

Abstract
Advanced therapies have transformed the treatment of inflammatory bowel disease; however, many patients fail to respond, highlighting the need for therapies tailored to the underlying cell and molecular disease drivers. The first-in-class oral molecule ABX464 (obefazimod), which selectively upregulates miR-124, has demonstrated its ability to be a well-tolerated treatment with rapid and sustained efficacy in patients with ulcerative colitis (UC). Here, we provide evidence that ABX464 affects the immune system in vitro, in the murine model of inflammatory bowel disease, and in patients with UC. In vitro, ABX464 treatment upregulated miR-124 and led to decreases in proinflammatory cytokines including interleukin (IL) 17 and IL6, and in the chemokine CCL2. Consistently, miR-124 expression was upregulated in the rectal biopsies and blood samples of patients with UC, and a parallel reduction in Th17 cells and IL17a levels was observed in serum samples. In a mouse model of induced intestinal inflammation with dextran sulfate sodium, ABX464 reversed the increases in multiple proinflammatory cytokines in the colon and the upregulation of IL17a secretion in the mesenteric lymph nodes. By upregulating miR-124, ABX464 acts as “a physiological brake” of inflammation, which may explain the efficacy of ABX464 with a favorable tolerability and safety profile in patients with UC.
KEYWORDS: ABX464 (obefazimod), microRNA, miR-124, ulcerative colitis, immune system
INTRODUCTION
Ulcerative colitis (UC) is a chronic disease that is characterized by a dysregulated immune response and chronic inflammation in the colonic mucosa (1). UC lesions are typically composed of resident innate cells in the lamina propria, including resident macrophages, innate-like lymphocytes, innate T cells, and inflammatory cells such as basophils and neutrophils, which collectively produce large amounts of interleukin (IL)-1β, IL6, IL8, and IL23. Next to this infiltrate reside activated adaptive immune cells, CD4+ and CD8+ cells, and a marked enrichment of Th17 cells and Th1 cells that produce tumor necrosis factor α (TNF-α) and interferon γ (IFNγ) (2).
More than 90% of patients with UC are treated with oral or rectal administration of 5-aminosalicylates shortly after disease diagnosis (3–5). If treatment is insufficient, corticosteroids are used as a rescue therapy during disease flares (6,7). Immunosuppressants are used to maintain remission in patients with UC and as a maintenance strategy after rescue therapy (4,6). Targeted strategies with biologic drugs (e.g., monoclonal antibodies, small molecules, and nucleic acids) and Janus kinase inhibitors are not effective in all patients or can lose efficacy with long-term use, and they may need close monitoring (8). Clearly, there are many gaps in the ability to effectively treat UC, and alternative therapies are needed in the management of the disease course.
ABX464 (obefazimod), a quinoline that induces the anti-inflammatory micro-RNA (miR)-124, has been proposed for the treatment of UC. ABX464 has demonstrated durable efficacy in patients with UC who were all intolerant and/or refractory to at least 1 approved treatment (steroids and biologic drugs) (9,10). Clinical findings showed that once-daily treatment with ABX464 (25, 50, or 100 mg) led to a significant reduction in the modified Mayo score at week 8. Significant improvements were seen in key secondary endpoints, including endoscopic improvements, clinical remission, clinical responses, and reductions in fecal calprotectin and rectal tissue inflammation compared with placebo (10). In addition to its efficacy, ABX464 may have a safety advantage over other nonconventional drugs, as in the phase 2b trial described above, ABX464-associated adverse events were mild, transient, dose-dependent, and similar to those in the placebo group at the lowest dose of 25 mg.
Selective upregulation of miR-124 by ABX464 can downregulate several inflammatory pathways (11). MiR-124 is a modulator of monocyte and macrophage activation (12,13) and plays a critical role in both the innate and adaptive immune responses (14–17) to establish and maintain tissue homeostasis. MiR-124 was previously shown to be a critical mediator of cholinergic anti-inflammatory effects through its reduction of IL6, TNF-α, and CCL2 (monocyte chemoattractant protein-1) production (18). In this study, we aimed to investigate the effects of upregulation miR-124 by ABX464 on immune cells and the immune system both in vitro, in the murine model of inflammatory bowel disease and in patients with UC. Our data show that ABX464 reverses the expression of several inflammatory cytokines triggered during inflammation and seems to be able to balance immune characteristics of inflammatory bowel disease that arise from abnormal responses of the innate and adaptive immune system.
METHODS
Isolation, culture, and treatment of PBMCs, purified CD4+ cells, and macrophages
Peripheral blood mononuclear cells (PBMCs), purified CD4+ cells, and macrophages were isolated from 11 to 16 healthy blood donors (anonymous blood bags were obtained by L'Etablissement Français du Sang). PBMCs were isolated by centrifugation on Ficoll, then resuspended at a density of 1.5 × 106 cells/mL in RPMI+ (RPMI with 10% fetal bovine serum and 1% penicillin/streptomycin) supplemented with IL2 (40 U/mL), and activated with PHA-L (5 μg/mL) for 48 hours; however, for dose-response experiments, PBMCs were activated with CD3+/CD28+ (T-cell TransAct 500x, Miltenyi Biotec) for 48 hours. CD4+ or CD14+ cells were isolated by CD4+ or CD14+ positive selection or directly from buffy coats (Miltenyi Biotec). After being counted, CD4+ cells were resuspended at a density of 1×106 cells/mL RPMI+ with 20 U/mL IL2 and were activated by CD3+/CD28+ (T-cell TransAct 500x). CD14+ cells were resuspended at 0.6 × 106 cells/mL in RPMI+ and were differentiated by granulocyte-macrophage colony-stimulating factor (GM-CSF) (100 ng/mL) for 6 days (37°C, 5% CO2), with the medium being refreshed on day 4. On day 7, macrophages were polarized into the M1 phenotype with lipopolysaccharide (100 ng/mL) and IFNγ (20 ng/mL). After activation, PBMCs and CD4+ cells were treated with ABX464 or DMSO for 6 days, with the medium being refreshed after 3 days of treatment, and macrophages were treated from days 4 to 8, with the medium being refreshed on days 6 and 7.
Generation of the DSS-induced colitis model and isolation of MLNs and colon cells
Age-matched and sex-matched C57BL6 mice (n = 26 to 39 per group) received 1 cycle of 2.5% (wt/vol) dextran sodium sulfate (DSS) administered through drinking water for 7 days, and then, depending on the extent of weight loss, a recovery period of 3–5 days was applied. DSS-induced mice were treated with ABX464 (40 mg/kg) daily by force-feeding (gavage) for the duration of the study. The control groups included mice receiving regular drinking water without colitis (the H2O group) and untreated mice with DSS (untreated DSS group).
At the end of each experiment, mice were euthanized, and mesenteric lymph nodes (MLNs) were individually collected from the mice in phosphate-buffered saline (PBS) and homogenized on a 40-μm cell strainer. MLN cells were washed with PBS and then subjected to staining for subsequent analysis of immune cell populations. In addition, colons were excised and longitudinally cut in ice-cold PBS. Each tissue was cultured with the villi facing upward in a 24-well plate in 500 μL of complete RPMI medium containing 10% fetal bovine serum, penicillin-streptavidin, 1× nonessential amino acids, 1× β-mercaptoethanol, 1× sodium pyruvate, 1× HEPES, and 1× L-glutamine. After 24 hours, the supernatants were collected, centrifuged, distributed in a 96-well plate, and frozen at −80°C until later use for cytokine titration assays.
Fluorescence-activated cell sorting analysis
Marker profile staining.
For intracellular staining, cells were stimulated with 100 ng/mL phorbol 12-myristate 13-acetate, 500 ng/mL ionomycin, and 1X brefeldin A (GolgiPlug, Becton Dickinson) for 4 hours at 37°C. Single staining for fluorescence-activated cell sorting compensation was performed on untreated cells. Surface markers and intracellular cytokine staining were performed using antibodies targeting i) the Th1/Th17 secretion panel (CD3+, CD4+, IL17a, and IFNγ) and Treg panel (CD3+, CD4+, CD25+, and FoxP3) for MLN cells and ii) the Th1/Th17 panel (CD4+, CD196+ [CCR6], and CD183+ [CXCR3]; IL17a; and IFNγ) for PBMCs and purified CD4+ cells. Fluorescence-activated cell sorting acquisition was performed on a MACSQuant Analyzer 10.
Secreted cytokine profile.
Cytokines secreted in the colon were analyzed with the LegendPlex multiplex assay (Biolegend) using a fluorescence-encoded bead staining procedure, and the data were acquired on a MACSQuant Analyzer 10. Mouse cytokines (IL1α, IL1β, IL10, IL12, IL17a, IL23, IL27, IFN-β, IFN-γ, TNF-α, and GM-CSF) and key targets involved in hematopoietic stem-cell differentiation and lineage-specific markers (IL34, IL5, M-CSF, TPO, IL6, GM-CSF, IL15, TGF-β1, IL3, LIF, SCF, EPO, and CXCL12) were simultaneously quantified. For PBMCs, purified CD4+ cells and macrophages, the levels of IL6, IL17a, TNF-α, and CCL2 were quantified by means of a staining procedure according to the Human MACSplex cytokine 12 KIT (Miltenyi Biotec) or the LegendPlex inflammation panel (Biolegend), and the data were acquired on a MACSQuant Analyzer 10. Cytokine profile analysis was performed with MACSQuantify (2.11) software (Miltenyi Biotec) or Quognit software (Biolegend).
Enzyme-linked immunosorbent assay analysis
IL6, IL22, and CCL2 levels were measured by enzyme-linked immunosorbent assay (ELISA) with adequate colon supernatant dilutions (IL6 and CCL2: ELISA MAX Deluxe Set Biolegend; IL22: Quantikine ELISA R&D Biosystems). Optical density was determined using the SPARK microplate reader (Tecan) set to 450 nm with wavelength correction set to 570 nm.
RNA extraction and RT-qPCR analysis of miR-124 copy number
The miRNAs were isolated from indicated cells and tissues with a Macherey-Nagel NucleoSpin miRNeasy kit. For PBMCs and purified CD4+ cells, cDNA templates were prepared using a miScript RT2 kit (Qiagen) starting from 12 μL of miRNA matrix and qPCR was performed using a miScript SybrGreen PCR kit (Qiagen). Specific primers for the miR-124 target (MS00006622), miR-26a (MS00029239), and miR191 (MS00003682) housekeeping gene were used to perform relative qPCR. PCR data were analyzed with Roche LightCycler software. Changes in miR-124 levels were quantified after normalization to the housekeeping genes miR-26a and miR-191. MiR-amplified cDNA templates were diluted at 1:10 for miR-124 and 1:100 for miR-26a and miR-191. For macrophages and PBMCs in the dose-response study, cDNA templates were prepared using a TaqMan Advanced miRNA cDNA Synthesis Kit (Applied Biosystems) starting with 2 μL of total RNA matrix, and qPCR was performed using TaqMan Advanced miRNA assays targeting miRNA124 (477879_mir), with miRNA26a (477995_mir) and miRNA191 (477952_mir) as references for expression data normalization. MiR-amplified cDNA templates were diluted 1:10.
Analysis of cytokine levels in blood samples from patients with UC
The serum samples from patients who received ABX464 (25, 50, or 100 mg) or placebo were collected during 5 visits (day 1, weeks 1, 4, 8, and 16) of a phase 2b trial (10). Nine biomarkers (IL1b, IL4, IL6, IL10, IL23, IFNγ, CCL2, TNF-α, and IL17a) were measured using the 8-plex assay on the ELLA Platform (Biotechne) and the MSD IL23 V-plex assay (MSD). All samples were tested in duplicate at the minimum required dilution.
Analysis of miR-124 copy number in rectal biopsies and blood samples from patients with UC
During the phase 2b trial (10), 2 rectal biopsies (plus optionally 2 sigmoidal biopsies, if the inflammation of the sigmoid was observed) were performed at screening, at day 57 (week 8), and at day 113 (week 16, if applicable). The 2 biopsies were sent to central laboratories for miRNA-124 determination and were stored in RNA later. Absolute quantification of the miR-124 copy number was performed on rectal biopsy samples and whole blood samples from 139 patients and in 1,104 from 229 patients using droplet digital PCR (ddPCR) technology (see below).
RNA and DNA extraction.
RNA and DNA were extracted concomitantly using the quick-DNA/RNA Magbead extraction kit (Zymo). Briefly, samples were lysed in 800 μL of DNA/RNA shield (Zymo) using TissueLyser LT (Qiagen), and then, the lysates were incubated with Proteinase K for 30 minutes at room temperature and frozen at −80°C. Bead-based RNA and DNA extraction from 100 μL of lysates was performed automatically using the Pureprep 96 Nucleic Acid Purification System (Magtivio). RNA and DNA concentrations were measured using the Qubit 2.0 fluorometer and the Qubit RNA (or DNA) High Sensitivity Kit (ThermoFisher).
cDNA synthesis and preamplification.
miRNAs in the extracted samples were specifically retrotranscribed from 10 ng of total RNA (2 μL at 5 ng/μL) using the TaqMan Advanced miRNA cDNA RNA Synthesis Kit (ThermoFisher), and following cDNA synthesis, preamplification was performed to ensure that miR-124 and miR16 were present. When RNA concentrations were lower than 5 ng/μL, no dilution was made, and a max input approach was used.
ddPCR.
ddPCR was performed on a QX200 droplet digital PCR system, and the data were analyzed with QuantaSoft Pro software version 1.7.4.0917. The preamplified cDNA was subjected to duplicate ddPCR measurements using 2 singleplex assays targeting miR-16 and miR-124. For each sample, miR-124 and miR-16 were measured on the same ddPCR plate in a singleplex reaction at a specified dilution (1:10 for miR-124; 1:2,000 for miR-16) and were analyzed together.
The total copy number per sample was calculated by considering the total dilution factor from lysis to ddPCR, the sample-specific dilution factor for cDNA synthesis, and the cycles of preamplification. The total copy number per cell was calculated based on the DNA concentrations by considering the total dilution factor during lysis and extraction and the hypothesis of a 3.3-pg total weight for 1 haploid genome. The normalized miR16 copy number was calculated using a sample-specific normalization factor that considers the relative expression of miR-16 per sample per batch.
Statistical analysis
The miR-124 modulation is expressed in fold change, while cellular population and cytokine secretion modulations are expressed in percentage of upregulation or downregulation, compared with untreated (DMSO) control. All data are presented as mean ± SEM except for the correlation between miR-124 and Th17 cells.
Residual normality of paired ANOVA data (cell frequencies or cytokine concentrations) was assessed by means of the Shapiro-Wilk test (less than 50 data points) or Kolmogorov-Smirnov (more than 50 data points) normality test using GraphPad Prism software. Nonparametric data were analyzed by Kruskal-Wallis tests with Dunn multiple comparison tests, and parametric data were analyzed by 1-way ANOVA with Dunnett multiple comparison tests. For dose-response experiments, correlations were analyzed with Spearman tests. The P-value threshold for statistically significant differences was P < 0.05.
IL-17A levels in UC patient serum were performed on rank, as Pearson residuals do not fit a normal distribution. Statistical analysis is performed on rank data transformation (nonparametric model).
Study approval
Mouse experiments were performed in strict accordance with the guidelines of the European Union (2010/63/EU) and French Décret No. 2013-118 published on February 1, 2013, at the Institute de Génétique Moléculaire de Montpellier (IGMM) animal facility (Agreement No. E34-172-16). The study plan was approved by the Institutional Review Board at the Animal Facility of IGMM and the Regional Ethics Committee for Animal Experimentation of Languedoc-Roussillon (Agreement No. CEEA-LR-12087).
The study was approved by the Institutional Review Board or Independent Ethics Committee at each center and conducted in accordance with the Good Clinical Practice, other relevant rules and regulations, and Helsinki declaration. All patients provided written informed consent.
RESULTS
To investigate the effects of ABX464 on the immune system, we first assessed the effect of ABX464 exposure on human PBMCs, purified CD4+ cells, and macrophages obtained from healthy donors and on serum samples from patients with UC by measuring secretion of cytokines. To confirm that ABX464 is affecting its predicted target, we measured the levels of miR-124.
Human PBMCs, CD4+ cells, and macrophages
The effect of ABX464 treatment was compared with a DMSO treatment used as control for miR-124 expression, CD4+ cell proportions, and secretion of cytokines in cell culture of PBMCs, purified CD4+ cells, and macrophages. In this analysis, ABX464 treatment resulted in significant upregulation of miR-124 levels; specifically, the increases in miR-124 levels were 9-fold in PBMCs (Figure 1a), 7-fold in purified CD4+ cells (Figure 1b), and 9-fold in macrophages (Figure 1c). Cytokine analysis revealed that after 6 days of ABX464 treatment, IL17a and IL6 secretion in PBMCs was significantly decreased (−61.6%, P < 0.0001 and −27.4%, P = 0.0002, respectively). A moderate but statistically significant increase was, however, observed for TNF-α secretion (+67.0%, P = 0.0004) (Figure 2a). Of note, ABX464 treatment caused an increase in 4 of 16 donors who cannot be explained at the moment, whereas for the 12 donors, ABX464 exerted no effect. We then analyzed the expression of CD183 and CD196 markers at the cellular membrane (Figure 2b) and the intracellular secretion of IFNγ and IL17a (Figure 2c) to determine the percentage of Th17+ (CD183− CD196+ and IL17a secretors only), Th1+ (CD183+ CD196− and IFNγ secretors only), and Th17+ like Th1+ (CD183+ CD196+ and both IL17a and IFNγ secretors) cells among PBMCs. ABX464 treatment did not change Th1+ cells but significantly decreased Th17 cells (−39.1%, P = 0.0002) or Th17+ like Th1+ (Figure 2b), confirmed by significant decreases (−50.8%, P < 0.0001) in IL17a-secreting cells (Figure 2c). Interestingly, dose-response curves of ABX464 showed that the increase in miR-124 and the increase in Th17 cell inhibition with increasing ABX464 dose presented parallel profiles (Figure 2d).
Figure 1.

Upregulation of miR-124 expression. Changes in miR-124 levels after 6 days of ABX464 5 μM treatment of (a) peripheral blood mononuclear cells (PBMCs), (b) CD4+ cells, and (c) macrophages. Fold change compared with DMSO-treated cells as a negative control. Dots represent blood samples from different donors, and mean ± SEM are reported.
Figure 2.

Effects of ABX464 on peripheral blood mononuclear cells (PBMCs). Cells were treated for 6 days with 5 μM ABX464. (a) Cytokine concentrations in the supernatant of PBMCs. (b) Th1/Th17 cells (CD196/CD183 expression) among viable CD4+ cells. (c) Intracellular IFNγ and IL17a expression among viable CD4+ cells. (d) Correlation between miR-124 modulation and Th17-cell inhibition in PBMCs related to ABX464 concentration. Cells were treated for 6 days with different concentrations of ABX464 (not above 10 μM to avoid a cytotoxic effect). Th17-cell inhibition and fold changes in miR-124 were determined in comparison with DMSO-treated cells as a negative control. Mean ± SD are reported. Results were obtained on PBMC isolated from 15 or 16 healthy blood donors and 5 different experiments (panel a), from 11 donors and 3 experiments (panel b), and from 16 donors and 5 experiments (panel c). Dots represent blood samples from different, and mean ± SEM are reported. Dunnett or Dunn test (ns = P > 0.05, *P < 0.05, ***P < 0.001, ****P < 0.0001).
To decipher the direct effect of ABX464 on CD4+ cells and avoid any effect mediated by interaction of CD4+ cells with other immune cells such as monocytes and macrophages present in PBMCs, CD4+ cells isolated from PBMCs were used. In isolated CD4+ cells, IL17a secretion was significantly decreased (−33.6%, P = 0.0019) by ABX464 treatment (Figure 3a); however, the levels of IL6 and TNF-α were not significantly changed. ABX464 treatment for 6 days significantly decreased the number of Th17 cells (−16%, P = 0.0368) (Figure 3b), the effect that is even more significant when looking for IL-17a-secreting cells (−25%, P = 0.0001), as well as the number of IL17a-secreting and IFNγ-secreting cells (−40.5%, P < 0.0001) (Figure 3c). However, ABX464 treatment did not change Th1+ or Th17+ like Th1+ cells (Figure 3b), which may be related to a more sensitive labeling of secreted cytokines vs extracellular labeling. As monocyte/macrophage can be part of PBMCs, the direct effect of 7-day treatment with ABX464 was tested on purified macrophages by analyzing secretion of cytokines. This analysis revealed significant decreases in CCL2, TNFα, and IL6 levels (−33.7%, −82.5%, and −93.3%, respectively; P < 0.05) (Figure 3d).
Figure 3.

Effects of ABX464 on CD4+ cells and macrophages. Cells were treated for 6 days with 5 μM ABX464. (a) Cytokine concentrations in the supernatant of CD4+ cells. (b) Th1/Th17 cells (CD196/CD183 expression) among viable CD4+ cells. (c) Intracellular interferon (IFN) γ and interleukin (IL) 17a expression among viable CD4+ cells. Dots represent blood samples from different donors, and mean ± SEM are reported. (d) Effects of ABX464 on macrophages. Cells were treated for 7 days with 5 μM ABX464. Results were obtained from 15 or 16 healthy blood donors and 5 different experiments (panel a), from 11 donors and 3 experiments (panel b), from 16 donors and 5 experiments (panel c), and from 7 donors and 3 experiments (panel d). Dots represent blood samples from different donors, and mean ± SEM are reported. Dunnett or Dunn test (ns = P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Overall, the most notable in vitro effect of ABX464 was the decrease in the Th17 population of PBMCs and of purified CD4+ cells. Th17 cells are known secretors of several cytokines, including IL-17A and IL-17F, IL-21, IL-22, IL-26, IL-8, IL-10, TNF-α, and GM-CSF, as well as the chemokine CXC ligands CXCL8 and CCL20 (19). IL17a, IFNγ (Th17+ like Th1+), and IL6 (through IL17 action on Th17 (20)), and the levels of these cytokines were decreased by ABX464 in PBMCs. The facts that ABX464 had modulatory effects on TNF-α and IL6 in PBMCs and macrophages (downregulation of TNF-α, IL6, and CCL2 levels) but not in purified CD4+ cells, and the greater magnitude of reduction in IL17 secretors in PBMCs compared with purified CD4+ cells may suggest a cooperative effect involving monocytes/macrophages and lymphocytes. The reduction in CCL2 production by macrophages in response to ABX464 supports the hypothesis that this chemoattractant is a target of miR-124 through which the effect of ABX464 is mediated. As ABX464 modulates both miR-124 (increase) and Th17/IL17 (decrease), one hypothesis is that miR-124 upregulation can affect the expression of cytokines by modulating signal transducer and activator of transcription 3 (STAT3) (18,21), a cytoplasmic signaling molecule that is central to many inflammatory processes (22), to ultimately dampen IL17a secretion by Th17 cells (see Discussion).
Patients with moderate-to-severe UC
To determine whether some of the effects observed in vitro can be found in the clinic, we assessed the expression of miR-124 in rectal biopsy and blood samples and we analyzed Th17 cells and relative concentrations of 9 cytokines (IL1b, IL4, IL6, IL10, IL23, IFNγ, CCL2, TNF-α, and IL17a) (see Supplementary Figure 1, Supplementary Digital Content 1, http://links.lww.com/CTG/A904) in the serum of patients with UC who were participating in a positive placebo-controlled clinical trial showing the efficacy of ABX464 (25, 50, or 100 mg) (10).
The number of miR-124 copies was determined by ddPCR, at baseline and after 8 weeks of treatment, in rectal biopsies and blood samples from patients who received ABX464 (25, 50, or 100 mg) or placebo during a phase 2b trial (10) (Figure 4). At baseline, there were no differences in the number of miR-124 copies in rectal biopsies in the placebo and treatment groups (the mean number of copies/cell: placebo, 685; ABX464 [25, 50, and 100 mg], 1,475, 1,417, and 1,029, respectively) (Figure 4a). At week 8, the number of miR-124 copies was significantly higher in the treatment groups than in the placebo group (the mean number of copies/cell: placebo, 746; ABX464 [25, 50, and 100 mg], 8,345, 8,347, and 9,171, respectively; Figure 4b). Among the treatment groups, there were no significant differences and no dose-dependent effects. The fold change in miR-124 was significant between the placebo and treated groups (P < 0.0001) (Figure 4c).
Figure 4.

MiR-124 modulation in rectal tissue (a–c) and blood (d–f) in patients with UC. The number of miR-124 copies was assessed by droplet digital PCR at baseline (a, d) and week 8 (b, e), and fold increase at week 8 compared with baseline (c, f). A total of 576 rectal biopsies from 139 patients and 1,104 blood samples from 229 patients who received ABX464 (25, 50, or 100 mg) or placebo were analyzed. Kruskal-Wallis test (***P < 0.001 vs placebo; ****P < 0.0001 vs placebo).
Since in vitro data showed an upregulation of miR-124 in human PBMCs, we assessed miR-124 expression in blood because it is expressed in both lymphocytes and monocytes. At baseline, there were no significant differences in the number of miR-124 copies in the serum samples of the placebo and treatment groups (the mean number of copies/cell: placebo, 32.27; ABX464 [25, 50, and 100 mg], 39.8, 19.42, and 48.20, respectively) (Figure 4d). At week 8, the number of miR-124 copies was significantly higher in each treatment group than in the placebo group (the mean number of copies/cell: placebo, 28.93; ABX464 [25, 50, and 100 mg], 4,598, 8,517, and 13,753, respectively). Significant dose-dependent upregulation was observed, as the number of miR-124 copies was significantly higher in the 50- and 100-mg groups than in the 25-mg group (P < 0.05 and P < 0.0001, respectively) (Figure 4e). The fold change in miR-124 was significant between the placebo and 25-mg groups (P < 0.001) and also between placebo and 50- and 100-mg groups (P < 0.0001) (Figure 4f).
For the cell and cytokine (IL1b, IL4, IL6, IL10, IL23, IFNγ, CCL2, TNF-α, and IL17a) analysis in serum samples from patients who received either ABX464 during the phase 2b trial (10), significant changes vs placebo were seen only for IL17a, at all doses, with no dose-dependent effect (Figure 5). Similarly, a reduction in Th17 population and IL17a expression was observed in serum samples, but it was not dose-dependent (see Discussion).
Figure 5.
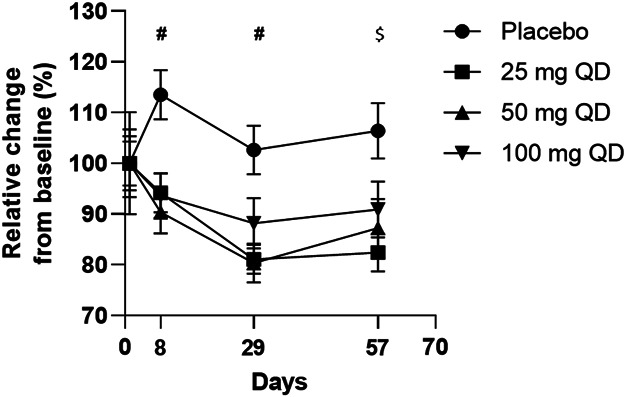
IL17a levels in serum from patients with UC treated with 25, 50, or 100 mg ABX464 daily or placebo for 8 weeks. A total of 1,185 serum samples (500 μL of serum per sample) from 254 patients who received either ABX464 (25, 50, or 100 mg) or placebo were assessed during 5 visits (day 1, weeks 1, 4, 8, and 16) of the phase 2b trial (10). ****P < 0.0001 vs placebo. #P < 0.01 for 25, 50, and 100 mg daily; $P < 0.01 for 25 and 50 mg daily. QD, every day (quaque die); UC, ulcerative colitis.
ABX464 reverses the increase in proinflammatory cytokine levels in the DSS colitis model
To gain a better understanding of the effect of ABX464 on the immune system at the onset of inflammation, we used a murine model of inflammation, the DSS model. In agreement with previously published data (23), DSS-induced weight loss was significantly reduced in mice receiving ABX464 (Figure 6). ABX464 exerts no effect on the immune system in the absence of inflammation (see Supplementary Figure 2, Supplementary Digital Content 1, http://links.lww.com/CTG/A904), confirming its mode of action as a modulator of immune cell activation during inflammation but not in its absence. After the establishment of colitis, the inflammation triggered by DSS in the colon significantly increased the secretion of a number of cytokines and chemokines (Figure 7a–c). Treatment with ABX464 significantly reversed the increases in several proinflammatory cytokines induced by DSS (GM-CSF, IL10, IL34, TPO, LIF, CXCL12, IL6, and CCL2) and maintained their levels close to those of healthy H2O-treated mice (Figure 7a–c). ABX464 treatment tended to reverse DSS-induced changes for IL17a, IL22, SCF, M-CSF, and IL1α (Figure 7a–c). However, IL12, IL15, TGFβ, IL3, and EPO were below the limit of detection in mouse colons of all groups analyzed.
Figure 6.
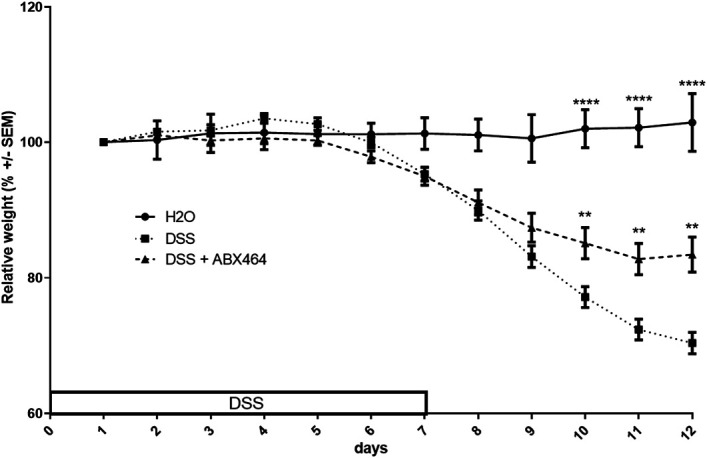
ABX464 treatment reduces disease severity in dextran sodium sulfate (DSS)-induced colitis. C57BL6 mice subjected to the DSS colitis protocol received orally once a day ABX464 (40 mg/kg) in methylcellulose (MC) or MC only through gavage. The weight profiles are representative of the curves obtained in all DSS experiments. They were obtained with 10 mice per group. **P < 0.01, ****P < 0.0001, 1-way ANOVA parametric test.
Figure 7.

Effects of ABX464 on cytokine secretion by colons of dextran sodium sulfate (DSS)-induced mice. The colons of untreated healthy mice (H2O group) or DSS-induced colitis mice treated by ABX464 (DSS + ABX464) or untreated (DSS) were cultured ex vivo at the end of an acute DSS experiment and assessed for cytokine secretions with (a) LegendPlex mu inflammation panel or (b) a LegendPlex mu HSC panel or (c) ELISA. Dots represent colons, and mean ± SEM are reported. The statistics are based on the fold change compared with DSS. Kruskal-Wallis with the Dunn multiple comparison test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). N = 26 mice per group.
Although T-cell modulations were observed in vitro, CD4+ T cells are not required for the induction of DSS colitis (24,25), but they do increase in number in the colon as the disease progresses (26). Importantly, changes in the percentage of T cells in spleen and MLNs can occur as early as day 1 exposure to DSS (26), indicating that during acute colitis, the adaptive immune system is already changing, although T cells are not required for induction. Given these findings, continued investigation into the role of CD4+ T cells in the DSS colitis model was warranted. Therefore, the effects of ABX464 on the MLN were examined. MLNs constitute a firewall that prevents live commensal intestinal bacteria from penetrating the systemic immune system (27). In mice, MLNs contribute to proinflammatory Th17-cell generation during inflammation in the small intestine (28). As expected, compared with healthy H2O-treated mice, mice with DSS-induced colitis showed significant increases in IL17a-secreting cells (+57.7%, P < 0.0001) (Figure 8a) and CD4+ Treg cells (+37.1%, P < 0.0001) (Figure 8c). ABX464 treatment reversed the DSS-induced upregulation of IL17a-secreting cells (−54.2%, P < 0.0001 vs untreated DSS mice) and maintained them at a level equivalent to that in healthy H2O-treated mice (P > 0.9999) (Figure 8a), which further support the effect of ABX464 on adaptive immunity through the reduction of the Th17-positive and CD4-positive subsets. In addition, MLNs from ABX464-treated DSS mice contained significantly fewer IFNγ-secreting CD4+ cells than untreated DSS mice (P < 0.0001), and ABX464 treatment decreased the ratio of IFNγ-secreting CD4+ cells in DSS mice to below that in healthy mice (P = 0.0004) (Figure 8b).
Figure 8.

Effects of ABX464 on CD4+ subsets in the mesenteric lymph nodes (MLNs) of dextran sodium sulfate (DSS)-induced mice. Cells in the MLNs of untreated healthy mice (H2O group), untreated mice with DSS-induced colitis (DSS group), or treated mice with DSS-induced colitis (DSS + ABX464) were collected and stained to determine proportions of (a) interleukin (IL) 17 secretors, (b) interferon (IFN) γ secretors, and (c) Treg cells. Dots represent samples from individual mice, and mean ± SEM are reported. Kruskal-Wallis tests with the Dunn multiple comparison test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
ABX464 counteracts the Th1 response that accompanies DSS exposure in mice and that is associated with a robust Th1-type immune response whose function is to eliminate infiltrating pathogens and promote tissue healing (29). Susceptibility to colonic inflammation may be linked to the intrinsic tendency of the immune system to generate Th1 responses, evidenced by the induction of IFN-γ production; however, Th17-mediated immunity is related to immune tolerance to the DSS challenge (30). This reduction in the Th1 response might increase the likelihood of the tissue becoming resistant to inflammatory responses, while a reduction in the Th17 response may confer protection. On the other hand, ABX464 treatment did not prevent the DSS-induced upregulation of CD4+ Treg cells (−13.1%, P = 0.2429) (Figure 8c). In certain patients, colitis involves a failure of immune regulation due to defects in Treg cells in MLNs (31); these defects could result from inherently defective Treg cells in the patient, or the causes of defective Treg cells could be a function of the abnormal intestinal environment and cytokine milieu in patients (32). The absence of a significant effect of ABX464 on CD4+ Treg cells may therefore contribute to attenuating DSS-induced colitis.
DISCUSSION
Collectively, the present findings provide evidence that upregulating miR-124 by ABX464 reverses the expression of several inflammatory cytokines triggered during inflammation. ABX464 may act as “a physiological brake” of inflammation, and it reverses the expression of several inflammatory cytokines but does not blunt the immune response altogether. This mechanism of action may explain why ABX464 provides a high remission rate at 1 year in UC, with a favorable tolerability and safety profile in patients with UC.
The reduction of Th17 cells is consistent with the anti-inflammatory effect of ABX464, as the inflamed gastrointestinal mucosa of patients with UC has a massive infiltration of Th17 cells (33,34) and Th17 cytokines are often increased in the inflamed intestinal mucosa of patients with active UC relative to unaffected regions and healthy controls (35–38). The absence of dose-dependent effect is more related to the mode of action of ABX464. Upregulation of miR-124 in CD4 naïve T cells can preferentially affect Th17 because STAT3, a crucial factor for Th17 differentiation, is a direct target of miR-124. For STAT3, miR-124 could adjust protein output in a manner that allows for customized expression in different CD4-cell types yet a more uniform level within each cell type while preventing Th17 differentiation which requires more STAT3 expression (39). In this context, even if the miR-124 upregulation is dose-dependent, the optimal STAT3 inhibition will not necessarily require the highest dose of miR-124 for Th17 differentiation.
ABX464 did not completely shut down the inflammatory response crucial to limit invasion by microorganisms but rather limits positive feedback amplification loops that promote immune cell activation that leads to significant tissue damage and evolution toward chronic disease. In this context, the effect of ABX464 on CCL2 secretion by activated macrophages, observed in vitro, should have a major effect on inflammation because this chemokine contributes to the restoration and resolution of tissue homeostasis by affecting the recruitment of naive T cells, monocytes, and neutrophils to the site of UC lesions. As these chemokines and other inflammatory cytokines are secreted locally in the gut, quantification of these cytokines/chemokines is currently investigated.
A previous study showed that ABX464 dampens DSS-induced colitis in mice and has long-term protective effects, and these effects are associated with a concomitant reduction in the production of several proinflammatory cytokines (23). Our findings that ABX464 treatment reduced established symptom of intestinal injury (weight loss) and reversed DSS-induced increases in multiple proinflammatory cytokines are consistent with those of previous studies (23). Among these proinflammatory markers, CCL2 is a target of miR-124, through which ABX464 mediates its effects. Our observation of the decreased levels of this chemoattractant in response to ABX464 is consistent with the previously demonstrated reduction in macrophage invasion in mucosal tissue (23). In addition, the observed decrease in the levels of IL-6 may be associated with the decreased numbers of proinflammatory macrophages observed in these tissues (23). A previous report showed that the interleukin-6 class cytokine LIF was markedly elevated in mouse colon tissue with DSS-induced peak disease severity due to bacterial invasion (40). The return of LIF to normal levels after ABX464 treatment is consistent with the finding that treatment-induced repair of the colon diminishes bacterial invasion (23). DSS disrupts the epithelial barrier and is associated with the infiltration of macrophages that express IL10 (41). As mentioned above, ABX464 reduces macrophage invasion in mucosal tissue (23), so the reduction in IL10 to normal levels was expected. On the other hand, there was no significant effect, but only a trend, of ABX464-mediated dampening of DSS-induced increases in several cytokines, including IL17a, suggesting that many interacting cellular components in the colon (e.g., epithelial and stromal cells) may contribute to different modulatory effects on cytokines. Indeed, although there is evidence showing a modulation of cytokine expression through epithelial and stromal cells (42,43), in vitro findings show that the effect of ABX464 on immune cells cannot be observed in epithelial and stromal cells (because miR-124 is not expressed in these cells). As a result, these cells may remain in an inflammatory state, with elevated cytokine expression. Alternatively, the lack of a significant decrease in these cytokines may be related to the time point of the analysis (D10), that is, 3 days after the end of the DSS administration. Although there is a clear anti-inflammatory effect of ABX464, exemplified by a significant reduction of DSS-induced weight loss, the weight of mice at D10 was not normalized to the weight of the non-DSS (noninflammatory) mice. It can be hypothesized that at the time of tissue collection to measure cytokine expression, recovery was not complete and cytokine levels were not normalized to the level of non-DSS mice. A kinetic analysis was performed to explore the variations of cytokines in the colon at the different time points of the DSS experiment (see Supplementary Figure 3, Supplementary Digital Content 1, http://links.lww.com/CTG/A904). The results show that for several cytokines (IL-1a, IL-1b, IFNγ, TNFα, TNFβ, and IL-17a) for which there were no changes at D10, significant differences were observed in the preceding days. The lack of significant effect at D10 may be explained by a decrease in cytokine levels in DSS mice from D8 to D10.
A large panoply of advanced therapies is available to patients with UC, but none offer deep remission to most patients (44,45), probably due to the complex nature of chronic inflammatory gastrointestinal diseases. Targeting IL17, as well as TNF-α, IL1, and IL6, using recombinant soluble receptors, antibodies, or inhibitors has demonstrated beneficial clinical outcomes in patients with autoimmune diseases that are refractory to glucocorticoid treatment. However, the long-term use of biologics raises safety concerns (46–48). There is still a need to improve the long-term clinical efficacy in UC while ensuring the safety of patients and preventing a refractory response to treatment. Among the ABX464-affected cytokines and chemokines in vitro, in patients with UC and the colon and MLNs of DSS-mice, IL17 and CCL2 are particularly notable because they are not known to be modulated by anti-inflammatory drugs. The effects of ABX464 on the levels of these 2 inflammatory mediators may be explained by ABX464-induced upregulation of miR-124. Indeed, as mentioned earlier, CCL2 is a known target of miR-124 (49) and the IL17-producing cells, Th17, are known to differentiate in response to IL1β, IL6, and IL23 through STAT3 signaling (19,50), which is also a known direct target of miR-124 (51). Given the key role attributed to Th17 cells in intestine inflammation, the effect of ABX464 on Th17/IL17a supports the efficacy of ABX464 as a potent anti-inflammatory drug candidate to treat patients with UC.
Targeting IL17 is a promising therapeutic approach for the treatment of UC (52). Still, to date, no clinical studies targeting the IL17 pathway are ongoing in UC, while in Crohn's disease, the immunomodulation attempt with the use of selective anti-IL17A antibody was ineffective and prematurely stopped for safety concerns (53). However, the ineffective results of clinical trials on the inhibition of IL17 in Crohn’s disease could be the clinical unmask of its context-dependent dual nature (54). IL17 is involved in the local control of barrier integrity and defense against extracellular pathogens, such as fungi and bacteria (55). In addition, the remaining cytokines of the IL17 family may take part in promoting inflammation in barrier organs or favoring repair of the gut mucosa after resolution of inflammation (56). An altered gut microbiota could be another possible explanation because gut microbiota has a pivotal role in the generation and functional training of innate and adaptive of immune cells, including Th17 cells, the most abundant CD4 T cells in mucosal tissues (57).
Unlike anti-17, ABX464 reduces the population of pathogenic Th17 population without affecting the repair process. Both in DSS mice and patients with UC, ABX464 treatment preserves the epithelium integrity. ABX464 induces a specific upregulation of miR-124 in naive T cells and affects proliferation of Th17 both in vitro, in treated patients with UC and in the DSS model. These effects can be explained by reduction of STAT3 protein, a key factor of Th17 proliferation/differentiation, whose encoding mRNA is a direct target of miR-124. ABX464, by upregulating miR-124 in macrophages, will also limit their activation and secretion of proinflammatory cytokines including IL23 and IL6. IL23 is an essential cytokine for TH17 proliferation; not surprisingly, several IL23-targeted biologics are currently in clinical trials. Reduction of IL23 is another way to explain the effect of ABX464 on the proliferation of TH17. In serum of patients with UC treated by ABX464, a significant reduction of IL23 was observed (Abivax data in file).
Compared with different strategies targeting Th17 including the blockade of Th17-cell differentiation and expansion (58), the neutralization of the cytokines produced by these cells (59), and the inhibition of the specific transcription factors required for Th17-cell function (19) which revealed in some cases the conflicting results, ABX464 introduces a novel anti-inflammatory mechanism to the field of UC treatments because it targets a functionally relevant and physiologically present miRNA, which selectively inhibits overexpression of multiple cytokines. Up to date, findings from the development program provide evidence that ABX464 dampens inflammation without impairing host defense, is safe, and does not allow the emergence of infection (Abivax, data in file). The results from the ongoing phase 3 trials of ABX464 (NCT05507203, NCT05507216, and NCT05535946), which are enrolling more than 1,200 patients with UC, are expected to confirm these statements.
CONFLICTS OF INTEREST
Guarantor of the article: Jamal Tazi, PhD.
Specific author contributions: C.A. and N.C. contributed equally to the experiments and should be considered as co-first authors. A.V., C.B.-P., L.L., A.G., and J.S. contributed to the experiments; A.G., J.S., and J.T. contributed to conducting experiments, acquiring data, analyzing data; A.G., J.S., D.S., P.G., H.E., and J.T. contributed to the conception of the manuscript; A.G., J.S., and J.T. contributed to the writing of the manuscript and literature review. The final version was approved by all authors. The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE).
Financial support: This study was supported by Abivax. The authors received no direct compensation related to the development of this manuscript.
Potential competing interests: C.A., N.C., A.V., L.L., D.S., P.G., A.G., J.S., H.E., and J.T. are employees at Abivax.
Supplementary Material
ACKNOWLEDGMENT
The authors thank P.A. Boyer on behalf of Abivax for his assistance in writing and preparing the manuscript.
Study Highlights.
WHAT IS KNOWN
✓ ABX464 (obefazimod) selectively upregulates miR-124 and has demonstrated durable efficacy in patients with ulcerative colitis.
WHAT IS NEW HERE
✓ ABX464 reverses the expression of several inflammatory cytokines triggered during inflammation.
✓ ABX464 acts as “a physiological brake” of inflammation and does not blunt the immune response.
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A904
Cécile Apolit and Noëlie Campos contributed equally to this work.
Contributor Information
Cécile Apolit, Email: Cecile.Apolit@abivax.com.
Noëlie Campos, Email: noelie.campos@abivax.com.
Audrey Vautrin, Email: Audrey.vautrin@abivax.com.
Christina Begon-Pescia, Email: christina.begon-pescia@umontpellier.fr.
Laure Lapasset, Email: laure.lapasset@abivax.com.
Didier Scherrer, Email: didier.scherrer@abivax.com.
Paul Gineste, Email: paul.gineste@abivax.com.
Hartmut Ehrlich, Email: hartmut.Ehrlich@abivax.com.
Aude Garcel, Email: aude.garcel@abivax.com.
Julien Santo, Email: julien.santo@abivax.com.
REFERENCES
- 1.Lee SH, Kwon Je, Cho ML. Immunological pathogenesis of inflammatory bowel disease. Intest Res 2018;16(1):26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med 2009;361(21):2066–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torres J, Mehandru S, Colombel JF, et al. Crohn's disease. Lancet 2017;389(10080):1741–55. [DOI] [PubMed] [Google Scholar]
- 4.Ungaro R, Mehandru S, Allen PB, et al. Ulcerative colitis. Lancet 2017;389(10080):1756–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mowat C, Cole A, Windsor A, et al. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011;60(5):571–607. [DOI] [PubMed] [Google Scholar]
- 6.Magro F, Cordeiro G, Dias AM, et al. Inflammatory bowel disease: Non-biological treatment. Pharmacol Res 2020;160:105075. [DOI] [PubMed] [Google Scholar]
- 7.Katz S, Liu Y. Challenges in the management of inflammatory bowel disease. In: Pitchumoni CS, Dharmarajan T. (eds.). Geriatric Gastroenterology. Springer: New York, NY, 2021, pp 1675–90. [Google Scholar]
- 8.Gordon JP, McEwan PC, Maguire A, et al. Characterizing unmet medical need and the potential role of new biologic treatment options in patients with ulcerative colitis and Crohn's disease: A systematic review and clinician surveys. Eur J Gastroenterol Hepatol 2015;27(7):804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vermeire S, Hebuterne X, Tilg H, et al. Induction and long-term follow-up with ABX464 for moderate-to-severe ulcerative colitis: Results of phase IIa trial. Gastroenterology 2021;160(7):2595–8.e3. [DOI] [PubMed] [Google Scholar]
- 10.Vermeire S, Sands BE, Tilg H, et al. ABX464 (obefazimod) for moderate-to-severe, active ulcerative colitis: A phase 2b, double-blind, randomised, placebo-controlled induction trial and 48 week, open-label extension. Lancet Gastroenterol Hepatol 2022;7(11):1024–35. [DOI] [PubMed] [Google Scholar]
- 11.Tazi J, Begon-Pescia C, Campos N, et al. Specific and selective induction of miR-124 in immune cells by the quinoline ABX464: A transformative therapy for inflammatory diseases. Drug Discov Today 2021;26(4):1030–9. [DOI] [PubMed] [Google Scholar]
- 12.Ponomarev ED, Veremeyko T, Barteneva N, et al. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat Med 2011;17(1):64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin Z, Wang PY, Su DF, et al. miRNA-124 in immune system and immune disorders. Front Immunol 2016;7:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma C, Li Y, Li M, et al. microRNA-124 negatively regulates TLR signaling in alveolar macrophages in response to mycobacterial infection. Mol Immunol 2014;62(1):150–8. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Qin Z, Li Q, et al. MicroRNA-124 negatively regulates LPS-induced TNF-α production in mouse macrophages by decreasing protein stability. Acta Pharmacol Sin 2016;37(7):889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin Y, Zhang S, Luo H, et al. Interleukin 7 up-regulates CD95 protein on CD4+ T cells by affecting mRNA alternative splicing: Priming for a synergistic effect on HIV-1 reservoir maintenance. J Biol Chem 2015;290(1):35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei J, Wang F, Kong LY, et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res 2013;73(13):3913–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y, Li Q, Gui H, et al. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res 2013;23(11):1270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddur MS, Miossec P, Kaveri SV, et al. Th17 cells: Biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol 2012;181(1):8–18. [DOI] [PubMed] [Google Scholar]
- 20.Ogura H, Murakami M, Okuyama Y, et al. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008;29(4):628–36. [DOI] [PubMed] [Google Scholar]
- 21.Cheng XJ, Li L, Xin BQ. MiR-124 regulates the inflammation and apoptosis in myocardial infarction rats by targeting STAT3. Cardiovasc Toxicol 2021;21(9):710–20. [DOI] [PubMed] [Google Scholar]
- 22.Hu YS, Han X, Liu XH. STAT3: A potential drug target for tumor and inflammation. Curr Top Med Chem 2019;19(15):1305–17. [DOI] [PubMed] [Google Scholar]
- 23.Chebli K, Papon L, Paul C, et al. The anti-HIV candidate Abx464 dampens intestinal inflammation by triggering Il-22 production in activated macrophages. Sci Rep 2017;7(1):4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Axelsson LG, Landstrom E, Goldschmidt TJ, et al. Dextran sulfate sodium (DSS) induced experimental colitis in immunodeficient mice: Effects in CD4(+)-cell depleted, athymic and NK-cell depleted SCID mice. Inflamm Res 1996;45(4):181–91. [DOI] [PubMed] [Google Scholar]
- 25.Dieleman LA, Ridwan BU, Tennyson GS, et al. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994;107(6):1643–52. [DOI] [PubMed] [Google Scholar]
- 26.Hall LJ, Faivre E, Quinlan A, et al. Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Dig Dis Sci 2011;56(1):79–89. [DOI] [PubMed] [Google Scholar]
- 27.Macpherson AJ, Smith K. Mesenteric lymph nodes at the center of immune anatomy. J Exp Med 2006;203(3):497–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawabe T, Suzuki N, Yamaki S, et al. Mesenteric lymph nodes contribute to proinflammatory Th17-cell generation during inflammation of the small intestine in mice. Eur J Immunol 2016;46(5):1119–31. [DOI] [PubMed] [Google Scholar]
- 29.Dieleman LA, Palmen MJHJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol 2001;114(3):385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang F, Wang D, Li Y, et al. Th1/Th2 balance and Th17/Treg-mediated immunity in relation to murine resistance to dextran sulfate-induced colitis. J Immunol Res 2017;2017:7047201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayne CG, Williams CB. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm Bowel Dis 2013;19(8):1772–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol 2010;10(12):849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weaver CT, Elson CO, Fouser LA, et al. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol Mech Dis 2013;8(1):477–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med 2009;15(5):199–207. [DOI] [PubMed] [Google Scholar]
- 35.Leal RF, Planell N, Kajekar R, et al. Identification of inflammatory mediators in patients with Crohn's disease unresponsive to anti-TNFα therapy. Gut 2015;64(2):233–42. [DOI] [PubMed] [Google Scholar]
- 36.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003;52(1):65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arijs I, Li K, Toedter G, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut 2009;58(12):1612–9. [DOI] [PubMed] [Google Scholar]
- 38.Seiderer J, Elben I, Diegelmann J, et al. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): Upregulated colonic IL-17F expression in active Crohn's disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis 2008;14(4):437–45. [DOI] [PubMed] [Google Scholar]
- 39.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004;116(2):281–97. [DOI] [PubMed] [Google Scholar]
- 40.Zhang YS, Xin DE, Wang Z, et al. STAT4 activation by leukemia inhibitory factor confers a therapeutic effect on intestinal inflammation. EMBO J 2019;38(6):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B, Alli R, Vogel P, et al. IL-10 modulates DSS-induced colitis through a macrophage-ROS-NO axis. Mucosal Immunol 2014;7(4):869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med 2017;49(5):e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y, Wang Y, Li Q, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol 2018;14(8):493–507. [DOI] [PubMed] [Google Scholar]
- 44.Maronek M, Link R, Ambro L, et al. Phages and their role in gastrointestinal disease: Focus on inflammatory bowel disease. Cells 2020;9(4):1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dal Buono A, Roda G, Argollo M, et al. Treat to target or 'treat to clear' in inflammatory bowel diseases: One step further? Expert Rev Gastroenterol Hepatol 2020;14(9):807–17. [DOI] [PubMed] [Google Scholar]
- 46.Nielsen OH, Ainsworth MA. Tumor necrosis factor inhibitors for inflammatory bowel disease. N Engl J Med 2013;369(8):754–62. [DOI] [PubMed] [Google Scholar]
- 47.van der Have M, Oldenburg B, Kaptein AA, et al. Non-adherence to anti-TNF therapy is associated with illness perceptions and clinical outcomes in outpatients with inflammatory bowel disease: Results from a prospective multicentre study. J Crohns Colitis 2016;10(5):549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vogelaar L, Spijker AV, van't Spijker A. The impact of biologics on health-related quality of life in patients with inflammatory bowel disease. Clin Exp Gastroenterol 2009;2:101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamachi Y, Kawano S, Takenokuchi M, et al. MicroRNA-124a is a key regulator of proliferation and monocyte chemoattractant protein 1 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum 2009;60(5):1294–304. [DOI] [PubMed] [Google Scholar]
- 50.Woś I, Tabarkiewicz J. Effect of interleukin-6, -17, -21, -22, and -23 and STAT3 on signal transduction pathways and their inhibition in autoimmune arthritis. Immunol Res 2021;69(1):26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Lu Y, Yue X, et al. MiR-124 suppresses growth of human colorectal cancer by inhibiting STAT3. PLoS One 2013;8(8):e70300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noviello D, Mager R, Roda G, et al. The IL23-IL17 immune axis in the treatment of ulcerative colitis: Successes, defeats, and ongoing challenges. Front Immunol 2021;12:611256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17a monoclonal antibody, for moderate to severe Crohn's disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012;61(12):1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaffen SL, Jain R, Garg AV, et al. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat Rev Immunol 2014;14(9):585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011;332(6025):65–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol 2008;1(5):339–49. [DOI] [PubMed] [Google Scholar]
- 57.Ivanov II, Frutos RdL, Manel N, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 2008;4(4):337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bettelli E, Korn T, Kuchroo VK. Th17: The third member of the effector T cell trilogy. Curr Opin Immunol 2007;19(6):652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guglani L, Khader SA. Th17 cytokines in mucosal immunity and inflammation. Curr Opin HIV AIDS 2010;5(2):120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.