

Abstract
Effective methods are needed for labelling acyclic ureas with carbon-11 (t1/2=20.4 min) as potential radiotracers for biomedical imaging with positron emission tomography (PET). Herein, we describe the rapid and high-yield syntheses of unsymmetrical acyclic [11C]ureas under mild conditions (room temperature and within 7 min) using no-carrier-added [11C]carbonyl difluoride with aliphatic and aryl amines. This methodology is compatible with diverse functionality (e.g., hydroxy, carboxyl, amino, amido, or pyridyl) in the substrate amines. The labelling process proceeds through putative [11C]carbamoyl fluorides and for primary amines through isolable [11C]isocyanate intermediates. Unsymmetrical [11C]ureas are produced with negligible amounts of unwanted symmetrical [11C]urea byproducts. Moreover, the overall labelling method tolerates trace water and the generally moderate to excellent yields show good reproducibility. [11C]carbonyl difluoride shows exceptional promise for application to the synthesis of acyclic [11C]ureas as new radiotracers for biomedical imaging with PET.
Keywords: [11C]carbonyl difluoride, carbon 11, positron emission tomography, radiochemistry, ureas
Introduction
Positron emission tomography (PET) uses well-designed radiotracers to image specific biological structures and processes in living animal and human subjects.[1] Consequently, PET has become an established molecular imaging modality for biomedical research,[1–2] medical diagnosis,[3] and drug development.[4] The short-lived positron-emitter carbon-11 (t1/2=20.4 min) is attractive for labelling PET radiotracers, mainly because this radionuclide may in principle replace non-radioactive carbon in any organic molecule.[5] Nonetheless, carbon-11 must always be produced on-site from a cyclotron at the time of need. Ensuing radiotracer synthesis, purification, and formulation for intravenous injection must also be completed rapidly, generally within only two or three half-lives of carbon-11 (i.e., in <1 h). The main cyclotron sources of carbon-11, from which all radiotracer syntheses must begin, are [11C]carbon dioxide and [11C]methane, each by the 14N(p,α)11C reaction on nitrogen doped with either oxygen or hydrogen, respectively.[5] Radiosyntheses must be reproducible and reliable, and amenable to automation within lead-shielded apparatus for personnel radiation protection. Nowadays, many secondary labelling agents may be prepared from the primary cyclotron sources for labelling various chemotypes.[5] Principal, among these secondary agents, is [11C]iodomethane for labelling at a non-metal heteroatom (e.g., N, O, and S) with a [11C]methyl group.[5c] [11C]Carbon monoxide has featured widely for labelling carbonyl functionality.[6] However, not all potential radiotracers have structures that may be labelled either efficiently or at all by existing methods.
Unsymmetrical acyclic ureido groups are prevalent in drugs and drug candidates,[7] and in known and prospective PET radiotracers.[6b,c, 8] Prior methods for labelling ureas with carbon-11 have been based on coupling amines with [11C]carbon dioxide,[9] [11C]carbon monoxide,[10] or [11C]phosgene[8] (Figure 1). Use of cyclotron-produced [11C]carbon dioxide for labelling unsymmetrical ureas requires the use of a strong base [e.g., 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP)[9a–c] or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)[9d]] to capture [11C]carbon dioxide in solution. A stoichiometric amount of phosphoryl chloride relative to the first amine is also required to dehydrate the derived intermediate [11C]carbamate to a [11C]isocyanate for reaction with a second amine. Because of the high reactivity of phosphoryl chloride, the dehydration step is incompatible with many functional groups and is vulnerable to moisture. [11C]Carbon dioxide may also be used to produce either symmetrical[9d] or unsymmetrical[9e] [11C]ureas by use of a pair of Mitsunobu reagents, di-tert-butyl azodicarboxylate and tributylphosphine. One primary amine plus one secondary amine must be used to achieve acceptable yields. Moreover, a strong base must still be used for initial [11C]carbon dioxide capture.[9e] The need for complete removal of the added base and byproducts from the Mitsunobu reagents from the labelled product by rapid single pass HPLC can be challenging. Broad substrate scope has not been shown for this method.
Figure 1.

Comparison of prominent methods for the synthesis of acyclic [11C]ureas from amines.
Palladium(II)-mediated conversion of [11C]carbon monoxide into [11C]ureas requires elevated temperature and produces mixtures of unsymmetrical and symmetrical [11C]ureas from dissimilar amines.[10] Agents for trapping [11C]carbon monoxide need to be present along with a quite large amount of each amine (e.g., 90 μmol), all of which may exacerbate labelled product isolation by single pass HPLC. Generally, at least one of the amines must be a primary amine. Substrate scope is therefore rather limited. This method has so far been little used for developing PET radiotracers.
[11C]Phosgene was first described in the 1970s[11] and has since proved to be a useful labelling agent.[8] Despite successive improvements,[12] all methods for [11C]phosgene synthesis remain complex, require highly specialized equipment, and must use hazardous chlorine gas. Therefore, the use of [11C]phosgene has not been adopted by many laboratories. Moreover, use of [11C]phosgene for [11C]urea synthesis has several limitations. Generation of a mixture of the desired unsymmetrical urea together with the undesired symmetrical urea often complicates purification and limits the yield. Furthermore, synthesis of [11C]isocyanates as intermediates for [11C]urea synthesis from [11C]phosgene and amines is very sensitive to moisture[13] and often requires heating in the presence of a tertiary amine catalyst.[8a,14] Attempts have been made to use alternative precursors, such as N-organosulfinylamines[13a] or tertiary N-benzylamines,[15] to control the reactivity of phosgene in urea syntheses, but these approaches have gained limited traction in PET radiotracer synthesis.
We recently reported a straightforward and efficient synthesis of a new labelling agent, [11C]carbonyl difluoride, from cyclotron-produced [11C]carbon dioxide by on-line conversion into [11C]carbon monoxide and then passage over silver(II) fluoride.[16] In this method, [11C]carbonyl difluoride is produced reliably in useful overall radiochemical yield and with high no-carrier-added (NCA) molar activity (>200 GBq/μmol). This new labelling agent has been shown to be very effective for 11C-carbonylation reactions giving intracyclic ureas, as especially exemplified by the synthesis of a β-adrenergic receptor imaging agent, [11C](S)-CGP12177.[16] Here we aimed to further explore NCA [11C]carbonyl difluoride as a labelling agent for acyclic ureas, especially unsymmetrical ureas.
Results and Discussion
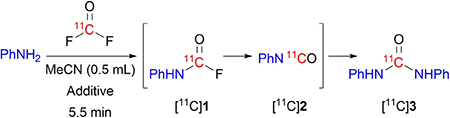
We started our study by investigating the reaction of NCA [11C]carbonyl difluoride with aniline (10 μmol) in acetonitrile (0.5 mL). We postulated that symmetrical [11C]1,3-diphenylurea ([11C]3) would be formed through successive formation of [11C]phenylcarbamoyl fluoride ([11C]1) and [11C]phenyl isocyanate ([11C]2). HPLC analysis of reaction mixtures after aqueous quench indeed revealed the formation of [11C]3 in moderately high yield (Table 1, entry 1) with only a trace of one of the postulated intermediates, the isocyanate [11C]2. By contrast, prompt analyses of reaction mixtures before any aqueous dilution gave substantial isolated yields of [11C]2 (39±8%; Table 1, entry 2). Peak streaking in the radiochromatograms (see Supporting Information, Figure S2) suggested that this yield was likely underestimated because of relatively slow hydrolysis of [11C]2 throughout the reversed phase gradient HPLC analysis, which used an aqueous-organic mobile phase (H2O-MeCN, initially 50: 50 v/v, and ending at 10: 90 v/v). We concluded that the homocoupling of aniline with [11C]carbonyl difluoride to form the urea [11C]3 via the isocyanate [11C]2 was mediated rapidly by water during the reaction quench. This conclusion accords with a study showing that ureas may be rapidly synthesized from isocyanates in high yields in cold (4–5°C) water,[17] and reports of the promoting effect of bulk[18] or trace water[19] on symmetrical urea synthesis from isocyanates, including that of [11C]1,3-bis(2-chloroethyl)urea.[13b]
Table 1.
Optimization of [11C]carbonyl difluoride trapping with aniline and [11C]3 synthesis.
| |||
|---|---|---|---|
| Entry | PhNH2 [μmol] | Additive [μmol] | Yield of [11C]3 [%]a |
|
| |||
| 1 | 10 | 0 | 64 ± 8b |
| 2 | 10 | 0 | 0 (39 ± 8 of [11C]2)c |
| 3 | 2 | Pyridine (1) | 43 ± 1 |
| 4 | 10 | Pyridine (0.1) | 76 ± 1 |
| 5 | 10 | Pyridine (1) | 84 ± 1 |
Yields are mean±range for n = 2.
The reaction mixture was quenched with water 1 minute before HPLC analysis.
Isolated yield after direct injection of the crude reaction mixture into HPLC. [11C]2 was likely continuously hydrolyzed during the analysis. Total synthesis time was 5.5 minutes from the release of [11C]carbon monoxide over silver(II) fluoride. Synthesis and cryogenic isolation of [11C]carbon monoxide itself required 12 min.
We surmised that [11C]2 was formed by spontaneous elimination of hydrogen fluoride from [11C]1. This rapid isocyanate formation was somewhat unexpected because analogous isocyanate synthesis from phosgene typically requires heating and a tertiary amine catalyst.[8a,14] This observation contrasts with those on reactions between anilines and excess carbonyl difluoride in chlorobenzene and dichloromethane, which exclusively yield the corresponding carbamoyl fluorides in high yields.[20] However, we did not test labelling reactions in either of these water-immiscible solvents. Water-miscible solvents are preferable in carbon-11 chemistry because the reaction mixtures can be diluted with water and then purified with rapid single-pass reversed phase HPLC without first spending time to remove the solvent. Moreover, an onerous need for automation of the solvent removal process is obviated.
Although there is as yet no prominent example, some symmetrical [11C]ureas could become candidate PET radiotracers, and efficient methods for their radiosynthesis is therefore of some interest. Tertiary amines and heterocyclic amines have been shown to improve the yields of symmetrical diaryl ureas from isocyanates.[19] Therefore, we investigated whether pyridine could promote homocoupling of aniline to produce symmetrical [11C]3. Use of 1 μmol of pyridine and 2 μmol of aniline in acetonitrile (0.5 mL) gave a moderate yield of [11C]3 (Table 1, entry 3). The yield increased substantially when using a lower amount of pyridine (0.1 μmol) and a higher amount of aniline (10 μmol) (Table 1, entry 4). Use of pyridine (1 μmol) and aniline (10 μmol) gave [11C]3 in yet higher yield (84%; Table 1, entry 5). These few experiments quickly confirmed the viability of reaction between [11C]carbonyl difluoride and anilines for synthesizing symmetrical [11C]ureas.
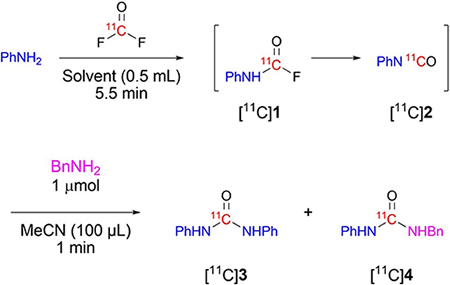
Our main interest was to develop methodology for the synthesis of potentially more useful unsymmetrical acyclic [11C]ureas. We considered that these might be accessed by treating intermediate isocyanates, generated from [11C]carbonyl difluoride and one amine, such as [11C]2, with a dissimilar amine. To test this possibility, we added benzylamine (1 μmol) after [11C]carbonyl difluoride had been fully introduced into a reaction mixture containing aniline (i.e., 5.5 minutes from [11C]carbon monoxide release). The desired unsymmetrical urea [11C]4 was formed in very high yield from [11C]2 after just 1 minute and gratifyingly without the symmetrical byproduct [11C]3 (Table 2, entry 1). A lower amount of aniline (5 μmol) gave a lower practical yield of [11C]4 (Table 2, entry 2) because of a lower trapping efficiency for the [11C]carbonyl difluoride. A higher amount of aniline (20 μmol) did not improve the trapping efficiency (Table 2, entry 3) and gave some of the undesired [11C]3. Inclusion of 1% water (5 μL, 0.28 mmol) did not lower the yield of [11C]4 (Table 2, entry 4), as would be the case with the dehydrative approaches to [11C]urea synthesis using [11C]carbon dioxide. Therefore, we considered that high humidity or the use of lower quality non-anhydrous solvents would be well tolerated. In this regard, a substantial yield of [11C]4 was obtained even in the presence of a much higher concentration (5% v/v) of water (added as 25 μL; 1.4 mmol) (Table 2, entry 5). These findings of water tolerance are important because they indicate the radiosynthetic procedure is robust and readily amenable to automation for routine PET radiotracer production.
Table 2.
Optimization of [11C]carbonyl difluoride trapping with aniline, followed by spontaneous [11C]isocyanate formation, and coupling with benzylamine.
| ||||
|---|---|---|---|---|
| Entry | PhNH2 [μmol] | Solvent [0.5 mL] | Yield of [11C]3 [%]a | Yield of [11C]4 [%]a |
|
| ||||
| 1 | 10 | MeCN | 0 ± 0b | 84 ± 9b |
| 2 | 5 | MeCN | 0 ± 0 | 68 ± 9 |
| 3 | 20 | MeCN | 2 ± 1b | 82 ± 9b |
| 4 | 10 | H2O/MeCN (1: 99 v/v) | 1 ± 0 | 91 ± 0 |
| 5 | 10 | H2O/MeCN (5: 95 v/v) | 1 ± 0 | 62 ± 2 |
| 6 | 10 | THF | 4 ± 1 | 83 ± 2 |
| 7 | 10 | DMSO | 20 ± 0 | 45 ± 1 |
| 8 | 10 | DMF | 7 ± 3 | 39 ± 5 |
Yields are mean±range for n = 2, unless otherwise indicated.
Yields are mean±SD for n = 3. Total synthesis time was 6.5 min from the release of [11C]carbon monoxide.
Use of a different dipolar aprotic non-nucleophilic solvent, namely THF, produced a high yield of the target unsymmetrical urea [11C]4 (Table 2, entry 6), but with some accompanying [11C]3. Two other weakly nucleophilic dipolar aprotic solvents, DMSO and DMF, gave lower yields of [11C]4 and more of the symmetrical byproduct [11C]3 (Table 2, entries 7 and 8). DMF is known to react with carbonyl difluoride to produce carbon dioxide in a difluoro-decarbonylation reaction.[21] Thus, difluoro-decarbonylation might be the cause for the lower yield of [11C]4 in DMF. Possibly DMSO is also reactive towards [11C]carbonyl difluoride. Consistent with these suggestions, a high proportion of radioactivity was retained in the reaction mixture as an unidentified polar byproduct when using either DMF or DMSO as solvent (see Supporting Information, Figures S13 and S14).
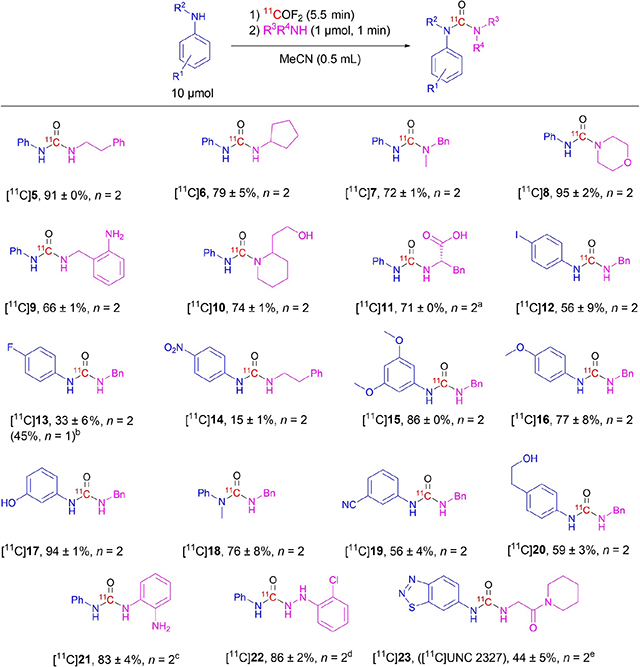
Having established a procedure for the synthesis of [11C]4 in high yield, we then explored whether this procedure might be applicable to the synthesis of unsymmetrical [11C]ureas from diverse amines, including some bearing various functional groups that would be incompatible with earlier methods. Treatment of the intermediate isocyanate [11C]2 with phenethylamine or cyclopentylamine, as examples of primary amines, gave excellent and high yields of the desired unsymmetrical [11C]ureas, [11C]5 and [11C]6, respectively (Table 3), and with no symmetrical [11C]urea byproduct. Likewise, the secondary amines, N-benzylmethylamine and morpholine, gave the unsymmetrical [11C]ureas [11C]7 and [11C]8 in high and excellent yields, respectively. Amines having a competing nucleophilic site, such as the arylamino group in 2-aminobenzylamine or the hydroxyl group in 2-(piperidin-2-yl)ethan-1-ol, gave the desired ureas, [11C]9 and [11C]10 in moderate and high yield, respectively, so demonstrating the preference of the intermediate to react with an aliphatic amino group over a less nucleophilic arylamino or hydroxy group. Treatment of the intermediate isocyanate [11C]2 in acetonitrile with the sodium salt of L-phenylalanine in water/acetonitrile (100 μL, 1: 1 v/v) produced the desired urea [11C]11 in high yield, but with some symmetrical byproduct, most likely due to water mediated homocoupling. Electron-deficient 4-iodoaniline instead of aniline for [11C]isocyanate formation and use of benzylamine as second amine gave a moderate yield of the urea [11C]12. Use of an arylamine with a very weak (p-fluoro) or strong (p-nitro) electron-withdrawing substituent for the [11C]isocyanate formation gave low and very low practical yields of the ureas, [11C]13 and [11C]14, respectively. These decreases were primarily due to lower trapping efficiencies for [11C]carbonyl difluoride. Use of 50 μmol of 4-fluoroaniline gave comparable yield of [11C]13, albeit with production of some symmetrical [11C]urea byproduct. Gratifyingly, more reactive electron-rich anilines, such as 3,5-dimethoxyaniline, p-anisidine, and 3-aminophenol, gave very high yields of the intermediate [11C]isocyanates, which reacted with benzylamine to give the respective [11C]ureas, [11C]15, [11C]16, and [11C]17 in high to excellent yields and without any symmetrical [11C]urea byproducts. Also a secondary aniline, N-methylaniline, reacted efficiently with [11C]carbonyl difluoride, presumably to form [11C]benzyl(methyl)carbamoyl fluoride, which upon treatment with a second nucleophilic amine, benzylamine, produced a high yield of the urea [11C]18. Reaction of [11C]carbonyl difluoride with one example of an aniline bearing a meta electron-withdrawing group, namely 3-cyanoaniline, gave a moderate yield of the desired urea [11C]19 after treatment of the labelled intermediate with benzylamine. An aniline having a competing nucleophilic hydroxyl group, namely 2-(4-phenylamino)-ethan-1-ol, was readily converted into the desired unsymmetrical urea [11C]20 in moderate yield.
Table 3.
Synthesis of unsymmetrical [11C]ureas by coupling anilines or aniline derivatives and aliphatic amines with [11C]carbonyl difluoride.
|
L-Phenylalanine in H2O-MeCN (1 : 1 v/v; 100 μL) with 1 equiv. of NaOH.
50 μmol 4-fluoroaniline was used, giving a 57 : 43 mix of [11C]13 to [11C]1,3-di-4- fluorophenyl urea.
50 μmol 1,2-diaminobenzene and 1 μmol pyridine were used.
HCl salt of hydrazine precursor in H2O-MeCN (1 : 1 v/v; 100 μL) and 1 equiv. of KOH were used.
50 μmol arylamine precursor and 10 μmol pyridine were used. Yields are decay-corrected and based on radioactivity recovered from the HPLC purification of a reaction aliquot (n = 2). The synthesis time was 6.5 min from release of [11C]carbon monoxide.
The synthesis of unsymmetrical [11C]ureas from weakly nucleophilic anilines can be challenging. Nonetheless, treatment of the isocyanate [11C]2 in situ with a five-fold excess of 1,2-diaminobenzene and a catalytic amount of pyridine gave the desired diaryl unsymmetrical urea [11C]21 in high yield with no symmetrical byproduct. As a further demonstration of the versatile reactivity of the [11C]isocyanate intermediates, treatment of [11C]2 with the free base of 2-chloro-phenylhydrazine (generated in situ) in water: acetonitrile (100 μL, 1: 1 v/v) gave [11C]22 in excellent yield and without any symmetrical byproducts. This reflects the high nucleophilicity of the hydrazine.
The synthesis of a labeled inhibitor of protein arginine methyltransferase 3, [11C]UNC 2327 ([11C]23),[22] a potential PET radiotracer, was selected to further show-case the broad utility of this new methodology. Outside its carbonyl group, 23 does not have other sites that are amenable to labelling with carbon-11 by other methodologies. Labelling in the carbonyl group is also very challenging because the radiotracer must be built from an electron-deficient arylamine (benzo[d][1,2,3]thiadiazol-6-amine) and an aliphatic amine (2-amino-1-(piperidin-2-yl) ethan-1-ol) holding an amide functionality. The amide functionality would be incompatible with any [11C]urea synthesis that depends on phosphoryl trichloride dehydration, as discussed earlier.
Use of our standard labelling conditions produced a very low yield (2%) of [11C]23, likely due to the low nucleophilicity of the bicyclic arylamine. Addition of pyridine (2 μmol) as catalyst improved the yield to 6%. The symmetrical product was not formed to any large extent, again reflecting low bicyclic amine nucleophilicity. A further five-fold increase of arylamine to 50 μmol and of pyridine to 10 μmol, while still retaining the very low amount of the aliphatic amine nucleophile (1 μmol) gave [11C]23 in a useful yield (44%).
To further explore the use of [11C]carbonyl difluoride for the synthesis of acyclic [11C]ureas, we attempted to couple two aliphatic amines. As expected, reaction between [11C]carbonyl difluoride and benzylamine (10 μmol) in acetonitrile (0.5 mL) did not stop at formation of the [11C]isocyanate, as with anilines, but proceeded to give the symmetrical 1,3-dibenzyl urea ([11C]24) in very high yield (Table 4, entry 1). Even with a lower amount of precursor (1 μmol), [11C]24 was produced in high yield (Table 4, entry 2). A further reduction of precursor, to achieve 1: 1 stoichiometry between labelling agent and precursor, is not feasible in routine NCA radiochemistry because of the variable and uncontrollable amount of carrier that arises in cyclotron carbon-11 production.[16] Instead, we attempted to control the reactivity of the first amine so that the reaction stopped at the intermediate [11C]carbamoyl fluoride or [11C]isocyanate. One approach that has been used with some success when using [11C]phosgene as labelling agent is to use a salt of the amine in suspension.[8a] Treatment of benzylammonium chloride (1 μmol) suspended in acetonitrile (0.5 mL) with [11C]carbonyl difluoride, produced the intermediate [11C]benzyl isocyanate in the absence of the symmetrical urea, [11C]24, as confirmed with HPLC analysis (Table 4, entry 3; also see Supporting Information, Figure S52).
Table 4.
Method development for synthesis of [11C]ureas from aliphatic amines.

| ||||||
|---|---|---|---|---|---|---|
| Entry | R1 in tosylate [μmol] | Additive [μmol] | TE [%](n) | R2 in amine [μmol] | R1NH11CONHR2 yield [%](n) | Product # |
|
| ||||||
| 1 | Bn (10)a | – | 93 ± 3(2) | – | 88 ± 1(2) | [11C]24 |
| 2 | Bn (1)a | – | 85(1) | – | 80(1) | [11C]24 |
| 3 | Bn (1)b,c | – | 62 ± 5(2) | – | NAd | – |
| 4 | Bn (1) | – | 65 ± 14(5) | c-Pen (10) | 63(1) | [11C]25 |
| 5 | Bn (2) | – | 63 ± 7(2) | – | NA | – |
| 6 | c-Pen (2) | – | 73 ± 15(2) | Bn (10) | 59 ± 13(2) | [11C]25 |
| 7 | c-Pen (1) | – | 74 ± 18(2) | Bn (10) | 62 ± 17(2) | [11C]25 |
| 8 e | Bn (3) | – | 77 ± 2(4) | c-Pen (30) | 61 ± 0(2) | [11C]25 |
| 9 | Bn (1) | DMAP·TsOH (1) | 43(1) | c-Pen (10) | 13(1) | [11C]25 |
| 10 | Bn (1) | Pyridine (1) | 72 ± 3(2) | c-Pen (10) | 60 ± 1(2) | [11C]25 |
| 11 | Bn (1) | Pyridine (2) | 87 ± 4(13) | c-Pen (10) | 83 ± 2(3) | [11C]25 |
10 μmol free base.
HCl salt.
Suspension.
Product is [11C]benzyl isocyanate.
1.5 mL MeCN. Synthesis time: 5.5 plus 1 min from release of [11C]carbon monoxide. TBAC = t-butylammonium chloride; TE= [11C]carbonyl difluoride trapping efficiency; NA= Not applicable; c-Pen=cyclopentyl. Superscript values in parentheses are numbers of experiments. Yields are mean±range for n = 2 and mean±SD for n ≥ 3.
However, alkylammonium chlorides have limited solubility in acetonitrile. Moreover, running carbon-11 chemistry in suspensions is undesirable because the amount of precursor in solution is unknown and because solids are ill-suited for easy product separation or analysis with HPLC. We therefore investigated the use of alkylammonium tosylates as much more soluble salts for these reactions. Treatment of benzylammonium tosylate (1 μmol) in acetonitrile with [11C]carbonyl difluoride resulted in high trapping efficiency for [11C]carbonyl difluoride and in [11C]benzyl isocyanate as the main product. Addition of cyclopentylamine in ten-fold excess then produced [11C]25 as the main product (Table 4, entry 4). Before investigating substrate scope in more detail, we wanted to improve trapping efficiency and [11C]isocyanate formation. We found that doubling the precursor concentration did not improve trapping efficiency (Table 4, entry 5). Reversing the order of amine addition and use of the tosylate salt of cyclopentylamine as trapping agent gave similar yield. Addition of 5- or 10-fold excess of benzylamine gave the unsymmetrical urea [11C]25 with good selectivity (Table 4, entries 6 and 7, respectively).
We hypothesized that the volatility of [11C]carbonyl difluoride (b.pt., −85°C) limits its time in solution for reaction with the first amine and that this could be the cause of lower yields from poorly nucleophilic amines. To increase the residence of [11C]carbonyldifluoride in solution, we increased the solvent volume three-fold to 1.5 mL while maintaining the precursor concentration at 2 μmol/mL. As a result, the trapping efficiency increased appreciably (Table 4, c.f., entries 8 and 4). However, these increased amounts of reagents might make purification of a PET radiotracer product more difficult. Instead, we saw the possibility of adding an agent that might sequester [11C]carbonyl difluoride in solution. We considered adding the common acylation catalyst, 4-N,N-dimethylaminopyridine (DMAP), but this would be expected to deprotonate the alkylammonium salt precursor and lead to formation of symmetrical products. Therefore, the tosylate of DMAP was tested, but this gave a low yield of [11C]25 (Table 4, entry 9). Pyridine is not basic enough to deprotonate aliphatic amines to a significant extent, whereas it would be expected to retain [11C]carbonyl difluoride in solution based on its transiently reversible reaction with acetyl groups.[23] Therefore, pyridine (1 μmol) was added to benzylammonium tosylate precursor and the [11C]carbonyl difluoride trapping efficiency improved appreciably (Table 4, entries 10). A further increase in amount of pyridine to 2 μmol increased trapping efficiency yet further and gave excellent yield of unsymmetrical [11C]25 (Table 4, entry 11).
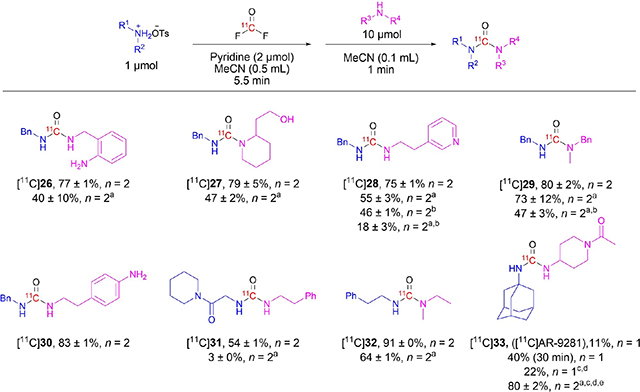
Having established a protocol for the synthesis of unsymmetrical [11C]ureas from aliphatic amines in the presence of pyridine, we explored substrate scope (Table 5). Addition of ambidently nucleophilic 2-aminobenzylamine to [11C]benzyl isocyanate intermediate produced a high yield of the unsymmetrical urea [11C]26 in which the arylamino group was retained. 2-aminobenzylamine had to be added in the second step because the reversed order of addition leads to intramolecular reaction to produce a cyclic [11C]urea in high yield.[16] Likewise, addition of ambident secondary amine, 2-(piperidin-2-yl)ethan-1-ol to [11C]benzyl isocyanate intermediate, produced the urea [11C]27, with a retained hydroxy group, in high yield. Pyridinyl and other heterocyclic groups are abundant among drug molecules. The unsymmetrical urea, [11C]28, which carries a terminal 3-pyridinyl group, was produced in high yield from [11C]benzyl isocyanate intermediate. Yield was progressively lowered by reversed addition of amine components, by omission of pyridine, and by both. Only primary amines can form isocyanates. Treatment of benzylammonium tosylate with [11C]carbonyl difluoride followed by excess N-benzylmethylamine gave [11C]29 in high yield. Nonetheless, reversed order of amine component addition, where the secondary ammonium tosylate salt was used, still gave a moderate yield. An excellent selectivity was observed in both cases showing that the reaction proceeds well through either [11C]isocyanate or [11C]carbamoyl fluoride intermediate. The highest yield of [11C]29 was however obtained in the presence of pyridine with N-benzylmethylamine as second amine component.
Table 5.
Synthesis of unsymmetrical [11C]ureas by coupling aliphatic amines with [11C]carbonyl difluoride.
|
No pyridine.
Reversed order of amine addition.
Three times the volume (1.5 mL) was used with retained concentration of precursor and reagents.
Heating for 5 min at 75 °C was used.
The second amine was added from H2O: MeCN (1 : 1 v/v; 300 μL). Each yield was determined after HPLC purification of a reaction aliquot.
The synthesis of the unsymmetrical ureas [11C]26-[11C]29 from primary benzylamine and secondary N-benzylmethylamine components was tested in the absence of pyridine and gave generally decreased yields. Yields were decreased yet further if pyridine was omitted in reversed amine addition mode.
[11C]30 was obtained in excellent yields from the intermediate [11C]benzyl isocyanate by treatment with 2-(4-aminophenyl) ethylamine, again showing the preference of the intermediate to react with an aliphatic amino group over a less nucleophilic arylamino group. In the attempted synthesis of [11C]31, the tosylate salt of the amine precursor in the absence of pyridine trapped [11C]carbonyl difluoride very inefficiently and produced only trace product. However, addition of pyridine increased the trapping efficiency markedly and also the yield of [11C]31. Likewise, an exceptionally high yield (91%) of the unsymmetrical urea [11C]32 was obtained as a result of efficient trapping of [11C]carbonyl difluoride into phenethylammonium tosylate solution in the presence of pyridine. Yield was much reduced in the absence of pyridine. Overall, for the syntheses of [11C]26-[11C]32, selectivity for the desired unsymmetrical [11C]ureas over the undesired symmetrical [11C]ureas exceeded 90%.
The soluble epoxide hydrolase inhibitor, [11C]AR-9281 ([11C]33), has potential for PET imaging in Alzheimer’s and Parkinson’s disease.[24] The urea carbonyl group of 33 is a potential site for labelling by using [11C]carbonyl difluoride, although challenging due to steric hindrance. The tosylate salt of 1-adamantylamine in the presence of pyridine retained 62% of the radioactivity introduced as [11C]carbonyl difluoride. After adding 1-acetyl-4-aminopiperidine for the usual 1-minute reaction time, [11C]33 was obtained in only 11% yield, but along with a 45% isolated yield of [11C]1-adamantyl isocyanate. A duplicate experiment with 30-minute reaction time gave an improved yield of [11C]33 (40%), but still with 29% intact [11C]isocyanate (co-injection in Supporting Information, Figure S51). Slow reaction had also been evident during the preparation of reference compound 33 (see the Supporting Information). Addition of a large quantity of 1-acetyl-4-aminopiperidine to speed up the reaction was impractical. Instead, reaction volume was increased to 1.5 mL with retained concentrations of precursor and reagents. This gave improved trapping efficiency (86%) in the presence of pyridine. Surprisingly, heating the reaction mixture at 75°C for 5 minutes only marginally improved the yield to 22%. The relatively minor influences of increased time and temperature suggested that [11C]urea formation accelerated after the aqueous quench and that water acts as a catalyst, as alluded to in the synthesis of [11C]3 (Table 1, entry 1). We considered that pyridine might not be an efficient acylation catalyst in this case and might only be serving to retain [11C]carbonyl difluoride in solution. Therefore, we excluded pyridine and added the second amine from water (150 μL) plus acetonitrile (150 μL). [11C]Carbonyl difluoride trapping efficiency was maintained. The reaction mixture was heated to 75°C for 5 minutes and gave the desired urea [11C]33 in high yield (80%) with only trace unreacted [11C]isocyanate. This again highlights that water is an effective promoter of [11C]urea formation.
Conclusion
The exploration of [11C]carbonyl difluoride for acyclic [11C]urea synthesis revealed wide substrate scope and good reproducibility. This new methodology is compatible with a range of functional groups and with both aliphatic amines and anilines. Reactions proceed via [11C]carbamoyl fluoride and for primary amines, [11C]isocyanate intermediates. Pyridine is effective for enhancing entrapment and utilization of [11C]carbonyl difluoride when using amine tosylates as soluble precursors. This methodology is highly promising for complex PET radiotracer production. Although elaborate amine substrates having limited reactivity may require some optimization of reaction conditions, such as reaction temperature, number of equivalents, and acylation catalyst or water addition, high yields are still attainable. These approaches to optimization are demonstrated here and their careful consideration should enable fast and trouble-free application. Moreover, the simplicity of this overall procedure should render it to be readily transferable to a GMP environment, although this has yet to be attempted.
Experimental Section
Full details are presented in Supporting Information. The production of [11C]carbon dioxide and the synthesis of [11C]carbon monoxide, [11C]carbonyl difluoride and the typical synthesis of an acyclic [11C]urea including yield measurements are as follows.
[11C]Carbon dioxide production:
[11C]carbon dioxide was prepared by the 14N(p,α)11C nuclear reaction by bombarding a nitrogen-1% oxygen gas target (2.1 MPa) with a beam of protons (16 MeV, 5 μA) from a cyclotron (PETrace; GE) for 5 min, typically generating 2 to 3 GBq of [11C]carbon dioxide with a molar activity between 18 and 45 GBq/μmol.
[11C]Carbon monoxide synthesis:
Cyclotron-produced [11C]carbon dioxide was converted into [11C]carbon monoxide using a modified Synthia radiosynthesis apparatus. [11C]Carbon dioxide was first trapped on molecular sieves (13X) from the released nitrogen-oxygen target gas. The sieves were then purged with helium to remove residual oxygen. The molecular sieve trap was then heated to 280°C while being purged with helium to release the [11C]carbon dioxide into a small stainless steel cryo-trap filled with silica immersed in liquid nitrogen (−196°C). The stainless steel cryo-trap was then heated to release the trapped [11C]carbon dioxide into a stream of helium that was passed through a heated molybdenum column (875°C). The generated [11C]carbon monoxide was separated from residual [11C]carbon dioxide by passage over sodium hydroxide coated on silica (ascarite) and then concentrated within a second stainless steel cryo-trap filled with silica immersed in liquid nitrogen. The full process typically required 11 to 12 min from the end of radionuclide production and gave [11C]carbon monoxide in 71±2% yield (mean ± SD; n=37), based on the proportion of the total radioactivity (decay-corrected) that was not immobilized on ascarite.
Synthesis of [11C]1-benzyl-3-phenylurea ([11C]4):
[11C]Carbon monoxide was passed over solid silver(II) fluoride and bubbled (5 mL/min) through a solution of aniline (10 μmol) in acetonitrile (0.5 mL) for 5.5 min. Benzylamine (1 μmol) in acetonitrile (0.1 mL) was added to the reaction mixture.
Radio-HPLC analysis and yield measurement:
An aliquot of the reaction mixture was diluted with water and injected onto HPLC. The amount of isolated radioactive product was compared to that of the decay-corrected amount of radioactivity injected onto HPLC to give the radiochemical purity of the labelled compound in the reaction mixture. This purity value was multiplied by the efficiency of the initial trapping of radioactivity in the reaction vial (decay-corrected and assumed to be all [11C]carbonyl difluoride), thus giving a measure of the radiochemical yield of the labelled compound based on [11C]carbon monoxide. Only negligible amounts (<0.1%) of radioactivity adhered to needles, stainless steel lines, and the silver(II) fluoride column. The full process from end of cyclotron irradiation to HPLC injection takes less than 20 minutes.
Supplementary Material
Acknowledgements
The authors acknowledge support from the Intramural Research Program of the National Institutes of Health (NIMH: Project number ZIA-MH002793). The authors are grateful to the NIH Clinical Center PET Department (Chief: Dr. P. Herscovitch) for cyclotron production of carbon-11.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is available on the WWW under https://doi.org/10.1002/chem.202100690
References
- [1].Phelps ME, Proc. Natl. Acad. Sci. USA 2000, 97, 9226–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Chua S, Groves A, Biomedical imaging: applications and advances, Elsevier, Cambridge, Waltham, 2014, p. 3–40; [Google Scholar]; b) Heurling K, Leuzy A, Jonasson M, Frick A, Zimmer ER, Nordberg A, Lubberink M, Brain Res. 2017, 1670, 220–234; [DOI] [PubMed] [Google Scholar]; c) Villemagne VL, Doré V, Burnham SC, Masters CL, Rowe CC, Nat. Rev. Neurol. 2018, 14, 225–236; [DOI] [PubMed] [Google Scholar]; d) Slifstein M, Abi-Dargham A, Semin. Nucl. Med. 2017, 47, 54–63. [DOI] [PubMed] [Google Scholar]
- [3].a) Herholz K, Heiss WD, Mol. Imaging Biol. 2004, 6, 239–269; [DOI] [PubMed] [Google Scholar]; b) Gallamini A, Zwarthoed C, Borra A, Cancers (Basel). 2014, 6, 1821–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) McCluskey SP, Plisson C, Rabiner EA, Howes O, Eur. J. Nucl. Med. Mol. Imaging 2019, 47, 451–489; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matthews PM, Rabiner EA, Passchier J, Gunn RN, Br. J. Clin. Pharmacol. 2011, 73, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Ametamey SM, Honer M, Schubiger PA, Chem. Rev. 2008, 108, 1501–1516; [DOI] [PubMed] [Google Scholar]; b) Pike VW, Curr. Med. Chem. 2016, 23, 1818–1869; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lu S, Simeon FB, Telu S, Cai L, Pike VW, The Chemistry of labelling heterocycles with carbon-11 and fluorine-18 for biomedical imaging, Advances in Heterocyclic Chemistry, volume 132, Scriven EFV, Ramsden CA (eds), Academic Press, Cambridge, 2020, p 241–384. [Google Scholar]
- [6].a) Rotstein BH, Liang SH, Placzek MS, Hooker JM, Gee AD, Dolle F, Wilson AA, Vasdev N, Chem. Soc. Rev. 2016, 45, 4708–4726; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Taddei C, Pike VW, EJNMMI Radiopharm. Chem. 2019, 4, 25; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Eriksson J, Antoni G, Långstrom B, Itsenko O, Nucl. Med. Biol. 2020, 92, 115–137. [DOI] [PubMed] [Google Scholar]
- [7].Ghosh AK, Brindisi M, J. Med. Chem. 2020, 63, 2751–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Roeda D, Dollé F, Curr. Top. Med. Chem. 2010, 10, 1680–1700; [DOI] [PubMed] [Google Scholar]; b) Fukumura T, Mori W, Ogawa M, Fujinaga M, Zhang MR, Nucl. Med. Biol. 2021, 92, 138–148. [DOI] [PubMed] [Google Scholar]
- [9].a) Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N, Bioorg. Med. Chem. Lett. 2012, 22, 2099–2101; [DOI] [PubMed] [Google Scholar]; b) Hicks JW, Parkes J, Tong J, Houle S, Vasdev N, Wilson AA, Nucl. Med. Biol. 2014, 41, 688–694; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wilson AA, Garcia A, Houle S, Sadovski O, Vasdev N, Chem. Eur. J. 2011, 17, 259–264; [DOI] [PubMed] [Google Scholar]; (d) Dheere AKH, Bongarzone S, Taddei C, Yan R, Gee AD, Synlett 2015, 26, 2257–2260; [Google Scholar]; (e) Dheere AKH, Yusuf N, Gee A, Chem. Commun. 2013, 49, 8193–8195. [DOI] [PubMed] [Google Scholar]
- [10].a) Kealey S, Husbands SM, Bennacef I, Gee AD, Passchier J, Labelled Compd J. Radiopharm. 2014, 57, 202–208; [DOI] [PubMed] [Google Scholar]; b) Roslin S, Brandt P, Nordeman P, Larhed M, Odell LR, Eriksson J, Molecules 2017, 22, 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brinkman GA, Hasslisewska I, Veenboer JT, Lindner L, Int. J. Appl. Radiat. Isot. 1978, 29, 701–702. [Google Scholar]
- [12].a) Landais P, Crouzel C, Int. J. Radiat. Appl. Instrum. Part A 1987, 38, 297–300; [Google Scholar]; b) Bramoulle Y, Roeda D, Dollé F, Tetrahedron Lett. 2010, 51, 313–316; [Google Scholar]; c) Ogawa M, Takada Y, Suzuki H, Nemoto K, Fukumura T, Nucl. Med. Biol. 2010, 37, 73–76. [DOI] [PubMed] [Google Scholar]
- [13].a) Brown GD, Henderson D, Steel C, Luthra S, Price PM, Brady F, Nucl. Med. Biol. 2001, 28, 991–998; [DOI] [PubMed] [Google Scholar]; b) Conway T, Diksic M, J. Nucl. Med. 1988, 29, 1957–1960; [PubMed] [Google Scholar]; c) Dollé F, Martarello L, Bramoulle Y, Bottlaender M, Gee AD, Labelled Compd J. Radiopharm. 2005, 48, 501–513. [Google Scholar]
- [14].Babad H, Zeiler AG, Chem. Rev. 1973, 73, 75–91. [Google Scholar]
- [15].Lemoucheux L, Rouden J, Ibazizene M, Sobrio F, Lasne MC, J. Org. Chem. 2003, 68, 7289–7297. [DOI] [PubMed] [Google Scholar]
- [16].Jakobsson JE, Lu S, Telu S, Pike VW, Angew. Chem. Int. Ed. 2020, 59, 7256–7260; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7323–7327. [Google Scholar]
- [17].Mane M, Balaskar R, Gavade S, Pabrekar P, Mane D, Arab. J. Chem. 2013, 6, 423–427. [Google Scholar]
- [18].Blanco JLJ, Barría CS, Benito JM, Mellet CO, Fuentes J, Santoyo-González F, Fernández JMG, Synthesis 1999, 1907–1914. [Google Scholar]
- [19].Perveen S, Hai SMA, Khan RA, Khan KM, Afza N, Sarfaraz TB, Synth. Commun. 2005, 35, 1663–1674. [Google Scholar]
- [20].a) Zhang N, Zhou XM, Quan HD, Sekiya A, J. Fluorine Chem. 2015, 178, 208–213; [Google Scholar]; b) Quan HD, Zhang N, Zhou XM, Qian H, Sekiya A, J. Fluorine Chem. 2015, 176, 26–30. [Google Scholar]
- [21].Fawcett FS, Coffman DD, Tullock CW, J. Am. Chem. Soc. 1962, 84, 4275–4285. [Google Scholar]
- [22].Liu F, Li F, Ma A, Dobrovetsky E, Dong A, Gao C, Korboukh I, Liu J, Smil D, Brown PJ, Frye SV, Arrowsmith CH, Schapira M, Vedadi M, Jin J, J. Med. Chem. 2013, 56, 2110–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fersht AR, Jencks WP, J. Am. Chem. Soc. 1970, 92, 5432–5422. [Google Scholar]
- [24].a) Du Y, Minn I, Foss C, Lesniak WG, Hu F, Dannals RF, Pomper MG, Horti AG, EJNMMI Res. 2020, 10, 67; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD, Bioorg. Med. Chem. Lett. 2006, 16, 5212–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.