

Abstract
The global burden of bacterial infections is rising due to increasing resistance to the majority of first-line antibiotics, rendering these drugs ineffective against several clinically important pathogens. Limited transport of antibiotics into cells compounds this problem for gram-negative bacteria that exhibit prominent intracellular lifecycles. Furthermore, poor bioavailability of antibiotics in infected tissues necessitates higher doses and longer treatment regimens to treat resistant infections. Although emerging antibiotics can combat these problems, resistance still may develop over time. Expanding knowledge of host-pathogen interactions has inspired research and development of host-directed therapies (HDTs). HDTs target host-cell machinery critical for bacterial pathogenesis to treat bacterial infections alone or as adjunctive treatment with traditional antibiotics. Unlike traditional antibiotics that directly affect bacteria, a majority of HDTs function by boosting the endogenous antimicrobial activity of cells and are consequently less prone to bacterial tolerance induced by selection pressure. Therefore, HDTs can be quite effective against intracellular cytosolic or vacuolar bacteria, which a majority of traditional antibiotics are unable to eradicate. However, in vivo therapeutic efficacy of HDTs is reliant on adequate bioavailability. Particle-based formulations demonstrate the potential to enable targeted drug delivery, enhance cellular uptake, and increase drug concentration in the host cell of HDTs. This review selected HDTs for clinically important pathogens, identifies formulation strategies that can improve their therapeutic efficacy and offers insights toward further development of HDTs for bacterial infections.
Keywords: Host-directed therapies, biomaterials, formulations, drug delivery
Introduction
Antimicrobial resistance is one of the biggest healthcare challenges of the 21st century, with close to 2.8 million resistant infections and 35,000 associated deaths occurring in the United States.1 These figures are likely higher in low and middle income countries where surveillance to resistant strains is difficult to account.1 Indeed, the WHO recently reported that close to 51% and 65% infections are resistant to common first and second line antibiotics, penicillin and ciprofloxacin, respectively, in several countries.2
While a variety of guiding factors are utilized to determine the choice of antibiotic for treating bacterial infections, initial treatment of common infections (e.g., tonsillitis, urinary tract infections, rhinosinusitis) involves use of first line antibiotics with broad-spectrum activity and a low toxicity profile. Several β lactams (e.g., penicillin, cephalosporin), macrolides (e.g., erythromycin and azithromycin) and tetracyclines (e.g., doxycycline, oxytetracycline) are among routinely used first line therapies. Second and third choice treatments use antibiotics deemed effective against specific or multi-drug resistant bacteria.3–5 Although, first-line antibiotics were the first class to be challenged with resistance, currently, several second- and third-line treatments are equally at risk.5 According to a recent WHO report, in addition to the tuberculosis (TB) causing microbe Mycobacterium tuberculosis (M. tb), some of the most commonly reported resistant bacteria include, Escherichia coli, and Salmonella spp.2 E. coli is an example of an extended-spectrum beta-lactamase producing bacteria classified as a serious threat by the CDC.1 Extended-spectrum beta-lactamase degrade common antibiotics such as penicillin and cephalosporin, making these bacterial infections extremely difficult to treat.6 Also, Salmonella serovars are a growing threat globally, with 10% and 74% of Non-Typhoidal Salmonella and Typhoidal Salmonella resistant to ciprofloxacin, respectively.5 While both gram-positive and gram-negative bacteria show antibiotic resistance, the gram-negative pathogens are more notorious to treat due to a cell wall that excludes amphipathic drugs as well as inner membranes and efflux pumps that exclude hydrophilic drugs. As a result, antibiotics often have to be administered at concentrations orders of magnitude higher than other therapeutics that target host cells, generating toxicity concerns.3 Moreover, several pathogens invade, proliferate and survive within host cells, making them even less accessible to treatment.7 The threat posed by the resistance of bacteria to a range of traditional antibiotics and the shortcomings of traditional antibiotic therapy for intracellular pathogens illustrates the urgent need for new treatment modalities.
While the development of new classes of antibiotics is one obvious path toward combating antibiotic resistance, host-directed therapies (HDTs) that target host-encoded functions necessary for bacterial infection, replication, virulence, and pathogenesis have the potential to be an alternative or adjunctive approach to conventional antimicrobials.2–4 In contrast to antibiotics that directly act on bacteria, HDTs enhance a potentially broad response against the bacteria, rendering them less prone to microbial tolerance. Bacteria can overcome the body’s natural defense systems, using countless different methods to escape recognition or clearance by the immune system. While bacteria-host interactions largely depend on the species, some bacteria (e.g., Salmonella spp., Francisella spp.) are facultative or obligate intracellular pathogens that reside in the host cell environment.8 In order to reside in the cell, often within acidic vesicles, the bacteria must usurp host cell processes such as autophagy. The vast knowledge of strategies used by pathogens to influence key machinery within host cells has inspired the development of HDTs. While a majority of these drugs are novel small molecules or peptides, several FDA-approved drugs developed for other indications have been repurposed as effective HDTs. Some examples include metformin, aspirin and glibenclamide.9 Additionally, emerging research indicates that HDTs might be most effective when co-administered with traditional antibiotics, and show potential to reduce the antibiotic dose necessary to treat drug resistant bacteria.10,11 Table 1 highlights HDTs identified against select pathogens of large clinical importance (Table 1). Although the role of HDTs as an adjunctive therapy against bacterial infections is being strongly recognized, the field is in its nascency and limited research groups have explored formulating HDTs and advanced delivery techniques for anti-bacterial applications (Table 2). Nonetheless, it is promising to note the progress made so far, especially by combination treatments of HDTs with standard antibiotics due to their potential for clinical translation. In this review, we discuss select HDTs that are being developed for clinically relevant bacteria and discuss the advantages of formulating HDTs, particularly for intracellular bacterial infections.
Table 1. Select HDTs developed for select intracellular bacteria.
HIF-1α, hypoxia inducible factor-1α; Akt, protein kinase B; TLR-3, toll-like receptor 3; Poly (I:C), Polyinosine:polycytosine; ROS, reactive oxygen species; ATRA, all-trans retinoic acid; NPC2, NPC intracellular cholesterol transporter; mTORC1, mammalian target of rapamycin complex 1
| Bacteria | HDTs | Antibacterial Mechanism | Ref |
|---|---|---|---|
| M. tb | Imatinib | Tyrosine kinase inhibitor | 12 |
| M. tb | Metformin | Mitochondrial ROS production | 13 |
| M. tb | Doxycycline | Matrix metalloproteinase inhibitor | 10 |
| M. tb | ATRA | Lysosomal acidification via NPC2 | 14 |
| M. tb | Nitazoxanide | Autophagy inducing via mTORC1 | 15 |
| M. tb | Dexamethasone | Unknown | 16 |
| E. coli | AKB 2924 | HIF-1α stabilizing agent | 17,18 |
|
E. coli
S. aureus |
Clavanin-MO | Leukocyte recruitment and immune mediator production | 19 |
| E. coli | Lactoferrin | Neutrophil recruitment | 20 |
|
S. Typhimurium S. Typhi F. tularensis |
AR-12 | Autophagy inducing | 21,22,24 |
| S. Typhimurium | Mibefradil Haloperidol |
Unknown | 23 |
| F. tularensis | Poly (I:C) | TLR3 agonist | 25 |
Table 2. Formulations of HDTs in development against select intracellular bacteria.
PLGA, poly lactic acid glycolic acid; Ace-DEX, acetalated dextran.
| Bacteria | Formulation | Fabrication Method | HDT | Drug Type | Formulation Delivery Route | Combination Therapy | Ref |
|---|---|---|---|---|---|---|---|
| M.tb | PLGA Particles | Spray drying | Nitazoxanide | Small molecule | Intranasal | Isoniazid and rifabutin | 26 |
| M. tb | PLGA Particles | Spray drying | All-trans retinoic acid | Small molecule | Intratracheal | Rifampicin | 27 |
| M. tb | PLGA Particles | Double emulsion, solvent evaporation | Magainin-I analog peptide | Peptide | Intranasal | Isoniazid | 28 |
| M. tb | PLGA Particles | Double emulsion, solvent evaporation | N/A | N/A | N/A | N/A | 29 |
| M. tb | Liposomes | Inverted emulsion | Phosphatidic acid | Lipids | Intranasal | Isoniazid | 30 |
|
E. Coli Klebsiella pneumoniae Pseudomonas aeruginosa Staphylococcus aureus Acinetobacter baumannii |
Liposomes | Inverted emulsion | Phosphatidylinositol-3-phosphate Phosphatidylinositol-5-phosphate Phosphatidic acid Arachidonic acid Sphingosine 1-phosphate Lysobisphosphatidic acid |
Lipids | N/A | N/A | 31 |
| E.coli | PLGA Particles | HHC10 | Peptide | N/A | N/A | 32 | |
| E.coli | Alginate Particles | Mannuronic acid in alginate | N/A | N/A | N/A | 33 | |
| F. tularensis | Ace-DEX Particles | Emulsion | AR-12 | Small molecule | Intranasal | Gentamicin | 34 |
| S. Typhimurium | Ace-DEX Particles | Emulsion | AR-12 | Small molecule | N/A | N/A | 35 |
| S. Typhi | Ace-DEX Particles | Emulsion | AR-12 | Small molecule | Intranasal | N/A | 36 |
Barriers to drug delivery: Need for HDTs formulations
Therapies delivered via oral, intravenous (i.v.) or intramuscular routes can achieve therapeutic serum concentrations; however, this does not ensure a sustained drug concentration in target tissues due to drug transport barriers.37 Therapies need to cross endothelial cells of the capillaries in addition to the epithelial cells of the GI tract (enteric infections), the tight cell layers of the stratum corneum (intradermal infections), blood brain barrier (meningeal infections) or the mucosa of the respiratory tract (pulmonary infections) to reach sites of infection.37,38 Furthermore, intracellular drug concentration also depends on the rate of drug excretion from the cells via efflux pumps and exocytosis.39 Formulating therapies to enable local and targeted delivery to the site of infection can help to greatly overcome these limitations. In general, drug formulations consist of the active drug and excipient carrier molecules that provide the packaging necessary to improve delivery of drugs to their site of action and impart the desired therapeutic effect. Formulating drugs into a delivery platform can significantly improve a variety of pharmacokinetic parameters, such as, maximum concentration (Cmax), time above minimum inhibitory concentration and the area under the curve for the drug (a measure of drug bioavailability and circulation time).40 Also they can offer protection for the drugs from physiological conditions, such as fluctuating pH and enzymatic activity. In addition to overcoming some of the above challenges, delivery platforms can enable controlled and targeted delivery of drugs as well as needle-free and/or local administration.40,41
Common drug delivery platforms
The site of infection and the length of treatment are factors that can influence the design of drug delivery platforms for bacterial infections. Accordingly, formulations can range from being multi-dose depots to drug loaded particulate systems that can protect the cargo while extending the circulating time in the body. Both organic (e.g. polymers, lipids) and inorganic (e.g. iron oxide, gold, silica) materials have been used to develop drug formulations.41 Considering a majority of inorganic formulations have been primarily the focus of cancer therapy and diagnostics, this review will focus on different types of organic formulations developed for HDTs.
Additionally, most formulations used for HDTs are nano- or microparticulate in structure. Particle-based delivery platforms are capable of passively targeting immune cells or the delivery location based on their size. As will be seen from example formulations highlighted in this review, delivery into the lungs can be achieved with larger porous microparticles (MPs), while particles with diameter in the 500 nm - 5 μm range are best suited to passively target phagocytes. Organic particulate formulation developed for HDTs can be grouped into two primary types: polymeric and liposomal.
Polymeric particles:
Most polymeric particles function by releasing therapeutics that are incorporated throughout the polymer matrix (Fig. 1). While drug release from slow degrading particles is initially by diffusion, the release rate can be controlled in many of the systems by tuning the polymer’s degradation rate. Polyesters, such as poly lactic-co-glycolic acid (PLGA), and polycaprolactone (PCL), are examples of slow release systems that have acidic by-products and degradation times on the order of months.42,43 In contrast, the biodegradable polymer acetalated dextran (Ace-DEX), formed from the FDA-approved polysaccharide dextran, has release rates on the order of days to months at neutral pH. Ace-DEX was synthesized by modifying the hydroxyl groups on dextran to create cyclic or acyclic acetals. The ratio between the two types of acetal groups determines the degradability of the polymer.44,45 Alginate is another biopolymer capable of forming ionically crosslinked microspheres and are typically utilized for faster drug release on the order of hours or days.46
Figure 1.
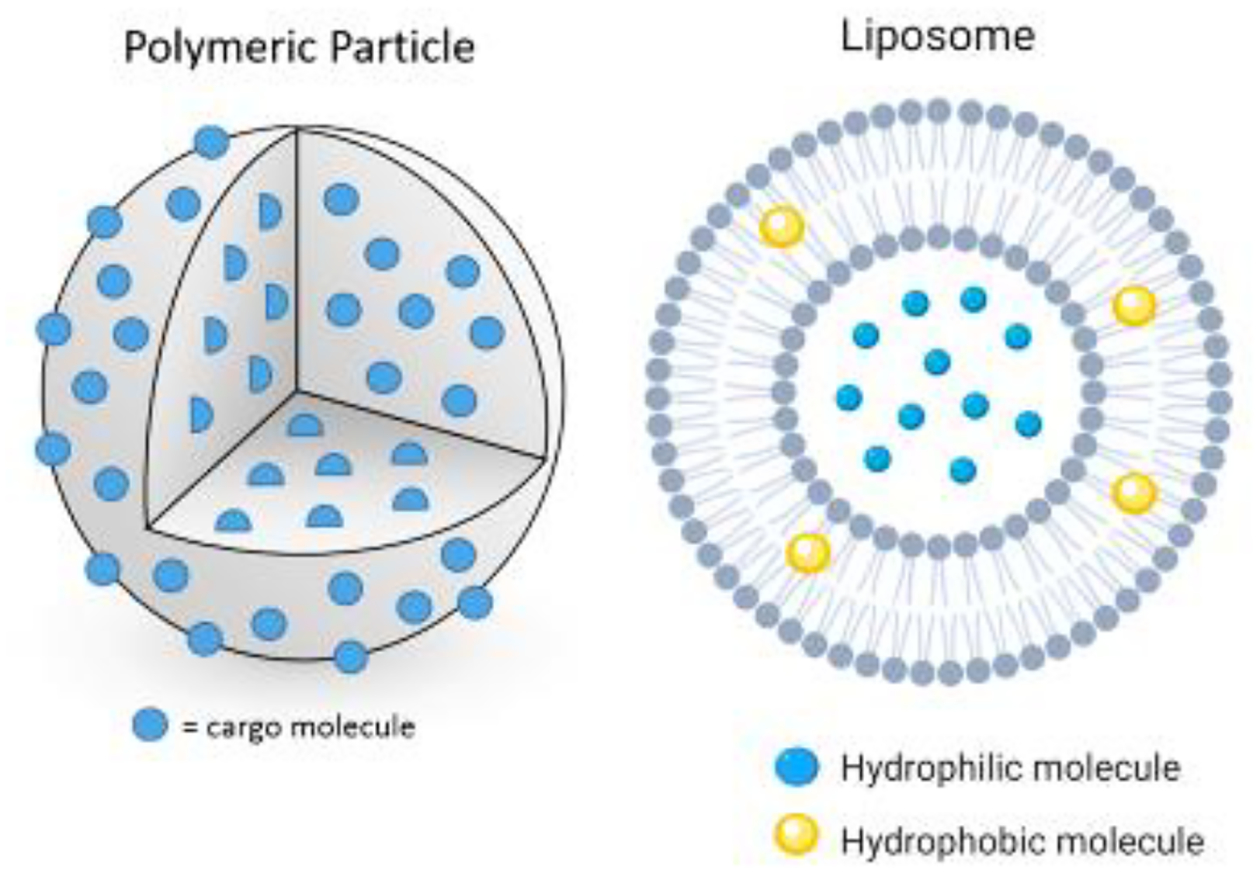
Schematic of common particle-based HDT delivery formulations. Pictured on the left is a solid polymer microparticle seeded with drug cargo (hydrophilic or hydrophobic). On the right is a liposome capable of carrying hydrophobic or hydrophilic drugs.
PLGA, PCL, Ace-DEX and alginate can be formulated into nano- or micro-particles using a variety of techniques.47,48 The most common method for industrial production of polymeric particles is coacervation, which relies on two immiscible phases that form colloidal droplets that solidify into polymeric MPs. Scalable liquid atomization techniques, such as electrospray or spray drying, are also commonly used to create nano- and micro-particles. Particles are created when polymer solutions in a volatile solvent are atomized under a high voltage, forming particles on the opposite side of the source upon solvent evaporation. Atomization techniques can be cost-effective and create particles of high encapsulation efficiency due to lack of an aqueous continuous phase. While spray drying is the more common of these methods, both techniques are widely reported to generate particles.47,48 At a smaller scale, particles can be fabricated using single and double emulsion techniques where the choice of oil-in-water or water-in-oil emulsion is made based on the hydrophilicity of the drug and the polymer system encapsulating it. Micro or nano-emulsions are created by applying high energy to the drug-polymer mixture with a homogenizer or sonicator.51 Numerous polymers, including PLGA and Ace-DEX, have been used to formulate particles via single or double emulsion techniques.52,53. For all of these techniques, parameters like molecular weight, polymer concentration, solvent physical properties, and flow-rate/mixing speed can influence the morphology and size of the particles.47 PLGA microparticulate formulations have been FDA approved since 1989 and include over ten formulations for applications ranging from antipsychotics (Risperidone Consta®) to cancer (Lupron Depot®)54. Many of these formulations are given in subcutaneous or intramuscular injections.
Liposomes:
Liposomes are particles formed from a phospholipid bilayer, capable of encapsulating hydrophilic or hydrophobic drugs (Fig. 1). They are comprised of phospholipids (e.g. phosphatidylserine, phosphatidylcholines) and cholesterol, with some containing polyethylene glycol (PEG) functionalized (PEGylated) lipids to improve circulation times. Ethanol injection is primarily used to create liposomes at an industrial scale, with methods like thin-film hydration used at the bench-scale for generation of liposomes.55,56 Compared to polymeric particles, liposomes are generally less stable during storage and injected via an intravenous route.57 These formulations have been widely applied in clinical settings for both cancer (e.g. Doxil®) and infectious diseases (e.g. AmBisome®).58 AmBisome®, a liposomal formulation of amphotericin B, is one example, developed for systemic fungal and parasitic infections. The liposome greatly enhances biodistribution of the free drug, which is otherwise known for its significant renal toxicity (nicknamed ampho-terrible for its effects).59 Another liposomal formulation that is in clinical trials is Combioxin SA’s CAL02, a sphingomyelin and cholesterol liposome used for toxin neutralization during pneumonia. It should be noted that none of the above drugs are HDTs.41
Formulation of select HDTs in development for bacterial infections
There are several clinically relevant bacteria that have been reported to be treated with formulated HDTs in pre-clinical models of infection. Bacteria that have the most significant infection in the lung (M. tb, F. tularensis), and gastrointestinal tract (S. enterica, Escherichia coli) are reviewed.
Mycobacterium tuberculosis:
M. tb is one of the most prevalent infectious diseases, particularly in developing countries, affecting close to 10.5 million people. Approximately 10% of M. tb infections are extensively drug resistant, meaning they are resistant to rifampicin (RPT), isoniazid (INH), any fluoroquinolone, and a second line injectable drug.10 The bacteria is transmitted via inhalation of droplets containing the pathogen, which readily enter alveolar macrophages upon entry into the lungs. Here, it arrests phagolysosome formation and establishes a niche to replicate in, subsequently leading to the development of the tuberculous granuloma, a hallmark of TB infection.60 HDTs for TB have been proposed to reduce treatment duration and disease-associated lung injury using molecules that promote autophagy and inhibit pathways that cause inflammation and matrix destruction.10,61 A variety of compounds, alone or in combination with standard anti-TB drugs, as well as repurposed, FDA-approved therapies are being evaluated and have been extensively reviewed previously.10,11 Some of these include imatinib (a tyrosine kinase inhibitor clinically approved for leukemia), metformin (a diabetes treatment), dexamethasone (corticosteroid used to reduce inflammation in TB meningitis), and doxycycline (a matrix metalloprotease inhibitor).10 Some compounds exhibit direct inhibitory effects on the pathogen as well as host directed activity towards clearing intracellular bacteria. Nitazoxanide is one such example, where its anti-mycobacterial effects are observed at concentrations ~50 μM, while the drug induces autophagy at lower concentrations (3 μM) via the mammalian target of rapamycin complex-1 (mTORC1) pathway in host macrophages and peripheral monocytes.15 Encouragingly, this compound is currently in phase II clinical trials in Haiti.61 Vitamin A has also been studied as a potential HDT against M.tb, based on clinical findings of vitamin A deficiency in TB patients. All-trans retinoic acid (ATRA), the active metabolite of vitamin A, significantly reduces mycobacterial growth in monocytes via lysosomal acidification and NPC intracellular cholesterol transporter 2 (NPC2), suggesting the role of cholesterol in ATRA-mediated antimicrobial activity.14 Although these HDTs have shown clinical promise, delivery to the affected lung tissue could help to penetrate the dense granulomas and help to successfully clear the infection.62 Thus, a majority of formulations for TB have been developed for delivery to the lungs, mostly through the intranasal (i.n.) or intratracheal route.
Sharma et al. fabricated porous aggregates of PLGA MPs to deliver magainin-I analog peptide (MIAP), an HDT found to have broad spectrum antimicrobial properties and potential host-directed activity against M. tb. MIAP was first encapsulated in PLGA NPs prepared using water-in-oil-in-water double emulsion technique. The NPs were then suspended in mannitol and spray freeze dried to create MPs (Fig. 2A). The resulting 4 μm diameter MPs possessed aerodynamic properties for maximum deposition in the lungs. In vitro characterization revealed that ~100% of the MIAP was released from the MPs over 10 days at physiological pH. Further, imaging studies depicted formation of phagolysosomes in cells treated with MIAP MPs, which were not observed in control cells treated with a standard anti-TB drug, INH (Fig. 2D). Additionally, MIAP MP treatment of M. tb-infected macrophages produced significantly more mitochondrial reactive oxygen species (ROS) and NO, both key actors of autophagy in macrophages, compared to controls. Cytotoxicity measurements revealed that soluble forms of MIAP reduced cell viability by 25%, while the encapsulated drug was able to maintain cell viability. Although the MIAP MPs were only evaluated in vitro, the report provided substantial mechanistic evidence of MIAP’s host-directed activity against M. tb in a formulated form.28
Figure 2.
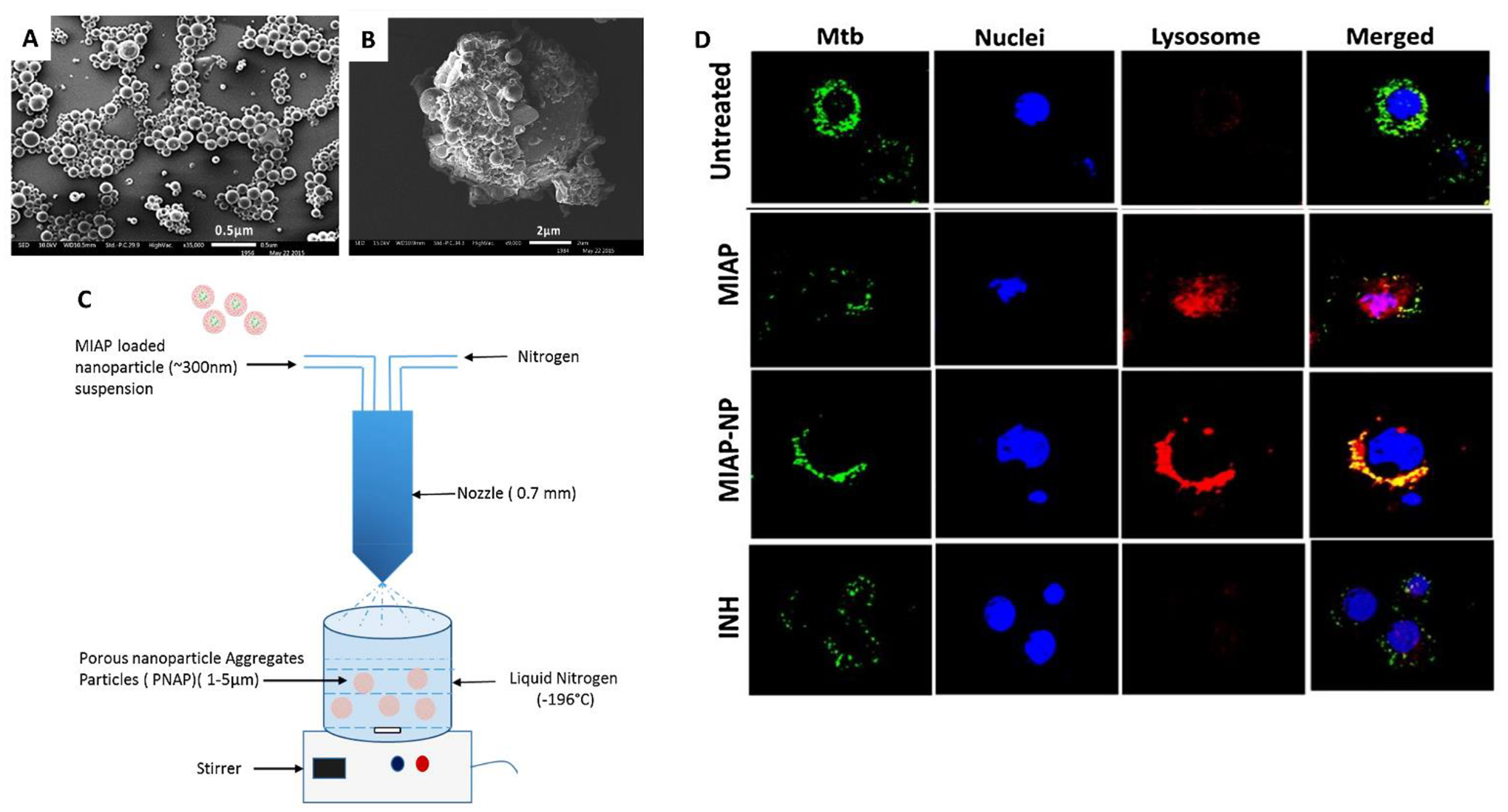
Sharma et al.28 formulated HDTs for tuberculosis. (A) Poly lactic-co-glycolic acid (PLGA) nanoparticles (NPs) encapsulating magainin-I analog peptide (MIAP) which are coated with mannitol to form larger (B) MIAP porous aggregates of PLGA NPs. (C) Schematic depicting fabrication of MIAP porous microparticles (MPs) via spray freeze drying. (D) Panel demonstrating co-localization of M. tb (green) with acidic lysosome (red) in MIAP and MIAP MP groups compared to controls (untreated, isoniazid (INH)) in RAW macrophages.28
There are several reports of carrier biomaterials eliciting an immunogenic response.63 Interestingly, Lawlor et al. reported findings that suggested that an hour-long treatment with empty PLGA MPs can inhibit intracellular M. tb growth via the NF-κB pathway; a result not achieved using control polystyrene MPs. Cytotoxicity evaluation of PLGA MPs at varying concentrations indicated no toxic effects on the cells, even at concentration 7.5 times the therapeutic concentration of PLGA MPs. Further, cell imaging studies and cytokine profiling demonstrated that PLGA MPs can induce NF-κB production with no detection of pro-inflammatory cytokines. This result was confirmed when bacterial clearance by PLGA MPs was blocked in the presence of NF-κB inhibitors. Although the authors note a similar NF-κB response with polystyrene MPs, no associated M. tb clearance was observed with them. This suggests that differences in the physicochemical properties of the two MPs could impact their intracellular bactericidal response. However, endotoxin levels in the particles were not evaluated in the study which could account for some of the noted differences.29
A recent study reported the formulation of ATRA to improve its therapeutic effect against M. tb, in vivo. Hydrophobicity, instability during storage and environmental sensitivity renders vitamin A difficult to deliver in vivo. Treatment of M. tb-infected macrophages with ATRA-loaded PLGA MPs (1–5 μm in diameter) demonstrated significant clearance of the intracellular bacteria. As was observed in this study, encapsulation tends to delay drug release. Additionally, in vitro cytokine profiling revealed a significant reduction in IL-10 and IL-6 post treatment with ATRA MPs, suggesting its role in phagolysosomal maturation. Intratracheal delivery of ATRA MPs demonstrated a significant reduction in bacterial load in mouse lung tissue compared to the vehicle control, but similar to soluble ATRA and a combination treatment with soluble RPT. Further, a transcriptional reduction in TNF-α and inducible nitric oxide synthase (iNOS) was observed with ATRA MP treatment but not with the soluble form, suggesting that the MP formulation improved pulmonary pathology. Overall, this study demonstrated the need to evaluate formulations for HDTs in vitro and in vivo to obtain a more accurate picture of the therapeutic potential of HDTs.27
HDTs are often evaluated in combination with standard drugs to identify synergistic effects that can potentially reduce dosing of standard therapies and render them more effective against drug-resistant strains. In this fashion Gupta et al. tested the efficacy of inhaled MPs of nitazoxanide alone or in combination with INH and rifabutin, two standard anti-TB therapies. Although the study successfully demonstrated greater intracellular concentration and biodistribution of the standard drugs when encapsulated in PLGA MPs compared to their soluble forms, host-directed activity of nitazoxanide against M. tb was not observed in soluble or encapsulated forms.26
Liposomes are another popular particulate platform investigated for delivery of HDTs.41 An study by Greco et al. describes the use of apoptotic body-like (ABL) liposomes as HDTs against M. tb. Here, the outer membrane was composed of phosphatidylserine (PS), a ubiquitous apoptosis marker, while the inner phase contained phosphatidic acid (PA), a second messenger lipid found to regulate phagolysosome maturation (Fig. 3A). Primary findings indicated that the presence of PS on the liposomal surface enhanced uptake by primary macrophages when compared to control liposomes coated with phosphatidylcholine (PC). Moreover, internalization of the liposome drastically reduced pro-inflammatory cytokines and significantly enhanced TGF-β production, in infected macrophages. Interestingly, the intracellular bacterial load reduced significantly with ABL/PA liposomal treatment and control (PC/PA) liposomes, both containing a PA core. This demonstrated that the PA modulated the anti-M. tb function of macrophages. Further, mechanistic studies using Ca2+ chelators and ROS inhibitors demonstrated that the antimycobacterial effect of the liposomes took place via a Ca2+ and ROS-dependent phagolysosome maturation. Stimulation of bronchoalveolar cells obtained from three TB patients with the liposomes also led to a strong reduction in bacterial load, delineating clinical applicability of this treatment. Lastly, a 4 week-long i.n. treatment of liposomes tested in combination with oral forms of anti-TB drug INH in a mouse model of the disease, led to a 100-fold reduction in lung bacterial load at 6 weeks post infection. In contrast, INH alone only showed a 2-fold reduction (Fig. 3C). Authors noted that this extent of bacterial clearance was not observed in the spleens, possibly highlighting the role of delivery routes on biodistribution.30
Figure 3.
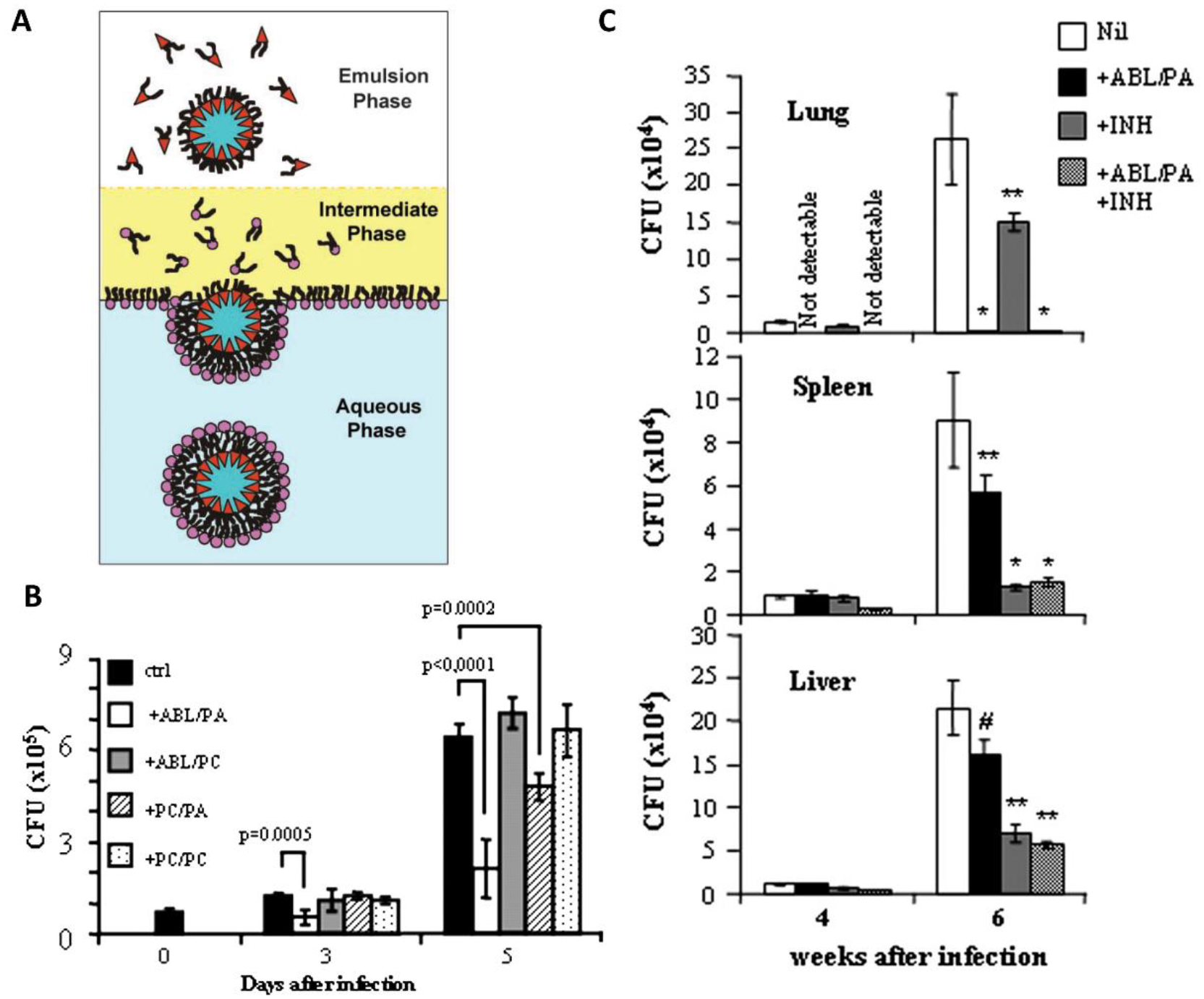
Greco et al.30 formulated liposomal HDTs for tuberculosis. (A) Schematic of asymmetric liposome fabrication64 used to create apoptotic body-like liposomes (ABL) carrying phosphatidic acid (PA) inside and phosphatidylserine on the outside. (B) Intracellular bacterial load in macrophages post treatment with test (ABL/PA) and control ((ABL/phosphatidylcholine (PC)), (PC/PA), (PC/PC)) liposomal formulations demonstrates the role of PA in inhibiting intracellular M.tb growth. (C) Bacterial organ load significantly drops with intranasal ABL/PA treatment in lungs, while bacterial clearance in spleen and liver requires systemic INH treatment. *P < 0.001, **P < 0.05, #P = not significant in comparison with infected control mice that received no treatment.30
Taking the above liposomal platform a step further, the same research group generated a library of PS-decorated liposomes encapsulating different bioactive lipids (PA, phosphatidylinositol-3-phosphate (PI3P), phosphatidylinositol-5-phosphate (PI5P)) that are known effectors of phagosome maturation. These revised liposomes were evaluated for their host-directed antibacterial effects on infection caused by Bacillus Calmette-Guerin (BCG), Pseudomonas aeruginosa, E. coli, and Klebsiella pneumonia. In vitro treatment of BCG infected macrophages with liposomes resulted in phagosome acidification and ROS generation across all lipid types encapsulated in the liposome. Interestingly, ROS generation peaked at 20 min in uninfected cells compared to 18 hours post infection, indicating the interference of ROS generation by BCG upon infection. Further, liposomes demonstrated significant antimicrobial killing in bronchoalveolar cells isolated from nine patients, irrespective of the bacterial species or their drug resistance status. Specifically, PA and PI5P-loaded liposomes were most effective on all pathogens, except E. coli. These findings delineate the broad-spectrum, host-directed activity of bioactive second messenger lipids, likely enhanced by the liposomal formulation. Although the ABL liposome strategy displays a lot of promise in effectively delivering HDTs across multiple pathogens, both studies did not include soluble forms of the lipid as controls for in vitro characterization, and hence do not fully elucidate the need for a liposomal formulation of the studied bioactive lipids.31
Escherichia coli:
Pathogenic strains of E. coli are most commonly associated with infections of the urinary and gastrointestinal (GI) tract. Although most E. coli are extracellular for a majority their lifecycle, invasive E. coli are thought to be facultative intracellular pathogens. Typically, E.coli infections are treated with β-lactams, cephalosporins and fluoroquinolones.1,3 Emerging literature points to stabilizing agents of hypoxia-inducing-factor 1α (HIF 1α),8 as a potential host targeted therapy against this pathogen. Low oxygen in inflamed tissue activates HIF-1α, which in turn drives the pro-inflammatory cytokine production necessary for macrophage aggregation and motility.65 A HIF-1α stabilizing agent, AKB-4924, caused a 10-fold drop in bladder colonization of uropathogenic E. coli in mice. A different study using an E. coli-initiated pneumonia mouse model also demonstrated the protective effects of AKB-4924 by HIF-1α stabilization in bone marrow-derived stem cells.18 Lactoferrin, an iron binding protein, is another molecule suggested to provide host-led protection against E. coli. A recent study demonstrated that pre-treatment of the urethra with exogenous lactoferrin can clear uropathogenic E. coli in vivo, likely via neutrophil recruitment. Considering lactoferrin does not exhibit any direct effect on E. coli in vitro and is naturally excreted by the body in exosomes at the onset of a urinary tract infections, this molecule seems to be the body’s natural protection against urinary tract infections.20 While a majority of HDTs are shown to have no direct bactericidal effects, some are reported to possess both host-directed and direct activity. A synthetic peptide derivative of clavanin A, Clavanin-MO, was shown to have direct bactericidal effects on E. coli and Staphylococcus aureus while also inducing leukocyte recruitment and production of immune mediators, GM-CSF, IFN-γ and MCP-1 in bacterial models of infection.19
Vaghasiya et al. investigated whether drug-free alginate MPs can induce a host directed bactericidal effect on intracellular E. coli. Here, MPs were created by a spraying alginate solution into a calcium chloride solution, resulting in ionically crosslinked MPs ~5 μm in diameter (Fig. 4A). Upon treating infected THP-1 macrophages with blank MPs, the authors noted a 11–16-fold increase in pro-inflammatory cytokines, IL-6 and IL-12, respectively. Interestingly, more MPs were phagocytosed by infected cells compared to uninfected ones. Flow cytometry analysis also revealed significant increase in ROS production with particle treatment in a concentration dependent manner (Fig. 4C). However, treatment with the highest concentration of MPs was accompanied with a ~20% loss in cell viability. The authors concluded that bactericidal effects observed in this study could be attributed to the mannuronic acid group on alginate, that has been shown to induce an innate immune response via TLR2 and TLR4 previously.33
Figure 4.
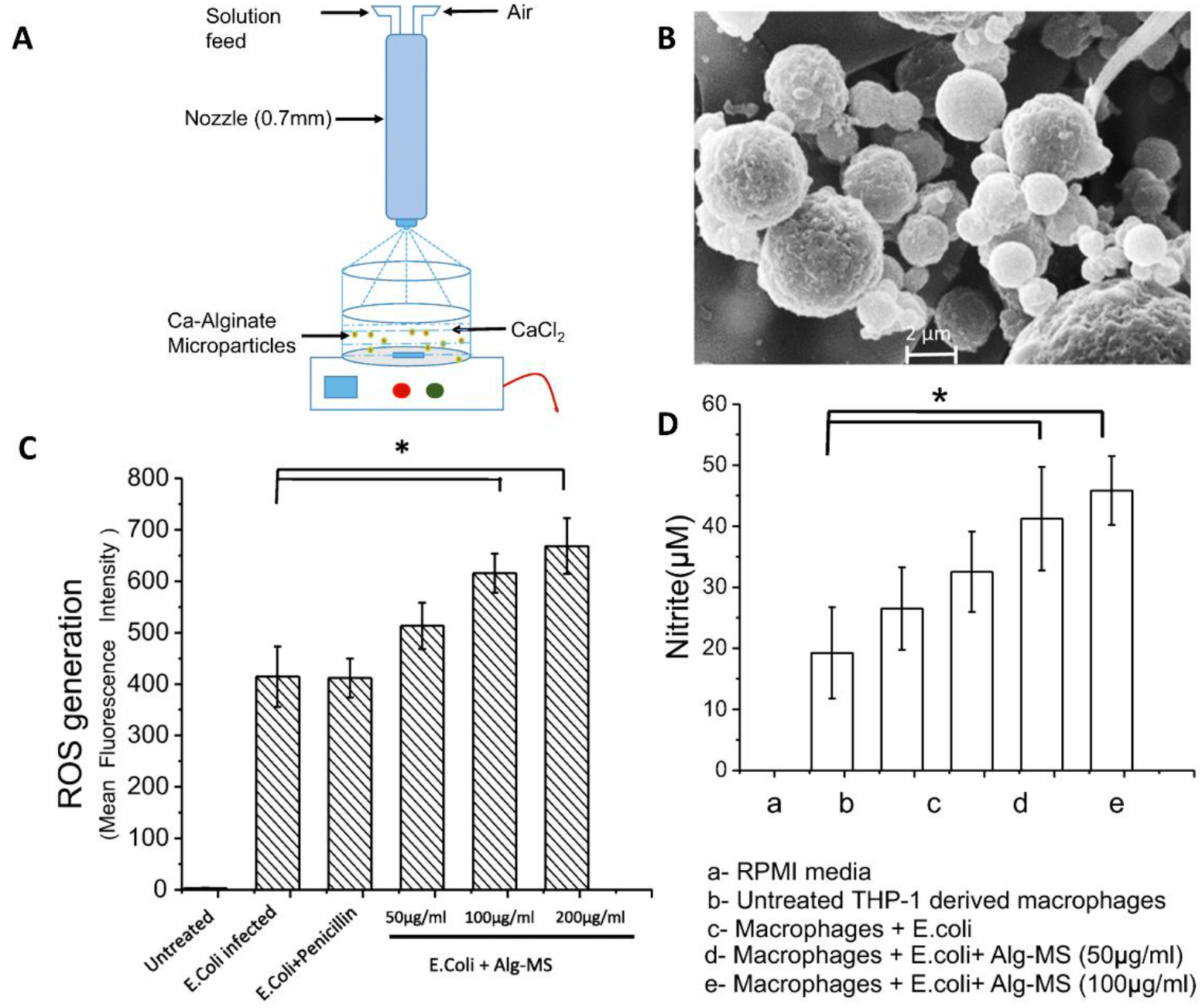
Vaghasiya et al.33 formulated drug-free alginate-based microparticles (MPs) for E. coli infections. (A) Schematic portraying alginate MP fabrication by spray congealing. (B) Scanning electron micrographs of the alginate MPs. Drug-free alginate MPs induce (C) reactive oxygen species (ROS) and (D) nitrite production in E. coli infected macrophages.33
Sharma et al. explored the use of PLGA NPs for protected delivery of HHC10, a broad spectrum HDT. HHC10-NPs, sized ~300 nm in diameter, were fabricated using a double emulsion solvent evaporation technique and evaluated for their effects directly on E. coli as well as infected RAW macrophages. Image-based analysis exhibited ~90% uptake of NPs over 24 hours (Fig. 5A, B). Further, flow cytometry assessment of infected cells treated with the HDT suggested that it induces apoptosis in infected cells. However, this apoptotic effect was significantly higher in the free drug treatment groups compared to the particle groups, possibly due to a delayed drug release from PLGA NPs. Overall, encapsulation in PLGA particles enabled peptide stability and maintained its activity against intracellular E. coli infections (Fig. 5C, D)32
Figure 5.
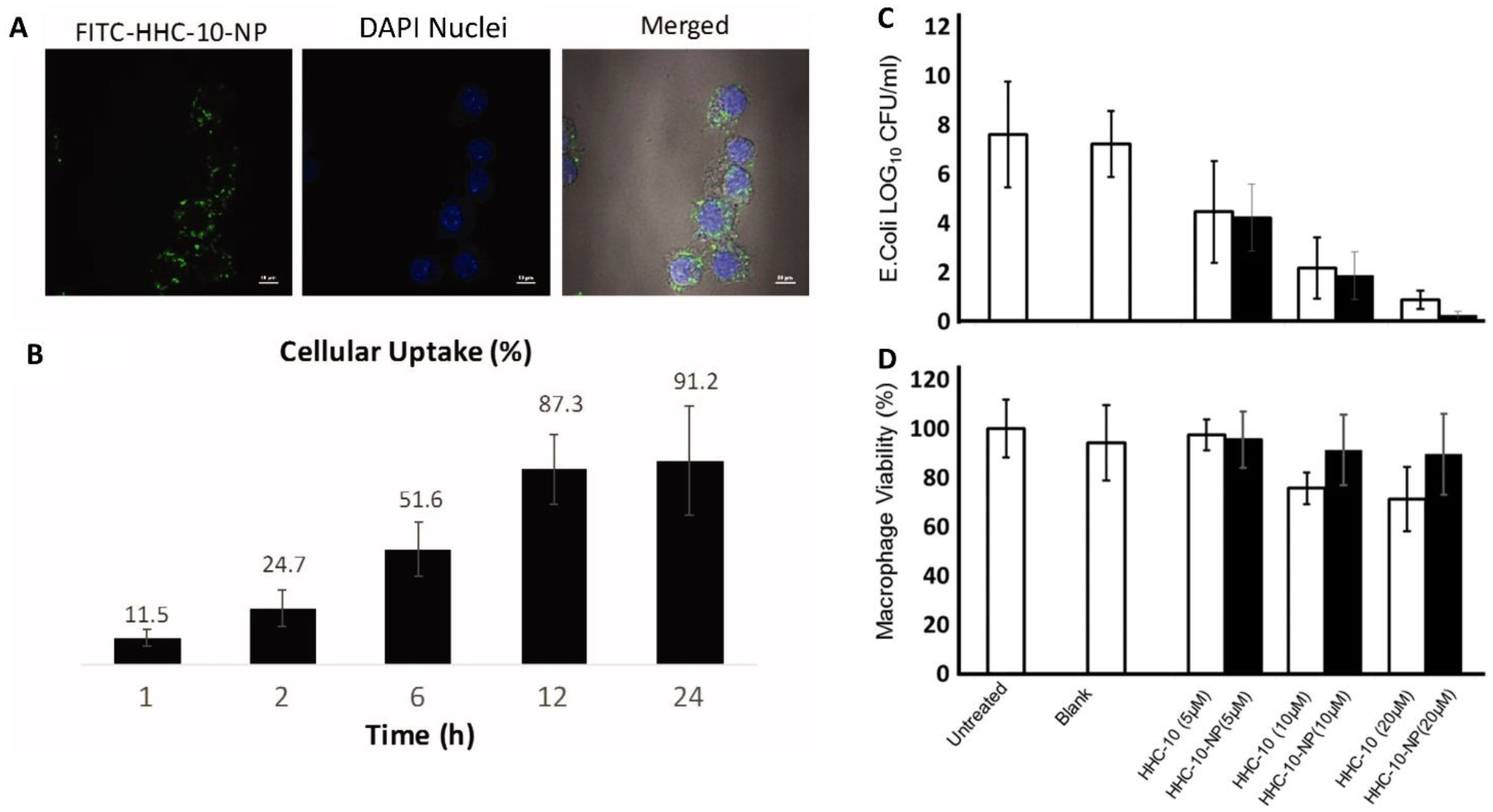
Sharma et al.32 formulated poly lactic-co-glycolic acid (PLGA) nanoparticles (NPs) encapsulating a host defense peptide for E. coli infections. (A) Fluorescence micrographs demonstrating uptake of NPs loaded with HHC-10. (B) A majority of the HHC-10 loaded NPs are phagocytosed over 24 hours. (C) HHC-10 reduces intracellular E. coli burden, as a free drug or encapsulated in PLGA NPs. (D) Formulating the drug in PLGA NPs enables cytocompatibility at higher concentrations.32
Salmonella enterica:
S. enterica serovar Typhimurium uses a wide array of mechanisms to evade host cell clearance. Within cells, the bacteria reside in Salmonella containing vesicles. Furthermore, the bacteria can evade clearance by the phagocyte to complex secretory systems and autophagy escaping factors.66 Salmonella infections in adults are routinely treated with fluoroquinolones1. As a HDT, several studies of a celecoxib derivative, AR-12 (also known as OSU-03012), exhibited significant reduction in S. Typhimurium burden and enhanced survival of mice post infection.21,22,35 Studies in a range of models have demonstrated that AR-12 induced autophagy via the Akt pathway and inhibited chaperone proteins, such as GRP 78.67 Additionally, co-administering AR-12 with aminoglycosides re-sensitized drug-resistant bacteria to the antibiotic.21,22 Another HDT study took a broader, multidisciplinary approach to screen a known library of receptor tyrosine kinase inhibitors against S. Typhimurium-infected cells using chemical genetics and bioinformatics data. They identified two potential HDTs, miberfradil - a calcium channel blocker, and haloperidol - a sigma receptor agonist. However, studies to elucidate their antibacterial mechanism of action were not performed.23
Hoang et al. used an intracellular infection model of S. enterica serovar Typhi to determine the efficacy of AR-12 encapsulated in Ace-DEX MPs for clearing the infection (Fig. 6B). AR-12 MPs enabled a three-fold increase in intracellular drug concentration (Fig. 6B) and readily lowered cytotoxicity of AR-12 towards human monocyte derived macrophages. Compared to the free drug, AR-12 MPs significantly reduced intracellular bacterial load and was effective at lower drug concentrations compared to the soluble drug, suggesting dose-sparing effects rendered by the AR-12 MP formulation. (Fig. 6C, E). Additionally, a strikingly higher percentage of AR-12-Ace-DEX MP treated cells exhibited co-localization of S. Typhi in autophagosomes, suggesting AR-12’s role in inducing autophagy (Fig. 6D).35 Trafficking studies of Ace-DEX MPs in mice via multiple routes of administration (i.n., i.v. and intraperitoneally) showed distribution of the particles in several organs until 6 hours and highest particle retention in liver, lungs and spleens over 10 days, which is far greater than the reported values for organ retention of PLGA particles. Encapsulation of AR-12 in Ace-DEX MPs significantly raised the maximum tolerated dose of AR-12 over the free drug when administered via i.n. and i.v. routes.36
Figure 6.
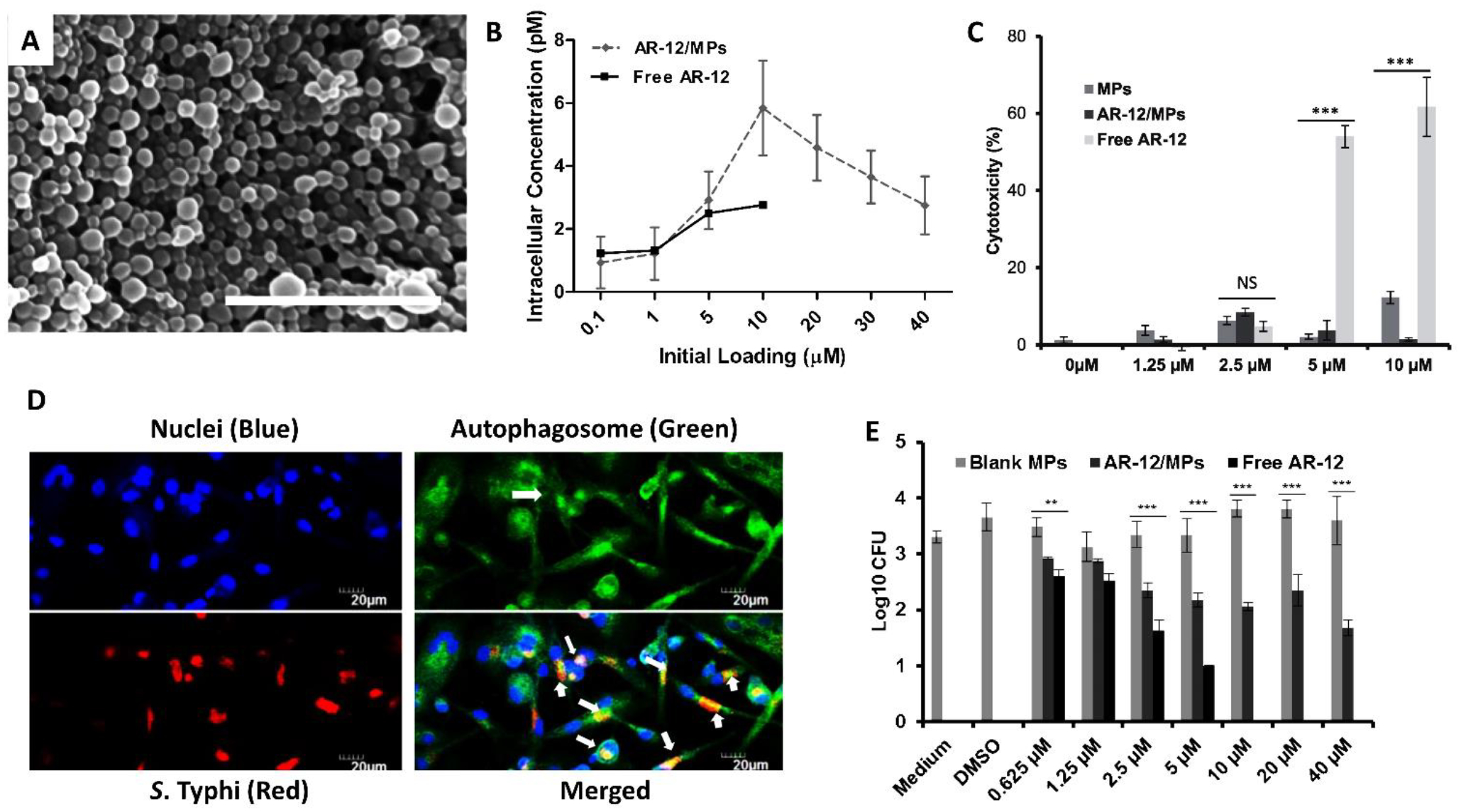
Acetalated dextran (Ace-DEX) formulations of AR-12 for Salmonella. (A) Ace-DEX microparticles (MPs) fabricated using an emulsion technique. Scale bar is 1μm.36 (B) Intracellular concentration of AR-12 increases dramatically when delivered via Ace-DEX MPs. (C) AR-12 cytotoxicity is significantly reduced upon encapsulation in Ace-DEX MPs. *** P, 0.001 (D) Epifluorescence micrographs of human monocyte derived macrophages infected with S. Typhi and treated with AR-12 MPs. (E) Intracellular bacterial load is significantly reduced with increasing AR-12 MPs concentration. ** P<0.01, *** P< 0.001.35
Francisella tularensis:
F. tularensis is a gram negative, facultative intracelluar bacterium causing fatal infections, especially when acquired via the pulmonary route. If untreated, these infections have a 60% mortality rate and treatment involves use of aminoglycosides, tetracyclines and fluoroquinolones. The pathogen is highly virulent and relies on infection and replication within macrophages.7 Similar to findings in S. Typhimurium, AR-12 also demonstrated host-directed activity in clearing F. tularensis from macrophages via an autophagy-induced mechanism.24 Pyles et al. evaluated a synthetic double-stranded RNA analog and toll-like receptor (TLR) 3 agonist, polyinosine:polycytosine (poly (I:C)), for its ability to clear F. tularensis infection in mice. Treatment with poly (I:C) for an hour before or after infection resulted in significantly lower lung bacterial load and increased neutrophil infiltration, consequently extending survival. These results were mimicked in vitro in human monocyte derived macrophages, which were accompanied with increased production of IL-8, TNF-α and IFN-γ. Although, the pre-treatment groups were found to be most effective in subverting the infection, an hour post infection was found to be an effective treatment window with poly (I:C).25
AR-12 loaded Ace-DEX MPs were also tested i.n. in mice infected with F. tularensis. When co-administered with gentamicin, mice treated with AR-12 MPs exhibited an 80% survival and survived significantly longer than gentamicin treatment alone (40% survival) with significantly lower bacterial load in the lungs and spleen by day 6 post infection (Fig. 7).34 The noted dose sparing observed with Ace-DEX MP treatment has been attributed to its acid-sensitivity, which results in enhanced drug delivery into macrophages.68
Figure 7.
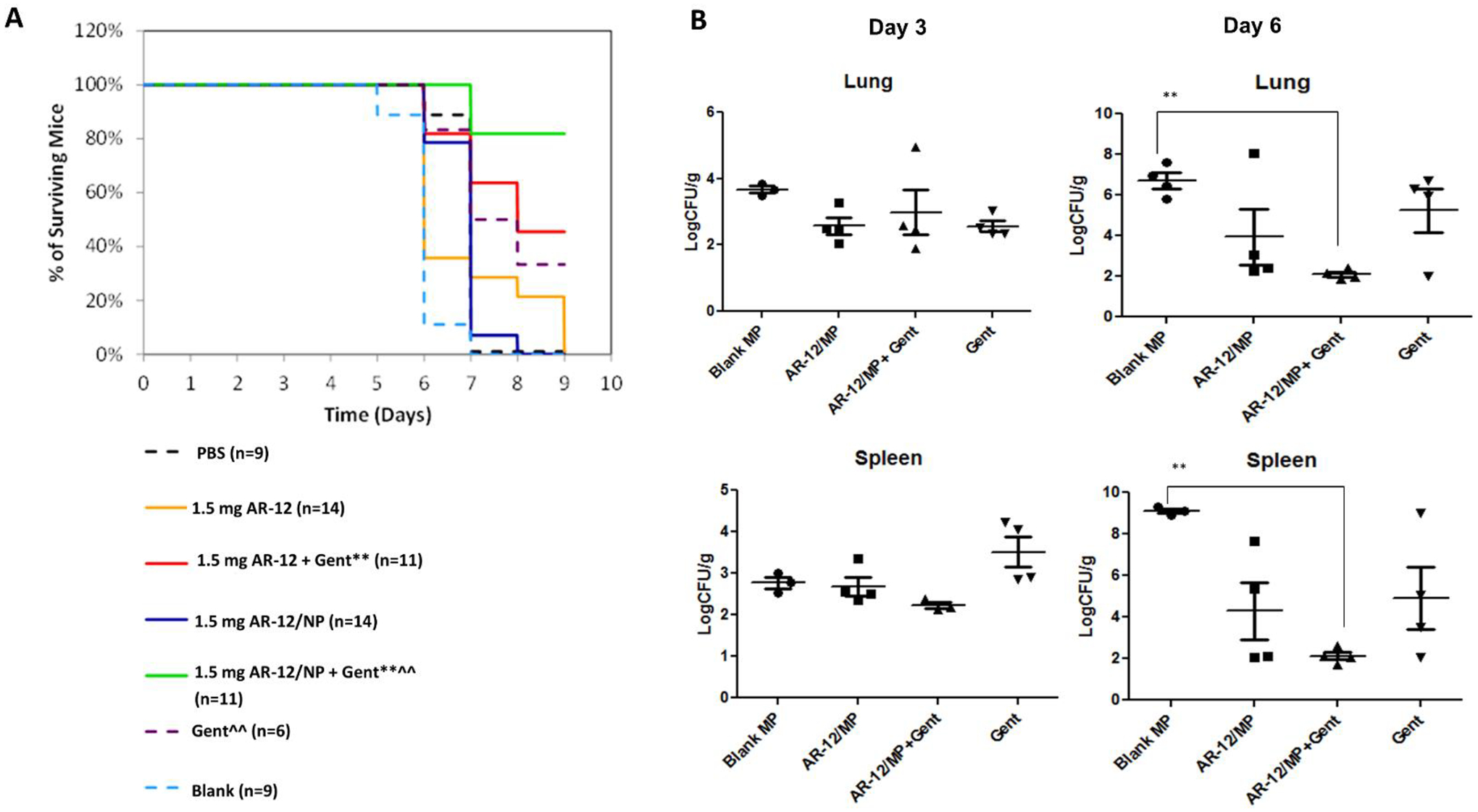
Acetalated dextran (Ace-DEX) formulation of HDTs for F. tularensis. (A) Co-treatment of AR-12 and Ace-DEX nanoparticles (NPs) (delivered intranasally) with gentamicin (Gent) (delivered intraperitoneally) doubles survival of mice infected with F. tularensis compared to gentamicin treatment alone. * P <0.05, **P<0.01 compared to control and blank. ^ P <0.05, ^^P<0.01 compared to AR-12. Gent = daily 0.5mg gentamicin/kg. (B) Significant reduction in bacterial load is observed with co-treatment of AR-12 encapsulated in Ace-DEX NPs (AR-12/NP) and gentamicin by Day 6 post infection in both lung and spleen tissues.34 ** P<0.01 compared to blank MP group.
Future Perspectives:
The overuse of antibiotics over many decades has created the widening problem of antibiotic resistance across the globe. Although this issue affects most bacterial infections, clinical treatment of pernicious, intracellular pathogens is particularly vulnerable, and novel therapies are needful to address this issue in a timely manner. Many of these novel methods, especially to treat TB, rely on formulation of HDTs into particle based therapies. Additionally, the increasing number of FDA-approvals and clinical trials of NP-based therapies sets regulatory precedent for formulated HDTs.54 In addition to the formulation strategies highlighted above, bioavailability and targeted delivery of HDTs can be improved by exploring a range of other formulation strategies and by extending beyond customary materials, such as PLGA.
Micelles are a well-established platform and offer an alternative formulation for HDT delivery (Fig. 8).69 They are self-assembled complexes comprised of amphiphilic molecules with hydrophilic tails and hydrophobic heads. Micelles are especially suited to deliver hydrophobic drugs. The clinical path of this technology is demonstrated by the number of formulations in clinical use or in trials, primarily for cancer treatment, which include formulations of chemotherapeutics paclitaxel (Cynviloq™, Genexol®PM) and docetaxel (Docetaxel-PM, CriPec.41 Hydrogels are another popular formulation for local and topical applications that necessitate steady release of drugs from a depot (Fig. 8).70 Several hydrogels composed of alginate, cellulose polymers and dextran have been developed for skin infections and oral formulations using photocrosslinking and redox-initiated reactions as methods of fabrication.70 These systems can be modified with specific chemical groups that enable bioadhesion and their degradation rate can be tuned based on their crosslinking density (Fig. 8). Several FDA-approved hydrogel products are being used routinely in the clinic, including Vantas®, a 12-month implant for prostate cancer treatment, Juvederm™ hyaluronic acid facial fillers, and Dilapan®, a hydrogel developed for pre-induction cervical ripening during labor.71–73
Figure 8.
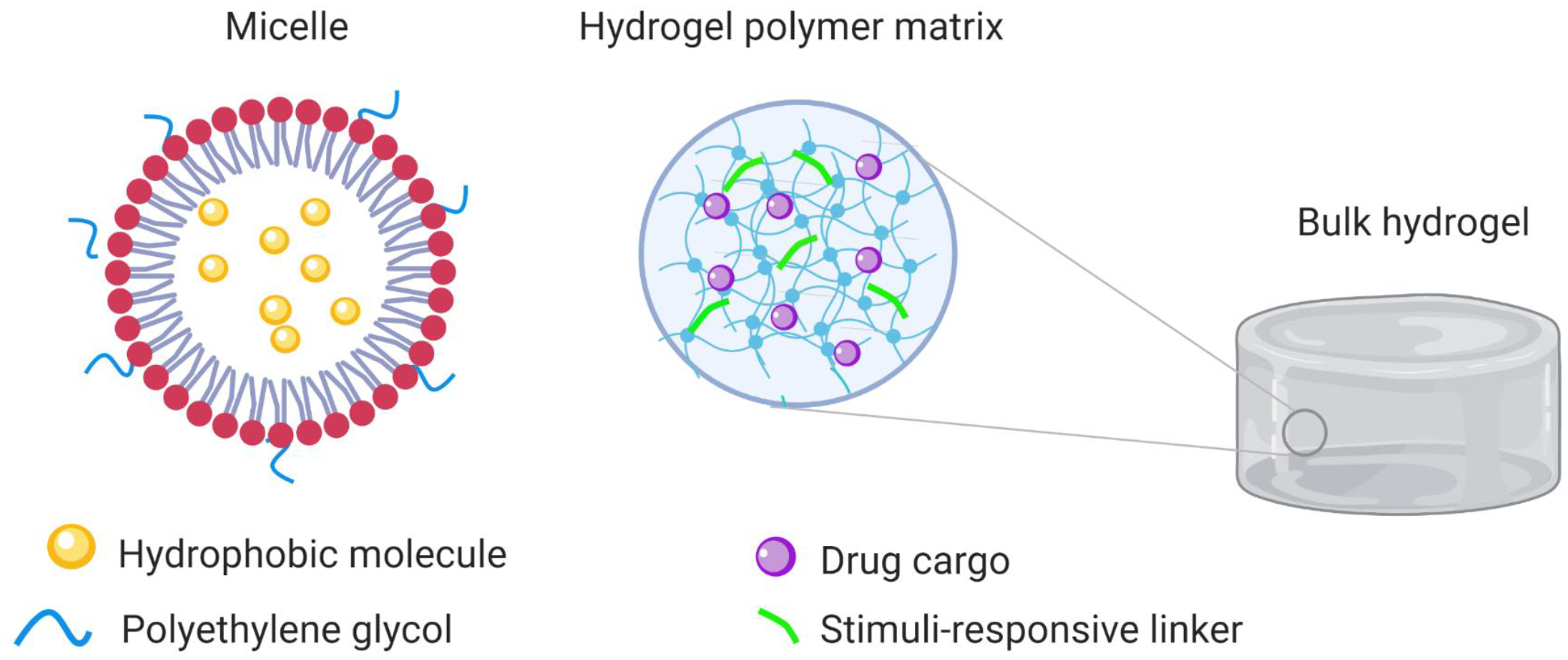
Schematic of potential HDTs delivery platforms. On the left is a micelle carrying a hydrophobic drug. The hydrophilic heads (red) of the micellar monomer can be modified with polyethylene glycol (PEG) to enhance circulation time in the body. Pictured in the center is the crosslinked polymer meshwork found within bulk hydrogels. Chemical linkers or pro-drugs can be incorporated into the polymer network to enable stimuli-responsive (e.g., enzymes, pH) drug release.
Particle-based formulations (e.g. polymeric particles, liposomes, micelles, particulate hydrogels) can be further advanced to enable targeted delivery and enhance circulation time by functionalizing them with receptor specific ligands or PEG chains, respectively (Fig 8). One such example is a cyclodextrin-PEG NPs labeled with a protein targeting transferrin receptor, recently developed by Calando Pharmaceuticals, and it is the first of its kind to deliver siRNA.41 PEGylation has been a successful strategy to increase circulation time in a range of platforms, including the first FDA approved liposome Doxil®. Particles can also be made stimuli-responsive by incorporating chemical entities that render those properties to the system, such as enzyme or pH sensitivity. As described previously, the acetal groups in Ace-DEX confers acid sensitivity to the material, enabling controlled and targeted drug release from Ace-DEX particles in the acidic phagolysosomal environment of a macrophage upon phagocytosis, making this system well-suited to treat intracellular bacterial infections.44
Selective absorption and extended retention of drugs in target tissues can vastly impact their intracellular concentration. Formulations can be chemically modified with cleavable linkers (e.g. pro-drugs) to target bacteria-specific effector molecules or metabolites, potentially increasing the drug concentration at the site of infection and in infected cells. Such strategies are being evaluated to deliver antibiotics into host cells.74–77 For instance, self-assembling amphiphilic vesicles with hydrogen sulfide-cleavable bonds were developed to deliver ciprofloxacin in Salmonella infected mice.75 Another group utilized arginine to direct delivery of cipro-loaded mesoporous silica NPs in mice infected with Salmonella.78 Macrophage targeting using mannose has been well studied for vaccine research and was recently evaluated to deliver RPT-loaded graphene oxide NPs to M. tb infected macrophages.74 Additionally, immunogenicity of bacterial membrane components can be used to activate the innate immune system and target intracellular delivery of antimicrobials.79
While the rise of formulated antibacterial HDTs is encouraging, clinical translation requires significantly more data. Several studies included in this review highlight the need for in vitro and in vivo evaluation of therapies, due to the large differences in these model systems. Additionally, it is important for researchers to define criteria of success to be able to compare therapeutic efficacies between strategies and enable clinical advancement of successful therapies. For example, selectivity of a drug towards a pathogen, as determined by its effect on the host cell versus the microbe, can be a good measure of its therapeutic efficacy. Further, characterization of formulations should include testing for endotoxins to avoid false positive immune responses from the delivery vehicles. Altogether, we have illustrated the potential for these therapies to improve the delivery of HDTs for treatment of bacterial infections, paving the way for these advanced therapies to be translated to the clinic.
Acknowledgements:
Funding is provided by NIH 5R01AI125147-02. Drs. Ainslie and Bachelder serve on the advisory board for IMMvention Therapeutix, Inc. Although a financial conflict of interest was identified for management based on the overall scope of the project and its potential benefit to IMMvention Therapeutix, Inc., the research findings included in this publication may not necessarily relate to the interests of IMMvention Therapeutix, Inc. Authors have disclosed potential conflicts of interest, read the journal’s policy on conflicts of interest and authorship agreement.
Abbreviations:
- ABL
Apoptotic body like
- Ace-DEX
Acetalated dextran
- ATRA
All-trans retinoic acid
- BCG
Bacillus Calmette-Guerin
- GI
Gastrointestinal
- HDTs
Host directed therapies
- HIF
Hypoxia inducing factor
- i.n.
Intranasal
- INH
Isoniazid
- iNOS
Inducible nitric oxide synthase
- i.v.
Intravenous
- MIAP
Magainin-I analog peptide
- MPs
Microparticles
- M. tb
Mycobacterium tuberculosis
- NO
Nitric oxide
- NPs
Nanoparticles
- PA
Phosphatidic acid
- PC
Phosphatidylcholine
- PCL
Polycaprolactone
- PI3P
Phosphatidylinositol-3-phosphate
- PI5P
Phosphatidylinositol-5-phosphate
- poly I:C
Polyinosine:polycytosine
- PLGA
Poly lactic-co-glycolic acid
- PNAP
Porous aggregates of PLGA nanoparticles
- PS
Phosphatidylserine
- ROS
Reactive oxygen species
- RPT
Rifampicin
- TB
Tuberculosis
- TLR
Toll-like receptor
References
- 1.CDC. Antibiotic resistance threats in the United States. Centers for Disease Control and Prevention. 2019. [Google Scholar]
- 2.WHO. Global Antimicrobial Resistance Surveillance System (GLASS) Report. Geneva, World Health Organization. WHO. 2017. [Google Scholar]
- 3.Fair RJ, Tor Y. Perspectives in Medicinal Chemistry Antibiotics and Bacterial Resistance in the 21st Century. Perspect Medicin Chem. 2014;25–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leekha S, Terrell CL, Edson RS. General principles of antimicrobial therapy. Mayo Clin Proc. 2011;86(2):156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Executive Summary: Report of the 22nd WHO Expert Committee on Selection and Use of Essential Medicines. 2019;(April):1–15. [Google Scholar]
- 6.Bush K Past and present perspectives on β-lactamases. Antimicrobial Agents and Chemotherapy. 2018. p. 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kingry LC, Petersen JM. Comparative review of Francisella tularensis and Francisella novicida. Front Cell Infect Microbiol. 2014;5(MAR):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salyer A, Whitt D. Bacterial Pathogenesis : A Molecular Approach. 2nd editio. 2002. [Google Scholar]
- 9.Zumla A, Rao M, Wallis RS, Kaufmann SHE, Rustomjee R, Mwaba P, Vilaplana C, Yeboah-Manu D, Chakaya J, Ippolito G, Azhar E, Hoelscher M, Maeurer M. Host-directed therapies for infectious diseases: Current status, recent progress, and future prospects. Lancet Infect Dis. 2016;16(4):e47–e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallis RS, Maeurer M, Mwaba P, Chakaya J, Rustomjee R, Migliori GB, Marais B, Schito M, Churchyard G, Swaminathan S, Hoelscher M, Zumla A. Tuberculosis-advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect Dis. 2016;16(4):e34–e46. [DOI] [PubMed] [Google Scholar]
- 11.Torfs E, Piller T, Cos P, Cappoen D. Opportunities for overcoming mycobacterium tuberculosis drug resistance: Emerging mycobacterial targets and host-directed therapy. Int J Mol Sci. 2019;20(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Napier RJ, Rafi W, Cheruvu M, Powell KR, Zaunbrecher MA, Bornmann W, Salgame P, Shinnick TM, Kalman D. Imatinib-Sensitive Tyrosine Kinases Regulate Mycobacterial Pathogenesis and Represent Therapeutic Targets against Tuberculosis. Cell Host Microbe. 2011;10(5):475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singhal A, Jie L, Kumar P, Hong GS, Leow MKS, Paleja B, Tsenova L, Kurepina N, Chen J, Zolezzi F, Kreiswirth B, Poidinger M, Chee C, Kaplan G, Wang YT, De Libero G. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6(263). [DOI] [PubMed] [Google Scholar]
- 14.Wheelwright M, Kim EW, Inkeles MS, De Leon A, Pellegrini M, Krutzik SR, Liu PT. All- Trans Retinoic Acid–Triggered Antimicrobial Activity against Mycobacterium tuberculosis Is Dependent on NPC2. J Immunol. 2014;192(5):2280–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam KKY, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, Anderson HJ, Anderson RJ, Av-Gay Y, Roberge M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thwaites GE, Bang ND, Dung NH, Quy HT, Oanh DTT, Thoa NTC, Hien NQ, Thuc NT, Hai NN, Lan NTN, Lan NN, Duc NH, Tuan VN, Hiep CH, Chau TTH, Mai PP, Dung NT, Stepniewska K, White NJ, Hien TT, Farrar JJ. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. 2004;351(17):1741–1752. [DOI] [PubMed] [Google Scholar]
- 17.Lin AE, Beasley FC, Olson J, Keller N, Shalwitz RA, Hannan TJ, Hultgren SJ, Nizet V. Role of Hypoxia Inducible Factor-1α (HIF-1α) in Innate Defense against Uropathogenic Escherichia coli Infection. PLoS Pathog. 2015;11(4):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta N, Nizet V. Stabilization of Hypoxia-inducible factor-1 alpha augments the therapeutic capacity of bone marrow-derived mesenchymal stem cells in experimental pneumonia. Front Med. 2018;5(MAY):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva ON, De La Fuente-Núñez C, Haney EF, Fensterseifer ICM, Ribeiro SM, Porto WF, Brown P, Faria-Junior C, Rezende TMB, Moreno SE, Lu TK, Hancock REW, Franco OL. An anti-infective synthetic peptide with dual antimicrobial and immunomodulatory activities. Sci Rep. 2016;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patras KA, Ha AD, Rooholfada E, Olson J, Ramachandra Rao SP, Lin AE, Nizet V. Augmentation of Urinary Lactoferrin Enhances Host Innate Immune Clearance of Uropathogenic Escherichia coli. J Innate Immun. 2019;481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu HC, Kulp SK, Soni S, Wang D, Gunn JS, Schlesinger LS, Chen CS. Eradication of intracellular Salmonella enterica serovar typhimurium with a small-molecule, host cell-directed agent. Antimicrob Agents Chemother. 2009;53(12):5236–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo JH, Kulp SK, Chen BCS, Chiua HC. Sensitization of intracellular salmonella enterica serovar Typhimurium to Aminoglycosides in vitro and in vivo by a host-targeted antimicrobial agent. Antimicrob Agents Chemother. 2014;58(12):7375–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korbee CJ, Heemskerk MT, Kocev D, Van Strijen E, Rabiee O, Franken KLMC, Wilson L, Savage NDL, Džeroski S, Haks MC, Ottenhoff THM. Combined chemical genetics and data-driven bioinformatics approach identifies receptor tyrosine kinase inhibitors as host-directed antimicrobials. Nat Commun. 2018;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiu HC, Soni S, Kulp SK, Curry H, Wang D, Gunn JS, Schlesinger LS, Chen CS. Eradication of intracellular Francisella tularensis in THP-1 human macrophages with a novel autophagy inducing agent. J Biomed Sci. 2009;16(110). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pyles RB, Jezek GE, Eaves-Pyles TD. Toll-like receptor 3 agonist protection against experimental Francisella tularensis respiratory tract infection. Infect Immun. 2010;78(4):1700–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta A, Meena J, Sharma D, Gupta P, Gupta UD, Kumar S, Sharma S, Panda AK, Misra A. Inhalable particles for “pincer therapeutics” targeting nitazoxanide as bactericidal and host-directed agent to macrophages in a mouse model of tuberculosis. Mol Pharm. 2016;13(9):3247–3255. [DOI] [PubMed] [Google Scholar]
- 27.O’Connor G, Krishnan N, Fagan-Murphy A, Cassidy J, O’Leary S, Robertson BD, Keane J, O’Sullivan MP, Cryan SA. Inhalable poly(lactic-co-glycolic acid) (PLGA) microparticles encapsulating all-trans-Retinoic acid (ATRA) as a host-directed, adjunctive treatment for Mycobacterium tuberculosis infection. Eur J Pharm Biopharm. 2019;134(July 2018):153–165. [DOI] [PubMed] [Google Scholar]
- 28.Sharma A, Vaghasiya K, Gupta P, Gupta UD, Verma RK. Reclaiming hijacked phagosomes: Hybrid nano-in-micro encapsulated MIAP peptide ensures host directed therapy by specifically augmenting phagosome-maturation and apoptosis in TB infected macrophage cells. Int J Pharm. 2018;536(1):50–62. [DOI] [PubMed] [Google Scholar]
- 29.Lawlor C, O’Connor G, O’Leary S, Gallagher PJ, Cryan SA, Keane J, O’Sullivan MP. Treatment of mycobacterium tuberculosis-infected macrophages with poly(lactic-co-glycolic acid) microparticles drives NFKB and autophagy dependent bacillary killing. PLoS One. 2016;11(2):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greco E, Quintiliani G, Santucci MB, Serafino A, Ciccaglione AR, Marcantonio C, Papi M, Maulucci G, Delogu G, Martino A, Goletti D, Sarmati L, Andreoni M, Altieri A, Alma M, Caccamo N, Di Liberto D, De Spirito M, Savage ND, Nisini R, Dieli F, Ottenhoff TH, Fraziano M. Janus-faced liposomes enhance antimicrobial innate immune response in Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A. 2012;109(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poerio N, Bugli F, Taus F, Santucci MB, Rodolfo C, Cecconi F, Torelli R, Varone F, Inchingolo R, Majo F, Lucidi V, Mariotti S, Nisini R, Sanguinetti M, Fraziano M. Liposomes loaded with bioactive lipids enhance antibacterial innate immunity irrespective of drug resistance. Sci Rep. 2017;7(March):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma A, Vaghasiya K, Ray E, Verma RK. Nano-encapsulated HHC10 host defense peptide (HDP) reduces the growth of Escherichia coli via multimodal mechanisms. Artif Cells, Nanomedicine Biotechnol. 2018;46(sup3):S156–S165. [DOI] [PubMed] [Google Scholar]
- 33.Vaghasiya K, Eram A, Sharma A, Ray E, Adlakha S, Verma RK. Alginate Microspheres Elicit Innate M1-Inflammatory Response in Macrophages Leading to Bacillary Killing. AAPS PharmSciTech. 2019;20(6):1–10. [DOI] [PubMed] [Google Scholar]
- 34.Hoang KV, Curry H, Collier MA, Borteh H, Bachelder EM, Schlesinger LS, Gunn JS, Ainslie KM. Needle-Free Delivery of Acetalated Dextran-Encapsulated AR-12 Protects Mice from Francisella tularensis Lethal Challenge. Antimicrob Agents Chemother. 2016;60(4):2052–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoang KV, Borteh HM, Rajaram MVS, Peine KJ, Curry H, Collier MA, Homsy ML, Bachelder EM, Gunn JS, Schlesinger LS, Ainslie KM. Acetalated dextran encapsulated AR-12 as a host-directed therapy to control Salmonella infection. Int J Pharm. 2014;477(1–2):334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson MM, Collier MA, Hoang KV, Pino EN, Graham-Gurysh EG, Gallovic MD, Zahid MSH, Chen N, Schlesinger L, Gunn JS, Bachelder EM, Ainslie KM. In Vivo and Cellular Trafficking of Acetalated Dextran Microparticles for Delivery of a Host-Directed Therapy for Salmonella enterica Serovar Typhi Infection. Mol Pharm. 2018;15(11):5336–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson C, Washington N, Washington C. Physiological Pharmaceutics, Barriers to drug absorption, 2nd Edition. Physiological Pharmaceutics. 2000. [Google Scholar]
- 38.Majumdar S, Mitra AK. Chemical modification and formulation approaches to elevated drug transport across cell membranes. Expert Opin Drug Deliv. 2006;3(4):511–527. [DOI] [PubMed] [Google Scholar]
- 39.Kamaruzzaman NF, Kendall S, Good L. Targeting the hard to reach: challenges and novel strategies in the treatment of intracellular bacterial infections. Br J Pharmacol. 2017;174(14):2225–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Onoue S, Yamada S, Chan HK. Nanodrugs: Pharmacokinetics and safety. Int J Nanomedicine. 2014;9(1):1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng Transl Med. 2016;1(1):10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu L, Peter SJ, D. Lyman M, Lai HL, Leite SM, Tamada JA, Uyama S, Vacanti JP, Langer Robert, AG Mikos. In vitro and in vivo degradation of porous poly(DL-lactic-co-glycolic acid) foams. Biomaterials. 2000;21(18):1837–1845. [DOI] [PubMed] [Google Scholar]
- 43.Sánchez-González S, Diban N, Urtiaga A. Hydrolytic degradation and mechanical stability of poly(ε-Caprolactone)/reduced graphene oxide membranes as scaffolds for in vitro neural tissue regeneration. Membranes (Basel). 2018;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bachelder EM, Beaudette TT, Broaders K, Frechet J, Albrecht M, Mateczun A, Ainslie KM, Pesce J, Keane-Myers A. In vitro analysis of acetalated dextran microparticles as a potent delivery platform for vaccine adjuvants. Mol Pharm. 2010;7(3):826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen N, Collier MA, Gallovic MD, Collins GC, Sanchez CC, Fernandes EQ, Bachelder EM, Ainslie KM. Degradation of acetalated dextran can be broadly tuned based on cyclic acetal coverage and molecular weight. Int J Pharm. 2016;512(1):147–157. [DOI] [PubMed] [Google Scholar]
- 46.Agüero L, Zaldivar-Silva D, Peña L, Dias M. Alginate microparticles as oral colon drug delivery device: A review. Carbohydr Polym. 2017;168:32–43. [DOI] [PubMed] [Google Scholar]
- 47.Peltonen L, Valo H, Kolakovic R, Laaksonen T, Hirvonen J. Electrospraying, spray drying and related techniques for production and formulation of drug nanoparticles. Expert Opin Drug Deliv. 2010;7(6):705–719. [DOI] [PubMed] [Google Scholar]
- 48.Steipel RT, Gallovic MD, Batty CJ, Bachelder EM, Ainslie KM. Electrospray for generation of drug delivery and vaccine particles applied in vitro and in vivo. Mater Sci Eng C. 2019;105(July):110070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nihant N, Grandfils C, Jérôme R, Teyssié P. Microencapsulation by coacervation of poly(lactide-co-glycolide) IV. Effect of the processing parameters on coacervation and encapsulation. J Control Release. 1995;35(2–3):117–125. [Google Scholar]
- 50.Lee P, Pokorski J. PLGA Devices: Production and Applications for Sustained Protein Delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2019;10(5):1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anton N, Benoit JP, Saulnier P. Design and production of nanoparticles formulated from nano-emulsion templates-A review. J Control Release. 2008;128(3):185–199. [DOI] [PubMed] [Google Scholar]
- 52.Chen N, Gallovic MD, Tiet P, Ting JPY, Ainslie KM, Bachelder EM. Investigation of tunable acetalated dextran microparticle platform to optimize M2e-based influenza vaccine efficacy. J Control Release. 2018;289(June):114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N, David CN, McGee CE, Sempowski GD, Shterev I, McKinnon K, Bachelder EM, Ainslie KM, Ting JPY. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J Control Release. 2018;270:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhong H, Chan G, Hu Y, Hu H, Ouyang D. A comprehensive map of FDA-approved pharmaceutical products. Pharmaceutics. 2018;10(4):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duong AD, Collier MA, Bachelder EM, Wyslouzil BE, Ainslie KM. One Step Encapsulation of Small Molecule Drugs in Liposomes via Electrospray-Remote Loading. Mol Pharm. 2016;13(1):92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M, Nejati-Koshki K. Liposome: Classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rideau E, Dimova R, Schwille P, Wurm FR, Landfester K. Liposomes and polymersomes: a comparative review towards cell mimicking. Chem Soc Rev. 2018;47(23):8572–8610. [DOI] [PubMed] [Google Scholar]
- 58.Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update. Bioeng Transl Med. 2019;4(3):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boswell G, Buell D, I. B. AmBisome (Liposomal A Comparative. J Clin Pharmacol. 1998;38(7):583–592. [DOI] [PubMed] [Google Scholar]
- 60.Chakravarty SD, Zhu G, Tsai MC, Mohan VP, Marino S, Kirschner DE, Huang L, Flynn JA, Chan J. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect Immun. 2008;76(3):916–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tiberi S, du Plessis N, Walzl G, Vjecha MJ, Rao M, Ntoumi F, Mfinanga S, Kapata N, Mwaba P, McHugh TD, Ippolito G, Migliori GB, Maeurer MJ, Zumla A. Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. The Lancet Infectious Diseases. 2018. p. 30110–5. [DOI] [PubMed] [Google Scholar]
- 62.Garcia Contreras L, Sung J, Ibrahim M, Elbert K, Edwards D, Hickey A. Pharmacokinetics of Inhaled Rifampicin Porous Particles for Tuberculosis Treatment: Insight into Rifampicin Absorption from the Lungs of Guinea Pigs. Mol Pharm. 2015;12(8):2642–2650. [DOI] [PubMed] [Google Scholar]
- 63.Andorko JI, Jewell CM. Designing biomaterials with immunomodulatory properties for tissue engineering and regenerative medicine. Bioeng Transl Med. 2017;2(2):139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pautot S, Frisken BJ, Weitz DA. Engineering asymmetric vesicles. Proc Natl Acad Sci U S A. 2003;100(19):10718–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chiang C-Y, Uzoma I, Moore R, Gilbert M, Duplantier A, Panchala R. Mitigating the Impact of Antibacterial Drug Resistance through Host-Directed Therapies: Current Progress, Outlook, and Challenges. MBio. 2018;9(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Biswas S, Paudyal N, Pan H, Li X, Fang W, Yue M. Antibiotic resistance in salmonella typhimurium isolates recovered from the food chain through national antimicrobial resistance monitoring system between 1996 and 2016. Front Microbiol. 2019;10(MAY):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Booth L, Roberts J, Ecroyd H, Reid SP, Proniuk S, Zukiwski A, Jacob A, Damonte E, Tuñón MJ, Dent P. AR-12 Inhibits Chaperone Proteins Preventing Virus Replication and the Accumulation of Toxic Misfolded Proteins. J Clin Cell Immunol. 2016;7(5):454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bachelder EM, Pino EN, Ainslie KM. Acetalated Dextran: A Tunable and Acid-Labile Biopolymer with Facile Synthesis and a Range of Applications. Chem Rev. 2017;117(3):1915–1926. [DOI] [PubMed] [Google Scholar]
- 69.Rösler A, Vandermeulen GWM, Klok HA. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv Drug Deliv Rev. 2012;64(SUPPL.):270–279. [DOI] [PubMed] [Google Scholar]
- 70.Li J, Mooney DJ. Designing hydrogels for controlled drug delivery. Nat Rev Mater. 2016;1(12):pii: 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saad AF, Villarreal J, Eid J, Spencer N, Ellis V, Hankins GD, Saade GR. A randomized controlled trial of Dilapan-S vs Foley balloon for preinduction cervical ripening (DILAFOL trial). Am J Obstet Gynecol. 2019;220(3):275.e1–275.e9. [DOI] [PubMed] [Google Scholar]
- 72.Shi Y, Li LC. Current advances in sustained-release systems for parenteral drug delivery. Expert Opin Drug Deliv. 2005;2(6):1039–1058. [DOI] [PubMed] [Google Scholar]
- 73.Allemann I, Baumann L. Hyaluronic acid gel (Juvéderm™) preparations in the treatment of facial wrinkles and folds. Clin Interv Aging. 2008;3(4):629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pi J, Shen L, Shen H, Yang E, Wang W, Wang R, Huang D, Lee BS, Hu C, Chen C, Jin H, Cai J, Zeng G, Chen ZW. Mannosylated graphene oxide as macrophage-targeted delivery system for enhanced intracellular M.tuberculosis killing efficiency. Mater Sci Eng C. 2019;103(April):109777. [DOI] [PubMed] [Google Scholar]
- 75.Mu H, Bai H, Sun F, Liu Y, Lu C, Qiu Y, Chen P, Yang Y, Kong L, Duan J. Pathogen-targeting glycovesicles as a therapy for salmonellosis. Nat Commun. 2019;10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kwok PCL, Grabarek A, Chow MYT, Lan Y, Li JCW, Casettari L, Mason AJ, Lam JKW. Inhalable spray-dried formulation of D-LAK antimicrobial peptides targeting tuberculosis. Int J Pharm. 2015;491(1–2):367–374. [DOI] [PubMed] [Google Scholar]
- 77.Vencken S, Foged C, Ramsey JM, Sweeney L, Cryan SA, MacLoughlin RJ, Greene CM. Nebulised lipid-polymer hybrid nanoparticles for the delivery of a therapeutic anti-inflammatory microRNA to bronchial epithelial cells. ERS Monogr. 2019;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mudakavi RJ, Vanamali S, Chakravortty D, Raichur AM. Development of arginine based nanocarriers for targeting and treatment of intracellular: Salmonella. RSC Adv 2017;7(12):7022–7032. [Google Scholar]
- 79.Lemmer Y, Kalombo L, Pietersen RD, Jones AT, Semete-Makokotlela B, Van Wyngaardt S, Ramalapa B, Stoltz AC, Baker B, Verschoor JA, Swai HS, De Chastellier C. Mycolic acids, a promising mycobacterial ligand for targeting of nanoencapsulated drugs in tuberculosis. J Control Release. 2015;211:94–104. [DOI] [PubMed] [Google Scholar]