

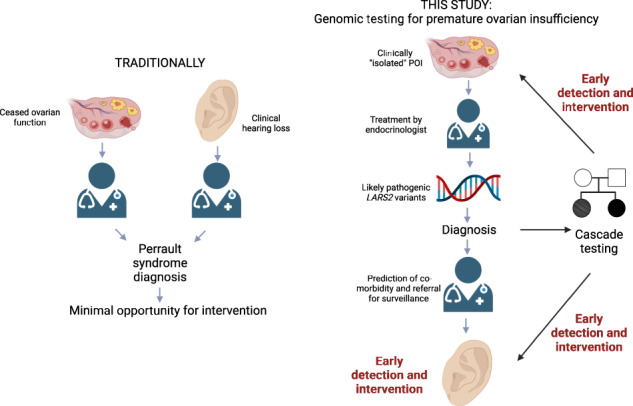
Abstract
Premature ovarian insufficiency (POI) affects 1 in 100 women and is a leading cause of female infertility. There are over 80 genes in which variants can cause POI, with these explaining only a minority of cases. Whole exome sequencing (WES) can be a useful tool for POI patient management, allowing clinical care to be personalized to underlying cause. We performed WES to investigate two French sisters, whose only clinical complaint was POI. Surprisingly, they shared one known and one novel likely pathogenic variant in the Perrault syndrome gene, LARS2. Using amino-acylation studies, we established that the novel missense variant significantly impairs LARS2 function. Perrault syndrome is characterized by sensorineural hearing loss in addition to POI. This molecular diagnosis alerted the sisters to the significance of their difficulty in following conversation. Subsequent audiology assessment revealed a mild bilateral hearing loss. We describe the first cases presenting with perceived isolated POI and causative variants in a Perrault syndrome gene. Our study expands the phenotypic spectrum associated with LARS2 variants and highlights the clinical benefit of having a genetic diagnosis, with prediction of potential co-morbidity and prompt and appropriate medical care, in this case by an audiologist for early detection of hearing loss.
Subject terms: Hypogonadism, Reproductive disorders, Genomics
Introduction
Premature ovarian insufficiency (POI) is characterized by amenorrhea (primary or secondary) and elevated follicle stimulating hormone (FSH) before the age of 40. It affects up to 1 in 100 women by age 40, but is rare in younger women with 1 in 1000 under the age of 30 affected (1). POI can be secondary to medical interventions such as cancer therapy or surgery or can result from genetic variation in one of at least 80 different genes [1, 2]. The known POI genes account for ~25% of cases [2]. Although it seems logical that a genetic diagnosis is more likely in the younger age-of-onset group of POI, this has not been yet demonstrated. Genetic diagnosis of POI patients, although not part of routine clinical care in most parts of the world, has demonstrated utility in improving patient management [3]. In France for example, a national network has been implemented to develop such genetic assessment through massive parallel sequencing (Plan France Médecine Génomique 2025 https://pfmg2025.aviesan.fr).
POI is not always an isolated condition but can present as a feature of several clinical syndromes, such as galactosemia, BPES syndrome, ovarioleukodystrophy [1]. Perrault syndrome (OMIM #233400) is one syndromic form of POI, being a rare autosomal recessive disorder characterized by sensorineural hearing loss in both males and females and POI in affected females. Some patients also have neurologic manifestations, including intellectual disability and cerebellar and peripheral nervous system involvement. It is difficult to comment on the prevalence of neurological symptoms as they can evolve in adult life and may not be evident before initial publication stating a diagnosis of Perrault syndrome. The first 15 patients from eight different families were reported to have Perrault syndrome Type 1 (no neurological involvement), but at least seven patients have since been published with Perrault syndrome Type 2 [4–6]. Interestingly, even within a single family there can be variability with respect to neurological involvement, with some individuals having Perrault syndrome Type 1 and relatives having ovarioleukodystrophy [5].
The association of gonadal dysgenesis and deafness was first reported in 1951, defining Perrault syndrome [7]. It is now known that Perrault syndrome has a broad genetic heterogeneity [8]: the identified causative genes include CLPP (OMIM *601119) required for the unfolded protein response and for mitochondrial ribosome biogenesis [9], ERAL1 involved in mitochondrial rRNA chaperoning [10], PRORP involved in mitochondrial tRNA processing [11], RMND1 (OMIM *614917) involved in mitochondrial translation [12], HARS2 (OMIM *600783) [13] and LARS2 (OMIM *604544) [14] responsible for charging mitochondrial tRNAs with histidine and leucine, respectively, HSD17B4 (OMIM *601860) involved in fatty-acid oxidation and steroid metabolism [15], PEX6 involved in peroxisomal biogenesis [8], GGPS1 required for C20-prenylation [8] and TFAM and TWNK (OMIM *606075) required for mtDNA replication and maintenance [8, 16, 17]. In ~60% of patients with Perrault syndrome, a molecular diagnosis cannot be made [18].
In this study, two pathogenic variants of LARS2 were identified in two young women, from the same French (Caucasian) family, presenting with POI but without known hearing deficit at the time of ovarian assessment. The LARS2 gene encodes mitochondrial leucyl-tRNA synthetase (LARS2), which belongs to the aminoacyl-tRNA synthetase (aaRS) family. AaRSs catalyze the esterification of amino acids on the 3′ end of their cognate transfer RNAs (tRNAs) and are essential for efficient translation of mRNAs into proteins [19]. Via amino-acylation assays, we established that the novel variant shared by these sisters has a significant impact on LARS2 function. The degree of LARS2 dysfunction cannot explain their relatively mild phenotype. These two cases and a review of the literature are used to explore the correlation between the phenotype and the LARS2 genotype.
Material and methods
Editorial policies and ethical considerations
Written informed consent was obtained from the patients. All procedures were in accordance with the ethical standards of the Ethics Committee of Rennes University Hospital and the French law.
Patients
The proband (III1) (Fig. 1A) experienced spontaneous puberty with her first menstruation at the age of 14 years. At the age of 20, she experienced secondary amenorrhea, and a hormonal evaluation was performed showing FSH: 64 IU/L (N: 3–9 IU/L), LH: 23 IU/L (N: 2–10 IU/L), estradiol <10 pg/mL (N: 27–161 pg/mL). Imaging via pelvic ultrasound and MRI showed a right ovary to be a fibrous band and two follicles in the left ovary. A second assessment was performed three months later and showed FSH: 64 IU/L (N: 3–9 IU/L), LH: 17 IU/L (N: 2–10 IU/L), estradiol <10 pg/mL (N: 27–161 pg/mL), and AMH: 0.01 ng/mL (N: 0.7–3.5 ng/mL). Anti-21-hydroxylase and anti-thyroid antibodies were not detected.
Fig. 1. Likely pathogenic LARS2 variants that co-segregate with POI.

A Pedigree of the family. III1 and arrow: proband, III2: affected sister, II1: father, II2: mother. Black: LARS2 variant segregation. Asterisk indicates individuals with reported infertility, but the nature of this infertility is not known. B Sanger validation of the LARS2 variants. III1: proband, III2: sister, II2: mother, II1: father. C Localization of LARS2 variants in the crystal structure of E. coli LARS ortholog (PDB: 4AQ7). The catalytic domain of the enzyme is shown in dark gray and additional domains (editing and tRNA binding) are indicated in light gray. The catalytic domain contains the active site (green) defined by two conserved motifs, HMGH and KMSKS, involved in the binding of the leucyl-adenylate (Leu-AMP). The formation of Leu-AMP is the first step of the amino-acylation reaction, indicated on the bottom of the structure. The second step consists in transferring the activated amino acid on the 3′ end of the tRNA molecule (not shown). Variants are indicated in purple. D Multi-species sequence alignment of LARS2 in metazoans. Primates (Homo sapiens, Macaca mulata, Mandrillus leucophaeus, Gorilla gorilla, Pongo abelii); other mammals (Trichechus manatus, Balaenoptera acutorostrata, Monodon monoceros, Orcinus orca); birds: (Apteryx rowi, Calidris pugnax, Aquila chrysaetos; Hirundo rustica); reptiles (Gopherus evgoodei, Platysternon megacephalum, Trachemys scripta elegans, Terrapene carolina triunguis) and fishes (Polypterus senegalus, Acipenser ruthenus, polyodon spathula, Lepisosteus oculatus) as well as in bacteria (Thermus thermophilus, Escherichia coli, Cyanobacteria bacterium J003, Fischerella thermalis). Alignments are restricted to the domains of eukaryotic LARS2 and bacterial LARS harboring the variants. Color codes are identical as those used in C.
The diagnosis of POI in the proband prompted evaluation of ovarian function in her younger sister (III2) (Fig. 1A), who had experienced spontaneous menarche at age 13 and had continued regular menses with hormonal assessment performed at age 16. Hormonal assessment revealed elevated FSH (80 IU/L) and LH (37 IU/L) along with low estradiol (<10 pg/mL) and AMH (<0.01 ng/mL). This hormone profile was confirmed 6 months later with FSH: 64 IU/L and estradiol <10 pg/mL. Anti-21-hydroxylase and anti-thyroid antibodies were not detected. Imaging showed streak ovaries. A marfanoid morphotype was noted.
For both patients, karyotypes and microarray analysis were normal. No premutation of the FMR1 gene was found.
Their parents (II1 and II2) were not related. Other more distant relatives reported infertility, including a maternal aunt of the mother as well as one of the daughters of this aunt and the son of a maternal uncle of the mother (Fig. 1A). Limited information is available on the nature of this familial infertility and DNA samples were not available for testing.
Whole-exome sequencing (WES)
DNA from patients and their parents were available allowing a quartet/quattro analysis.
Exome capture was performed using the SureSelect Human All Exon V7 kit (Agilent Technologies Inc, Santa Clara, CA, USA) and sequenced on a NextSeq500 instrument (Illumina, San Diego, CA, USA). WES data were processed using C-GeVarA pipeline (Constitutional Genetic Variant Analysis, Bioinformatics Department, Rennes University Hospital) and deposited into SeqR for analysis (https://seqr.broadinstitute.org/).
We performed two phases of analysis—the first focused on gene priority and the second focused on variant priority, as previously described [20]. Recessive mode of inheritance was prioritized (homozygous variants or compound heterozygous). Given there are extended family members with infertility, autosomal dominant inheritance was also explored. Allele frequency and tolerance of genes to missense and/or loss-of-function (LoF) variation were assessed in the public database, gnomAD (https://gnomad.broadinstitute.org/). Variant pathogenicity was predicted in silico using VarSome (https://varsome.com) which gives an overall view on prediction scores including Mutation Taster, Polyphen-2, SIFT, PROVEAN, REVEL, and CADD for example. The conservation of affected residues was assessed by Multiz Alignments of 100 vertebrates (UCSC Genome Browser https://genome.ucsc.edu/). The novel LARS2 variant has been submitted to ClinVar (SCV002099518 and SCV002099519). Variants were confirmed by targeted sequencing with the Sanger method and are described according to the hg19/GrCH37 version of the genome.
Functional studies
Recombinant LARS2 proteins were produced to assess amino-acylation efficiency of the identified variant. Wild-type and variant LARS2 proteins were expressed and purified as previously described [5, 21] in the presence of protease inhibitor cocktail (cOmpleteTM tablets EDTA-free, Roche, Merck KGaA, Darmstadt, Germany). In addition to the LARS2 c.1670A>C, p.(Tyr557Cys) patient variant, a c.351G>C, p.(Met117Ile) variant was produced and assayed as it was the only variant that has also been reported in compound heterozygosity with the p.(Thr522Asn) variant which has not been functionally evaluated [22]. E. coli tRNA5Leu transcript was prepared according to Perret et al. [23]. In vitro leucylation of E. coli tRNALeu transcript was performed according to Sohm et al. [24] in the presence of 20 µM L-[14C]leucine at 37 °C. Apparent kinetic parameters were determined from Lineweaver–Burk plots with 10 nM of enzyme (LARS2 WT, LARS2 Met117Leu or LARS2 Tyr557Cys) and 0.2–2 µM tRNALeu transcript. Data were expressed as averages of at least three independent experiments. A t test was performed to compare kcat/Km ratio of Wt and variant LARS2.
Results
WES identified compound heterozygous missense variants in the LARS2 gene
Variant filtration in the proband and her sister revealed two stand-out candidate variants in the LARS2 gene. No other candidate variant was identified. The first variant (NM_015340.3:c.1565C>A) was inherited from the mother (rs199589947) (Fig. 1B). This variant leads to the substitution of a threonine to asparagine (NP_056155.1:p.(Thr522Asn)) in the catalytic domain of the protein (Fig. 1C) and affects an amino acid that is highly conserved between mammals, birds, reptiles and fishes (Fig. 1D). This variant has previously been associated with Perrault syndrome in a homozygous or compound heterozygous state and functional analyses have proven its deleterious effect on the activity of the protein [14, 22]. It is also reported in ClinVar as pathogenic/likely pathogenic. It was classified as a pathogenic variant (class 5) according to the American College of Medical Genetics and Genomics (ACMG) [25] (Supplementary Table S1).
The second variant (NM_015340.3:c.1670A>G) was inherited from the father (rs1269970737) (Fig. 1B). It leads to the substitution of a tyrosine with a cysteine (NP_056155.1:p.(Tyr557Cys)) in the catalytic domain of the protein (Fig. 1C). It is a very rare variant with a MAF < 0.00001 in gnomAD and not referenced in ClinVar. At the protein level, the variant lies within a highly conserved region, close to the active site (Fig. 1C), and is thus probably damaging, which is in accordance with the in silico predictions (almost all pathogenic/disease causing/deleterious). It was classified as a likely pathogenic variant (class 4) according to ACMG [25] (Supplementary Table S1).
These two variants are compound heterozygous, consistent with an autosomal recessive mode of inheritance, likely explaining the POI phenotype. Following the molecular diagnosis, the proband and her sister reported some difficulties in hearing conversation, which had not been perceived as significant. The absence of obvious hearing loss at initial clinical assessment expands the phenotypic spectrum of patients with variants in this gene, to include isolated POI, although mild hearing loss may be missed, and progressive hearing loss may occur.
The pathogenic LARS2 variants were associated with mild hearing loss
The proband did not report any hearing difficulties during the initial genetic consultation. Once the molecular diagnosis was disclosed, however, she reported mild difficulties following conversation and when watching television. Prior to genetic diagnosis, these difficulties were not perceived as significant by the patient and did not prompt her to seek medical investigation. Subsequent audiological assessment showed a mild bilateral hearing loss at 250–500 Hz and normal hearing thresholds from 1000 to 8000 Hz (Fig. 2). Tympanometry was normal (Type A) bilaterally.
Fig. 2. Audiological data of the proband and her sister.

Hearing thresholds at standard audiometric frequencies are shown for air conduction (cross: left ear, open circle: right) and masked bone conduction (open square to left: left ear, open square to right: right ear) of the proband (A) and her sister (B). Speech audiometry shows the percentage of correct word perception at varying presentation level (dB HL, hearing level) in the proband (C) and her sister (D). Tympanometry in the sister (E) demonstrates a Type C profile (conductive hearing loss) in the right ear, and Type A (normal) in the left ear. MEP: middle ear pressure, ECV: ear canal volume, SC: static compliance.
Her sister similarly retrospectively reported some mild difficulties in hearing (requiring people to repeat themselves during conversation) that had not been of personal concern. Subsequent audiological assessment revealed a mild mixed bilateral hearing loss in the low frequencies at 125–1000 Hz. Hearing thresholds were found to be within the normal range bilaterally from 1000 to 8000 Hz. Tympanometry results were within normal limits for the left ear (Type A). A Type C tympanogram was found for the right ear, supportive of a middle ear component to the hearing loss. Speech audiometry was consistent with a mild hearing loss with greater amplification required to reach 100% correct speech understanding. Review of audiological history identified a similar level of hearing loss detected at 9 years of age after recommendation for assessment by the school nurse, however this had not been recalled by the patient and regular follow-up had not been pursued. Had genetic analysis been performed at the time, an early diagnosis of Perrault syndrome could have been achieved and the patient would have been monitored for decline in hearing as well as ovarian function.
The novel p.(Tyr557Cys) LARS2 variant is associated with a reduction in LARS2 function
The effect of the LARS2 p.(Tyr557Cys) variant on leucylation was investigated by generating a recombinant mutant enzyme and measuring its ability to attach radiolabeled leucine to an E. coli tRNALeu transcript in vitro, as we have performed previously [5]. The LARS2 p.(Tyr557Cys) variant had a 12.5-fold loss in catalytic efficiency compared to WT LARS2, mainly due to a decreased kcat value (Table 1). This in-vitro evidence of a deleterious impact of the variant escalated the variant from a “likely pathogenic” annotation to a “pathogenic” annotation (Supplementary Table S1) and is in keeping with the ninefold loss in aminoacylation efficiency previously determined for the LARS2 p.(Thr522Asn) variant [26].
Table 1.
Kinetic parameters for leucylation of E. coli tRNA5Leu transcript by LARS2 wild-type and variant recombinant proteins.
| LARS2 | Km (µM) | kcat (min−1) | kcat/Km (min−1 µM−1) | Loss of catalytic efficiencya |
|---|---|---|---|---|
| Wt | 2.0 (0.3) | 13.7 (0.8) | 6.85 (1.38) | 1 |
| Tyr557Cys | 2.6 (0.4) | 1.5 (0.3) | 0.55 (0.02; P = 0.024) | 12.5 |
| Met117Ile | 2.0 (0.5) | 10.1 (0.9) | 5.05 (0.74; P = 0.045) | 1.4 |
Values are mean (SEM), n ≥ 3. P values are shown for kcat/Km ratio compared to Wt.
Wt wildtype.
aLoss of efficiency is calculated as a fold-change relative to wild-type (Wt) LARS2.
These two amino acids, Thr 522 and Tyr557 are both highly conserved, even in bacterial leucyl-tRNA synthetase alignments (Fig. 1D) and located in the active site, in spatial proximity to the leucyl-AMP binding pocket (Fig. 1C).
The LARS2 p.(Met117Ile) variant was previously reported in compound heterozygosity with p.(Thr522Asn) in a family with Perrault syndrome characterized by severe hearing loss [22]. In contrast to Thr522 and Tyr557, the Met117 is located away from the active site (Fig. 1C), and as expected p.(Met117Ile) variant had only a marginal effect on amino-acylation efficiency in our conditions (Table 1).
Discussion
Our study describes the identification of one known and one novel LARS2 variant in association with apparently isolated POI. To date, 24 pathogenic or likely pathogenic single nucleotide variants (SNVs) or small insertions or deletions (indels) have been identified in association with LARS2—Perrault syndrome (https://www.ncbi.nlm.nih.gov/clinvar/, https://databases.lovd.nl/shared/genes/LARS2, http://www.hgmd.cf.ac.uk/ac/index.php) and no LARS2 variants have been associated with isolated POI (Fig. 3). A homozygous 4 kb gene deletion has also been recently reported in a patient with hearing loss [27]. While LARS2 variants were first identified in patients with Perrault syndrome, the phenotypic spectrum now extends from Perrault syndrome presenting initially as apparently isolated POI to more severe phenotypes (Perrault syndrome with neurological symptoms; hydrops, lactic acidosis, and sideroblastic anemia).
Fig. 3. Pathogenic or likely pathogenic variants reported in ClinVar, LOVD, or HGMD Pro with DOG tool [34].

Green: Perrault syndrome, Blue: hydrops, lactic acidosis, and sideroblastic anemia, Orange: mitochondrial myopathy, Pink: non-syndromic hearing loss and deafness, Light blue: rare genetic deafness, Gray: Neu-Laxova syndrome, Red: no phenotypic information, Black: VUS, Star: variants carried by the patients of this study, Underscored: novel variant LARS2 protein with domains, from left to right: The mitochondrial targeting signal (MTS), the catalytic domain (61–243 and 444–649) in the middle of which is inserted the editing domain (244–443) and followed by the tRNA binding domains (650–903). The color code used in Fig. 1C is conserved.
The LARS2 protein is responsible for loading leucine onto its cognate tRNALeu within mitochondria to enable mitochondrial protein synthesis. The human mitochondrial genome is limited in size and contains genes encoding only 22 tRNAs, two rRNAs and 13 polypeptides, the latter of which are all subunits of the mitochondrial respiratory chain complexes. Mitochondrial translation is therefore necessary for efficient energy generation. Dysfunctional LARS2 can impair mitochondrial protein synthesis leading to energy deficit and increased reactive oxygen species, which can in turn damage oocytes and cause other manifestations of mitochondrial disease. Impaired mitochondrial function has been demonstrated in yeast models harboring LARS2 variants [14]. Impaired mitochondrial translation in mice has been shown to cause hearing loss due to tissue-specific apoptosis [28]. Similarly, variants in LARS2 and other genes that disrupt mitochondrial translation, lead to failed ovarian development/function [10, 12, 29]. Clearly, loss of LARS2 function leading to energy deficit and increased reactive oxygen species has the potential to disrupt both ovarian function and hearing. The mechanism by which variants present first as an isolated POI are less clear. Hearing loss can be influenced by environment as well as genetics eg. drug exposure, loud noise exposure, low birth weight. It is possible that the sisters were fortunate to avoid significant environmental insult, protecting their hearing until a later age. The ovaries have a particularly high energy demand, and it remains possible that the level of LARS2 activity is enough to support hearing but not enough for protecting development and integrity of the oocyte pool. Although we measured the level of LARS2 activity in vitro, mitochondrial deficiency is known to have high variability and tissue specificity. It could be that LARS2 function and subsequent mitochondrial OXPHOS activity is more severely reduced in the ovaries of this patient.
Here we describe the first cases of variants in a known Perrault syndrome gene, LARS2, with POI as an isolated clinical concern on initial consultation. Unlike other LARS2 cases, the molecular diagnosis for these patients was obtained during the assessment of what had been diagnosed as “isolated” POI instead of the assessment of hearing loss. LARS2 patients show highly variable bilateral hearing loss, ranging from mild to profound, and sometimes progressive, particularly between the ages of 6 and 25 [30]. The age of onset also varies from congenital to early adulthood in women [31]. Of note, all individuals with the p.(Thr522Asn) LARS2 variant had Perrault syndrome with sensorineural hearing loss at low frequencies [22]. Audiograms of LARS2 patients have a distinct upward sloping shape [14]. Such audiograms offer clues to a possible Perrault phenotype in affected girls. Conclusive diagnosis, however, can be achieved via genetic analysis.
The p.(Thr522Asn) LARS2 variant has now been reported in ten individuals from five families (Table 2). The severity of hearing loss varies across these families, and cannot solely be explained by the variant in trans as phenotypic variability is seen even between families homozygous for the p.(Thr522Asn) variant. We have functionally evaluated the in vitro amino-acylation efficiency of the p.(Thr522Asn) variant and variants found in trans with it ([26] and this study). The amino-acylation efficiency of the variants does not appear to correlate with disease severity (Table 2) indicating that there may be genetic and/or environmental modifiers of LARS2-associated disease. This has been observed in other mitochondrial aminoacyl-tRNA synthetase disorders [32].
Table 2.
Clinical features of patients that harbor at least one LARS2 p.(Thr522Asn) variant (adapted from ref. [22]).
| ID | III-1 (This study) | III-2 (This study) | P3:II-1 [22] | P3:II-2 [22] | P2:II-1 [22] | P2:II-2 [22] | F1:II-2 [14] | F1:II-1 [14] | F1:II-3 [14] | Proband [26] |
|---|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | French | French | British | British | Argentinian | Argentinian | Palestinian | Palestinian | Palestinian | Pakistani |
| Sex | F | F | F | M | F | M | F | M | M | F |
| Karyotype | 46, XX | 46, XX | 46, XX | NR | 46, XX | NR | 46, XX | NR | NR | 46, XX |
| Age at genetic diagnosis (y) | 21 | 16 | 25 | 26 | 30 | 27 | 30 | 32 | 27 | Post-mortem |
| LARS2 variants | p.[(Thr522Asn)]; [(Tyr557Cys)] | p.[(Thr522Asn)]; [(Tyr557Cys)] | p.[(Thr522Asn)]; [(Met117Ile)] | p.[(Thr522Asn)]; [(Met117Ile)] | p.[(Thr522Asn)]; [(Thr522Asn)] | p.[(Thr522Asn)]; [(Thr522Asn)] | p.[(Thr522Asn)]; [(Thr522Asn)] | p.[(Thr522Asn)]; [(Thr522Asn)] | p.[(Thr522Asn)]; [(Thr522Asn)] | p.[(Thr522Asn)]; [(Ala430Val)] |
| Loss of amino-acylation efficiency for each variant (fold-change) | 9; 12.5 | 9; 12.5 | 9; 1.4 | 9; 1.4 | 9; 9 | 9; 9 | 9; 9 | 9; 9 | 9; 9 | 9; 18 |
| Sensorineural hearing loss | ||||||||||
| Age at hearing loss diagnosis (y) | 23 | 19 | 2.5 | 2.5 | 8 | 26 | 3–5 | 3–5 | 3–5 | NR |
| Degree of hearing loss | Sub-clinical | Sub-clinical | Severe/profound | Severe/profound | Moderate | Mild / moderate |
Mild / moderate / severe Right ear more affected than left |
Severe at low frequency Less severe at high frequency |
Severe at low frequency Less severe at high frequency |
NR |
| Gonadal dysfunction | ||||||||||
| Pelvic ultrasound/ MRI | Right ovary = fibrous band, two follicles on the left ovary | Streak ovaries | Small uterus and ovaries | NA | Small uterus and ovaries | NA | Small uterus, ovaries not detected | NA | NA | NR |
| Menarche | Yes | Yes | Yes | NA | No | NA | No | NA | NA | NA |
| POI | Yes | Yes | Yes | NA | Yes | NA | Yes | NA | NA | NA |
| FSH IU/L | 64 | 80 | 74 (≤30) | 3.1 (1–11) | 99.6 (2.3–29) | NR | 76.9 | NR | NR | NA |
| LH IU/L | 23 | 37 | 63 (≤30) | 3.9 (1–11) | 48.0 (1.7–52) | NR | 30.3 | NR | NR | NA |
| Estradiol pg/mL | <10 | <10 | 24.8 (>49) | NA | 7.0 (10–388) | NA | NR | NA | NA | NA |
| Neurological features | No | No | No | No | No | No | No | NR | NR | Seizures |
| Other features | No | Marfanoid habitus | Hemidystrophy, mild facial dysmorphism | Hypospadias, mild facial dysmorphism | No | No | No | NR | NR | Hydrops, lactic acidosis, sideroblastic anemia. Multi-system failure. Death at 5 days. |
It remains unclear whether LARS2 can be responsible for truly isolated POI, or whether all patients will experience some form of hearing loss. Audiology follow-up is essential to elucidate hearing ability over time in these individuals. Our patients do, however, demonstrate that LARS2-related Perrault syndrome can present as an isolated clinical concern, broadening the phenotypic spectrum associated with this gene. Our results highlight the potential for genetic diagnosis of POI patients to provide insight into future health risks, such as hearing loss. Audiological examination is not recommended for the routine care of all cases of apparently isolated POI [33] as the vast majority have a truly isolated POI, but with genetic indication audiological examination is imperative. Early management of POI-related co-morbidities can improve long-term outcomes. Cascade testing can allow early detection of hearing loss in family members, including male relatives. Conversely, genetic diagnosis of pre-pubertal girls with hearing loss can identify genetic Perrault syndrome. This allows prediction of future ovarian dysfunction and early interventions for improved clinical outcomes [31].
Conclusion
Here we described a new presentation of LARS2-related disease of POI without perceived hearing loss. A new likely pathogenic variant of LARS2 was identified in two affected women, and we demonstrated that it leads to relatively severe functional impairment of the variant protein in vitro. This novel variant has further expanded the existing genotypic spectrum of LARS2 variants, and our results can aid in the molecular diagnosis and genetic counseling of patients with hearing loss and/or POI.
Our cases show that genomic testing is beneficial for the diagnosis of POI, particularly in young women. We demonstrate new considerations for genetic counseling of families carrying LARS2 variants. Early genetic diagnosis allows prevention or early detection and improved management by pre-emptive screening for associated pathologies (such as hearing loss or neurological pathologies). Preservation of female fertility (ovarian stimulation followed by puncture and oocyte vitrification) can also be proposed to patients and their relatives if the parameters of the ovarian reserve allow it.
Supplementary information
Acknowledgements
We thank the Bioinformatic department of CHU Rennes (UF Bioinformatique et Génétique Computationnelle, Service de Génétique Moléculaire et Génomique, Pr M. De Tayrac and Dr W. Carré) for helpful advice and technical assistance. Dr A. Meyer (otorhinolaryngologist at Polyclinique Sévigné, Cesson-Sévigné) performed audiology assessment. Some figures were created with BioRender.com.
Author contributions
EJT and SJ contributed to conception and design of the study. MABR, AHS, SJ, and EJT participated in the project supervision. ASN, MB, LA, and CR were involved in patients’ medical care. JRT, MF, LGR, and JC contributed to functional work. All authors contributed to the acquisition and/or analysis of data. ASN, EJT, DG, and SJ wrote the manuscript. All authors revised it critically and approved the final version.
Funding
This work was supported by a CHU Rennes grant (Appel à Projets Innovations 2019 to SJ), an Australian NHMRC program grant (1074258 to AHS), NHMRC fellowships (1054432 to EJT, 1062854 to AHS) and an Australian Mito Foundation grant (EJT). Part of this work was performed under the Rare Diseases Functional Genomics program, supported by the Luminesce Alliance—Innovation for Children’s Health, a not for profit cooperative joint venture between the Sydney Children’s Hospitals Network, the Children’s Medical Research Institute, and the Children’s Cancer Institute. It has been established with the support of the NSW Government to coordinate and integrate pediatric research. Luminesce Alliance is also affiliated with the University of Sydney and the University of New South Wales Sydney (LGR). The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian government’s operational infrastructure support program. The Chair in Genomic Medicine awarded to JC is generously supported by The Royal Children’s Hospital Foundation.
Data availability
The datasets generated and/or analyzed during the current study are contained within the manuscript or available from the corresponding author on reasonable request and within ethical constraints. The LARS2 variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, SCV002099518 and SCV002099519).
Competing interests
The authors declare no competing interests.
Ethical approval
Written informed consent was obtained from the patients. All procedures were in accordance with the ethical standards of the Ethics Committee of Rennes University Hospital and the French law.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Elena J. Tucker, Sylvie Jaillard.
Contributor Information
Elena J. Tucker, Email: elena.tucker@mcri.edu.au
Sylvie Jaillard, Email: sylvie.jaillard@chu-rennes.fr.
Supplementary information
The online version contains supplementary material available at 10.1038/s41431-022-01252-1.
References
- 1.Tucker EJ, Grover SR, Bachelot A, Touraine P, Sinclair AH. Premature ovarian insufficiency: new perspectives on genetic cause and phenotypic spectrum. Endocr Rev. 2016;37:609–35. doi: 10.1210/er.2016-1047. [DOI] [PubMed] [Google Scholar]
- 2.Qin Y, Jiao X, Simpson JL, Chen ZJ. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update. 2015;21:787–808. doi: 10.1093/humupd/dmv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tucker EJ, Grover SR, Robevska G, van den Bergen J, Hanna C, Sinclair AH. Identification of variants in pleiotropic genes causing “isolated” premature ovarian insufficiency: implications for medical practice. Eur J Hum Genet. 2018;26:1319–28. doi: 10.1038/s41431-018-0140-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosaki R, Horikawa R, Fujii E, Kosaki K. Biallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms. Am J Med Genet A. 2018;176:404–8. doi: 10.1002/ajmg.a.38552. [DOI] [PubMed] [Google Scholar]
- 5.Riley LG, Rudinger-Thirion J, Frugier M, Wilson M, Luig M, Alahakoon TI, et al. The expanding LARS2 phenotypic spectrum: HLASA, Perrault syndrome with leukodystrophy, and mitochondrial myopathy. Hum Mutat. 2020;41:1425–34. doi: 10.1002/humu.24050. [DOI] [PubMed] [Google Scholar]
- 6.van der Knaap MS, Bugiani M, Mendes MI, Riley LG, Smith DEC, Rudinger-Thirion J, et al. Biallelic variants in LARS2 and KARS cause deafness and (ovario)leukodystrophy. Neurology. 2019;92:e1225–e1237. doi: 10.1212/WNL.0000000000007098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perrault M, Klotz B, Housset E. [Two cases of Turner syndrome with deaf-mutism in two sisters] Bull Mem Soc Med Hop Paris. 1951;67:79–84. [PubMed] [Google Scholar]
- 8.Tucker EJ, Rius R, Jaillard S, Bell K, Lamont PJ, Travessa A, et al. Genomic sequencing highlights the diverse molecular causes of Perrault syndrome: a peroxisomal disorder (PEX6), metabolic disorders (CLPP, GGPS1), and mtDNA maintenance/translation disorders (LARS2, TFAM). Hum Genet. 2020;139:1325–43. [DOI] [PubMed]
- 9.Jenkinson EM, Rehman AU, Walsh T, Clayton-Smith J, Lee K, Morell RJ, et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet. 2013;92:605–13. doi: 10.1016/j.ajhg.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatzispyrou IA, Alders M, Guerrero-Castillo S, Zapata Perez R, Haagmans MA, Mouchiroud L, et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum Mol Genet. 2017;26:2541–50. doi: 10.1093/hmg/ddx152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochberg I, Demain LAM, Richer J, Thompson K, Urquhart JE, Rea A, et al. Bi-allelic variants in the mitochondrial RNase P subunit PRORP cause mitochondrial tRNA processing defects and pleiotropic multisystem presentations. Am J Hum Genet. 2021;108:2195–204. [DOI] [PMC free article] [PubMed]
- 12.Demain LA, Antunes D, O’Sullivan J, Bhaskhar SS, O’Keefe RT, Newman WG. A known pathogenic variant in the essential mitochondrial translation gene RMND1 causes a Perrault-like syndrome with renal defects. Clin Genet. 2018;94:276–7. doi: 10.1111/cge.13255. [DOI] [PubMed] [Google Scholar]
- 13.Pierce SB, Chisholm KM, Lynch ED, Lee MK, Walsh T, Opitz JM, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA. 2011;108:6543–8. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pierce SB, Gersak K, Michaelson-Cohen R, Walsh T, Lee MK, Malach D, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet. 2013;92:614–20. doi: 10.1016/j.ajhg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierce SB, Walsh T, Chisholm KM, Lee MK, Thornton AM, Fiumara A, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome. Am J Hum Genet. 2010;87:282–8. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominguez-Ruiz M, Garcia-Martinez A, Corral-Juan M, Perez-Alvarez AI, Plasencia AM, Villamar M, et al. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: overlap of TWNK-related recessive disorders. J Transl Med. 2019;17:290. doi: 10.1186/s12967-019-2041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ullah F, Rauf W, Khan K, Khan S, Bell KM, de Oliveira VC, et al. A recessive variant in TFAM causes mtDNA depletion associated with primary ovarian insufficiency, seizures, intellectual disability and hearing loss. Hum Genet. 2021;140:1733–51. [DOI] [PubMed]
- 18.Pan Z, Xu H, Tian Y, Liu D, Liu H, Li R, et al. Perrault syndrome: Clinical report and retrospective analysis. Mol Genet Genom Med. 2020;8:e1445. doi: 10.1002/mgg3.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubio Gomez MA, Ibba M. Aminoacyl-tRNA synthetases. RNA. 2020;26:910–36. doi: 10.1261/rna.071720.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tucker EJ, Jaillard S, Grover SR, van den Bergen J, Robevska G, Bell KM, et al. TP63-truncating variants cause isolated premature ovarian insufficiency. Hum Mutat. 2019;40:886–92. [DOI] [PubMed]
- 21.Yao YN, Wang L, Wu XF, Wang ED. Human mitochondrial leucyl-tRNA synthetase with high activity produced from Escherichia coli. Protein Expr Purif. 2003;30:112–6. doi: 10.1016/S1046-5928(03)00097-4. [DOI] [PubMed] [Google Scholar]
- 22.Demain LA, Urquhart JE, O’Sullivan J, Williams SG, Bhaskar SS, Jenkinson EM, et al. Expanding the genotypic spectrum of Perrault syndrome. Clin Genet. 2017;91:302–12. doi: 10.1111/cge.12776. [DOI] [PubMed] [Google Scholar]
- 23.Perret V, Garcia A, Grosjean H, Ebel JP, Florentz C, Giege R. Relaxation of a transfer RNA specificity by removal of modified nucleotides. Nature. 1990;344:787–9. doi: 10.1038/344787a0. [DOI] [PubMed] [Google Scholar]
- 24.Sohm B, Frugier M, Brule H, Olszak K, Przykorska A, Florentz C. Towards understanding human mitochondrial leucine aminoacylation identity. J Mol Biol. 2003;328:995–1010. doi: 10.1016/S0022-2836(03)00373-5. [DOI] [PubMed] [Google Scholar]
- 25.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riley LG, Rudinger-Thirion J, Schmitz-Abe K, Thorburn DR, Davis RL, Teo J, et al. LARS2 variants associated with hydrops, lactic acidosis, sideroblastic anemia, and multisystem failure. JIMD Rep. 2016;28:49–57. doi: 10.1007/8904_2015_515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan A, Lenarduzzi S, Spedicati B, Cattaruzzi E, Murru FM, Pelliccione G, et al. Lights and shadows in the genetics of syndromic and non-syndromic hearing loss in the Italian population. Genes. 2020;11. [DOI] [PMC free article] [PubMed]
- 28.Someya S, Yamasoba T, Kujoth GC, Pugh TD, Weindruch R, Tanokura M, et al. The role of mtDNA mutations in the pathogenesis of age-related hearing loss in mice carrying a mutator DNA polymerase gamma. Neurobiol Aging. 2008;29:1080–92. doi: 10.1016/j.neurobiolaging.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen A, Tiosano D, Guran T, Baris HN, Bayram Y, Mory A, et al. Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum Mol Genet. 2018;27:1913–26. doi: 10.1093/hmg/ddy098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lerat J, Jonard L, Loundon N, Christin-Maitre S, Lacombe D, Goizet C, et al. An application of NGS for molecular investigations in Perrault syndrome: study of 14 families and review of the literature. Hum Mutat. 2016;37:1354–62. doi: 10.1002/humu.23120. [DOI] [PubMed] [Google Scholar]
- 31.Carminho-Rodrigues MT, Klee P, Laurent S, Guipponi M, Abramowicz M, Cao-van H, et al. LARS2-Perrault syndrome: a new case report and literature review. BMC Med Genet. 2020;21:109. doi: 10.1186/s12881-020-01028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez-Serrano LE, Chihade JW, Sissler M. When a common biological role does not imply common disease outcomes: disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J Biol Chem. 2019;294:5309–20. doi: 10.1074/jbc.REV118.002953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.European Society for Human R, Embryology Guideline Group on POI. Webber L, Davies M, Anderson R, Bartlett J, et al. ESHRE guideline: management of women with premature ovarian insufficiency. Hum Reprod. 2016;31:926–37. doi: 10.1093/humrep/dew027. [DOI] [PubMed] [Google Scholar]
- 34.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19:271–3. doi: 10.1038/cr.2009.6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are contained within the manuscript or available from the corresponding author on reasonable request and within ethical constraints. The LARS2 variants have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, SCV002099518 and SCV002099519).