

Abstract
The contrasting actions of mechanical forces and glucocorticoids (GC) on bone have been long recognized. However, the cellular and molecular mechanisms by which these stimuli impact the skeleton remain only partially known. Recent evidence gained from studies on bone cell apoptosis has revealed that mechanical forces and GC exhibit converse effects on osteocyte and osteoblast survival resulting from divergent actions on the focal adhesion kinases FAK and Pyk2, molecules that regulate integrin-dependent interactions between bone cells and the extracellular matrix (ECM). This prospect reviews these findings and poses the possibility that similar opposing effects on kinase signaling are responsible for other actions of mechanical forces and GC on the skeleton, in particular on bone formation and the Wnt signaling pathway.
Keywords: MECHANICAL FORCES, GLUCOCORTICOIDS, FAK, Pyk2, OSTEOCYTES, BONE FORMATION, Wnt PATHWAY
Mechanical forces enhance bone mass and strength, whereas chronic GC excess decreases bone formation and increases bone fragility. Bone anabolism induced by mechanical stimuli is associated with increased pre-osteoblast proliferation, acceleration of osteoblast differentiation, and inhibition of osteoblast and osteocyte apoptosis [Aguirre et al., 2006; Robling and Turner, 2009; Turner et al., 2009]. Remarkably, all these actions are also promoted by activation of the Wnt pathway [Bodine and Komm, 2006; Bodine, 2008]. Moreover, bone loading activates Wnt-dependent transcription in osteocytes [Robinson et al., 2006] and downregulates the Wnt antagonists sclerostin expressed by osteocytes and Dkk1 expressed by osteoblasts and osteocytes [Robling et al., 2008]. Furthermore, the Wnt co-receptor LRP5 is required for a full anabolic response to mechanical loading [Sawakami et al., 2006], strongly suggesting that Wnt signaling is a crucial component of mechanotransduction in bone. In contrast, GC excess induces a profound decrease in bone formation resulting at least partially from inhibition of osteoblast differentiation and induction of osteoblast apoptosis [Weinstein, 2001]. The increase in bone fragility induced by GC is also associated with enhanced prevalence of osteocyte apoptosis. GC also inhibit β-catenin-dependent transcription, stimulate Forkhead box O3a (FOXO) transcription, increase Dkk1 expression, and induce reactive oxygen species (ROS) [Canalis et al., 2007]—all events that counteract the Wnt signaling pathway.
Recent in vitro studies revealed that the converse effects of mechanical forces and GC on apoptosis of osteoblasts and osteocytes result from opposing actions on kinases of the focal adhesion family focal adhesion kinase (FAK) and proline-rich tyrosine kinase 2 (Pyk2) and downstream signaling pathways. Thus, mechanical stimuli prevent apoptosis of osteocytes and osteoblasts by a mechanism that requires activation of FAK and extracellular signals regulated kinases (ERKs) [Plotkin et al., 2005]. On the other hand, GC oppose these survival signals by activating the pro-apoptotic kinases Pyk2 and c-Jun N-terminal kinase (JNK) [Plotkin et al., 2007a]. Remarkably, FAK/ERK activation and anti-apoptosis induced by mechanical stimulation in osteocytes is abolished by interfering with the Wnt signaling pathway [Martin-Millan et al., 2008]; and conversely, Pyk2-dependent apoptosis by GC is counteracted by Wnts [Almeida et al., 2005]. These findings give credence to the notion that there is an antagonistic interplay between mechanical forces and GC governed by FAK/Pyk2 signaling that impinges on the Wnt pathway and regulates osteocyte and osteoblast survival. Whether similar opposing effects on kinase signaling are responsible for other contrasting actions of mechanical forces and GC on the skeleton, in particular on bone formation, awaits elucidation.
THE FAK/Pyk2 PATHWAY AND OUTSIDE-IN AND INSIDE-OUT SIGNALING MEDIATED BY INTEGRINS
Cells are surrounded by a complex protein network, the ECM, which exerts a tight control on cellular functions profoundly affecting proliferation, differentiation and survival. Most effects of the ECM are mediated by integrins, cell surface receptors responsible for attachment of cells to the matrix and for conveying chemical and mechanical signals from it. Integrins interact with ECM proteins and with intracellular structural and catalytic molecules at sites on the cell surface called focal adhesions [Clark and Brugge, 1995]. Because they lack intrinsic enzymatic activity, signaling through integrins requires their association with molecules capable of signal transduction. This phenomenon is facilitated by integrin location within caveolae, specialized plasma membrane microdomains rich in caveolin-1. Direct interaction of integrin β1 with caveolin-1 results in association of the integrin with Src kinases and phosphorylation of downstream substrates including Shc46/52 [Plotkin et al., 2005]. A molecule crucial for integrin signaling is FAK. Integrin engagement induces FAK autophosphorylation followed by its interaction with Src, which in turn further phosphorylates FAK leading to ERK activation. Due to their unique ability to convey information from the extracellular environment as well as from the intracellular space, signaling mediated by integrins is bidirectional [Hynes, 2002]. Hence, binding of ECM proteins to integrin receptors triggers intracellular signaling, a process called outside-in signaling. Conversely, intracellular signals or changes in the composition of the focal adhesions regulate the extracellular binding activity of integrins to matrix proteins, referred to as inside-out signaling.
Accumulating evidence demonstrates that mechanical stimulation of osteoblastic and osteocytic cells induced by substrate stretching or fluid flow renders cells that are protected from apoptosis [Pavalko et al., 2003; Bakker et al., 2004; Plotkin et al., 2005]. Anti-apoptosis induced by mechanical forces requires activation of ERKs, but not phosphatidylinositol-3 kinase (PI3K) or p38 [Plotkin et al., 2005]. This ERK-activating pro-survival effect requires nuclear translocation of the kinases and new gene transcription and it is transmitted by integrins β1, α5, and α2 and a signalsome comprising actin filaments, microtubules, caveolae, and Src kinases [Plotkin et al., 2005]. Furthermore, protection from apoptosis requires activation of FAK, since a dominant negative autophosphorylation-deficient mutant Y397F FAK abolished the survival effect of stretching. FAK has been also shown to be required for the induction of mechanoresponsive genes [Young et al., 2009]. These findings support the notion that transduction of mechanical forces into prosurvival signaling is exerted by outside-in signaling mediated by integrins and a signalsome comprising the focal adhesion kinase FAK and Src kinases; resulting in activation of the ERK pathway (Fig. 1).
Fig. 1.
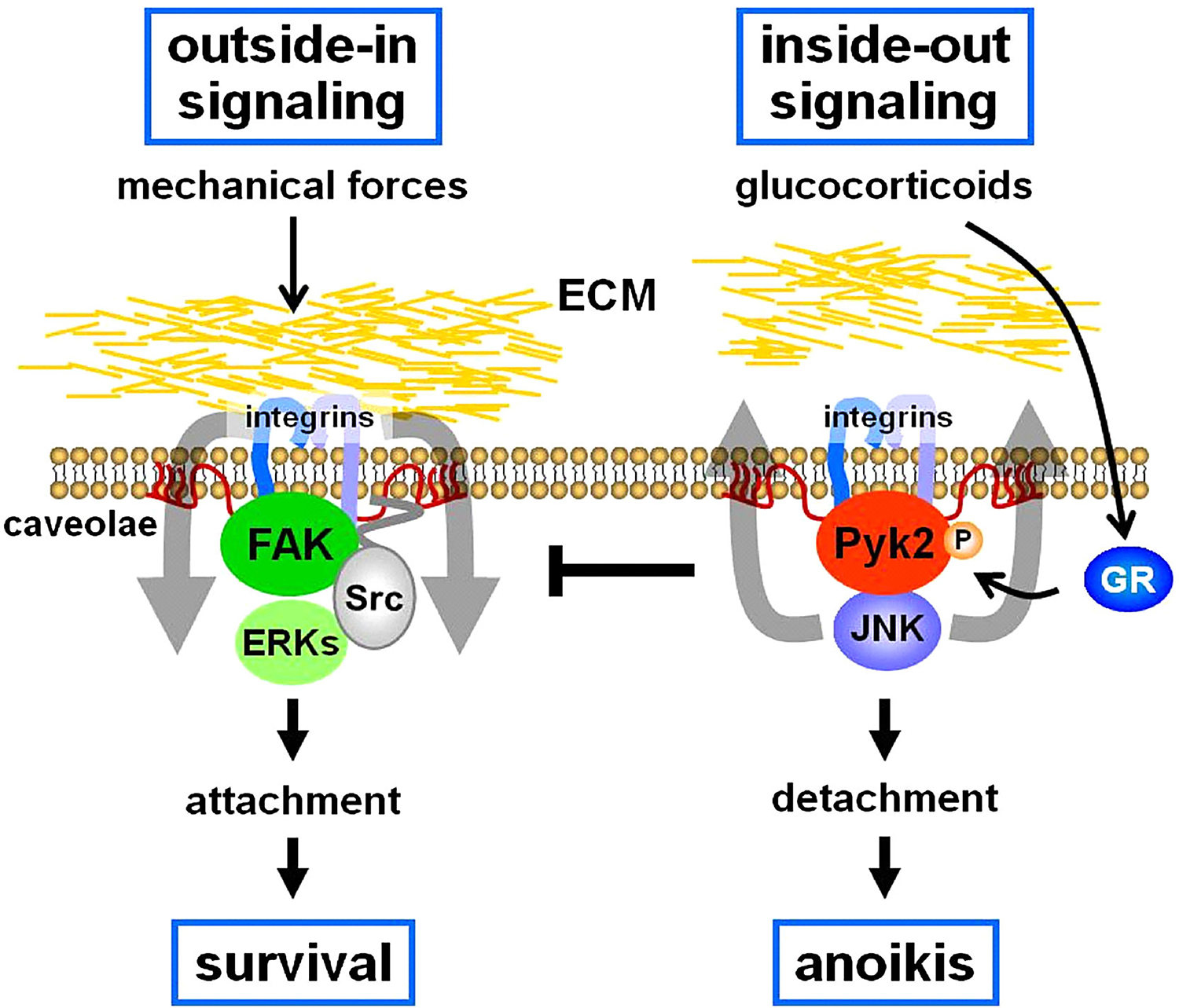
Opposing actions of mechanical stimuli and GC on the FAK/Pyk2 pathway regulate osteocyte and osteoblast apoptosis. GR:GC receptor.
The in vivo relevance of FAK for bone anabolism induced by mechanical forces however has not been addressed. FAK null mice exhibit embryonic lethality [Furuta et al., 1995], precluding the study of the skeletal phenotype. Mice in which FAK is lacking only in osteoblastic cells by virtue of its deletion using Cre-lox approaches (FAKflox/flox;Col1a1–2.3kb-Cre mice) are born normal and without skeletal abnormalities [Kim et al., 2007]. Although basal osteoblast differentiation is not affected, matrix deposition is poor leading to delayed bone healing. Furthermore, ectopic matrix deposition induced by controlled movement of implants in the marrow cavity is defective in these mice [Leucht et al., 2007]. These findings suggest that the response of marrow osteoprogenitors to mechanical stimulation might be impaired in the absence of FAK, but do not provide a direct proof that FAK is required for mechanotransduction in vivo. Future experiments using recognized models of mechanical stimulation, such as the murine forelimb-loading model, would be required to address directly the role of FAK-mediated outside-in signaling in mechanotransduction in bone. In addition, to dissect the individual contributions of osteoblasts versus osteocytes to loading-induced osteogenesis, bone cell specific FAK deletion should be pursued using the available osteoblast or osteocyte specific promoters driven Cre recombinase.
Pyk2 is another member of the FAK family of non-receptor tyrosine kinases, also called related adhesion focal tyrosine kinase (RAFTK), cellular adhesion kinase (CAKβ), or calcium-dependent tyrosine kinase (CADTK). Although Pyk2 and FAK are highly homologous, they exhibit opposite effects on cell fate. Thus, whereas FAK activation leads to cell spreading and survival, Pyk2 induces reorganization of the cytoskeleton, cell detachment, and apoptosis [Xiong and Parsons, 1997]. We have shown that the pro-apoptotic effect of GC on osteocytes is due to interference with FAK-mediated survival signals, through activation of Pyk2 [Plotkin et al., 2007a] (Fig. 1). Hence, osteocytic cells lacking Pyk2 or overexpressing FAK are protected from GC-induced apoptosis [Plotkin et al., 2005, 2007a]. Whether Pyk2 plays a role in the effects of GC on the skeleton remains to be investigated. Specifically, future studies are required to establish whether animals lacking Pyk2 are refractory to the deleterious effects of GC on osteocyte and osteoblast viability, as well as on osteoblastogenesis and bone formation; and whether in the absence of FAK in vivo there is an enhanced response to GC due to unopposed Pyk2 signaling.
MECHANOTRANSDUCTION: REQUIREMENT OF FAK AND THE Wnt/β-CATENIN PATHWAY
Skeletal loading is essential for the maintenance of weight-bearing bones. Bone strength is plastic and can be modulated in adults, as illustrated by increased bone mass in the playing arms of athletes. We have shown that mechanical loading improves bone strength by inducing bone formation in regions of high strain energy [Turner et al., 2009], demonstrating the existence of a mechanosensing apparatus that targets osteogenesis to where bone is needed. The most likely sensors of mechanical forces are the osteocytes, which through interaction with the ECM enhance their biological outcomes in response to increasing loading frequencies.
The precise mechanism of bone anabolism by mechanical stimulation remains uncertain. However, the reduced expression of the Wnt antagonists sclerostin and Dkk1 [Robling et al., 2008] associated with rapid activation of the Wnt pathway in osteocytes upon loading [Robinson et al., 2006] as well as the dependence on LRP5 expression to achieve a full anabolic response to mechanical stimulation [Sawakami et al., 2006] strongly suggest that the Wnt pathway is essential for mechanotransduction. Because changes in cytosolic calcium, ATP, prostaglandins and NO accompany sclerostin downregulation [Bonewald and Johnson, 2008], it is likely that other intracellular mediators participate in the skeletal response to loading. Furthermore, the demonstration that outside-in signaling mediated by FAK is crucial for osteocyte survival induced by mechanical stimulation in vitro [Plotkin et al., 2005] together with evidence that mechanically-induced FAK/ERK activation and anti-apoptosis is abolished by Dkk1 or degradation of β-catenin [Martin-Millan et al., 2008], suggest that ECM-integrin/FAK signaling is linked with the Wnt/β-catenin pathway. Another molecule required for mechanotransduction is the estrogen receptor (ER). Thus, osteoblastic cells derived from mice lacking ERα/β or osteocytic cells in which the ERs are silenced are not protected from apoptosis by mechanical stimuli, and the response is rescued by transfection of either receptor [Aguirre et al., 2007]. The ERs interact with caveolin and with Src kinases, and an ERα unable to bind caveolin or to locate in the cell membrane fails to confer the antiapoptotic effect of mechanical stimulation. Consistent with these in vitro findings on cell survival, mice lacking ERα or ERβ do not exhibit the normal anabolic response to loading [Lee et al., 2003, 2004]. Remarkably, the ERα is also required for β-catenin nuclear accumulation and Wnt-dependent transcription in response to loading in vitro [Armstrong et al., 2007]. These pieces of evidence support the hypothesis that membrane-associated forms of the ER collaborate with FAK in transducing mechanical signals into osteocyte/osteoblast survival. Further studies are needed to directly test this possibility.
SKELETAL ACTIONS OF GLUCOCORTICOIDS: Pyk2 REQUIREMENT AND INHIBITION OF THE Wnt/β-CATENIN PATHWAY
GC, produced and released by the adrenal glands in response to stress, regulate numerous physiological processes in a wide range of tissues. Among many other effects, these hormones exert profound immunosuppressive and anti-inflammatory actions and induce apoptosis of many cell types, including T lymphocytes and monocytes. Because of these properties, GC are extensively used for the treatment of immune and inflammatory conditions, the management of organ transplantation, and as components of chemotherapy regimens for hematological cancers. However, long-term use of GC is associated with severe adverse side effects manifested in several organs. In particular, prolonged use of these drugs leads to a dramatic loss of bone mineral and strength, similar to endogenous elevation of GC.
Bone loss induced by GC excess has two phases [Weinstein, 2001; Canalis et al., 2007]. The initial loss results from a transient increase in resorption due to delayed osteoclast apoptosis. This is followed by a sustained and profound reduction in bone formation and turnover resulting from decreased osteoblast and osteoclast generation and increased osteoblast apoptosis. GC also increase apoptosis of osteocytes, which contributes to bone fragility. The pro-apoptotic effect of GC results from direct actions on these cells. Thus, apoptosis is readily demonstrable in cultured osteocytes and osteoblasts (Plotkin et al., 2007a); and, furthermore, transgenic mice overexpressing the enzyme that inactivates GC 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) in osteocytes and osteoblasts are protected from GC-induced bone fragility (O’Brien et al., 2004).
The mechanism of glucocorticoid action involves binding to the GC receptor (GR), conformational changes and nuclear translocation of the ligand-bound receptor, followed by cis or trans interactions with DNA and thereby induction or repression of gene transcription. In addition, glucocorticoids exert actions independently of changes in gene transcription. Such actions include modulation of the activity of intracellular kinases like ERKs, JNK, and Pyk2. Our findings demonstrate that apoptosis of osteoblastic cells by GC, although mediated by the GC receptor, is independent of new gene transcription and results from rapid Pyk2 and JNK activation [Plotkin et al., 2007a]. This is consistent with evidence supporting a role for Pyk2 on GC-induced apoptosis of myeloma cells [Chauhan et al., 1999]. More recently, we found that the small GTPase RhoA, and its target Rock, are involved in actin reorganization leading to anoikis induced by GC [Plotkin et al., 2007b]. RhoA/Rock might be activated directly by Pyk2 or indirectly by ROS elevation induced by GC [Ohtsu et al., 2005; Dada et al., 2007] (Fig. 2). A role of kinase-mediated effects is supported by findings in which transgenic mice expressing a dimerization-deficient GC receptor lacking DNA binding ability maintain a response to GC in bone [Tuckermann et al., 2005]. The in vivo relevance of the Pyk2/JNK/RhoA/Rock signaling could be probed by examining whether pharmacological or genetic interference with this pathway prevents or at least ameliorates the deleterious effects of GC on the skeleton.
Fig. 2.
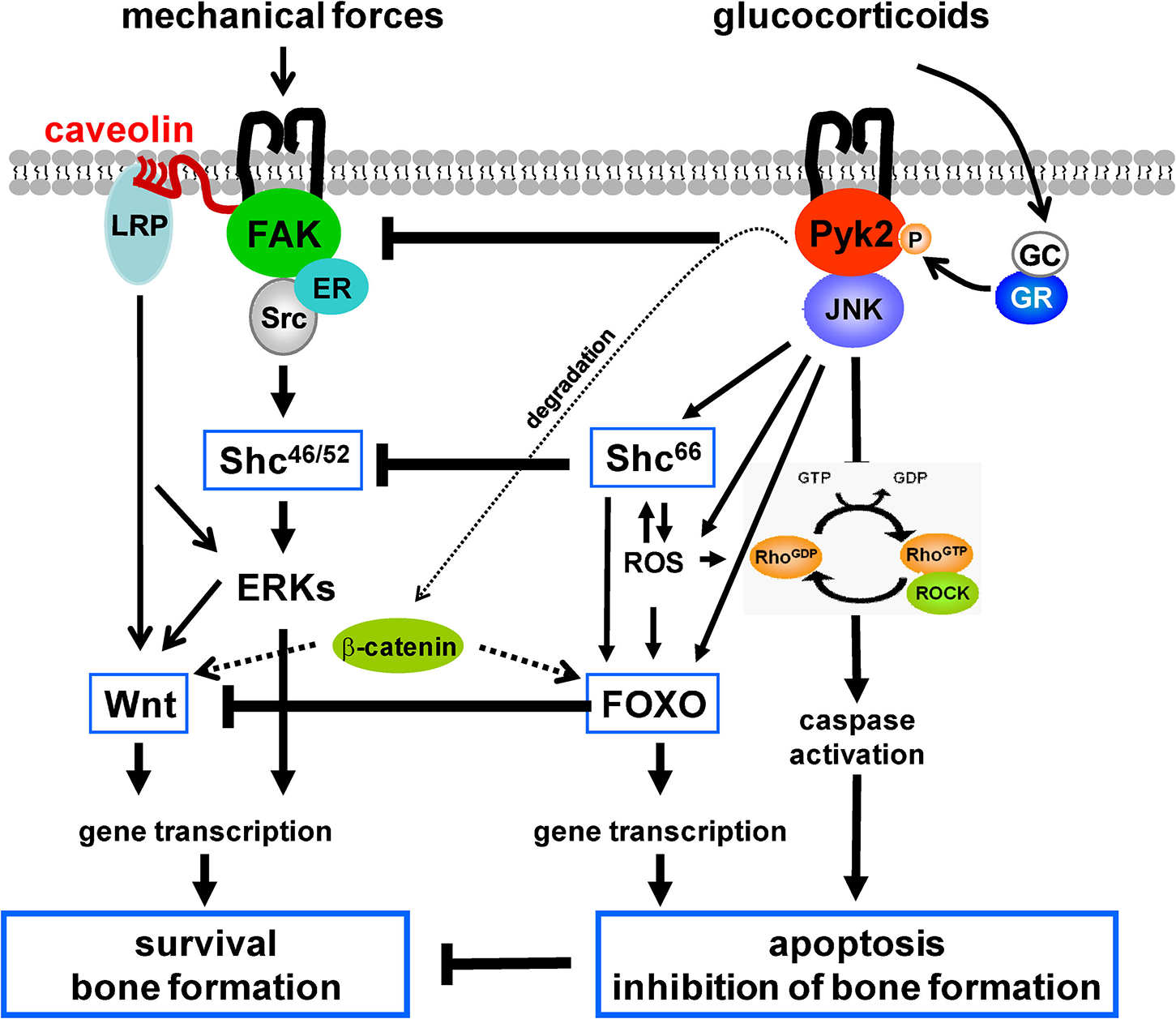
Working model: Antagonism between mechanical forces and GC governed by FAK/Pyk2 signaling, regulating the Wnt/β-catenin pathway, bone formation, and osteocyte and osteoblast survival.
GC might promote apoptosis of osteoblasts by a similar mechanism than the one we described for osteocytes. Indeed, survival of both osteoblasts and osteocytes is maintained by their interaction with ECM, as neutralizing antibodies to fibronectin or inhibitors of metaloproteinases (MMP) induce osteoblast apoptosis [Globus et al., 1998; Karsdal et al., 2002, 2004]. Moreover, transgenic mice expressing collagenase-resistant collagen type-I exhibit increased osteoblast and osteocyte apoptosis [Zhao et al., 2000]; and, osteocyte apoptosis is also elevated in MMP2 null mice [Inoue et al., 2006]. Therefore, cellular interactions with cryptic domains of ECM proteins exposed by MMPs appear to be required for osteocyte and osteoblast viability. Pyk2 involvement in cell-ECM interactions controlling osteoblast as well as osteocyte apoptosis by GC remains to be determined.
Increasing evidence indicate that GC inhibit Wnt signaling. GC increase expression of the antagonists Dkk1 and secreted frizzled-related protein 1 (SFRP1) and inhibit βcatenin-dependent transcription [Ohnaka et al., 2004, 2005; Wang et al., 2005; Leclerc et al., 2008]. In addition, GC activate Shc66 [Almeida et al., 2008], a redox enzyme of the same family that Shc46/52 but with pro-apoptotic properties due to its ability to generate mitochondrial ROS. Shc66 is itself a key component of the signaling cascade activated by ROS inducing apoptosis, thereby contributing to perpetuating pro-apoptotic signaling [Giorgio et al., 2005]. Shc66 might be a key player in Wnt inhibition by GC. Thus, Shc66 activates Forkhead box O (FOXO) transcription factors, either directly or through ROS [Essers et al., 2005]. Because β-catenin is required for FOXO-mediated transcription, there is a competition between FOXO- and the β-catenin activated transcription factor T-cell factor- (TCF) mediated transcription [Essers et al., 2005]. In osteoblastic cells, ROS antagonizes the skeletal effects of Wnt/β-catenin by diverting β-catenin from TCF- to FOXO-mediated transcription [Almeida et al., 2007]. In addition, we found that indeed ROS-dependent activation of FOXO is required for GC-induced apoptosis of osteocytes and osteoblasts [Almeida et al., 2008]. Remarkably, Pyk2 activates GSK3β leading to β-catenin degradation in several cell models, including CHO and neuronal cells [Hartigan et al., 2001; Sayas et al., 2006], suggesting an additional mechanism by which GC could inhibit Wnt/β-catenin signaling [Yun et al., 2008].
Increased endogenous GC may contribute to the bone fragility associated with old age. Thus, similar to patients treated with GC, older individuals are approximately 10 times more likely to suffer fractures than younger individuals with the same mineral density [Hui et al., 1988]. Similar to GC excess, aging in mice is associated with increased osteocyte and osteoblast apoptosis and decreased bone strength that is not completely explained by a reduction in bone mineral density [Weinstein et al., 2009]. Although Pyk2 null mice do not show gross anatomical alterations, recent studies revealed that these animals exhibit higher bone mass. This phenotype appears to result from defective osteoclast attachment and resorption leading to osteopetrosis [Gil-Henn et al., 2007] combined with increased bone formation and improved bone structure [Buckbinder et al., 2007]. Whether the response to GC is altered in Pyk2 deficient mice or whether Pyk2 null mice are protected from aging-induced bone fragility warrants further investigations.
CONCLUDING REMARKS
In closing, mounting evidence supports the hypothesis that GC antagonize bone anabolism promoted by physiological bone loading at different levels downstream of FAK/Pyk2 signaling, by favoring activation of Pyk2 versus FAK, Shc66 versus Shc46/52, and FOXO- versus Wnt/ERK-dependent transcription (Fig. 2). Whether these opposing actions do indeed stem from divergent activation of rapid kinase signaling or are independent effects of GC remains to be directly investigated. Advancing our knowledge of the cellular and molecular mechanisms by which mechanical forces and GC act on bone has the potential of improving therapeutic interventions attempting to preserve or restore skeletal health in patients treated with GC or exhibiting endogenous GC elevation such, as during aging.
REFERENCES
- Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T. 2006. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Min Res 21:605–615. [DOI] [PubMed] [Google Scholar]
- Aguirre JI, Plotkin LI, Gortazar AR, O’Brien CA, Manolagas SC, Bellido T. 2007. A novel ligand-independent function of the estrogen receptor is essential for osteocyte and osteoblast mechanotransduction. J Biol Chem 282:25501–25508. [DOI] [PubMed] [Google Scholar]
- Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. 2005. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem 280:41342–41351. [DOI] [PubMed] [Google Scholar]
- Almeida M, Han L, Martin-Millan M, O’Brien CA, Manolagas SC. 2007. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem 282:27298–27305. [DOI] [PubMed] [Google Scholar]
- Almeida M, Ambrogini E, Martin-Millan M, Han L, Warren A, Shelton RS, Plotkin LI, Bellido T, O’Brien CA, Jilka RL, Weinstein RS, Manolagas SC. 2008. Induction of oxidative stress and diversion of β-catenin from TCF- to FOXO-mediated transcription by glucocorticoids or TNFα in osteoblastic cells. J Bone Min Res 23:S170 (Abstr.). [Google Scholar]
- Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, Lanyon LE. 2007. Wnt/β-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor α. J Biol Chem 282:20715–20727. [DOI] [PubMed] [Google Scholar]
- Bakker A, Klein-Nulend J, Burger E. 2004. Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem Biophys Res Commun 320:1163–1168. [DOI] [PubMed] [Google Scholar]
- Bodine PV. 2008. Wnt signaling control of bone cell apoptosis. Cell Res 18:248–253. [DOI] [PubMed] [Google Scholar]
- Bodine PV, Komm BS. 2006. Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord 7:33–39. [DOI] [PubMed] [Google Scholar]
- Bonewald LF, Johnson ML. 2008. Osteocytes, mechanosensing and Wnt signaling. Bone 42:606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckbinder L, Crawford DT, Qi H, Ke HZ, Olson LM, Long KR, Bonnette PC, Baumann AP, Hambor JE, Grasser WA, et al. 2007. Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci USA 104:10619–10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canalis E, Mazziotti G, Giustina A, Bilezikian JP. 2007. Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporos Int 18:1319–1328. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Hideshima T, Pandey P, Treon S, Teoh G, Raje N, Rosen S, Krett N, Husson H, Avraham S, et al. 1999. RAFTK/PYK2-dependent and -independent apoptosis in multiple myeloma cells. Oncogene 18:6733–6740. [DOI] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. 1995. Integrins and signal transduction pathways: The road taken. Science 268:233–239. [DOI] [PubMed] [Google Scholar]
- Dada LA, Novoa E, Lecuona E, Sun H, Sznajder JI. 2007. Role of the small GTPase RhoA in the hypoxia-induced decrease of plasma membrane Na,K-ATPase in A549 cells. J Cell Sci 120:2214–2222. [DOI] [PubMed] [Google Scholar]
- Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. 2005. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308:1181–1184. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Ilic D, Kanazawa S, Takeda N, Yamamoto T, Aizawa S. 1995. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene 11:1989–1995. [PubMed] [Google Scholar]
- Gil-Henn H, Destaing O, Sims NA, Aoki K, Alles N, Neff L, Sanjay A, Bruzzaniti A, De Camilli P, Baron R, et al. 2007. Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(−/−) mice. J Cell Biol 178:1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, et al. 2005. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122:221–233. [DOI] [PubMed] [Google Scholar]
- Globus RK, Doty SB, Lull JC, Holmuhamedov E, Humphries MJ, Damsky CH. 1998. Fibronectin is a survival factor for differentiated osteoblasts. J Cell Sci 111:1385–1393. [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Xiong WC, Johnson GV. 2001. Glycogen synthase kinase 3β is tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun 284:485–489. [DOI] [PubMed] [Google Scholar]
- Hui SL, Slemenda CW, Johnston CC Jr. 1988. Age and bone mass as predictors of fracture in a prospective study. J Clin Invest 81:1804–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R 2002. Integrins: Bidirectional, allosteric signaling machines. Cell 110:673–687. [DOI] [PubMed] [Google Scholar]
- Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, Park JS, Onodera T, Krane SM, Noda M, et al. 2006. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem 281:33814–33824. [DOI] [PubMed] [Google Scholar]
- Karsdal MA, Larsen L, Engsig MT, Lou H, Ferreras M, Lochter A, Delaisse JM, Foged NT. 2002. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J Biol Chem 277:44061–44067. [DOI] [PubMed] [Google Scholar]
- Karsdal MA, Andersen TA, Bonewald L, Christiansen C. 2004. Matrix metalloproteinases (MMPs) safeguard osteoblasts from apoptosis during transdifferentiation into osteocytes: MT1-MMP maintains osteocyte viability. DNA Cell Biol 23:155–165. [DOI] [PubMed] [Google Scholar]
- Kim JB, Leucht P, Luppen CA, Park YJ, Beggs HE, Damsky CH, Helms JA. 2007. Reconciling the roles of FAK in osteoblast differentiation, osteoclast remodeling, and bone regeneration. Bone 41:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc N, Noh T, Cogan J, Samarawickrama DB, Smith E, Frenkel B. 2008. Opposing effects of glucocorticoids and Wnt signaling on Krox20 and mineral deposition in osteoblast cultures. J Cell Biochem 103:1938–1951. [DOI] [PubMed] [Google Scholar]
- Lee K, Jessop H, Suswillo R, Zaman G, Lanyon L. 2003. Endocrinology: Bone adaptation requires oestrogen receptor-alpha. Nature 424:389. [DOI] [PubMed] [Google Scholar]
- Lee KC, Jessop H, Suswillo R, Zaman G, Lanyon LE. 2004. The adaptive response of bone to mechanical loading in female transgenic mice is deficient in the absence of oestrogen receptor-alpha and -beta. J Endocrinol 182:193–201. [DOI] [PubMed] [Google Scholar]
- Leucht P, Kim JB, Currey JA, Brunski J, Helms JA. 2007. FAK-Mediated mechanotransduction in skeletal regeneration. PLoS ONE 2:e390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Millan M, Plotkin LI, Vyas K, Frera G, Gortazar AR, Almeida M, Manolagas SC, Bellido T. 2008. Kinase activation and osteocyte survival promoted by mechanical stimulation require LRP5/6 signaling and β-catenin accumulation, but not β-catenin/TCF-dependent transcription. J Bone Min Res 23:S400. (Abstr.). [Google Scholar]
- O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. 2004. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 145:1835–1841. [DOI] [PubMed] [Google Scholar]
- Ohnaka K, Taniguchi H, Kawate H, Nawata H, Takayanagi R. 2004. Glucocorticoid enhances the expression of dickkopf-1 in human osteoblasts: Novel mechanism of glucocorticoid-induced osteoporosis. Biochem Biophys Res Commun 318:259–264. [DOI] [PubMed] [Google Scholar]
- Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. 2005. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun 329:177–181. [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Mifune M, Frank GD, Saito S, Inagami T, Kim-Mitsuyama S, Takuwa Y, Sasaki T, Rothstein JD, Suzuki H, et al. 2005. Signal-crosstalk between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration of vascular smooth muscle cells stimulated by angiotensin II. Arterioscler Thromb Vasc Biol 25:1831–1836. [DOI] [PubMed] [Google Scholar]
- Pavalko FM, Gerard RL, Ponik SM, Gallagher PJ, Jin Y, Norvell SM. 2003. Fluid shear stress inhibits TNF-alpha-induced apoptosis in osteoblasts: A role for fluid shear stress-induced activation of PI3-kinase and inhibition of caspase-3. J Cell Physiol 194:194–205. [DOI] [PubMed] [Google Scholar]
- Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T. 2005. Mechanical stimulation prevents osteocyte apoptosis: Requirement of integrins, Src kinases and ERKs. Am J Physiol Cell Physiol 289:C633–C643. [DOI] [PubMed] [Google Scholar]
- Plotkin LI, Manolagas SC, Bellido T. 2007a. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival: Evidence for inside-out signaling leading to anoikis. J Biol Chem 282:24120–24130. [DOI] [PubMed] [Google Scholar]
- Plotkin LI, Vyas K, Manolagas SC, Bellido T. 2007b. The small GTPase RhoA and its effector kinase ROCK mediate actin cytoskeleton reorganization leading to osteocyte anoikis by glucocorticoids. J Bone Min Res 22:S107 (Abstr.). [Google Scholar]
- Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, Kharode Y, Sauter L, Babij P, Brown EL, et al. 2006. WNT/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem 281:31720–31728. [DOI] [PubMed] [Google Scholar]
- Robling AG, Turner CH. 2009. Mechanical signaling for bone modeling and remodeling. Crit Rev Eukaryot Gene Expr 19:319–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MJ, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido T, Harris SE, et al. 2008. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283:5866–5875. [DOI] [PubMed] [Google Scholar]
- Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, et al. 2006. The WNT co-receptor LRP5 is essential for skeletal mechanotransduction, but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem 281:23698–23711. [DOI] [PubMed] [Google Scholar]
- Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH. 2006. GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell 17:1834–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckermann J, Schilling AF, Priemel M, Stride B, Kirilov M, Wintermantel T, Tronche F, Amling M, Schultz G. 2005. Glucocorticoid induced osteoporosis requires the glucocorticoid receptor in osteoblasts and does not depend on DNA binding of the receptor. J Bone Min Res 20:S27 (Abstr.). [Google Scholar]
- Turner CH, Warden SJ, Bellido T, Plotkin LI, Kumar N, Jasiuk I, Danzig J, Robling AG. 2009. Mechanobiology of the skeleton. Sci Signal 2:t3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang FS, Lin CL, Chen YJ, Wang CJ, Yang KD, Huang YT, Sun YC, Huang HC. 2005. Secreted frizzled-related protein 1 (SFRP1) modulates glucocorticoid attenuation of osteogenic activities and bone mass. Endocrinology 146:2415–2423. [DOI] [PubMed] [Google Scholar]
- Weinstein RS. 2001. Glucocorticoid-induced osteoporosis. Rev Endocr Metab Disord 2:65–73. [DOI] [PubMed] [Google Scholar]
- Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O’Brien CA, Thostenson J, Roberson PK, Boskey AL, Clemens TL, et al. 2009. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in 21-month-old mice. Aging Cell 9:147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Parsons JT. 1997. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J Cell Biol 139:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SR, Gerard-O’Riley R, Kim JB, Pavalko FM. 2009. Focal adhesion kinase is important for fluid shear stress-induced mechanotransduction in osteoblasts. J Bone Miner Res 24:411–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SI, Yoon HY, Jeong SY, Chung YS. 2009. Glucocorticoid induces apoptosis of osteoblast cells through the activation of glycogen synthase kinase 3β. J Bone Miner Metab 27(2):140–148. [DOI] [PubMed] [Google Scholar]
- Zhao W, Byrne MH, Wang Y, Krane SM. 2000. Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest 106:941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]