

ABSTRACT
Prodrugs reliant on microbial activation are widely used but exhibit a range of efficacies that remain poorly understood. The anti-inflammatory compound 5-aminosalicylic acid (5-ASA), which is packaged in a variety of azo-linked prodrugs provided to most Ulcerative Colitis (UC) patients, shows confounding inter-individual variabilities in response. Such prodrugs must be activated by azo-bond reduction to form 5-ASA, a process that has been attributed to both enzymatic and non-enzymatic catalysis. Gut microbial azoreductases (AzoRs) are the first catalysts shown to activate azo-linked drugs and to metabolize toxic azo-chemicals. Here, we chart the scope of the structural and functional diversity of AzoRs in health and in patients with the inflammatory bowel diseases (IBDs) UC and Crohn’s Disease (CD). Using structural metagenomics, we define the landscape of gut microbial AzoRs in 413 healthy donor and 1059 IBD patient fecal samples. Firmicutes encode a significantly higher number of unique AzoRs compared to other phyla. However, structural and biochemical analyses of distinct AzoRs from the human microbiome reveal significant differences between prevalent orthologs in the processing of toxic azo-dyes, and their generally poor activation of IBD prodrugs. Furthermore, while individuals with IBD show higher abundances of AzoR-encoding gut microbial taxa than healthy controls, the overall abundance of AzoR-encoding microbes is markedly low in both disease and health. Together, these results establish that gut microbial AzoRs are functionally diverse but sparse in both health and disease, factors that may contribute to non-optimal processing of azo-linked prodrugs and idiopathic IBD drug responses.
KEYWORDS: Azoreductase, microbial metabolism, Irritable Bowel Disease, Sulfasalazine, Structural Metagenomics, Multi-omics, metagenomics, Structural Biology
Introduction
Compounds with azo linkages (R-N=N-R) are remarkably stable and prevalent in nature; more than 120 unique azo-linked natural products have been isolated to date, with sources ranging from fungi and plants to marine invertebrates1. A range of azo-linked small molecules also reach the human gut, including natural and synthetic dyes, numerous consumer products, and several pharmaceuticals2–5. The reduction of the azo-bond to amines catalyzed by bacterial azo-reductase (AzoR) enzymes was one of the first examples of human gut microbial xenobiotic metabolism6–11. Because of their stability, nitroaromatics are a favorable scaffold for the synthesis of optically active compounds12,13. Indeed, derivatives of azobenzene and related compounds form the azo-linked dyes that span the visible electromagnetic spectrum, many of which are common in the cosmetic and pharmaceutical industries and account for 65% of global synthetic food pigments (Supplemental Figure S1a)3,14. Despite recent regulation to reduce their prevalence, azo dyes have been linked to a range of disorders including neurotoxicity, genotoxicity, carcinogenicity, and colitis15–18.
Gut microbial azo-reduction has also been used in pharmaceutical development as a mechanism for colonic drug delivery (Supplemental Figure S1b)5,6. The frontline treatment for ulcerative colitis (UC), the chronic mucosal inflammation of the colon, is the anti-inflammatory drug 5-aminosalicylic acid (5-ASA)5,6,19,20. 5-ASA confers a topical effect on the gut epithelial mucosal layer and is administered as an azo-linked prodrug designed to be activated by gut bacterial azoreductase enzymes (AzoRs)3–5,20–23. Over 90% of UC patients receive such drugs, which include sulfasalazine (AzulfidineⓇ), balsalazide (ColazideⓇ), and olsalazine (DipentumⓇ) (Supplemental Figure S1b), although they often require gram-scale doses daily for efficacy20,22,24. Even with these high dosages, 50% of patients experience treatment failure and much progress to corticosteroids and/or immunosuppression20,22,24.
Azo-linked prodrug activation was long thought to rely on protein catalysts, although non-enzymatic activation has recently been highlighted25. Microbial AzoRs are considered to be important drivers for the intestinal activation of 5-ASA near the colonic mucosal layer2,5. Bacterial AzoRs are oxygen insensitive, flavin mononucleotide (FMN)-requiring homo- and heterodimers that employ NAD(P)H as sources of hydrides5,18,26,27. They follow a bi-bi ping-pong mechanism where the hydride donor reduces the active site flavin to FMNH26,27–29. The oxidized nicotinamide cofactor then leaves the active site and is replaced by the azo-linked substrate, which is reduced to a hydrazo-intermediate27,30,31. The process then repeats itself to generate two primary amine-containing products (Supplemental Figure S1c). Over 84 strains of bacteria have been shown to perform this reaction in cell-based assays with 5-ASA-linked prodrugs5,6,28,32. However, few AzoRs derived from commensal gut microbes have been characterized, and the majority of relevant studies have focused on enzymes from Proteobacteria and their processing of azo-linked dyes7–11,14,18,33–35. Indeed, only a very small number of AzoRs from Firmicutes and no AzoRs from Bacteroides, the two dominant gut microbial phyla, have been examined to date8,9,27–29,34,36–39. Thus, the current data on microbial AzoR structure-function likely falls short of capturing the diversity of enzyme orthologs present in the human gut microbiome40,41.
Defining the scope of gut microbial AzoRs may facilitate the improved treatment of IBD3 and/or the management of disorders caused by azo dye metabolism14–18. Therefore, we examined AzoRs in the human gut microbiome by employing several datasets, including samples collected longitudinally over one year from patients with IBD and healthy individuals42. Using bioinformatics paired with kinetics analysis and structural biology, we characterized the landscape of gut microbial AzoRs across the human gut microbiome. We find that the majority of unique AzoRs in the human microbiome are derived from Firmicutes and that structural features common to Firmicutes AzoRs explain poor processing of the IBD drug sulfasalazine. We also find that the abundance of AzoR-producing microbes is markedly low in both health and disease. Thus, gut microbial AzoRs are sparse and poorly active at prodrug processing, factors that provide a molecular rationale for the idiopathic IBD drug responses and a new understanding of the limitations of enzyme-mediated therapeutic activation in the human gut.
Results
Structural metagenomics defines an atlas of human gut microbial AzoRs
We applied a structural metagenomics approach to begin to characterize the landscape of AzoRs across the human microbiome. To define functionally relevant structural features conserved across microbial AzoRs, we examined four bacterial AzoRs with known crystal structures: Escherichia coli (EcAzoR), Pseudomonas putida (PpAzoR), Pseudomonas aeruginosa (PaAzoR), and Klebsiella pneumoniae (KpAzoR) (Figure 1a). We identified five functionally relevant residues conserved in all four of these proteins, as shown in E. coli AzoR (Figure 1a ; PDB: 2D5I)43. Three directly interact with FMN (S16, P95, and G142) and reside within a loop that “cradles” the cofactor in the active site37,44,45. The side chains of residues 97 and 98 point away from FMN but their main chain amides form hydrogen bonds with the isoalloxazine ring. These amino acids were not conserved and were therefore not included, but we noted that no prolines were present. We also identified two conserved residues that form a salt bridge at the dimer interface, K105 and D109 (Figure 1a) and we included them, allowing for K/R and D/E substitutions.
Figure 1.
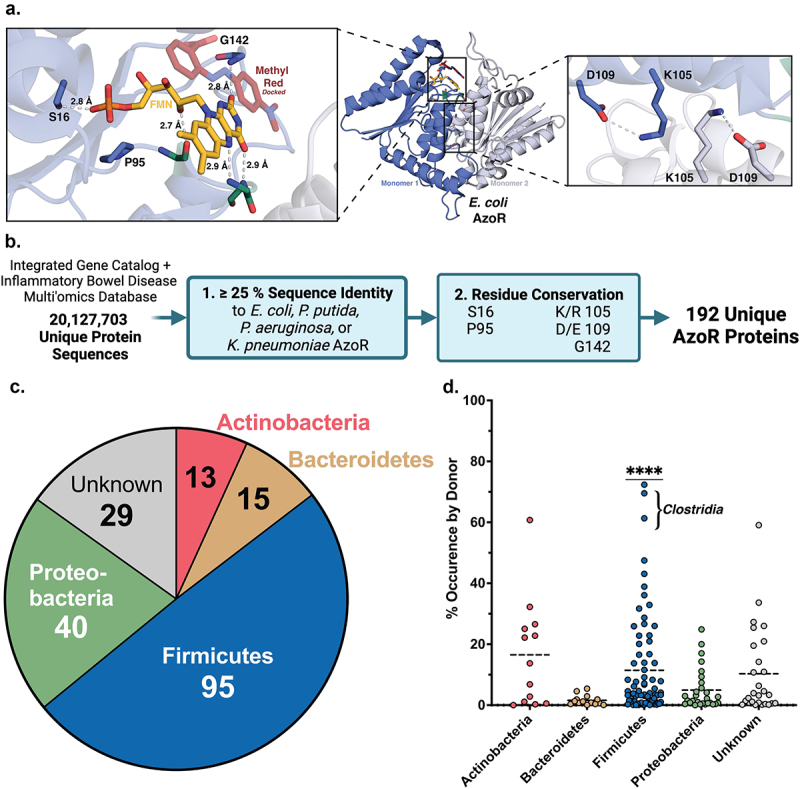
Structural metagenomics pipeline used to identify gut bacterial AzoR Enzymes. (a) Conserved residues (blue) used to define two-step rubric to identify AzoR proteins shown in EcAzoR (PDB: 2D5I), showing placement of FMN (yellow) and methyl red (red) docked as an example ligand. (b) Workflow of structure-guided rubric used to identify AzoR proteins across the IGC and IBDMDB. (c) Phylogenetic distribution of unique putative AzoR proteins across IGC and IBDMDB. (d) Occurrence for each AzoR sequence per Individual across IGC arranged by phyla, ****P < 0.0001 by a one sample t test comparing Firmicutes to all other groups.
These five residues were then employed to identify AzoR proteins from the human gut microbiome (Figure 1b). The Integrative Human Microbiome Project (HMP2) obtained metagenomic sequencing data collected from 130 individuals, including 103 patients diagnosed with IBD, over the course of one year to form a set of 1,472 unique metagenomic samples (after filtering for antibiotic use) as a component of the Irritable Bowel Disease Multi-Omic Database (IBDMDB)42. The Integrated Gene Catalog (IGC) combines metagenomic sequencing data from 249 human samples examined in the Metagenomics of the Human Intestinal Tract (MetaHit) project along with 1,018 collections of extant human gut metagenomics data, including samples collected from individuals across three continents, as well as near-complete genomes for many commensal gut microbes46. We combined all predicted proteins derived from both the IBDMDB and the IGC then filtered for redundancies at 100% sequence identity to form a set of 20,127,703 unique sequences that were queried in two steps for putative AzoR proteins47. First, sequences with E-values≤0.05 and sequence identities≥25% by a pairwise alignment to any of the AzoRs of known structure were obtained. Second, sequences that then contained the five residues outlined above were selected. Binning sequences with≥95% identity reduced our collection of AzoRs to a final set of 196 unique proteins including the four reference sequences (Figure 1b)45–51. A multiple sequence alignment then revealed an additional residue conserved in>95% of sequences. An aromatic side chain from the adjacent monomer within the homodimer is consistently positioned above the substrate binding area (F119 in EcAzoR). Such a residue may function to align NADH/NADPH at the active site for hydride transfer to FMN and then the azo substrate for reduction. In summary, we employ a structure-guided analysis of gut metagenomics to define an atlas of 196 unique AzoR proteins in the human microbiome that help to expand our understanding of active site function.
Phylogenetic diversity of AzoRs across the gut microbiome
To begin exploring the diversity across these 196 unique AzoRs, we identified their taxonomic origins. Nearly half were from Firmicutes, followed by Proteobacteria (20%), and~15% from either Actinobacteria or Bacteroidetes; the taxonomic identities of the remaining 15% were uncertain at a 95% confidence level (Figure 1c ; Supplemental Table S2). We assessed the occurrence frequency of AzoR sequences within a donor’s gut microbiome provided by the metadata provided for genes within the IGC cohort and found that Firmicute-derived AzoRs are significantly more common than those from other phyla (P < 0.0001; Figure 1d; Supplemental Table S2). Notably, no sequences in the collection contained signal peptides, indicating that all putative AzoRs are likely maintained intracellularly, as expected for these two-redox cofactor utilizing enzymes. This finding, paired with the narrow distribution of the length of sequences across both databases (Supplemental Figure S2), support the conclusion that the 196 proteins identified belong to a single enzyme family.
To explore the sequence diversity across our collection in the context of IBD, we used EFI-EST to create a sequence similarity network (SSN), which highlighted several additional features (Figure 2a)48,49. First, Firmicutes produce the most diverse set of AzoR proteins, as sequences belonging to this phylum are separated into several clades across the SSN. Second, 70% of unique sequences in our atlas were not detected across the larger IBDMDB dataset and are instead detected exclusively in the IGC (Figure 2a). While this result is somewhat unsurprising given that samples collected from patients with IBD typically reflect lower alpha diversity and demonstrated lower sequencing depth, it also highlights the increased diversity across an enzyme family that can be uncovered using deeper strain-level sequencing, such as those used to curate the IGC42. Third, using a sequence search with a low identity threshold combined with specific structural motifs identified putative AzoRs not annotated as such, suggesting some of these orphan enzymes may exhibit functional promiscuity or were otherwise mis-annotated (Supplemental Table S1)50. Indeed, when annotated with “evolutionary genealogy of genes: Non-supervised Orthologous Groups” (eggNOG v5.0), only 35% of the proteins in our atlas were pinpointed as AzoRs (Supplemental Table S1, Supplemental Figure S3). Collectively, these results highlight that active site-directed structural analyses, even with relatively few available structures, can inform and refine the functional annotations of metagenomic sequencing data.
Figure 2.
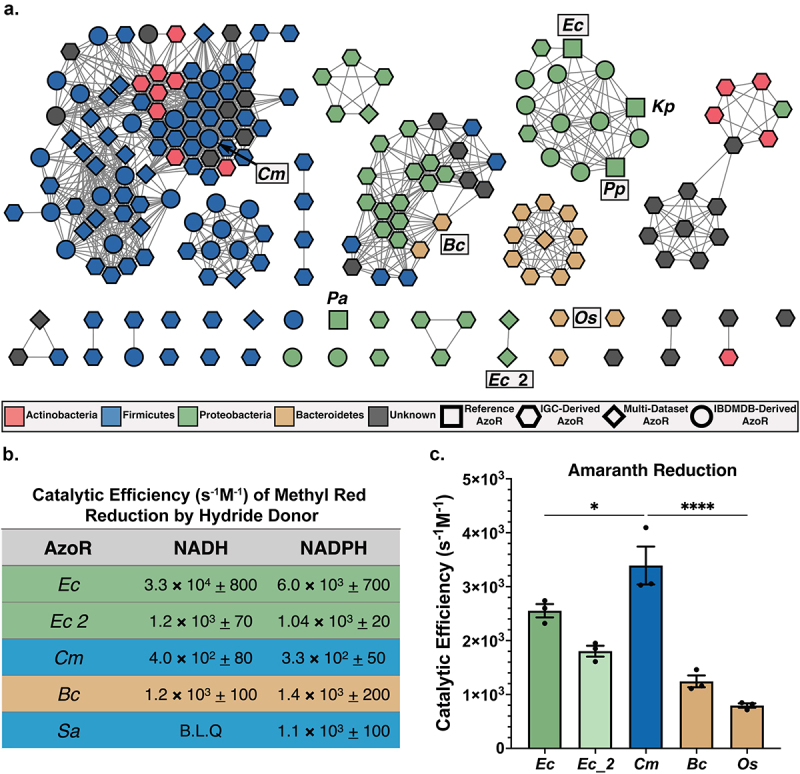
Diversity across atlas of bacterial AzoRs and activity of purified proteins. (a) Sequence Similarity Network of AzoR proteins across IGC and IBDMDB including reference sequences used to identify putative AzoRs colored according to Phyla, with abbreviations shown for proteins used in this study (E=1×10−40). (b) Catalytic efficiencies for Methyl Red azo reduction by purified AzoR enzymes as a function of hydride donor present in reaction. (c) Catalytic efficiencies for Amaranth azo reduction by purified AzoR enzymes. Statistical comparisons reflect activity of CmAzoR compared to other enzymes and were calculated using Tukey’s multiple comparisons test. *P < 0.05, ****P < 0.0001.
Carcinogenic dye processing by gut microbial AzoRs
To explore how the vast sequence diversity across our AzoR atlas (Figure 2a) translates to differences in function, and to confirm that these novel proteins do indeed exhibit AzoR activity, we selected representative AzoR proteins and assessed their ability to reduce azo-linked dyes. We first selected three AzoRs from our SSN for purification (Figure 2a). We chose a Firmicutes protein from Clostridium sp. Marseille-P7770 (CmAzoR) that was prevalent in both healthy donors and IBD patients, was from the genus Clostridia that occurred in the most individuals across the IGC (69%, Figure 1a), and was representative of the largest clade of our SSN (Figure 2a). We also chose a Bacteroidetes protein from Bacteroides cellulosilyticus (BcAzoR) representative of the second largest clade in the SSN, and a novel, second E. coli AzoR (Ec_2AzoR) (Figure 2a). We examined the cofactor utilization preferences of these novels recombinantly expressed and purified enzymes with the optically active azo reporter substrate Methyl Red (Supplemental Figure S1a). We compared them to two controls: EcAzoR, which utilizes both NADH and NADPH as a hydride donor, and Staphylococcus aureus AzoR (SaAzoR), which exclusively uses NADPH5,6,8. We found that the novel AzoR enzymes were able to use both NADH and NADPH as cofactors (Figure 2b). While E. coli and Bacteroides proteins exhibited greater catalytic activities with Methyl Red than the Firmicutes enzymes, these results confirm that the gut microbial proteins examined here are azoreductases and that they are not selective for nicotinamide cofactor hydride donor.
We next assessed activities with the larger and more clinically significant azo dye Amaranth (FD&C Rd No. 2; Supplemental Figure S1A), a compound banned in the U.S. but still employed elsewhere in food and cosmetics. It is known to induce genotoxicity, cytotoxicity and to be carcinogenic16. We found that EcAzoR exhibited markedly reduced efficiency with Amaranth compared to Methyl Red, and that the Firmicute CmAzoR protein exhibited significantly higher activity with Amaranth than the other enzymes (Figure 2c; p < 0.05). Indeed, while the CmAzoR catalytic efficiency with Amaranth was 8-fold higher than with Methyl Red, catalytic efficiencies for BcAzoR were comparable between the two substrates (Figure 2b,c). To confirm these results, we also purified a second Bacteroidetes AzoR from our SSN, from Odoribacter splanchnicus, (OsAzoR), and found that this protein was similarly inefficient with Amaranth (Figure 2c). Given the extent to which CmAzoR exhibits significantly higher activity with Amaranth than other AzoRs, we sought to identify the structural features enabling its efficiency.
Crystal structure of clostridium sp. Marseille AzoR
To examine the molecular basis for differential CmAzoR activities (Figure 2c), we determined the crystal structure of the enzyme at 1.4 Å resolution (Figure 3a ; Supplemental Table S1). CmAzoR and EcAzoR share a root mean-square deviation of 3.3 Å across 201 equivalent Cα positions with 25% sequence identity (Supplemental Figure S4). Modeling Amaranth into the active site using a co-crystal structure of Methyl Red bound to PaAzoR as a guide (PDB: 2V9C) highlights features distinct in CmAzoR (Figure 3b)38. First, two structured loop-turn-helices that frame the hydrophobic EcAzoR are replaced by polar loops in CmAzoR that create a larger and more hydrophilic active site (Figure 3b). Second, two cationic residues, R13 and R17, in one of the CmAzoR loops are uniquely positioned to form electrostatic interactions with the sulfates of Amaranth; these residues are replaced with alanine and glutamine within the more sterically occluded active site of EcAzoR. Thus, compared to EcAzoR, the active site of CmAzoR provides a more open hydrophilic active site with complementary electrostatic contacts that explain its efficient processing of the clinically relevant azo-dye Amaranth. Furthermore, of the 80 Firmicutes AzoRs in the main clade of the SSN in Figure 2a, 76 have an R at the position 13 equivalent and an R or K at the position 17 equivalent, and 67 have Rs in both positions. Arginines in both positions are also conserved in the top four most abundant Firmicutes AzoRs in Figure 1d. As such, we conclude that Firmicutes AzoRs, which are the most prevalent in the human gut microbiome, are likely to be efficient activators of the azo-toxin Amaranth.
Figure 3.
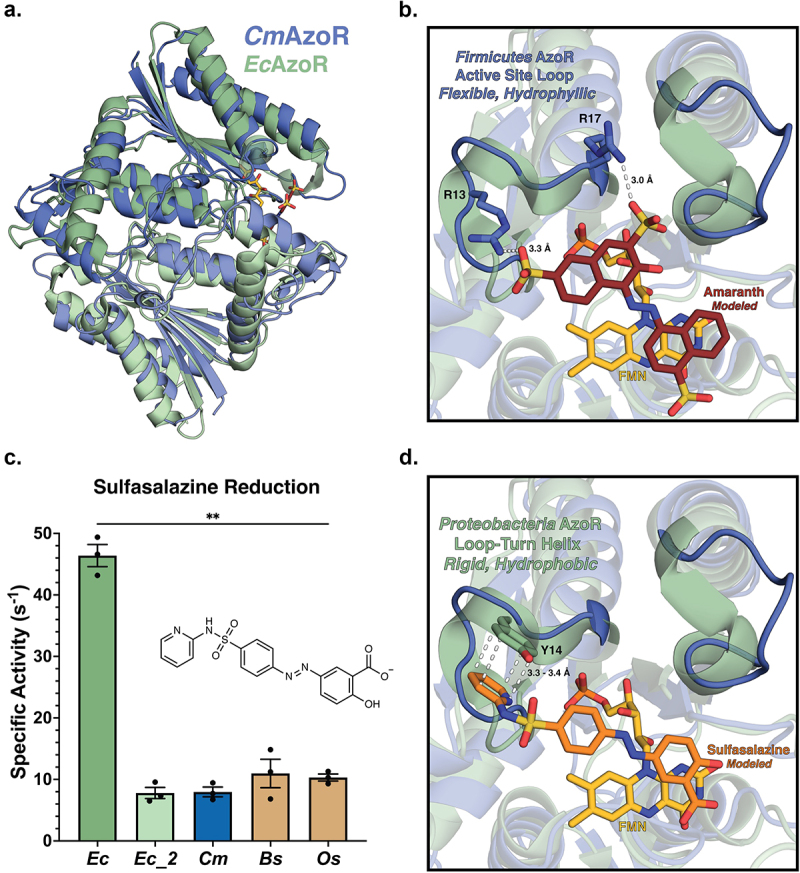
AzoR active site environments point to substrate specificity. (a) Homodimers of crystal structures of EcAzoR (green) and CmAzoR (blue) overlaid and analysis of active site structural disparities. FMN (yellow) is located according to the structure of EcAzoR (PDB: 2D5I). Amaranth (red) was modeled into the active site using methyl red in the crystal structure of PaAzoR (PDB: 2V9C) as a reference for ligand placement. (b) Active site docked models for Amaranth (B) and Sulfasalazine (d) azo reduction by purified AzoR enzymes. Ligands were docked into active sites of crystal structures to point toward structural differences driving differential enzyme activity. Error bars represent SEM across three biological replicates. Catalytic efficiencies and active site docked models for Sulfasalazine (c) azo reduction by purified AzoR enzymes. Statistical comparisons reflect activity of CmAzoR compared to other enzymes and were calculated using Tukey’s multiple comparisons test, **P < 0.01.
Sulfasalazine processing by gut microbial AzoRs
To provide context to our representative AzoRs in the context of the landscape of AzoRs across the IBDMDB, we examined the activation of the IBD drug Sulfasalazine (Supplemental Figure S1B) to 5-ASA by gut microbial AzoRs. Using MS/MS to determine apparent rates (Supplemental Figure S5; Supplemental Table S3), we found that EcAzoR was dramatically more efficient at Sulfasalazine activation, exhibiting a significant threefold higher specific activity than the other enzymes in our panel (Figure 3c; p < 0.01). Modeling Sulfasalazine into the active sites of CmAzoR and EcAzoR revealed key structural features underlying these differences (Figure 3d). The hydrophobic turns adjacent to the EcAzoR active site form a pocket amenable to Sulfasalazine binding that is enhanced by the presence of a unique aromatic side chain capable of pi-stacking interaction with this drug (Figure 3d). We further note that R13 in CmAzoR would clash with Sulfasalazine in the EcAzoR active site (Figure 3d). Note that all Firmicutes AzoRs identified in our main SSN clade have an R in the position equivalent to R13 in CmAzoR (Figure 2a), including the most prevalent in Figure 1d. These observations support the conclusion that Firmicutes AzoRs, the most common in the human gut microbiome, are likely to be both efficient activators of azo-toxins and poor activators of azo-linked prodrugs like sulfasalazine, factors that may influence the etiology and treatment of IBD.
Gut microbial AzoRs are sparse in health and disease
To assess the prevalence and distribution of AzoRs in health and disease, we determined relative abundance of microbes that produce AzoRs in the metagenomes of 1,472 human samples collected from 413 healthy donor samples and 1,059 IBD patient samples over the course of one year in the IBDMDB42. First, we assessed the relative abundances of the 41 strains in the IBDMDB denoted as AzoR-encoding by Enzyme Commission (EC) annotations. More than 80% of individuals, regardless of health or disease, were predicted to have<1% abundance of AzoR-encoding microbes, and that 30–35% had no such microbes (Supplemental Figure S6a). These trends remained in place when taxa from the Proteobacteria and Firmicute phyla were examined separately, with 40–50% and nearly 60% showing no AzoR-encoding microbes, respectively (Supplemental Figure S6b,c).
We found in creating the AzoR atlas that many taxa encoding AzoR proteins that were not defined as such when predicting function using eggNOG or EC annotation. Thus, we reasoned that the absence and low relative abundance of AzoR-encoding microbes across the IBDMDB could be a consequence of incomplete functional annotation. Therefore, we performed a more integrative analysis in which we added to the 41 EC-annotated strains examined above, including all microbes for which AzoR activity has been reported in the literature, either for purified proteins or instances in which strain isolates were shown to metabolize IBD prodrugs (including those that are not canonically considered commensal gut microbes)5,6,32,51,52. To those, we added the AzoR-encoding species identified using our structural metagenomics approach (see Figure 1a). Eliminating cross-dataset redundancies formed a comprehensive set of 159 unique bacterial strains either suggested or confirmed to exhibit AzoR activity (Supplemental Table S2). A cladogram of these taxa is shown in Figure 4a, along with their average abundances in the IBDMDB metagenomics dataset normalized relative to other AzoR-producing strains (Supplemental Table S2). We found that nearly half of these strains were absent across the cohort, the majority of those present were only sparsely abundant, and that only three taxa (one from each major phylum) stood out as notably abundant relative to the other strains (Figure 4a). As expected, the Proteobacterium E. coli was detected in all samples. In the Firmicutes, the AzoR-encoding species Flavonifractor plautii was abundant, and its AzoR (FpAzoR) belongs to the same clade as the Clostridium sp. Marseille-P7770 protein (CmAzoR) examined above. An AlphaFold model of FpAzoR reveals that it shares 1.8 Å root-mean-square deviation in Cα positions with CmAzoR (Supplemental Figure S7), indicating that these proteins are highly similar. The Bacteroidetes AzoR-encoding species of greatest relative abundance was Bacteroides cellulosilyticus, which encodes the BcAzoR examined above (Figure 4a).
Figure 4.
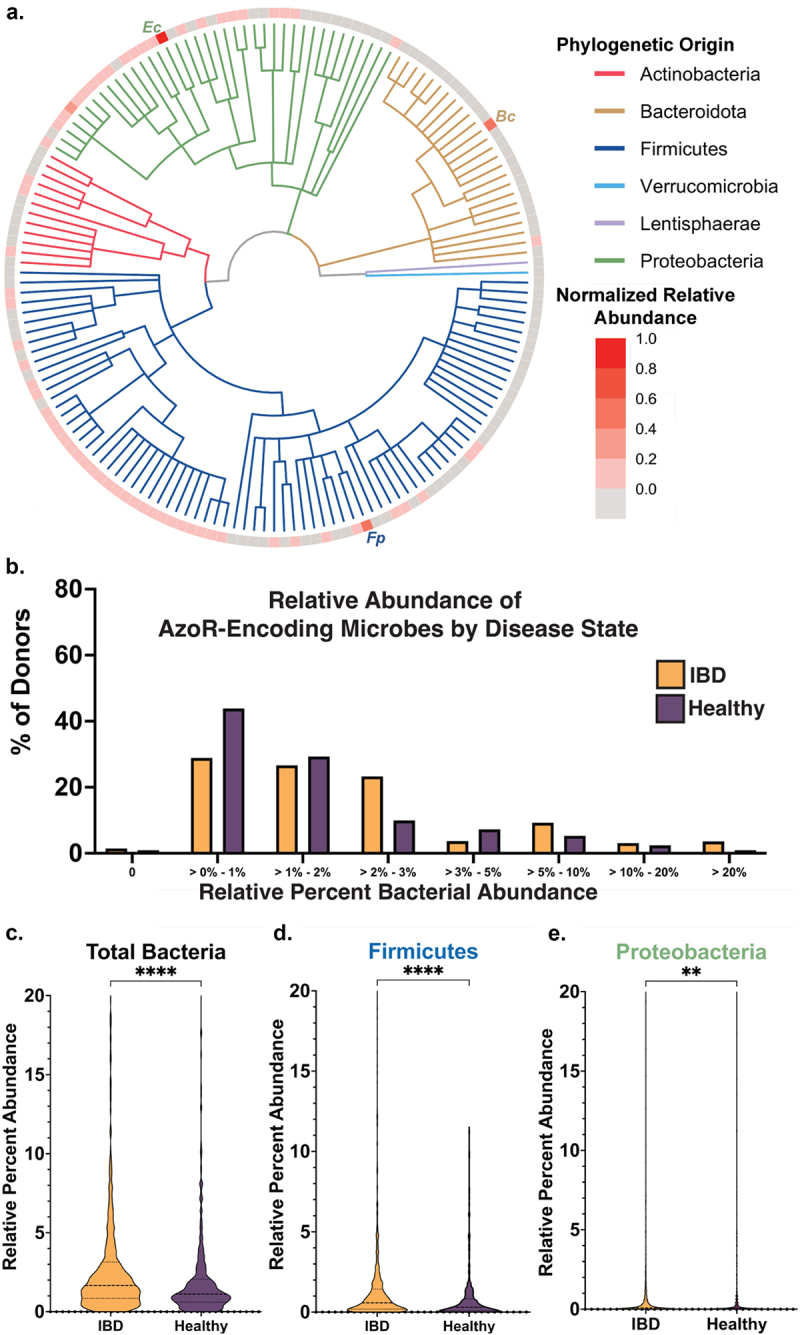
Azo—encoding microbes are sparse in health and disease. (a) Cladogram reflecting microbes used for integrative analysis query, with abundances for each relative to other AzoR-encoding strains. Percent of percent of donors containing AzoR-encoding microbes as determined by Integrated Search space combining EC Annotations, extant AzoR-encoding microbes reported in literature, and species identified using structural metagenomics in this study for total bacterial abundance. (b) Abundance of Percent of percent of donors containing AzoR-producing microbes at a range of relative abundance levels across the x-axis, queried using an Integrated Search space combining EC Annotations, extant AzoR-producing microbes reported in literature, and species identified using structural metagenomics in this study for total bacterial abundance. Violin plots comparing AzoR abundances between IBD patients and healthy individuals across the IBDMDB, comparing relative % abundances for all microbes (c), Firmicutes (d), and Proteobacteria (e) that produce AzoR genes as annotated by Integrative Analysis used in this manuscript. ****P < 0.0001, **P < 0.01 by an unpaired t test.
Using this more complete set of AzoR-encoding taxa generated via an integrated approach, we reassessed again relative abundances in the IBDMDB data. We now found that nearly all samples were predicted to have bacteria with the potential to produce AzoRs, (Supplemental Figure S6d,e). We further found that while nearly 30% of donor samples lacked Proteobacteria AzoR-encoding taxa, nearly all donor samples contained Firmicutes taxa with the capacity to produce AzoRs (Supplemental Figures S6e,f). Although most donor samples contained microbes capable of producing AzoRs, the relative abundance of these microbes was remarkably low. Most healthy and IBD samples had less than 2% relative abundance of AzoR-encoding taxa, and a relative abundance exceeding 10% was only observed in 3% of healthy samples and 7% of IBD patient samples (Figure 4b). These findings indicate that the relative abundance of AzoR-encoding taxa is low in health and in IBD, with only a small percentage of samples across the cohort harboring a substantial relative abundance of AzoR-encoding microbes (Figure 4b).
However, we noted that IBD patient samples were enriched in AzoR-encoding taxa (Figure 4c; Supplemental Figure S8A). Indeed, when compared side-by-side, we found that AzoR-encoding microbes are significantly higher abundance in IBD patients than healthy individuals (P < 0.0001; Figure 4c). Similarly, IBD patients also show significantly higher abundances of Firmicutes (P < 0.01; Figure 4d; Supplemental Figure S8B) and Proteobacteria (P < 0.0001; Figure 4e ; Supplemental Figure S8C) with AzoR genes. These low abundance trends are similarly reflected in the transcriptomics data for the IBDMDB, wherein only 5% of samples contain any AzoR protein transcripts, 0.5% of which originate from Proteobacteria (Supplemental Table S4)42. Finally, for completeness, we also sought to examine the metaproteomics data in the IBDMDB for AzoR proteins. None of the 472 proteomic samples contained a bacterial AzoR, likely due to the low abundance of these enzymes42. Together, these results show that protein structure-informed approaches directed toward gut metagenomic data analyses can encompass a more comprehensive set of clinically relevant enzymes within a given enzyme family, assigning functions with greater accuracy than EC annotations. They further support the conclusion that azo-linked prodrugs may show idiopathic efficacies in IBD because of the relatively low levels of AzoR enzymes and their differential functional landscapes in health and disease1,3,12,15.
Discussion
Although azo-linked compounds are widely prevalent across xenobiotics, particularly synthetic dyes and drugs used to treat IBD, the diversity of the microbial enzymes that metabolize these compounds or the abundance of AzoR-encoding microbes has not been explored8,18,27,29,34,53. While the AzoR-mediated reduction of azo dyes is associated with a myriad of cellular defects ranging from neurotoxicity to colitis, the same enzyme-mediated reaction chemistry is the singular method of delivery for certain widely used IBD prodrugs19. AzoR proteins derived from several species of Proteobacteria have been shown to be capable of reducing azo dyes and activating IBD prodrugs; however, this phylum accounts for a remarkably small portion of the total bacterial composition of the gut19,37,38,44. Thus, we sought to define the metagenomic landscape of AzoR proteins across the human gut microbiome, and to explore their differences in health and disease.
We used a structural metagenomics approach (Figure 1a,b) to identify putative AzoR proteins across the IGC and IBDMDB, which collectively contain over 20 million unique predicted protein sequences derived from gut microbes46. Nearly half of the 196 unique AzoR sequences we identified from our analysis originated from Firmicutes (Figure 1b,c), with several from Clostridia being present in the microbiomes of >70% of individuals across the IGC (Figure 1d). Importantly, several clades of putative AzoRs distinct from the Proteobacteria-derived reference AzoRs were identified (Figure 1c). Furthermore, Proteobacteria AzoRs overall accounted for less than 20% of unique sequences across the IGC. While these sequences are simply the genes present within an individual sample and do not necessarily reflect the presence or abundance of proteins, these findings suggest that the field’s focused characterization of AzoRs from Proteobacteria has overlooked functionally diverse proteins from Firmicutes, which appear more prevalent in the gut.
To begin to assess the functional diversity of gut microbial AzoRs, we purified three putative AzoRs spanning the diversity of the IGC and IBDMDB (Figure 2a), and then examined their cofactor utilization preferences and enzyme activity. All purified AzoRs were active azoreductases capable of reducing Methyl Red and utilizing either NADH or NADPH as hydride donors (Figure 2c). Given that the purified AzoRs were less efficient in reducing the small azo dye Methyl Red compared to EcAzoR, we sought to further delineate substrate preferences by assessing enzyme activities with the larger azo dye Amaranth. CmAzoR from Firmicutes was a significantly more efficient reducer of Amaranth compared to either EcAzoR or other purified AzoRs, however the protein was relatively inefficient with the smaller azo dye Methyl Red (Figure 2b,c, Supplemental Figure S1). These findings suggest that AzoRs derived from Firmicutes may be uniquely efficient reducers of relatively large, hydrophilic scaffolds commonly observed across azo-linked dyes. Structural features unique to CmAzoR support this conclusion (Figure 3a,b), showing that this enzyme contains hydrophilic regions at its active site amenable to Amaranth binding but not to the IBD drug sulfasalazine, which is favored by EcAzoR and its more hydrophobic motifs (Figure 3c,d)43. Most Firmicutes AzoRs in our atlas maintain the structural features present in CmAzoR that appear to drive its poor activation of sulfasalazine. It is possible that EcAzoR, which is relatively efficient with sulfasalazine, led to the expectation that such prodrugs would be efficiently processed by all AzoR isoforms. However, this assumption could be challenged with this new appreciation of AzoR diversity in the gut microbiome.
EC and eggNOG annotations are an automatic and often convenient way to assign putative functions to genes sequenced by metagenomics efforts. Once rubric criteria are defined, a structural metagenomics approach is also automated and can refine functional annotations for a range of gut bacterial protein families23,49,54–56. We show here that an integrated approach extending beyond EC classifications to include structural metagenomics approaches and examples in extant literature provides a much richer view of enzyme families, which in our case defines a set of 159 unique bacterial strains either suggested or confirmed to contain AzoR genes (Figure 4a). While IBD patients contained more AzoR-encoding bacteria overall than healthy individuals, a trend also reflected in Proteobacteria and Firmicutes, total relative abundances for the majority of individuals still fell below 3% and were often only a fraction of a percent (Figure 4b–e). Such abundance values fall below the lower thresholds typically considered in gut metagenomic studies and are often excluded from such analyses57. We therefore conclude that AzoRs are sparse in the gut microbiome in both health and disease, which is further supported by the lack of AzoRs in the complementary metatranscriptomics and metaproteomics datasets. Together, these findings suggest that azo-linked prodrugs may show poor efficacies and/or idiopathic responses in IBD due in part to low levels of AzoR enzymes and microbial taxa capable of producing the proteins. As shown recently, there are also non-enzymatic processes that reduce azo linkages in the gut in a manner dependent on microbial hydrogen sulfide, further suggesting that the clinical significance of bacterial AzoR enzymes may indeed be limited25.
In summary, we define a set of 196 intracellular AzoRs associated with the human gut microbiome and highlight the structural basis for functional differences in the processing of small and large azo-dyes and an azo-linked IBD prodrug. The differential and sparse landscape of these enzymes in the human gut in health and disease suggests that relying on azo-linked prodrug activation for the colonic delivery of therapeutics is likely to remain problematic, even given the higher abundance of AzoR-encoding bacteria in IBD patients compared to healthy controls. It is possible, however, that integrating our growing understanding of enzymatic and non-enzymatic processes associated with azo-linked compounds may lead to improved chemotypes and/or approaches that deliver more effective and less idiopathic treatments for IBD and other disorders.
Methods details
Identification and characterization of AzoR sequences
Metagenomic amino acid sequences of the IGC were each aligned pairwise to four representative AzoR enzymes with reported crystal structures (Ec PDB:2D5I, Pa PDB: 2V9C, Pp PDB: 4C0W, Kp PDB: 6DXP) using Protein–Protein BLAST (BLASTP v2.5.0+) as described in Figure 1b58. Candidate sequences with ≥ 25% identity to any representative AzoR enzyme were then assessed for the presence of five conserved residues (Figure 1b)56. Sequences that both met the identity threshold and contained all five conserved residues were accepted as AzoR enzymes. Accepted sequences were filtered for redundancies at a sequence identity threshold of 95% using CD-HIT (v4.8.1). This representative set of AzoR sequences used to determine the phylogenetic distribution of unique AzoR sequences across the IGC, which was represented using Graphpad PRISM (v9.3.1)47. Taxonomy was assigned to representative AzoR sequences using BLASTP (v2.5.0+) as reported previously, and taxonomic identifiers were used to rename these sequences56,58. FASTA headers of AzoR sequences were cross-referenced with extant IGC population metadata to determine AzoR occurrence per individual by phyla, which was represented using Graphpad PRISM (v9.3.1). Representative sequences were clustered and represented as sequence similarity networks (Supplemental Figure S5) using the Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST), which was also used to determine the sequence length that was represented as a histogram using R (v4.1.2), as shown in Figure 4c and Supplemental Figure S859–61. The predicted function of putative AzoR proteins was determined using the “evolutionary genealogy of genes: Non-supervised Orthologous Groups” (eggNOG v5.0) database62.
IBDMDB data retrieval
Data associated with analysis of the IBDMDB, including all data shown in Supplemental Figure S5, was directly retrieved from https://ibdmdb.org/tunnel/public/summary.html42, then filtered for EC number 1.7.1.6 (azobenzene reductase).
IBDMDB metaproteomic analysis
Processed IBDMDB metaproteomics data did not contain AzoR proteins when protein function was assigned using EC annotations42. To confirm the absence of AzoR proteins, raw IBDMDB metaproteomics files (directly retrieved from https://ibdmdb.org/tunnel/public/summary.html) were re-analyzed using all AzoR protein sequences identified using structural metagenomics as the reference database using proteomics data analysis methods as described previously55,63.
Protein expression and purification
All AzoR genes were codon-optimized for E. coli expression, synthesized, and ligated into a pLIC-His vector, then purchased from Bio Basic. The vectors were each transformed into chemically competent BL21-Gold (DE3) E. coli cells then grown on LB agar with ampicillin (100 μg/mL) at 37°C overnight. A single colony was selected and grown overnight in 100 mL of LB broth with ampicillin (100 μg/mL) at 37°C and shaking at 215 RPM. After reaching saturation, 50 mL of the culture was added to 1 L of LB broth with ampicillin (100 μg/mL), ~3 μL Antifoam 204, and 500 μM FMN. The culture was incubated at 37°C and 215 RPM until it reached an OD of 0.6 at 600 nm. After reaching the target OD, 1-thio-β-D-galactopyranoside (IPTG; 100 μM) was added to induce protein expression, the temperature was lowered to 18°C, and the culture was incubated overnight. The cells were collected by centrifugation at 4,500 × g at 4°C in a Sorvall (model RC-3B). Cell pellets were resuspended in 35 mL Purification Buffer A (20 mM Potassium Phosphate, 50 mM imidazole, 500 mM NaCl, 50 μM FMN, pH 8.0) with DNase, lysozyme, and a complete-EDTA-free protease inhibitor tablet (Roche). Resuspended cells were sonicated and clarified via centrifugation at 17,000 × g for 60 min in a Sorvall (model RC-5B). The lysate was flowed over a Ni-NTA HP column (GE Healthcare) then loaded onto the Aktaxpress FPLC system (Amersham Bioscience) and washed with Purification Buffer A. Protein was eluted with Buffer B (20 mM Potassium Phosphate, 250 mM Imidazole, 500 mM NaCl, 50 μM FMN, pH 8.0). Fractions containing the protein of interest were concatenated then passed through a HiLoad 16/60 Superdex 200 gel-filtrationcolumn (GE Life Sciences). Protein was eluted in S200 buffer (20 mM HEPES, 50 mM NaCl, 50 μM FMN, pH 8.0). Fractions containing the protein of interest were analyzed via SDS-PAGE, then those with>95% purity were combined and concentrated to ~10 mg/mL using 20 kDa cutoff molecular weight centrifuge concentrators (EMDMillipore). Samples were snap-frozen using liquid nitrogen and stored at −80°C.
CmAzoR crystallography
Crystals of CmAzoR were obtained via the hanging-drop vapor diffusion method. Crystals were produced in a condition containing 0.1 M HEPES pH 7.5 and 30% PEG 8000, and CmAzoR diluted in assay buffer (20 mM Tris-HCl, pH 8.0) to a concentration of 10 mg/mL before combining with the crystallization condition. Trays were incubated at 20°C, and crystals formed after four days. Crystals were cryo-protected in the crystallization condition with 20% glycerol. Diffraction data for the crystals were collected at 100 K at GM-Ca-CAT 23-ID-B (Advanced Photon Source, Argonne National Laboratory). CCP4i was used to scale the raw data to 1.4 Å resolution, and Phenix (v.1.17) was used to refine the model to the statistics shown in Supplemental Figure S8. Interestingly, an aromatic tyrosine residing in the N-terminal Histidine affinity tag used to purify CmAzoR was observed to have swapped from a neighboring molecule into the active site in our refined structure. To confirm that the affinity tag did not interfere with enzyme activity in vitro, we cleaved the tag using an established tobacco etch virus (TEV) protease system64. We then repurified the enzyme and found that the catalytic efficiencies for the TEV-cleaved version of the enzyme were equivalent to those of the enzyme with the affinity tag. Thus, the presence of the affinity tag at the active site appears to be a crystal packing artifact (Supplemental Table S1).
CmAzoR affinity tag removal
CmAzoR was purified as described in “Protein Expression and Purification” up to the point which protein was eluted with Buffer B (20 mM Potassium Phosphate, 250 mM Imidazole, 500 mM NaCl, 50 μM FMN, pH 8.0). Fractions of protein were pooled and then placed in dialysis tubing with recombinant Tobacco Etch Virus Protease (rTEV; 100 μL rTEV/1 mL CmAzoR). The dialysis tubing apparatus was placed in 200 mL rTEV cleavage buffer (20 mM Tris-HCl, 50 mM NaCl, 50 mM Imidazole, 3 mM β-mercaptoethanol, pH 8.0), which was incubated at 4°C overnight while stirring. The following day, CmAzoR was separated from rTEV using size-exclusion chromatography and eluted in S200 buffer (20 mM HEPES, 50 mM NaCl, 50 μM FMN, pH 8.0). Fractions containing the protein of interest were analyzed via SDS-PAGE to confirm the absence of rTEV, then those with >95% purity were combined and concentrated to ~10 mg/mL using 20 kDa cutoff molecular weight centrifuge concentrators (EMDMillipore). Samples were snap-frozen using liquid nitrogen and stored at −80°C.
CmAzoR and EcAzoR Ligand modeling
The completed structure of CmAzoR was aligned to EcAzoR (PDB: 2D5I) and PaAzoR (PDB: 2V9C) using the cealign plugin in PyMOL (v2.5.2)65. Amaranth and Sulfasalazine were manually docked into the enzyme active site in PyMOL using the crystal structure of PaAzoR bound to Methyl Red as a reference scaffold, as there are no extant structures of amaranth or sulfasalazine bound to AzoR proteins.
Azo dye kinetic assays
Continuous Absorbance assays were conducted in 96-well UV-transparent plates (Corning) using a CLARIOstar Plus Microplate Reader at 37°C. Initial conditions were NAD(P)H (1 mM), assay buffer (Tris-HCl 20 mM, pH 8.0), AzoR (500 nM), and substrate (20–50 μM suspended in 50% DMSO). Total reaction volume was 200 μL, and reaction mixtures excluding Azo dye were incubated for 10 min in the plate reader at 37°C to establish a baseline absorbance. Azo dye was added to initiate the reactions. Absorbance of methyl red was detected at 410 nm, and absorbance of amaranth was detected at 520 nm. After the reactions plateaued, recording ceased. Absorbances were converted to concentrations using a standard curve, and the rates of initial loss of substrate absorbance were determined. These velocities at a single substrate concentration were transformed into apparent kcat (s−1) values, then the experiment was conducted for at least three additional substrate concentrations. Rate was plotted as a function of substrate concentration, the slope of which represents catalytic efficiency (kcat/KM in units of s−1M−1).
Sulfasalazine kinetic assays
Sulfasalazine was purchased as a solid (VWR; CAS #: 599-79-1) and suspended in 100% DMSO to a concentration of 10 mM. Assays were conducted in 96-well, clear bottom assay plates (Costar) at a total volume of 50 μL. Reactions consisted of NADH (1 mM), assay buffer Tris-HCl 20 mM, pH 8.0), AzoR (500 nM), and substrate (50 μM suspended in 50% DMSO). After the addition of substrate, reactions were incubated at 37°C. Five wells were prepared in total, which were individually quenched with an equivalent volume of 25% trichloroacetic acid every 15 min over the course of 1 hr, beginning at 0 min. After all wells were quenched, reactions were transferred to epi-tubes then centrifuged at 13,000 × g for 20 min. 80 μL of the supernatant was subjected to analysis via liquid chromatography-mass spectrometry (LC-MS/MS). Analysis of the reaction mixture was conducted on an Agilent 1260 Infinity II liquid chromatography system, and separated with an Agilent InfinityLab Poroshell 120 C18 column (4.6 × 100 mm, 2.7-μm particle size) at 38°C. The flow rate was 0.6 ml/min with an injection volume of 5 μL. Reverse-phase gradient elution was conducted using the following solvents: 98% water with 0.012% formic acid and 5 mM ammonium acetate (Solvent A), and methanol (HPLC-grade) with 0.012% formic acid and 5 mM ammonium acetate (Solvent B). Elution gradient was as follows: 90% A/10% B, 0–5 min: linearly ramp to 10% A/90% B; 5–8 min linearly ramp to 90% A/10% B, 8–9 min: 90% A/10% B. Sulfasalazine eluted at 5.6 min and was quantified using the acquisition parameters and m/z quantification values shown in Supplemental Table S3. Sulfasalazine was detected using MS/MS in multiple reaction monitoring mode using positive polarity. The source gas temperature was 325ºC at 10 L/min flow rate. The sheath gas temperature was 400ºC at 12 L/min. The capillary voltage was −3500 V. Nebulizer pressure was 45 psi. The AUC for Sulfasalazine was converted to the concentration of analyte from a standard curve collected before the start of the reaction (0–100 μM of Sulfasalazine in assay buffer). Rate of loss of Sulfasalazine over the course of an hour was determined in units of 1/s. The final value represents the average of three biological replicates. No degradation of Sulfasalazine was observed in the presence of heat-killed enzyme.
Supplementary Material
Acknowledgments
We like to thank current and previous Redinbo lab members for helpful discussions. Figure 1a,b were created with BioRender.com. We would also like to acknowledge that a manuscript analyzing AzoR enzymes was published while this manuscript was under review, titled “Gut microbiome-wide search for bacterial azoreductases reveals potentially uncharacterized azoreductases encoded in the human gut microbiome” DOI: 10.1124/DMD.122.000898.
Funding Statement
This work was supported by NIH grants GM137286 and GM135218 (M.R.R.) and by NSF DGE-1650116 (J.B.S).
Disclosure statement
M.R.R. is a founder of Symberix, Inc., which is developing microbiome-targeted therapeutics. M.R.R. also receives research funding from Merck and Eli Lilly.
Author contributions
J.B.S., B.S.C., and M.R.R. conceptualization; J.B.S., B.S.C, and J.J.S. data curation; J.B.S. formal analysis; J.B.S., B.S.C., and J.J.S. investigation; J.B.S., B.S.C. and M.R.R. writing – original draft; J.B.S., B.S.C., J.J.S. and M.R.R. writing – review and editing; J.B.S. and M.R.R. supervision; J.B.S., B.S.C., and J.J.S. methodology; J.B.S. and M.R.R. funding acquisition; M.R.R. project administration.
Data availability statement
The datasets supporting the conclusions of this article are included within the article and its additional files.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2203963
References
- 1.Dembitsky VM, Gloriozova TA, Poroikov V.. Pharmacological and predicted activities of natural azo compounds. Nat Prod Bioprospect. 2017;7(1):151–17. doi: 10.1007/s13659-016-0117-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sousa T, Yadav V, Zann V, Borde A, Abrahamsson B, Basit AW. On the colonic bacterial metabolism of azo-bonded prodrugs of 5-aminosalicylic acid. J Pharm Sci. 2014;103(10):3171–3175. doi: 10.1002/JPS.24103. [DOI] [PubMed] [Google Scholar]
- 3.Koppel N, Rekdal VM, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science (1979). 2017;356(6344):1246–1257. doi: 10.1126/science.aag2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan AKA, Piris J, Truelove SC. AN experiment to determine the active therapeutic moiety of sulphasalazine. Lancet. 1977;310(8044):892–895. doi: 10.1016/S0140-6736(77)90831-5. [DOI] [PubMed] [Google Scholar]
- 5.Misal SA, Gawai KR. Azoreductase: a key player of xenobiotic metabolism. Bioresour Bioprocess [Internet]. 2018;5(1). doi: 10.1186/s40643-018-0206-8. [DOI] [Google Scholar]
- 6.Ryan A. Azoreductases in drug metabolism. Br J Pharmacol. 2017;174(14):2161–2173. doi: 10.1111/bph.13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Campos Ventura-Camargo B, M AMM. 2013. Azo dyes: characterization and toxicity-a review. [place unknown]. www.tlist-journal.org
- 8.Chung KT, Stevens SE, Cerniglia CE. The reduction of azo dyes by the intestinal microflora. Crit Rev Microbiol. 1992;18(3):175–190. doi: 10.3109/10408419209114557. [DOI] [PubMed] [Google Scholar]
- 9.Sun J, Jin J, Beger RD, Cerniglia CE, Chen H. Evaluation of metabolism of azo dyes and their effects on Staphylococcus aureus metabolome. J Ind Microbiol Biotechnol [Internet]. 2017. 2022 May11;44(10):1471–1481. doi: 10.1007/s10295-017-1970-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zahran SA, Ali-Tammam M, Hashem AM, Aziz RK, Ali AE. Azoreductase activity of dye-decolorizing bacteria isolated from the human gut microbiota. Sci Rep [Internet]. 2019;9(1):1–14. doi: 10.1038/s41598-019-41894-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rafii F, Cerniglia CE. Reduction of azo dyes and nitroaromatic compounds by bacterial enzymes from the human intestinal tract. Environ Health Perspect. 1995;103(SUPPL. 5):17–19. doi: 10.1289/ehp.95103s417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crespi S, Simeth NA, König B. Heteroaryl azo dyes as molecular photoswitches. Nat Rev Chem [Internet]. 2019. 2022 Nov 1;33(33):133–146. doi: 10.1038/s41570-019-0074-6. [DOI] [Google Scholar]
- 13.Jerca FA, VV J, Hoogenboom R. Advances and opportunities in the exciting world of azobenzenes. Nat Rev Chem [Internet]. 2021. 2022 Nov 1;66(11):51–69. doi: 10.1038/s41570-021-00334-w. [DOI] [PubMed] [Google Scholar]
- 14.Ahlström L-H, Sparr Eskilsson C, Björklund E. Determination of banned azo dyes in consumer goods. TrAc Trends Anal Chem. 2005;24(1):49–56. doi: 10.1016/j.trac.2004.09.004. [DOI] [Google Scholar]
- 15.Nagaraja TN, Desiraju T. Effects of chronic consumption of metanil yellow by developing and adult rats on brain regional levels of noradrenaline, dopamine and serotonin, on acetylcholine esterase activity and on operant conditioning. Food Chem Toxicol. 1993;31(1):41–44. doi: 10.1016/0278-6915(93)90177-Z. [DOI] [PubMed] [Google Scholar]
- 16.Mpountoukas P, Pantazaki A, Kostareli E, Christodoulou P, Kareli D, Poliliou S, Mourelatos C, Lambropoulou V, Lialiaris T. Cytogenetic evaluation and DNA interaction studies of the food colorants amaranth, erythrosine and tartrazine. Food Chem Toxicol. 2010;48(10):2934–2944. doi: 10.1016/j.fct.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 17.Khehra MS, Saini HS, Sharma DK, Chadha BS, Chimni SS. Biodegradation of azo dye C.I. acid red 88 by an anoxic - Aerobic sequential bioreactor. Dyes and Pigments. 2006;70(1):1–7. doi: 10.1016/j.dyepig.2004.12.021. [DOI] [Google Scholar]
- 18.Pinheiro HM, Touraud E, Thomas O. Aromatic amines from azo dye reduction: status review with emphasis on direct UV spectrophotometric detection in textile industry wastewaters. Dyes and Pigments. 2004;61(2):121–139. doi: 10.1016/j.dyepig.2003.10.009. [DOI] [Google Scholar]
- 19.Wang CJ, Laurieri N, Abuhammad A, Lowe E, Westwood I, Ryan A, Sim E. Role of tyrosine 131 in the active site of paAzor1, an azoreductase with specificity for the inflammatory bowel disease prodrug balsalazide. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;66(1):2–7. doi: 10.1107/S1744309109044741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen NH, Fumery M, Dulai PS, Prokop LJ, Sandborn WJ, Murad MH, Singh S. Comparative efficacy and tolerability of pharmacological agents for management of mild to moderate ulcerative colitis: a systematic review and network meta-analyses. Lancet Gastroenterol Hepatol. 2018;3(11):742–753. doi: 10.1016/S2468-1253(18)30231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ervin SM, Redinbo MR. The gut microbiota impact cancer etiology through “Phase IV Metabolism” of Xenobiotics and endobiotics. Cancer Prev Res (Phila). 2020;13(8):635–642. doi: 10.1158/1940-6207.CAPR-20-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.le Berre C, Roda G, Nedeljkovic Protic M, Danese S, Peyrin-Biroulet L. Modern use of 5-aminosalicylic acid compounds for ulcerative colitis. Expert Opin Biol Ther. 2020;20(4):363–378. doi: 10.1080/14712598.2019.1666101. [DOI] [PubMed] [Google Scholar]
- 23.Walker ME, Simpson JB, Redinbo MR. A structural metagenomics pipeline for examining the gut microbiome. Curr Opin Struct Biol Internet. 2022;75:102416. doi: 10.1016/j.sbi.2022.102416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural history of adult ulcerative colitis in population-based cohorts: a systematic review. Clin Gastroenterol Hepatol. 2018;16(3):343–356.e3. doi: 10.1016/j.cgh.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolfson SJ, Hitchings R, Peregrina K, Cohen Z, Khan S, Yilmaz T, Malena M, Goluch ED, Augenlicht L, Kelly L. Bacterial hydrogen sulfide drives cryptic redox chemistry in gut microbial communities. Nat Metabolism [Internet]. 2022. Oct 24;44(10):10–1270 1260–1270. doi: 10.1038/s42255-022-00656-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui K, Lu AYH, Yang CS. Subunit functional studies of NAD(P)H: quinone oxidoreductase with a heterodimer approach. Proc Natl Acad Sci U S A. 1995;92(4):1043–1047. doi: 10.1073/pnas.92.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryan A, Laurieri N, Westwood I, Wang CJ, Lowe E, Sim E. A novel mechanism for azoreduction. J Mol Biol. 2010;400(1):24–37. doi: 10.1016/j.jmb.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Ryan A, Wang C, Laurieri N, Westwood I, Sim E. Reaction mechanism of azoreductases suggests convergent evolution with quinone oxidoreductases. Protein Cell. 2010;1(8):780–790. doi: 10.1007/s13238-010-0090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan A, Kaplan E, Laurieri N, Lowe E, Sim E. Activation of nitrofurazone by azoreductases: multiple activities in one enzyme. Sci Rep. 2011;1(1):1–5. doi: 10.1038/srep00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bin Y, Jiti Z, Jing W, Cuihong D, Hongman H, Zhiyong S, Yongming B. Expression and characteristics of the gene encoding azoreductase from Rhodobacter sphaeroides AS1.1737. FEMS Microbiol Lett [Internet]. 2004. 2022 Oct 31;236(1):129–136. doi: 10.1111/j.1574-6968.2004.tb09638.x. [DOI] [PubMed] [Google Scholar]
- 31.Song ZY, Zhou JT, Wang J, Yan B, Du CH. Decolorization of azo dyes by Rhodobacter sphaeroides. Biotechnol Lett. 2003;25(21):1815–1818. doi: 10.1023/A:1026244909758. [DOI] [PubMed] [Google Scholar]
- 32.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature [Internet]. 2019. December 2018;1–8. doi: 10.1038/s41586-019-1291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He Z, Chen L, Catalan-Dibene J, Bongers G, Faith JJ, Suebsuwong C, DeVita RJ, Shen Z, Fox JG, Lafaille JJ, et al. Food colorants metabolized by commensal bacteria promote colitis in mice with dysregulated expression of interleukin-23. Cell Metab. 2021;33(7):1358–1371.e5. doi: 10.1016/j.cmet.2021.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng J, Cerniglia CE, Chen H. Toxicological significance of azo dye metabolism by human intestinal microbiota. [place unknown]. [DOI] [PMC free article] [PubMed]
- 35.Yamjala K, Nainar MS, Ramisetti NR. Methods for the analysis of azo dyes employed in food industry - a review. Food Chem. 2016;192:813–824. doi: 10.1016/j.foodchem.2015.07.085. [DOI] [PubMed] [Google Scholar]
- 36.Yu J, Ogata D, Gai Z, Taguchi S, Tanaka I, Ooi T, Yao M. Structures of AzrA and of AzrC complexed with substrate or inhibitor: insight into substrate specificity and catalytic mechanism. Acta Crystallogr D Biol Crystallogr. 2014;70(2):553–564. doi: 10.1107/S1399004713030988. [DOI] [PubMed] [Google Scholar]
- 37.Gonçalves AMD, Mendes S, de Sanctis D, Martins LO, Bento I. The crystal structure of Pseudomonas putida azoreductase - the active site revisited. FEBS J. 2013;280(24):6643–6657. doi: 10.1111/febs.12568. [DOI] [PubMed] [Google Scholar]
- 38.Wang C, Hagemeier C, Rahman N, Lowe E, Noble M, Coughtrie M, Sim E, Westwood I. Molecular cloning, characterisation and ligand-bound structure of an azoreductase from Pseudomonas aeruginosa. J Mol Biol. 2007;373(5):1213–1228. doi: 10.1016/j.jmb.2007.08.048. [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Wang RF, Cerniglia CE. Molecular cloning, overexpression, purification, and characterization of an aerobic FMN-dependent azoreductase from Enterococcus faecalis. Protein Expr Purif. 2004;34(2):302–310. doi: 10.1016/j.pep.2003.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med [Internet]. 2016;8(1):1–11. doi: 10.1186/s13073-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knight R, Callewaert C, Marotz C, Hyde ER, Debelius JW, Mcdonald D, Sogin ML. The Microbiome and Human Biology. Annu Rev Genomics Hum Genet. 2017;18:65–86. doi: 10.1146/annurev-genom-083115-022438. [DOI] [PubMed] [Google Scholar]
- 42.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito K, Nakanishi M, Lee WC, Sasaki H, Zenno S, Saigo K, Kitade Y, Tanokura M. Three-dimensional structure of AzoR from Escherichia coli: an oxidereductase conserved in microorganisms. J Biol Chem. 2006;281(29):20567–20576. doi: 10.1074/jbc.M513345200. [DOI] [PubMed] [Google Scholar]
- 44.Agarwal R, Bonanno JB, Burley SK, Swaminathan S. Structure determination of an FMN reductase from Pseudomonas aeruginosa PA01 using sulfur anomalous signal. Acta Crystallogr D Biol Crystallogr. 2006;62(4):383–391. doi: 10.1107/S0907444906001600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tedeschi G, Deng PSK, Chen HH, Forrest GL, Massey V, Chen S. A site-directed mutagenesis study at Lys-113 of NAD(P)H: quinone-acceptor oxidoreductase: an involvement of Lys-113 in the binding of the flavin adenine dinucleotide prosthetic group. Arch Biochem Biophys. 1995;321(1):76–82. doi: 10.1006/abbi.1995.1370. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32(8):834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 47.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 48.Biernat KA, Pellock SJ, Bhatt AP, Bivins MM, Walton WG, Tran BNT, Wei L, Snider MC, Cesmat AP, Tripathy A, et al. Structure, function, and inhibition of drug reactivating human gut microbial β-glucuronidases. Sci Rep. 2019;9(1):1–15. doi: 10.1038/s41598-018-36069-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ervin SM, Simpson JB, Gibbs ME, Creekmore BC, Lim L, Walton WG, Gharaibeh RZ, Redinbo MR. Structural insights into endobiotic reactivation by human gut microbiome-encoded sulfatases. Biochemistry. 2020;59(40):3939–3950. doi: 10.1021/acs.biochem.0c00711. [DOI] [PubMed] [Google Scholar]
- 50.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res [Internet]. 2016. 2022 Nov 1;44(D1):D457–462. doi: 10.1093/NAR/GKV1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye J, Yang HC, Rosen BP, Bhattacharjee H. Crystal structure of the flavoprotein ArsH from Sinorhizobium meliloti. FEBS Lett. 2007;581(21):3996–4000. doi: 10.1016/J.FEBSLET.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Binter A, Staunig N, Jelesarov I, Lohner K, Palfey BA, Deller S, Gruber K, Macheroux P. A single intersubunit salt bridge affects oligomerization and catalytic activity in a bacterial quinone reductase. FEBS J [Internet]. 2009. 2022 Sep 14;276(18):5263–5274. doi: 10.1111/j.1742-4658.2009.07222.x. [DOI] [PubMed] [Google Scholar]
- 53.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science (1979) [Internet]. 2012. 2022 May 11;336(6086):1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 54.Simpson JB, Redinbo MR. Multi-omic analysis of host-microbial interactions central to the gut-brain axis. Mol Omics [Internet] 2022. 2022 Oct 24;18(10):896–907. doi: 10.1039/D2MO00205A. [DOI] [PubMed] [Google Scholar]
- 55.Simpson JB, Sekela JJ, Graboski AL, Borlandelli VB, Bivins MM, Barker NK, Sorgen AA, Mordant AL, Johnson RL, Bhatt AP, et al. Metagenomics combined with activity-based proteomics point to gut bacterial enzymes that reactivate mycophenolate. Gut Microbes [Internet]. 2022. 2022 Sep 12;14(1): 10.1080/19490976.2022.2107289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pollet RM, D’Agostino EH, Walton WG, Xu Y, Little MS, Biernat KA, Pellock SJ, Patterson LM, Creekmore BC, Isenberg HN, et al. An Atlas of β-Glucuronidases in the human intestinal microbiome. Structure [Internet]. 2017;25(7):967–977.e5. doi: 10.1016/j.str.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de CJ, Zhang J, Deng D, Damé-Teixeira N, Do T. Low-abundant microorganisms: the human microbiome’s dark matter, a scoping review. Front Cell Infect Microbiol. 2021;11:11. doi: 10.3389/fcimb.2021.689197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009;10:10. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wickham H. Ggplot2: elegant graphics for data analysis. place unknown: Springer-Verlag; 2016. [Google Scholar]
- 60.Yu G, Smith DK, Zhu H, Guan Y, Lam TTY, McInerny G. Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8(1):28–36. doi: 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- 61.Kanz C, Aldebert P, Althorpe N, Baker W, Baldwin A, Bates K, Browne P, van den Broek A, Castro M, Cochrane G, et al. The EMBL nucleotide sequence database. Nucleic Acids Res. 2005;33(DATABASE ISS.):D29–33. doi: 10.1093/nar/gki098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al. EggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jariwala PB, Pellock SJ, Cloer EW, Artola M, Simpson JB, Bhatt AP, Walton WG, Roberts LR, Davies GJ, Overkleeft HS, et al. Discovering the microbial enzymes driving drug toxicity with activity-based protein profiling. ACS Chem Biol. 2020;15(1):217–225. doi: 10.1021/acschembio.9b00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raran-Kurussi S, Cherry S, Zhang D, Waugh DS. Removal of affinity tags with TEV protease. Methods in Molecular BiologyVol. 1586, [place unknown], Humana Press Inc.;2017. pp. 221–230. 10.1007/978-1-4939-6887-9_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pellock SJ, Walton WG, Ervin SM, Torres-Rivera D, Creekmore BC, Bergan G, Dunn ZD, Li B, Tripathy A, Redinbo MR. Discovery and characterization of FMN-Binding β-Glucuronidases in the human gut microbiome. J Mol Biol. 2019;431(5):970–980. doi: 10.1016/j.jmb.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.