [Google Scholar]
- 58.Reading PC, and Smith GL (2003). Vaccinia virus interleukin-18-binding protein promotes virulence by reducing gamma interferon production and natural killer and T-cell activity. J. Virol. 77, 9960–9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dupont CD, Christian DA, and Hunter CA (2012). Immune response and immunopathology during toxoplasmosis. Semin. Immunopathol. 34, 793–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Belkaya S, Michailidis E, Korol CB, Kabbani M, Cobat A, Bastard P, Lee YS, Hernandez N, Drutman S, de Jong YP, et al. (2019). Inherited IL-18BP deficiency in human fulminant viral hepatitis. J. Exp. Med. 216, 1777–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robertson MJ, Mier JW, Logan T, Atkins M, Koon H, Koch KM, Kathman S, Pandite LN, Oei C, Kirby LC, et al. (2006). Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 12, 4265–4273. [DOI] [PubMed] [Google Scholar]
- 62.Tarhini AA, Millward M, Mainwaring P, Kefford R, Logan T, Pavlick A, Kathman SJ, Laubscher KH, Dar MM, and Kirkwood JM (2009). A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer 115, 859–868. [DOI] [PubMed] [Google Scholar]
- 63.Chiossone L, Audonnet S, Chetaille B, Chasson L, Farnarier C, Berda-Haddad Y, Jordan S, Koszinowski UH, Dalod M, Mazodier K, et al. (2012). Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front. Immunol. 3, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Freeman BE, Hammarlund E, Raué HP, and Slifka MK (2012). Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc. Natl. Acad. Sci. USA 109, 9971–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sturge CR, and Yarovinsky F (2014). Complex immune cell interplay in the gamma interferon response during toxoplasma gondii infection. Infect. Immun. 82, 3090–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dupont CD, Harms Pritchard G, Hidano S, Christian DA, Wagage S, Muallem G, Tait Wojno ED, and Hunter CA (2015). Flt3 ligand is essential for survival and protective immune responses during toxoplasmosis. J. Immunol. 195, 4369–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stumhofer JS, Silver JS, and Hunter CA (2013). IL-21 is required for optimal antibody production and T cell responses during chronic toxoplasma gondii infection. PLoS One 8, e62889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kugler DG, Mittelstadt PR, Ashwell JD, Sher A, and Jankovic D (2013). CD4+ T cells are trigger and target of the glucocorticoid response that prevents lethal immunopathology in toxoplasma infection. J. Exp. Med. 210, 1919–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kühn R, Müller W, Trinchieri G, and Sher A (1996). In The absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157, 798–805. [PubMed] [Google Scholar]
- 70.Villarino A, Hibbert L, Lieberman L, Wilson E, Mak T, Yoshida H, Kastelein RA, Saris C, and Hunter CA (2003). The IL-27r (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity 19, 645–655. [DOI] [PubMed] [Google Scholar]
- 71.Hauser WE Jr., Sharma SD, and Remington JS (1983). Augmentation of NK cell activity by soluble and particulate fractions of Toxoplasma gondii. J. Immunol. 131, 458–463. [PubMed] [Google Scholar]
- 72.Hunter CA, Ellis-Neyer L, Gabriel KE, Kennedy MK, Grabstein KH, Linsley PS, and Remington JS (1997). The role of the CD28/B7 interaction in the regulation of NK cell responses during infection with Toxoplasma gondii. J. Immunol. 158, 2285–2293. [PubMed] [Google Scholar]
- 73.Wherry JC, Schreiber RD, and Unanue ER (1991). Regulation of gamma interferon production by natural killer cells in scid mice: roles of tumor necrosis factor and bacterial stimuli. Infect. Immun. 59, 1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication under GEO Accession # GSE207173. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not generate original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.