

Abstract
Background
Endothelial to mesenchymal transition (EndMT) is a dynamic process in which endothelial cells acquire mesenchymal properties and in turn contribute to tissue remodeling and growth. Previously, we found EndMT associated with mitral valve adaptation after myocardial infarction. Furthermore, mitral valve endothelial cells (VECs) collected at 6 months post-MI expressed the pan-leukocyte marker CD45 as well as EndMT markers. Additionally, mitral VECs induced to undergo EndMT with transforming growth factor (TGF)β1 strongly co-expressed CD45, but not CD11b or CD14. Pharmacologic inhibition of the CD45 protein tyrosine phosphatase domain in mitral VECs blocked TGFβ-induced EndMT. This prompted us to speculate that, downstream of TGFβ, CD45 induces EndMT.
Methods
We activated the endogenous CD45 promoter in human endothelial colony forming cells (ECFCs) using CRISPR/inactive Cas9 transcriptional activation. Bulk RNA-Seq was carried out on control ECFCs and CD45-positive ECFCs to identify transcriptomic changes. Three functional assays – cellular migration, collagen gel contraction, and trans endothelial electrical resistance (TEER) – were conducted to assess mesenchymal properties in CD45-positive ECFCs.
Results
Activation of the endogenous CD45 promoter in ECFC and 3 additional sources of endothelial cells induced expression of several genes implicated in EndMT. In addition, CD45-positive ECFCs showed increased migration, a hallmark of EndMT, increased collagen gel contraction, a hallmark of mesenchymal cells, and decreased cell-cell barrier integrity, indicating reduced endothelial function.
Conclusions
CD45 is sufficient to incite an EndMT phenotype and acquisition of mesenchymal cell properties in normal human ECFCs. We speculate that CD45, through its C-terminal protein tyrosine phosphatase domain, initiates signaling events that drive EndMT.
Keywords: endothelial-to-mesenchymal transition, CD45, endothelial colony forming cells
Graphical Abstract
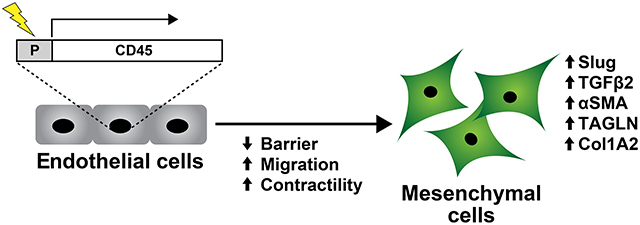
INTRODUCTION
Endothelial to mesenchymal transition (EndMT) occurs during embryonic heart development and in some instances re-emerges in adult life associated with cardiovascular diseases such as ischemic mitral regurgitation, pulmonary arterial hypertension, and atherosclerosis1, 2,. EndMT consists of loosening of endothelial cell-cell contacts, endothelial cell migration, and acquisition, of mesenchymal cell markers and properties3. We showed previously the leukocyte marker CD45 is expressed in mitral valve endothelium at 6 months post-myocardial infarction (MI) and also in mitral valve endothelial cells (VECs) induced to undergo EndMT with TGFβ14. TGFβ1 induction of EndMT in mitral VECs could be blocked by a small molecule inhibitor of CD45 protein tyrosine phosphatase5. In contrast, carotid artery endothelial cells did not undergo TGFβ1-induced EndMT and did not express CD45. This led us to speculate that CD45 drives EndMT in certain settings4.
CD45 protein tyrosine phosphatase receptor type C is a transmembrane glycoprotein expressed on all hematopoietic cells except mature red blood cells and platelets. It is best known for regulating T and B cell antigen receptor-mediated activation6 and has been implicated in migration of hematopoietic cells7. Its cytoplasmic domain dephosphorylates a negative regulatory phospho-tyrosine on Src family kinases, which activates the kinase. CD45 can also dephosphorylate activating phospho-tyrosine sites and thereby dampen signaling pathways8. CD45 knockout mice show effects on T cell maturation and function9, altered Src kinase activity and increased ERK phosphorylation10. Alternative splicing of exons 4, 5, and 6 generates different isoforms of CD45; all isoforms have the protein tyrosine phosphatase domain6.
Here, we asked if CD45 is sufficient to induce EndMT in normal human endothelial colony forming cells (ECFC), which are isolated from umbilical cord blood and sometimes referred to as blood outgrowth endothelial cells. ECFC maintain an endothelial phenotype, are highly proliferative in vitro, and form perfused human blood vessels when implanted into immune-deficient mice11. Importantly ECFC do not express CD4511, 12, do not undergo EndMT or express CD45 in response to TGFβ1 (Figure S1A–D. These properties make ECFC suitable for evaluating our hypothesis, which we confirmed in human umbilical vein endothelial cells (HUVEC), human dermal microvascular endothelial cells (HDMEC) and ovine carotid artery endothelial cells (CAEC).
METHODS
Data Availability Statement
Data are available from the corresponding author upon request. RNA-Seq data is available at the Gene Expression Omnibus archive at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/geo/), with the accession number GSE207837.
Cell Culture
Human ECFC were isolated as described11 and expanded in culture by plating on fibronectin (0.1 μg/cm2)-coated dishes at 15,000 – 20,000 cells/cm2 in Endothelial Cell Growth Medium-2 (Lonza) which consists of Endothelial Cell Growth Basal Medium-2 (EBM-2), SingleQuot supplements (all except hydrocortisone and GA-1000), 20% heat-inactivated fetal bovine serum (FBS, Cytiva) and 1X glutamine-penicillin-streptomycin (GPS, ThermoFisher). HUVEC, HDMEC, and CAEC were grown in the same manner.
Mitral VEC and mitral valve interstitial cells (VIC) were plated on 1% gelatin-coated dishes in EBM-2 media,10% heat-inactivated FBS, 1% GPS, and 2 ng/mL basic fibroblast growth factor (R&D Systems), designated EBM-B media. Mitral VEC served as positive control in the TEER assay and mitral VIC served as positive control in the collagen gel contraction assay.
CRISPR/dCas9 Transcriptional Activation of CD45
The endogenous CD45 promoter in ECFC was activated using a CRISPR/inactive Cas9 synergistic activation mediator (SAM) transcriptional activation system13 (Santa Cruz Biotechnology, CD45 Lentiviral Activation Particles (h2)) and Control Lentiviral Activation Particles). The SAM system consists of three lentiviral activation particles: one encodes a guide RNA specific to the human CD45 promoter, the second encodes a fusion protein that increases transcription, and the third a deactivated Cas9. Each construct has a different antibiotic resistance gene allowing for selection of transduced cells. ECFC were selected for puromycin and hygromycin resistance, but blasticidin was omitted because of toxicity. Flow cytometry of fixed and permeabilized cells showed 50–60% of the cells expressed CD45 and over 90% of the cells expressed VE-cadherin (Figure S2). These cells are termed ECFC-CD45. ECFC-control cells were subjected to the same protocol, but a non-specific 20 nucleotide guide RNA was used instead of the CD45 promoter guide RNA. SAM transcriptional activation of the CD45 promoter in HUVECs, HDMEC and CAECs was conducted in the same manner. Please see Major Resources table for antibodies. The APC-conjugated mouse anti-human CD45 recognizes all isoforms of CD45.
RNA-Seq
Total RNA was extracted from ECFC-control and ECFC-CD45 with a RNeasy Micro extraction kit (Qiagen). Five replicates of each were prepared on different days. RNA libraries were prepared from each replicate using a NEBNExt Ultra II directional RNA library prep kit and sequenced by Illumina HiSeq PE150 (Novogene). Data were analyzed by the following workflow. The raw reads were mapped onto the human genome (version hg19) using STAR (version 2.7.3a)14 with the parameters --readFilesCommand zcat --outFilterType BySJout --outFilterMultimapNmax 20 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonical --outSAMtype BAM SortedByCoordinate. Next, differentially expressed genes were detected by Cuffdiff (version 2.2.1)15. Cuffdiff computes the logarithm of the ratio of the normalized read counts by FPKM (fragments per kilobase of transcript per million fragments mapped) between two conditions. Then it uses the delta method to estimate the variance of the log odds. This can be transformed into a test statistic T = E[log(y)]/Var[log(y)] by normalizing the log ratio with the variance, where y is the ratio of the normalized read counts between two conditions. This ratio approximately follows a normal distribution; thus a one-sided t-test is used to calculate the P-value for differential expression. Finally, the Benjamini-Hochberg correction is used for multiple testing to control the Type I errors. Genes with mean expression level larger than 1, log2 fold change larger than 0.5 and adjusted P-value less than 0.05 were identified as significantly differentially expressed genes (DEG). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was conducted with the R package clusterProfiler (version 3.18.1)16.
qPCR
Reverse transcriptase reactions were performed using an iScript™cDNA synthesis kit (BioRad,). qPCR was performed using KAPA SYBR® FAST ABI Prism 2x qPCR Master Mix (KAPA BioSystems). Amplification was carried out on a QuantStudio 6 Flex System (Applied Biosystems). A relative standard curve for each gene amplification was generated to determine efficiency; two technical replicates for each amplification reaction were performed Fold increases were calculated according to two delta CT method. Gene expression was normalized to ECFC-Control. ATP5B was used as housekeeping gene reference. Primer sequences are shown in Major Resources Table.
Silencing Interleukin 1 beta (IL1β) by siRNA
ECFC-Control and ECFC-CD45 cells were transfected with ON-TARGET plus siRNA against human IL-1β (NM_000576, Dharmacon) using lipofectamine RNAiMAX transfection reagent. ECFCs were seeded in 6-well plates at 80–90% confluence; the medium was replaced with medium without antibiotics before transfection. Transfection reagent and siRNA products were incubated in EBM-2 separately at room temperature for 5 min. Mixtures were combined, incubated another 20 min, and added to cells at 2 μl/ml transfection reagent and 25 nM siRNAs. Reagents were removed 48h post-transfection, serum-free media was added for one hour, and cells were lysed for Western Blot with anti-ICAM-1, anti-TAGLN, anti-actin, anti-p-ERK1/2 and anti-ERK1/2. Two independent experiments were conducted.
Immunostaining
ECFC-Control and ECFC-CD45 on chamber slides were fixed in 4% paraformaldehyde for 15 minutes, permeabilized with 0.1% Triton-X for 15 minutes, and blocked with 5% BSA/PBS for 1 hour, each step at room temperature. Immunostaining was done with rabbit anti-human VE-cadherin (Clone D87F2) and mouse anti-human α-smooth muscle actin (αSMA) (Clone 1A4). Secondary antibodies were Alexa Fluor 568 goat anti-rabbit IgG and donkey anti-mouse IgG Alexa Fluor 488. See Major Resources table. Mounting was performed with ProLong Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Cell Signaling). Fluorescent images were taken on an upright Zeiss Laser Scanning 880 confocal microscope.
Migration Assay
ECFC-Control and ECFC-CD45 cells were treated for 30 minutes ± CD45-selective PTPase inhibitor (N-(9,10-dioxo-9,10-dihydro-phenanthren-2-yl)-2,2-dimethyl propionamide) at 1μmol/L prior to trypsinization. 20,000 cells in 0.1% BSA/EBM-2 ± the CD45-selective PTPase inhibitor were placed in the upper chamber of 6.5mm Transwells containing fibronectin-coated (0.2 ug/cm2) polycarbonate membranes with 8.0μm pores. The lower chambers contained either 0.1% BSA/EBM-2 or 10% FBS/EBM-2. Cells were allowed to migrate across the membrane for 8 hours at 37°C. Cells that migrated through the pores were fixed with methanol and Eosin-Y, Azure A and Methylene Blue for visualization and quantification using Three Step Stain Set (VWR,). Three technical replicates were performed with five images from each Transwell analyzed in each replicate. Data are representative of three independent experiments.
Collagen Gel Assay
Cells were trypsinized, counted, and seeded in collagen gels (300,000 cells/0.5mL) in 24 well plates using a Collagen-based Cell Contraction Assay (Cell Biolabs). The cell/collagen gel suspensions, two technical replicates for each condition, were allowed to polymerize for 1 h at 37 °C. EBM-2 growth media was added to each well, and gels were released from the sides of the plate with a 27-gauge needle to create free-floating gels. At 0, 8, 24, and 36 hours, the gels were photographed, and gel areas calculated using ImageJ. Data are representative of three independent experiments.
Trans Endothelial Electrical Resistance (TEER) Assay
For ECFC-Control and ECFC-CD45, 40,000 cells/well were seeded on fibronectin-coated (0.1mg/cm2) 96 well array with 10 interdigitated electrodes per well (96W10idfPET ECIS array, Applied BioPhysics, NY) in Endothelial Cell Growth Media-2. Once cells adhered and formed a monolayer (~3 hour), media was switched to reduced serum media (2% FBS) and monitored for ~20 hours. For mitral VEC, 10,000 cells/well were seeded on 1% gelatin coated 96W10idfPET ECIS array. When maximum TEER was reached (~23hours), TGFβ1 (1ng/ml) was added, and monitoring continued for ~55 hours. TEER of either ECFCs or mitral VECs was measured at multiple frequencies (62.5–64,000 Hz) in real-time using an ECIS™ Zθ system (Applied BioPhysics). All ECIS measurements are reported at 4,000 Hz, which is the optimal resistance for endothelial cells. The percent change in barrier was quantified compared to appropriate control cells at indicated timepoints. Four technical replicates were included for each condition; data are representative of three independent experiments.
Statistics
Shapiro-Wilk tests for normality and Brown Forsythe tests for equal variance were applied but not as a precondition for using parametric analyses. TEER data passed the Shapiro Wilk but migration and contractility data not. Migration assay data were analyzed by one-way ANOVA followed by Dunnett’s T3 post hoc test. Welch’s correction was applied.
The time course experiments for contractility and endothelial barrier were analyzed by two-way-ANOVA for repeated measures followed by Tukey’s post-hoc test. Two-way ANOVA followed by Tukey’s post-hoc test was performed in contractility assays in presence of CD45 PTPase inhibitor. Two-tailed student’s t-tests were applied to TEER data between ECFC-Control and ECFC-CD45 at 20 hours. Significance considered as p<0.05. Data are reported as mean±SD. All tests were run with GraphPad Prism 9.4.1 Software.
RESULTS
We induced expression of endogenous CD45 in ECFCs using a CRISPR/inactive Cas9 transcriptional activation system13. Flow cytometry showed 50–60% of the cells expressed CD45 and over 90% expressed VE-cadherin (Figure S2A, B). We termed these cells ECFC-CD45; ECFC-Control cells were subjected to the same protocol with a non-specific 20 nucleotide guide RNA. ECFC-CD45 expressed the CD45R0 isoform, shown by PCR amplification using exon 2 and exon 7 primers, which produced an amplicon of ~143 base pairs (data not shown), consistent with lack of exons 4, 5 and 617. ECFC-Control display a cobblestone morphology, whereas ECFC-CD45 display a spindle morphology (Figure S2C),
Bulk RNA-Seq was carried out on the 5 independent replicates of ECFC-CD45 and ECFC-Control shown in Figure S2A–B. The cells were seeded at 20,000 cells/cm2 and grown for three days before isolating RNA. Differentially expressed genes (DEG) are shown in the Volcano plot in Fig1A. We found 1760 genes upregulated and 2716 genes down regulated by the expression of CD45 in ECFC. DEG, Gene Ontology and KEGG analyses are provided in Tables S1, S2 and S3. Fig1B shows the log2 fold changes and adjusted p-values for several transcripts encoding proteins involved in EndMT and mesenchymal markers. The upregulation of six of these was verified by qPCR (Figure S3 A). Fold increases aligned well with increases seen by bulk RNA-Seq. Western blot analyses showed that TAGLN, ICAM1 and phosphorylated ERK, which is involved in TGFβ-induced EndMT18, were increased in ECFC-CD45 compared to ECFC-control (Fig 1C). Knockdown of endogenous interleukin-1β (IL1β) with siRNA resulted in decreased TAGLN, ICAM1, and phosphor-ERK, indicating IL1β, which has been implicated in EndMT19, 20, plays a role in CD45-induced EndMT. Co-expression of αSMA and VE-cadherin was seen in a subset of ECFC-CD45 but there was no αSMA detected in ECFC-Control (Fig1D). The greater than 4-fold increases in EndMT markers TAGLN, TGFβ2, αSMA, and SNAI2/SLUG, each with adjusted p-values (q) of 0.0002, suggests CD45 is sufficient to drive EndMT in human ECFCs. Further, the co-expression of CD45 and α-SMA in ECFC-CD45 but not in ECFC-control indicates CD45 involvement in EndMT (Figure S2D).
Figure 1. Bulk RNA-Seq of ECFC-CD45 versus ECFC-Control.
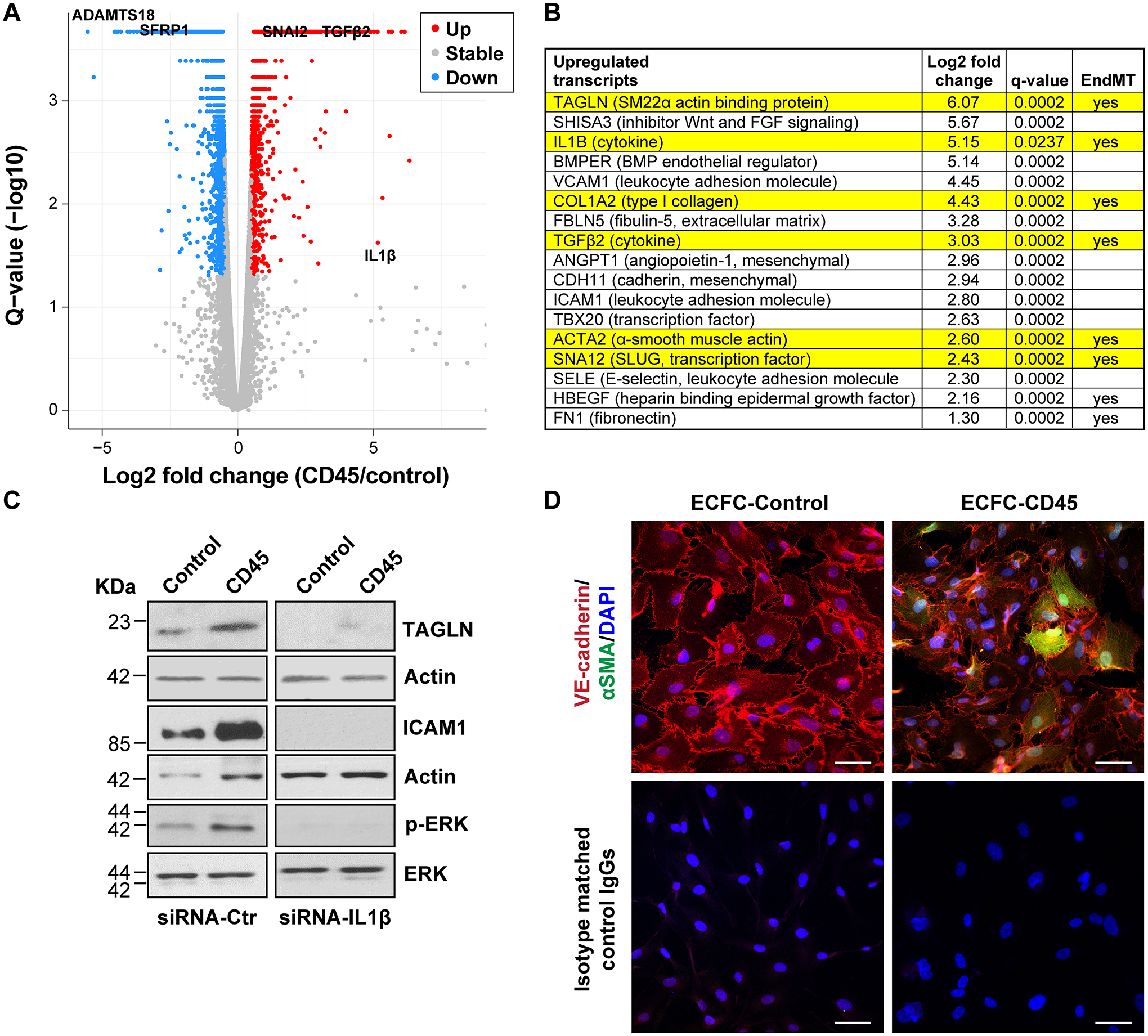
(A) Differential q value plotted against fold change of gene expression determined by RNA-Seq. Names of four genes are labeled in the plot. (B) Log2 fold changes and adjusted p-values (q-values) for a sample of genes upregulated in ECFC-CD45 relative to ECFC-Control. (C) Western blot of ECFC-Control and ECFC-CD45 treated with siRNA-control or siRNA-IL1β for 48 hours. (D) Endothelial cells co-stained with anti-VE-cadherin (red) and anti-αSMA (green) and DAPI (blue). Bottom panels show staining with isotype matched control IgGs, the corresponding fluorescently tagged secondary antibodies, and DAPI. Scale bar, 50 μm.
We used the same SAM transcriptional activation system to induce CD45 in three distinct primary endothelial cells: HUVEC, HDMEC and ovine CAEC. Induction of CD45 was detected by flow cytometry and qPCR (Figure S3B, C). EndMT markers αSMA, SLUG, TGFβ2, COL1A2 and TAGLN were increased in all three (Figure S3C). Thus, CD45 increased EndMT markers in four types of primary endothelial cells.
We next assessed whether ECFC-CD45 exhibit mesenchymal cell properties, which would indicate functional changes consistent a mesenchymal phenotype. Increased migration, a hallmark of EndMT, enables cells to migrate away from the endothelium, move into the subendothelial space, and begin to affect tissue remodeling and repair. We measured migration of ECFC-Control and ECFC-CD45 across the Transwell membrane towards 10% FBS, which contains endothelial chemoattractants such as platelet-derived growth factor-B, compared to 1% BSA. We tested each condition without (black symbols) or with (red symbols) the CD45 PTPase inhibitor (Fig2A). ECFC-Control migrated towards 10% FBS as expected but there was no significant inhibition when the CD45 inhibitor was included. ECFC-CD45 migrated towards 10% FBS at significantly increased level compared to ECFC-Control (p<0.0001), which was significantly reduced when the CD45 PTPase inhibitor was included (p<0.0001). Representative images of FBS-induced migrated ECFC-Control versus ECFC-CD45 are shown in Fig2B. Results in Fig2A, B demonstrate the increased migratory activity of ECFC-CD45 compared to ECFC-Control and suggest the phosphatase activity of CD45 is required for eliciting the increased migration.
Figure 2. Functional Assays.
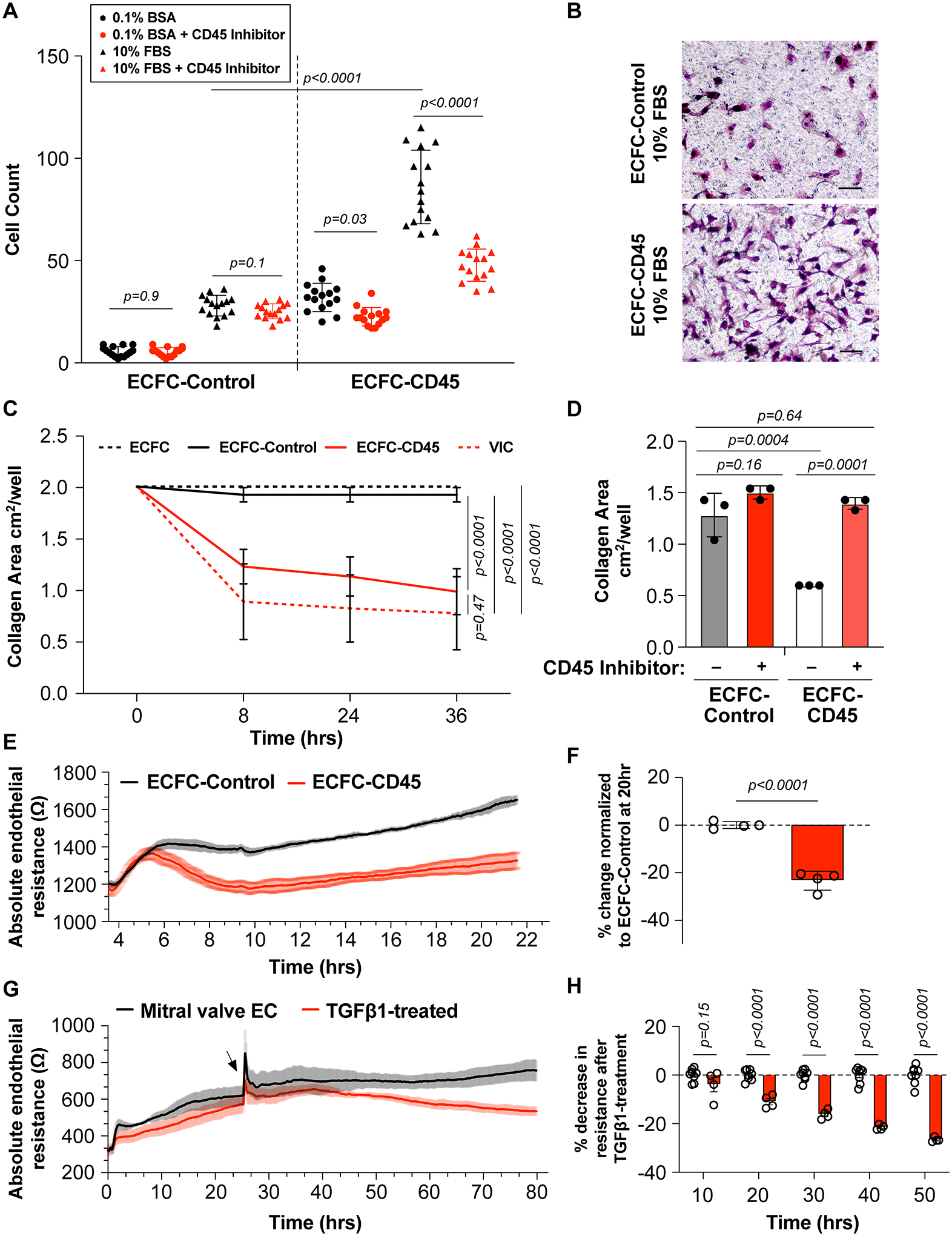
(A) ECFC-Control (left panel) and ECFC-CD45 (right panel) were treated without (black symbols) or with the CD45 PTPase inhibitor (red symbols). After migration towards 1% BSA (circles) versus 10% FBS (triangles) for 8 hours, cells on the underside of the Transwell membrane were quantified. (B) Representative view of migrated ECFC-Control and ECFC-CD45. Scale bar, 50μm. (C) Cells in free floating type I collagen gels were allowed to contract, and gel sizes were measured at the indicated time points. (D) Collagen contraction in ECFC-Control and ECFC-CD45 in presence of the CD45 PTPase inhibitor (red bars). Representative images of collagen gels at 8 hours shown in Figure S4. (E) Endothelial resistance (Ω) of ECFC-Control (black) and ECFC-CD45 (red) over 22 hours. Shading represents the variations. (F) Percent change in resistance in ECFC-CD45 normalized to ECFC-Control at 20 hours. (G) Endothelial resistance (Ω) of mitral VECs treated with 1ng/mL TGFβ1 (red) starting at 24 hours versus non-treated (black). (H) Percent change in endothelial resistance over time of TGFβ treatment. Arrow indicates time of TGFβ1 addition.
Mesenchymal cells, particularly in an activated state, contract type I collagen gels, indicative of their ability to modulate the extracellular matrix. Contractility can be assessed by suspending cells in type I collagen followed by transition from acidic to neutral pH to polymerize the cell/collagen suspension into a 3-dimensional gel. We tested ovine mitral VICs as a positive control21, ECFC-CD45, ECFC-Control, and naïve ECFC as negative a negative control21. Collagen gel area was reduced by mitral VIC and ECFC-CD45 at 8, 24 and 36 hours whereas collagen gel area was unchanged in naïve ECFC and ECFC-Control gels. Significant differences in collagen gel area were seen between ECFC-Control and ECFC-CD45 at 8 (p=0.028), 24 (p=0.031) (Table S4) and 36 hours (p=0.033) (Fig 2C). ECFC-Control and naïve ECFC showed no contraction of the collagen gels, while ECFC-CD45 contracted the gels to an extent similar to ovine mitral VICs. Representative images of the collagen gels at 8 hours are shown in Figure S4 along with p values for 8- and 24- hour time points in Table S4. The CD45 PTPase inhibitor blocked collagen contraction by ECFC-CD45 but had no effect on ECFC-Control cells (Figure 2D)
A characteristic of endothelial cells is the ability to form cell junctions to create a barrier that separates circulating blood from the parenchyma. The integrity of the endothelial barrier can be assayed in vitro by measuring TEER, reduction in which is attributed to loosened cell-cell junctions and increased permeability. As such, TEER has been used to assess onset of EndMT in response to cytokines22. We measured TEER in ECFC-Control and ECFC-CD45 plated at high density so that cell-cell contacts would form without need for proliferation. ECFC-Control rapidly formed an electrically resistant barrier that increased steadily over 22 hours. In contrast, ECFC-CD45 formed a reduced barrier (Fig2E). Quantification at 20 hours showed a significant, 23% reduction in TEER in ECFC-CD45 compared to ECFC-Control (Fig2F).
As TEER is not routinely used to functionally assess EndMT, we validated the assay using ovine mitral VECs that consistently undergo EndMT when treated with TGFβ14, 18. Mitral VECs were plated at 30,000 cells/cm2, treated with TGFβ1 at 24 hours (a small spike in TEER occurs with media change) and monitored for another 56 hours (Fig 2G). TGFβ1-treated mitral VECs showed a steady decrease in TEER compared to non-treated mitral VECs, with significant reduction detected at 20 hours after TGFβ1 addition, increasing to 26% reduction at 50 hours (Fig2H). In summary, the decrease in TEER in ECFC-CD45 cells compared to ECFC-Control is consistent with a loosening of cell-cell junctions, which allows endothelial cells to detach from the endothelium and migrate into the sub endothelial space, critical steps in both partial and full EndMT3.
DISCUSSION
Here we show that transcriptional activation of PTPRC and resultant expression of CD45 protein tyrosine phosphatase receptor type C, in human ECFC is sufficient to induce robust expression of several EndMT markers and acquisition of mesenchymal properties. CD45 expression in three additional primary endothelial cells – HUVECs, HDMEC, and CAECs – also resulted in expression of several EndMT markers. The ECFC-CD45 showed significant increases in cellular migration, acquisition of collagen gel contractility, and reduced electrically resistant cell-cell contacts. The increased cellular migration and contractility seen in ECFC-CD45 was reduced by the CD45 PTPase inhibitor. Our results are consistent with our previous studies wherein the same CD45 phosphatase inhibitor blocked TGFβ1 induction of EndMT markers and increased cellular migration in mitral VECs expressing CD454. CD45 expression in mitral valve endothelium post-MI and in TGFβ1-treated VECs was a not predicted or expected, but instead discovered during survey of post-MI mitral valve leaflets for leukocyte infiltration. The novelty of the finding prompted a direct test of CD45 in EndMT. Experiments herein demonstrate that CD45 is sufficient to induce EndMT in human endothelial cells and the phosphatase activity of CD45 plays a role. We anticipate that while CD45 is sufficient to induce EndMT, there are likely other mediators and pathways that induce EndMT in certain settings. For example, we did not find CD45 co-expressed in α-SMA-positive/VE-cadherin-positive mitral VECs at 8–10 days post-MI23 suggesting that expression of CD45 in vivo may develop over a longer time period, as we originally observed4. The context(s) under which CD45 is implemented to drive EndMT will require further studies.
The bulk RNA-seq analysis provides an unbiased overview of how the endothelial transcriptome changes upon CD45 expression. Indeed, PTPRC (CD45) was the most highly upregulated (log2 fold change = 6.32; adjusted p value 0.0038). Known drivers of EndMT – interleukin-1β (IL1β), TGFβ2, and SNAI2/SLUG were increased in ECFC-CD45 cells, along with EndMT markers TAGLN, α-SMA, CDH11, COL1A2 and fibronectin (FN1) (Fig1B, Figure S3). The strong increase in endogenous TGFβ2 and IL1β indicates a potential for cells to autonomously perpetuate EndMT. And indeed, TGFβ-receptors - TGFβR1, TGFβR2 and TGFβR3 - are significantly increased in ECFC-CD45 (Table S1). TGFβR1, also known as ALK5, was recently shown to be responsible for shear stress induced EndMT that occurs in atherosclerosis24 Interestingly, the leukocyte adhesion molecules ICAM1, VCAM1 and E-selectin were also increased, perhaps due to increased IL1β, which would signal to NF-κB, the transcription factor that controls expression of these leukocyte adhesion molecules. Indeed, siRNA knockdown of endogenous IL1β resulted in decreased ICAM1 (Fig 1C). This suggests that perhaps CD45 regulates EndMT in inflammatory settings.
Potentially novel EndMT-associated genes identified by bulk RNA-Seq are SHISA3 and BMPER, both found strongly upregulated in ECFC-CD45. Shisa proteins inhibit FGF signaling by retaining FGF-receptors in the endoplasmic reticulum25, which, if this occurred in human endothelial cells, would facilitate EndMT as FGF signaling has been shown to block EndMT26. BMPER (BMP-binding endothelial regulator) has been shown to inhibit BMP-4 induced endothelial differentiation in murine embryonic stem cells27 and thus one might speculate that its increase in ECFC-CD45 would negatively affect the endothelial phenotype of these cells.
In addition, many genes were significantly downregulated in ECFC-CD45 compared to ECFC-Control. Noteworthy is the strong downregulation of secreted frizzled related protein-1 (SFRP1) (log2fold −3.5 adjusted p = 0.0002), an inhibitor of Wnt signaling. Recently, we showed SFRP3 in plasma blocks EndMT in mitral VECs23. However, SFRP3 transcripts were not detected in the bulk RNA-seq analysis; SRFP2, 4 and 5 were detected but not differentially expressed. ADAMTS18, recently implicated in angiogenesis28 was also strongly down-regulated in ECFC-CD45 cells compared to ECFC-Control (log2fold decrease of −4.48; adjusted p= 0.0002), which could indicate a loss of endothelial function. These interesting genes that are up or down regulated in response to endothelial expressed CD45 may be considered in future studies.
LIMITATIONS
The first limitation is that due to toxicity of blasticidin, we could not generate endothelial cell populations in which 100% of the cells express CD45. A second limitation is that while we show induction of EndMT markers in CD45 induced HUVEC, HDMEC and CAEC, we restricted our RNA-Seq and functional assays to ECFC-Control and ECFC-CD45 cells. A third limitation is that we did not examine reversibility of CD45-driven EndMT.
CONCLUSIONS
Transcriptional activation of the endogenous CD45 expression in endothelial cells is sufficient to induce functional hallmarks of EndMT and expression of genes associated with EndMT such as TGFβ2, SNAI2/SLUG, TAGLN and α-SMA. Bulk RNA-Seq of five independent replicates of ECFC-CD45 versus ECFC-Control provides a valuable resource for further investigation of CD45-driven EndMT.
Supplementary Material
Highlights.
CD45 expression in human endothelial cells increased EndMT markers
Functional assays show CD45 expression in human endothelial cells increased mesenchymal cell properties and reduced endothelial cell properties
Bulk RNA-Seq identified novel transcripts differentially regulated by CD45 expression
Sources of Funding
Research reported in this manuscript was supported by the National Heart, Lung, and Blood Institute, part of the National Institutes of Health, under Award Number 5R01HL141917-04 (J.B.) and 5R01HL141853-02 (H.C.). K.C. was supported in part by R01GM125632 and R01HL148338. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Non-standard abbreviations
- BSA
bovine serum albumin
- CAEC
carotid artery endothelial cells
- DAPI
4′,6-diamidino-2-phenylindole
- DEG
differentially expressed genes
- ECFC
endothelial colony forming cells
- EndMT
endothelial to mesenchymal transition
- FBS
fetal bovine serum
- GPS
glutamine-penicillin-streptomycin
- HUVEC
human umbilical vein endothelial cells
- HDMEC
male human dermal microvascular endothelial cells
- ICAM1
intracellular adhesion molecule 1
- MI
myocardial infarction
- PTPase
protein tyrosine phosphatase
- αSMA
α-smooth muscle actin
- sFRP
secreted frizzled related protein
- TEER
trans endothelial electrical resistance
- TGFβ
transforming growth factor-β
- VEC
valve endothelial cell
- VIC
valve interstitial cell
Footnotes
References
- 1.Dal-Bianco JP, Levine RA, Hung J. Mitral regurgitation postinfarction: The mitral valve adapts to the times. Circ Cardiovasc Imaging. 2020;13:e012130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu Y, Kovacic JC. Endothelial to mesenchymal transition in health and disease. Annu Rev Physiol. 2022 [DOI] [PubMed] [Google Scholar]
- 3.Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2021;41:2357–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bischoff J, Casanovas G, Wylie-Sears J, Kim DH, Bartko PE, Guerrero JL, Dal-Bianco JP, Beaudoin J, Garcia ML, Sullivan SM, et al. Cd45 expression in mitral valve endothelial cells after myocardial infarction. Circ Res. 2016;119:1215–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panchal RG, Ulrich RL, Bradfute SB, Lane D, Ruthel G, Kenny TA, Iversen PL, Anderson AO, Gussio R, Raschke WC, et al. Reduced expression of cd45 protein-tyrosine phosphatase provides protection against anthrax pathogenesis. J Biol Chem. 2009;284:12874–12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al Barashdi MA, Ali A, McMullin MF, Mills K. Protein tyrosine phosphatase receptor type c (ptprc or cd45). J Clin Pathol. 2021;74:548–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai JC, Wlodarska M, Liu DJ, Abraham N, Johnson P. Cd45 regulates migration, proliferation, and progression of double negative 1 thymocytes. Journal of immunology. 2010;185:2059–2070 [DOI] [PubMed] [Google Scholar]
- 8.Saunders AE, Johnson P. Modulation of immune cell signalling by the leukocyte common tyrosine phosphatase, cd45. Cell Signal. 2010;22:339–348 [DOI] [PubMed] [Google Scholar]
- 9.Kishihara K, Penninger J, Wallace VA, Kundig TM, Kawai K, Wakeham A, Timms E, Pfeffer K, Ohashi PS, Thomas ML, et al. Normal b lymphocyte development but impaired t cell maturation in cd45-exon6 protein tyrosine phosphatase-deficient mice. Cell. 1993;74:143–156 [DOI] [PubMed] [Google Scholar]
- 10.Shivtiel S, Kollet O, Lapid K, Schajnovitz A, Goichberg P, Kalinkovich A, Shezen E, Tesio M, Netzer N, Petit I, et al. Cd45 regulates retention, motility, and numbers of hematopoietic progenitors, and affects osteoclast remodeling of metaphyseal trabecules. The Journal of experimental medicine. 2008;205:2381–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melero-Martin JM, Khan ZA, Picard A, Wu X, Paruchuri S, Bischoff J. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood. 2007;109:4761–4768 [DOI] [PubMed] [Google Scholar]
- 12.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. Crispr rna-guided activation of endogenous human genes. Nat Methods. 2013;10:977–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. Star: Ultrafast universal rna-seq aligner. Bioinformatics. 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu G, Wang LG, Han Y, He QY. Clusterprofiler: An r package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothstein DM, Saito H, Streuli M, Schlossman SF, Morimoto C. The alternative splicing of the cd45 tyrosine phosphatase is controlled by negative regulatory trans-acting splicing factors. J Biol Chem. 1992;267:7139–7147 [PubMed] [Google Scholar]
- 18.Wylie-Sears J, Levine RA, Bischoff J. Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-beta-induced phosphorylation of erk. Biochem Biophys Res Commun. 2014;446:870–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E, Fiocchi C. Inflammation-induced endothelial-to-mesenchymal transition: A novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maleszewska M, Moonen JR, Huijkman N, van de Sluis B, Krenning G, Harmsen MC. Il-1beta and tgfbeta2 synergistically induce endothelial to mesenchymal transition in an nfkappab-dependent manner. Immunobiology. 2013;218:443–454 [DOI] [PubMed] [Google Scholar]
- 21.Shapero K, Wylie-Sears J, Levine RA, Mayer JE Jr., Bischoff J. Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelial-to-mesenchymal transition and myofibroblastic activation. J Mol Cell Cardiol. 2015;80:175–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krizbai IA, Gasparics A, Nagyoszi P, Fazakas C, Molnar J, Wilhelm I, Bencs R, Rosivall L, Sebe A. Endothelial-mesenchymal transition of brain endothelial cells: Possible role during metastatic extravasation. PLoS One. 2015;10:e0123845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvandi Z, Nagata Y, Passos LSA, Hashemi Gheinani A, Guerrero JL, Wylie-Sears J, Romero DC, Morris BA, Sullivan SM, Yaghoubian KM, et al. Wnt site signaling inhibitor secreted frizzled-related protein 3 protects mitral valve endothelium from myocardial infarction-induced endothelial-to-mesenchymal transition. J Am Heart Assoc. 2022;11:e023695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehta V, Pang KL, Givens CS, Chen Z, Huang J, Sweet DT, Jo H, Reader JS, Tzima E. Mechanical forces regulate endothelial-to-mesenchymal transition and atherosclerosis via an alk5-shc mechanotransduction pathway. Sci Adv. 2021;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto A, Nagano T, Takehara S, Hibi M, Aizawa S. Shisa promotes head formation through the inhibition of receptor protein maturation for the caudalizing factors, wnt and fgf. Cell. 2005;120:223–235 [DOI] [PubMed] [Google Scholar]
- 26.Chen PY, Qin L, Tellides G, Simons M. Fibroblast growth factor receptor 1 is a key inhibitor of tgfbeta signaling in the endothelium. Sci Signal. 2014;7:ra90 [DOI] [PubMed] [Google Scholar]
- 27.Moser M, Binder O, Wu Y, Aitsebaomo J, Ren R, Bode C, Bautch VL, Conlon FL, Patterson C. Bmper, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol Cell Biol. 2003;23:5664–5679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mushimiyimana I, Niskanen H, Beter M, Laakkonen JP, Kaikkonen MU, Yla-Herttuala S, Laham-Karam N. Characterization of a functional endothelial super-enhancer that regulates adamts18 and angiogenesis. Nucleic Acids Res. 2021;49:8078–8096 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon request. RNA-Seq data is available at the Gene Expression Omnibus archive at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/geo/), with the accession number GSE207837.