

Abstract
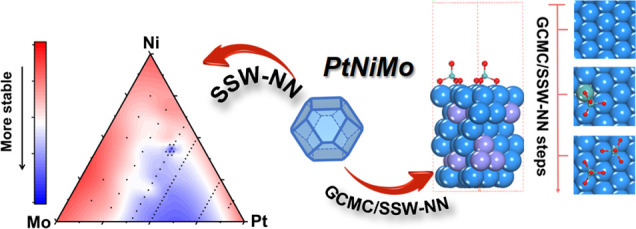
Pt–Ni alloy is by far the most active cathode material for oxygen reduction reaction (ORR) in the proton-exchange membrane fuel cell, and the addition of a tiny amount of a third-metal Mo can significantly improve the catalyst durability and activity. Here, by developing machine learning-based grand canonical global optimization, we are able to resolve the in situ structures of this important three-element alloy system under ORR conditions and identify their correlations with the enhanced ORR performance. We disclose the bulk phase diagram of Pt–Ni–Mo alloys and determine the surface structures under the ORR reaction conditions by exploring millions of likely structure candidates. The pristine Pt–Ni–Mo alloy surfaces are shown to undergo significant structure reconstruction under ORR reaction conditions, where a surface-adsorbed MoO4 monomer or Mo2Ox dimers cover the Pt-skin surface above 0.9 V vs RHE and protect the surface from Ni leaching. The physical origins are revealed by analyzing the electronic structure of O atoms in MoO4 and on the Pt surface. In viewing the role of high-valence transition metal oxide clusters, we propose a set of quantitative measures for designing better catalysts and predict that six elements in the periodic table, namely, Mo, Tc, Os, Ta, Re, and W, can be good candidates for alloying with PtNi to improve the ORR catalytic performance. We demonstrate that machine learning-based grand canonical global optimization is a powerful and generic tool to reveal the catalyst dynamics behavior in contact with a complex reaction environment.
Keywords: oxygen reduction reaction, SSW-NN, GCMC, PtNiMo alloy, machine learning
1. Introduction
Pt3Ni is an active oxygen reduction reaction (ORR) electrocatalyst for the cathode of fuel cells1−5 but suffers from stability problems. Ni leaching under the operating conditions leads to the rapid decay of current density from 2.70 mA/cm2 in an as-prepared sample to 0.20 mA/cm2 after 10,000 cyclic voltammetry (CV) cycles.2,6−8 To solve this problem, further alloying Pt3Ni with a third transition-metal element is a natural choice to improve the durability and activity of the catalysts.2,6,7,9 To date, many transition metals, e.g., 3d (V, Cr, Mn, Fe, Co, and Cu10−12), 4d (Mo10 and Rh13), and 5d (Re and W10) metals, have been tested. Among them, the low-content (∼1%) Mo-doped Pt3Ni (Pt3Ni-Mo) exhibits the best ORR activity (9.7 mA/cm2, 10-fold higher than pristine Pt3Ni) with high stability (very low Ni leaching, only 1%, after 8000 CV cycles).10,14,15 Even after 15,000 CV cycles, the activity remains at 6.4 mA/cm2.16 However, there is great uncertainty about the atomic structure of Mo species and the catalytic role of the Mo dopant under ORR conditions due to the lack of experimental data on the Mo dopant. As a result, it is still unclear why such a small amount of Mo can significantly suppress Ni leaching and meanwhile notably boost the ORR.
It is now known that Ni leaching can occur on all PtNi alloy surfaces, regardless of the Pt:Ni ratio. This leaching is caused by surface oxidation as described by the following steps. First, the *O adatom is generated on the catalyst’s surface via the reaction of * + H2O → *O + 2H+ + 2e– at the elevated electrochemical potentials.17−19 With the presence of *O, the Ni atoms in the catalyst may then migrate to the surface due to their strong binding strength toward *O. These newly exposed Ni atoms, in turn, extract more *O atoms from the aqueous electrolyte20−23 and from O2 dissociation.17 Ni leaching will continue until few Ni atoms are left around the surface, which therefore leads to the quick decay of ORR activity2,7 since Pt is catalytically less active than PtNi alloy.
In PtNiMo alloy, the Ni leaching kinetics and, thus, the ORR activity appear to be significantly affected by the Pt:Ni ratio under the same low Mo content according to different experimental data with a wide alloy composition from Pt-rich to Ni-rich conditions. For the Pt-rich case, Huang et al. reported that the Ni loss for Pt73.4Ni25.0Mo1.6 is only 1.0% even after 8000 (CV) cycles,10 and the original cuboctahedral shape of the catalyst can be maintained during the reaction. A high current density of 9.7 mA/cm2 can still be retained after this long-time test, even higher than the as-prepared Pt74.3Ni25.7, that is, 2.70 mA/cm2. In contrast, the Ni-rich samples (e.g., Pt44.0Ni55.0Mo1.0, Pt27.0Ni71.4Mo1.6, and Pt25.0Ni73.3Mo1.7) show severe Ni leaching under the same conditions, resulting in a significant change in the morphology of the nanoparticles, from a cuboctahedron to an approximate sphere or concave polyhedron.15 After 10,000 CV cycles, up to 70.2% Ni are dissolved, which transforms the sample to a Pt-rich phase, e.g., Pt79.4Ni20.6Mo1.7. The activity of this deformed Pt-rich PtNiMo (3.08 mA/cm2) is notably lower than that of the best Pt-rich catalyst, indicating that the local structure of the catalyst, not merely the Pt:Ni ratio, also plays an important role in catalysis.
To date, the reported best ORR catalysts adopt exclusively the recipe with a trace of Mo content despite the fact that high Mo-content PtMo materials, such as Pt80.0Mo20.024 and Pt40.0Mo60.0,25 were synthetically feasible. It implies that Mo should not be the catalytic active site and the high Mo content will deteriorate the ORR activity. Therefore, the research efforts have been mainly focused on the location and the anti-leaching role of Mo under ORR conditions. Using kinetic Monte Carlo and cluster expansion calculations, Mueller and Cao showed that the Mo atoms in Pt73.4Ni25.0Mo1.6 prefer to locate at the subsurface layer around the corners of nanoparticles.26 In this configuration, the formation energies of Pt and Ni vacancies on the surface (the initial step of Ni leaching) increase by 0.25–0.50 eV compared to the reference Pt75.0Ni25.0. Therefore, they suggested that the low leaching of Ni atoms in the Pt73.4Ni25.0Mo1.6 alloy is attributed to the presence of subsurface Mo atoms around the corner.16,26 This finding is consistent with the early thermodynamics analysis for PtM binary alloys, where the subsurface Mo is shown to increase the anticorrosion ability of surface Pt.27 However, the recent in situ Mo K-edge X-ray absorption near edge structure (XANES) spectra identify a gradual shift toward higher energy as the electrode potential increases, reflecting that the oxidation state (OS) of Mo increases, and beyond 0.54 V, the spectra converge to those coinciding with the MoO3 solid.16 Therefore, XANES suggests that doped Mo is likely to be fully oxidized, instead of being metallic inside PtNi alloy.
With the above puzzles in the literature, it would be desirable to track the dynamical structural evolution of the PtNiMo electrocatalyst under ORR conditions from theory since the initial state of PtNi alloy with low doping Mo is more or less certain. The complexity arises in three aspects, including (i) the initial Mo positions in PtNi alloy, (ii) the Ni leaching-induced surface and bulk reconstruction, and (iii) the presence of surface O species generated under the electrochemical conditions. To properly take into all these dynamic structural complexities, the grand canonic ensemble with flexible Pt, Mo, and O atom exchange between the catalyst and the solution seems to be essential. Considering the quaternary Pt–Ni–Mo–O elements and the low doping of Mo (large supercell required), such a variable-composition simulation is far beyond the current ability of first principles-based atomic simulation. It is no wonder that all previous simulations for ORR on the PtNiMo alloy were performed on a perfect (111) planar surface with fixed *O coverages,17,21,26 in which the surface reconstruction and composition change are totally neglected.
By using the global neural network potential (G-NN) method developed by our group in recent years, here, we develop a variable-composition global structural search method, namely, GCMC/SSW-NN, that integrates G-NN potential, stochastic surface walking (SSW) global optimization, and grand canonical Monte Carlo (GCMC) in one framework. A Pt–Ni–Mo–O–H five-element G-NN potential is generated for the first time, which can treat the complex potential energy surface (PES) in between the PtNiMo metal electrode and the aqueous solution. Provided more than four orders of magnitude speed-up in G-NN calculations compared to conventional DFT calculations, we are able to perform a large-scale GCMC/SSW global search for thousands of compositions and follow the structural evolution of major low-energy surfaces of the PtNiMo nanoparticles, namely, (111), (100), (211), (221), and (210), under ORR conditions. We clarify how the Mo dopant suppresses Ni leaching and, importantly, how it can simultaneously enhance the ORR activity. The puzzles on the oxidation state of Mo are finally resolved.
2. Calculation Details
2.1. SSW-NN Method and G-NN Potential
The SSW-NN method combines the SSW global PES exploration with the G-NN potential for PES evaluation. The method is extensively utilized in this work for first generating Pt–Ni–Mo–O–H G-NN potential and then for exploring global PES of different PtNiMo catalyst compositions.28−30 All SSW-NN simulations are performed using the Large-scale Atomic Simulation with neural network Potential (LASP) code31 (http://www.lasphub.com) developed in our group.32,33 More details on the SSW-NN method can be found in our previous publications and also in the Supporting Information.
The Pt–Ni–Mo–O–H five-element G-NN potential is developed by self-learning the SSW global PES dataset from a wide range of structures at different Pt–Ni–Mo–O–H compositions with varied supercells up to 206 atoms/cell. The dataset contains different structure types (clusters, layers, and bulks) and full combinations of the four elements, e.g., pure metals (Pt, Ni, and Mo), oxides (PtOx, NiOx, MoOx, PtNixOy, PtMoxOy, NiMoxOy, and PtNixMoyOz), hydrides (PtHx, NiHx, and MoHx), and hydroxides (PtOxHy, NiOxHy, PtNixOyHz, and PtNiwMoxOyHz). This dataset information is provided in Table S1 of the Supporting Information. The as-trained G-NN potential can be applied generally to different systems with Pt–Ni–Mo–O–H elements. The training is iteratively carried out to make the G-NN potential robust enough to describe the global PES quantitatively. More than 107 structures in total were generated during G-NN training, and 89,250 of them were selected by multiple rounds (>100) of iterative self-learning of SSW global PES. The details of G-NN potential training can be found in our previous works.30,31,34,35 Briefly, the initial structures are selected randomly from a DFT-based SSW global search, which is utilized to construct the first NN potential. The subsequent self-learning cycles will continuously perform SSW-NN calculations and collect structures from the global PES. These structures are either randomly selected or screened out for new structural patterns (according to the structural descriptors of the NN input layer). The NN potential is repeatedly trained using the updated dataset. In the production stage where a G-NN potential is available for target systems, additional Bayesian optimization cycles (>5 cycles) are utilized to confirm the global minima (GM) predicted from SSW-NN being consistent with DFT calculations (also see the Supporting Information for details).
During DFT calculations, we utilize the higher accuracy DFT setups, i.e., an automatic Monkhorst-Pack k-point mesh by 25 times the reciprocal lattice vectors and a plane-wave-basis set cutoff energy of 450 eV, which is enough to achieve consistent energy between structures. To pursue a high accuracy of PES, a large set of power-type structural descriptors (PTSDs)30,31 is adopted for each element, including 216 two-body ones, 156 three-body ones, and 54 four-body ones. The root-mean-square errors (RMSE) of final G-NN potential for the energy and force are 4.832 meV/atom and 0.134 eV/Å, respectively, which is accurate enough to describe the Pt–Ni–Mo–O–H global PES quantitatively. The final G-NN potential is openly accessed in LASP website (see Web page link36). The benchmark data between G-NN calculations and DFT calculations are given in Table S2.
2.2. GCMC/SSW-NN Simulations
To understand the dynamically structural evolution under reaction conditions, we combine GCMC and SSW-NN to realize the global structure search with variable number of atoms. In the GCMC/SSW-NN simulation, we perform a GCMC move after every 10 SSW-NN steps, where the GCMC will exchange atoms in the simulation supercell with the environment pinned by the chemical potential of the exchangeable species. Following the GCMC scheme, we define the free energy of a surface system as G(N1, N2, ..., Nk) with k species, and the chemical potential of a species i (μi) is given by
| 1 |
where Ni is the number of atoms for species i. In the grand canonical ensemble, all μi reach the equilibrium with the chemical potential of environment (μext), i.e., {μext – μi,ext}, in average being 0. The minimization of the chemical potential difference can be achieved via the Metropolis Monte Carlo algorithm to make the state selection according to the probability (P).
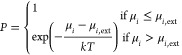 |
2 |
In each trial GCMC move, we randomly add/remove an O or Pt/Mo atom on the surface, calculate the corresponding free energy change using G-NN potential, and finally utilize the Metropolis MC to decide whether the trial move is accepted or not. Other details for GCMC move, including the definition of the chemical potentials for Pt/Mo and O, are given in Section 3.2. A grand canonical structure evolution trajectory can finally be recovered by grouping structures at the same composition and ordering the GM of each composition according to the composition. Such a GCMC/SSW-NN trajectory is continuous in composition and can represent a thermodynamically favored evolution pathway under reaction conditions.
2.3. DFT Calculations
All low-energy structures identified from GCMC/SSW-NN simulations and the ORR reaction pathways are finally converged using DFT calculations. All DFT calculations were carried out within the periodic plane wave framework as implemented in the Vienna Ab initio Simulation Package (VASP).37 All DFT results are obtained by spin-polarized calculations with a PBE functional. The interaction between the ion and inner electrons was represented by the projector augmented wave (PAW)-type pseudopotential, while the valence electrons (5d and 6s for Pt; 3d and 4s for Ni; 4d and 5s for Mo) are considered explicitly. The kinetic energy cutoff of the plane wave was set as 500 and 450 eV for surface and bulk calculations, respectively. The first Brillouin zone k-point sampling utilizes the Monkhorst-Pack scheme with an automated mesh determined by 25 times the reciprocal lattice vectors. The initial on-site spins for Pt, Ni, and Mo atoms are set to the ferromagnetic state during calculations. We have tested other spin configurations and confirmed that this setup can converge to the ground state. The energy and force criteria for the convergence of the electron density and structure optimization are set at 10–5 eV and 0.01 eV/Å, respectively. Zero-point energy (ZPE) is calculated from the phonon spectra using DFT. The solvation effect was modeled by a periodic continuum solvation model with the modified Poisson–Boltzmann equation (CM-MPB).38−40
The free energy profiles for ORR are calculated by the computational hydrogen electrode (CHE) method, which takes into account the electrode potential effect during calculations (see the Supporting Information for details). The transition states (TS) for the rate-determining step are determined by the constrained Broyden dimer (CBD)28,29,41 method. To derive the free energy reaction profile, we first obtain the reaction energy of each elementary step from DFT total energy after zero-point energy (ZPE) correction. Then, for reactions involving gaseous or liquid molecules, such as oxygen, hydrogen, and water, we further add the free energy contributions from temperature and pressure by utilizing the standard thermodynamics data at the standard state.42
2.4. Nomenclature and Computational Models
We represent the compositions of PtNiMo alloy in this paper by the percentages of content: PtxNiyMoz, where x + y + z = 100. For bulk models, we use a 48-atom unit cell to explore the phase diagram. For surface models, we adopt a series of six-atomic-layer slab models with dimensions from (7.77 Å × 7.77 Å) to (9.52 Å × 8.24Å). During simulations, all atoms were relaxed to explore the GM structure in the SSW-NN and GCMC/SSW-NN. The electrode potential (U) used in GCMC/SSW-NN and ORR profiles is 0.9 V versus reversible hydrogen electrode (RHE), which is the typical working potential for ORR activity measurement in the experimental literature.10 The free energy of charged species in the aqueous solution is derived from their experimental standard electrode potentials (see the Supporting Information for more details).43,44
3. Results
This work contains four parts, from bulk to surface and to reactions, as outlined in Figure 1. First, the phase diagram of bulk PtNiMo alloy is determined using SSW-NN. Then, the surface structures at different PtNiMo compositions (from Pt66.7Ni30.6Mo2.7 to Pt83.5Ni13.8Mo2.7) are explored using GCMC/SSW-NN and the most stable alloy surface structures are determined. Next, using the most stable surface structures, the dynamic structural evolution of the PtNiMo surfaces is examined using GCMC/SSW-NN where the O atom concentration varies according to the electrode potential. The ORR activities of the stable PtNiMo surfaces at a given reaction condition are finally resolved.
Figure 1.

Overall scheme to resolve PtNiMo structures and activity from bulk to surface and to ORR activity. (a) Ternary phase diagram of the PtNiMo bulk alloy. Region I for fcc; region II for hcp; region III for bcc; region IV for mixed structure. (b) Thermodynamic convex hull diagram of Pt75.0Ni25.0–xMox alloys with reference to Pt75.0Ni25.0 and Pt75.0Mo25.0. The color bar represents the energy spectrum. (c) GM structures for two convex compositions Pt75.0Ni23.0Mo2.0 and Pt75.0Ni15.0Mo10.0. (d) GCMC/SSW-NN results of Pt75.0Ni25.0(111) under ORR reaction conditions (0.9 V), showing the Ni surfacing and NiO2 formation after 35,217 steps. Structure and top 3 layer Pt:Ni content of the final state Pt75.0Mo25.0 (0.75 ML *O). (e) Structure and top 3 layer Pt:Ni content of the final state of Pt75.0Ni22.3Mo2.7(111) under reaction conditions (0.67 ML *O). (f, g) Energy profile (f) from the GCMC/SSW-NN trajectory of Pt75.0Ni22.3Mo2.7(111) and the corresponding structures at different O coverages. The atoms above the surface plane are depicted in the ball-and-stick style, while other atoms are depicted in the CPK style. The subsurface and bulk layer are in the gray area. Colors in panels (c)–(g): blue balls and bars are Pt; violet balls and bars are Ni; cyan balls and bars are Mo.
3.1. Ternary Diagram for Bulk PtNiMo Alloy
To probe the Mo location and thus identify the stable bulk PtNiMo alloy structures, we have performed SSW-NN simulation for 83 different PtNiMo compositions as depicted by the black points in the PtNiMo ternary phase diagram in Figure 1a. In total, 830,000 structures were explored with at least 104 minima for each composition. The GM structure is thus obtained for each composition, and the total energy is utilized to plot Figure 1a.
By examining the GM structure for 83 PtNiMo compositions, we found that the whole phase diagram involves three different crystal structures, denoted as regions I–III in Figure 1a. The GM structure in region I is the fcc crystal as pure Pt and Ni, whose Mo composition is below 37.5%. Region II is hcp, whose Mo composition is in the range from 39.5 to 50.0%. Region III is a bcc crystal, the same as pure Mo metal, whose Mo composition is above 66.7%.
The thermodynamically most favored compositions can be identified from Figure 1a, as shown by the blue region enclosed by the compositions Pt39.6Ni43.7Mo16.7 (the lowest energy alloy composition), Pt39.0Mo61.0, and Pt75.0Mo25.0 (which is the triangle point). In this region, the alloy formation is exothermic by at least 0.25 eV/atom with respect to the pure metal phases. Notably, the Mo content in these more favored compositions is at least 17%, not low at all, indicating that Mo-rich PtNiMo alloy is in fact thermodynamically more stable. This may not be surprising since a number of Mo-rich alloys, i.e. Pt42.8Ni42.8Mo14.4,45 Pt40.0Mo60.0,25 and Pt80.0Mo20.0,24 were synthesized in experiments, whose compositions are close to the boundaries of the blue region, confirming the theoretical predictions from the phase diagram. The Mo-lean alloy composition (Mo contents <2.0%) nevertheless still has negative formation energies (<−0.09 eV/atom). Therefore, it is expected that Mo-lean alloys utilized for the ORR electrode are a choice for ORR reaction kinetics, and the Mo atoms should have a thermodynamic tendency toward agglomeration.
Specifically, for the lowest energy alloy Pt39.5Ni43.7Mo16.7, it adopts the fcc crystal with the average bond lengths of Pt–Ni, Pt–Mo, and Ni–Mo being 2.69, 2.71, and 2.65 Å, respectively, and the average pair numbers of them being 5.89, 3.16, and 2.86, respectively. For the highest Mo content PtNiMo alloy (Pt12.5Ni12.5Mo75.0), it is a bcc alloy with the average bond lengths of Pt–Ni, Pt–Mo, and Ni–Mo being 2.65, 2.79, and 2.71 Å, respectively, and the average pair numbers of them being 1.50, 7.67, and 6.67, respectively. This suggests that Mo always prefers to bond with Pt in bulk alloys.
Among the Mo-lean compositions, Pt75.0Ni25.0–xMox (x < 2.0) with a high Pt content of 75% are commonly utilized in experiments for ORR,10,46 since it is known that Pt is the active site of ORR and the presence of Ni can modify the electronic structure of Pt. By focusing on the compositions with 75% Pt, we plot the thermodynamic convex hull diagram for Pt75.0Ni25.0–xMox in Figure 1b. It shows that four convex points are present, corresponding to Pt75.0Ni23.0Mo2.0, Pt75.0Ni15.0Mo10.0, Pt75.0Ni13.0Mo12.0, and Pt75.0Ni8.0Mo17.0, indicating Pt75.0Ni23.0Mo2.0 is thermodynamically stable along the 75% Pt content line.
Figure 1c illustrates the GM structures for two of these convex points, Pt75.0Ni23.0Mo2.0 and Pt75.0Ni15.0Mo10.0, while others are given in Figure S2. These two low-Mo alloys are fcc packing without the presence of Mo–Mo bonds in bulk. For Pt75.0Ni23.0Mo2.0, the average pair numbers (with cutoff = 3.0 Å) of Pt–Ni, Pt–Mo, and Ni–Mo are 3.67, 0.33, and 0.00, respectively. Importantly, the Mo atoms disperse well in the supercell (only at a Mo content higher than 10.0%, Pt75.0Ni12.0Mo13.0; two Mo atoms can be neighboring). The shortest distance between two disperse Mo atoms is 4.04 Å. For each Mo, their average pair numbers of Mo–Pt and Mo–Ni are 9.14 and 1.71, respectively, indicating that the Mo atoms are mostly surrounded by Pt atoms.
The lack of Mo–Mo bonds in Pt75.0Ni25.0–xMox alloys can be understood as follows. First, the Pt75.0Ni25.0–xMox alloys are in the fcc lattice and, thus, the Mo–Mo bonding in the fcc lattice is intrinsically less favorable compared to Mo–Mo bonding in the bcc lattice (the bcc Mo metal is 0.42 eV/atom more stable than the fcc Mo metal). Second, Mo prefers to form bonding with Pt. The Pt–Mo bond distance (2.6 Å) is even shorter than the Pt–Ni bond distance (2.7 Å), suggesting the strong d–d hybridization between Pt and Mo. As a fact, the Pt75.0Mo25.0 fcc bulk is 1.7% smaller in volume compared to the Pt75.0Ni25.0 fcc bulk.
3.2. PtNiMo Surfaces under Reaction Conditions
3.2.1. Clean Surfaces
Before we investigated the PtNiMo surfaces under reaction conditions, we first explored the structure for a series of clean, low-Miller index Pt–Ni–Mo surfaces, including (111), (100), (211), (221), and (210). All simulations utilize a six-layer slab model where the Mo atoms were initially randomly placed in the middle layers (see in Figure S3). For each surface, four Pt:Ni compositions with the ratios being 66.7: 30.6, 75.0: 22.3, 80.6: 16.7, and 83.5: 13.8 were considered, where the Mo content is kept the same at 2.7%. These surfaces are, hereafter, denoted as Pt66.7Ni30.6Mo2.7, Pt75.0Ni22.3Mo2.7, Pt80.6Ni16.7Mo2.7, and Pt83.5Ni13.8Mo2.7. Note that the 2.7% Mo content used here is slightly higher than that reported in the experiment for the overall composition (1.6%). This is because the Mo atoms tend to aggregate on the catalyst’s surface under both resting and reactive conditions (see results below). Moreover, all our initial models are then verified by the variable-composition GCMC/SSW-NN method, which can adjust the composition until the equilibrium is reached. Therefore, the surface model with a slightly higher Mo content should in fact be closer to the real surface composition in experiments.
The GM structures are obtained from the SSW-NN structural search, and the general features of the clean surfaces are summarized as follows.
-
(1)
The surface layer is always fully covered by Pt atoms. This leads to the depletion of Pt in the sublayer regions. As exampled in Figure S2, the content of Pt is 100% in the first layer as “Pt-skin” but reduces markedly in the second and third layers.
-
(2)
The Mo atoms are present dominantly at the sublayer to maximally form bonding with Pt atoms. The Mo atom at the sublayer is ∼0.1 eV lower than those at the bulk layers (the third layer) in Pt75.0Ni22.3Mo2.7, and there is no direct Mo–Mo bond (Mo–Mo distance >4.75 Å). The Mo–Ni bonds are present only when the Ni content is above 16.7% (Pt80.6Ni16.7Mo2.7).
-
(3)
The Ni atoms are present both at the sublayer and the bulk layers (the third layer and beneath). The Ni–Ni bonds are present above the Ni content of 16.7% (Pt80.6Ni16.7Mo2.7). The ratio of Ni:Mo of the sublayer decreases with the decrease in Ni content, i.e., 2:1 in Pt66.7Ni30.6Mo2.7 and 1:1 in Pt83.5Ni13.8Mo2.7. In the sublayer of Pt75.0Ni22.3Mo2.7, each Pt is surrounded by 1.5 Mo, 2.5 Ni, and 2.0 Pt on average and each Ni is surrounded by 0.75 Mo and 3.75 Pt on average.
-
(4)
The Pt–Mo bond distance increases slightly with the decrease in Ni content (2.70 Å in Pt66.7Ni30.6Mo2.7, 2.73 Å in Pt75.0Ni22.3Mo2.7, and 2.75 Å in Pt83.5Ni13.8Mo2.7), suggesting that the presence of Ni weakens the Pt–Mo bonding.
3.2.2. PtNiMo Surfaces under ORR Conditions
In the presence of surface O atoms as generated during ORR, a marked change both in the surface morphology and in the compositions was observed from our GCMC/SSW-NN simulations. Our GCMC/SSW-NN simulations started from the above-mentioned clean surfaces, namely, (111), (100), (221), (211), and (210), with the electrode potential being held at 0.9 V. Although it should, in principle, allow all elements (Pt, Ni, Mo, and O) to change during GCMC/SSW-NN, we in practice found that the Ni content variation leads to a very fast Ni loss in many compositions and introduces unnecessary complexity in the phase space. Instead, we choose to perform several independent GCMC/SSW-NN simulations with differently fixed Ni contents, namely, Pt75.0Ni25.0, Pt83.5Ni13.8Mo2.7, Pt80.6Ni16.7Mo2.7, Pt75.0Ni22.3Mo2.7, and Pt66.7Ni30.6Mo2.7, and allow the composition to vary for the other three elements. We also restricted the surface Mo coverage to be less than 0.2 monolayer (ML) for two reasons. First, a fully variable Mo content GCMC/SSW-NN simulation showed that the surface will be fully terminated by Mo species (MoOx) and there is no active Pt exposed. Second, the Mo coverage from experiment is known to be less than 0.2 ML as estimated from a Pt73.4Ni25.0Mo1.6 nanoparticle with a diameter of 4.1 nm (a typical size observed in the experiment;10 also see the Supporting Information on Mo upper limit discussion).
In the GCMC/SSW-NN simulations, the key parameters are the chemical potentials of the variable elements, which should reflect the experimental conditions (such as atomic percent, concentration, and electrode potential) and control the final structure/composition of the system. To accurately calculate the chemical potentials, we considered all possible sources that may appear in nature, either neutral metal atoms in the bulk phase or free ions/molecules in the electrolyte. The source with the most positive μ is used as the final chemical potential. With this approach, the chemical potentials of Pt, Mo, and O are determined by the following chemical reactions at 0.9 V (see the Supporting Information for more details).
Chemical potential for Pt(μPt)
| 3 |
| 4 |
Chemical potential for Mo (μMo):
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
| 11 |
Chemical potential for O (μo):
| 12 |
| 13 |
[Pt], [Mo], and [O] represent an atom in the reservoir, and G(nPt-Ni), G[(n – 1)Pt-Ni], G(PtNi-nMo), and G[PtNi-(n – 1) Mo] are energies of GM for the corresponding bulk phase determined by SSW-NN (see detailed information in the Supporting Information). G(Mo) are the Gibbs free energy of pure Mo metals. E0Mo3+/Mo and E0H2MoO4/Mo are standard electrode potentials from experiments.43,44 The concentration of ions (c in eqs 8 and 10) is set as c = 10–4 mol/L.47 The e is the elemental charge, and U is the electrode potential.
With this approach, we determined that the reference μPt, μMo, and μo are −6.21, −13.23, and – 5.62 eV, respectively, where μMo is determined by eq 6 (see the Supporting Information for more details). We note that the different choices of PtNiMo composition (ranging from Pt75.0Ni24.4Mo0.6 to Pt75.0Ni22.9Mo2.1) will only change the μMo by less than 0.15 eV (see Table S4 in the Supporting Information). According to the formula of μPt and μMo, only μO is explicitly correlated to the electrode potential by the eU term (see eq 13). Therefore, we can perform the GCMC/SSW-NN process at a constant electrode potential U by controlling μO.
For comparison, we first overview the GCMC/SSW-NN results on the zero-Mo catalyst, Pt75.0Ni25.0(111), which is obtained from a GCMC/SSW-NN trajectory of a total of 350,000 steps (see Figure 1d and the detailed reaction evolution trajectory in Figure S4). We found that at 51,282 steps, the 0.17 ML O adatom adsorbs on the surface hollow site, and then the Ni atom starts to get exposed on the surface lattice site by exchanging its position with the surface Pt atom (see 0.17 ML state in Figure 1d). This step is exothermic by 0.54 eV. At 154,810 steps, the O adatom coverage increases to 0.58 ML (see 0.58 ML state in Figure 1d), which leads to 44.4% Ni atoms exposed on the surface. A large fraction of surface Ni atoms (33.3%) aggregate to [−NiO2−] chains covering the surface at an interval of 2.75 Å, which further gains energy by 3.90 eV. After 300,681 steps, the surface O coverage reaches to 0.75 ML and the surface structure no longer changes (see 0.75 ML state in Figure 1d). It should be mentioned that these [−NiO2−] chains are in fact highly unstable under ORR conditions: by dissolving these surface Ni into solution, *Ni → * + Ni2+(aq) + 2e–, the energy can further drop by 1.19 eV. The NiO2 dissolution to Ni2+ is also thermodynamically feasible, which can release 3.35 eV per Ni atom. Therefore, the Ni leaching process can be decomposed into a series of Ni migration steps, where Ni inside Pt75.0Ni25.0(111) first exchanges to the surface, then forms [−NiO2−] chains, and finally dissolves into solution as Ni2+(aq). The surface Ni is thus an early sign of Ni leaching. The other Pt75.0Ni25.0 surfaces yield a similar Ni leaching picture, and the results are given in Figures S5–S7.
In the presence of
Mo, we note that Ni leaching can be prevented
by inhibiting Ni surfacing and the formation of [−NiO2−] chains. Taking Pt75.0Ni22.3Mo2.7(111) as an example, we show the structure snapshots and
the energy profile for the trajectory of 500,000-step GCMC/SSW-NN
simulation in Figure 1e,f. In the initial state, the Pt75.0Ni22.3Mo2.7(111) surface is clean, where all Mo atoms locate
on the subsurface. As early as 728 steps, 0.17 ML O adatoms (add two
O atoms in each unit cell) appear on the surface to bond with surface
Mo atoms, which come from the exchange between the subsurface Mo atoms
and the surface Pt atoms. It is noted that the O stands on the top
site of Mo, forming a terminal Mo=O bond with a bond length
of 1.7 Å. The Mo=O moieties are not far with each other
with the distance being 4.8 Å. This step is exothermic by 2.19
eV per unit cell ( surface
slab). At 3577 steps, the O coverage
increases to 0.25 ML and two MoO join to form to a MoO2-MoO complex. At this stage, because the Mo atom of MoO2 moves above the surface to bond from the Mo2O3 dimer, the surface has one lattice vacancy. This step is exothermic
by 2.76 eV. At 271,176 steps, the O coverage increases to 0.33 ML
(see 0.33 ML state in Figure 1f). The MoO-MoO2 is oxidized to MoO-MoO3, and the surface vacancy is already filled by a Pt atom from the
bulk phase. This step is exothermic by 3.25 eV. At 25,444 steps, the
O coverage increases to 0.58 ML (add three O adatoms; see 0.58 ML
state in Figure 1f).
The MoO-MoO3 is oxidized to a MoO4-MoO3 complex. Similar to the MoO2 group on the 0.25 ML O-covered
surface, the newly formed MoO4 lifts up from the surface,
leaving a vacancy on the surface. This vacancy is healed later by
another Pt atom from the bulk phase. The MoO4 group is
in the tetrahedral coordination (see 0.67 ML state in Figure 1f), with three O atoms linking
to three anchoring surface Pt atoms and one terminal O pointing upright
to the solution. The Mo atom is directly above the hollow site of
three anchoring Pt atoms with a distance of 4.54 Å above the
surface. This step is strongly exothermic by 7.31 eV, which reaches
the lowest energy in the trajectory. It should be emphasized that
in the GM (0.67 ML state), all O atoms are bonded with Mo and no O
adatoms are present on the surface.
surface
slab). At 3577 steps, the O coverage
increases to 0.25 ML and two MoO join to form to a MoO2-MoO complex. At this stage, because the Mo atom of MoO2 moves above the surface to bond from the Mo2O3 dimer, the surface has one lattice vacancy. This step is exothermic
by 2.76 eV. At 271,176 steps, the O coverage increases to 0.33 ML
(see 0.33 ML state in Figure 1f). The MoO-MoO2 is oxidized to MoO-MoO3, and the surface vacancy is already filled by a Pt atom from the
bulk phase. This step is exothermic by 3.25 eV. At 25,444 steps, the
O coverage increases to 0.58 ML (add three O adatoms; see 0.58 ML
state in Figure 1f).
The MoO-MoO3 is oxidized to a MoO4-MoO3 complex. Similar to the MoO2 group on the 0.25 ML O-covered
surface, the newly formed MoO4 lifts up from the surface,
leaving a vacancy on the surface. This vacancy is healed later by
another Pt atom from the bulk phase. The MoO4 group is
in the tetrahedral coordination (see 0.67 ML state in Figure 1f), with three O atoms linking
to three anchoring surface Pt atoms and one terminal O pointing upright
to the solution. The Mo atom is directly above the hollow site of
three anchoring Pt atoms with a distance of 4.54 Å above the
surface. This step is strongly exothermic by 7.31 eV, which reaches
the lowest energy in the trajectory. It should be emphasized that
in the GM (0.67 ML state), all O atoms are bonded with Mo and no O
adatoms are present on the surface.
The results for all Pt97.3–xNixMo2.7 (x is the Ni content) surfaces are summarized in Figure 2, and the final state from GCMC/SSW-NN simulation is elaborated in the following.
Figure 2.

In situ structures of PtNiMo surfaces from GCMC/SSW-NN for different surfaces at different compositions (Pt97.3–xNixMo2.7, x = 13.8, 16.7, 22.3, and 30.6). The orange solid line box outlines the stable surfaces at different compositions that can prohibit Ni leaching under ORR conditions. The atoms above the surface plane are depicted in the ball-and-stick style, while other atoms are depicted in the CPK style. The subsurface layer atoms are in gray. Colors in the figure: blue balls are Pt; violet balls are Ni; cyan balls are Mo; red balls are O.
3.2.3. Pt83.5Ni13.8Mo2.7 Surfaces under ORR Conditions
All surfaces at this composition are composed of a Pt-skin surface layer with on-top MoO4, Mo2O8, or Mo2O9 (MoxOy) groups. The Pt-skin layer fully covers the (111) and (100) surfaces, while it has vacancies under the MoxOy groups on the stepped (221), (211), and (210) surfaces (see Figure 2), and the exposed sublayer atoms within the vacancies are all Pt atoms. These surface structures suggest the high stability of the Pt83.5Ni13.8Mo2.7 surfaces. The (111), (100), and stepped (211) surfaces only have separated MoO4 groups, while the stepped (210) and (221) surfaces consist of Mo2O8 and Mo2O9 dimers, respectively. The MoO5 of the Mo2 dimers has a pyramid configuration surrounded by five O atoms, which can utilize one or two of its O atoms to link with another MoO5 or MoO4.
3.2.4. Pt80.6Ni16.7Mo2.7 Surfaces under ORR Conditions
Only (111) and (100) surfaces can form the full Pt-skin surface layer with on-top (MoxOy) groups, and all the other stepped surfaces contain Ni atoms on the surface. These Ni atoms are all at the lattice sites and bond with the O atoms of MoxOy groups. In particular, (210) and (211) surfaces have a high surface Ni content (0.2 ML).
3.2.5. Pt75.0Ni22.3Mo2.7 Surfaces under ORR Conditions
Only (111) surfaces can form the full Pt-skin surface layer with on-top MoO4 groups. For the other surfaces, the surfaces generally have vacancies due to the surface reconstruction of Ni and Mo exchanging to the surface layer. Ni atoms locate on the surface lattice sites that bond with MoxOy groups.
3.2.6. Pt66.7Ni30.6Mo2.7 Surfaces under ORR Conditions
All surfaces have Ni exposed on the surface. On the (111) surface, two MoO4 groups link with one NiO, forming a Mo2NiO9 cluster. All the other surfaces contain MoO4 groups to bond with surface Ni atoms. In addition, an O adatom appears on the (100) surface, which bonds with three surface Ni surface atoms.
After examining a series of GM structures of Pt97.3–xNixMo2.7 from GCMC/SSW-NN, we find that the Mo atoms have a strong tendency to segregate on the catalyst’s surface under ORR conditions (ΔG ∼ −4 eV for each Mo; see Figure 1f), leading to an evident Mo coverage (∼0.16 ML) on the surface. It is also quite clear that the presence of Mo can suppress the formation of *O adatom and the coming O atoms instead form MoxOy groups (e.g., MoO4 or Mo2O8) on the surface. We note that the adsorbed MoO4 species are not water-soluble under acidic conditions (whereas MoO4 species can be water-soluble under alkaline conditions; see the Supporting Information for more discussions).48−50 We have evaluated the dissolution free energy of the adsorbed MoO4 by using two references H2MoO4(aq) and Mo3+(aq):
| 14 |
| 15 |
We find that the ΔG calculated with H2MoO4(aq) is +0.16 eV, while ΔG calculated with Mo3+(aq) is +1.60 eV. Therefore, the adsorbed MoO4 species are stable under ORR conditions. In addition, the surface MoO4 has a much lower ability to bring out the bulk Ni atoms compared to *O. As a result, Ni leaching tends to occur only at the high Ni content and on the high-index stepped surface. From our simulations, eight surfaces, namely, Pt75.0Ni22.3Mo2.7(111), Pt80.6Ni16.7Mo2.7(111), Pt80.6Ni16.7Mo2.7(100), Pt83.5Ni13.8Mo2.7(111), Pt83.5Ni13.8Mo2.7(100), Pt83.5Ni13.8Mo2.7(221), Pt83.5Ni13.8Mo2.7(211), and Pt83.5Ni13.8Mo2.7(210), are stable under ORR conditions, as judged by the lack of surface Ni atoms at the final state Pt-skin with on-top MoxOy groups.
It is of interest to compare our results with the in situ experiment measurement. Jia et al., using an in situ Mo K-edge XANES experiment, showed that the PtNiMo catalyst (Pt74.6Ni25.0Mo0.4) under reaction conditions exhibits a pattern that coincides with the MoO3 solid,16 which is however not present in the as-prepared alloy catalysts. This result suggests that Mo is likely to be fully oxidized to Mo(VI) oxides. Since O atoms are unlikely to be present in the bulk of Pt-based metal (as indeed found in our calculations), these Mo oxides are expected to be on the surface. Consistently, the Mo in *MoO4 or *Mo2O8 on the PtNiMo surfaces shows a positive Bader charge, e.g., 2.30e on Pt75.0Ni22.3Mo2.7(111), from our calculations, which is very close to that of Mo in MoO3, i.e., 2.34e. This indicates that both Mo atoms are at the highest formal oxidation state of +6 according to the Mo valence shell (Mo 4d55s1). The consistency between theory and experiment indicates that MoxOy clusters are indeed generated under the ORR conditions, which act as the anticorrosion layer on Pt surfaces to prevent Ni leaching.
To provide deeper insights on why *MoO4 does not lead to Ni leaching but *O does, we compare the electronic structures of Pt75.0Ni22.3Mo2.7(111) under ORR conditions (0.67 ML state in Figure 1f) with Pt75.0Ni25.0(111)-0.16 ML O (a metastable structure where two *O adatoms are manually added on the Pt-skin of the clean Pt75.0Ni25.0(111) unit cell), as shown in Figure 3. For Pt75.0Ni22.3Mo2.7(111), both Mo 4d and O 2p states show they are largely closed-shell species, as evidenced by diminished unoccupied states across the Fermi level, indicating that the dangling bonds of the *MoO4 group are well passivated by the Pt-skin. In contrast, the *O adatom of Pt75.0Ni25.0(111)-0.16 ML O is largely an open-shell species, where the unoccupied states across the Fermi level are huge in intensity. This result indicates that the *O adatom on the Pt-skin still has dangling bonds and therefore prefers to further bond with other coming metals (e.g., Ni). This causes the subsurface Ni to break the Pt-skin surface and the subsequent Ni leaching.
Figure 3.
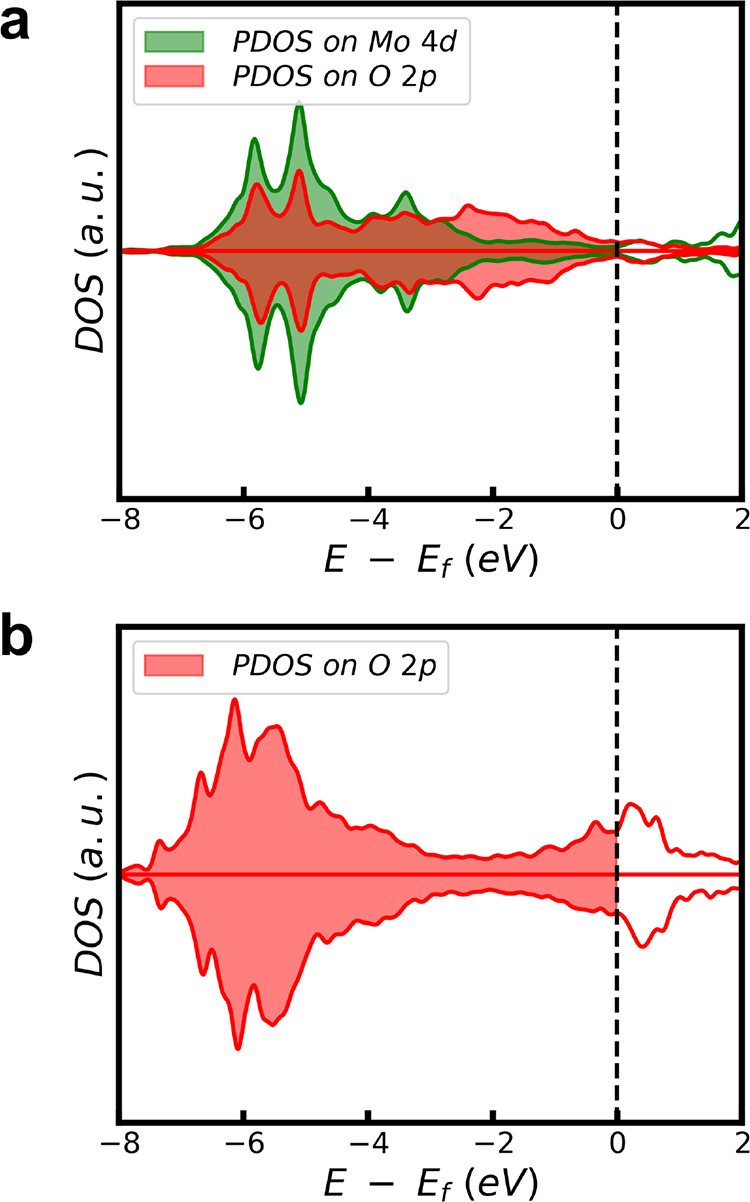
(a) Projected density of states (PDOS) of the in situ Pt75.0Ni22.3Mo2.7(111)-0.67 ML O and (b) Pt75.0Ni25.0(111)-0.16 ML O under ORR conditions. Green lines: PDOS of Mo 4d states; red lines: PDOS of O 2p states. The dashed line is the Fermi level.
The combination of GCMC with SSW-NN allows us to disclose the dynamic reconstruction of the PtNiMo surface under ORR conditions. Our key finding is the accumulation of *MoO4-based Mo-oxide moieties on the Pt-skin surface, which act as an O-atom reservoir to suppress the formation of *O. A single *MoO4 group is a divalent anion as *MoO42–, similar to an adsorbed *O. Due to electrostatic repulsion between anions, *MoO4 and *O compete with each other for the surface sites. Obviously, the GCMC/SSW-NN trajectories, e.g., Figure 1f for Pt75.0Ni22.3Mo2.7(111), indicate that *MoO4 anions are much preferable under ORR conditions compared to the surface *O. This leads to the depletion of *O and the prevention of Ni surfacing, [NiO2] formation, and Ni leaching.
Based on the role of Mo (O-atom reservoir), we can understand the influence of the electrode potential on the stability of the PtNiMo surface. Using Pt75.0Ni22.3Mo2.7(111) as an example (see Figure S29 in the Supporting Information), we find that at low electrode potential (e.g., 0.8 and 0.9 V), there are few O atoms on the surface, where all of them are fixed in the MoOx clusters and therefore the catalyst is stable. On the contrary, at high electrode potential (e.g., 1.2 V), a large number of O atoms exist on the surface, which exceeds the limit of the MoOx clusters that it can contain. As a result, the *O adatom and surface Ni start to appear and the catalyst starts to degrade.
3.3. ORR Activities on PtNiMo Surfaces
Next, we investigated the ORR activity on all PtNiMo surfaces that are stable under ORR conditions according to the above GCMC/SSW-NN simulations. These surfaces are GM from the dynamically reconstructed Pt75.0Ni22.3Mo2.7(111), Pt83.5Ni13.8Mo2.7(111), Pt83.5Ni13.8Mo2.7(100), Pt83.5Ni13.8Mo2.7(211), Pt83.5Ni13.8Mo2.7(221), and Pt83.5Ni13.8Mo2.7(210) surfaces, as shown in Figure 2. The ORR activity on Pt75.0Ni25.0(111)-0.16 ML O was also computed for comparison. For ORR, two mechanisms, the “OOH-dissociation” and “OO-dissociation”, were considered, as described by eqs 16 and 17.
“OOH-dissociation” path:
| 16 |
“OO-dissociation” path:
| 17 |
Our results show that the “OOH-dissociation” path is always more favorable than the “OO dissociation” path on all surfaces investigated. Therefore, only the “OOH-dissociation” path is discussed in the following, while the “O2-dissociation” path can be found in Figures S33–S35. Figure 4 shows the energetic profiles of ORR on the Pt75.0Ni22.3Mo2.7(111), Pt83.5Ni13.8Mo2.7(111), and Pt75.0Ni25.0(111) surfaces and the key intermediate states of Pt75.0Ni22.3Mo2.7(111), while the results of Pt83.5Ni13.8Mo2.7(100), Pt83.5Ni13.8Mo2.7(211), Pt83.5Ni13.8Mo2.7(221), and Pt83.5Ni13.8Mo2.7(210) are briefly summarized in Figure 4c.
Figure 4.

ORR mechanisms on Pt75.0Ni25.0(111), Pt75.0Ni22.3Mo2.7(111), and Pt83.5Ni13.8Mo2.7(111). (a) Free energy profiles of ORR on three (111) surfaces at different PtNiMo compositions. (b) Structures of key reaction intermediates on Pt75.0Ni22.3Mo2.7(111). The atoms above the surface plane are depicted in the ball-and-stick style, while other atoms are depicted in the CPK style. Blue balls are Pt; violet balls are Ni; cyan balls are Mo; red balls are O; white balls are H. (c) Overall free energy barrier (ΔG) for ORR on five different surfaces of Pt83.5Ni13.8Mo2.7.
Figure 4a highlights the reaction pathway on the in situ Pt75.0Ni22.3Mo2.7(111) surface, which has the lowest overall barrier, 0.60 eV at 0.9 V. As the first step, an O2 molecule adsorbs on a bridging site of the exposed Pt-skin (see *O2 state in Figure 4b) and becomes a superoxo anion, O2–, as evident by the feature O–O bond length of 1.34 Å. The free energy change of the adsorption is small, being only −0.10 eV. Next, *O2 is reduced to a bridging *OOH by a proton-coupled electron transfer (PCET) step (see *OOH state), which has a free energy change of 0.36 eV. Then, *OOH dissociates to a pair of *O and *OH and the reaction barrier is only 0.24 eV (see TS). Subsequently, both *OH and *O are reduced to *H2O via three successive hydrogenation steps. The rate-determining step (rds, the slowest elementary step) is at the *OOH dissociation, which has the highest energy TS along the reaction pathway. The overall barrier is 0.60 eV with respect to the adsorbed O2 state.
The ORR on Pt83.5Ni13.8Mo2.7(111) and Pt75.0Ni25.0(111) follows the same pathway, but the overall barriers are 0.01 and 0.13 eV higher than that of Pt75.0Ni22.3Mo2.7(111), respectively. By comparing their energetic profiles, we found that the key to lower the barrier is to increase the adsorption free energy of the O2 molecule: on Pt75.0Ni22.3Mo2.7(111) and Pt83.5Ni13.8Mo2.7(111), the values are −0.1 eV, but it is −0.29 eV on Pt75.0Ni25.0(111). This is because the energetics of both adsorbed *OOH and the O–OH dissociation TS states are basically the same across different surfaces.
The weakened O2 adsorption on Pt75.0Ni22.3Mo2.7(111) and Pt83.5Ni13.8Mo2.7(111) can be understood by the fact that the adsorbed *O2 is a superoxo anion, O2–, and therefore, its stability should be sensitive to its nearby charged species. In particular, Pt75.0Ni25.0(111) differs from Pt75.0Ni22.3Mo2.7(111) in the nearby species of O2, which changes from adsorbed *O atoms to *MoO4 groups in the presence of Mo. It is expected that these anions are repulsive with *O2. For the *MoO4 group, its size is large and is close to *O2, leading to high electrostatic repulsion and, therefore, weak O2 adsorption. Indeed, the ORR barrier is sensitive to the O2 distance with respect to the nearby MoO4 group on Pt75.0Ni22.3Mo2.7(111). It is 0.60 eV when the distance is 4.2 Å and increases to 0.71 eV when it is shortened to 3.4 Å. On the contrary, the size of *O is small and is far away from *O2 (from our calculations, the distance is 3.3 Å between adsorbed O2 and the nearby O*), which incurs less electrostatic repulsion.
For the other minority surfaces, the ORR barrier increases in general. For Pt83.5Ni13.8Mo2.7, where all surfaces are stable under ORR conditions, the reaction barriers on (100), (221), (211), and (210) are 1.84, 1.17, 0.99, and 1.28 eV, respectively, being significantly higher than that of Pt83.5Ni13.8Mo2.7(111) (see in Figure 4c). This indicates that ORR occurs exclusively on the (111) surface. The minority surfaces, although not reactive, determine the stability of the catalyst and therefore need to be protected to maintain the correct Pt:Ni:Mo ratio.
It is of interest to compare our results with previous experimental and theoretical results. Huang et al.’s experiment shows that Pt73.4Ni25.0Mo1.6 after 8000 CV cycles still exhibits higher activity (9.7 mA/cm2) than the as-prepared Pt75.0Ni25.0 (2.7 mA/cm2).10 The presence of Mo can increase the rate by three times. From our results, the overall barrier between the two systems is 0.14 eV, suggesting that the rate difference can be 2 orders of magnitude. The high enhancement from our results suggests that the proton-coupled electron transfers and the O2 adsorption (mass transport) can also be important in experiments. The previous theoretical calculations were typically carried out on the unrealistic clean Pt or PtNi surfaces (zero O precoverage).19,51 For example, Duan and Wang51 by using DFT CI-NEB methods also found that ORR on Pt75.0Ni25.0(111) has a low barrier in O–OH bond breaking (0.15 eV) and our value for Pt75.0Ni25.0(111) is 0.25 eV, mainly due to the 0.25 ML O coverage considered in this work. In addition, Yang et al.52 utilized the simple adsorption descriptors to estimate the ORR-calculated limiting step potential for compare the PtNi catalysts and ternary metal PtNiFe catalysts as it is difficult to obtain the total energy barrier for a ternary catalyst with an ambiguous in situ structure under ORR conditions.
With the calculated ORR activities and Ni leaching tendencies, we can finally understand the different ORR activities observed in experiments for PtNiMo nanoparticles at other Ni contents. From our results, in Ni-rich PtNiMo, the GM structures (as exemplified by Pt66.7Ni30.6Mo2.7) reflect that Ni leaching occurs on all surfaces during ORR. Considering that the ORR has the highest activity on the most stable (111) facet, Ni leaching on the (111) surface will lead to surface roughing and directly cause the activity drop. Therefore, for the PtNiMo nanoparticles at high Ni contents, even after Ni leaching, the surface is no longer (111) dominant and the activity is still low. By contrast, for the Pt-rich PtNiMo, the nanoparticles can maintain their thermodynamically stable morphology, e.g., the cuboctahedral shape, that should be dominated by the flat (111) surface. The activity is thus high as Ni leaching does not occur appreciably.
4. Discussions on Catalyst Screening
We have shown that Mo doping leads to the in situ production of surface MoO42– groups under ORR conditions, which can not only suppress Ni leaching but also decrease the adsorption of O2. As a result, Mo doping simultaneously enhances the activity and durability of Pt75.0Ni25.0 alloy-based ORR catalysts. Naturally, one would wonder whether other elements in the periodic table can perform equally well or even better than Mo since the tetrahedral coordinated metal-oxo species similar to MoO42– are not uncommon for elements. In experiment, W, Re, and Tc elements have been reported as an effective doping element to the PtNi catalyst, and interestingly, we notice that the related CrO42–, ReO4–, and TcO4– anions (hereafter, these anions are referred to as MO4)53−56 were known to be stable in chemistry. This inspires us to design a set of quantitative criteria to screen transition metal elements by assessing the feasibility of forming stable *MO4 groups on the Pt-skin (111) surface, the Ni surfacing ability, and the O2 adsorption tendency nearby *MO4 group. The four quantities are listed as follows.
4.1. Formation Free Energy of *MO4 (ΔGf)
ΔGf is the free energy change in forming a MoO4 group at 0.9 V with respect to the initial unreconstructed clean alloy surface. Taking Pt75.0Ni22.3Mo2.7(111) as an example, ΔGf can be computed using eqs 18 and 19,
| 18 |
| 19 |
where nMo is the number of Mo atoms in each unit cell. For other metals, we replace the Mo atom by the other transition metals in both nPt-Ni-Mo and (n + 2)Pt-Ni-2MoO4, and the ΔGf is estimated by the energy difference of eq 18.
4.2. Exchanging Free Energy Change (ΔGex)
ΔGex is defined as the free energy change for exchanging a surface Pt atom (that directly bonds with MO4 in *Pt56Ni16-2MO4) with a subsurface Ni atom. A positive ΔGex indicates that *MO4 will not lead to Ni leaching (the bond between *MO4 and Pt is stronger than that with Ni), and vice versa.
4.3. Dissolution Free Energy of *MO4 (ΔGdiss)
ΔGdiss is defined as the free energy change for the dissolution energy of *MO4 by the following reactions.
| 20 |
| 21 |
where the *MO4 group can be left from the surface by the forms of MO4n– (VO2+, MnO4–, and TcO4–), HnMO4 (H2MoO4 and HCrO4–), or Mn+ (Fe3+, Nb3+, Ta3+, W3+, Ru2+, Os2+, and Re3+). If a metal has multiple ionic forms (e.g., Mn2+, Mn3+, HMnO4–, and MnO42–), then the most negative ΔGdiss is utilized for comparison.
4.4. O2 Adsorption Free Energy (ΔGads)
The Gads of O2 on the *MO4-covered surface is utilized as the simple descriptor to judge the ORR activity (data shown in the Supporting Information).
The ΔGf can be utilized to first screen all 3d, 4d, and selected 5d transition metals, and the results are listed in Figure 5a (see the blue bar in Figure 5a). For 3d metals, we can find that ΔGf from Sc to Fe (Sc, Ti, V, Cr, Mn, and Fe) is negative (−0.79 to −0.19 eV), indicating that the formation of surface metal-oxo complex *MO4 for these elements is thermodynamically favorable. Meanwhile, for the late transition metals from Co to Zn, their ΔGf’s are positive (0.36 to 3.38 eV), indicating that these elements cannot form surface *MO4. Similarly, for 4d metals, the ΔGf’s of Zr to Ru (Zr, Nb, Mo, Tc, and Ru) are negative (−1.47 to −4.11 eV), while ΔEf’s of Rh to Cd are positive (0.76 to 4.28 eV). For 5d metals, the ΔGf’s of W to Os (W, Re, and Os) are negative (−2.50 to −4.67 eV), while the ΔGf’s of Au are positive (3.83 eV). Our results are consistent with the “oxo-wall” theory, which states that the transition metals to the right of Fe-Ru-Os cannot form a stable metal-oxo complex in the tetragonal coordination.
Figure 5.

(a) Formation free energy of *MO4 (ΔGf) and exchanging free energy change (ΔGex) to screen transition metal candidates for doping with PtNi alloy. (b) Projected DOS of Pt-Ni(111)-*WO4 showing the similar quenched O 2p states at the Fermi level for O in WO4. (c) Dissolution free energy of *MO4 (ΔGdiss) of the promising transition metal candidates from panel (a) showing that six elements (Mo, Tc, Os, Ta, Re, and W) have the positive ΔGdiss.
We emphasize that the above analysis is to fast screen the likely transition metals as good ORR catalysts. From our results, it appears that our criterion of forming MO4 is largely valid for the transition metals left to Fe-Ru as they do have a negative formation energy of *MO4 (see Figure 5a) and therefore should induce a similar surface reconstruction to that of Mo under ORR conditions. For the transition metals right to Fe-Ru, they will not form *MO4 under ORR conditions (see Figure 5a) and we can deduce that these metals cannot stabilize the catalyst in the same mechanism as Pt–Ni–Mo.
For the elements left to the Fe-Ru-Os wall (Sc, Ti, V, Cr, Mn, and Fe for the 3d metal; Zr, Nb, Mo, Tc, and Ru for the 4d metal; W, Re, and Os for the 5d metal), their computed ΔGex’s are shown in Figure 5a (see the orange bar). We found that the ΔGex’s for Sc, Ti, Y, and Zr are negative (−0.82, −0.38V, −0.94, and −0.59 eV, respectively), indicating that even in the presence of MO4 groups on the surface, Ni can still emerge to the surface and thus they are not a valid dopant. All the other metals yield the positive ΔGex and thus survive in the ΔGex screening. It might be mentioned that the electronic structures of these adsorbed MO4 groups are quite similar to that of MoO4, in which the O atom in MO4 is largely saturated. Figure 5b shows the PDOS of Pt75.0Ni22.3W2.7 (WO4 group-covered Pt-skin surface). The O 2p states in the *WO4 group also have a very low population across the Fermi level, indicating that the O atom in *WO4 can only weakly bond with the surface Ni when present.
The stability of MO4 groups on the surface needs to be further compared with the M-containing species in solutions, as described by ΔGdiss. The computed ΔGdiss’s for the 11 elements (V, Cr, Mn, Fe, Nb, Tc, Ru, Os, Ta, Re, and W) are shown in Figure 5c. We found that six elements, namely, Mo, Tc, Os, Ta, Re, and W, have the positive ΔGdiss, indicating that their dissolution is thermodynamically unfavorable. It is interesting to note that although the Mo element has a ΔGdiss of only +0.16 eV, the surface MoO4 may further form Mo2O8 clusters, which can further increase the stability of surface Mo species. Therefore, the ΔGdiss in Figure 5c mainly reflects the stability tendency for the elements investigated. The elements with large ΔGdiss, namely, Tc, Os, Ta, Re, and W, are promising candidates possessing highly stable surface MO4 groups.
Finally, we also evaluated the ΔGads of O2 nearby *MO4 on the Pt-skin surface and found that the adsorption of O2 on all surfaces is weak (Gads ≈ −0.1 eV). This may not be surprising since O2 does not bond directly with the MO4 group and the electrostatic repulsions between O2 and different MO4 anions are similar.
To sum up, our catalyst screening predicts that Mo, Tc, Os, Ta, Re, and W are good candidates for alloying with PtNi to improve the ORR catalytic performance. In this list, four elements, Mo, Os,57 Re,10 and W10 dopants, were reported to enhance ORR, while the two less common elements, Tc and Ta, were not reported yet, and they are worthy of experimental verification in the future.
5. Conclusions
This work represents a comprehensive survey of ternary PtNiMo alloy metal structure and its catalytic activity under electrochemical ORR conditions. A general variable-composition global structure search method under the grand canonical ensemble, namely, GCMC/SSW-NN, is developed, which combines the GCMC, SSW global structural search, and G-NN potential methods. It allows the identification of the in situ active catalyst structure by following the structure dynamics under reaction conditions. We demonstrate that even the low-Mo-doped Pt–Ni alloy has a dramatic structure change under ORR conditions, and the in situ active sites correspond to the Mo1–2Ox cluster-covered Pt-skin (111) surface. The dynamics of alloy surface structures and the anticorrosion mechanism revealed in this work should benefit the design of better alloy materials in a wide range of applications. Our key findings are summarized as follows:
-
(1)
The PtNiMo ternary bulk phase diagram is established, which shows that Mo-rich alloy is in fact the most stable alloy composition and the Mo-lean alloy (<1.6%) utilized in the experiment is metastable. The Mo-lean alloy as chosen in the experiment can avoid too high coverages of nascent MoOx species on the surface that could block all exposed Pt sites. The as-synthesized Mo-lean alloy surfaces share the same Pt-skin surface structure, and Mo atoms tend to segregate in the subsurface layer due to the preference to form Mo–Pt bonds instead of Mo–Ni bonds.
-
(2)
The structures of a series of Mo-lean PtNiMo surfaces are explored using GCMC/SSW-NN, all exhibiting significant reconstruction under ORR conditions (∼0.9 V vs RHE). In general, Mo atoms migrate to the surface and are oxidized to various MoOx clusters, including MoO4, Mo2O8, and Mo2O9. For Ni-rich surfaces (Pt:Ni < 2:1), a large amount of Ni will leach from the surface, which breaks the Pt-skin and leads to a Pt-rich surface with Pt:Ni ranging between 3:1 and 5:1. For the Pt-rich surfaces that end up with MoO4 clusters on the surface, Ni leaching is totally suppressed due to the lack of adsorbed O atoms, and the Pt-skin is maintained.
-
(3)
The ORR activity of the Pt–Ni–Mo alloy arises from the reconstructed MoO4-covered (111) surface. The overall barrier for ORR on Pt75.0Ni22.3Mo2.7(111) is 0.61 eV, which is 0.13 eV lower than that of the pristine Pt75.0Ni25.0(111). The reaction follows the OOH-dissociation mechanism.
Acknowledgments
This work received financial support from the National Key Research and Development Program of China (2018YFA0208600), the National Science Foundation of China (12188101, 22033003, 91945301, 91745201, 92145302, 22122301, and 92061112), the Tencent Foundation for XPLORER PRIZE, and Fundamental Research Funds for the Central Universities (20720220011).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00038.
SSW-NN method; benchmark of G-NN against DFT; GCMC/SSW-NN method; GCMC/SSW-NN trajectory of the Pt75.0Ni25.0 surface; GCMC/SSW-NN trajectory of the PtNiMo surface; ORR activity on Pt75.0Ni22.3Mo2.7(111), Pt83.5Ni13.8Mo2.7(111), and Pt75.0Ni25.0(111)-0.16 ML O; dissolution free energy of *MO4; and the specific activities of ORR catalysts at different CV cycles from the experimental literature (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Stamenkovic V. R.; Fowler B.; Mun B. S.; Wang G.; Ross P. N.; Lucas C. A.; Markovic N. M. Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 2007, 315, 493–497. 10.1126/science.1135941. [DOI] [PubMed] [Google Scholar]
- Cui C.; Gan L.; Heggen M.; Rudi S.; Strasser P. Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 2013, 12, 765–771. 10.1038/nmat3668. [DOI] [PubMed] [Google Scholar]
- Stamenkovic V. R.; Mun B. S.; Arenz M.; Mayrhofer K. J. J.; Lucas C. A.; Wang G.; Ross P. N.; Markovic N. M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. 10.1038/nmat1840. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Polani S.; Luo F.; Ott S.; Strasser P.; Dionigi F. Advancements in cathode catalyst and cathode layer design for proton exchange membrane fuel cells. Nat. Commun. 2021, 12, 5984. 10.1038/s41467-021-25911-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeley J.; Stephens I. E. L.; Bondarenko A. S.; Johansson T. P.; Hansen H. A.; Jaramillo T. F.; Rossmeisl J.; Chorkendorff I.; Nørskov J. K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. 10.1038/nchem.367. [DOI] [PubMed] [Google Scholar]
- Gocyla M.; Kuehl S.; Shviro M.; Heyen H.; Selve S.; Dunin-Borkowski R. E.; Heggen M.; Strasser P. Shape Stability of Octahedral PtNi Nanocatalysts for Electrochemical Oxygen Reduction Reaction Studied by in situ Transmission Electron Microscopy. ACS Nano 2018, 12, 5306–5311. 10.1021/acsnano.7b09202. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang H.; Fang J.; Zou S. Synthesis and Oxygen Reduction Activity of Shape-Controlled Pt3Ni Nanopolyhedra. Nano Lett. 2010, 10, 638–644. 10.1021/nl903717z. [DOI] [PubMed] [Google Scholar]
- Liu F.; Sun K.; Rui Z.; Liu J.; Juan T.; Liu R.; Luo J.; Wang Z.; Yao Y.; Huang L.; Wang P.; Zou Z. Highly Durable and Active Ternary Pt–Au–Ni Electrocatalyst for Oxygen Reduction Reaction. ChemCatChem 2018, 10, 3049–3056. 10.1002/cctc.201800360. [DOI] [Google Scholar]
- Antolini E. The oxygen reduction on Pt-Ni and Pt-Ni-M catalysts for low-temperature acidic fuel cells: A review. Int. J. Energy Res. 2018, 42, 3747–3769. 10.1002/er.4134. [DOI] [Google Scholar]
- Huang X.; Zhao Z.; Cao L.; Chen Y.; Zhu E.; Lin Z.; Li M.; Yan A.; Zettl A.; Wang Y. M.; Duan X.; Mueller T.; Huang Y. High-performance transition metal-doped Pt3Ni octahedra for oxygen reduction reaction. Science 2015, 348, 1230–1234. 10.1126/science.aaa8765. [DOI] [PubMed] [Google Scholar]
- Li Y.; Quan F.; Chen L.; Zhang W.; Yu H.; Chen C. Synthesis of Fe-doped octahedral Pt3Ni nanocrystals with high electro-catalytic activity and stability towards oxygen reduction reaction. RSC Adv. 2014, 4, 1895–1899. 10.1039/C3RA46299D. [DOI] [Google Scholar]
- Cao L.; Zhao Z.; Liu Z.; Gao W.; Dai S.; Gha J.; Xue W.; Sun H.; Duan X.; Pan X.; Mueller T.; Huang Y. Differential Surface Elemental Distribution Leads to Significantly Enhanced Stability of PtNi-Based ORR Catalysts. Matter 2019, 1, 1567–1580. 10.1016/j.matt.2019.07.015. [DOI] [Google Scholar]
- Beermann V.; Gocyla M.; Willinger E.; Rudi S.; Heggen M.; Dunin-Borkowski R. E.; Willinger M.-G.; Strasser P. Rh-Doped Pt–Ni Octahedral Nanoparticles: Understanding the Correlation between Elemental Distribution, Oxygen Reduction Reaction, and Shape Stability. Nano Lett. 2016, 16, 1719–1725. 10.1021/acs.nanolett.5b04636. [DOI] [PubMed] [Google Scholar]
- George M.; Zhang G.-R.; Schmitt N.; Brunnengräber K.; Sandbeck D. J. S.; Mayrhofer K. J. J.; Cherevko S.; Etzold B. J. M. Effect of Ionic Liquid Modification on the ORR Performance and Degradation Mechanism of Trimetallic PtNiMo/C Catalysts. ACS Catal. 2019, 9, 8682–8692. 10.1021/acscatal.9b01772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polani S.; MacArthur K. E.; Klingenhof M.; Wang X.; Paciok P.; Pan L.; Feng Q.; Kormányos A.; Cherevko S.; Heggen M.; Strasser P. Size and Composition Dependence of Oxygen Reduction Reaction Catalytic Activities of Mo-Doped PtNi/C Octahedral Nanocrystals. ACS Catal. 2021, 11, 11407–11415. 10.1021/acscatal.1c01761. [DOI] [Google Scholar]
- Jia Q.; Zhao Z.; Cao L.; Li J.; Ghoshal S.; Davies V.; Stavitski E.; Attenkofer K.; Liu Z.; Li M.; Duan X.; Mukerjee S.; Mueller T.; Huang Y. Roles of Mo Surface Dopants in Enhancing the ORR Performance of Octahedral PtNi Nanoparticles. Nano Lett. 2018, 18, 798–804. 10.1021/acs.nanolett.7b04007. [DOI] [PubMed] [Google Scholar]
- Wei G.-F.; Fang Y.-H.; Liu Z.-P. First Principles Tafel Kinetics for Resolving Key Parameters in Optimizing Oxygen Electrocatalytic Reduction Catalyst. J. Phys. Chem. C 2012, 116, 12696–12705. 10.1021/jp3034616. [DOI] [Google Scholar]
- Sha Y.; Yu T. H.; Merinov B. V.; Shirvanian P.; Goddard W. A. III Mechanism for Oxygen Reduction Reaction on Pt3Ni Alloy Fuel Cell Cathode. J. Phys. Chem. C 2012, 116, 21334–21342. 10.1021/jp303966u. [DOI] [Google Scholar]
- Duan Z.; Wang G. Comparison of Reaction Energetics for Oxygen Reduction Reactions on Pt(100), Pt(111), Pt/Ni(100), and Pt/Ni(111) Surfaces: A First-Principles Study. J. Phys. Chem. C 2013, 117, 6284–6292. 10.1021/jp400388v. [DOI] [Google Scholar]
- Wei G.-F.; Liu Z.-P. Optimum nanoparticles for electrocatalytic oxygen reduction: the size, shape and new design. Phys. Chem. Chem. Phys. 2013, 15, 18555–18561. 10.1039/c3cp53758g. [DOI] [PubMed] [Google Scholar]
- Cao L.; Mueller T. Rational Design of Pt3Ni Surface Structures for the Oxygen Reduction Reaction. J. Phys. Chem. C 2015, 119, 17735–17747. 10.1021/acs.jpcc.5b04951. [DOI] [Google Scholar]
- Ma Y.; Balbuena P. B. Surface Properties and Dissolution Trends of Pt3M Alloys in the Presence of Adsorbates. J. Phys. Chem. C 2008, 112, 14520–14528. 10.1021/jp8046888. [DOI] [Google Scholar]
- Yang Z.; Wang J.; Yu X. The adsorption, diffusion and dissociation of O2 on Pt-skin Pt3Ni(111): A density functional theory study. Chem. Phys. Lett. 2010, 499, 83–88. 10.1016/j.cplett.2010.09.021. [DOI] [Google Scholar]
- Liu Z.; Hu J. E.; Wang Q.; Gaskell K.; Frenkel A. I.; Jackson G. S.; Eichhorn B. PtMo Alloy and MoOx@Pt Core–Shell Nanoparticles as Highly CO-Tolerant Electrocatalysts. J. Am. Chem. Soc. 2009, 131, 6924–6925. 10.1021/ja901303d. [DOI] [PubMed] [Google Scholar]
- Shapovalov S. S.; Mayorova N. A.; Modestov A. D.; Shiryaev A. A.; Egorov A. V.; Grinberg V. A. Pt-Mo/C, Pt-Fe/C and Pt-Mo-Sn/C Nanocatalysts Derived from Cluster Compounds for Proton Exchange Membrane Fuel Cells. Catalysts 2022, 12, 225. 10.3390/catal12030255. [DOI] [Google Scholar]
- Cao L.; Mueller T. Theoretical Insights into the Effects of Oxidation and Mo-Doping on the Structure and Stability of Pt-Ni Nanoparticles. Nano Lett. 2016, 16, 7748–7754. 10.1021/acs.nanolett.6b03867. [DOI] [PubMed] [Google Scholar]
- Wei G. F.; Liu Z. P. Towards active and stable oxygen reduction cathodes: a density functional theory survey on Pt2M skin alloys. Energy Environ. Sci. 2011, 4, 1268–1272. 10.1039/c0ee00762e. [DOI] [Google Scholar]
- Shang C.; Liu Z.-P. Stochastic Surface Walking Method for Structure Prediction and Pathway Searching. J. Chem. Theory Comput. 2013, 9, 1838–1845. 10.1021/ct301010b. [DOI] [PubMed] [Google Scholar]
- Shang C.; Zhang X.-J.; Liu Z.-P. Stochastic surface walking method for crystal structure and phase transition pathway prediction. Phys. Chem. Chem. Phys. 2014, 16, 17845–17856. 10.1039/C4CP01485E. [DOI] [PubMed] [Google Scholar]
- Huang S.-D.; Shang C.; Zhang X.-J.; Liu Z.-P. Material discovery by combining stochastic surface walking global optimization with a neural network. Chem. Sci. 2017, 8, 6327–6337. 10.1039/C7SC01459G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-D.; Shang C.; Kang P.-L.; Zhang X.-J.; Liu Z.-P.. LASP: Fast global potential energy surface exploration. WIREs Comput. Mol. Sci. 2019, 9 (), 10.1002/wcms.1415. [DOI] [Google Scholar]
- Ma S.; Huang S.-D.; Liu Z.-P. Dynamic coordination of cations and catalytic selectivity on zinc-chromium oxide alloys during syngas conversion. Nat. Catal. 2019, 2, 671–677. 10.1038/s41929-019-0293-8. [DOI] [Google Scholar]
- Ma S.; Shang C.; Liu Z.-P. Heterogeneous catalysis from structure to activity via SSW-NN method. J. Chem. Phys. 2019, 151, 050901. 10.1063/1.5113673. [DOI] [Google Scholar]
- Ma S.; Liu Z.-P. Machine Learning for Atomic Simulation and Activity Prediction in Heterogeneous Catalysis: Current Status and Future. ACS Catal. 2020, 10, 13213–13226. 10.1021/acscatal.0c03472. [DOI] [Google Scholar]
- Kang P.-L.; Shang C.; Liu Z.-P. Large-Scale Atomic Simulation via Machine Learning Potentials Constructed by Global Potential Energy Surface Exploration. Acc. Chem. Res. 2020, 53, 2119–2129. 10.1021/acs.accounts.0c00472. [DOI] [PubMed] [Google Scholar]
- Shang C.; Liu Z.-P., LASP G-NN Datasets. http://www.lasphub.com/supportings/PtNiMoOH.pot (accessed2023–0104), PtNiMoOH dataset download 2023.
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Fang Y.-H.; Liu Z.-P. Mechanism and Tafel Lines of Electro-Oxidation of Water to Oxygen on RuO2(110). J. Am. Chem. Soc. 2010, 132, 18214–18222. 10.1021/ja1069272. [DOI] [PubMed] [Google Scholar]
- Li Y.-F.; Liu Z.-P.; Liu L.; Gao W. Mechanism and Activity of Photocatalytic Oxygen Evolution on Titania Anatase in Aqueous Surroundings. J. Am. Chem. Soc. 2010, 132, 13008–13015. 10.1021/ja105340b. [DOI] [PubMed] [Google Scholar]
- Fattebert J.-L.; Gygi F. First-principles molecular dynamics simulations in a continuum solvent. Int. J. Quantum Chem. 2003, 93, 139–147. 10.1002/qua.10548. [DOI] [Google Scholar]
- Shang C.; Liu Z.-P. Constrained Broyden Minimization Combined with the Dimer Method for Locating Transition State of Complex Reactions. J. Chem. Theory Comput. 2010, 6, 1136–1144. 10.1021/ct9005147. [DOI] [Google Scholar]
- Norskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jonsson H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
- Bard Allen J.; Faulkner Larry R., Electrochemical methods: fundamentals and applications. Wiley; New York: 2001. [Google Scholar]
- Bosch R.-W.; Féron D.; Celis J.-P., Electrochemistry in light water reactors: reference electrodes, measurement, corrosion and tribocorrosion issues. Elsevier: 2007. [Google Scholar]
- Feng S.; Lu J.; Luo L.; Qian G.; Chen J.; Abbo H. S.; Titinchi S. J. J.; Yin S. Enhancement of oxygen reduction activity and stability via introducing acid-resistant refractory Mo and regulating the near-surface Pt content. J. Energy Chem. 2020, 51, 246–252. 10.1016/j.jechem.2020.03.063. [DOI] [Google Scholar]
- Mao J.; Chen W.; He D.; Wan J.; Pei J.; Dong J.; Wang Y.; An P.; Jin Z.; Xing W.; Tang H.; Zhuang Z.; Liang X.; Huang Y.; Zhou G.; Wang L.; Wang D.; Li Y.. Design of ultrathin Pt-Mo-Ni nanowire catalysts for ethanol electrooxidation. Sci. Adv. 2017, 3 (), 10.1126/sciadv.1603068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.-F.; Hutchison M.; Santucci R. Jr.; Scully J. R.; Rondinelli J. M. J. T. J. Improved electrochemical phase diagrams from theory and experiment: the Ni–water system and its complex compounds. J. Phys. Chem. C 2017, 121, 9782–9789. 10.1021/acs.jpcc.7b02771. [DOI] [Google Scholar]
- Saji V. S.; Lee C.-W. Molybdenum, Molybdenum Oxides, and their Electrochemistry. ChemSusChem 2012, 5, 1146–1161. 10.1002/cssc.201100660. [DOI] [PubMed] [Google Scholar]
- Jiang K.; Zhao D.; Guo S.; Zhang X.; Zhu X.; Guo J.; Lu G.; Huang X. Efficient oxygen reduction catalysis by subnanometer Pt alloy nanowires. Sci. Adv. 2017, 3, e1601705. 10.1126/sciadv.1601705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Zhao Z.; Cheng T.; Fortunelli A.; Chen C.-Y.; Yu R.; Zhang Q.; Gu L.; Merinov B. V.; Lin Z.; Zhu E.; Yu T.; Jia Q.; Guo J.; Zhang L.; Goddard W. A.; Huang Y.; Duan X. Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction. Science 2016, 354, 1414–1419. 10.1126/science.aaf9050. [DOI] [PubMed] [Google Scholar]
- Duan Z.; Wang G. A first principles study of oxygen reduction reaction on a Pt(111) surface modified by a subsurface transition metal M (M = Ni, Co, or Fe). Phys. Chem. Chem. Phys. 2011, 13, 20178–20187. 10.1039/c1cp21687b. [DOI] [PubMed] [Google Scholar]
- Yang W.; Gong W.; Shi Y.; Wang X.; Wang Y.; Qiao J.; Zeng S.; Di J.; Li Q. PtNiFe nanoalloys with co-existence of energy-optimized active surfaces for synergistic catalysis of oxygen reduction and evolution. J. Mater. Chem. A 2021, 9, 16187–16195. 10.1039/D1TA03048E. [DOI] [Google Scholar]
- Herber R. H.; Johnson D. Lattice dynamics and hyperfine interactions in M2FeO4 (M = potassium(1+), rubidium(1+), cesium(1+)) and M’FeO4 (M’ = strontium(2+), barium(2+)). lnorg. Chem. 1979, 18, 2786–2790. 10.1021/ic50200a030. [DOI] [Google Scholar]
- Brunold T. C.; Güdel H. U. Absorption and Luminescence Spectroscopy of MnO42--Doped Crystals of BaSO4. lnorg. Chem. 1997, 36, 1946–1954. 10.1021/ic9611711. [DOI] [PubMed] [Google Scholar]
- Refaey S. A. M.; Abd El-Rehim S. S.; Taha F.; Saleh M. B.; Ahmed R. A. Inhibition of chloride localized corrosion of mild steel by PO43–, CrO42–, MoO42–, and NO2– anions. Appl. Surf. Sci. 2000, 158, 190–196. 10.1016/S0169-4332(00)00016-7. [DOI] [Google Scholar]
- Best P. E. Electronic Structure of the MnO4–, CrO42—, and VO43— Ions from the Metal K X-Ray Spectra. J. Chem. Phys. 1966, 44, 3248–3253. 10.1063/1.1727219. [DOI] [Google Scholar]
- Zhang W.; Yang Y.; Huang B.; Lv F.; Wang K.; Li N.; Luo M.; Chao Y.; Li Y.; Sun Y.; Xu Z.; Qin Y.; Yang W.; Zhou J.; Du Y.; Su D.; Guo S. Ultrathin PtNiM (M = Rh, Os, and Ir) Nanowires as Efficient Fuel Oxidation Electrocatalytic Materials. Adv. Mater. 2019, 31, 1805833. 10.1002/adma.201805833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.