

OVERVIEW
The rapidly changing landscape of oncology has brought new light, and with it, new challenges to optimizing therapeutic strategies for patients. Although the concept of patient heterogeneity is well known to any practicing clinician, a more detailed understanding of the biologic changes that underscore the clinical picture is beginning to emerge. Thus, tumor heterogeneity has come to encompass more than just the clinical picture and can represent both intratumor and intertumor differences. Within the fields of thoracic oncology and melanoma, the discovery of key molecular drivers has resulted in landmark breakthroughs in therapy. However, the complexities of tumor genetics and the interaction within the environment continue to drive the search for better therapies. Ongoing challenges include the accurate and timely assessment of genetic changes as well as the development of resistance and the resultant compensatory mechanisms. Novel technologies, including commercially available next-generation sequencing, have allowed for a greater breadth and depth of information to be gained from a single pathologic specimen, and it is now being incorporated into routine clinical practice. Translational advances have subsequently provided valuable insight into mechanisms of resistance, with the development of novel treatment strategies. Future work will focus on novel diagnostic techniques and adaptive mechanisms that can ultimately drive the development of the next generation of cancer therapy.
In an address to the New Haven Medical Association in 1903, Sir William Osler, an icon of modern medicine, declared, “Variability is the law of life… no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”1 Certainly, all clinicians involved in the care of patients with cancer have a deep understanding of this sentiment. In our day-to-day practices, we are accustomed to the variability, or heterogeneity, we observe among different patients diagnosed with the same type of cancer. This heterogeneity comes from differences in age, gender, and comorbid conditions as well as numerous other factors that influence disease course and treatment decisions. Although this demographic heterogeneity is part- and-parcel to the daily practice of oncologists, there is increasing necessity to incorporate tumor heterogeneity into our daily practice and decision-making plans.
Tumor heterogeneity is not one distinct term but rather encompasses several facets that render tumors unique.2,3 At the highest level, there is variability both within an individual tumor (intratumor heterogeneity) and across several different tumors (intertumor heterogeneity; Fig. 1). Just as no two patients are the same, no two tumors are the same, even if they carry the identical histopathologic diagnosis. Tumors arising from the same cell type in different patients may share some common attributes (for example, expression of a particular cell surface protein) but are not identical. This intertumor heterogeneity is also referred to as population heterogeneity.4
FIGURE 1. Intertumor and Intratumor Heterogeneity.
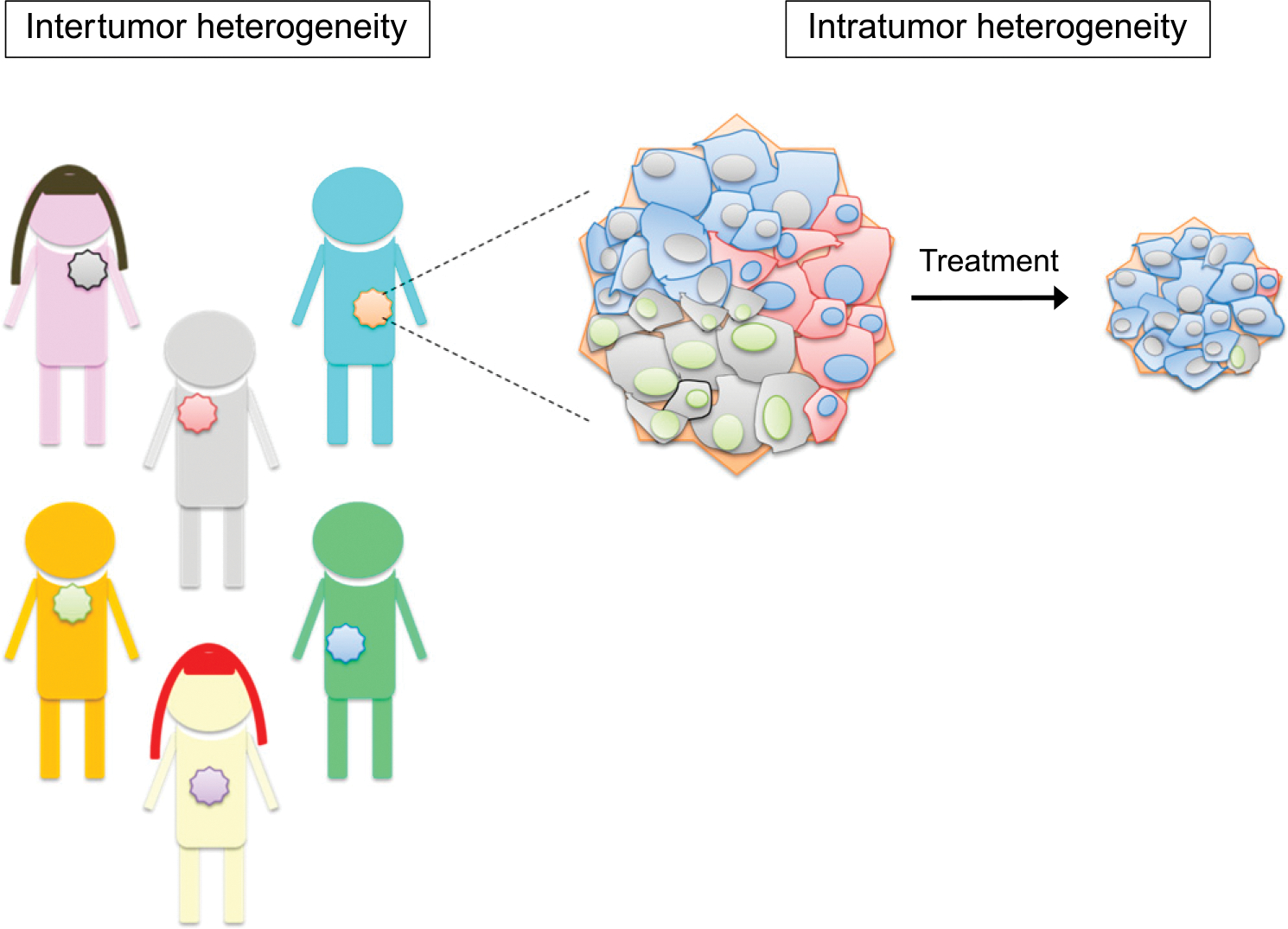
Intertumor heterogeneity results from variability across different tumors from different individuals, even with the same histopathologic diagnosis. Intratumor heterogeneity results from variability within an individual tumor. Subpopulations exist within a given tumor, as represented by the different colored cells (red, blue, and gray) shown within the tumor. These subpopulations, often referred to as tumor clones, may differ in cell morphology, genetic makeup, metabolism, proliferation rate, and, ultimately, response to therapy.
Conversely, intratumor heterogeneity refers to the presence of different cell subpopulations within a given tumor sample. These subpopulations, which are often referred to as tumor clones, may differ in cell morphology, genetic makeup, metabolism, proliferation rate, and metastatic potential.2,3,5 Indeed, intratumor heterogeneity exists at several levels. Most commonly, intratumor heterogeneity is equated with genetic and epigenetic variability. Recent advances in sophisticated “-omic”-based technologies have resulted in detailed analyses of tumor samples, even to the single cell level. These types of analyses have characterized the molecular variations between different tumor samples (intertumor heterogeneity) and between different cells in the same tumor, overall leading to an increasingly detailed and complex view of tumor cell biology. For example, deep sequencing of tumor cells has revealed differences in present mutations and genes expressed within a given tumor sample, which have therapeutic implications for the patient.
Molecular variabilities, or tumor intrinsic properties, are not the only source of tumor heterogeneity. Extrinsic properties of the tumor cell—including interactions with surrounding cells and proximity to blood vessels—can influence the tumor cell phenotype, leading to what is termed positional heterogeneity.4 For example, tumor cells can interact with other tumor cells, the extracellular matrix, surrounding stromal cells, and immune cells. These cell-to-cell communications affect the signals the tumor cell receives, thereby modulating properties such as metabolism, motility, and drug responsiveness. The location of the tumor can also influence the availability of nutrients, oxygen, and growth factors, all of which can modulate tumor cell growth.2,3,5 Furthermore, temporal heterogeneity refers to the concept that tumors are constantly changing under various selective pressures. Patient-specific factors, such as medications, concurrent illnesses, nutrition, and hormonal status, as well as the cancer therapies themselves, may influence the evolution of a tumor.4
What is the origin of intratumor heterogeneity? Two models have been postulated to explain this phenomenon.3,4 First, the cancer stem cell model proposes that a small subpopulation of cells with self-renewal properties drive tumor progression. The differentiation of these cancer stem cells may generate the variability observed within a tumor and may also be responsible for the repopulation of a tumor after treatment. Second, the clonal evolution model proposes that premalignant or malignant cells accumulate genetic changes over time due to inherent genomic instability. These changes may confer selective advantages or disadvantages for the cell, and, over time, the cells that acquire advantageous genetic changes are selected in a Darwinian-like evolutionary process. The cells continue to accumulate genetic changes, thereby driving the diversification of the tumor and leading to the phenotypes observed in an advanced cancer—abnormal proliferation, invasion, evasion of apoptosis, and drug resistance.
It is important to note that these models are not mutually exclusive. Several studies have now demonstrated that clonal evolution may occur within the cancer stem cell compartment. For example, Notta et al showed that samples obtained at the time of diagnosis from patients with BCR-ABL lymphoblastic leukemia contain multiple genetically distinct leukemia-initiating stem cell subclones.6
INTEGRATING THE HETEROGENEITY OF CANCER IN CLINICAL DECISION MAKING
Intertumor and intratumor heterogeneity are important obstacles to overcome when designing the most effective therapeutic strategies for patients with cancer. The genotypic and phenotypic variability of tumors can have important consequences for diagnosis, prognosis, and treatment. However, our ability to interrogate the underlying heterogeneity present within a given tumor sample is limited by several factors. First, analysis of a single core biopsy sample or fine needle aspirate—techniques that are commonly used for the diagnosis of advanced cancers—may not be an accurate representation of a given tumor site or of the tumor as a whole for an individual patient. The full spectrum of distinct clones may not be captured within one small region from a tumor biopsy specimen.5,7 Therefore, the treatment selected based on a single biopsy site may not result in response in all areas of the tumor. In addition, a biopsy is only reflective of a single moment in time within the tumor’s evolution, and there is no standard way to clinically predict how a tumor will evolve over time. A repeat biopsy of the tumor at the time of disease progression during treatment with a given therapy to assess changes in genetic and phenotypic makeup has not been routine practice until recently, and, even now, this practice is not the standard of care for all tumor types.
As diagnostic platforms advance and clinical treatment paradigms progress, our ability to effectively tackle the problem of tumor heterogeneity increases. This clinical relevance of this evolution is highlighted here using examples from the fields of lung cancer and melanoma. In lung cancer, initial trials of the EGFR tyrosine kinase inhibitors (TKIs), erlotinib and gefitinib, in an unselected patient population yielded less than promising results, with only a small subset of patients who experienced a benefit, albeit dramatically. Subsequent research efforts focused on identifying molecular markers of response leading to the identification of activating mutations within the EGFR tyrosine kinase domain in the tumor samples from those patients who derived substantial antitumor benefit.8,9 This finding took a large heterogeneous group of patients with lung cancer, encompassing variability in clinical factors and tumor histology, and narrowed down drug selection to a specific tumor genotype. Conversely, in melanoma, a relevant genetic target in BRAF was identified in 2002; approximately half of all melanomas carry an activating mutation at amino acid position 600 (V600E),10 yet therapeutic agents with appropriate selectivity did not exist. Sorafenib, initially developed as a RAF-1 inhibitor, failed to benefit patients with BRAF-mutated advanced melanoma.11,12 The subsequent development of the highly selective BRAF inhibitors vemurafenib and dabrafenib resulted in meaningful clinical benefit in large randomized studies, completely altering the treatment landscape for this subset of patients.13,14 However, even BRAF, a validated oncogenic target that has been successfully targeted in melanoma, is clearly not one-size-fits-all. The BRAF gene is mutated in 1% to 3% of non–small cell lung cancers (NSCLCs), 10% of colorectal cancers, 50% of papillary thyroid cancers, and in varying degrees in other malignancies.15–18 However, although early-phase studies in lung and papillary thyroid cancers yielded promising clinical activity, responses are rarely seen in BRAF-mutated colorectal cancer.19–21
ADVANCES IN GENETIC TESTING
Next-generation sequencing (NGS) has facilitated remarkable advances in terms of the breadth and depth of potential information that can now be discerned from a single tumor biopsy. The molecular profile of lung cancer is highly heterogeneous, and research continues to discover new markers to characterize and target. One study evaluated the molecular profile of patients with a broad number of cancer histologies.22 Although this particular study characterized the molecular profile of this cohort with great detail, only 7% of the cohort had lung cancer. Vigneswaran et al collected and characterized the genomic profiles of 364 patients with NSCLC whose tumor biopsy samples were analyzed by NGS over 5 years at a single institution.23 Out of those 364 patients, 289 (75%) patients were diagnosed with adenocarcinoma. Most of the patients (99.4%) had genomic alterations with an average of 10.8 alterations per sample. In the molecular profile of the adenocarcinoma cohort, 23.3% had EGFR alterations, 32.5% had KRAS alterations, 7.5% had ALK alterations, 10.8% had RET alterations, 5.0% had MET alterations, and 0.8% had ROS1 alterations. Of the samples with KRAS mutations, 90% were from smokers, which included current and former smokers. Furthermore, 46% of EGFR and 61% of ALK alterations were detected in never smokers.
Kris et al evaluated the genomic profiles of 733 patients with lung cancer as part of the multi-institutional Lung Cancer Mutation Consortium (LCMC) study. An oncogenic driver mutation was found in 466 of 733 patients (64%).24 Among the 733 tumors, the distribution of known driver mutations was 182 (25%) in KRAS, 122 (17%) TKI-sensitizing in EGFR, 57 (8%) ALK rearrangements, 29 (4%) other EGFR mutations, 24 (3%) with two or more concurrent alterations, 19 (3%) in ERBB2, 16 (2%) in BRAF, six (less than 1%) in PIK3CA, five (less than 1%) MET amplifications, five (less than 1%) in NRAS, one (less than 1%) in MEK1, and none in AKT1. Results were used to select a targeted therapy or trial among 275 of 1,007 patients (28%).
Despite the numerous advantages, however, traditional platforms still require the availability of tumor tissue, and thus an invasive procedure, which limits the ability to obtain multiple samples in real time. Most recently, the development and clinical implementation of sophisticated NGS technologies that can interrogate for genomic alterations in plasma-derived cell-free tumor DNA are being used with greater frequency. These so-called liquid biopsies have been shown to track the evolution of resistance mutations, such as EGFR T790M and EGFR C797S, during therapy over a patient’s disease course, thereby giving clinicians a way to monitor tumor clones and assess genetic heterogeneity.25,26 For patients with melanoma, detection of BRAF mutations in circulating tumor DNA has been shown to be feasible and may have some predictive value.27,28 Importantly, additional limited and defined mutations in RAF, as well as mutations in RAS, have been described as resistance mechanisms to RAF/MEK inhibitors; these alterations can also be assessed through sensitive measures of circulating cell-free tumor DNA.
Most importantly, the ability to understand the heterogeneity leading to drug resistance and the ability to monitor this heterogeneity over time has led to the development of more refined treatment strategies that account for the heterogeneity, and, hopefully, it will result in better outcomes for patients.
ACQUIRED RESISTANCE
Unfortunately, even patients who derive substantial initial benefit from targeted therapy will eventually experience disease progression, a concept referred to as acquired resistance. Acquired resistance simply refers to genotypic and/or phenotypic changes within the tumor that alter drug sensitivity, the natural selection of drug-tolerant clones. Understanding that tumors evolve over time under selective pressures such as drug therapy, the study of acquired resistance to EGFR TKI therapy necessitated the acquisition of a fresh biopsy specimen at the time of disease progression. Analysis of these tumors revealed the presence of a second site mutation within EGFR, T790M, which is attributed to the development of approximately 50% of cases of acquired resistance to EGFR TKI therapy.29 Other pathways also play a role in the development of resistance, including MET, in which amplification has been shown to cause resistance in approximately 5% of patients.30,31 Tumor rebiopsy at the time of acquired resistance is now the accepted standard of care for patients with EGFR-mutant lung cancer. Tumor rebiospy allows for evaluation of T790M and several other potential resistance mechanisms, such as transition to small cell lung cancer histology, all of which have important therapeutic implications.
Acquired resistance to the EGFR TKIs, erlotinib and gefitinib, can also be driven by activation of several receptor tyrosine kinases, including MET,32,33 HER2,34 HER3,35 IGF-1R,36 and AXL.37 In addition, activation of the MAPK pathway can drive EGFR TKI resistance in vitro and in vivo.29,30
In targeting BRAF, the picture of acquired resistance is similarly complex, and a deeper understanding has led to important clinical developments. Early on, the recognition that MAPK pathway reactivation was a major cause of resistance to BRAF inhibitor therapy led to the development of combined BRAF/MEK inhibition. However, the heterogeneity of resistance mechanisms presents a challenge in creating a rationally designed combination strategy that will be applicable for all patients. In one of the largest series to date, 132 samples obtained at disease progression during treatment with a BRAF inhibitor were analyzed using whole-exome sequencing (WES). A defined mechanism of resistance was identified in only 58%, with the most common being NRAS or KRAS mutations. Other alterations included BRAF splice variants, BRAF amplification, MEK1 and MEK2 mutations, and non-MAPK mechanisms. Most striking was the marked heterogeneity in patients who had more than one biopsy at progression; 18 of 19 patients had distinct mechanisms identified in each sample.38 Earlier work has identified other pathways, including PDGFR, IGF-1R, PI3K, and EGFR, among others.12 Although dual BRAF/MEK inhibition has resulted in improved response rates and, in some studies, improved survival for patients with BRAF-mutated melanoma, progression remains a concern.39–41 Early analyses to assess for mechanisms of resistance suggest that many overlapping pathways may still play a role.32,42 The collection of tissue in future studies will remain critical to further elucidate additional mechanisms of resistance and to define optimal therapeutic combinations.
Assessing the importance of compensatory pathways within the kinome may also provide additional insight into the degree of heterogeneity seen in tumors driven by the RAS/RAF/MAPK pathway.23,43 Inhibition of the RAS/RAF/MAPK pathway has been shown to upregulate many receptor tyrosine kinase pathways, particularly the PDGFR, VEGF, and EGFR pathways. Profiling of compensatory pathways has been studied in only a limited fashion to date among patients with cancer.44–46 Relatedly, RAS/RAF/MEK inhibition has been shown to modulate the host immune response, including alteration of T-cell responses and downregulation of mediators such as PD-L1, IL-1, and IL-8.47,48 These factors can directly modulate tumor heterogeneity, in part by modulating the micro-environment and immune checkpoint expression.47,48 In one of the few reports on this topic, changes in the inflammatory mediators IGFBP3 and PDGF-BB were correlated with benefit from trametinib in a randomized phase II study of this agent in advanced pancreatic cancer.49
REPORTS
Here, we highlight representative examples from our own clinical practice in which patients either have well-documented genomic alterations and are receiving straightforward treatment, or uncharacterized alterations, which demand a multifaceted approach.
Case 1
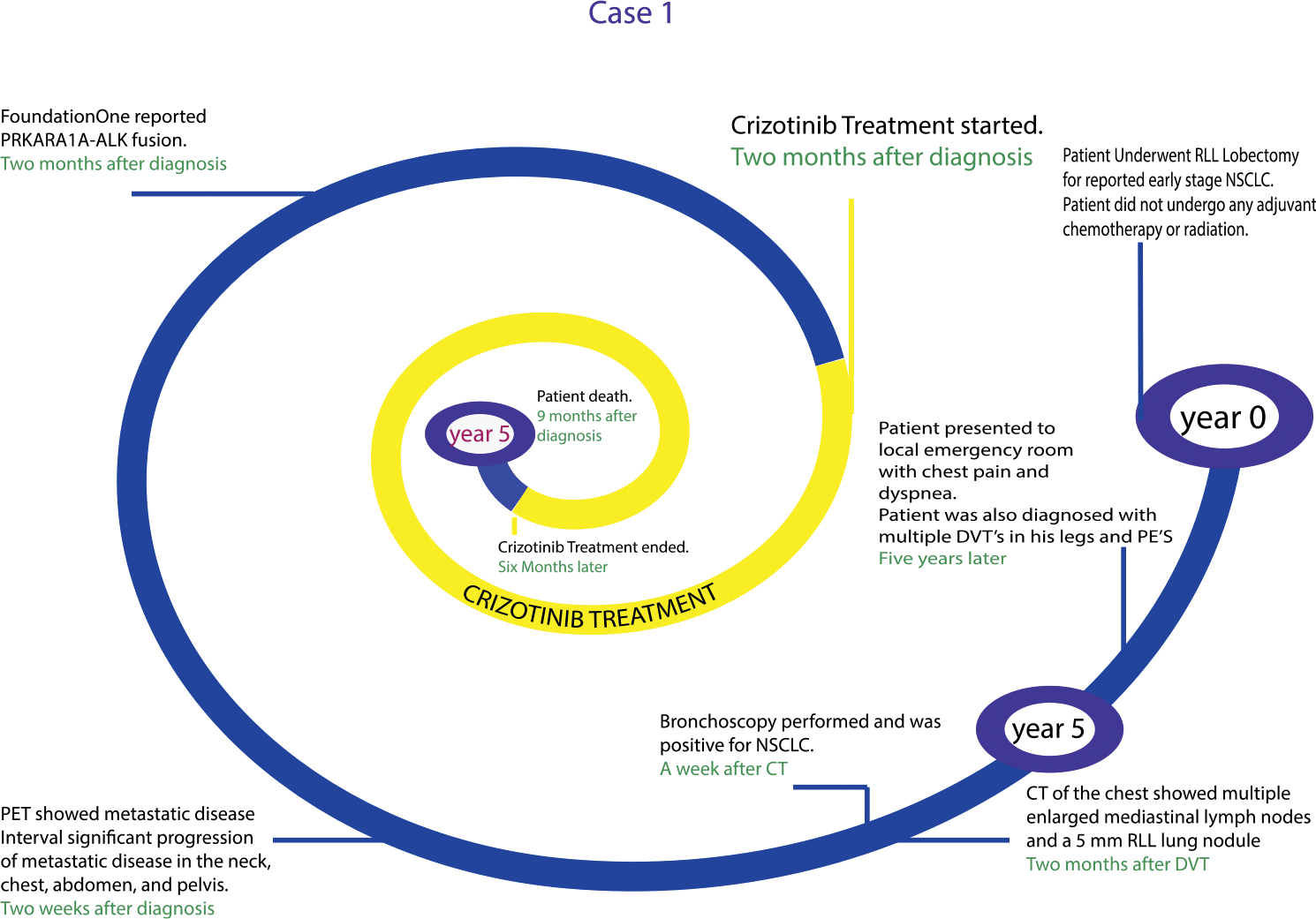
A 68-year-old white male who was a never-smoker initially presented with early-stage T1N0MX NSCLC and underwent right lower lobe resection with no adjuvant therapy (Fig. 2). Thereafter, he was monitored as an outpatient with chest x-rays performed every 6 months. He did well for 5 years with surveillance, but then suddenly developed chest pain and dyspnea. A CT scan of the chest showed multiple enlarged mediastinal lymph nodes and a 5-mm right lower lobe lung nodule. A subsequent bronchoscopy with biopsy was positive for NSCLC. A PET scan showed metastatic disease in the neck, chest, abdomen, and pelvis. Next-generation sequencing analysis of the tumor biopsy sample revealed a PRKAR1A-ALK fusion. As a result, the patient was treated with the ALK TKI, crizotinib (250 mg orally), and he had a partial response (PR) by Response Evaluation Criteria in Solid Tumors (RECIST) criteria.
FIGURE 2. Overview of the Timeline From Diagnosis to the Development of Metastatic Disease for a Patient With Lung Cancer Harboring an ALK Translocation.
Issues for consideration.
As NGS has become common in our analysis of metastatic lung cancer, we can identify known and novel genetic alterations. This patient responded well to a known TKI (crizotinib) that targets the ALK translocation. Of note for this case, the patient’s tumor did not harbor an EML4-ALK translocation, which is typically the ALK translocation found in lung cancer. Other ALK fusions partners, such as PRKAR1A, have been described, albeit with decreased frequency compared with EML4-ALK translocations in lung cancer. Nonetheless, the PRKAR1A-ALK translocation was therapeutically targetable with the ALK-targeted TKI, crizotinib.
Case 2
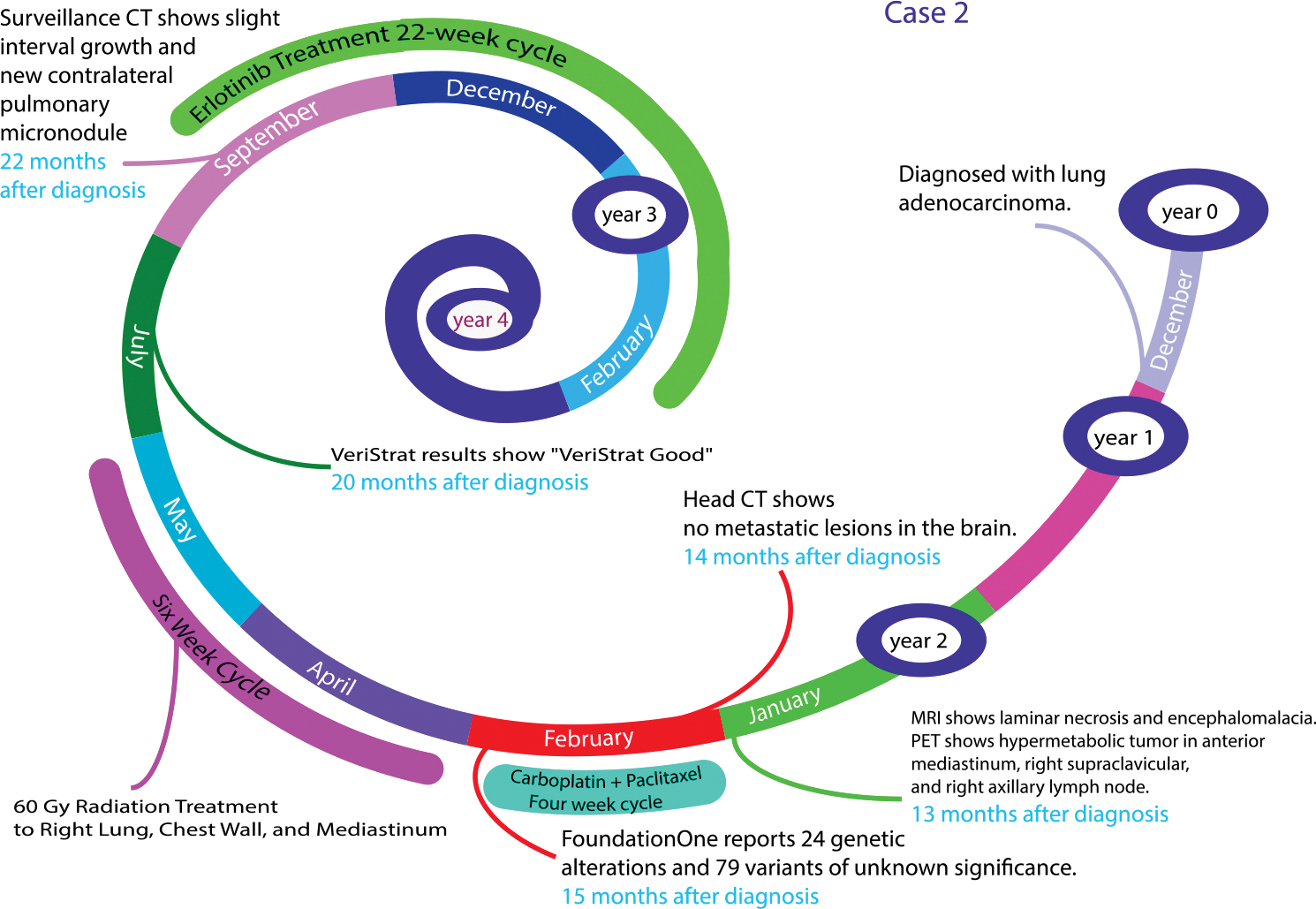
A 67-year-old black male who was a current smoker was diagnosed with lung adenocarcinoma (Fig. 3). The patient was lost for follow-up until more than a 1 year later. Thirteen months after his initial diagnosis, the patient underwent an MRI, which showed laminar necrosis and encephalomacia. A PET scan performed on the same day also showed hypermetabolic tumor in the anterior mediastinum, right supraclavicular, and the right axillary lymph node. A CT of the head was done a week later and showed no metastatic lesions in the brain. The patient then began to receive treatment with carboplatin and paclitaxel for 4 weeks. Next-generation sequencing analysis of the patient’s tumor sample revealed 24 genetic alterations and 79 variants of unknown significance, one of which was the EGFR R1068* mutation. There was no data to suggest that the EGFR R1068* mutation was sensitive to EGFR TKI therapy. The patient was initially treated with 60 Gy of radiation to the right lung, chest wall, and mediastinum over the course of 6 weeks. During that time, results received from the VeriStrat test (which is a proteomic test designed to predict benefit from EGFR TKI therapy) demonstrated a “VeriStrat good” score, indicating a favorable tumor signature for response to EGFR TKI therapy. Shortly thereafter, the patient began erlotinib therapy.
FIGURE 3. Overview of the Timeline From Diagnosis to the Development of Metastatic Disease for a Patient With Lung Cancer With a Number of Genetic Alterations.
Issues for consideration.
Even with the most available tools for precision medicine, we have still a long way to go for therapeutic decision making. Our traditional therapies are still of value for better outcomes for complex patients. As our patients respond to therapy and therefore survive longer, we have to consistently consider repeat analysis of tumor burden, the nature of genetic/proteomic changes, and further therapies.
Case 3
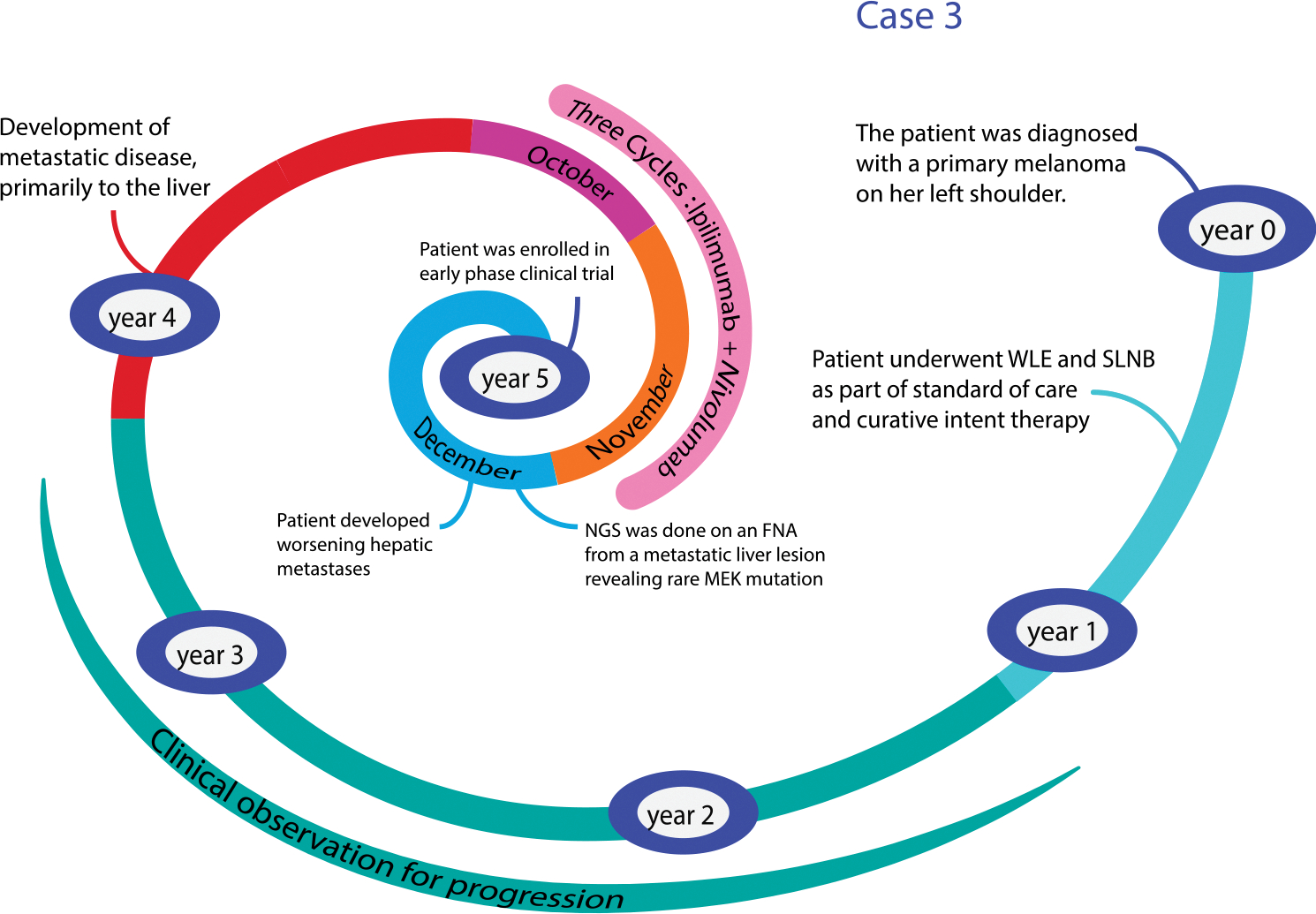
A 58-year-old woman presented to discuss treatment options for metastatic melanoma (Fig. 4). She had initially been diagnosed with a melanoma on the left shoulder (Breslow 3.2 mm, Clark level IV with ulceration) 4 years prior. After her initial wide local excision and sentinel lymph node biopsy, which was negative, she was observed clinically with routine dermatologic examinations. She was in her usual state of health until she presented with dyspepsia and abdominal pain. A CT scan of the chest, abdomen, and pelvis performed at that time showed multiple hepatic masses, with no other sites of disease. An MRI of the brain was negative for intracranial metastases. An ultrasound-guided liver biopsy was positive for metastatic melanoma. She was initially treated with combined ipilimumab (CTLA-4 inhibitor) and nivolumab (PD-1 inhibitor). After three cycles of therapy, she developed worsening symptoms, including abdominal pain and decreased appetite. A CT scan indicated disease progression with multiple new and enlarging hepatic metastases. Next-generation sequencing was also performed and was negative for BRAF, KIT, and NRAS mutations; however, an MEK K57N mutation was found. Because there was no agent approved by the U.S. Food and Drug Administration (FDA) that targeted this specific gene mutation, she elected to undergo screening for an early-phase clinical trial.
FIGURE 4. Overview of the Timeline From Diagnosis to the Development of Metastatic Disease for a Patient With Metastatic Melanoma Harboring a MEKK57N Mutation.
Issues for consideration.
This case highlights several important factors regarding the novelty and complexity of information obtained from tumor NGS assays for the practicing clinician. In melanoma, selective inhibitors are readily available for more common mutations such as BRAF, and clinical trials show evidence of clinical activity with agents targeted to KIT or NRAS.43,44 However, we have little data to guide us regarding less common variants. MEK K57N is a rare mutation that has previously been identified in melanomas.45 It has also been identified in other tumor types including NSCLC and neuroblastoma.46–48 The K57N mutation occurs in the nonkinase portion of the protein, and preclinical data suggest that this variant can induce MAPK pathway upregulation that may be potentially sensitive to MEK inhibition.46 Pursuing off-label treatment with a commercially available MEK inhibitor would be one option, although in our experience this can result in substantial delays in the administration of therapy, primarily related to coverage given the high cost of these agents. Newer trial designs, centered around specific genetic abnormalities (as opposed to a specific disease subtype), such as the NCI-Molecular Analysis for Therapy Choice (NCI-MATCH, NCT02465060) may allow for more therapeutic options for patients with rare alterations.
FUTURE RESEARCH DIRECTIONS FOR ASSESSING HETEROGENEITY: FRACTALS AND CHAOS
Fractals are mathematical constructs that show self-similarity over a range of scales and noninteger fractal dimensions. The important feature of fractals within the context of lung cancer heterogeneity is that small differences in initial conditions can yield widely diverging outcomes by which the cancer cells grow and mutate in seemingly random patterns. This process of deterministic nature and unpredictability can be studied mathematically through the use of chaos theory, where behavior of a system can be predicted in principle. One such method is through the use of fractal dimensions, which aim to calculate the changes in shape and surface area, and offer another alternative to liquid biopsies by observing the irregular patterns present in lung cancer growth over space and time. By measuring the changes in fractal dimension and lacunarity (texture) of lung cancer growth and regression, we hope to create a prognostic model that predicts the rate of growth and the change in the shape of the tumor. In one analysis, Lennon et al (also a good reference article for the topic presented here) evaluated the fractal analysis of lung cancer histology by analyzing representative hematoxylin and eosin–stained tissue slides for normal lung and common lung cancer histologies (adenocarcinoma, large-cell carcinoma, and small cell carcinoma).49 The fractal dimension and lacunarity of the lung tissue samples was then calculated using imaging software. All of the lung tumor tissues exhibited an increase in fractal dimension and a decrease in lacunarity showing marked clusters. In contrast, normal lung tissue showed stable levels of fractal dimension and lacunarity. This type of image analysis, together with chromatin, cellular, and radiologic analyses, could be useful in clinical diagnosis and classification of patients with lung cancer. Thus, fractal dimension and lacunarity of lung cancer, as measured in radiologic images or even pathologic slides, could someday potentially be used as biomarkers. Ultimately, heterogeneity can be measured at the genomic, proteomic, cellular, organoid, and metastatic levels. Unique biomarkers can be developed, as well as potential for therapeutics.
CONCLUSION
An increased understanding of the genetic underpinnings of cancer over the past several decades has led to remarkable advances in the field of oncology, affecting everything from the way cancers are classified to the way they are treated. A view that one gene, even a powerful oncogenic driver, would have the same effect across diseases or that one type of treatment will produce predictable results has long been a vestige of the past. Tumor heterogeneity has many facets—within a disease, within a patient, even within the tumor itself. With this evolution, however, new questions have emerged regarding the complex interactions of these systems and the development of resistance to therapy. Careful collaboration will be required to further elucidate these processes, with the hope that this will translate into better outcomes for patients.
KEY POINTS.
Tumor heterogeneity is not one distinct term but rather encompasses several facets that make tumors unique. Intra-tumor heterogeneity results from variability within an individual tumor. Intertumor heterogeneity results from variability across different tumors from different individuals, even within the same histopathological diagnosis.
Intratumor heterogeneity may result from genetic and epigenetic variability, interactions of tumor cells with surrounding cells, proximity to blood vessels, and presence of tumor-infiltrating lymphocytes. Each of these properties can influence the tumor cell phenotype and therefore lead heterogeneity.
Two models have been proposed to explain the origin of heterogeneity, the cancer stem cell model and the clonal evolution model. These models are not mutually exclusive.
Advances in sophisticated omic-based technologies have resulted in a greater understanding of intra- and intertumor heterogeneity.
Acquired therapeutic resistance refers to genotypic and/or phenotypic changes within the tumor that alter drug sensitivity, the natural selection of drug tolerant clones. Intensive research efforts are underway to better understand acquired resistance and to develop strategies for this important clinical problem.
Representative patient cases are presented herein to highlight the complexity of heterogeneity in clinical decision making.
ACKNOWLEDGMENT
Dr. Lovly and Dr. Salama contributed equally to this article.
Footnotes
Disclosures of potential conflicts of interest provided by the authors are available with the online article at asco.org/edbook.
References
- 1.Osler W On the educational value of the medical society. Yale Medical Journal. 1903;9:325. [Google Scholar]
- 2.Hölzel M, Bovier A, Tüting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer. 2013;13:365–376. [DOI] [PubMed] [Google Scholar]
- 3.Michor F, Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res (Phila). 2010;3:1361–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welch DR. Tumor heterogeneity-A ‘contemporary concept’ founded on historical insights and predictions. Cancer Res. 2016;76:4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zellmer VR, Zhang S. Evolving concepts of tumor heterogeneity. Cell Biosci. 2014;4:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2001;469:362–367. [DOI] [PubMed] [Google Scholar]
- 7.Andor N, Graham TA, Jansen M, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. [DOI] [PubMed] [Google Scholar]
- 9.Hanna N, Shepherd FA, Fossella FV, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol. 2004;22:1589–1597. [DOI] [PubMed] [Google Scholar]
- 10.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. [DOI] [PubMed] [Google Scholar]
- 11.Hauschild A, Agarwala SS, Trefzer U, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27:2823–2830. [DOI] [PubMed] [Google Scholar]
- 12.Salama AK, Flaherty KT. BRAF in melanoma: current strategies and future directions. Clin Cancer Res. 2013;19:4326–4334. [DOI] [PubMed] [Google Scholar]
- 13.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. [DOI] [PubMed] [Google Scholar]
- 14.Chapman PB, Hauschild A, Robert C, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAFV600E-mutated melanoma. J Clin Oncol. 2012;30 (suppl; abstr 8502). [Google Scholar]
- 15.Xing M BRAF mutation in thyroid cancer. Endocr Relat Cancer. 2005;12:245–262. [DOI] [PubMed] [Google Scholar]
- 16.Nagasaka T, Sasamoto H, Notohara K, et al. Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol. 2004;22:4584–4594. [DOI] [PubMed] [Google Scholar]
- 17.Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen D, Zhang LQ, Huang JF, et al. BRAF mutations in patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS One. 2014;9:e101354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Planchard D, Kim TM, Mazieres J, et al. Dabrafenib in patients with BRAF V600E-mutant advanced non-small cell lung cancer (NSCLC): a multicenter, open-label, phase II trial (BRF113928). Ann Oncol. 2014;25 (suppl 4; abstr LBA38_PR). [Google Scholar]
- 20.Falchook GS, Millward M, Hong D, et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid. 2015;25:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33:4032–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson DB, Dahlman KH, Knol J, et al. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. Oncologist. 2014;19:616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vigneswaran J, Tan Y, Murgu S, et al. Comprehensive genetic testing identifies targetable genomic alterations in most patients with non-small cell lung cancer, specifically adenocarcinoma, single institute investigation. Oncotarget. Epub 2016 Feb 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piotrowska Z, Niederst MJ, Karlovich CA, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015;5:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinozaki M, O’Day SJ, Kitago M, et al. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin Cancer Res. 2007;13:2068–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Board RE, Ellison G, Orr MC, et al. Detection of BRAF mutations in the tumour and serum of patients enrolled in the AZD6244 (ARRY-142886) advanced melanoma phase II study. Br J Cancer. 2009;101:1724–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long GV, Fung C, Menzies AM, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. [DOI] [PubMed] [Google Scholar]
- 33.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L, Mayes PA, Eastman S, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21:1639–1651. [DOI] [PubMed] [Google Scholar]
- 36.Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–1394. [DOI] [PubMed] [Google Scholar]
- 37.Heymach J, Tran HT, Nixon AB, et al. Circulating baseline plasma cytokines and angiogenic factors (CAF) as markers of clinical response in the study of trametinib (T) plus gemcitabine (G) vs placebo (P) plus gemcitabine for patients (pts) with untreated metastatic adenocarcinoma of the pancreas (MEK113487). J Clin Oncol. 2013;31 (suppl; abstr 4042). [Google Scholar]
- 38.Johnson DB, Menzies AM, Zimmer L, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51:2792–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–1876. [DOI] [PubMed] [Google Scholar]
- 40.Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386:444–451. [DOI] [PubMed] [Google Scholar]
- 41.Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. [DOI] [PubMed] [Google Scholar]
- 42.Wagle N, Van Allen EM, Treacy DJ, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–256. [DOI] [PubMed] [Google Scholar]
- 45.Murugan AK, Dong J, Xie J, et al. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle. 2009;8:2122–2124. [DOI] [PubMed] [Google Scholar]
- 46.Marks JL, Gong Y, Chitale D, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arcila ME, Drilon A, Sylvester BE, et al. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin Cancer Res. 2015;21:1935–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shukla N, Ameur N, Yilmaz I, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res. 2012;18:748–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lennon FE, Cianci GC, Cipriani NA, et al. Lung cancer-a fractal viewpoint. Nat Rev Clin Oncol. 2015;12:664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]