

Abstract
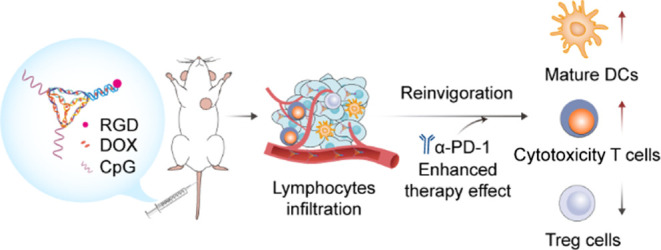
The low response rate and serious side effects of cancer treatment pose significant limitations in immunotherapy. Here, we developed a multifunctional tetrahedral DNA framework (TDF) as a drug carrier to recruit chemotherapeutants and trigger immunogenic cell death (ICD) effects, which could turn tumors from cold to hot to boost the efficacy of antitumor immunotherapy. A tumor-targeting peptide RGD was modified on the TDF to increase the delivery efficiency, and the chemotherapeutant doxorubicin (DOX) was loaded to induce ICD effects, which were assisted by the immune adjuvant of CpG immunologic sequences linked on TDF. We demonstrated that the multifunctional TDF could suppress 4T1 breast tumor growth by increasing tumor infiltration of CD8+ T cells, upregulating granzyme B and perforin expressions to twice as much as the control group, and decreasing 30% CD25+ Treg cells. Furthermore, the combination of α-PD-1 could inhibit the growth of distant tumor and suppressed tumor recurrence in a bilateral syngeneic 4T1 mouse model; the distant tumor weight inhibition rate was about 91.6%. Hence, through quantitatively targeting the delivery of DOX to reduce the side effects of chemotherapy and sensitizing the immune response by ICD effects, this multifunctional TDF therapeutic strategy displayed better treatment effect and a promising clinical application prospect.
Keywords: tetrahedral DNA framework, drug delivery, tumor targeting, immunogenic cell death, immunotherapy
1. Introduction
As the most common malignant tumor, breast cancer (BC) is a significant threat to the health and wellness of females.1 Despite the fact that traditional therapies, such as surgery, chemotherapy, and radiotherapy, have an increased 5 year relative survival rate, numerous patients still suffer from the low efficacy due to tumor development and metastasis.2,3 Recent advances in cancer immunotherapy have enabled innovative treatment of BC.4 However, the “cold tumor” microenvironment with insufficient processing and presenting antigens substantially limits the initiation of an immune response.5−7 These critical facts remind us of the urgent need for better innovative immunotherapy to prolong the survival of patients.
To overcome the low immune response of cold tumors, diverse strategies have been developed to induce immunogenic cell death (ICD) by promoting the uptake of dying tumor cells and fostering dendritic cell (DC) maturation,8 such as exposure of CRT, release of adenosine triphosphate (ATP), and excretion of high-mobility group protein 1 (HMGB1).9−11 Doxorubicin (DOX), as a traditional chemotherapeutic drug, could serve as a typical ICD-induced drug12 to suppress the growth of cancer cells and simultaneously elicit antitumor immune responses. However, the cardiotoxicity, inadequate half-life, poor tumor penetration, and drug resistance of DOX limit its clinical therapeutic dosage and efficacy.13,14 Besides, the immune responses induced by DOX are also restricted to the number of mature DCs and the inefficient activation of naive T cells in the tumor microenvironment.12 To break through those challenges, targeted DOX delivery and addition of immune adjuvants are required for synergy in tumor therapy.
Framework nucleic acid (FNA), a new type of artificial DNA nanostructures formed by self-assembly of DNA strands, has been widely explored in biosensors, biomedicine, and bioimaging due to their biocompatibility, versatility, and spatial structural controllability.15−18 Recently, more and more attention has been paid to the application of FNA in immunomodulatory therapy.19 Herein, we constructed a multifunctional tetrahedral DNA framework (TDF) with both tumor targeting and immunotherapeutic functions, as shown in Scheme 1. Specifically, this TDF modified with tumor-targeting RGD peptides could increase the accumulation in solid tumors of mouse tumor models.20,21 CpG oligonucleotide, which is known as a potent Toll-like receptor-9 (TLR9) agonist,22 recruited in TDF, could initiate immune responses.23 Moreover, DOX, which was inserted into FNA, inhibited tumor proliferation and induced ICD effects to activate immunotherapy. This well-designed functional TDF incorporates multiple tumor treatment strategies that could facilitate better tumor treatment outcomes.
Scheme 1. Schematic Illustration of Multifunctional TDF-Assisted Breast Tumor Treatment by Combination of Targeted Therapy and Immunotherapy.

Multifunctional TDF targeted the 4T1 tumor and released DOX to induce ICD effects. Then, DAMP signals and CpG motifs promoted immature DCs to turn into mature DCs. The recruited cytotoxic T cells inhibited the growth of the primary tumor and also suppressed tumor recurrence by combination with anti-PD-1 antibody.
2. Results and Discussion
2.1. Preparation and Characterization of Multifunctional TDF
In this work, CpG-modified TDF (CpG-T) and RGD/CpG-modified TDF (RGD-CpG-T) were assembled by four precisely designed single-stranded DNA oligonucleotides (Table S1), according to previous methods.23 Two CpG motifs were designed and linked on TDF. RGD tumor-targeted peptide was linked to ssDNA by click reaction (named RGD-ssDNA) and then modified to TDF by the base complementary pairing principle. DOX was inserted into the double helix of CpG-T and RGD-CpG-T to form CpG-TDOX and RGD-CpG-TDOX, respectively (Figure 1a). RGD-modified ssDNA was characterized in nondenaturing polyacrylamide gel electrophoresis (PAGE) shown in Figure S1a, where the single band of RGD-ssDNA indicated a slower mobility than ssDNA. After verification of the synthesis of RGD-ssDNA, various types of TDF were synthesized accordingly, and then excess DOX was mixed with TDF to form CpG-TDOX and RGD-CpG-TDOX, respectively. Subsequently, excess DOX was removed by ultrafiltration, and 8% PAGE was used to verify the structures of CpG-TDOX and RGD-CpG-TDOX. The indicative bands of multifunctional TDF are shown in Figure 1b, where single bands of CpG-TDOX and RGD-CpG-TDOX were displayed in lane 7 and lane 9, respectively. Furthermore, the hydrodynamic diameter of CpG-TDOX and RGD-CpG-TDOX measured by dynamic light scattering (DLS) was ∼11.7 and ∼13.5 nm, respectively (Figure 1d), which was slightly bigger than TDF (∼10.1 nm, Figure S1b). Furthermore, the morphologies of CpG-TDOX and RGD-CpG-TDOX were evaluated by atomic force microscopy (AFM) imaging (Figure 1c).
Figure 1.

Characterization of CpG-TDOX and RGD-CpG-TDOX. (a) Synthesis scheme of RGD-ssDNA and RGD-CpG-TDOX. (b) PAGE result of identification for TDF. Lane 1: DNA marker, lane 2: S1, lane 3: S1 + S2, lane 4: S1 + S2 + S3, lane 5: S1 + S2 + S3 + S4, lane 6: CpG-T, lane 7: CpG-TDOX, lane 8: RGD-CpG-T, lane 9: RGD-CpG-TDOX. (c) AFM imaging of RGD-CpG-TDOX. Scale bar: 100 nm. (d) DLS result of CpG-TDOX and RGD-CpG-TDOX. (e) UV–vis–near-IR spectra of DOX, CpG-T, RGD-CpG-T, RGD-CpG-TDOX, and CpG-TDOX. (f) PAGE result of CpG-TDOX and RGD-CpG-TDOX after incubating with 10% FBS for 0, 4, 8, 12, and 20 h. (g) Structural stability analysis of CpG-TDOX and RGD-CpG-TDOX by semiquantitative analysis of the relative band intensity.
The loading capacity of DOX in TDF was calculated via the UV–vis absorbance spectrum. CpG-T and RGD-CpG-T had a significant absorbance peak at 260 nm due to the characteristic absorption of DNA. After DOX loading, in addition to the absorbance peak at 260 nm, CpG-TDOX and RGD-CpG-TDOX both had a typical absorbance peak at 480 nm due to the characteristic absorption of DOX (Figure 1e), which indicated that DOX was successfully loaded in CpG-T and RGD-CpG-T. According to the concentration-absorption standard curve (Figure S1d), the ratio of DOX/TDF was 41:1 for both CpG-T and RGD-CpG-T.
The structural stability of CpG-TDOX and RGD-CpG-TDOX was then interrogated in serum. CpG-TDOX and RGD-CpG-TDOX were first incubated with 10% FBS for 0, 4, 8, 12 and 20 h, respectively. After incubation, PAGE was performed to investigate their structural stability. As shown in Figure 1f, both CpG-TDOX and RGD-CpG-TDOX had clear single bands after 20 h of incubation, which shown the toleration of TDF. With the semiquantitative analysis of the relative band intensity (Figure 1g), about 50% TDF remained intact after 20 h of incubation, which provided a prerequisite for further studies in vivo.
2.2. Tumor Targeting Capacity and Cytotoxicity of Multifunctional TDF
We then investigated the tumor-targeting capacity of RGD-CpG-TDOX. RGD is a short peptide (arginine–glycine–aspartic acid) recognition motif for integrins, which are highly expressed on tumor cell surfaces.24 Several reports indicated that RGD peptide-modified drugs had better tumor targeting ability.25 In our design, RGD peptide modification will enhance the internalization of DOX-loaded TDF in tumors.
First, cellular uptake ability of DOX-loaded TDF with or without RGD modification in 4T1 cells was observed by a confocal laser scanning microscope (CLSM). Cy5-labeled CpG-TDOX and Cy5-labeled RGD-CpG-TDOX were separately incubated with 4T1 cells. After incubation, CLSM analysis was performed, and the images are collected in Figure 2a. 4T1 cells incubated with RGD-CpG-TDOX showed more stronger Cy5 fluorescence in the cytoplasm than the CpG-TDOX group, indicating that the RGD modification enhanced the internalization of TDF by tumor cells. The same conclusion was demonstrated by the statistical analysis of their corresponding fluorescence intensity in CLSM (Figure S2).
Figure 2.

Tumor targeting ability of multifunctional TDF in vitro and in vivo. (a) CLSM images of cellular uptake of CpG-TDOX and RGD-CpG-TDOX in 4T1 cells. Blue: nucleus, green: lysosome, red: Cy5-labeled nanostructures. Scale bar: 10 μm. (b) Cytotoxicity of DOX and DNA nanoparticles. (c) Animal in vivo fluorescent images after injection of CpG-TDOX or RGD-CpG-TDOX in 4T1 tumor-bearing mice. (d) Major organ and tumor fluorescent images after injection of CpG-TDOX or RGD-CpG-TDOX in 4T1 tumor-bearing mice. (e) Relative fluorescent intensity of CpG-TDOX and RGD-CpG-TDOX in tumors. Data were presented as mean ± S.D., *p < 0.05, **p < 0.01, and ***p < 0.001.
Moreover, we found that the LysoTracker green probe could colocalize with Cy5 fluorescence (Figure 2a). It indicated that both CpG-TDOX and RGD-CpG-TDOX were taken up by 4T1 cells and accumulated in lysosomes, and the DOX which intercalated in the DNA helix could be released from CpG-TDOX or RGD-CpG-TDOX after the DNA skeleton degraded by enzymes of lysosomes. Meanwhile, more DOX entered the nucleus in the RGD-CpG-TDOX-treated group than in other treatment groups (Figure S3), implying that targeting accumulation of RGD-CpG-TDOX in 4T1 cells. Subsequently, the tumor cell killing effect of RGD-CpG-TDOX was tested by the cell counting kit-8 (CCK-8). The cytotoxicity results (Figure 2b) showed RGD-CpG-TDOX displayed greater lethality of 4T1 cells than free DOX and CpG-TDOX, indicating that RGD-CpG-TDOX can improve the sensitivity of 4T1 cells to DOX.
To further investigate the tumor-targeting capacity of RGD-CpG-TDOX in mice, Alexa Fluor 680-labeled RGD-CpG-TDOX was injected into 4T1 tumor-bearing mice via the tail vein, and images of ex vivo major organs and tumors were performed. As shown in Figure 2c, the tumor site of mouse injected with RGD-CpG-TDOX exhibited a more stronger fluorescence signal than the CpG-TDOX group in vivo. Also, the ex vivo tumor tissue of mice injected with RGD-CpG-TDOX exhibited a 3-fold stronger fluorescence signal than the CpG-TDOX group (Figure 2d,e), which indicated that RGD-CpG-TDOX had better tumor targeting ability and accumulation.
2.3. ICD Inducing and Immunostimulatory Activity of Multifunctional TDF In Vitro
After establishment of the tumor targeting and tumor cell killing effects of RGD-CpG-TDOX, the immunostimulatory activity of RGD-CpG-TDOX was assessed in vitro. DOX can cause ICD effects by inducing dying tumor cells to expose “eat me” and DAMP signals.12 To investigate the ICD inducing ability of RGD-CpG-DOX, the ICD hallmarks, such as exposure of CRT, excretion of HMGB1, and release of ATP, were monitored by immunofluorescence, ELISA, and luciferase reporting systems, respectively. Results in Figure 3a, much stronger intensity of green fluorescence in 4T1 cells of the RGD-CpG-TDOX group was observed, indicating that increased exposure of CRT to release more “eat me” signals for attracting DCs. Besides, RGD-CpG-TDOX also triggered intense excretion of HMGB1 and release of ATP from 4T1 cells (Figure 3b,c), which could further promote DC maturation and antitumor immune activity.
Figure 3.

ICD inducing and immunostimulatory activity of RGD-CpG-TDOX in vitro. (a) CRT immunofluorescence images of 4T1 cells treated with different preparations. Scale bar: 50 μm. Green: CRT, blue: nucleus. (b) Quantitative measurement of HMGB1 release in 4T1 cells treated with different preparations (n = 3). (c) Quantification measurement of ATP secretion in 4T1 cells treated with different preparations (n = 3). (d) Representative flow cytometry images of DC maturation (CD80+ CD86+) in different groups (gated on CD11c+). (e) Percentages of CD11c+, CD80+, CD86+ cells in different groups (n = 3). Data were presented as mean ± S.D., *p < 0.05, **p < 0.01, and ***p < 0.001.
Subsequently, the function of RGD-CpG-TDOX to induce ICD on DC maturation was investigated. 4T1 cells were treated with RGD-CpG-TDOX, then the dying cells and culture media were collected to coculture with immature bone marrow-derived dendritic cells (BMDCs). The results in Figure 3d,e showed that RGD-CpG-T slightly enhanced DC maturation (CD11c+, CD80+, CD86+ DCs) from 28.5 to 36.4%; this simulating activity might be caused by the CpG motif released from RGD-CpG-T by lysosomal degradation of 4T1 tumor cells. Moreover, 4T1 cells treated with RGD-CpG-TDOX had a more robust promoting effect on DC maturation; the maturity ratio increased from 28.5 to 72.0%, compared with 60.7% of DOX treatment (Figure 3d,e). This robust effect might be attributed to the synergistic action of ICD and CpG in promoting DC maturation.
2.4. Antitumor Efficacy of Multifunctional TDF In Vivo
To evaluate the antitumor efficacy of RGD-CpG-TDOX in vivo, a therapeutic study was performed in 4T1 tumor-bearing mice with an intact immune system. The therapeutic schedule is illustrated in Figure 4a. 4T1 cells were inoculated in BALB/c mice. After the tumor grew to 100 mm3, different preparations were intravenously administrated every 2 days for a total of five injections. As shown in Figures S4a and 4c, RGD-CpG-TDOX obviously suppressed the growth of the tumor; the tumor volume was reduced up to 50% compared with the control group, while the free DOX group with the same injection dose had no significant suppression for tumor growth. In addition, the direct visualization of tumor tissue size showed that RGD-CpG-TDOX could noticeably suppress the growth of tumor (Figure 4b), and the tumor weights also verified the inhibitory effect of RGD-CpG-TDOX on the 4T1 tumor (Figure 4d). We also discussed body weight changes of mice as an indicator to evaluate the systemic toxicity. As shown in Figure S4b, the body weights of mice in each group had no significant difference during the treatment period, indicating that RGD-CpG-TDOX had no obvious systemic toxicity. Moreover, H&E staining in Figure S4c was employed to test the cardiotoxic properties of TDF, where no obvious histopathological changes were found, indicating that a therapeutic dose of RGD-CpG-TDOX did not cause cardiotoxicity. Conclusively, RDG-CpG-TDOX presented an efficient therapeutic effect for 4T1 tumor, which was mainly attributed to the ability of tumor targeting and immune stimulation of multifunctional TDF.
Figure 4.

Antitumor efficacy and immune responses induced by multifunctional TDF in 4T1 tumor-bearing mice. (a) Therapeutic schedule of the antitumor therapy in 4T1 tumor-bearing mice. (b) Tumor tissue images of mice treated by different preparations (n = 5). (c) Tumor growth curves of mice in different treatment groups (n = 5). (d) Tumor weight of mice in different treatment groups (n = 5). (e) CD8 and CD4 immunohistochemical staining of tumor tissues. Scale bar: 50 μm. (f) Quantitative analysis of mature DCs in TDLN of different groups. (g) Quantitative analysis of mature DCs in the spleen of different groups. (h,i) Quantitative analysis of perforin (h) and granzyme B (i) in tumor of different groups. (j) Quantitative analysis of Tregs (CD25+) in tumor of different groups. Data were presented as mean ± S.D., *p < 0.05, **p < 0.01, and ***p < 0.001.
2.5. Immunostimulatory Activity of Multifunctional TDF In Vivo
The mechanism of the antitumor effect of RGD-CpG-TDOX was further investigated. From the results of CD8 and CD4 immunohistochemical staining of tumor tissues (Figure 4e), we found that there were much more CD8 and CD4 positive cells in the RGD-CpG-TDOX treatment group than in other groups, indicating that the antitumor therapeutic effect of RGD-CpG-TDOX was related to its immunostimulatory activity. To further clarify the immunostimulatory activity of RGD-CpG-TDOX, the maturity of DCs in the tumor draining lymph node (TDLN) and spleen were separately assessed. As shown in Figure S5a,b, the percentage of matured DCs (CD11c+, CD80+, CD86+) in TDLN and spleen of the RGD-CpG-TDOX treatment group was 25.60 and 13.80%, respectively, which were obviously higher than the control group (11.00 and 7.79%, Figure 4f,g), indicating that RGD-CpG-TDOX can promote DC maturation after inducing ICD effects of tumor cells.
In addition, T cells in the tumor were also analyzed. Except for the tumor infiltration of CD8+ T cells by RGD-CpG-TDOX treatment, the expressions of perforin and granzyme B, which were secreted by CD8+ T cells to kill tumor cells, were enhanced to 16.50 and 9.02%, almost 2-fold higher than other treatment groups (Figures S5c,d and 4h,i). Also, CD4+ CD25+ regulatory T cells (Tregs), which were considered as suppressor T cells, were evaluated by flow cytometry (FCM) analysis. Results (Figures S5e and 4j) demonstrated that CD4+ CD25+ Tregs decreased in tumor with RGD-CpG-TDOX treatment; the percentage of CD4+ CD25+ Tregs in tumor decreased by 30% compared with other treatment groups, indicating that RGD-CpG-TDOX treatment reversed the tumor-associated immunosuppression. To sum up, the antitumor mechanism of RGD-CpG-TDOX was a combined effect of both chemotherapy and immunotherapy.
2.6. Abscopal Effect of Multifunctional TDF Combined with α-PD-1
After verification of antitumor and immunostimulatory effects, we explored whether RGD-CpG-TDOX could be used to potentiate immune checkpoint blockade (ICB) therapy to improve adaptive antitumor immunity and further suppress tumor recurrence. A bilateral mouse tumor model of 4T1 cells was established for evaluating the anti-metastasis capacity of RGD-CpG-TDOX and α-PD-1. The therapeutic schedule is illustrated in Figure 5a.
Figure 5.

Abscopal efficacy in 4T1 bilateral tumor-bearing mice. (a) Therapeutic schedule of the abscopal therapy in 4T1 bilateral tumor-bearing mice. (b) Distant tumor volumes of mice in different treatment groups (n = 5). (c) Distant tumor weights of mice in different treatment groups (n = 5). (d) Percentages of CD3+ and CD8+ cells in different groups (n = 3). (e) Representative flow cytometry images of CD3+ and CD8+ T cells in different groups. Data were presented as mean ± S.D., *p < 0.05, **p < 0.01, and ***p < 0.001.
First, the primary tumors were subcutaneously implanted in the left flank regions of BALB/c mice. After primary tumors grew to 50 mm3, different preparations were intravenously administrated every 2 days for a total of five injections. Except for the injection of RGD-CpG-TDOX, mice in the combined treatment group (RGD-CpG-TDOX and α-PD-1) also received injection of α-PD-1 antibody additionally every 2 days for a total of four injections. Subsequently, the secondary tumors were subcutaneously implanted in the right flank regions of BALB/c mice to be monitored as distant tumors for 2 weeks. As the results shown in Figure 5b, the distant tumor volumes of RGD-CpG-TDOX showed smaller tumor growth (71.06 ± 37.76 mm3) compared to the control group (263.34 ± 94.90 mm3), indicating that RGD-CpG-TDOX could inhibit the recurrence of tumor effectively. Noticeably, the combined treatment group exhibited higher tumor growth inhibition compared with the RGD-CpG-TDOX groups; the distant tumor volume was only 20.72 ± 20.55 mm3, and no distant tumors were detected in two of five mice. Moreover, the statistical results of distant tumor weights also confirmed the ability of inhibiting tumor recurrence of RGD-CpG-TDOX and combined treatment, tumor weights decreased from 0.467 ± 0.185 g (control group) to 0.160 ± 0.089 g (RGD-CpG-TDOX group) and 0.039 ± 0.036 g (combined treatment group), especially the tumor weights of the combined treatment group were much lighter than the RGD-CpG-TDOX group and α-PD-1 group (Figure 5c), suggesting that combination with anti-PD-1 antibody can boost the antitumor therapeutic effect of RGD-CpG-TDOX and immune therapeutic effect of α-PD-1 ICB therapy. In addition, the combination treatment also extended the median survival time of the mice (Figure S6).
The FCM results (Figure 5d,e) demonstrated that the percentage of CD8+ T cells in distant tumors of RGD-CpG-TDOX and combined treatment groups were 3.29 and 3.98%, respectively, which were much higher than other treatment groups (control 1.59%, RGD-CpG-T 2.00%, free DOX 1.79%, and α-PD-1 2.23%), suggesting the mechanism of recurrence inhibition was related to the increase of systemic T-cell immunity and tumor immune infiltration. Besides, the body weight of mice in different groups had no obvious changes during the treatment period (Figure S7), indicating the well biocompatibility of RGD-CpG-TDOX.
3. Conclusions
In summary, we have developed a multifunctional TDF for cancer immunotherapy, which included a tumor-target RGD peptide and a CpG immunologic sequence and loaded DOX in DNA nanostructures. RGD peptide contributed to targeting of TDF in tumor sites, and alleviating the side effects of loaded DOX. The killing effect of DOX to cancer cells further triggered an anti-cancer immune response through ICD effects with the help of CpG and reversed the “cold tumor”. In vitro and in vivo assays also confirmed that RGD-CpG-TDOX robustly induced ICD response, promoted DC maturation, and improved the intratumoral CTL infiltration. Moreover, the combination of RGD-CpG-TDOX with PD-1 antibody significantly suppressed the growth of distant tumor and prevented tumor recurrence by recruiting CD8+ T cells infiltrating intratumor. Conclusively, a rationally designed multifunctional TDF could be easily assembled to facilitate chemotherapy and immunotherapy simultaneously and could be potentially developed as a synergist for ICB immunotherapy.
4. Methods
4.1. Preparation and Characterization of DNA Structures
DNA sequence information is detailed in Table S1. Equivalent amounts of four corresponding ssDNA (1 μM each) were mixed in 100 μL TM buffer (10 mM Tris–HCl, 5 mM MgCl2, pH 8.0), heating to 95 °C for 10 min, and then quickly cooling to 4 °C. RGD-ssDNA was obtained by connecting alkynyl-modified RGD and A20-N3 by click reaction, and then was purified by Zeba spin desalting columns (Thermo Fisher Scientific). CpG-T was composed of S1, S2, S3, and S4. RGD-CpG-T was synthesized by mixing of CpG-T and RGD-ssDNA at 37 °C for 1 h. RGD-CpG-TDOX was obtained by incubating RGD-CpG-T with DOX solution at a molar ratio of 1:100 at 37 °C for 6 h. Because of the tetracyclic region, DOX can intercalate in the DNA base pairs. 30 kDa ultra-filtration tube (Millipore) was used to remove excess DOX. CpG-TDOX was synthesized in a similar way to RGD-CpG-TDOX. To make the DOX loading consistent for CpG-TDOX and RGD-CpG-TDOX, the aim chain of CpG-T was hybridized with the RGD-free A20 chain.
15% nondenaturing PAGE and 8% PAGE were used to identify the structures of RGD-ssDNA and TDF, respectively. After loading samples, electrophoresis was taken in 1 × TAE buffer (4 mM Tris–HCl, 2 mM acetic acid, 0.2 mM EDTA, and 1.25 mM MgAc2) for 1 h, and then gel was stained with sliver solution or GelRed to indicate samples.
The hydrodynamic size of TDF was measured by Zetasizer Nano ZS (Malvern). The morphologies of CpG-TDOX and RGD-CpG-TDOX were evaluated by AFM (Bruker). The loading of DOX in the material is determined by a UV–vis spectrometer (Cary100, Agilent). Standard curves were made according to the absorption values of different concentrations of DOX (7, 15, 30, 60, and 100 μM) at 480 nm.
The structural stability of CpG-TDOX and RGD-CpG-TDOX in serum was detected by PAGE. CpG-TDOX and RGD-CpG-TDOX were incubated with 10% FBS at 37 °C for 0, 4, 8, 12 and 20 h, then samples were detected by 6% PAGE.
4.2. Cell Uptake of DNA Structures
4T1 cells were seeded in confocal culture plates and cultured for 24 h at 37 °C. Afterward, Cy5-labeled CpG-TDOX and Cy5-labeled RGD-CpG-TDOX were separately incubated with 4T1 cells for 6 h. After incubation, cells were washed three times with phosphate buffer (PBS) and incubated with LysoTracker Green for 1 h at 37 °C. Subsequently, cells were washed three times with PBS, fixed with 4% paraformaldehyde, and stained with Hoechst 33258. Then cells were observed by confocal microscope setup (Leica TCS SP8).
4.3. Tumor Targeting Assay
4T1 tumor-bearing models were used for the imaging experiment. Alex Fluor 680-labeled CpG-TDOX and RGD-CpG-TDOX were separately injected via tail vein of 4T1 tumor-bearing mice. Mice were anesthetized and taken for imaging after injection by IVIS Spectrum in vivo imaging system (PerkinElmer). After in vivo imaging was finished, heart, liver, spleen, lung, kidney, and tumor of mice were harvested and taken for imaging.
4.4. Antitumor Activity In Vivo
To evaluate the in vivo antitumor efficacy of nanostructures, 1 × 106 4T1 cells were subcutaneously inoculated into female BALB/c mice. When the tumor volume achieved approximately 100 mm3, tumor-bearing mice were randomly divided into four groups (n = 5 per group), and different preparations (saline, RGD-CpG-T, DOX, and RGD-CpG-TDOX) were intravenously administrated every 2 days for a total of five injections with an equivalent DOX dosage. The tumor sizes and body weights of mice in each group were monitored every 4 days. Tumor sizes were measured by a digital vernier caliper, and the volume was calculated by the formula of length × width2/2.
Mice were sacrificed and main organs were harvested. To investigate the cardiotoxicity of nanostructures, the hearts of each mouse were fixed with 4% paraformaldehyde for H&E staining. Meanwhile, tumor tissues were harvested and weighted. A portion of tumor tissue was taken for immunohistochemical analysis with anti-CD4 and anti-CD8 antibody staining to identify the immune activation effect of nanostructures.
To further clarify the immunostimulatory activity of DNA nanostructures, DCs and tumor-infiltrated T lymphocytes were analyzed by FCM. The tumor draining lymph node (TDLN) and spleen were harvested and made single cell suspensions, then stained with PE-conjugated anti-CD11c, PE/Cyanine7-conjugated anti-CD86, and PerCP/Cyanine5.5-conjugated anti-CD80 antibody for 20 min for DC analysis by FCM. Tumor tissues were cut into small pieces and incubated with digestion solution for 1 h to make single-cell suspensions. Subsequently, tumor cells were blocked with anti-CD16/32 antibody, and then stained with APC-conjugated anti-CD3, Alexa Fluor 488-conjugated anti-CD8a, PE/Cyanine7-conjugated anti-granzyme B antibody for analyzing granzyme B+ CD8+ T cells, stained with APC-conjugated anti-CD3, Alexa Fluor 488-conjugated anti-CD8a, PE-conjugated anti-perforin antibody for analyzing perforin+ CD8+ T cells, and stained with APC-conjugated anti-CD3, PerCP/Cyanine5.5-conjugated anti-CD4, PE-conjugated anti-CD25 antibody for analyzing CD25+ CD4+ T cells by FCM.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81801818, 82002241, 82273897, 81922070, 81973286, and 82104281), the 2022 Shanghai “Science and Technology Innovation Action Plan” Fundamental Research Project (22JC1401202), the Science Foundation of the Shanghai Municipal Science and Technology Commission (20ZR1425600), and the Foundation of National Facility for Translational Medicine (Shanghai) (TMSK-2020-010).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00099.
Experimental details, PAGE image for identifying RGD-ssDNA, AFM image of CpG-TDOX, DLS image of TDF, CLSM images of cellular uptake, tumor growth curves, body weight curves, H&E staining images, representative FCM images, survival rates image, and a list of DNA sequences (PDF)
Author Contributions
⊥ T.T. and B.Z. contributed equally. The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Giaquinto A. N.; Sung H.; Miller K. D.; Kramer J. L.; Newman L. A.; Minihan A.; Jemal A.; Siegel R. L. Breast Cancer Statistics, 2022. Ca-Cancer J. Clin. 2022, 72, 524–541. 10.3322/caac.21754. [DOI] [PubMed] [Google Scholar]
- Miller K. D.; Nogueira L.; Devasia T.; Mariotto A. B.; Yabroff K. R.; Jemal A.; Kramer J.; Siegel R. L. Cancer treatment and survivorship statistics, 2022. Ca-Cancer J. Clin. 2022, 72, 409–436. 10.3322/caac.21731. [DOI] [PubMed] [Google Scholar]
- Pedersen R. N.; Esen B. O.; Mellemkjær L.; Christiansen P.; Ejlertsen B.; Lash T. L.; Norgaard M.; Cronin-Fenton D. The Incidence of Breast Cancer Recurrence 10-32 Years After Primary Diagnosis. JNCI, J. Natl. Cancer Inst. 2022, 114, 391–399. 10.1093/jnci/djab202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S.; Gatti-Mays M. E.; Kalinsky K.; Korde L. A.; Sharon E.; Amiri-Kordestani L.; Bear H.; McArthur H. L.; Frank E.; Perlmutter J.; Page D. B.; Vincent B.; Hayes J. F.; Gulley J. L.; Litton J. K.; Hortobagyi G. N.; Chia S.; Krop I.; White J.; Sparano J.; Disis M. L.; Mittendorf E. A. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol. 2019, 5, 1205–1214. 10.1001/jamaoncol.2018.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan T. E.; Tolaney S. M. Role of Immunotherapy in Triple-Negative Breast Cancer. J. Natl. Compr. Cancer Network 2020, 18, 479–489. 10.6004/jnccn.2020.7554. [DOI] [PubMed] [Google Scholar]
- Esteva F. J.; Hubbard-Lucey V. M.; Tang J.; Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019, 20, E175–E186. 10.1016/s1470-2045(19)30026-9. [DOI] [PubMed] [Google Scholar]
- Emens L. A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. 10.1158/1078-0432.ccr-16-3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmeerbeek I.; Sprooten J.; De Ruysscher D.; Tejpar S.; Vandenberghe P.; Fucikova J.; Spisek R.; Zitvogel L.; Kroemer G.; Galluzzi L.; Garg A. D. Trial watch: chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. 10.1080/2162402x.2019.1703449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid M.; Tesniere A.; Ghiringhelli F.; Fimia G. M.; Apetoh L.; Perfettini J.-L.; Castedo M.; Mignot G.; Panaretakis T.; Casares N.; Metivier D.; Larochette N.; van Endert P.; Ciccosanti F.; Piacentini M.; Zitvogel L.; Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- Garg A. D.; Krysko D. V.; Verfaillie T.; Kaczmarek A.; Ferreira G. B.; Marysael T.; Rubio N.; Firczuk M.; Mathieu C.; Roebroek A. J. M.; Annaert W.; Golab J.; de Witte P.; Vandenabeele P.; Agostinis P. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko D. V.; Garg A. D.; Kaczmarek A.; Krysko O.; Agostinis P.; Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- Casares N.; Pequignot M. O.; Tesniere A.; Ghiringhelli F.; Roux S.; Chaput N.; Schmitt E.; Hamai A.; Hervas-Stubbs S.; Obeid M.; Coutant F.; Metivier D.; Pichard E.; Aucouturier P.; Pierron G.; Garrido C.; Zitvogel L.; Kroemer G. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacar O.; Sriamornsak P.; Dass C. R. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2012, 65, 157–170. 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- Singal P. K.; Iliskovic N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. 10.1056/nejm199809243391307. [DOI] [PubMed] [Google Scholar]
- Chen L.; Zhang J.; Lin Z.; Zhang Z. Y.; Mao M.; Wu J. C.; Li Q.; Zhang Y. Q.; Fan C. H. Pharmaceutical applications of framework nucleic acids. Acta Pharm. Sin. B 2022, 12, 76–91. 10.1016/j.apsb.2021.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Li J.; Dong B. J.; Wang F.; Fan C. H.; Zuo X. L. DNA nanotechnology-empowered nanoscopic imaging of biomolecules. Chem. Soc. Rev. 2021, 50, 5650–5667. 10.1039/d0cs01281e. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Tian T. R.; Lin Y. F. Functionalizing Framework Nucleic-Acid-Based Nanostructures for Biomedical Application. Adv. Mater. 2022, 34, e2107820 10.1002/adma.202107820. [DOI] [PubMed] [Google Scholar]
- Mao X.; Liu M.; Li Q.; Fan C.; Zuo X. DNA-Based Molecular Machines. JACS Au 2022, 2, 2381–2399. 10.1021/jacsau.2c00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y.; Shen F.; Zhang Z.; Wang Q.; Huang H.; Xu Y.; Li Q.; Zhu X.; Sun L. Applications of Functional DNA Materials in Immunomodulatory Therapy. ACS Appl. Mater. Interfaces 2022, 14, 45079–45095. 10.1021/acsami.2c13768. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Sun Y.; Li J.; Li Q.; Lv M.; Zhu B.; Tian T.; Cheng D.; Xia J.; Zhang L.; Wang L.; Huang Q.; Shi J.; Fan C. Multiple-Armed Tetrahedral DNA Nanostructures for Tumor-Targeting, Dual-Modality in Vivo Imaging. ACS Appl. Mater. Interfaces 2016, 8, 4378–4384. 10.1021/acsami.5b10792. [DOI] [PubMed] [Google Scholar]
- Tian T.; Li J.; Xie C.; Sun Y.; Lei H.; Liu X.; Xia J.; Shi J.; Wang L.; Lu W.; Fan C. Targeted Imaging of Brain Tumors with a Framework Nucleic Acid Probe. ACS Appl. Mater. Interfaces 2018, 10, 3414–3420. 10.1021/acsami.7b17927. [DOI] [PubMed] [Google Scholar]
- Krieg A. M. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002, 20, 709–760. 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- Li J.; Pei H.; Zhu B.; Liang L.; Wei M.; He Y.; Chen N.; Li D.; Huang Q.; Fan C. Self-Assembled Multivalent DNA Nanostructures for Noninvasive Intracellular Delivery of Immunostimulatory CpG Oligonucleotides. ACS Nano 2011, 5, 8783–8789. 10.1021/nn202774x. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- Danhier F.; Le Breton A.; Preat V. RGD-Based Strategies To Target Alpha(v) Beta(3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharm. 2012, 9, 2961–2973. 10.1021/mp3002733. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.