

Abstract
Many diseases, especially cancer, are caused by the abnormal expression of non-coding microRNAs (miRNAs), which regulate gene expression, leading to the development of miRNA-based therapeutics. Synthetic miRNA inhibitors have shown promising efficacy in blocking the activity of aberrant miRNAs that are upregulated in disease-specific pathologies. On the other hand, miRNAs that aid in preventing certain diseases and are reduced in expression in the disease state need different strategies. To tackle this, miRNA mimics, which mimic the activity of endogenous miRNAs, can be delivered for those miRNAs downregulated in different disease states. However, the delivery of miRNA mimics remains a challenge. Here, we report a cationic polylactic-co-glycolic acid (PLGA)-poly-L-histidine delivery system to deliver miRNA mimics. We chose miR-34a mimics as a proof of concept for miRNA delivery. miR-34a-loaded PLGA-poly-L-histidine nanoparticles (NPs) were formulated and biophysically characterized to analyze the structural properties of miRNA mimic-loaded NPs. In vitro efficacy was determined by investigating miR-34a and downstream target levels and performing cell viability and apoptosis assays. We confirmed in vivo efficacy through prolonged survival of miR-34a NP-treated A549-derived xenograft mice treated intratumorally. The results of these studies establish PLGA-poly-L-histidine NPs as an effective delivery system for miRNA mimics for treating diseases characterized by downregulated miRNAs.
Keywords: miRNA mimic, PLGA, PLGA-poly-L-histidine, miRNA mimic delivery, miR-34a mimic
Graphical abstract
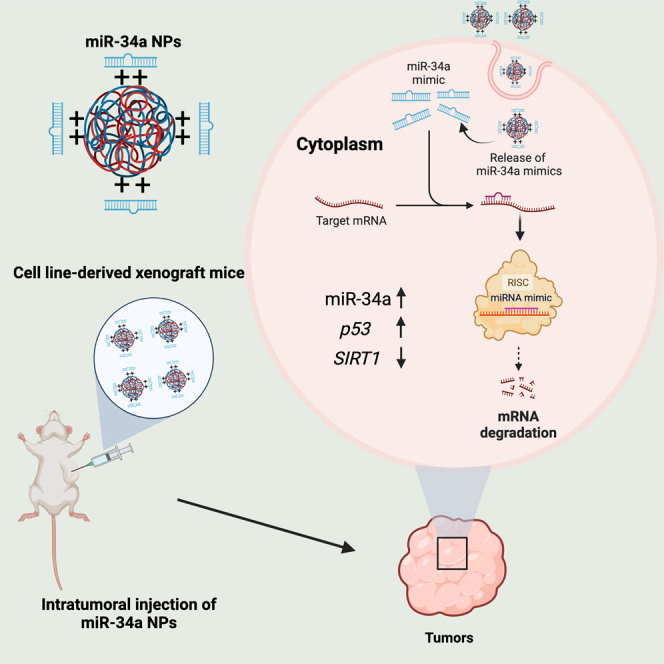
Raman Bahal and colleagues report a novel cationic PLGA-poly-L-histidine nanoparticle delivery platform for the delivery of miR-34a mimics as a proof of concept for miRNA mimic delivery. Herein, they demonstrate efficacy through robust biophysical, cell culture, and in vivo studies to highlight the therapeutic potential of PLGA-poly-L-histidine nanoparticles for miRNA mimic delivery.
Introduction
The RNA therapeutics field has taken off over the past decade as several new nucleic acid-based drugs have entered the market to treat various disorders through RNA silencing or processing. Currently, 14 antisense oligonucleotide drugs and 4 siRNA drugs have been approved for clinical application.1,2,3 Although targeting a messenger RNA (mRNA) of interest has been a successful strategy for treating different diseases, targeting non-coding RNAs, such as microRNAs (miRNAs), still presents a challenge. miRNAs are short, non-coding RNAs (∼22–23 nt in length) that are critical players in post-transcriptional gene regulation, as they activate RNA silencing through the RNA-induced silencing complex (RISC).4 Although it is a promising area of RNA therapeutics, there are currently no Food and Drug Administration-approved miRNA-based therapeutics.5,6 Two forms of miRNA-based therapy can treat miRNA dysregulation in disease: antimiRs and miRNA mimics. AntimiRs have been shown to target upregulated miRNAs in various disease conditions, especially in cancer. AntimiR-155 is under investigation to treat diffuse large B cell lymphoma by silencing miR-155.7,8,9,10 For treating cutaneous T cell lymphoma, cobomarsen (antimiR-155 drug candidate) is being investigated by Viridian Therapeutics in phase 2 clinical trials.11
miRNA mimics are a promising technology with therapeutic potential to treat numerous diseases caused by depleted levels of specific miRNAs. miRNA mimics are synthetic double-stranded nucleic acids that mimic the activity of endogenous miRNAs through the activation of the RISC via the Argonaute 2 protein.12 The synthetic nucleic acid design consists of a double-stranded structure with guide and passenger strands. Upon entering the cytoplasm and activation of the RISC, the passenger strand degrades, and the miRNA guide strand binds to the target RNA strand through Watson-Crick base pairing.13 Given the negative charge of miRNA mimics and their susceptibility to enzymatic degradation, successful delivery of miRNA mimics is challenging. A positively charged delivery system is essential for the delivery of negatively charged oligonucleotides.14 However, many currently existing highly cationic delivery systems, such as cationic lipids, can cause an adverse reaction that limits their translation to the clinic.15
Here, we established polylactic-co-glycolic acid (PLGA)-poly-L-histidine (His) nanoparticles (NPs) containing a moderate cationic charge as a potential nanocarrier to successfully deliver miRNA mimics both in vitro and in vivo. To test the PLGA-poly-L-His delivery platform for miRNA mimics, we used miR-34a mimics for a proof-of-concept study. miR-34a is a potent tumor suppressor miRNA that inhibits various cancer-causing pathways, including the epithelial to the mesenchymal transition state,16 and is downregulated in many solid tumors, including lung adenocarcinomas.17,18,19,20,21 This loss can be combatted through the delivery of miR-34a mimics to increase miR-34a activity with the desired target response. However, successfully translating miRNA mimic technology to the clinic has remained challenging. Mirna Therapeutics, now Synlogic, developed a liposomal-miRNA mimic formulation to target miR-34a (MRX34), which was tested in phase 1 clinical trials.22 This formulation used a cationic liposome to encapsulate miR-34a mimics. However, the trial was halted after severe adverse events, such as cytokine release, hypoxia, and hepatic failure, were noted.23 Therefore, there is a need for a safe and biocompatible nanocarrier for the delivery of miRNA mimics.
In this study, we developed an effective delivery vehicle for miR-34a mimics using PLGA-poly-L-His NPs. We initially performed a thorough biophysical characterization to confirm the stability of our formulation. Small-angle X-ray scattering (SAXS) analysis revealed the structural arrangement of miRNA mimics within the PLGA-poly-L-His NPs. To establish the proof of concept in vitro, we tested the miR-34a NPs in cell culture in A549 cells, which are a lung adenocarcinoma epithelial cell line. We evaluated cellular uptake and the route of endocytosis. We also assessed miR-34a and p53 mRNA and protein levels using gene expression and western blot analysis. We evaluated A549 cell survival using cell viability and apoptosis-based assays. We then demonstrated in vivo efficacy by testing the miR-34a NPs intratumorally in A549-derived xenograft mice. Overall, we created an miRNA mimic-based therapeutic utilizing a safe and effective delivery system based on PLGA-poly-L-His formulations.
Results
miR-34a NP formulations and physicochemical characterization
Here, we utilized a PLGA-poly-L-His NP delivery system to circumvent some of the present challenges and achieve optimal loading during formulation. PLGA-poly-L-His NPs are a unique cationic delivery system in which a PLGA NP core contains patches of His residues on the surface to give the particles a cationic charge that can be fine-tuned based on the amount of His present during formulation.24 Prior studies established that PLGA-poly-L-His (w/w 4.9/0.1) can deliver cargo without toxicity.24 Initially, we tested if PLGA NPs can encapsulate the miR-34a mimic. However, we did not observe significant loading of the miR-34a mimic (not shown), which could be due to the negative charge on the PLGA surface that hinders the loading of the negatively charged miRNA mimic.
Hence, we formulated PLGA-poly-L-His NPs using the double emulsion solvent evaporation technique to encapsulate miRNA mimics (Figure 1A). We prepared NPs without miRNA mimic (blank NPs), miR-34a mimic-containing NPs (miR-34a NPs), and NPs containing a scramble sequence mimic (Scr-34a NPs). To investigate the cellular uptake of mimics, an NP formulation with miR-34a mimics covalently conjugated to fluorescein isothiocyanate (miR-34a-FITC NPs) was used.
Figure 1.

Formulation of PLGA-poly-L-His-miR-34a nanoparticles containing miR-34a mimic and their biophysical characterization
(A) Schematic showing PLGA-poly-L-His (PH) NP formulation using double emulsion solvent evaporation. PLGA and poly-L-histidine (PLGA-poly-L-His) were solubilized at a 4.9/0.1 w/w ratio in acetone:DCM (2:1) organic solvent. (B) SEM and TEM images of miR-34a-loaded PLGA-PH NPs. The respective size distribution was calculated from the SEM image using ImageJ software. The scale bar represents 100 nm. (C) NP hydrodynamic size (in nm), polydispersity index (PDI), and surface charge density (in mV) of blank, miR-34a, and Scr-34a NPs. Data represent the mean of three batches (n = 3) ± standard deviation. Data for individual batches are shown in Figure S1. (D) Loading (percentage of initial loading/mg NP) and release kinetics as percentage cumulative release of miR-34a and Scr-34a NPs in phosphate-buffered saline. Data represent the mean (n = 3) ± standard deviation. (E) In vitro release analysis to determine the miRNA mimic integrity using polyacrylamide gel electrophoresis. (Top) Schematic showing the workflow for release of miRNA mimic from NPs followed by PAGE. (Bottom) Gel shift assay to analyze the stability of released miRNA mimic stability. Lane 1 contains the miRNA mimic stock at 1 μM concentration. Lanes 2–4 contain the released miRNA mimic (equivalent concentration) from the NPs. Data show samples from three separate NP batches.
We utilized electron microscopy and dynamic light scattering to conduct thorough physicochemical testing, including morphology, particle size/polydispersity, and surface charge of the formulations. Visually, the blank NPs and miR-34a NPs were spherical in morphology and uniform in size (∼90 nm), as shown by scanning electron microscopy (SEM) images (Figure 1B). In addition, we performed transmission electron microscopy (TEM) and observed uniform morphology and stable morphological structure of NPs containing miR-34a mimics (Figure 1B). Further, we measured the hydrodynamic size and polydispersity index (PDI) of the NP formulations using dynamic light scattering. Dynamic light scattering is used to quantify the Brownian motion of NPs in a solvent, revealing hydrodynamic size. We confirmed that the hydrodynamic size was ∼200 nm for all formulations, with a PDI of 0.10–0.20, indicating a minimal change in particle size and distribution when loaded with miR-34a mimics (Figure 1C). We also assessed the surface charge density via zeta potential. We noted zeta potentials (mV) of +4.29 ± 1.195 mV for blank NPs, −21 ± 5.60 mV for miR-34a NPs, and −20 ± 2.73 mV for Scr-34a NPs (Figure 1C). The change in surface charge density from positive to negative when loaded with the miRNA mimic is attributed to the interactions of the negatively charged miRNA mimic with the NP formulation. We also confirmed NP formulations of different batches, where we observed minimal differences in particle size, PDI, and surface charge among three batches when testing blank NPs, miR-34a NPs, and Scr-34a NPs (Figure S1).
Loading and release kinetics of mimic-loaded NPs
Next, to understand the quantity of miRNA mimic present in the NP formulation and determine the release of miRNA mimic, we quantified the loading and percentage cumulative release for both miR-34a and Scr-34a NPs. We performed a solvent extraction technique for the loading study and quantified the miRNA mimic in the aqueous buffer layer. The loading was estimated to be ∼15%–20% for miR-34a and Scr-34a NPs, respectively, based on the initial loading dose of 1 nmol/mg NP (Figure 1D). We also investigated the extraction efficiency of miRNA mimic from the dichloromethane (DCM)/sodium acetate mixture. For this study, we used blank NPs and added 150 pmol of miR-34a mimic. DCM was then added to the blank NPs and miR-34a mimic, samples were shaken for 24 h, and the sodium acetate buffer was added to extract the miR-34a mimic into the aqueous layer. The extraction efficiency was quantified to be 80%–100% (Figure S2).
We further quantified the release of miRNA mimic over a 48 h period in phosphate-buffered saline (PBS) at physiological pH (7.4). Here, we noted an initial burst release at 15 min, followed by 50%–70% release by 1 h, and a complete overall release by 24 h (Figure 1D). The release profile is consistent with the presence of the miRNA mimic on the surface of the NP, as shown by SAXS results (Figure 2). At pH 7.4, the His on the NP surface becomes un-ionized, reducing the affinity for the miRNA mimic.
Figure 2.

Structural arrangement of miR-34a NPs using small-angle X-ray scattering
(A) The SAXS patterns of PLGA-poly-L-His NPs with and without miR-34a mimics. (Table 1) The best-fitted results from the core-shell spherical model with a power law. (B) Schematic showing the arrangement of miR-34a mimics and PLGA-poly-L-His NPs. The miR-34a mimic is shown to be binding around the surface of the PLGA-poly-L-His NP based on the SAXS outcomes.
RNA integrity of the miR-34a mimic in the NP formulation
miRNA mimics have the potential to degrade both during the formulation process and in storage due to structural changes and RNase-based contamination. To test the stability of the miR-34a mimics in our formulation, we performed an in vitro release assay of the NP formulation (Figure 1E). Here, we resuspended both miR-34a and Scr-34a NPs in PBS at physiological temperature (37°C) for 48 h to release the mimic from the formulation. We then loaded the released miR-34a mimic onto a 5% polyacrylamide gel electrophoresis (PAGE) gel to evaluate RNA stability. We loaded the 5% PAGE gel with a 1 μM miRNA mimic stock as a control, and the released miRNA mimic was also loaded in the adjacent wells. In this study, we tested samples from three NP batches of both miR-34a and Scr-34a NP formulations. The PAGE gel demonstrates the stability of both miR-34a and Scr-34a mimics by the presence of similar band intensities between the miRNA mimic stock in lane 1 and the released mimic in lanes 2–4 (Figure 1E). In addition, an intense single band in lanes 2–4 shows minimal impurities or degradation products in the formulation. Therefore, we established that the miR-34a mimic cargo is stable in the NP and does not degrade during the formulation process.
Structural characterization of miR-34a NPs using small-angle X-ray scattering
In previous work, it has been shown that the PLGA-poly-L-His NPs have a core-shell spherical structure by small-angle neutron scattering (SANS).24 Using SANS, we also established that PLGA-poly-L-His NPs consist of a PLGA core with His patches on the surface, giving the NPs a cationic charge.24 We adopted the same morphological model to fit the SAXS data of PLGA-poly-L-His NPs in the presence and absence of miR-34a mimics to understand how miR-34a mimics associate with the NPs (Figure 2A). The low q regime for both BL NPs and miR-34a NPs shows q−4 decays, indicating that the particle size is more significant than 800 Å, outside the SAXS probing range. We observed subtle differences in the high q regime (>0.1 Å−1), with higher scattering intensity and slower decay of miR-34a NPs than those of BL NPs. The differences suggest structural variation at a length scale smaller than 60 Å. We utilized the core-shell spherical model as the best-fitting parameters, which show the most significant difference to be the shell thickness (T), varying from 98 ± 1 Å for BL NPs to 106 ± 1 Å after the association with miR-34a mimics (Figure 2, table, and supplemental information). Based on the SAXS results, we attribute the scattering patterns to the binding of miR-34a mimics onto the surface of PLGA-poly-L-His NPs (Figure 2B). Further, consistent with our hypothesis, quantitatively, miR-34a NPs possess a negative surface charge due to the miR-34a mimics present on the NP surface.
Cellular uptake of miR-34a NPs
After understanding our formulation’s physicochemical properties, we evaluated the functional efficacy in cell culture in A549 cells. The A549 cell line is a lung adenocarcinoma cell line with reduced miR-34a and p53 levels, promoting cell proliferation. p53 is the downstream factor of choice as it functions as a major tumor suppressor gene directly associated with miR-34a in a positive feedback loop. We also tested levels of direct targets of miR-34a, such as SIRT1, MYC, and BCL2. Initially, we performed cellular uptake analysis of the formulations in the A549 cells. We loaded miR-34a mimic covalently conjugated to FITC into the NPs for cellular uptake and endocytosis studies. We treated A549 cells for 24 h with the miR-34a-FITC NPs at a 2 mg/mL NP dose as PLGA-poly-L-His NPs. We analyzed cellular uptake using confocal microscopy and then quantified the uptake in a separate study with the same treatment conditions using flow cytometry. We observed high cellular uptake as the NPs localized near the nucleus and distributed in the cytoplasm (Figure 3A). We also compared the cellular distribution of the miR-34a mimic when transfected with Lipofectamine and when encapsulated in NPs at an equivalent NP dose of 300 pmol of mimic. Delivery by NPs exhibited higher cellular distribution for miR-34a mimic (Figure S3).
Figure 3.

Functional analysis of miR-34a NPs in A549 cell culture studies
(A) Cellular uptake of miR-34a-FITC NPs in A549 cells using confocal microscopy. Cells were treated at a 2 mg/mL NP dose for 24 h. White arrows point to green puncta, representing miR-34a-FITC NPs undergoing cellular uptake. Blue represents nuclei. The image was taken at 100× original magnification. The scale bar represents 50 μm. (B) Stacked histogram of flow cytometry analysis to quantify cellular uptake of miR-34a-FITC NPs after 24 h in A549 cells. Cells were also treated with miR-34a-FITC transfected with Lipofectamine (Lipo) as a positive control and blank NPs as a negative control. PBS was used as the untreated control. The quantification of internalization is represented in the bar graph on the right. The data are represented as mean ± standard deviation. (C) Normalized miR-34a fold change of RNA isolated from A549 cells after being treated with Scr-34a NPs and miR-34a NPs for 24 h at a 2 mg/mL dose. Data represent the mean (n = 3) ± standard deviation. A 1% agarose gel of amplified PCR product is shown above the dataset. (D) p53 gene expression in A549 cells after being treated with Scr-34a NPs and miR-34a NPs for 24 h at a 2 mg/mL dose. Data represent the mean (n = 3) ± standard deviation. A 1% agarose gel of amplified PCR product is shown above the dataset. (E) Western blot analysis of p53 protein from A549 cells treated with Scr-34a NPs and miR-34a NPs for 24 h at a 2 mg/mL dose. p53 protein levels are normalized to vinculin levels. Data represent the mean (n = 3) ± standard deviation. Protein blots indicate protein intensity based on pixels per band, quantified using ImageJ. (F) p53 protein levels of A549 cells treated with Scr-34a NPs and miR-34a NPs for 24 h (2 mg/mL dose) and incubated under hypoxic conditions for 24 h. Data represent the mean (n = 3) ± standard deviation. Protein blots indicate protein intensity based on pixels per band, quantified using ImageJ. The p53 protein levels are normalized to vinculin levels. Unpaired t test was used to determine statistical significance; ∗p < 0.05; ∗∗∗p < 0.001.
We further quantified the uptake by flow cytometry, which also confirms a higher FITC signal when treated with miR-34a-FITC NPs compared with Lipofectamine-delivered miR-34a-FITC at an equivalent dose of 300 pmol as the positive control for this study (Figure 3B). From this study, we observed that the PLGA-poly-L-His NP formulation is effective at undergoing significant cellular uptake at a 2 mg/mL dose. Approximately 30% of the cell population internalized the miR-34a-FITC when delivered using PLGA-poly-L-His NPs (Figure 3B).
Using confocal microscopy and flow cytometry, we also investigated the endocytosis route using endocytosis inhibitors (Figure S4). Chlorpromazine inhibits clathrin-mediated endocytosis, genistein inhibits caveolae-mediated endocytosis, and amiloride inhibits micropinocytosis.25 We noted less uptake in cells pre-treated with chlorpromazine to conclude that miR-34a NPs most likely undergo clathrin-mediated endocytosis (Figure S4A). We also quantified the uptake using flow cytometry, as we noted less FITC signal in cells treated with chlorpromazine compared with the other inhibitors. Based on the flow cytometry results, we established that clathrin-mediated endocytosis is a prominent pathway for cellular uptake of PLGA-poly-L-His NPs containing miRNA mimics (Figure S4B).
We visualized cellular uptake over time using confocal microscopy to understand the kinetics of miR-34a NPs in cells (Figure S5). Here, we noted a substantial increase in uptake at 8–24 h of incubation followed by complete uptake by 24 h (Figure S5). We determined a time-dependent effect in miR-34a-FITC NP uptake and hence kept the treatment durations of 24 h.
Functional analysis in A549 cells treated with miR-34a NPs
Next, we performed a series of gene expression studies to confirm the upregulation of miR-34a and p53 after transfection with miR-34a NP formulations. A549 cells were treated with Scr-34a NPs and miR-34a NPs. After 24 h, there was a statistically significant 5-fold increase in miR-34a levels when treated with miR-34a NPs compared with Scr-34a NPs (Figures 3C and S6). We also observed the significant upregulation of p53 by 50% when treated with miR-34a NPs (Figure 3D). In addition, we tested the miR-34a and p53 levels when the miR-34a mimic was transfected with Lipofectamine. We transfected the cells with miR-34a mimic and noticed a significant increase in the miR-34a level and a moderate increase in the p53levels (Figures S7A and S7B). Although we used Lipofectamine for the comparative analysis, it is challenging to perform the in vivo studies with Lipofectamine due to the side effects associated with its lipid content.26
In addition, we tested direct targets of miR-34a, such as SIRT1, MYC, and BCL2, during in vitro studies. SIRT1 is a p53 deacetylating gene, and we hypothesized it would be downregulated when miR-34a was upregulated.27 MYC is an oncogene that regulates cell proliferation and is often highly upregulated in cancer.28 BCL2 is another antiapoptotic gene highly expressed with the inactivation of miR-34a in cancers.29 In the gene expression analysis, we noticed an ∼50% decrease in SIRT1, MYC, and BCL2 when the A549 cells were treated with miR-34a NPs, confirming the tumor-suppressive effects of miR-34a NPs (Figure S8).
After assessing p53 levels through gene expression, we corroborated these results on the protein level using western blot. Vinculin was used as the endogenous control, and p53 protein levels were normalized to Scr-34a NPs. After 24 h, we confirmed an ∼1.5-fold increase in p53 protein levels in miR-34a NP-treated cells (Figures 3E and S9). We also analyzed the p53 protein levels when transfected with Lipofectamine, where there was a slight increase (nonsignificant) when transfected with 300 pmol (Figure S7C). Hypoxic conditions have been found to lower p53 levels in A549 cells, contributing to tumor proliferation.30 We investigated p53 protein levels under hypoxic conditions (1% O2 in nitrogen) and compared them with normoxia conditions (20% O2, 5% CO2). Baseline p53 protein levels in hypoxia were reduced by 60% compared with normal incubation conditions (Figure S10). To evaluate if p53 levels could be further upregulated after treatment with miR-34a NPs under hypoxic conditions, we treated cells with miR-34a NPs under hypoxia for 24 h. We observed a 25% increase in p53 protein levels compared with Scr-34a NPs (Figure 3F). This equates to an ∼80% increase in p53 protein from the baseline levels in A549 cells. As the tumor microenvironment is hypoxic, we confirmed that the miR-34a NPs can increase p53 levels, suggesting this as a possible mechanism for the tumor-suppressor effects of miR-34a.
Apoptosis of A549 cells when treated with miR-34a NPs
As miR-34a’s primary function is to regulate genes that control apoptosis and cell growth, we initially wanted to test the extent of apoptosis in A549 cells treated with miR-34a NP. Here, we utilized the Annexin-V apoptosis assay through flow cytometry and fluorescence microscopy (Figure 4A). We treated the cells with Annexin-V, which binds to phosphatidylserine exposed on the cell membrane of apoptotic cells. Further, we stained with 7-aminoactinomycin D (7-AAD), which stains necrotic cells. We observed an ∼40% increase in apoptosis in miR-34a NP- versus Scr-34a NP-treated cells (∼15% of the total cell population was in the apoptotic state compared with 10% for Scr-34a NP-treated control). We also utilized Annexin-V covalently conjugated to FITC to visualize the apoptotic cell population using fluorescence microscopy with the same NP treatment conditions. We noticed more cells in the apoptotic state, indicated by green puncta, when treated with miR-34a NPs compared with the Scr-34a NP treatment group (Figure 4B). Therefore, we established that the miR-34a NPs induce apoptosis in A549 cells. We also evaluated cell-cycle changes in A549 cells by propidium iodide staining following treatment with miR-34a NPs. We did not see any significant changes in cell-cycle phases (G0–G1, S, and G2–M) for miR-34a NPs or BL NPs, as seen in earlier reported studies (Figure S11).31
Figure 4.

Cell viability and proliferation analysis of miR-34a NP-treated A549 cells
(A) Apoptosis of miR-34a NP-treated A549 cells using an Annexin-V/7-aminoactinomycin D (7-AAD)-based assay. Cells were treated with miR-34a NPs and Scr-34a NPs for 24 h at a 2 mg/mL dose. The cells undergoing apoptosis and necrosis were stained with Annexin labeled with phycoerythrin (PE) and 7-AAD, which stains necrotic and dead cells. The quadrants of the dot plots represent necrotic (Q1), late-stage apoptotic (Q2), early apoptotic (Q3), and live cells (Q4). The data were quantified using FlowJo to calculate the total percentage of apoptotic cells. Data represent the mean (n = 3) ± standard deviation. (B) Apoptosis of A549 cells using confocal microscopy. A549 cells were treated with miR-34a and Scr-34a NPs at a 2 mg/mL NP dose for 24 h. Apoptotic cells were then stained with Annexin-V and conjugated to FITC to visualize the apoptotic cell populations. The cells were imaged at 10× original magnification. Images shown are cells treated with Scr-34a NPs and miR-34a NPs. The green puncta represent apoptotic cells. The scale bar represents 50 μm. (C) A549 colony-forming efficiency of miR-34a NP-treated cells. Cells were treated with miR-34a NPs or Scr-34a NPs for 24 h at a 2 mg/mL dose. The crystal violet stain represents the number of colonies. The bar graph shows data where the number of colonies is normalized with respect to Scr-34a NP-treated cells. Data represent the mean (n = 3) ± standard deviation. (D) Cell viability using trypan blue assay. Data represent normalized cell viability with respect to blank NP. Data represent the mean (n = 3) ± standard deviation. Unpaired t test was used to determine statistical significance; ∗p < 0.05; ∗∗p < 0.01.
Colony-forming efficiency when treated with miR-34a NPs
In our studies, we expect reduced cell viability of A549 cells through the effective upregulation of miR-34a with our NP formulation. We tested the colony-forming efficiency of cancer cell lines to indicate whether the miR-34a NPs caused reduced proliferation, signified by fewer cell colonies. We treated A549 cells with miR-34a NPs and Scr-34a NPs. After 13 days, we noted that the colony survival was reduced by 60% when the cells were treated with miR-34a NPs compared with Scr-34a NP treatment, indicated by a lower number of colonies in the miR-34a NP treatment group (Figure 4C). Based on the results, we showed that miR-34a NPs effectively reduce the potential for A549 cell proliferation.
Cell viability using trypan blue assay
We also performed a trypan blue assay to further confirm cell viability reduction. The trypan blue assay is a cell staining assay that aids in differentiating between the living and the dead cell populations. We treated the cells for 24 h with Scr-34a NPs and miR-34a NPs and noticed a decrease in cell viability of approximately 50% after treatment with miR-34a NPs compared with Scr-34a NPs (Figure 4D). We demonstrated that miR-34a NPs reduce cell viability, indicated by an increase in the dead cell population after miR-34a NP treatment.
In vivo intratumoral efficacy of miR-34a NPs
To confirm the effect of miR-34a NPs in vivo, we tested the miR-34a NPs in A549-derived xenograft mice. A549 cells were cultured and implanted via subcutaneous injections along the flank region in female immunocompromised NOD-SCID mice. Initially, we evaluated the intratumoral biodistribution of the miR-34a NPs at 8 and 24 h to confirm their delivery. We treated mice with miR-34a NPs labeled with 6-carboxyfluorescin (FAM) at a 3 mg dose per mouse intratumorally. We noted the distribution of the miR-34a-FAM NPs in the tumor at 8 h with continued accumulation until 24 h by IVIS imaging (Figures 5A and S12). In the tumor images taken by confocal microscopy, we observed that miR-34a-FAM NPs localized in the tumor at both 8 and 24 h, which gives a good indication of tumor retention of the NP formulation.
Figure 5.

Intratumoral in vivo efficacy studies
(A) Cryosection images after intratumoral injection of miR-34a-FAM NPs at 8 and 24 h. Tumors were injected with 3 mg of NPs. Images were taken at 40× original magnification. Scale bar represents 100 μm. Blue fluorescence represents cell nuclei stained with DAPI, and green puncta represent miR-34a-FAM NPs. (B) Survival curve for tumors treated with PBS, miR-34a NPs, and Scr-34a NPs. The survival endpoint was a tumor volume of 2,000 mm3 and plotted against the number of days (n ≥ 4 mice in each treatment group). Log-rank Mantle-Cox test was performed for statistical significance of percentage survival. (C) Histological analysis of A549 xenograft tumors. Histology section images of staining with Ki-67. Tumors were stained with Ki-67 to mark cell proliferation. The brown color indicates proliferative cells. Scale bar represents 100 μm. Quantification of total positive Ki-67 signal was done using ImageJ software. The data shown are the averages of n = 6 section areas ± standard deviation and normalized to Scr-34a NPs. (D) miR-34a gene expression in tumor samples. Data are plotted normalized to Scr-34a NP-treated tumors. Data are shown as the mean of n ≥ 5 ± standard deviation. (E) p53 and SIRT1 protein analysis by western blot. Vinculin was used as the endogenous control for measuring p53 levels (left blot images). GAPDH was used as the endogenous control for measuring SIRT1 levels (right blot images). Band intensity was normalized to Scr-34a NPs and was quantified with ImageJ software. (F) Quantification of protein blot intensity. (Left) p53 protein levels were normalized to Scr-34a NPs. (Right) SIRT1 levels were normalized to Scr-34a NPs. Unpaired t test was used to determine statistical significance; ∗p < 0.05; ∗∗p < 0.01.
We tested the survival of A549-derived xenograft mice when treated with miR-34a NPs. We injected the mice intratumorally with PBS, miR-34a NPs, or Scr-34a NPs at a dose of 3 mg NPs (Figure S13). The mice were euthanized once the tumor volume reached 2,000 mm3, and percentage survival was plotted. We noted that the average survival of mice treated with PBS was 17 days. Compared with the Scr-34a NP treatment group, we noticed a significant survival of mice treated with miR-34a NPs (20 days) (Figure 5B). Next, we assessed the extent of cell proliferation in the tumors by Ki-67 staining of tumor sections (Figure 5C). As expected, we observed a significant reduction in Ki-67-positive-marked cells when treated with miR-34a NPs compared with PBS and Scr-34a NPs. We quantified the reduction in proliferative cells by ImageJ software to compare the relative number of Ki-67-positive-marked cells when treated with Scr-34a NPs and miR-34a NPs. We confirmed an ∼60% reduction in proliferative cells when treated with miR-34a NPs intratumorally compared with Scr-34a NPs. Next, to verify the functionality of miR-34a NPs in vivo, we also assessed miR-34a levels in the tumor samples using gene expression analysis. In addition, we used western blot studies to measure the amount of p53 and SIRT1 protein levels in the tumor samples. SIRT1 is a direct miR-34a target involved in p53 deacetylation, contributing to tumor progression. Here, we expected the levels of miR-34a and p53 to increase and SIRT1 to be downregulated. We observed that the miR-34a levels increased by 2-fold (Figure 5D). On the protein level, we determined that p53 was increased by 1.5-fold (Figures 5E and 5F). We also established a 0.60-fold decrease in SIRT1 (Figures 5E and 5F). Also, we did not observe any adverse effects on the liver, kidney, and spleen when treated with miR-34a NPs, as shown by the H&E histological staining of organ sections (Figure S14). Hence, we established prolonged survival and increased miR-34a levels attributed to the efficacy of miR-34a NPs in vivo.
Discussion
Non-coding RNAs such as miRNAs, lncRNAs, and circRNAs have been identified for their role in gene expression regulation through mRNA stability, chromatin regulation, and RNA sponging, respectively.32 miRNAs have been established as potential drug targets for various diseases and are being further explored for therapeutic purposes. Here, we have focused our studies on delivering miRNA mimics, as it has been well established that the downregulation of specific miRNAs is a cause of many diseases, including cancer.
To target aberrantly upregulated miRNAs, oligonucleotides can be designed to be complementary to an miRNA sequence to block the miRNA from binding to its target RNA sequence. AntimiRs bind to the target miRNA through Watson-Crick base pairing and inhibit the miRNA activity through steric hindrance.33 Several miRNA inhibitor-based drug candidates are being tested in pre-clinical and clinical studies. Cobomarsen is a locked nucleic acid inhibitor of miR-155 (antimiR-155) developed by miRagen Therapeutics (now acquired by Viridian Therapeutics). It is currently being tested in phase 2 clinical trials to treat cutaneous T cell lymphoma.34 In addition, this locked nucleic acid inhibitor has also shown efficacy in blocking miR-155 in diffuse large B cell lymphoma cell lines.10 Moreover, tiny locked nucleic acids and peptide nucleic acids are also being explored to target miRNAs’ seed region (8 bp functionally active stretch).7,35
On the other hand, miRNA mimics effectively replenish miRNAs that are downregulated in diseases such as cancer by mimicking endogenous miRNA activity. However, the delivery of miRNA mimics has remained challenging due to various factors, including the negatively charged phosphodiester backbone. Another challenge to consider is the double-stranded structure that can trigger the release of pro-inflammatory cytokines such as IFN-α, IFN-γ, and IL-1.36
A cationic delivery system can improve the loading of negatively charged cargo. However, there are challenges, as highly cationic delivery systems are known to cause systemic toxicity in vivo.37 Delivery systems with modest positive charges would reduce potential toxicity. Liposomal delivery, specifically amphoteric liposomes, had been a promising route for the delivery of miRNA mimics, as they were effectively delivered systemically for liver cancer treatment in pre-clinical mouse studies.19,38 However, in phase 1 clinical trials, side effects were reported when liposomal delivery was used in humans. For the MRX34 clinical trial (Synlogic), these side effects included liver failure, hypoxia, and cytokine release, which can all be attributed to the use of charged lipids and off-target delivery. However, amphoteric lipids’ composition helps alleviate issues with loading and encapsulating miRNA mimic payloads. Furthermore, the scale-up of liposomal formulations containing nucleic acids is challenging, as observed in lipid-based vaccines. A polymeric nanodelivery system is a safer alternative because the polymer breaks down into biocompatible metabolites when administered. PLGA is an FDA-approved, biodegradable polymer that has been shown to be effective in the delivery of nucleic acids.39 For the delivery of negatively charged miRNA mimics, using a cationic delivery system is optimal to improve loading through ionic interaction.
In our current studies, we have shown that the use of PLGA-poly-L-His NPs is beneficial for the delivery of miR-34a, standing as a proof of concept for the delivery of other miRNA mimics for the treatment of other diseases. Previously, a PLGA-poly-L-His NP system has been shown to exhibit optimal cellular uptake properties when loaded with peptide nucleic acids and paclitaxel.24 Through endocytosis inhibitor studies, we established that miRNA mimic-loaded NPs undergo clathrin-mediated endocytosis. The details of this uptake mechanism will need to be explored further to better understand the different receptors and proteins involved in the cellular uptake of miRNA mimics. This has been previously established for siRNA; thus, investigating the mechanism for miRNA mimics can further enhance their functionality.40 We confirmed efficacy in vitro through thorough gene expression and western blot analysis to corroborate the functional activity of the formulation. Our results indicated upregulation of both miR-34a and its downstream tumor-suppressor transcription factor, p53, on both the gene and the protein levels in vitro. By testing miR-34a direct target genes such as SIRT1, MYC, and BCL2, we support that miR-34a NPs effectively target the miR-34a downstream gene network. This is accompanied by a significant reduction in the A549 cell survival through increased apoptosis, as shown by Annexin-V-based apoptosis assays.
Understanding the structural characterization of miRNA mimics within the PLGA-poly-L-His NPs, using SAXS, allows us to fine-tune our formulation to increase the payload. SAXS was used in identifying the structures of different delivery systems for improved delivery of nucleic acid analogs. Previously, SAXS has determined the structure of lipid bicelles loaded with peptide nucleic acids (PNAs).41 In addition, nano- and self-assemblies of gamma-PNAs have also been structurally defined by SAXS.42 In our studies, the SAXS patterns show increased shell thickness of NPs with the association of miR-34a mimics compared with the BL NPs, suggesting that miR-34a mimics presumably coat the NP surface, consistent with the observed release profile.
Studies have shown that other oligonucleotides, such as PNAs, undergo exocytosis.43 Although we have investigated the endocytosis of miRNA mimics, the mechanism of exocytosis has not been explored. This mechanistic understanding would shed light on the amount of miRNA mimic present and the amount excreted out of the cells over time to determine the amount of miRNA mimics available in the cytoplasm.
In addition to investigating cellular trafficking, the stability of the mimic in systemic delivery must be explored, given the structural arrangement of the formulation. PLGA-poly-L-His NPs have moderate bioavailability, and increasing the circulation time using polyethylene glycol (PEG) in the formulation would improve the retention after systemic treatment. In addition, miRNA mimics are prone to enzymatic degradation in systemic circulation. Improved stability can also result from modifying the backbone of the miRNA mimic. Phosphorothioate modifications have shown increased longevity of antisense oligonucleotides in systemic circulation, given their ability to bind to serum proteins compared with oligonucleotides containing the phosphodiester backbone.44
In in vivo studies, we have shown that PLGA-poly-L-His NPs effectively deliver miR-34a mimics to prolong the survival of A549-derived xenograft mice via intratumoral injection. Further, we increased miR-34a levels in A549 tumors while upregulating the p53 protein and downregulating the SIRT1 protein, which plays roles in tumor suppression and regulation of apoptosis, respectively. In the future, systemic delivery must be explored to open the doors for translating this technology to the clinic. With systemic delivery, it would be possible to target other malignant cancers. It would be interesting further to understand the kinetics and distribution of miRNA mimics when delivered systemically to help expand this technology to target other organs.
Our current results show the potential of PLGA-poly-L-His NPs for the delivery of miR-34a mimics. miRNA mimic-based therapeutic research is currently on the rise and should be explored further, as many miRNA targets are emerging as prime factors in disease onset. By pinpointing miRNA dysregulation in diseases, we can halt the aberrant mRNA network that becomes activated, preventing disease progression.
Materials and methods
Hsa-miR-34a-5p (no. MC11030), hsa-Scr-miR-34a mimic (no. MC10340) and Lipofectamine (RNAiMax) were commercially purchased from Thermo Fisher Scientific. miR-34a-FITC mimic was custom designed and purchased from Midland (Midland, TX). miR-34a-FAM was custom designed and purchased from Integrated DNA Technologies (Coralville, IA). PLGA(50:50)-ester-terminated polymer was purchased from Lactel Absorbable Polymers at a 0.39 g/dL viscosity grade. Poly-L-His and organic solvents such as acetone, DCM, and sucrose were purchased from Sigma-Aldrich (St. Louis, MO).
Synthesis of PLGA-poly-L-histidine NPs
To formulate miR-34a-loaded NPs, a double emulsion solvent evaporation technique was used. Acetone and DCM were used as the organic solvent, containing 60 mg of PLGA and poly-L-His at a 4.9/0.1 w/w ratio in 750 μL of acetone:DCM (2:1) solution. miR-34a mimic, dissolved in water (1 mM, 1 nmol/mg), was added dropwise to the organic phase while vortexing to form a w/o emulsion. This single emulsion was then sonicated using a probe sonicator (Sonics VibraCell, Newtown, CT) for 10 s in three pulses. The single emulsion was added to 1.5 mL of 5% w/v polyvinyl alcohol (Sigma-Aldrich, St. Louis, MO) solution to form a w/o/w double emulsion. The double emulsion was sonicated using the probe sonicator for 10 s in three pulses. The double emulsion was added dropwise to 15 mL of 0.3% w/v polyvinyl alcohol solution while stirring. The suspension was stirred overnight at room temperature. The nanosuspension was centrifuged using an ultracentrifuge (Beckman-Coulter Optima XPN-100) at 20,000 rpm for 20 min cycles and washed with water three times. After the third cycle, the resulting pellet was resuspended in a 5 mg/mL sucrose solution at a 1/1 PLGA/sucrose w/w ratio. The NPs were then lyophilized overnight. Using the same method, Scr-34a mimics and miR-34a-FITC mimics were loaded into PLGA-poly-L-His NPs.
Biophysical characterization of miR-34a NPs
Dynamic light scattering was performed using the Zetasizer Nano ZS (Malvern Panalytical, Westborough, MA) to measure the hydrodynamic diameter and the PDI. Zeta potential was evaluated using the laser Doppler microelectrophoresis technique at 25°C. Each sample consisted of three measurements, of which the average was taken.
Loading analysis
The loading of miRNA mimics in PLGA-poly-L-His NPs was quantified by solvent extraction. Briefly, 200 μL of DCM was added to lyophilized miR-34a NPs, followed by shaking at 37°C at 1,000 rpm for 24 h. After 24 h, 100 μL of sodium acetate buffer (pH 5.8) was added, and NPs were allowed to shake at 37°C at 1,000 rpm for 1 h. The NP tube was centrifuged at 10,000 rpm for 5 min, and the aqueous supernatant containing the miRNA mimic was collected in another Eppendorf tube. The absorbance of the aqueous supernatant was then measured at 260 nm using Nanodrop One (Thermo Fisher, Waltham, MA). The loading was then calculated in pmol/mg of NPs based on the initial amount of 1 nmol/mg NPs. The same procedure was followed for quantifying the loading of Scr-34a NPs. To measure the extraction efficiency using this method, we used BL NPs and resuspended in DCM containing 150 pmol of miR-34a mimic. After 24 h, sodium acetate was added to extract the miRNA mimic stock into the aqueous phase. Absorbance at 260 nm was measured, and the efficiency based on the starting quantity was calculated.
Release kinetics of miR-34a NPs
PBS (300 μL, pH 7.4) was added to miR-34a NPs and vortexed until NPs were resuspended and shaken at 300 rpm at 37°C for 15 min. After 15 min, the NPs were centrifuged for 10 min at 15,000 rpm, and the absorbance of the supernatant was taken at 260 nm using Nanodrop One (Thermo Fisher Scientific, Waltham, MA). The NPs were then resuspended in 300 μL of fresh PBS and shaken until the next time point. This was repeated for each time point thereafter (1, 2, 4, 6, 8, 12, 24, 48 h). The percentage cumulative release was plotted against time.
Scanning and transmission electron microscopy of miR-34a NPs
A small amount (<0.5mg) of lyophilized NPs without cryoprotectant was placed on a double-sided carbon tape and sputter coated. The images were taken at 30,000× using the FEI Nova NanoSEM 450 and quantified using ImageJ. For TEM, lyophilized NPs with sucrose cryoprotectant were dispersed in water. They were then added to TEM carbon grids and stained with 1% uranyl acetate for 5 min, and an FEI Tecnai TEM was used at 80 kV for imaging.
In vitro miRNA mimic integrity assay
miR-34a NPs were resuspended in PBS and shaken at 300 rpm at 37°C for 48 h. The NPs were then centrifuged at 4,000 rpm for 5 min. The absorbance of the supernatant was taken at 260 nm using Nanodrop One, and the concentration was calculated. The sample was then diluted to 1 μM and loaded in a 5% PAGE gel, followed by SYBR Gold staining (Invitrogen, Waltham, MA). The gel was imaged using the Bio-Rad Gel-Doc imager.
Structural characterization of miR-34a NPs
SAXS experiments were performed using the 16ID-LiX Beamline at the National Synchrotron Light Source II at Brookhaven National Laboratory (Upton, NY). The concentration of the samples was 4 mg/mL. The solution was loaded in a sample cell sandwiched by two mica windows with a gap of ∼2 mm and with an X-ray energy of 13.5 keV. The intensity was quantified with respect to the scattering vector, q defined as , where θ is the scattering angle, and λ is the wavelength. The data covered a q range from 0.005 to 2.5 Å−1. Jupyter Notebook was used for the radial averaging and q conversion of data.45 The background subtraction and transmission correction were performed to reduce the intensity of the hydrogen bond from water at ∼2.0 Å−1. The absolute intensity was derived by comparing it against the water incoherent scattering intensity Iwater as reported in the literature.
Cell culture experiments
For cell culture studies, A549 cells (CCL-185; ATCC, Manassas, VA) were grown in Eagle’s minimum essential medium (EMEM) (30-2003; ATCC, Manassas, VA) supplemented with fetal bovine serum albumin (FBS) (30-2020; ATCC, Manassas, VA) and PenStrep (Gibco, Grand Island, NY) at 37°C and 5% CO2. Cells were grown in a 10 cm Petri dish and were passaged at 80%–90% confluency. Cells from passages 1–11 were used for experiments.
Cellular uptake using confocal microscopy
Using a 12-well plate, 150,000 A549 cells were seeded for two treatment groups in triplicate. Cells were treated with BL NPs and miR-34a-FITC NPs. A 2 mg/mL NP dose was used and treated for 24 h. The cells were then washed with PBS, fixed with 4% paraformaldehyde (PFA) and permeabilized using 0.1% Triton-X. The cells were then mounted onto microscope slides using Prolong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, Waltham, MA) and imaged using the Nikon A1R microscope at 100× magnification. For time-dependent cellular uptake analysis, 100,000 cells were seeded in a 24-well plate and treated with miR-34a-FITC NPs for 2, 4, 8, and 24 h. After the respective time points, the cells were washed with PBS three times, fixed using 4% PFA and permeabilized using 0.1% Triton-X. The cells were then mounted onto microscope slides and imaged using the confocal microscope (Nikon A1R).
Cellular uptake using flow cytometry
Using a 12-well plate, 150,000 A549 cells were seeded for four treatment groups in triplicates. Cells were treated with PBS, BL NPs, and miR-34a-FITC NPs. A 2 mg/mL NP dose and an equivalent 300 pmol of miR-34a-FITC were transfected with Lipofectamine using forward transfection. After 24 h, the cells were washed with PBS, trypsinized, and then transferred to Eppendorf tubes. The cells were centrifuged at 2,000 rpm for 4 min and washed with PBS. The final pellet was resuspended in 300 μL of PBS and passed through filtered fluorescence-activated cell sorting (FACS) tubes. The intracellular FITC signal was quantified using the LSR Fortessa X-20 cell analyzer and FlowJo. To investigate the route of endocytosis, endocytosis inhibitors such as chlorpromazine (10 μg/mL; Sigma-Aldrich, St. Louis, MO), genistein (200 μM; Sigma-Aldrich, St. Louis, MO), and amiloride (1 mM; Sigma-Aldrich, St. Louis, MO) were used to pre-treat A549 cells. The cells were pre-treated with the inhibitors for 30 min and then incubated with miR-34a-FITC NPs (2 mg/mL) for 4 h. Flow cytometry was then performed to quantify cellular uptake. The same experimental setup was used for imaging the cells using fluorescence microscopy. However, the cells were treated with miR-34a-FITC NPs for 8 h.
RT-PCR gene expression analysis
A549 cells (200,000) were seeded in a 12-well plate and treated with Scr-34a NPs and miR-34a NPs at a 2 mg/mL dose. The next day the cells were trypsinized and centrifuged, and the total RNA was extracted using a Qiagen RNeasy kit. For miR-34a expression, the cDNA synthesis kit (Invitrogen) along with RT primer for miR-34a (4331182; Thermo Fisher, Waltham, MA) and U6 (001973; Thermo Fisher, Waltham, MA) were used to generate the cDNA. The cDNA synthesis kit was also used for cDNA synthesis when measuring p53 levels. The samples were incubated under specified temperature conditions for the cDNA using a Bio-Rad C1000 Touch thermal cycler. PCR amplification was done using the TM primers, RNase-free water, and Universal Mastermix II with UNG (Invitrogen, Waltham, MA) in the Bio-Rad CFX-Connect Real-Time PCR instrument. Samples were incubated under the specified temperature conditions. Primers for p53 (Hs01034249; Thermo Fisher, Waltham, MA) and GAPDH (Hs0275889; Thermo Fisher, Waltham, MA) were used, where GAPDH served as the endogenous control. In addition, SIRT1 (Hs01009006; Thermo Fisher, Waltham, MA), MYC (Hs01562521; Thermo Fisher, Waltham, MA), and BCL2 (Hs00153350; Thermo Fisher, Waltham, MA) primers were also used for gene expression analysis. The amplified PCR product samples were run on a 1% agarose gel at 120 V for 20 min and stained with ethidium bromide (Sigma-Aldrich, St. Louis, MO). For Lipofectamine-treated groups we used a dose of 300–600 pmol.
Western blot for in vitro study
A549 cells (200,000) were seeded in a 12-well plate and treated with Scr-34a NPs and miR-34a NPs at a 2 mg/mL NP dose. Cells were then trypsinized and pelleted down at 2,000 rpm for 4 min. The proteins were extracted from the cell pellet with 1× RIPA buffer (Cell Signaling Technology, Danvers, MA) with 1× Protease inhibitor (Thermo Fisher, Waltham, MA). The pellets were kept in ice and vortexed every 10 min, three times, to ensure efficient lysis. BSA standards were used to measure the concentration of each protein sample by developing a standard curve based on absorbance. Twenty micrograms of protein was added into each well of a 4%–15% Mini-Protean TGX Stain Free 50 μL gel (Bio-Rad, Hercules, CA). The protein samples were run at 200 V for 40 min and transferred to a polyvinylidene fluoride (PVDF) membrane at 110 V for 90 min. The proteins on the membrane were blocked using 5% milk in 1× Tris-buffered saline-Tween (TBST) buffer and shaken for 1 h. The blots were then washed with 1× TBST buffer and cut according to the molecular weights of vinculin (124 kDa) and p53 (53 kDa). For Lipofectamine-treated groups we used a dose of 300–600 pmol.
Vinculin was used as the endogenous control. The p53 (1:500) (9282; Cell Signaling) and Vinculin (1:1,000) (13901S; Cell Signaling Technology, Danvers, MA) antibodies were added to the specific blots and shaken overnight at 4°C. The anti-rabbit IgG horseradish peroxidase (HRP)-linked secondary antibody (7074; Cell Signaling Technology, Danvers, MA) at 1:2,000 dilution was then added to probe for the primary antibody. The blots were then submerged in chemiluminescence HRP substrate (P90719; Millipore Sigma, St. Louis, MO) and imaged using a Bio-Rad ChemiDoc Imaging instrument. The band intensity was quantified using ImageJ software.
Hypoxia cell culture studies
A549 cells were grown in EMEM at 37°C. For baseline p53 level studies, one 12-well plate was seeded with 200,000 cells (n = 3) and placed under normoxia conditions. Another 12-well plate was seeded with 200,000 cells (n = 3) and placed under hypoxia conditions (1% O2 in nitrogen). After 24 h, the cells were centrifuged down, and protein extraction was performed on cell pellets for western blot analysis to measure p53 protein levels. For NP treatment, cells were treated with either Scr-34a or miR-34a NPs and placed under hypoxia conditions (1% O2 in nitrogen) for 24 h. After 24 h of treatment, proteins from cell pellets were extracted for western blot analysis to measure p53 protein levels.
Cell viability: Trypan blue assay
A549 cells (10,000) were seeded in a 96-well plate and were treated with Scr-34a NPs and miR-34a NPs at a 2 mg/mL dose for 24 h. The cells were then stained with trypan blue dye (Bio-Rad, Hercules, CA) and counted using an automated Bio-Rad cell counter.
Cell-cycle arrest assay
Using a 12-well plate, 150,000 A549 cells were seeded for three treatment groups in triplicates. Cells were treated with BL NPs, miR-34a-FITC NPs, or PBS at a 2 mg/mL NP dose for 24 h. After 24 h, the cells were washed with PBS, followed by trypsinization, and then transferred to Eppendorf tubes. The cells were centrifuged at 2,000 rpm for 4 min and washed with PBS. The final pellet was resuspended in 1,000 μL of absolute ethanol and kept at −20°C for 20 min for fixing. The cells were centrifuged at 2,000 rpm for 4 min to remove ethanol and incubated with PBS for 15 min for rehydration. Finally, the cells were pelleted following centrifugation at 2,000 rpm for 4 min, stained with 500 μL of FxCycle PI/RNase staining solution for 45 min, and passed through filtered FACS tubes. The propidium iodide cellular staining was quantified using the LSR Fortessa X-20 cell analyzer at the phycoerythrin (PE)-Texas red channel and FlowJo.
Cell viability: Clonogenic assay
A549 cells (50,000 cells/well) were seeded in a 24-well plate and were treated with either miR-34a NPs or Scr-34a NPs at a 2 mg/mL NP equivalent dose for 24 h. The cells in each well were then counted, and 100 cells/well were reseeded in a 6-well plate. The cells were then incubated for 13 days to allow the treated cells to form colonies. When there were 30–50 cells per colony, the cells were washed with PBS and fixed with 4% PFA for 10 min. The cells were then washed to remove the PFA and stained with 1 mL of 1% w/v crystal violet solution (Sigma-Aldrich, St. Louis, MO) for 24 h. After 24 h, the stained cells were washed with water until all residual stain was removed, and the plates were allowed to dry. After drying, each individual colony was counted.
Annexin-V FACS assay
A549 cells (200,000) were seeded in a 12-well plate and treated with miR-34a NPs and Scr-34a NPs at a 2 mg/mL NP dose for 24 h. The cells were trypsinized, centrifuged at 2,000 rpm for 4 min, and then resuspended in Annexin-V binding buffer. One hundred thousand cells were taken to be stained with Annexin-V dyes and passed through the filtered FACS tube. Staining was performed using 7.5 μL of Annexin-V-PE (BD Biosciences, USA) and 7.5 μL of 7-AAD (BD Biosciences, USA). The tubes were kept in the dark for 15 min. The stained cells were then diluted in Annexin-V binding buffer and quantified using the LSR-Fortessa X-20 instrument.
Annexin-V FITC apoptosis assay
A549 cells (10,000) were seeded in a 96-well plate and treated with either miR-34a NPs or Scr-34a NPs at a 2 mg/mL NP dose for 24 h. Annexin-V FITC was added to 1× Annexin V binding buffer, and 100 μL of the diluted stock was added to each well. The plate was kept in the dark for 15 min and imaged using the Keyence fluorescence microscope at 10× magnification.
In vivo efficacy studies
The in vivo studies were performed at University of Connecticut, Storrs campus under compliance and approval by the Institutional Animal Care and Use Committee (IACUC) under protocol A21-041. Fourteen female NOD-SCID mice (NOD.Cg-PrkdcscidIl2rgtm1wjl/Szj, strain 00557) at 5 weeks of age were purchased from The Jackson Laboratory and maintained in a pathogen-free environment with standard food and water per IACUC guidelines. A549 cells were expanded for tumor implantation. Each mouse was injected with 1 × 107 cells on both flanks subcutaneously. Tumors were monitored every 2 days. Once the tumor reached 150 mm3, the mice were randomly split into three groups (n ≥ 4): PBS, miR-34a NP, and Scr-34a NP. PBS-treated mice were injected with 80 μL intratumorally, and NP treatment groups were dosed intratumorally with 3 mg NPs in 80 μL of PBS. Based on the prior work and considering the limit of intratumoral injection volume, we limited the dose of NP to 3 mg in 80 μL.8 NPs were vortexed and sonicated using a bath sonicator before intratumoral injections to resuspend the particles. Doses were given on days 1, 5, 9, and 13. Tumor dimensions were taken daily to calculate the tumor volume. When the tumors reached a volume of 2,000 mm3, the mice were euthanized, and tumors, heart, lungs, liver, kidneys, and spleen were harvested. Tumors were dissociated using dispase (STEMCELL Technologies, Vancouver, BC, Canada) and collagenase (Worthington Biochemical, NJ). The A549 cells were isolated from mouse cells using a mouse cell depletion kit (Miltenyi Biotech, CA). Cell pellets were stored at −80°C for further protein extraction for western blots. The p53 (1:500) (9282; Cell Signaling Technologies, Danvers, MA) and SIRT1 (1:500) (80-110; Cell Signaling Technologies, Danvers, MA) antibodies were used. GAPDH (1:1,000) (5174S; Cell Signaling Technologies, Danvers, MA) was used as the endogenous control for SIRT1, and vinculin (1:1,000) (13901S; Cell Signaling Technologies, Danvers, MA) was used as the endogenous control for p53. The anti-rabbit IgG HRP-linked secondary antibody (7074; Cell Signaling Technologies, Danvers, MA) at 1:2,000 dilution was then added to probe for the primary antibody. Protein blot intensity was quantified using ImageJ software. Complete blood count (CBC) analysis was done on whole blood samples using a Sysmex CBC analyzer. Ki-67 staining was performed for tumor samples, and H&E staining was performed for liver, kidney, and spleen samples.
For biodistribution studies, tumors were implanted subcutaneously on the right flank. Once the tumor reached ∼600 mm3, the mice were randomly split into three treatment groups (n = 2): PBS, miR-34a NP 8 h, and miR-34a NP 24 h. NP-treated mice were treated with miR-34a NPs labeled with FAM fluorophore. After the specified time, the mice were euthanized, and the tumors, heart, lungs, kidneys, and spleen were harvested and imaged using SpectrumCT IVIS. Tumors were cryosectioned and imaged using the Keyence microscope at 40× magnification.
Data availability
There are no sequencing data that have been deposited in a data repository. All data will be made available upon request to the corresponding author.
Acknowledgments
This work was supported by the V Foundation award, NIH R35CA232105 to F.J.S., and an NIH R01 (1R01CA241194-01A1) grant to R.B. and F.J.S.
Author contributions
R.B. and V.K. designed the research; V.K., A.W., CH.L., and L.Y. performed the research and analyzed the data; R.B., V.K., A.W., CH.L., L.Y., MP.N., and F.J.S. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.03.015.
Supplemental information
References
- 1.Zogg H., Singh R., Ro S. Current advances in RNA therapeutics for human diseases. Int. J. Mol. Sci. 2022;23:2736. doi: 10.3390/ijms23052736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullard A. FDA approves fifth RNAi drug - alnylam's next-gen hATTR treatment. Nat. Rev. Drug Discov. 2022;21:548–549. doi: 10.1038/d41573-022-00118-x. [DOI] [PubMed] [Google Scholar]
- 3.Moumné L., Marie A.C., Crouvezier N. Oligonucleotide therapeutics: from discovery and development to patentability. Pharmaceutics. 2022;14:260. doi: 10.3390/pharmaceutics14020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pratt A.J., MacRae I.J. The RNA-induced silencing complex: a versatile gene-silencing machine. J. Biol. Chem. 2009;284:17897–17901. doi: 10.1074/jbc.R900012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasinski A.L., Slack F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 7.Malik S., Lim J., Slack F.J., Braddock D.T., Bahal R. Next generation miRNA inhibition using short anti-seed PNAs encapsulated in PLGA nanoparticles. J. Control. Release. 2020;327:406–419. doi: 10.1016/j.jconrel.2020.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Babar I.A., Cheng C.J., Booth C.J., Liang X., Weidhaas J.B., Saltzman W.M., Slack F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA. 2012;109:E1695–E1704. doi: 10.1073/pnas.1201516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng C.J., Bahal R., Babar I.A., Pincus Z., Barrera F., Liu C., Svoronos A., Braddock D.T., Glazer P.M., Engelman D.M., et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–110. doi: 10.1038/nature13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anastasiadou E., Seto A.G., Beatty X., Hermreck M., Gilles M.E., Stroopinsky D., Pinter-Brown L.C., Pestano L., Marchese C., Avigan D., et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, slows DLBCL tumor cell growth in vitro and in vivo. Clin. Cancer Res. 2021;27:1139–1149. doi: 10.1158/1078-0432.CCR-20-3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seto A.G., Beatty X., Lynch J.M., Hermreck M., Tetzlaff M., Duvic M., Jackson A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018;183:428–444. doi: 10.1111/bjh.15547. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z. The guideline of the design and validation of MiRNA mimics. Methods Mol. Biol. 2011;676:211–223. doi: 10.1007/978-1-60761-863-8_15. [DOI] [PubMed] [Google Scholar]
- 13.O'Brien J., Hayder H., Zayed Y., Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018;9:402. doi: 10.3389/fendo.2018.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y., Miao L., Satterlee A., Huang L. Delivery of oligonucleotides with lipid nanoparticles. Adv. Drug Deliv. Rev. 2015;87:68–80. doi: 10.1016/j.addr.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lv H., Zhang S., Wang B., Cui S., Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 16.Kawami M., Takenaka S., Akai M., Yumoto R., Takano M. Characterization of miR-34a-induced epithelial-mesenchymal transition in non-small lung cancer cells focusing on p53. Biomolecules. 2021;11:1853. doi: 10.3390/biom11121853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slabáková E., Culig Z., Remšík J., Souček K. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis. 2017;8:e3100. doi: 10.1038/cddis.2017.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi X., Kaller M., Rokavec M., Kirchner T., Horst D., Hermeking H. Characterization of a p53/miR-34a/CSF1R/STAT3 feedback loop in colorectal cancer. Cell. Mol. Gastroenterol. Hepatol. 2020;10:391–418. doi: 10.1016/j.jcmgh.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasinski A.L., Kelnar K., Stahlhut C., Orellana E., Zhao J., Shimer E., Dysart S., Chen X., Bader A.G., Slack F.J. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene. 2015;34:3547–3555. doi: 10.1038/onc.2014.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasinski A.L., Slack F.J. miRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53-induced mouse model of lung adenocarcinoma. Cancer Res. 2012;72:5576–5587. doi: 10.1158/0008-5472.CAN-12-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anastasiadou E., Stroopinsky D., Alimperti S., Jiao A.L., Pyzer A.R., Cippitelli C., Pepe G., Severa M., Rosenblatt J., Etna M.P., et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia. 2019;33:132–147. doi: 10.1038/s41375-018-0178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong D.S., Kang Y.K., Borad M., Sachdev J., Ejadi S., Lim H.Y., Brenner A.J., Park K., Lee J.L., Kim T.Y., et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer. 2020;122:1630–1637. doi: 10.1038/s41416-020-0802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beg M.S., Brenner A.J., Sachdev J., Borad M., Kang Y.K., Stoudemire J., Smith S., Bader A.G., Kim S., Hong D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs. 2017;35:180–188. doi: 10.1007/s10637-016-0407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wahane A., Malik S., Shih K.C., Gaddam R.R., Chen C., Liu Y., Nieh M.P., Vikram A., Bahal R. Dual-modality poly-l-histidine nanoparticles to deliver peptide nucleic acids and paclitaxel for in vivo cancer therapy. ACS Appl. Mater. Interfaces. 2021;13:45244–45258. doi: 10.1021/acsami.1c11981. [DOI] [PubMed] [Google Scholar]
- 25.Chang C.C., Wu M., Yuan F. Role of specific endocytic pathways in electrotransfection of cells. Mol. Ther. Methods Clin. Dev. 2014;1:14058. doi: 10.1038/mtm.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dokka S., Toledo D., Shi X., Castranova V., Rojanasakul Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm. Res. 2000;17:521–525. doi: 10.1023/a:1007504613351. [DOI] [PubMed] [Google Scholar]
- 27.Yamakuchi M., Lowenstein C.J. MiR-34, SIRT1 and p53: the feedback loop. Cell Cycle. 2009;8:712–715. doi: 10.4161/cc.8.5.7753. [DOI] [PubMed] [Google Scholar]
- 28.Yamamura S., Saini S., Majid S., Hirata H., Ueno K., Deng G., Dahiya R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS One. 2012;7:e29722. doi: 10.1371/journal.pone.0029722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin X., Guan H., Huang Z., Liu J., Li H., Wei G., Cao X., Li Y. Downregulation of Bcl-2 expression by miR-34a mediates palmitate-induced Min6 cells apoptosis. J. Diabetes Res. 2014;2014:258695. doi: 10.1155/2014/258695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang H., Zhang Z., Xu Y., Shao J. Expression of p53, p21 in human lung adenocarcinoma A549 cell strains under hypoxia conditions and the effect of TSA on their expression. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2003;23:359–361. doi: 10.1007/BF02829417. [DOI] [PubMed] [Google Scholar]
- 31.Lai M., Du G., Shi R., Yao J., Yang G., Wei Y., Zhang D., Xu Z., Zhang R., Li Y., et al. MiR-34a inhibits migration and invasion by regulating the SIRT1/p53 pathway in human SW480 cells. Mol. Med. Rep. 2015;11:3301–3307. doi: 10.3892/mmr.2015.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Statello L., Guo C.-J., Chen L.-L., Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021;22:96–118. doi: 10.1038/s41580-020-00315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Malik S., Suh H.W., Xiao Y., Deng Y., Fan R., Huttner A., Bindra R.S., Singh V., Saltzman W.M., Bahal R. Anti-seed PNAs targeting multiple oncomiRs for brain tumor therapy. Sci. Adv. 2023;9:eabq7459. doi: 10.1126/sciadv.abq7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.miRagen. Miragen Announces Internal Review of Preliminary Topline Data for the Phase 2 Solar Clinical Trial of Cobomarsen in Patients with Cutaneous T-cell Lymphoma (CTCL). GlobalNewswire; 2020.
- 35.Zhang Y., Roccaro A.M., Rombaoa C., Flores L., Obad S., Fernandes S.M., Sacco A., Liu Y., Ngo H., Quang P., et al. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood. 2012;120:1678–1686. doi: 10.1182/blood-2012-02-410647. [DOI] [PubMed] [Google Scholar]
- 36.Sivori S., Falco M., Della Chiesa M., Carlomagno S., Vitale M., Moretta L., Moretta A. CpG and double-stranded RNA trigger human NK cells by Toll-like receptors: induction of cytokine release and cytotoxicity against tumors and dendritic cells. Proc. Natl. Acad. Sci. USA. 2004;101:10116–10121. doi: 10.1073/pnas.0403744101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kedmi R., Ben-Arie N., Peer D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials. 2010;31:6867–6875. doi: 10.1016/j.biomaterials.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 38.Daige C.L., Wiggins J.F., Priddy L., Nelligan-Davis T., Zhao J., Brown D. Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther. 2014;13:2352–2360. doi: 10.1158/1535-7163.MCT-14-0209. [DOI] [PubMed] [Google Scholar]
- 39.Malik S., Bahal R. Investigation of PLGA nanoparticles in conjunction with nuclear localization sequence for enhanced delivery of antimiR phosphorothioates in cancer cells in vitro. J. Nanobiotechnology. 2019;17:57. doi: 10.1186/s12951-019-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma D. Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale. 2014;6:6415–6425. doi: 10.1039/c4nr00018h. [DOI] [PubMed] [Google Scholar]
- 41.Tahmasbi Rad A., Malik S., Yang L., Oberoi-Khanuja T.K., Nieh M.P., Bahal R. A universal discoidal nanoplatform for the intracellular delivery of PNAs. Nanoscale. 2019;11:12517–12529. doi: 10.1039/c9nr03667a. [DOI] [PubMed] [Google Scholar]
- 42.Malik S., Kumar V., Liu C.H., Shih K.C., Krueger S., Nieh M.P., Bahal R. Head on comparison of self- and nano-assemblies of gamma peptide nucleic acid amphiphiles. Adv. Funct. Mater. 2022;32:2109552. doi: 10.1002/adfm.202109552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malik S., Saltzman W.M., Bahal R. Extracellular vesicles mediated exocytosis of antisense peptide nucleic acids. Mol. Ther. Nucleic Acids. 2021;25:302–315. doi: 10.1016/j.omtn.2021.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crooke S.T., Vickers T.A., Liang X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020;48:5235–5253. doi: 10.1093/nar/gkaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L. Using an in-vacuum CCD detector for simultaneous small- and wide-angle scattering at beamline X9. J. Synchrotron Radiat. 2013;20:211–218. doi: 10.1107/S0909049512048984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are no sequencing data that have been deposited in a data repository. All data will be made available upon request to the corresponding author.