

Abstract
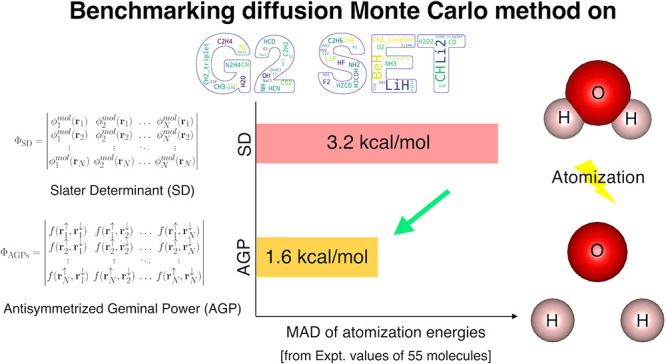
Herein, we report accurate atomization energy calculations for 55 molecules in the Gaussian-2 (G2) set using lattice regularized diffusion Monte Carlo (LRDMC). We compare the Jastrow–Slater determinant ansatz with a more flexible JsAGPs (Jastrow correlated antisymmetrized geminal power with singlet correlation) ansatz. AGPs is built from pairing functions, which explicitly include pairwise correlations among electrons, and hence, this ansatz is expected to be more efficient in recovering the correlation energy. The AGPs wave functions are first optimized at the variational Monte Carlo (VMC) level, which includes both the Jastrow factor and the nodal surface optimization. This is followed by the LRDMC projection of the ansatz. Remarkably, for many molecules, the LRDMC atomization energies obtained using the JsAGPs ansatz reach chemical accuracy (∼1 kcal/mol), and for most other molecules, the atomization energies are accurate within ∼5 kcal/mol. We obtained a mean absolute deviation of 1.6 kcal/mol with JsAGPs and 3.2 kcal/mol with JDFT (Jastrow factor + Slater determinant with DFT orbitals) ansatzes. This work shows the effectiveness of the flexible AGPs ansatz for atomization energy calculations and electronic structure simulations in general.
1. Introduction
Ab initio quantum Monte Carlo (QMC) techniques,1 such as variational Monte Carlo (VMC) and diffusion Monte Carlo (DMC), have been used to compute accurate many-body wave functions (WF) for atomic, molecular, simple crystal, and even complex electronic systems.2−4 Benchmark results are of great interest for QMC methods because these methods have the capability to match the accuracy of quantum chemical methods and also scale more favorably than several wave function based methods.1 A WF-based quantum chemical method, such as the coupled-cluster with single and double and perturbative triple excitations (CCSD(T)), scales as O(N7), where N is the number of electrons.5,6 In contrast, DMC at most scales as O(N4) and is closer to the computational scaling of O(N3) (for N < 1000–2000)7 of the more traditional mean-field methods like DFT. Since there is a large prefactor involved in the DMC method, the WF-based method is often more favorable for smaller systems than DMC. However, as the system size increases, the scaling becomes the dominating factor in their computational costs. It limits the application of the conventional CCSD(T) to systems of up to ∼80–90 electrons,8 while recent implementations extend this limit further, e.g., up to ∼300 electrons for molecules9 and ∼100 electrons for solids.10 On the other hand, for QMC methods, system sizes up to ∼2000 electrons are accessible, which makes them invaluable for condensed matter calculations.7 Thus, making QMC methods more accurate and efficient using various approaches (like using the more flexible WF ansatz) is an active area of research.
A widely used set of benchmark data is the atomization energies of the Gaussian-2 (G2) set of molecules.11 It contains 55 small molecules composed of elements from the first, second, and third rows of the periodic table. Accurate experimental values for the atomization energies of the G2 set of molecules are available, which makes it very attractive for benchmarking. Another way to benchmark is to compare the computed total energies with the estimated exact energies. However, to evaluate the error cancellation, it is important to benchmark the atomization energies.
The G2 set benchmarks have been used to test several state-of-the-art ab initio computational methods. Chemical accuracy (deviation of 1 kcal/mol from experimental estimates of atomization energies) has often been used as the target accuracy. The benchmarking relative to molecular atomization energies of DFT methods in previous studies resulted in a relatively large mean absolute deviation (MAD) value of ∼40 kcal/mol for local density approximation (LDA) and ∼2.5 kcal/mol for the hybrid B3LYP functional.12 In another study, B3LYP on an extended G2 set gave an MAD of 3.11 kcal/mol,13 with a maximum deviation of ∼20 kcal/mol. This large deviation suggests that DFT methods are not systematically accurate, and there might be significant variation in the accuracy across various systems. Efforts have been made to improve the DFT estimates by determining correction factors from fitting to experimental data;14 however, these reduce the prediction ability of the theory. Coupled cluster theory based methods such as CCSD(T) have been largely regarded as the “gold standard” for accuracy in quantum chemistry. Several CCSD(T) studies have shown that the method can achieve subchemical accuracy if large enough basis sets and multiple corrections are used.5,15−18
QMC methods lie on the sweet spot of accuracy and computational cost. They are far more accurate than mean-field methods (no inherent approximation like the XC functional) and have a more favorable scaling than the WF-based quantum chemical methods. CCSD(T)/aug-cc-pVQZ gave an MAD of 2.8 kcal/mol for the G2 set atomization energies. When extrapolated to the complete basis set limit, the MAD value was reduced to 1.3 kcal/mol.19 Several FN-DMC benchmark tests (using single SD ansatz) for the G2 set atomization energies have obtained MAD values close to 3 kcal/mol. For instance, Nemec et al.20 used all-electron FN DMC on Slater determinant (SD) WF to obtain the atomization energies of the G2 set to an MAD of 3.2 kcal/mol. Similar accuracy in atomization energies was also obtained previously in DMC pseudopotential calculations.21 However, these simple DMC approaches (i.e., Jastrow + SD) have not been able to achieve chemical accuracy or subchemical accuracy due to the residual FN errors.
In this paper, we present FN DMC benchmark results using the so-called AGPs (antisymmetrized geminal power with singlet correlation) ansatz(22) along with a Jastrow factor (JF). Combined with VMC optimization, the more flexible AGPs ansatz leads to improved nodal surfaces. The main outcome of this work is that the combination of a more flexible ansatz (AGPs) and nodal surface optimization leads to a much better quality many-body WF, which in turn leads to better DMC energies. This is very important because, AGPs (even though being multiconfigurational in nature4) in practice is as efficient as a single determinant ansatz and thus can be extended to much larger systems, even within the computationally demanding QMC methods. QMC also provides an added advantage of the near ideal parallel scaling of QMC algorithms.4
2. Computational Details
The TURBORVB(4) QMC package was used for all calculations. It employs resonating valence bond (RVB23) WF and allows one to choose a more flexible ansatz than the SD ansatz and includes correlation effects beyond the standard SD.
2.1. Wave Functions
The choice of the WF ansatz plays an important role in determining the accuracy and the computational cost of QMC calculations. A many-body WF ansatz can be written as the following product
| 1 |
where exp J is the JF, and ΦAS is the antisymmetric part that satisfies the antisymmetry condition for fermions. Generally, a single SD is used for the antisymmetric part in QMC calculations. SD is simply an antisymmetrized product of single particle electron orbitals and does not include any electron correlation by itself.
2.1.1. Jastrow Factor
The Jastrow factor is a multiplier term which improves the quality of a many-body WF by providing a significant portion (≈70%) of correlation energy and is necessary for fulfilling Kato’s cusp conditions.24 The JF used here comprises three terms: one-body, two-body, and three/four-body terms (J = J1 + J2 + J3/4). The one-body term is necessary to satisfy the electron–ion cusp condition. A separate one-body term is used for each element present in the molecule. It consists of the so-called homogeneous part
| 2 |
and the so-called inhomogeneous part
| 3 |
where ri denotes the electron coordinates, Ra represents the atomic positions, Za denotes the corresponding atomic numbers, Nat is the number of nuclei, χa,l represents a Gaussian-type atomic orbital l centered over atom a, and Ma,l denotes the corresponding variational parameters. The function ua is defined as
| 4 |
where bea is a variational parameter that depends on each nucleus a. The homogeneous one-body term was carefully optimized at the DFT level before final optimization at the VMC level. The two-body term is necessary to satisfy the electron–electron cusp condition and is optimized at the VMC level
| 5 |
The function v has the following form
| 6 |
where ri,j = |ri – rj|, and bee is a single variational parameter. The three/four-body Jastrow term is defined as
| 7 |
where M{a,l},{b,m} represents the variational parameters, and l and m indicate orbitals centered on atomic sites a and b, respectively. In the current work, the four-body Jastrow term (i.e., a ≠ b) was not used.
2.1.2. Antisymmetrized Geminal Power (AGP)
One way to improve the description of electron correlations and move beyond the SD is to explicitly include pairwise correlation among electrons. This fairly general and flexible ansatz is called the Pfaffian WF.25 The Pfaffian WF is constructed from pairing functions (known as geminals). When only the singlet electron pairing terms are considered, we get the AGPs ansatz.22 A generic pairing function for the AGPs, f(ri, rj), can be written as
| 8 |
For a simpler unpolarized case, where the number of electrons N is even and N↑ = N↓, all possible combinations of singlet pairs can be written in the form of a matrix:
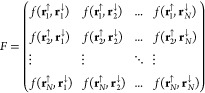 |
9 |
The determinant of matrix F gives the AGPs WF:
| 10 |
In the case of an open-shell chemical system (N↑ ≠ N↓), matrix F consisting of singlet terms only would become rectangular matrix. To convert matrix F into a square matrix, additional unpaired molecular orbitals (Θi(r)) are added to matrix F. Suppose N↑ > N↓, N↑ – N↓ unpaired spin-up molecular orbitals are added to matrix F:
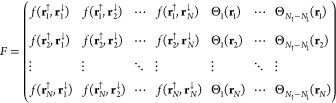 |
11 |
det F now gives the antisymmetric WF.
The singlet electron pairing terms are represented by
| 12 |
where ϕa and ϕb represent atomic orbitals centered on atoms a and b, respectively, and the indices l and m indicate different orbitals centered on atoms a and b, respectively. The elements of matrix λ are the coefficients or the variational parameters of the WF. An important advantage of the AGPs ansatz is that it is equivalent to a linear combination of SDs (or multideterminants) while maintaining the computational cost of a single determinant ansatz.4 Hence, this flexible ansatz (greater variational freedom) could be an effective way to improve the quality of the many-body WF.
2.2. Computational Workflow
The equilibrium geometries of the G2 set molecules were taken from previous benchmark studies13,17,26 (see Table S2 in the Supporting Information). The pairing function was expanded using the triple-ζ atomic basis sets obtained from the Basis Set Exchange library.27 Larger exponents greater than 8Z2 (where Z is the atomic number) were removed from the basis set to avoid numerical instabilities. The large exponent orbitals cut from the basis set are implicitly included by utilizing the one-body Jastrow term.28,29 The basis sets used for the determinant and Jastrow expansion are listed in Table. 1. The same basis sets were used for the VMC and LRDMC calculations.
Table 1. Basis set orbitals used for the determinant and Jastrow expansion.
| element | det. basis | jas. basis |
|---|---|---|
| H | 4s, 2p, 1d | 4s, 2p |
| Li | 8s, 5p, 2d, 1f | 8s, 5p, 2d |
| C | 8s, 5p, 2d, 1f | 8s, 5p, 2d |
| N | 8s, 5p, 2d, 1f | 8s, 5p, 2d |
| O | 7s, 5p, 2d, 1f | 7s, 5p, 2d |
| F | 7s, 5p, 2d, 1f | 7s, 5p, 2d |
| Na | 11s, 10p, 2d, 1f | 11s, 10p, 2d |
| Si | 11s, 9p, 2d, 1f | 11s, 9p, 2d |
| P | 11s, 9p, 2d, 1f | 11s, 9p, 2d |
| S | 11s, 9p, 2d, 1f | 11s, 9p, 2d |
| Cl | 11s, 9p, 2d, 1f | 11s, 9p, 2d |
The trial WF for the Jastrow single
determinant ansatz was obtained from DFT calculations
using the TURBORVB DFT
module. To improve the efficiency of the DFT calculations, we utilized
the double-grid DFT algorithm, which used a finer DFT mesh when in
the vicinity of the nuclei.29 For the JDFT
(JF + SD with DFT orbitals) ansatz, the JF was optimized
at the VMC level, which was followed by the LRDMC projection. In LRDMC,
instead of the conventional time discretization30 of the continuous Hamiltonian, the regularization of the
original Hamiltonian is done over the lattice with a step size a, such that 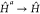 for a → 0.31−33 Further details on the
VMC and LRDMC algorithms can be found in
refs (34−38). The target error bar for the DMC and VMC energies was taken as
≈0.3 mHa. For the VMC optimization, we used the linear39−41 and stochastic reconfiguration methods.42,43 The JDFT WF ansatz was then converted into JsAGPs.
No information loss occurs during this conversion because we are rewriting
the SD ansatz into a more flexible AGPs ansatz, and maximum overlap between the two WFs is ensured. The JsAGPs
was then optimized at the VMC level, including both JF and nodal surface
optimization. This was followed by the LRDMC projection and extrapolation
to zero lattice space. The complexity of VMC optimization can be roughly
estimated by the number of variational parameters to be optimized.
For instance, the number of variational parameters used for a simple
system like BeH were 306 and 735 for JDFT and JsAGPs ansatzes, respectively. For a complex system like Si2H6, the number of variational parameters optimized were 872 and 6422
for JDFT and JsAGPs ansatzes, respectively. The variational
energy, E[α], as well as the maximum value
of the signal-to-noise ratio for forces (termed as devmax) was monitored
for a → 0.31−33 Further details on the
VMC and LRDMC algorithms can be found in
refs (34−38). The target error bar for the DMC and VMC energies was taken as
≈0.3 mHa. For the VMC optimization, we used the linear39−41 and stochastic reconfiguration methods.42,43 The JDFT WF ansatz was then converted into JsAGPs.
No information loss occurs during this conversion because we are rewriting
the SD ansatz into a more flexible AGPs ansatz, and maximum overlap between the two WFs is ensured. The JsAGPs
was then optimized at the VMC level, including both JF and nodal surface
optimization. This was followed by the LRDMC projection and extrapolation
to zero lattice space. The complexity of VMC optimization can be roughly
estimated by the number of variational parameters to be optimized.
For instance, the number of variational parameters used for a simple
system like BeH were 306 and 735 for JDFT and JsAGPs ansatzes, respectively. For a complex system like Si2H6, the number of variational parameters optimized were 872 and 6422
for JDFT and JsAGPs ansatzes, respectively. The variational
energy, E[α], as well as the maximum value
of the signal-to-noise ratio for forces (termed as devmax) was monitored
| 13 |
where fk and 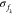 denote force and the corresponding error
bar, respectively. It has been observed that when devmax stabilizes
to values < ∼4, an energy minimum with repect to the variational
parameters is being approached.4 The same
criteria was used (in addition to checking a general convergence of
energy and one-/two-body Jastrow parameters) in general to decide
when to stop VMC optimization. For instance, for Si2H6 VMC optimization was carried out for 4000 optimization steps
using the linear method. For most of the molecules considered, the
linear method was used for VMC optimization. For SiH3,
C2H2, CH2 (triplet), and CH3, the stochastic method was used, requiring ∼15000–20000
optimization steps.
denote force and the corresponding error
bar, respectively. It has been observed that when devmax stabilizes
to values < ∼4, an energy minimum with repect to the variational
parameters is being approached.4 The same
criteria was used (in addition to checking a general convergence of
energy and one-/two-body Jastrow parameters) in general to decide
when to stop VMC optimization. For instance, for Si2H6 VMC optimization was carried out for 4000 optimization steps
using the linear method. For most of the molecules considered, the
linear method was used for VMC optimization. For SiH3,
C2H2, CH2 (triplet), and CH3, the stochastic method was used, requiring ∼15000–20000
optimization steps.
The majority of the calculations was performed on the supercomputer Fugaku using 2304 CPU cores distributed across 48 nodes. To improve the efficiency of the calculations, we used TURBOGENIUS, a python-based wrapper for TURBORVB, which is useful in performing high-throughput calculations.4
3. Results
3.1. JsAGPs for the N2 Molecule
To validate the methodology and verify whether the basis sets used were good enough to approach chemical accuracy, the energies of the N2 molecule and N atom were compared with previous benchmark tests and experimental values (see Table 2).
Table 2. Nitrogen Energies.
| method | basis set | atom (Ha) | molecule (Ha) | atomization energy (eV) | |
|---|---|---|---|---|---|
| JDFT-VMC | cc-pVTZ | –54.5543(2) | –109.4522(3) | 9.35(1) | |
| JDFT-DMC | cc-pVTZ | –54.5765(3) | –109.5068(3) | 9.61(2) | |
| JsAGPs-VMC | cc-pVTZ | –54.5614(1) | –109.4702(5) | 9.45(1) | |
| current work (TURBORVB) | JsAGPs-DMC | cc-pVTZ | –54.5785(4) | –109.5165(3) | 9.92(1) |
| JHF-DMCa | QZ4P | –54.5765(2) | –109.5065(4) | 9.61(2) | |
| CCSD(T)b | – | – | – | 9.85(1) | |
| Fermi netc | – | –54.58882(6) | –109.5388(1) | 9.828(5) | |
| JSD-DMC pseudopotentiald | cc-pV5Z | – | – | 9.573(4) | |
| estimated exacte | – | –54.5892 | –109.5427 | 9.91 | |
| previous reports | experimentalf | – | – | – | 9.91 |
The JsAGPs-DMC atomization energy shows excellent agreement with the experimental value.47 It is better than the value computed using CCSD(T)17 and the value computed recently using a neural network based ansatz (called the Fermi net) .44 The correlation energies recovered for the N2 molecule at the JsAGPs-DMC level and the JDFT-DMC level were ≈95% and ≈93%, respectively. At the VMC level, for JsAGPs, ≈86% percent correlation energy was recovered. The computed JDFT-DMC energies are in excellent agreement with the ones computed by Nemec et al.20 Clearly, in the case of nitrogen, a triple-ζ basis set for orbital and Jastrow expansion was good enough. The best total (closest to the estimated exact) energies are the ones computed by Pfau et al. using Fermi net.44 The JsAGPs atomization energy, however, is more accurate than that obtained by Pfau et al. This shows that the JsAGPs ansatz allows remarkable cancellation of errors when the difference between the molecular and atomic energies is computed. Interestingly, the N2 atomization energy computed using the Jastrow Slater determinant (JSD) ansatz by Petruzielo et al.45 could not approach CCSD(T) level accuracy. Hence, for the JSD ansatz, Jastrow and nodal surface optimizations are not sufficient for improving the quality of the WF ansatz. Thus, the standard JSD might be inadequate for this purpose.
3.2. Application to the G2 Set
Our JDFT-DMC atomization energies (Figure 1) were first compared with the ones obtained by Nemec et al.20 (Jastrow Hartee Fock (JHF)-DMC, QZ4P STO basis set) and Petruzielo et al.45 (JSD-DMC, 5z basis set). The results obtained for the JDFT-DMC atomization energies are in good agreement with the ones obtained by Nemec et al. Most atomization energies obtained using the JDFT ansatz were within a deviation of ±0.25 eV (±3.0 kcal/mol) from the experimental values although very few were in the chemical accuracy range. The JDFT atomization energies had an MAD of ≈3.2 kcal/mol, which is quite close to the value of ≈3.13 kcal/mol reported by Nemec et al. An MAD of 2.9 kcal/mol was reported in another FN DMC (atomic cores treated with pseudopotentials) G2 set benchmark by Grossman.21 This overall agreement with previous benchmark tests points out that FN DMC provides “near chemical accuracy”, and the primary sources of error are the fixed (unoptimized) nodes. There could be other sources of error, such as the basis set used for orbital expansion. However, it can be ruled out based on the fact that Nemec et al. used a larger basis set (Qz) than that used in the current study. Nonetheless, the errors in the atomization energies are quite similar. Comparison of the JDFT atomization energies with the JSD atomization energies (obtained by Petruzielo et al.) shows that optimizing the nodal surfaces improves the DMC atomization energy estimates over the ones obtained using DFT or mean-field nodal surfaces.
Figure 1.
Deviation of the DMC atomization energies from the experimentally obtained values for the JDFT ansatz with the triple-ζ basis set. Zero point energies and relativistic + spin orbit were corrected before computing the deviations between the DMC and experimental values.20 Values obtained by Nemec et al.20 (JHF-QZ4P) and Petruzielo et al.45 (JSD-5z) are also plotted for comparison. The MAD for the JDFT ansatz is ≈3.2 kcal/mol, and the MAD values obtained by Nemec et al. and Petruzielo et al. are 3.13 and 2.1 kcal/mol, respectively.
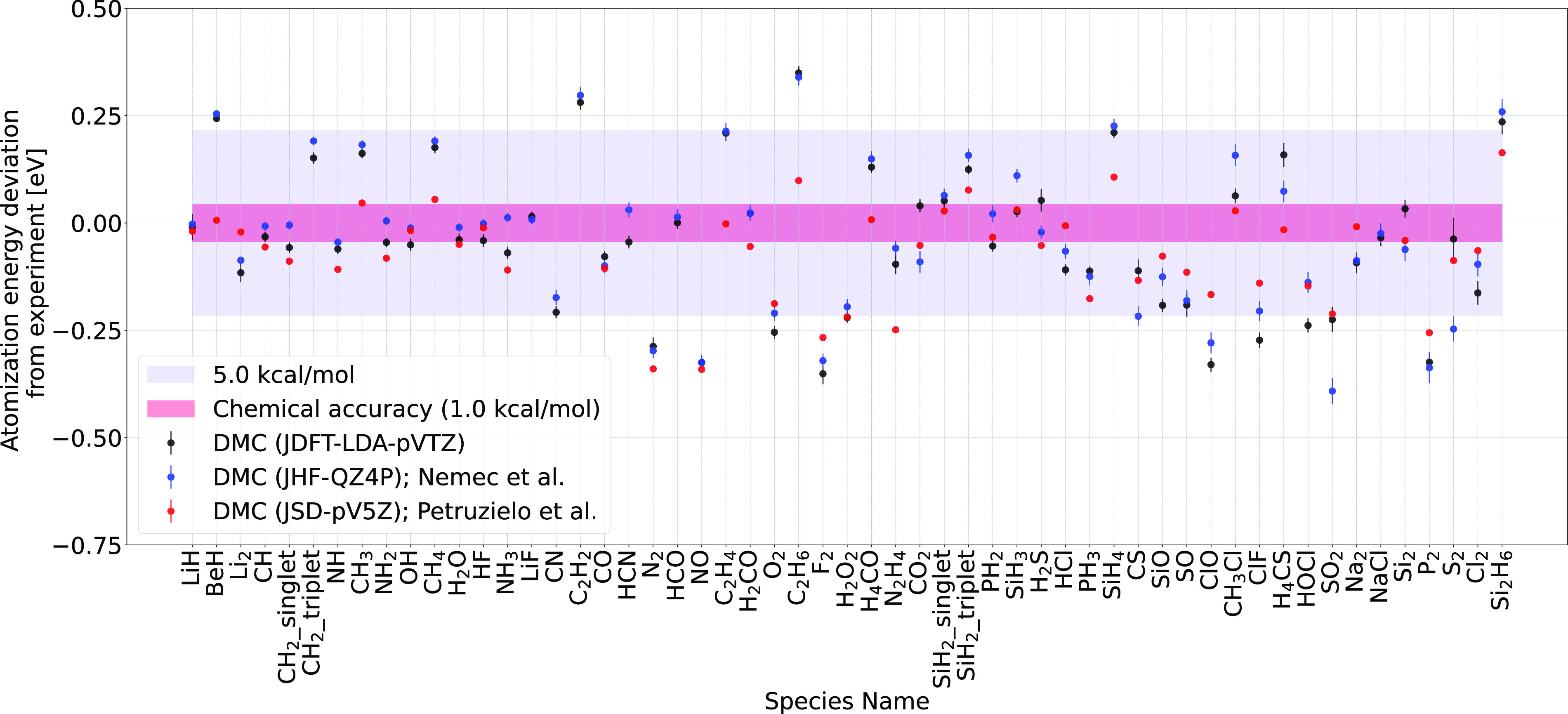
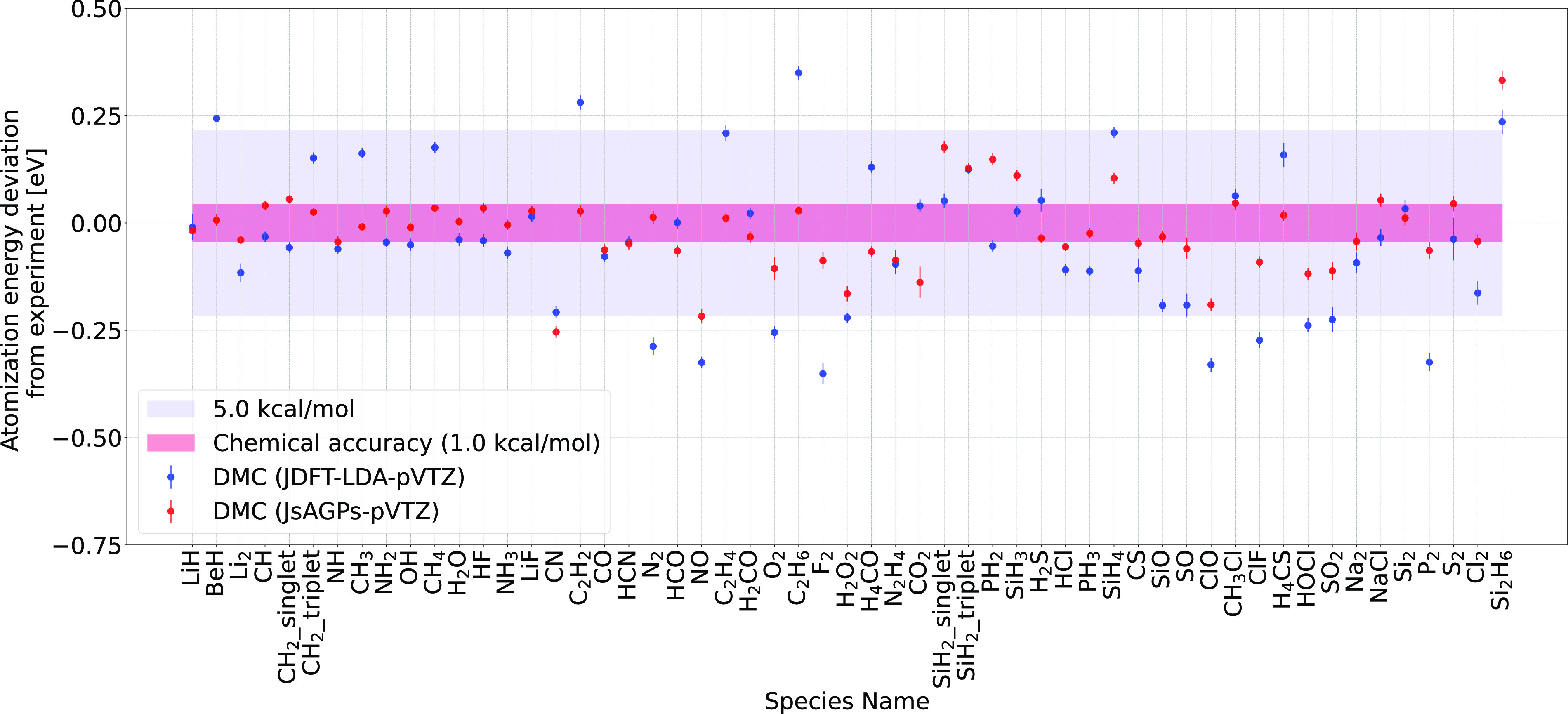
AGPs, which is a more flexible ansatz, allows for better nodal surfaces. Figure 2 shows a comparison of the DMC atomization energy between the JDFT and the more flexible JsAGPs ansatz. Total energies are shown in Figure 3 and Figure 4. Clearly, the best variational energies were obtained when LRDMC was applied to the JsAGPs ansatz both for the atoms and the molecules. This means that the nodal surfaces of JsAGPs WF are better than those of JDFT, and hence, considerably more correlation energy is recovered. For example, in the case of atoms, an average of 95.6% correlation energy was recovered at the DMC level for the JsAGPs WF, and 93.7% correlation energy was recovered using the JDFT. In the case of atomization energies, JsAGPs is clearly better than JDFT. The MAD using the JsAGPs and JDFT ansatzes was 1.6 and 3.2 kcal/mol, respectively, demonstrating a clear superiority of the JsAGPs. For almost all the molecules, the error was within 5.0 kcal/mol, and chemical accuracy was achieved for 26 molecules in the G2 set. These results are not only better than the ones obtained using JDFT (with nodal surface from DFT) but also better than the case wherein the SD nodal surfaces were optimized by Petruzielo et al.45 The better atomization energies indicate that JsAGPs not only provides better variational energies but also improves error cancellation.
Figure 2.
Deviation of the DMC atomization energies from the experimentally obtained values for the JDFT and JsAGPs ansatzes. The error bars are shown within the markers. The MAD from the experiment for the JDFT and JsAGPs ansatzes is ≈3.2 kcal/mol and ≈1.6 kcal/mol, respectively. Zero point energies and relativistic + spin orbit were corrected before computing the deviations between the DMC and experimental values.20
Figure 3.
Deviation of the VMC and DMC total energies from the estimated exact energies. Values obtained by Nemec et al.20 are plotted for comparison.
Figure 4.
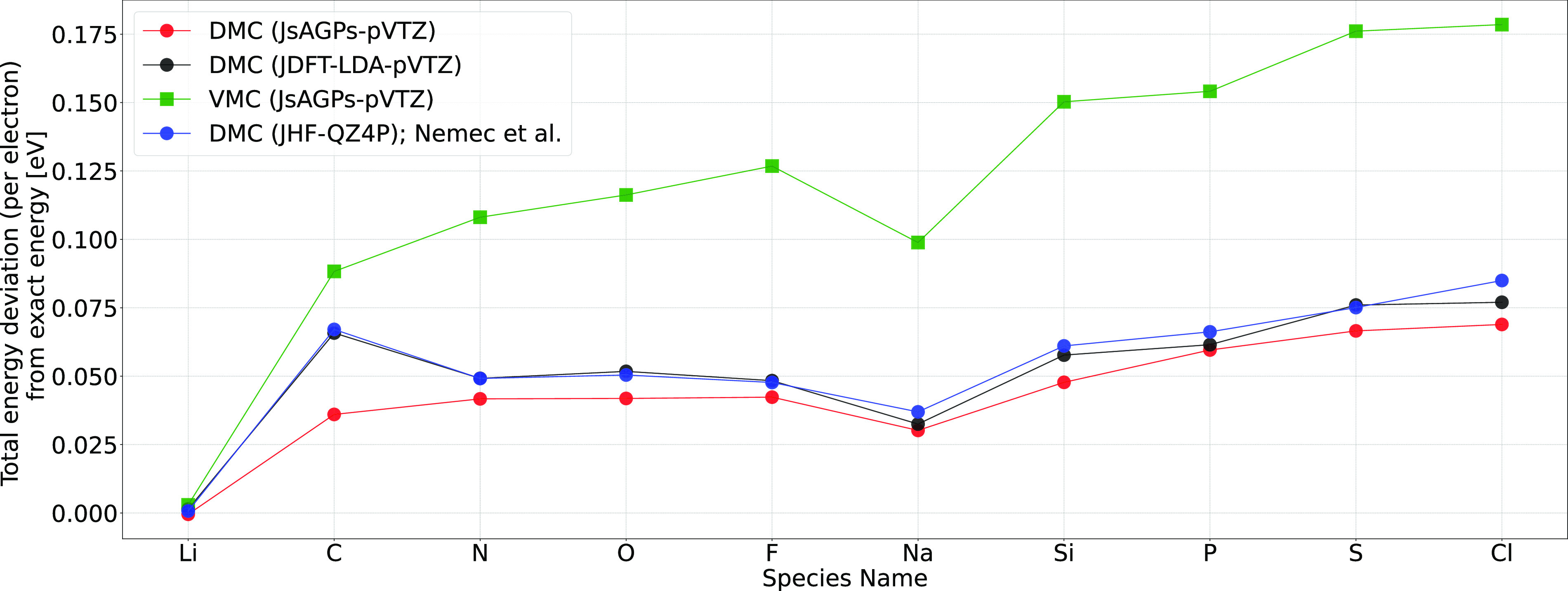
Deviation of the VMC and DMC total energies of atoms from the estimated exact energy. Values obtained by Nemec et al.20 are plotted for comparison.
4. Discussion
It is interesting to note that for a few molecules (e.g., Si2H6, CO2), the JsAGPs atomization energies are worse than their JDFT counterparts. It turns out that although AGPs leads to better nodal surfaces (and lower DMC energies) than JDFT for all molecules and atoms, the quality of nodal surface depends upon the chemical structure of molecules and atoms, and hence, the error cancellation is not always predictably better.
In this study, we tried to reduce the FN error by optimizing the nodal surfaces at the variational level with the single-determinant ansatz (JsAGPs). It is important to compare our QMC results with those obtained with the multideterminant ansatz that is promising in terms of accuracy. Petruzielo et al.45 showed that DMC with the multideterminant ansatz constructed from multiconfigurational self-consistent field theory (MCSCF) calculations achieves the MAD of 1.2 kcal/mol. Morales et al.48 were able to achieve subchemical accuracy (MAD of 0.8 kcal/mol) in atomization energies of the G2 set. In another study, chemical accuracy (and almost exact energies) could be achieved for the ionization potentials for several atoms from Li to Mg using full-configuration interaction.49 Yao et al. used the recently developed semistochastic heat-bath configuration interaction method to obtain excellent atomization energies for the G2 set with an MAD of 0.46 kcal/mol.50 Scemama et al.51 have recently developed a way to combine short-range XC functionals (from DFT) with a selected configuration interaction, which led to trial WFs with lower FN energies with compact multideterminant trial wave functions. The least MAD obtained reached 2.06 kcal/mol. Thus, the MADs obtained with the multideterminant approach are compatible with or better than the value obtained using the JsAGPs ansatz in this study.
The advantage of the multideterminant approach is that one can systematically improve the accuracy by increasing the number of SDs, and in theory, it can describe any ground state exactly with a sufficient number of SDs.52 However, the number of SDs that should be considered scales exponentially as the system size increases, which makes the approach computationally demanding for larger systems. While, in recent years, there have been successful efforts to reduce the number of determinants required for accurate calculations by truncating less important determinants,53,54 establishing a technique to apply the multideterminant ansatz to large systems with high accuracy is still an active field of research.51,55 Instead, the single-determinant approach we employed in this study enables the ansatz to have a large variational freedom, while keeping the computational cost lower, which allows one to tackle large molecules.52 It is an advantage of our single-determinant approach from the practical viewpoint. However, it is worth mentioning that a flexible single-determinant ansatz like the AGPs might suffer from its own limitations. For instance, the ansatz cannot be improved systematically unlike in the multideterminant approach. Thus, one should devise an appropriate ansatz from the physics viewpoint. For instance, Claudio et al.52,56 demonstrated that the atomization energy of the carbon dimer, for which spin fluctuations should be considered, is significantly improved by using the Pfaffian ansatz.25 Another limitation is that the optimization of a large number of nonlinear variational parameters in the single determinant ansatz should be handled efficiently. More specifically, due to the large number of variational parameters contained in the single determinant ansatz, the optimization may fall into physically incorrect local minima, or the optimization itself may diverge. To prevent them, in practice, one needs to prepare a good initial estimate and/or reduce the number of variational parameters by applying constraints in a physically meaningful way, but there are no general guidelines for these. Thus, applying QMC methods to large-scale systems beyond the fixed-node obtained by DFT or HF still requires developing new techniques.
5. Conclusions
To conclude, we have demonstrated the effectiveness of the AGPs ansatz (built from electron pairing functions or geminals) in VMC and LRDMC calculations using the TURBORVB QMC package. Using AGPs ansatz with the cc-pVTZ basis set, the LRDMC calculations for atoms recovered 95.6% correlation energy. The atomization energies computed using AGPs had an MAD of ∼1.6 kcal/mol from the experimental values. Chemical accuracy was achieved for several molecules, and the error was within ±5 kcal/mol for almost all the molecules. These results are quite encouraging as they show that allowing more variational freedom by utilizing a more flexible ansatz is a viable path toward more accurate QMC calculations. We believe that our work represents an important step toward showing that a combination of more flexible single determinant ansatz (JsAGPs) and nodal surface optimization can match the accuracy of quantum chemistry calculations, with the added advantages of excellent scaling and lower computational cost.
Acknowledgments
The computations in this work were mainly performed using the supercomputer Fugaku provided by RIKEN through the HPCI System Research Projects (Project IDs: hp210038 and hp220060). The authors are also grateful for the computational resources at the Research Center for Advanced Computing Infrastructure at the Japan Advanced Institute of Science and Technology (JAIST). A.R. is grateful to MEXT Japan for the support through the MEXT scholarship. R.M. is grateful for financial support from MEXT-KAKENHI (22H05146, 21K03400, and 19H04692) and from JSPS Bilateral Joint Projects (JPJSBP120197714). K.H. is grateful for the financial support from MEXT-KAKENHI, Japan (JP19K05029, JP21K03400, JP21H01998, and JP22H02170). K.N. acknowledges financial support from the JSPS Overseas Research Fellowships, from Grant-in-Aid for Early Career Scientists (Grant No. JP21K17752), from Grant-in-Aid for Scientific Research (Grant No. JP21K03400), and from MEXT Leading Initiative for Excellent Young Researchers (Grant No. JPMXS0320220025). This work was partly supported by the European Centre of Excellence in Exascale Computing (TREX-Targeting Real Chemical Accuracy at the Exascale). This project received funding from the European Union’s Horizon 2020 Research and Innovation program under Grant Agreement No. 952165.
Data Availability Statement
TURBORVB is available from the web site [https://turborvb.sissa.it] upon request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.2c01141.
Computed total energies and references for molecular geometries (PDF)
The authors declare no competing financial interest.
Dedication
Finally, we dedicate this paper to the memory of Prof. Sandro Sorella (SISSA), who passed away during this collaboration. We remember him as one of the most influential contributors to the QMC community in the past century, and in particular for deeply inspiring this work with the development of the ab initio QMC code, TURBORVB.
Supplementary Material
References
- Foulkes W.; Mitas L.; Needs R.; Rajagopal G. Quantum Monte Carlo simulations of solids. Rev. Mod. Phys. 2001, 73, 33. 10.1103/RevModPhys.73.33. [DOI] [Google Scholar]
- Needs R.; Towler M.; Drummond N.; Lopez Rios P.; Trail J. Variational and diffusion quantum Monte Carlo calculations with the CASINO code. J. Chem. Phys. 2020, 152, 154106. 10.1063/1.5144288. [DOI] [PubMed] [Google Scholar]
- Kent P. R. C.; Annaberdiyev A.; Benali A.; Bennett M. C.; Landinez Borda E. J.; Doak P.; Hao H.; Jordan K. D.; Krogel J. T.; Kylänpää I.; Lee J.; Luo Y.; Malone F. D.; Melton C. A.; Mitas L.; Morales M. A.; Neuscamman E.; Reboredo F. A.; Rubenstein B.; Saritas K.; Upadhyay S.; Wang G.; Zhang S.; Zhao L. QMCPACK: Advances in the development, efficiency, and application of auxiliary field and real-space variational and diffusion quantum Monte Carlo. J. Chem. Phys. 2020, 152, 174105. 10.1063/5.0004860. [DOI] [PubMed] [Google Scholar]
- Nakano K.; Attaccalite C.; Barborini M.; Capriotti L.; Casula M.; Coccia E.; Dagrada M.; Genovese C.; Luo Y.; Mazzola G.; et al. TurboRVB: A many-body toolkit for ab initio electronic simulations by quantum Monte Carlo. J. Chem. Phys. 2020, 152, 204121. 10.1063/5.0005037. [DOI] [PubMed] [Google Scholar]
- Feller D.; Peterson K. A.; Crawford T. D. Sources of error in electronic structure calculations on small chemical systems. J. Chem. Phys. 2006, 124, 054107. 10.1063/1.2137323. [DOI] [PubMed] [Google Scholar]
- Gyevi-Nagy L.; Kállay M.; Nagy P. R. Accurate Reduced-Cost CCSD(T) Energies: Parallel Implementation, Benchmarks, and Large-Scale Applications. J. Chem. Theory Comput. 2021, 17, 860–878. 10.1021/acs.jctc.0c01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needs R.; Towler M.; Drummond N.; Lopez Rios P.; Trail J. Variational and diffusion quantum Monte Carlo calculations with the CASINO code. J. Chem. Phys. 2020, 152, 154106. 10.1063/1.5144288. [DOI] [PubMed] [Google Scholar]
- Ramabhadran R. O.; Raghavachari K. Extrapolation to the Gold-Standard in Quantum Chemistry: Computationally Efficient and Accurate CCSD (T) Energies for Large Molecules Using an Automated Thermochemical Hierarchy. J. Chem. Theory Comput. 2013, 9, 3986–3994. 10.1021/ct400465q. [DOI] [PubMed] [Google Scholar]
- Gyevi-Nagy L.; Kállay M.; Nagy P. R. Accurate Reduced-Cost CCSD (T) Energies: Parallel Implementation, Benchmarks, and Large-Scale Applications. J. Chem. Theory Comput. 2021, 17, 860–878. 10.1021/acs.jctc.0c01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsoulis T.; Sakong S.; Groß A.; Grüneis A. Reaction energetics of hydrogen on Si (100) surface: A periodic many-electron theory study. J. Chem. Phys. 2018, 149, 244105. 10.1063/1.5055706. [DOI] [PubMed] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Trucks G. W.; Pople J. A. Gaussian-2 theory for molecular energies of first-and second-row compounds. J. Chem. Phys. 1991, 94, 7221–7230. 10.1063/1.460205. [DOI] [Google Scholar]
- Bauschlicher C. W. Jr A comparison of the accuracy of different functionals. Chem. Phys. Lett. 1995, 246, 40–44. 10.1016/0009-2614(95)01089-R. [DOI] [Google Scholar]
- Curtiss L. A.; Raghavachari K.; Redfern P. C.; Pople J. A. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J. Chem. Phys. 1997, 106, 1063–1079. 10.1063/1.473182. [DOI] [Google Scholar]
- Grindy S.; Meredig B.; Kirklin S.; Saal J. E.; Wolverton C. Approaching chemical accuracy with density functional calculations: Diatomic energy corrections. Phys. Rev. B 2013, 87, 075150. 10.1103/PhysRevB.87.075150. [DOI] [Google Scholar]
- Feller D.; Dixon D. A. Extended benchmark studies of coupled cluster theory through triple excitations. J. Chem. Phys. 2001, 115, 3484–3496. 10.1063/1.1388045. [DOI] [Google Scholar]
- Feller D.; Peterson K. A. Probing the limits of accuracy in electronic structure calculations: Is theory capable of results uniformly better than “chemical accuracy”?. J. Chem. Phys. 2007, 126, 114105. 10.1063/1.2464112. [DOI] [PubMed] [Google Scholar]
- Feller D.; Peterson K. A.; Dixon D. A. A survey of factors contributing to accurate theoretical predictions of atomization energies and molecular structures. J. Chem. Phys. 2008, 129, 204105. 10.1063/1.3008061. [DOI] [PubMed] [Google Scholar]
- Haunschild R.; Klopper W. New accurate reference energies for the G2/97 test set. J. Chem. Phys. 2012, 136, 164102. 10.1063/1.4704796. [DOI] [PubMed] [Google Scholar]
- Feller D.; Peterson K. A. An examination of intrinsic errors in electronic structure methods using the Environmental Molecular Sciences Laboratory computational results database and the Gaussian-2 set. J. Chem. Phys. 1998, 108, 154–176. 10.1063/1.475370. [DOI] [Google Scholar]
- Nemec N.; Towler M. D.; Needs R. Benchmark all-electron ab initio quantum Monte Carlo calculations for small molecules. J. Chem. Phys. 2010, 132, 034111. 10.1063/1.3288054. [DOI] [PubMed] [Google Scholar]
- Grossman J. C. Benchmark quantum Monte Carlo calculations. J. Chem. Phys. 2002, 117, 1434–1440. 10.1063/1.1487829. [DOI] [Google Scholar]
- Casula M.; Sorella S. Geminal wave functions with Jastrow correlation: A first application to atoms. J. Chem. Phys. 2003, 119, 6500–6511. 10.1063/1.1604379. [DOI] [Google Scholar]
- Anderson P. W. Resonating valence bonds: A new kind of insulator?. Mater. Res. Bull. 1973, 8, 153–160. 10.1016/0025-5408(73)90167-0. [DOI] [Google Scholar]
- Kato T. On the eigenfunctions of many-particle systems in quantum mechanics. Commun. Pure Appl. Math. 1957, 10, 151–177. 10.1002/cpa.3160100201. [DOI] [Google Scholar]
- Bajdich M.; Mitas L.; Drobný G.; Wagner L. K.; Schmidt K. E. Pfaffian pairing wave functions in electronic-structure quantum Monte Carlo simulations. Phys. Rev. Lett. 2006, 96, 130201. 10.1103/PhysRevLett.96.130201. [DOI] [PubMed] [Google Scholar]
- O’eill D. P.; Gill P. M. Benchmark correlation energies for small molecules. Mol. Phys. 2005, 103, 763–766. 10.1080/00268970512331339323. [DOI] [Google Scholar]
- Pritchard B. P.; Altarawy D.; Didier B.; Gibson T. D.; Windus T. L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. 10.1021/acs.jcim.9b00725. [DOI] [PubMed] [Google Scholar]
- Mazzola G.; Helled R.; Sorella S. Phase diagram of hydrogen and a hydrogen-helium mixture at planetary conditions by Quantum Monte Carlo simulations. Phys. Rev. Lett. 2018, 120, 025701. 10.1103/PhysRevLett.120.025701. [DOI] [PubMed] [Google Scholar]
- Nakano K.; Maezono R.; Sorella S. All-electron quantum Monte Carlo with Jastrow single determinant ansatz: Application to the sodium dimer. J. Chem. Theory Comput. 2019, 15, 4044–4055. 10.1021/acs.jctc.9b00295. [DOI] [PubMed] [Google Scholar]
- Umrigar C. Accelerated metropolis method. Phys. Rev. Lett. 1993, 71, 408. 10.1103/PhysRevLett.71.408. [DOI] [PubMed] [Google Scholar]
- Ten Haaf D. F.; Van Bemmel H. J.; Van Leeuwen J. M.; Van Saarloos W.; Ceperley D. M. Proof for an upper bound in fixed-node Monte Carlo for lattice fermions. Phys. Rev. B 1995, 51, 13039–13045. 10.1103/PhysRevB.51.13039. [DOI] [PubMed] [Google Scholar]
- Calandra Buonaura M.; Sorella S. Numerical study of the two-dimensional Heisenberg model using a Green function Monte Carlo technique with a fixed number of walkers. Phys. Rev. B 1998, 57, 11446–11456. 10.1103/PhysRevB.57.11446. [DOI] [Google Scholar]
- Sorella S.; Capriotti L. Green function Monte Carlo with stochastic reconfiguration: An effective remedy for the sign problem. Phys. Rev. B 2000, 61, 2599–2612. 10.1103/PhysRevB.61.2599. [DOI] [Google Scholar]
- Casula M.; Filippi C.; Sorella S. Diffusion Monte Carlo method with lattice regularization. Phys. Rev. Lett. 2005, 95, 100201. 10.1103/PhysRevLett.95.100201. [DOI] [PubMed] [Google Scholar]
- Casula M.; Sorella S.; Senatore G. Ground state properties of the one-dimensional Coulomb gas using the lattice regularized diffusion Monte Carlo method. Phys. Rev. B 2006, 74, 100201. 10.1103/PhysRevB.74.245427. [DOI] [Google Scholar]
- Casula M.; Moroni S.; Sorella S.; Filippi C. Size-consistent variational approaches to nonlocal pseudopotentials: Standard and lattice regularized diffusion Monte Carlo methods revisited. J. Chem. Phys. 2010, 132, 154113. 10.1063/1.3380831. [DOI] [PubMed] [Google Scholar]
- Becca F.; Sorella S.. Quantum Monte Carlo approaches for correlated systems; Cambridge University Press, 2017. [Google Scholar]
- Nakano K.; Maezono R.; Sorella S. Speeding up ab initio diffusion Monte Carlo simulations by a smart lattice regularization. Phys. Rev. B 2020, 101, 155106. 10.1103/PhysRevB.101.155106. [DOI] [Google Scholar]
- Sorella S. Wave function optimization in the variational monte carlo method. Phys. Rev. B 2005, 71, 241103. 10.1103/PhysRevB.71.241103. [DOI] [Google Scholar]
- Toulouse J.; Umrigar C. J. Optimization of quantum Monte Carlo wave functions by energy minimization. J. Chem. Phys. 2007, 126, 084102. 10.1063/1.2437215. [DOI] [PubMed] [Google Scholar]
- Umrigar C.; Toulouse J.; Filippi C.; Sorella S.; Hennig R. G. Alleviation of the fermion-sign problem by optimization of many-body wave functions. Phys. Rev. Lett. 2007, 98, 110201. 10.1103/PhysRevLett.98.110201. [DOI] [PubMed] [Google Scholar]
- Sorella S. Green function Monte Carlo with stochastic reconfiguration. Phys. Rev. Lett. 1998, 80, 4558. 10.1103/PhysRevLett.80.4558. [DOI] [Google Scholar]
- Sorella S.; Casula M.; Rocca D. Weak binding between two aromatic rings: Feeling the van der Waals attraction by quantum Monte Carlo methods. J. Chem. Phys. 2007, 127, 014105. 10.1063/1.2746035. [DOI] [PubMed] [Google Scholar]
- Pfau D.; Spencer J. S.; Matthews A. G. D. G.; Foulkes W. M. C. Ab initio solution of the many-electron Schrödinger equation with deep neural networks. Phys. Rev. Research 2020, 2, 033429. 10.1103/PhysRevResearch.2.033429. [DOI] [Google Scholar]
- Petruzielo F. R.; Toulouse J.; Umrigar C. Approaching chemical accuracy with quantum Monte Carlo. J. Chem. Phys. 2012, 136, 124116. 10.1063/1.3697846. [DOI] [PubMed] [Google Scholar]
- Bytautas L.; Ruedenberg K. Correlation energy extrapolation by intrinsic scaling. IV. Accurate binding energies of the homonuclear diatomic molecules carbon, nitrogen, oxygen, and fluorine. J. Chem. Phys. 2005, 122, 154110. 10.1063/1.1869493. [DOI] [PubMed] [Google Scholar]
- Feller D.; Peterson K. A. Re-examination of atomization energies for the Gaussian-2 set of molecules. J. Chem. Phys. 1999, 110, 8384–8396. 10.1063/1.478747. [DOI] [Google Scholar]
- Morales M. A.; McMinis J.; Clark B. K.; Kim J.; Scuseria G. E. Multideterminant wave functions in quantum Monte Carlo. J. Chem. Theory Comput. 2012, 8, 2181–2188. 10.1021/ct3003404. [DOI] [PubMed] [Google Scholar]
- Booth G. H.; Alavi A. Approaching chemical accuracy using full configuration-interaction quantum Monte Carlo: A study of ionization potentials. J. Chem. Phys. 2010, 132, 174104. 10.1063/1.3407895. [DOI] [PubMed] [Google Scholar]
- Yao Y.; Giner E.; Li J.; Toulouse J.; Umrigar C. Almost exact energies for the Gaussian-2 set with the semistochastic heat-bath configuration interaction method. J. Chem. Phys. 2020, 153, 124117. 10.1063/5.0018577. [DOI] [PubMed] [Google Scholar]
- Scemama A.; Giner E.; Benali A.; Loos P.-F. Taming the fixed-node error in diffusion Monte Carlo via range separation. J. Chem. Phys. 2020, 153, 174107. 10.1063/5.0026324. [DOI] [PubMed] [Google Scholar]
- Genovese C.; Shirakawa T.; Nakano K.; Sorella S. General Correlated Geminal Ansatz for Electronic Structure Calculations: Exploiting Pfaffians in Place of Determinants. J. Chem. Theory Comput. 2020, 16, 6114–6131. 10.1021/acs.jctc.0c00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giner E.; Scemama A.; Caffarel M. Fixed-node diffusion Monte Carlo potential energy curve of the fluorine molecule F2 using selected configuration interaction trial wavefunctions. J. Chem. Phys. 2015, 142, 044115. 10.1063/1.4905528. [DOI] [PubMed] [Google Scholar]
- Caffarel M.; Applencourt T.; Giner E.; Scemama A. Communication: Toward an improved control of the fixed-node error in quantum Monte Carlo: The case of the water molecule. J. Chem. Phys. 2016, 144, 151103. 10.1063/1.4947093. [DOI] [PubMed] [Google Scholar]
- Benali A.; Gasperich K.; Jordan K. D.; Applencourt T.; Luo Y.; Bennett M. C.; Krogel J. T.; Shulenburger L.; Kent P. R.; Loos P.-F.; et al. Toward a systematic improvement of the fixed-node approximation in diffusion Monte Carlo for solids—A case study in diamond. J. Chem. Phys. 2020, 153, 184111. 10.1063/5.0021036. [DOI] [PubMed] [Google Scholar]
- Genovese C.; Sorella S. The nature of the chemical bond in the dicarbon molecule. J. Chem. Phys. 2020, 153, 164301. 10.1063/5.0023067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TURBORVB is available from the web site [https://turborvb.sissa.it] upon request.