

Summary
Proliferating cells exhibit a metabolic phenotype known as “aerobic glycolysis”, which is characterized by an elevated rate of glucose fermentation to lactate irrespective of oxygen availability. Although several theories have been proposed, a rationalization for why proliferating cells seemingly waste glucose carbon by excreting it as lactate remains elusive. Using the NCI-60 cell lines, we determined that lactate excretion is strongly correlated with the activity of mitochondrial NADH shuttles, but not proliferation. Quantifying the fluxes of the malate-aspartate shuttle (MAS), the glycerol 3-phosphate shuttle (G3PS), and lactate dehydrogenase under various conditions demonstrated that proliferating cells primarily transform glucose to lactate when glycolysis outpaces the mitochondrial NADH shuttles. Increasing mitochondrial NADH shuttle fluxes decreased glucose fermentation but did not reduce the proliferation rate. Our results reveal that glucose fermentation, a hallmark of cancer, is a secondary consequence of MAS and G3PS saturation rather than a unique metabolic driver of cellular proliferation.
Graphical Abstract
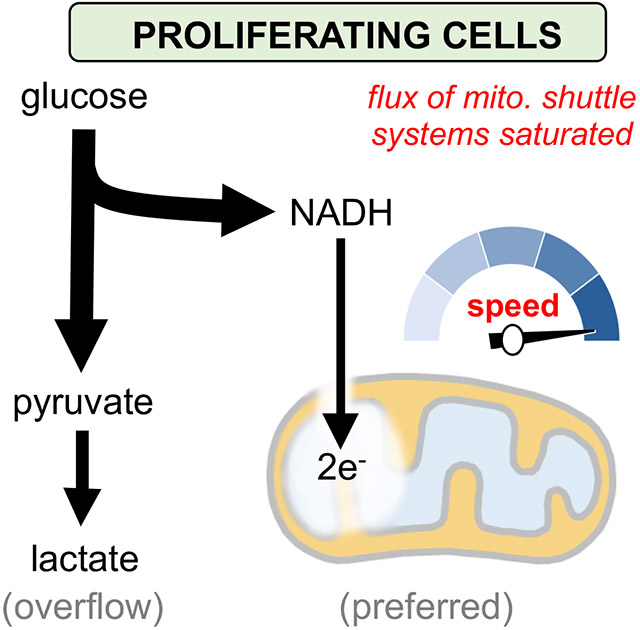
eTOC Blurb
Oxygenated cancer cells excrete much of the glucose they consume as lactate, a seemingly wasteful process that has been challenging to rationalize. Here, Wang et al. demonstrate that proliferating cells prefer to oxidize glucose. Lactate excretion primarily occurs because glycolysis produces NADH faster than shuttles can transport electrons into mitochondria.
Introduction
Aerobic glycolysis, also known as the Warburg effect, is characterized by a high rate of glucose fermentation to lactate irrespective of oxygen availability (Vander Heiden et al., 2009). This metabolic phenotype has long been associated with rapidly dividing cancer cells (Warburg, 1956). More recently, aerobic glycolysis has been identified as a general feature of most proliferating cells including lymphocytes, stem cells, and fibroblasts (Brand et al., 1986; Gerriets and Rathmell, 2012; Hedeskov, 1968). Although it seems counterintuitive that proliferating cells would waste glucose carbon by excreting it as lactate, a number of theories have been proposed to explain why aerobic glycolysis may be a metabolic driver of cellular proliferation. One suggestion is that the rate of ATP production by fermentation may be faster than the rate of ATP production by mitochondrial respiration (Pfeiffer et al., 2001). Another possibility is that aerobic glycolysis allows cells to divert glucose carbon to glycolytic branch points, thereby driving the flux of biosynthetic pathways essential for proliferation, such as one-carbon metabolism and the pentose phosphate pathway (Cairns et al., 2011; Vander Heiden et al., 2009). An alternative consideration is that the limited physical volume of a cell may make it more spatially efficient to produce energy from glycolysis, compared to oxidative phosphorylation (Vazquez et al., 2010). Even though these rationalizations and others are intriguing, they have raised additional questions, and efforts to better understand aerobic glycolysis continue.
In glycolysis, glyceraldehyde 3-phosphate (GAP) is oxidized to 1,3-bisphosphoglycerate by the enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH). This transformation is coupled with the reduction of cytosolic NAD+ to cytosolic NADH. Regenerating cytosolic NAD+ is required to maintain GAPDH activity and continue glycolysis. Although NADH can be oxidized to NAD+ by the electron transport chain, a process that drives ATP production via oxidative phosphorylation, that reaction occurs in mitochondria and neither NAD+ nor NADH can cross the inner mitochondrial membrane. There are therefore three different mechanisms mainly used to oxidize NADH in the cytosol, thereby regenerating NAD+ for GAPDH. The first two mechanisms rely on the cytosolic enzymes malate dehydrogenase 1 (MDH1) and glycerol 3-phosphate dehydrogenase 1 (GPD1) (Hanse et al., 2017; Ou et al., 2006), which are components of the malate-aspartate shuttle (MAS) and glycerol 3-phosphate shuttle (G3PS) respectively. We note that some cells express glycerol 3-phosphate dehydrogenase 1-Like (GPD1L) protein in the cytosol, which has a function redundant with GPD1(Kelly et al., 2011). The third mechanism to oxidize cytosolic NADH is to reduce pyruvate to lactate with lactate dehydrogenase (LDH). The MAS and G3PS are however more energetically efficient ways to regenerate NAD+, compared to LDH, because they allow glycolysis-derived pyruvate to be oxidized in mitochondria. Additionally, the shuttles translocate reducing equivalents produced during glycolysis across the inner mitochondrial membrane where they can then be used to drive ATP production via oxidative phosphorylation. By contrast, LDH regenerates cytosolic NAD+ at the cost of transforming glucose carbon into lactate, which is generally assumed to be excreted from the cell.
The reducing equivalents transferred from cytosolic NADH to mitochondrial NAD+ by the MAS and G3PS are ultimately used to reduce molecular oxygen at complex IV of the electron transport chain. Thus, when oxygen is limiting, the only mechanism for regenerating cytosolic NAD+ is by using LDH to produce lactate. This phenomenon, known as anaerobic glycolysis, results in the accumulation of lactate. In aerobic glycolysis, however, LDH is still used to regenerate cytosolic NAD+, even though oxygen is readily available. The main question we aimed to answer here is why lactate accumulates in oxygenated cells.
Increasing evidence indicates that mitochondria are highly active during proliferation (Ahn and Metallo, 2015; Yao et al., 2019). Furthermore, lactate production in proliferating cells was recently associated with an insufficient ATP demand to regenerate mitochondrial NAD+ (Fernandez-de-Cossio-Diaz and Vazquez, 2017; Luengo et al., 2021). Building on these findings, we sought to evaluate the possibility that mitochondrial oxidation of glucose is maximized when cancer cells are rapidly dividing. We speculated that the rate of NADH production by glycolysis may outpace the maximum rate at which the MAS and/or G3PS can translocate reducing equivalents across the inner mitochondrial membrane in proliferating cells, thereby driving LDH activity in the presence of oxygen. By applying a quantitative method that combines stable isotope labeling and mathematical modeling to a panel of different cancer cell lines, we compared the flux of glycolysis with the maximum flux of MDH1 and GPD1/GPD1L. Our results show that MDH1 and GPD1/GPD1L become saturated during rapid proliferation. Only after MDH1 and GPD1/GPD1L are saturated does lactate accumulation occur, demonstrating that fermentation is not the preferred metabolic phenotype of cancer cells.
Results
High lactate excretion is strongly correlated with MAS flux, but not proliferation, in cancer cells
Most proliferating cancer cells excrete lactate, both in vitro and in vivo. Whether the amount of lactate excreted varies as a function of tumor source or oncogene expression, however, is not well established. We assessed the lactate-excretion rate for the NCI-60 cell lines in culture as Lac/(2 x Glc), where Lac is lactate excreted to the media and Glc is glucose consumed over the same time integral (Zielinski et al., 2017). Interestingly, the NCI-60 cell lines displayed a wide range of lactate-excretion rates that varied by nearly 5-fold (Figure 1A), showing that different cancer cell lines excrete different amounts of glucose carbon as lactate.
Figure 1: Lactate excretion is correlated with malate-aspartate shuttle flux, but not proliferation in cancer cells.

(A) Histogram showing the distribution of lactate-excretion rates across the NCI-60 panel of cell lines.
(B) No significant correlation was observed between lactate-excretion rate and doubling times for cancer cell lines from the NCI-60 panel. A correlation coefficient (r) and a p value as determined by a Pearson correlation are provided.
(C) Complementary cumulative distribution function (CCDF) for Pearson correlations between metabolic fluxes and the lactate-excretion rate for cancer cell lines from the NCI-60 panel. Fluxes highlighted in red correspond to reactions involved in the malate-aspartate shuttle. We note that the glycerol 3-phosphate shuttle was not included in the metabolic model used for flux balance analysis.
(D-I) Plot of malate-aspartate shuttle fluxes as a function of lactate-excretion rate for cancer cell lines from the NCI-60 panel. Correlation coefficients (r) and p values as determined by a Pearson correlation are provided.
Results are from the analysis of the NCI-60 panel of cancer cell lines. Lac, lactate; Glc, glucose.
We next sought to determine whether the lactate-excretion rate is correlated with proliferation rate. For the NCI-60 cell lines, doubling times range from 17.4 to 79.5 hours. Surprisingly, no significant correlation was observed between the lactate-excretion rate and doubling time (r= 0.099, p=0.45) (Figure 1B). This result suggests that the amount of glucose excreted as lactate is not directly linked to cancer cell proliferation.
Given the lack of correlation between the lactate-excretion rate and proliferation, we next searched for metabolic fluxes that may be associated with increased glucose fermentation. From flux balance analysis results (Zielinski et al., 2017), using a model of human metabolism derived from Recon 2, we determined that two metabolic processes were most strongly correlated with the NCI-60 lactate-excretion rate: pyruvate mitochondrial transport (PYRt2m) and all MAS components including MDH1 (cytosolic), AKGMALtm (transport, mitochondrial), ASPTAm (mitochondrial), ASPTA (cytosolic), ASPGLUm (transport, mitochondrial), and MDHm (mitochondrial) (Figures 1C-1I and S1; Table S1). This result raised the possibility that limitations on the transport of glycolytic products into mitochondria drive lactate production. There are multiple well-established mechanisms to import carbon produced during glycolysis into mitochondria. By contrast, for many cells, the MAS is the only major transport system to import reducing equivalents produced during glycolysis into mitochondria, with the G3PS generally being recognized as a minor contributor (Safer et al., 1971). Thus, we focused our attention on the possibility that lactate production is driven by limitations on the transport of reducing equivalents by the NADH shuttles.
MDH1 and GPD1/GPD1L activities regulate lactate production
We were interested in determining whether a change in the activity of the MAS or G3PS is in itself sufficient to alter lactate production in proliferating cancer cells. Given that MDH1 and GPD1/GPD1L are the reactions in the MAS and G3PS systems that regenerate cytosolic NAD+, we focused on modulating the activities of these enzymes in the prostate cancer cell line (PC3) from the NCI-60 panel. First, we overexpressed MDH1 and GPD1/GPD1L in separate experiments (Figure 2A). To confirm that MDH1 and GPD1/GPD1L overexpression resulted in increased enzyme activity in PC3 cells, we performed isotope-tracer analysis with [4-2H] glucose (Hanse et al., 2017). Metabolism of [4-2H] glucose through glycolysis produces [1-2H] GAP. GAPDH then transfers the deuterium label from GAP to NADH. MDH1 and GPD1/GPD1L subsequently transfer the deuterium label from NADH to malate and glycerol 3-phosphate (G3P), producing [2-2H] malate and [2-2H] G3P respectively (Figure S2A). It is also possible for [1-2H] GAP to transfer its deuterium label to dihydroxyacetone phosphate (DHAP) via triose phosphate isomerase, thereby producing [1-2H] DHAP. When GPD1/GPD1L uses deuterated DHAP and deuterated NADH, it yields [1,2-2H] G3P. The fraction of [2-2H] malate normalized to the fraction of [1-2H] GAP can be used to assess the relative activity of MDH1 (i.e., M+1 malate / M+1 GAP; where M+1 represents a molecule with one deuterium). The fraction of [1,2-2H] G3P normalized to the fraction of [1-2H] DHAP and [4-2H] NADH can be used to assess the relative activity of GPD1/GPD1L (i.e., M+2 G3P/ (M+1 NADH × M+1 DHAP); where M+1 and M+2 represent molecules with one and two deuterium labels, respectively). As expected, we found that MDH1 and GPD1/GPD1L overexpression did increase the rate at which cytosolic NAD+ was regenerated by these enzymes (Figure 2B and 2C). Further, we found that MDH1 and GPD1/GPD1L overexpression decreased the lactate-excretion rate (Figure 2D). Despite the decrease in lactate excretion, however, the proliferation rate of PC3 cells did not decrease (Figure 2E). To the contrary, by increasing the activities of MDH1 and GPD1/GPD1L, the proliferation rate of PC3 cells actually increased. Next, to decrease enzyme activity, we knocked down MDH1 and GPD1L by using siRNA (Figure 2F). We then performed an isotope-tracer analysis with [4-2H] glucose to confirm that knockdown decreased MDH1 activity and GPD1/GPD1L activity (Figure 2G and 2H). The data revealed that decreasing MDH1 or GPD1/GPD1L activities leads to an increased lactate-excretion rate (Figure 2I and 2J). Expressing siRNA-resistant MDH1 (MDH1res) or GPD1L (GPD1Lres) in MDH1 or GPD1L knockdown cells restored protein levels and decreased the lactate-excretion rate (Figure S2B-S2C). Notably, although MDH1 and GPD1L knockdown increased the lactate-excretion rate, it decreased the cellular proliferation rate (Figure 2K and 2L, S2D-S2F). Taken together, our results show that a change in MDH1 or GPD1/GPD1L activity is sufficient to alter lactate production and that lactate production is not positively correlated with proliferation rate.
Figure 2: MDH1 activity and GPD1/GPD1L activity regulate lactate-excretion rate.

(A) Western blots for MDH1, GPD1, and GPD1L in PC3 cancer cells. Overexpression increases protein levels. Control cells overexpressed green fluorescent protein (GFP) instead of a dehydrogenase.
(B and C) Overexpressing MDH1 (B), GPD1, and GPD1L (C) in PC3 cancer cells increases MDH1 and GPD1/GPD1L activities. Control cells overexpressed GFP instead of a dehydrogenase. MDH1 activities was determined by the fraction of [2-2H] malate normalized to the fraction of [1-2H] GAP. GPD1/GPD1L activities was determined by the fraction of [1,2-2H] G3P normalized to the fraction of [1-2H] DHAP and [4-2H] NADH. n=3 replicates per group. p values were determined by using a two-tailed Student’s t test.
(D) Lactate-excretion rates decrease when MDH1 or GPD1/GPD1L are overexpressed. n=3 replicates per experimental group. p values were determined by using a one-way analysis of variance (ANOVA) followed by Dunnett’s test.
(E) Proliferation rate of PC3 cells before and after overexpression of MDH1 or GPD1/GPD1L. n=3 replicates per experimental group. p values were determined by using a one-way ANOVA followed by Dunnett’s test.
(F) Western blots for MDH1 and GPD1L in PC3 cells after MDH1 or GPD1L knockdown. GPD1 is not shown because its levels were below detection prior to knockdown.
(G and H) Knocking down MDH1 (G) or GPD1L (H) in PC3 cells decreases MDH1 and GPD1/GPD1L activities. MDH1 and GPD1/GPD1L activities were determined as in (B) and (C).
(I and J) Lactate-excretion rates in PC3 cells are increased after MDH1 (I) or GPD1L (J) knockdown. Control cells were administered scrambled siRNA. n=9 replicates per experimental group for panel (I). n=10 replicates per experimental group for panel (J). p values were determined by using a two-tailed Student’s t test.
(K and L) Proliferation rate of PC3 cells decreases when MDH1(K) and GPD1L (L) is knocked down. n=3 replicates per experimental group. p values were determined by using a two-tailed Student’s t test.
(M) Lactate-excretion rate decreases when PC3 cells are given 0.5 mM AKB. n=3 replicates per experimental group. p values were determined by using a two-tailed Student’s t test.
(N) Proliferation rate increases when PC3 cells are given 0.5 mM AKB. n=3 replicates per experimental group. p values were determined by using a two-tailed Student’s t test.
Error bars denote standard error. *p≤0.05, **p≤0.001, ***p≤0.001, n.s.>0.05 Ctrl, control; OE, overexpression; KD, knockdown; Lac, lactate; Glc, glucose; GAP, glyceraldehyde 3-phosphate; G3P, glycerol 3-phosphate; DHAP, dihydroxyacetone phosphate; AKB, alpha-ketobutyrate.
We speculated that treating PC3 cells with an exogenous electron acceptor capable of regenerating cytosolic NAD+ may lead to the same effects as increased MDH1 and GPD1/GPD1L activities, namely a decreased rate of lactate production and an increased rate of cell proliferation. To test this prediction, we treated cells with the electron acceptor alpha-ketobutyrate (AKB). Reduction of AKB to alpha-hydroxybutyrate (AHB) is coupled with the oxidation of NADH to NAD+ (Sullivan et al., 2015). As expected, treating PC3 cells with AKB decreased lactate excretion and increased cellular proliferation (Figure 2M and 2N), confirming that lactate production is not essential for cancer cells to divide, as long as they have access to other electron acceptors for regenerating cytosolic NAD+.
We then evaluated whether modulating MDH1 or GPD1/GPD1L expression affects the rate at which lactate is excreted from other cell types. We overexpressed MDH1 and GPD1 in two additional cell lines from the NCI-60 panel: human colorectal carcinoma cells (HCT116) and human breast cancer cells (BT549). Overexpression of MDH1 and GPD1 decreased the lactate-excretion rate (Figure S3A-S3F), but it did not lead to a decrease in proliferation rate (Figure S3G and S3H). Moreover, treating HCT116 and BT549 cells with AKB decreased lactate excretion without impairing cellular proliferation (Figure S3I-S3L). These data are consistent with our results from PC3 cells, indicating that the activity of MDH1 and GPD1/GPD1L can alter lactate production independent of proliferation in multiple cell types.
To evaluate the effect of MDH1 overexpression in vivo, we implanted PC3 cells overexpressing MDH1 or control PC3 cells into nude mice and compared the tumors that formed (Figure S3M). In agreement with our in vitro results, we found that overexpression of MDH1 led to increased tumor growth (Figure S3N). To assess whether overexpression of MDH1 affected glucose fermentation to lactate, we provided mice with an intratumoral infusion of [U-13C] glucose. After 1 h, the tumors were harvested and analyzed by mass spectrometry-based metabolomics. We found that the amount of 13C-labeled lactate relative to the amount of 13C-labeled pyruvate was lower in tumors overexpressing MDH1 (Figure S3O). Additionally, the amount of 13C-labeled citrate relative to the amount of 13C-labeled pyruvate was higher in tumors overexpressing MDH1 (Figure S3P). These data are consistent with less fermentation of glucose to lactate and instead more oxidation of glucose by the tricarboxylic acid (TCA) cycle in tumors overexpressing MDH1. These findings are in line with other studies showing that the TCA cycle plays an important role in supporting tumor growth (Hensley et al., 2016; Marin-Valencia et al., 2012)
Saturating MDH1 and GPD1/GPD1L activities drives lactate production
Most cancer cell lines in the NCI-60 panel have more than one oncogenic mutation (Ikediobi et al., 2006). To test whether lactate production in transformed cells with a single oncogene is similarly regulated by MDH1 activity and GPD1/GPD1L activity, we expressed H-Ras (G12V) in 3T3 cells. Upon transformation with H-Ras (Figure 3A), 3T3 cells are no longer sensitive to contact inhibition (Figure 3B). As with PC3 cells, we modulated MDH1 and GPD1/GPD1L activities in H-Ras 3T3 cells by overexpression and siRNA. Using [4-2H] glucose as a tracer, we first confirmed that overexpression increased MDH1 and GPD1/GPD1L activities (Figures 3C, 3D, and S4A, and S4B). We then determined that increased MDH1 or GPD1/GPD1L activities in H-Ras 3T3 cells decreased the lactate-excretion rate without impairing cellular proliferation (Figures 3E and S4C). By contrast, decreased enzyme activity resulting from MDH1 or GPD1L knockdown increased the lactate-excretion rate and had no effect on cellular proliferation (Figures S4D-S4J). Expressing siRNA-resistant MDH1 (MDH1res) or GPD1L (GPD1Lres) in MDH1 or GPD1L knockdown cells restored protein levels and decreased the lactate-excretion rate (Figure S4K-S4N). Additionally, treating H-Ras 3T3 cells with AKB to provide an alternative mechanism to regenerate cytosolic NAD+ decreased lactate excretion without impairing cellular proliferation (Figures S4O and S4P). These results reveal that modulating MDH1 and GPD1/GPD1L activities is sufficient to change lactate production in transformed 3T3 cells harboring the H-Ras oncogene.
Figure 3: Saturating MDH1 and GPD1/GPD1L drives lactate production.

(A) Transfecting H-Ras (G12V) into non-transformed 3T3 cells results in detectable H-Ras (G12V) protein expression.
(B) Non-transformed 3T3 cells are contact inhibited when grown to confluency, but H-Ras 3T3 cells are no longer sensitive to contact inhibition. n=3 replicates per group.
(C and D) Overexpressing MDH1 (C) and GPD1 (D) in H-Ras 3T3 cells increases MDH1 and GPD1/GPD1L activities, respectively. Control cells overexpressed GFP instead of a dehydrogenase. MDH1 activities was determined by the fraction of [2-2H] malate normalized to the fraction of [1-2H] GAP. GPD1/GPD1L activities was determined by the fraction of [1,2-2H] G3P normalized to the fraction of [1-2H] DHAP and [4-2H] NADH. n=3 replicates per group. p values were determined by using a two-tailed Student’s t test.
(E) Lactate-excretion rates decrease when MDH1 or GPD1 is overexpressed in H-Ras 3T3 cells. Control cells overexpressed GFP instead of a dehydrogenase. n=3 replicates per experimental group. p values were determined by using a one-way ANOVA followed by Dunnett’s test.
(F) Relative glycolytic activity in H-Ras 3T3 cells as a function of 2DG concentration. Glycolytic activity was determined by the fraction of M+1 GAP during [4-2H] glucose labeling. n=3 replicates per 2DG concentration.
(G) Relative fluxes of MDH1, GPD1/GPD1L, and LDH as a function of glycolytic activity in H-Ras 3T3 cells. Glycolytic activities were derived from (F). MDH1 activity was determined by the fraction of [2-2H] malate normalized to the fraction of [1-2H] GAP. GPD1/GPD1L activity was determined by the fraction of [1,2-2H] G3P normalized to the fraction of [1-2H] DHAP and [4-2H] NADH. LDH activity was determined by the amount of lactate excreted over time. Data were fit with a logistic function. The R2 value is the coefficient of determination. n = 3 replicates per experimental group.
(H) First derivatives of MDH1, GPD1/GPD1L, and LDH activities in H-Ras 3T3 cells. Calculations were based on the fitted equations shown in (G).
(I) Relative glycolytic activity in non-transformed proliferating 3T3 cells as a function of 2DG concentration. Glycolytic activity was determined by the fraction of M+1 GAP during [4-2H] glucose labeling. n=3 replicates per 2DG concentration.
(J) Relative fluxes of MDH1, GPD1/GPD1L, and LDH as a function of glycolytic activity in non-transformed proliferating 3T3 cells. Glycolytic activities were derived from (I). MDH1 activity was determined by the fraction of [2-2H] malate normalized to the fraction of [1-2H] GAP. GPD1/GPD1L activity was determined by the fraction of [1,2-2H] G3P normalized to the fraction of [1-2H] DHAP and [4-2H] NADH. LDH activity was determined by the amount of lactate excreted over time. Data were fit with a logistic function. The R2 value is the coefficient of determination. n=3 replicates per experimental group.
Error bars denote standard error. *p≤0.05, **p≤0.001, ***p≤0.001, n.s.>0.05.
Ctrl, control; OE, overexpression; KD, knockdown; Lac, lactate; Glc, glucose; GAP, glyceraldehyde 3-phosphate; G3P, glycerol 3-phosphate; DHAP, dihydroxyacetone phosphate.
Given that MDH1 and GPD1/GPD1L provide more energetically efficient mechanisms for regenerating cytosolic NAD+, compared with a reduction of pyruvate to lactate, we wished to consider why proliferating cells rely heavily upon LDH. We hypothesized that proliferating cells prefer to use MDH1 and GPD1/GPD1L to regenerate cytosolic NAD+. We surmised that when glycolysis outpaces maximal MDH1 and GPD1/GPD1L activities, however, LDH becomes critical to replenish cytosolic NAD+. To test this hypothesis, we aimed to manipulate glycolytic flux and monitor accompanying changes in MDH1, GPD1/GPD1L, and LDH activities. We expected cells to predominantly use MDH1 and GPD1/GPD1L to regenerate cytosolic NAD+ at low glycolytic fluxes and then to increase LDH activity as MDH1 and GPD1/GPD1L became saturated at higher rates of glycolysis.
To tune glycolytic flux, we treated H-Ras 3T3 cells with different concentrations of the glycolytic inhibitor 2-deoxyglucose (2DG). 2DG is a glucose analog that is taken up by glucose transporters and phosphorylated by hexokinase to yield 2-deoxyglucose 6-phosphate (2DG 6-P). Unlike glucose 6-phosphate, 2DG 6-P cannot be further metabolized by the glycolytic enzyme phosphoglucose isomerase. Administration of 2DG therefore leads to the accumulation of 2DG 6-P within the cell, which then suppresses glycolytic activity by competitively inhibiting hexokinase. The larger the concentration of 2DG administered, the more that 2DG 6-P accumulates and the greater the inhibition of glycolysis. To quantitate the inhibitory effect of 2DG on glycolytic activity, we cultured cells with [4-2H] glucose and different concentrations of 2DG. We then measured M+1 GAP, a glycolytic intermediate containing one deuterium, by liquid chromatography/mass spectrometry (LC/MS). As expected, the labeling percentages of GAP gradually decreased as the concentration of 2DG increased (Figure 3F). When H-Ras 3T3 cells were treated with 1 mM 2DG, glycolytic activity was decreased by ~20%. Further increasing the concentration of 2DG up to 20 mM resulted in a ~75% decrease in glycolytic activity. To assess the relative activities of MDH1 and GPD1/GPD1L at low and high glycolytic fluxes, we applied the method described above for PC3 cells, using [4-2H] glucose as a tracer. To assess the relative activities of LDH at low and high glycolytic fluxes, we measured the rate of lactate excretion. As glycolytic flux was increased by administering lower concentrations of 2DG, we determined that MDH1 and GPD1/GPD1L activities gradually increased. When glycolytic flux was above 60% of its unattenuated value, we found that MDH1 and GPD1/GPD1L activities reached a plateau (Figure 3G), indicating that they had been saturated. As glycolytic flux continued to increase, however, LDH activity then greatly increased without reaching a plateau. The relationship between MDH1, GPD1/GPD1L, and LDH activities is reflected by the first derivative of the changes in relative flux (Figure 3H). As glycolytic activity increases to its maximum value, the changes in MDH1 and GPD1/GPD1L fluxes approach zero because enzyme activity is saturated. LDH flux, on the other hand, accelerates at a much faster rate as glycolysis approaches its maximum activity. We also note that increasing the oxygen consumption rate with a mitochondrial uncoupler did not significantly increase MDH1 or GPD1/GPD1L activities beyond their saturation values (Figure S5). These data indicate that H-Ras 3T3 cells prefer to use the MAS and G3PS to regenerate cytosolic NAD+ until glycolysis outpaces the maximum flux of MDH1 and GPD1/GPD1L, at which point LDH becomes the primary mechanism of NADH oxidation.
MDH1 and GPD1/GPD1L are also saturated in non-transformed proliferating cells
Although aerobic glycolysis was originally observed in tumors, non-transformed proliferating cells exhibit a similar metabolic phenotype that is characterized by elevated glucose consumption and lactate excretion (Gerriets and Rathmell, 2012; Medzhitov, 2015; Yao et al., 2016). To determine whether saturation of MDH1 and GPD1/GPD1L also drives lactate production in these non-transformed proliferating cells, we repeated the experiments above where we measured the activities of MDH1, GPD1/GPD1L, and LDH as a function of glycolytic flux in non-transformed proliferating 3T3 cells. Similar to H-Ras transformed cells, MDH1 and GPD1/GPD1L activities increased with increasing glycolytic flux initially (Figures 3I and 3J). At ~60% of the unattenuated flux of glycolysis, however, both MDH1 and GPD1/GPD1L became saturated. Further increases in glycolysis were supported by using increased LDH activity to regenerate cytosolic NAD+. These data suggest that the MAS and G3PS are not saturated due to oncogenic transformation, but rather as a result of cellular proliferation.
Increased lactate dehydrogenase A (LDHA) expression supports elevated lactate production when MDH1 and GPD1/GPD1L are saturated
We next sought to understand how LDH is activated upon saturation of MDH1 and GPD1/GPD1L. Given that transcriptional regulation is known to be an important mechanism to control LDHA activity (Valvona et al., 2016), we performed qPCR to examine enzyme expression levels in H-Ras 3T3 cells and non-transformed proliferating 3T3 cells (Figure 4). When glycolytic flux was above 60% of its unattenuated value, we observed only minimal changes in MDH1, GPD1, and GPD1L. The data are consistent with MDH1 and GPD1/GPD1L activities being saturated during these experiments. The expression level of LDHA, on the other hand, was significantly increased with higher glycolytic fluxes. These results suggest that LDH activity is at least partially activated upon saturation of MDH1 and GPD1/GPD1L by increasing LDHA expression in both transformed and non-transformed proliferating cell lines.
Figure 4: Increased LDHA expression supports elevated lactate production when MDH1 and GPD1/GPD1L are saturated.

Levels of MDH1 (A and E), GPD1 (B and F), and GPD1L (C and G) mRNA are minimally changed when glycolytic flux is above 60% of its unattenuated value. By contrast, the expression level of LDHA (D and H) is significantly increased over the same conditions. n=6 replicates per experimental group in panel (A)-(D). n=3 replicates per experimental group in panel (E)-(H). p values were determined by using a one-way ANOVA followed by Dunnett’s test. Data are shown for H-Ras transformed 3T3 cells (A-D) and non-transformed proliferating 3T3 cells (E-H).
Error bars denote standard error. *p≤0.05, ****p≤0.0001, n.s.>0.05.
Absolute quantitation of NAD+ regeneration fluxes
We wished to determine the percentage of glycolysis-derived NADH that is recycled to NAD+ by each dehydrogenase reaction as a function of increasing glycolytic flux. Accordingly, we developed an approach to quantitate MDH1, GPD1/GPD1L, and LDH fluxes by measuring the labeling kinetics of malate, G3P, and lactate in cells cultured with [4-2H] glucose. The basis of our approach is the model depicted in Figure 5A, which illustrates the fates of deuterium label derived from [4-2H] glucose. After being taken up by cells, [4-2H] glucose produces [1-2H] GAP. GAPDH transfers the deuterium from GAP to NADH with a flux that we designate as f1. The deuterium label is then transferred to malate, G3P, or lactate with fluxes that we designated as f3, f4, and f6, respectively. The fluxes of reactions that do not contribute deuterium labels to the pools of malate, G3P, lactate, and NADH are designated as f2, f5, f7 and f8, respectively. The consumption fluxes for malate, G3P, lactate, and NADH are designated as f9, f10, f11, and f12, respectively. We use [GAP], [NADH], [Malate], [G3P], and [Lactate] to represent the intracellular concentrations of each metabolite. Equations 1, 2, 3, and 4 represent a complete model of the labeling dynamics. Additional details are provided in the STAR Methods. While we only use this model to investigate aerobic glycolysis in proliferating cells here, we note that it is broadly applicable to the study of dehydrogenase fluxes in any biological system.
Figure 5: Absolute quantitation of NAD+ regeneration fluxes.

(A) Schematic of our model used to calculate fluxes from [4-2H] glucose labeling.
(B) Fraction of labeled GAP in H-Ras 3T3 cells as a function of time after introducing [4-2H] glucose. n=3 replicates per time point.
(C) Fraction of labeled M+1 G3P, labeled M+2 G3P, labeled M+1 malate, and labeled M+1 lactate in H-Ras 3T3 cells as a function of time after introducing [4-2H] glucose. n=3 replicates per time point.
(D) Fraction of unlabeled malate, G3P, and lactate in H-Ras 3T3 cells as a function of time after introducing [4-2H] glucose. The measured metabolite concentrations, the rate of glucose consumption, the rate of lactate excretion, and Equations 1, 2, 3, and 4 were used to find the best model fit from which the activities of MDH1, GPD1/GPD1L, and LDH were determined. n=3 replicates per experimental group.
(E) Proportion of glycolysis-derived NADH oxidized by MDH1, GPD1/GPD1L, and LDH in H-Ras 3T3 cells as a function of glycolytic activity. Calculations were based on the fitted curve shown in Figure S6B.
(F) Proportion of glycolysis-derived NADH oxidized by MDH1, GPD1/GPD1L, and LDH in multiple cell lines. Actual flux values of MDH1, GPD1/GPD1L, and LDH were determined by the measured metabolite concentrations, the rate of glucose consumption, the rate of lactate excretion, and Equations 1, 2, 3, and 4.
Error bars denote standard error.
GAP, glyceraldehyde 3-phosphate; G3P, glycerol 3-phosphate; DHAP, dihydroxyacetone phosphate.
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
We first applied our approach to H-Ras 3T3 cells labeled with [4-2H] glucose. We point out that, compared with H-Ras 3T3 cells cultured with [U-13C6] glucose, deuterium labeling of GAP and other downstream metabolites is diluted by triose phosphate isomerase activity (Figures 5B, 5C, and S6A). After transferring H-Ras 3T3 cells to a medium containing [4-2H] glucose, the fraction of unlabeled malate, G3P, lactate, and NADH exponentially decreased until an isotopic steady state was reached. Based on these dynamic labeling data, metabolite concentrations, and observed rates of glucose consumption and lactate excretion, we calculated f3 (MDHl), f4 (GPD1/GPD1L), and f6 (LDH) in H-Ras 3T3 cells to be 2.23, 0.03, and 18.23 fmol/cell/min, respectively (Figure 5D; Tables S2 and S3). Consistent with the results shown in Figures 3G and 3H, when glycolytic activity was not attenuated by 2DG, LDH flux was considerably higher than MDH1 and GPD1/GPD1L. Under these conditions, LDH oxidized 89% of the total NADH that was produced by glycolysis, whereas MDH1 and GPD1/GPD1L only oxidized 11%.
We then repeated the same set of experiments in H-Ras 3T3 cells, but we administered different concentrations of 2DG to quantitate changes at lower glycolytic activities (Figures 5E, S6B, and S6C; Tables S2 and S3). Under low glycolytic activity, quantitative analysis revealed that MDH1 and GPD1/GPD1L oxidize ~60% of the total NADH produced by glycolysis, whereas LDH only oxidizes 40%. The results are consistent with cells preferentially regenerating NAD+ by using MDH1 and GPD1/GPD1L when glycolytic activity is low. When glycolytic activity is elevated, however, the fluxes of MDH1 and GPD1/GPD1L become saturated, and cells increase LDH to regenerate cytosolic NAD+ faster so that glycolysis is not impaired.
The saturation flux of MDH1 and GPD1/GPD1L is cell-type dependent
We aimed to determine whether the amount of glycolysis-derived NADH oxidized by MDH1 and GPD1/GPD1L in other proliferating cells is comparable to H-Ras 3T3 cells, where we determined it to be just 11%. We therefore assessed the flux of MDH1, GPD1/GPD1L, and LDH in PC3 cells, BT549 cells, non-transformed proliferating 3T3 cells, and transformed rat astrocytes (DITNC1). To quantitate each flux, we cultured cells with [4-2H] glucose and then evaluated the fraction of 2H-labeled malate, G3P, and lactate. We calculated the fluxes of MDH1, GPD1/GPD1L, and LDH on the basis of 2H-labeling, metabolite concentrations, and rates of glucose consumption and lactate excretion without administration of 2DG, when glycolysis was unimpaired. Interestingly, the proportion of glycolysis-derived NADH that was oxidized by MDH1 and GPD1/GPD1L varied across cell lines (Figure 5F). The amount of glycolysis-derived NADH that was oxidized by MDH1 and GPD1/GPD1L in 3T3, DITNC1, and BT549 cells was relatively low, ranging from 11-42%. In PC3 cells, on the other hand, 75% of the NADH produced by glycolysis was oxidized by MDH1 and GPD1/GPD1L. A comparison of protein levels between PC3 and BT549 cells revealed changes in GPD1L expression (Figure S6D).
MDH1 is not saturated in non-proliferating cells
To evaluate the activity of the NADH shuttles in non-proliferating cells, we exploited the contact-inhibition property of 3T3 cells. When 3T3 cells reach 100% confluency in culture, they transition from a proliferative state to a quiescent state (Green and Kehinde, 1975), thereby allowing direct comparison. As expected, based on prior work (Yao et al., 2019), proliferating 3T3 cells consume more glucose and excrete more lactate than quiescent 3T3 cells (Figure 6A). Accordingly, the lactate-excretion rate is nearly 2-fold higher during proliferation, compared with the quiescent state (Figure 6B). We have already established that MDH1 and GPD1/GPD1L are saturated in proliferating 3T3 cells (Figure 3J). To evaluate whether MDH1 and GPD1/GPD1L are also saturated in quiescent 3T3 cells, we manipulated glycolytic flux with 2DG and calculated MDH1, GPD1/GPD1L, and LDH fluxes by culturing cells in [4-2H] glucose (Figures 6C and 6D). We found that the flux of GPD1/GPD1L remained at low values over all experimental conditions tested, indicating that the G3PS does not play an important role in quiescent 3T3 cells. The flux of MDH1 and LDH, however, increased as glycolysis increased, without reaching a plateau. Contrary to proliferating cells, these results demonstrate that MDH1 is not saturated in non-proliferating cells.
Figure 6: MDH1 is not saturated in non-proliferating cells.

(A) Comparison of the rate of glucose consumption and lactate excretion between proliferating and quiescent 3T3 cells. n=3 replicates for each group. Error bars denote standard error. p values were determined by using a two-tailed Student’s t test. ***p≤0.001.
(B) Comparison of lactate-excretion rates between proliferating and quiescent 3T3 cells. n=3 replicates per group. Error bars denote standard error. p values were determined by using a two-tailed Student’s t test. ***p≤0.001.
(C) Relative glycolytic activity in quiescent cells as a function of 2DG concentration. Glycolytic activity was determined by the fraction of M+1 GAP during [4-2H] glucose labeling. n=3 replicates per 2DG concentration. Error bars denote standard error.
(D) Fluxes of MDH1, GPD1/GPD1L, and LDH as a function of glycolytic activity in quiescent cells. Glycolytic activities were derived from (C). Flux values of MDH1, GPD1/GPD1L, and LDH were determined by the measured metabolite concentrations, the rate of glucose consumption, the rate of lactate excretion, and Equations 1, 2, 3, and 4. Data were fit with a logistic function. Error bars show the interquartile range of the Monte Carlo simulated fluxes.
Lac, lactate; Glc, glucose.
Discussion
Cancer cells reprogram their metabolism to support the demands of rapid proliferation. A well-established metabolic adaptation is increased consumption of glucose, much of which is excreted as lactate even in the presence of oxygen. This phenomenon, referred to as aerobic glycolysis, is considered to be a hallmark of cancer (Hanahan and Weinberg, 2011). Nearly a hundred years after first being described by Otto Warburg, there continues to be a lack of clarity about why proliferating cells engage in aerobic glycolysis.
The perceived relationship between lactate production and the metabolic state of mitochondria in cancer cells has evolved over the last century. Warburg originally postulated that cancer cells activate glycolysis because they have defective mitochondria, thereby limiting their capacity for respiration (Warburg, 1956). We now know that the mitochondria of most cancers are not only functional but also active. Indeed, isotope-tracing experiments have confirmed that glucose is oxidized by the TCA cycle of both transformed and non-transformed proliferating cells in culture as well as human tumors in patients (Fan et al., 2009; Johnston et al., 2021; Yao et al., 2019). Beyond providing energy, recent evidence further demonstrates that mitochondrial metabolism plays a key role in synthesizing building blocks that are essential to cell proliferation (Ahn and Metallo, 2015; Birsoy et al., 2015; Sullivan et al., 2015; Zong et al., 2016).
Despite an increasing appreciation for the importance of mitochondria in cancer, questions remain about the relative activity of mitochondrial metabolism as compared with that of glycolysis. Proliferating cells are often described as having “switched” to glycolysis, with the implication that fermentation of glucose is preferred over oxidation of glucose in the TCA cycle. Here, building upon prior work establishing that the flux of mitochondrial pathways is elevated during proliferation (Ahn and Metallo, 2015; Yao et al., 2019), we explored the alternative possibility that oxidation of glucose carbon by the TCA cycle is preferred over fermentation. In this model, the accumulation of lactate occurs as a compensatory mechanism when the rate of cytosolic NADH generation exceeds the maximum rate at which NADH can be oxidized in mitochondria.
Our model is supported by an analysis of the NCI-60 panel of human cancer cell lines. The data reveal that the amount of glucose excreted as lactate in cancer cells is not correlated with the rate of proliferation but rather strongly correlated with the activity of the MAS, which is recognized as a main shuttle used to transport glycolytic reducing equivalents from the cytosol to the mitochondrial matrix (Borst, 2020). To evaluate whether lactate production in proliferating cells is a secondary consequence of saturating the activities of the MAS and G3PS, we tuned glycolytic flux in proliferating cells by administering different concentrations of 2DG and then quantitated the activities of the dehydrogenase reactions that oxidize cytosolic NADH, namely LDH, MDH1, and GPD1/GPD1L. At low glycolytic fluxes, we found that most of the NADH produced by GAPDH in proliferating cells was oxidized by the MDH1 and GPD1/GPD1L reactions of the MAS and G3PS, respectively. Only after glycolysis increased and the rate of cytosolic NADH oxidation by electron shuttles reached a maximum did LDH become highly activated. Moreover, increasing the capacity of MDH1 or GPD1/GPD1L by overexpression decreased lactate production without decreasing the rate of cell proliferation. In some cases, it even increased the speed of proliferation. Taken together, these findings suggest that fermentation occurs in proliferating cells under oxygenated conditions because glycolysis outpaces electron transport into mitochondria.
The notion that lactate production occurs in the presence of oxygen due to saturation of the MAS and G3PS has physiological precedent. During periods of exercise, when adequate oxygen is available to metabolize pyruvate in mitochondria, the rate of NADH produced by glycolysis in human skeletal muscle can exceed the ability of the shuttle systems to transport reducing equivalents into mitochondria and result in the generation of lactate (Phypers and Pierce, 2006; Spriet et al., 2000). A similar phenomenon occurs in heart tissue where, despite sufficient myocardial blood flow and oxygenation, lactate is produced at elevated workloads as a consequence of limited MAS activity (Kobayashi and Neely, 1979; O'Donnell et al., 2004). Lactate is also produced in oxygenated brain tissue, particularly during periods of neural activity when glycolysis increases and mitochondrial oxidation of cytosolic NADH can be limited by MAS activity (Contreras and Satrústegui, 2009; Fellows et al., 1993; Fox et al., 1988). Our results indicate that the role of glucose fermentation in proliferating cancer cells is analogous to that in healthy tissues during normal physiology. In all cases, glucose fermentation is a secondary mechanism to replenish cytosolic NADH when the mitochondrial shuttle systems are insufficient.
The proposed explanation for aerobic glycolysis is attractive because it does not require proliferating cells to favor a metabolic program that is inefficient in terms of energy production and carbon utilization, which has historically been difficult to rationalize (Liberti and Locasale, 2016). To the contrary, our data indicate that both transformed and non-transformed proliferating cells prefer to oxidize glucose carbon in mitochondria. An interesting question is why proliferating cells increase glycolytic flux beyond the capacity of the mitochondrial shuttle systems, thereby causing LDH to become activated. In healthy tissues such as muscle and brain, fermentation is thought to provide a complementary source of energy beyond what can be produced from the maximum rate of oxidizing glucose in mitochondria (Spriet et al., 2000). Proliferating cells may employ a similar mechanism. Another possibility is that high rates of glucose metabolism in proliferating cells promote anabolic reactions branching from glycolysis to drive production of biomass, as has been suggested previously (Vander Heiden et al., 2009). Additionally, we note that phosphoinositide 3-kinase signaling is highly integrated with glycolysis (Xu et al., 2021). Thus, high glycolytic activity in cancer cells may also provide a mechanism to control growth signaling networks.
Currently, our understanding of the processes that regulate the MAS and G3PS is incomplete. Data from this study and others support that the flux of MDH1 and GPD1/GPD1L is at least partially affected by protein levels (Kozak, 1985; Oh et al., 2016; Ratner et al., 1981), but additional mechanisms for mediating shuttle activity have been implicated. Prior work has suggested that the MAS is regulated by expression of the aspartate-glutamate antiporter (Alkan et al., 2018; Rabinovich et al., 2020), calcium levels (Contreras et al., 2007), and post-translational modifications (Wang et al., 2018; Yang et al., 2015). The activity of the MAS and G3PS may also be controlled by the rate of ATP production in mitochondria. Without ATP synthesis by complex V, coupled mitochondria cannot use the electron transport chain to regenerate NAD+, and the function of the NADH shuttle systems will be compromised. It is particularly compelling to consider this connection between NADH shuttle activity and ATP synthesis in light of recent work demonstrating that insufficient turnover of ATP limits mitochondrial NAD+ regeneration in proliferating cells (Luengo et al., 2021).
Limitations of study
To understand the relationship between dehydrogenase fluxes during aerobic glycolysis, we developed a quantitative model for rapidly proliferating cancer cells. Our model is based on labeling from [4-2H] glucose, metabolite pool sizes, glucose consumption, and lactate excretion. Although such measurements are readily accomplished for cells in culture, extension to an in vivo system is complicated by several challenges such as assessing metabolite consumption and excretion fluxes from cancer cells in a tumor. Here, we validated a key conclusion from our study, namely that increasing mitochondrial shuttle activity decreases glucose fermentation, by implanting genetically engineered PC3 cancer cells into mice. Future work, however, would benefit from more quantitative analyses in a larger number of in vivo systems, including human tumors. Of particular interest is whether the microenvironment of a tumor influences the activity of the MAS or G3PS. In this study, we demonstrated that specific cell lines activate the MAS and G3PS to different extents under standard cell-culture conditions. Further investigation is needed to better understand whether nutrient availability may affect each shuttle’s activity.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Gary J Patti (gjpattij@wustl.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Original Western Blot images have been deposited in Mendeley and are publicly available as of the date of publication. The link is listed in the key resources table.
All original code has been deposited in Github and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze data reported in this study is available from the lead contact upon reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MDHC (H-6) | Santa Cruz Biotechnology | Cat# sc-166879, RRID:AB_10609257 |
| GPD1 Polyclonal antibody | proteintech | Cat# 27943-1-AP, RRID:AB_2881016 |
| GPD1L Polyclonal antibody | proteintech | Cat# 17263-1-AP, RRID:AB_2112359 |
| β-tubulin antibody (HRP conjugated) | Cell signaling | Cat# 5346 RRID:AB_1950376 |
| Goat anti-rabbit secondary antibodies (HRP conjugated) | LI-COR | Cat# 926-80011 RRID:AB_2721264 |
| Anti-mouse secondary antibody (m-IgGκ BP-HRP) | Santa Cruz Biotechnology | Cat# sc-516102, RRID:AB_2687626 |
| Rabbit anti-Ras (G12V), monoclonal | Cell Signaling | Cat.#: 14412, RRID:AB_2714031 |
| Chemicals, peptides, and recombinant proteins | ||
| Sodium 2-oxobutyrate (AKB) | Sigma | K0875 |
| Sodium pyruvate | Sigma | P5280 |
| Critical commercial assays | ||
| BCA protein assay | Thermo Fisher | Cat.#: 23225 |
| SuperScript III First-Strand Synthesis SuperMix | Invitrogen | Cat#18080400 |
| Maxima SYBR Green/ROX qPCR Master Mix (2X) | Thermo Scientific | K0221 |
| NAD/NADH Quantitation Colorimetric Kit | Biovision | Cat#K337 |
| Deposited Data | ||
| Raw image data | This manuscript | DOI: 10.17632/btc9bbcch8.1 |
| Original code | This manuscript | https://github.com/e-stan/NADH_KFP |
| Experimental models: Cell lines | ||
| 3T3 | American Type Culture Collection | RRID:CVCL_0123 |
| PC-3 | American Type Culture Collection | RRID:CVCL_0035 |
| DITNC1 | American Type Culture Collection | |
| BT549 | American Type Culture Collection | RRID:CVCL_1092 |
| HCT 116-Luc2 | American Type Culture Collection | Cat# CCL-247-LUC2, RRID:CVCL_VU38 |
| Experimental Models: Organisms/Strains | ||
| Mouse: 43-56 days old female nude mice | Charles River Laboratories | 490CRATHHO |
| Oligonucleotides | ||
| Negative Control DsiRNA | IDT | Cat.#: 51-01-14-04 |
| DsiRNA: GPD1L (human) | IDT TriFECTa DsiRNA Kit | hs.Ri.GPD1L.13 |
| DsiRNA: MDH1(human) | IDT TriFECTa DsiRNA Kit | hs.Ri.MDH1.13 |
| DsiRNA: GPD1L(mouse) | IDT TriFECTa DsiRNA Kit | mm.Ri.Gpd1l.13 |
| DsiRNA: MDH1(mouse) | IDT TriFECTa DsiRNA Kit | mm.Ri.Mdh1.13 |
| MDH1 primer 1: Sequence: GGT TAC ACC GAG TTT AAG AGC A | IDT PrimeTime® qPCR Primers | Mm.PT.58.5174962 |
| MDH1 primer 2: Sequence: CCA ATA CGA ACT GCC TGA CA | IDT PrimeTime® qPCR Primers | Mm.PT.58.5174962 |
| GPD1 primer 1: Sequence: TGT CAC CGA AGC CAA GC | IDT PrimeTime® qPCR Primers | Mm.PT.56a.32463908 |
| GPD1 primer 2: Sequence: ACC CAA CTT TCG CAT CAC T | IDT PrimeTime® qPCR Primers | Mm.PT.56a.32463908 |
| GPD1L primer 1: Sequence: TCT TGT GGA TGA ACT GGT GAG | IDT PrimeTime® qPCR Primers | Mm.PT.58.7971254 |
| GPD1L primer 2: Sequence: AAC GTG AAA TAT CTC CCA GGA C | IDT PrimeTime® qPCR Primers | Mm.PT.58.7971254 |
| LDHA primer 1: Sequence: CCC TTG AGT TTG TCT TCC ATG A | IDT PrimeTime® qPCR Primers | Mm.PT.58.29860774 |
| LDHA primer 2: Sequence: CTC CCC AGA ACA AGA TTA CAG T | IDT PrimeTime® qPCR Primers | Mm.PT.58.29860774 |
| B2M primer 1: Sequence: GGG TGG AAC TGT GTT ACG TAG | IDT PrimeTime® qPCR Primers | Mm.PT.39a.22214835 |
| B2M primer 2: Sequence: TGG TCT TTC TGG TGC TTG TC | IDT PrimeTime® qPCR Primers | Mm.PT.39a.22214835 |
| Recombinant DNA | ||
| Mdh1_OMu15660D_pcDNA3.1+/C-(K)-DYK | Genscript | Clone ID:OMu15660D ORF Clones (Accession No.):NM_008618.3 (ORF Sequence),1002 bp Vector:pcDNA3.1+/C-(K)-DYK |
| Gpd1_OMu18825D_pcDNA3.1+/C-(K)-DYK | Genscript | Clone ID:OMu18825D ORF Clones (Accession No.):NM_010271.3 (ORF Sequence),1047 bp Vector:pcDNA3.1+/C-(K)-DYK |
| MDH1_OHu13813D_pcDNA3.1+/ C-(K)-DYK | Genscript | Clone ID:OHu13813D ORF Clones (Accession No.):NM_005917.4 (ORF Sequence),1002 bp Vector:pcDNA3.1+/C-(K)-DYK |
| GPD1_OHu20325D_pcDNA3.1+/C-(K)-DYK | Genscript | Clone ID:OHu20325D ORF Clones (Accession No.):NM_005276.4 (ORF Sequence),1047 bp Vector:pcDNA3.1+/C-(K)-DYK |
| Human MDH1res | Genscript | codon-optimized cDNA of human MDH1 in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| Mouse MDH1res | Genscript | codon-optimized cDNA of mouse MDH1 in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| Human GPD1Lres | Genscript | codon-optimized cDNA of human GPD1L in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| Mouse GPD1Lres | Genscript | codon-optimized cDNA of mouse GPD1L in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| GFP_pcDNA3.1+/C-(K)-DYK | Genscript | Vector name: pcDNA3.1+/C-(K)-DYK |
| pCMV-VSV-G | (Yao et al., 2019) | Addgene plasmid # 8454 |
| pCMV delta R8.2 | (Yao et al., 2019) | Addgene plasmid # 12263 |
| pLVX-HRasV12- hygromycin | (Yao et al., 2019) | |
| Purified lentiviral particles for human MDH1 (NM_005917.3) in pReceiver-Lv105 with CMV promoter, puromycin | GeneCopoeia | LPP-C0441-Lv105-050-S |
| Lentifect™ Purified Lentiviral Particles for Negative Control for pReceiver-Lv105 | GeneCopoeia | LP105-025 |
| Software and algorithms | ||
| Python (v3.7) | NA | www.python.org |
| NumPy (v1.19.5) | NA | www.numpy.org |
| Pandas (v1.0.1) | NA | www.pandas.pydata.org |
| SciPy (v1.4.1) | NA | www.scipy.org |
| Matplotlib (v3.1.3) | NA | www.matplotlib.org |
| Other | ||
| Lipofectamine™ RNAiMAX Transfection Reagent | Thermo Fisher | Cat.#: 13778075 |
| Lipofectamine™ 3000 Transfection Reagent | Thermo Fisher | Cat.#: L3000001 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Fisher | Cat.#: 11668019 |
Experimental model and subject details
Cell culture
The following cell lines were maintained in high-glucose Dulbecco’s Modified Eagle’s Medium (Life Technologies) containing 10% FBS (Life Technologies) and 1% penicillin/streptomycin (Life Technologies) at 37 °C with 5% CO2: 3T3 (male), PC-3 (male), DITNC1 (unspecified), BT549 (female), and HCT 116-Luc2 (male). Glucose consumption, lactate excretion, flux balance analysis results, and doubling times for the NCI-60 cell lines were derived from a prior report (Zielinski et al., 2017) and the Developmental Therapeutics Program at the National Cancer Institute (cancer.gov).
Mice
Animals in this study were female nude mice (43-56 days old) purchased from Charles River Laboratories. All mouse procedures were approved by the Washington University Institutional Animal Care and Use Committee.
Method Details
H-Ras transformation
H-Ras (HRASV12) was transfected into 3T3 cells as described previously (Yao et al., 2019). Briefly, HEK293T cells were co-transfected with pCMV-VSV-G, pCMV delta R8.2, and pLVX-HRasv12- hygromycin constructs with Lipofectamine 2000 reagent. Media were then collected for infection of 3T3 cells.
Proliferation assay
To assess proliferation, cells were grown under various experimental conditions for 48–72 hours. Cells were then collected and counted in trypan blue with a hemocytometer.
Stable overexpression of MDH1 in PC3 cells
PC3 cells were plated in 24-well plates. The next day, cells were transfected with either negative control lentifect™ purified lentiviral particles (GeneCopoeia) or purified lentiviral particles for human MDH1 (NM_005917.3) in pReceiver-Lv105 by using the CMV promoter, puromycin (GeneCopoeia). Media were also supplemented with polybrene (8 μg/mL) to increase the infection efficiency. After 24 hours, the media were then refreshed, and cells were selected by 1 μg/mL puromycin.
Mouse xenograft experiments
To form xenografts, 4 million PC3 cells in 1:1 DMEM:matrigel were injected into the left flank of female nude mice (Charles River Laboratories). Tumor volume was measured with calipers and calculated by using the formula V = (1/2) (L*W2), where L and W are length and width, respectively. To assess glucose metabolism in the tumors, uniformly 13C-labeled ([U-13C6]) glucose was introduced via intratumoral infusion. A standard solution of [U-13C6] glucose was first prepared by suspending 200 mg [U-13C6] glucose into 1 mL of 0.9% saline. The amount of [U-13C6] glucose solution injected into each mouse tumor was normalized by individual tumor volumes (40 μL of stock [U-13C6] glucose per 200 mm3 tumor volume). One hour after [U-13C6] glucose labeling, tumors were collected and stored at −80°C until extraction for LC/MS analysis.
Transient overexpression of MDH1 and GPD1
PC3 cells were plated in 6-well plates. The next day, cells were transduced with plasmids MDH1_OHu13813D_pcDNA3.1+/ C-(K)-DYK (Genscript) or GPD1_OHu20325D_pcDNA3.1+/C-(K)-DYK (Genscript) by using Lipofectamine 3000 for 24 hour. The media were then refreshed, and cells were assayed within 48 hours after plasmid transduction. The control vector was GFP_pcDNA3.1+/C-(K)-DYK (GenScript), which expresses GFP instead of MDH1 or GPD1. The same protocol was used for H-Ras 3T3 cells, but with plasmids Mdh1_OMu15660D_pcDNA3.1+/C-(K)-DYK (Genscript), or Gpd1_OMu18825D_pcDNA3.1+/C-(K)-DYK (Genscript).
siRNA-mediated knock-down of MDH1 and GPD1L
MDH1 and GPD1L knockdown in PC3 cells was achieved by using a validated pool of small interfering RNA (siRNA) duplexes directed against human MDH1 or GPD1L (Trifekta Kit, IDT) and Lipofectamine RNAiMAX Transfection Reagent according to the manufacturer’s instructions. MDH1 and GPD1L knockdown in H-Ras 3T3 cells was achieved by using a validated pool of small interfering RNA (siRNA) duplexes directed against mouse MDH1 or GPD1L (Trifekta Kit, IDT) and Lipofectamine RNAiMAX Transfection Reagent according to the manufacturer’s instructions. Cells given scrambled siRNA were used as a negative control.
Transient overexpression of siRNA-resistant MDH1 (MDH1res) and GPD1L (GPD1Lres) in MDH1 and GPD1L knockdown cells
To express MDH1 and GPD1L in MDH1 and GPD1L knockdown cells, an siRNA-resistant cDNA that expresses human MDH1 (human MDH1res) or human GPD1L (human GPD1Lres) was first cloned separately in the pcDNA3.1+/C-(K)-DYK vector (GenScript). The codon was optimized to be resistant to the siRNA added. Next, MDH1 or GPD1L was knocked down in PC3 cells by treatment with human MDH1 or human GPD1L siRNA for 24 hours. The cells were then transfected with human MDH1res or human GPD1Lres by using Lipofectamine 3000 as described above. The same protocol was used for H-Ras 3T3 cells, but with mouse siRNAs and with mouse MDH1res or mouse GPD1Lres plasmids. Sequences for the DsiRNA are provided in Table S4. Sequences for MDH1res and GPD1Lres are provided in Data S1 and S2.
Nutrient-consumption/excretion analysis
After incubating cells in fresh media for 24 hours or 48 hours, the spent media was collected and extracted with methanol/acetonitrile (ACN)/water (2:2:1) containing 13C-labeled internal standards ([U-13C6] glucose, [U-13C3] lactate, and [U-13C5] glutamine; Cambridge Isotope Laboratories). Samples were vortexed for 30 s, incubated in liquid nitrogen for 1 min, and then sonicated for 10 min. Following a 1 hour incubation at −20 °C, the samples were centrifuged and the supernatant was collected for LC/MS analysis.
The concentrations of glucose and lactate were determined by using the ratio between the fully unlabeled peak from samples and the fully labeled peak from standards. The amount of glucose consumed over time and the amount of lactate excreted over time (x) were normalized by cell growth over the experimental time period according to the following equation where N0 is the starting cell number, t is the incubation time, DT is the doubling time, and Y is the change in nutrient level:
| (Equation 5) |
The lactate-excretion rate was determined from the amount of lactate excreted normalized by the amount of glucose consumed (i.e. Lac/(2 x Glc).
Isotope labeling and metabolite extraction
Cells were cultured in 6-well plates containing complete medium and allowed to adhere overnight. To modulate glycolytic flux, cells were treated with vehicle control (water) or 2DG (Sigma) for 24 hours. Next, the media were refreshed to introduce 25 mM [4-2H] glucose (Cambridge Isotope Laboratories). After labeling for the indicated time, cells were harvested by aspirating media and washing with PBS three times. After a final wash with HPLC-grade water, the metabolic activity was quenched with ice-cold HPLC-grade methanol. Cells were scraped from the plate and collected in methanol before being pelleted and dried via a SpeedVac. Dried cell pellets were extracted with 1 mL methanol/ACN/water (2:2:1). Samples were then vortexed for 30 s and incubated for 1 min in liquid nitrogen before being sonicated for 10 min. Following a 1 hour incubation at −20 °C, the samples were centrifuged and the supernatant was collected. The supernatant was dried via a SpeedVac and reconstituted in acetonitrile/water (1:1) for LC/MS analysis.
pHILIC column separation
Metabolites were separated by using a ZIC-pHILIC column (100 × 2.1 mm, 5 μm, polymer; Merck-Millipore) with a ZIC-pHILIC guard column (20 × 2.1 mm, 5 μm, polymer, Merck-Millipore). The column compartment temperature was 40 °C. Mobile phase A was 95% water and 5% ACN with 20 mM ammonium bicarbonate, 0.1% ammonium hydroxide solution, and 2.5 μM medronic acid. The ammonium hydroxide solution was purchased as 25% ammonia in water (Honeywell). Mobile phase B was 95% ACN and 5% water with 2.5 μM medronic acid. While we used medronic acid in mobile phase B in this study, we do not recommend doing so because it can cause issues with the pump over time. Thus, we recommend adding 5 μM medronic acid to the aqueous mobile phase A, but no medronic acid in B. Separation was carried out with a flow rate of 250 μL min−1 and the following linear gradient: 90% B from 0 to 1 min, 90% to 25% B from 1 to 14 min, 25% B from 14 min to 14.5 min, and 25% to 90% B from 14.5 to 15 min. The column was equilibrated with 90% B for 5 min at a flow rate of 400 μL min−1 and 2 min at a flow rate of 250 μL min−1.
Luna column separation
Metabolites were separated by using a Luna aminopropyl column (100 × 2 mm, 3 μm, 100 Å; Phenomenex). The column compartment temperature was 25 °C. Mobile phase A was 95% water, 5% ACN, 10 mM ammonium hydroxide, and 10 mM ammonium acetate. Mobile phase B was 95% ACN and 5% water. Separation was achieved with a flow rate of 200 μL min−1 and the following linear gradient: 95% B from 0 to 2 min, 95% to 0% B from 0 to 18 min, and 0% B from 18 to 25 min. Between runs, the column was washed and equilibrated with a flow rate of 300 μL min−1 and the following gradients: 0% to 95% B from 0 to 5 min followed by 95% B for 5 min.
Analysis with an Orbitrap mass spectrometer
A Dionex Ultimate 3000 UHPLC system was coupled to a Thermo Scientific Q Exactive Plus Orbitrap mass spectrometer. Full MS scan mode in negative polarity was used at a resolution of 140,000 with the following settings: automatic gain control (AGC) target 1e6, maximum injection time 200 ms, sheath gas flow rate 45 arbitrary units (Arb), auxiliary gas flow rate 10 Arb, sweep gas 2 Arb, spray voltage 2.8 kV, capillary temperature 250 °C, and auxiliary gas heater temperature 350 °C.
Analysis with a QTOF mass spectrometer
An Agilent 1290 UHPLC system was coupled to an Agilent 6540 quadruple time-of-flight (Q-TOF) mass spectrometer. Full MS scan mode in negative polarity was used with the following settings: gas temperature 200 °C, sheath gas temperature 300 °C, drying gas flow rate 10 L min−1, sheath gas flow rate 12 L min−1, nebulizer pressure 44 psi, capillary voltage 3000 V, nozzle voltage 2000 V, fragmentor voltage 100 V, skimmer voltage 65 V, and scan rate 1 spectrum s−1.
Analytical configurations
Metabolites extracted from cells labeled with [4-2H] glucose were analyzed by using a pHILIC column and an Orbitrap mass spectrometer. Media metabolites were analyzed to quantitate glucose consumption and lactate excretion. For overexpression and AKB-treated conditions, media metabolites were analyzed by using a pHILIC column and a Q-TOF mass spectrometer. After GPD1L knockdown, metabolites from the media of PC3 cells and H-Ras 3T3 cell were analyzed by using a pHILIC column and an Orbitrap or Q-TOF mass spectrometer, respectively. After MDH1 knockdown in PC3 cells, metabolites from media were analyzed by using a pHILIC column and an Orbitrap mass spectrometer. After MDH1 knockdown in H-Ras 3T3 cells, metabolites from media were analyzed by using a Luna column and a Q-TOF mass spectrometer. For overexpression of siRNA-resistant MDH1 (MDH1res) and GPD1L (GPD1Lres) in MDH1 or GPD1L knockdowns, media metabolites were analyzed by using a pHILIC column and an Orbitrap mass spectrometer.
Analysis of relative MDH1 and GPD1/GPD1L activity
To assess the relative activity of MDH1 and GPD1/GPD1L, cells were cultured with 25 mM [4-2H] glucose for 10 min and metabolites were extracted and analyzed by using a pHILIC column and an Orbitrap mass spectrometer as detailed above. The relative MDH1 activity was determined by the fraction of [2-2H] malate (M+1 malate) normalized to the fraction of [1-2H] GAP (M+1 GAP). To assess the relative GPD1/GPD1L activity, it is important to note that both [2-2H] G3P and [1-2H] G3P can give rise to M+1 G3P. [2-2H] G3P can be generated from [4-2H] NADH and unlabeled DHAP. [1-2H] G3P can be generated from unlabeled NADH and [1-2H] DHAP. Thus, the fraction of M+1 G3P normalized to the fraction of M+1 GAP cannot be used to assess the relative GPD1/GPD1L activity. Instead, the relative GPD1/GPD1L activity was determined by the fraction of [1,2-2H] G3P normalized to the fraction of [1-2H] DHAP and [4-2H] NADH (i.e., M+2 G3P/ (M+1 NADH × M+1 DHAP)).
GAP and DHAP cannot be resolved with the LC/MS method used here. Considering that DHAP and GAP are in equilibrium in mammalian cells (triosephosphate isomerase, ΔG ≈ 0 kJ/mol) (Park et al., 2016), we assumed the same labeling pattern for GAP and DHAP. To determine the amount of deuterium labeled NADH (M+1 NADH), we had to correct for natural abundance 13C. There are two sources of 13C natural abundance. First, because NAD+ and NADH were not chromatographically separated with the pHILIC method used in this study, we had to remove the natural-abundance 13C peaks arising from NAD+ that were not mass resolved from unlabeled NADH and deuterated NADH. Second, natural abundance 13C from NADH itself was not mass resolved from deuterated NADH and required a second correction.
Naturally occurring NAD+ with two 13C atoms (m/z = 664.1086) is within 20 ppm of the m/z of unlabeled NADH (m/z = 664.1175). Additionally, naturally occurring NAD+ with three 13C atoms (m/z = 665.1119) is within 20 ppm of the m/z of M+1 NADH (m/z = 665.1238). We corrected for each NAD+ interference by subtracting the theoretical 13C natural abundance NAD+ signal from the corresponding NADH peak area (either unlabeled NADH or M+1 labeled NADH). To account for the 13C natural abundance of NADH itself, we performed another correction to remove the naturally occurring 13C contribution to the deuterated NADH signal. The code for performing this correction is available on GitHub (https://github.com/e-stan/NADH_KFP).
As an independent method to confirm the amount of deuterated NADH in our samples, we also applied a second strategy. Deuterated NADH at isotopic steady state can be calculated from the isotopic steady state labeling of G3P and DHAP (see Equations 11, 12, and 13 below). For H-Ras 3T3 cells, the percentage of NADH with a deuterium label was calculated to be 29.1% with this method. For the same H-Ras 3T3 cells, the mean value for the percentage of NADH labeled with deuterium at isotopic steady state was determined to be 28.6% when using NADH data corrected for 13C natural abundance. Thus, irrespective of which method we use, we obtain consistent results for the amount of NADH that is labeled with deuterium.
Metabolite quantitation with a QqQ mass spectrometer
Quantitation of malate, G3P, and lactate was achieved by using a multiple reaction monitoring (MRM) method on a TSQ Altis triple quadrupole (QqQ) mass spectrometer (Thermo Scientific). To find the optimal fragments and collision energies used for MRM transitions, separate unlabeled standard solutions (malate, G3P, and lactate) and 13C-labeled internal standards ([U-13C4] malate, [U-13C3] G3P, and [U-13C3] lactate (Cambridge Isotope Laboratories) were prepared and introduced by flow injection. The lists of MRM transitions with precursor ions, product ions (quantifier and qualifier), and corresponding collision energies are shown in Table S5.
In preparation for analysis by QqQ mass spectrometry, cells were cultured in 6-well plates for 48 hours and then harvested by aspirating media and washing with PBS three times. After a final wash with HPLC-grade water, metabolic activity was quenched with ice-cold HPLC-grade methanol containing 13C-labeled internal standards ([U-13C4] malate, [U-13C3] G3P, and [U-13C3] lactate; Cambridge Isotope Laboratories). Metabolites were extracted as described above. For calibration purposes, standard solutions of different concentrations of unlabeled malate (0.3 μM to 120 μM), G3P (0.2 μM to 40 μM), and lactate (2 μM to 520 μM) spiked with the corresponding 13C-labeled internal standards were measured (5 μM for 13C-malate and 13C-G3P, and 10 μM for 13C-lactate). The same concentrations of 13C-labeled standards were spiked into research samples. The absolute concentrations of malate, G3P, and lactate in research samples were then determined from the ratio of the unlabeled signal to the labeled signal.
The TSQ Altis QqQ was coupled with a Vanquish UHPLC system. Metabolites were separated on a pHILIC column as described above. Mass spectrometry detection was accomplished in a negative ion mode with the following source settings: sheath gas flow 50 Arb, auxiliary gas flow 10 Arb, sweep gas flow 1 Arb, ion transfer tube temperature 325 °C, and vaporizer temperature 350 °C. Negative electrospray ionization with 2.5 kV spray voltage was applied. The collision gas pressure was set to 1.5 mTorr.
Western blots
Cells were lysed with RIPA buffer (Thermo Fisher Scientific) in the presence of a protease inhibitor and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Lysates were separated by SDS–PAGE under reducing conditions, transferred to a nitrocellulose membrane, and analyzed by immunoblotting. MDHC (H-6) (Cat# sc-166879, RRID:AB_10609257) was purchased from Santa Cruz Biotechnology. GPD1 polyclonal antibody (Cat# 27943-1-AP, RRID:AB_2881016) and GPD1L polyclonal antibody (Cat# 17263-1-AP, RRID:AB_2112359) were purchased from Proteintech. Rabbit anti-Ras (G12V) monoclonal antibody (Cat.#: 14412, RRID:AB_2714031) was purchased from Cell Signaling. Goat anti-rabbit secondary antibodies (HRP conjugated) and anti-mouse secondary antibodies were purchased from LI-COR and Santa Cruz Biotechnology, respectively. Immunoblotting for β-tubulin by rabbit anti-β-tubulin antibody (HRP conjugated) (Cat# 5346, RRID:AB_1950376) (Cell Signaling) was used as a loading control. Signal was detected by using WesternSure premium chemiluminescent substrate and the C-Digit Blot Scanner (LI-COR) according to the manufacturer’s instructions.
Oxygen consumption assays
The oxygen consumption rate (OCR) of whole cells was determined by using a Seahorse XFp Extracellular Flux Analyzer (Seahorse Bioscience). Cells were trypsinized and plated on a miniplate 24 hours prior to the Seahorse assay. For 2DG experiments, the OCR was normalized to the final cell number determined by manual cell counting. For DNP experiments, the OCR was monitored upon injections of 20 μM DNP or 100 μM DNP.
Real-time PCR analysis
RNA was extracted by using Trizol (Invitrogen). cDNA was synthesized with the Superscript III First-Strand Synthesis SuperMix following the manufacturer’s guidelines. Real-time reverse-transcription polymerase chain reaction was performed by using the Maxima SYBR Green/ROX qPCR Master Mix (2X) with a StepOnePlus Real-Time PCR system (Applied Biosystems) according to the manufacturer’s guidelines. Primers were purchased from IDT and sequences are listed in the key resources table. The results were normalized to a housekeeping gene, B2M, and further analyzed by using the 2-ΔΔCt method.
Measurement of NAD+/NADH ratio
To measure the NAD+/NADH ratio after MDH1 or GPD1L knockdown, PC3 cells were treated with human MDH1 or human GPD1L siRNA for 24 hours. Cells given scrambled siRNA were used as a negative control. The next day, cells were washed with cold PBS and the NAD+/NADH ratio was measured by using an NAD/NADH Quantification Colorimetric Kit (BioVision; Cat#K337) according to the manufacturer’s instructions. To measure the NAD+/NADH ratio after MDH1 or GPD1 overexpression, PC3 cells were transduced with plasmids MDH1_OHu13813D_pcDNA3.1+/ C-(K)-DYK (Genscript) or GPD1_OHu20325D_pcDNA3.1+/C-(K)-DYK (Genscript) for 24 hours. Cells given plasmid GFP_pcDNA3.1+/C-(K)-DYK (GenScript) were used as a control. The next day, cells were washed with cold PBS and the NAD+/NADH ratio was measured by using an NAD/NADH Quantification Colorimetric Kit (BioVision; Cat#K337).
Kinetic flux profiling
The mathematical model presented in Equations 1, 2, 3, and 4 was used to compute MDH1, G3P1/GPD1L, and LDH fluxes (f3, f4, and f6, respectively). The following measurements were used as input: pool-size concentrations of malate ([Malate]), G3P ([G3P]), and lactate ([Lactate]); labeling dynamics of GAP, DHAP, malate, G3P, and lactate; the rate of glucose consumption; and the rate of lactate excretion. The labeling dynamics of malate, G3P, and lactate were used to fit the unknown parameters of the model (fi and [NADH]).
By assuming metabolic steady state, we can relate the consumption and production terms of the model, Equations 6, 7, 8, 9, and 10.
| (Equation 6) |
| (Equation 7) |
| (Equation 8) |
| (Equation 9) |
| (Equation 10) |
At isotopic Steady State , additional relationships can be derived from the isotopic steady state unlabeled fractions of GAP, DHAP, malate, lactate, G3P, as well as the M+2 and M+1 fractions of G3P . First, we can relate steady state NADH labeling and the relative value of f5 to the total production flux (f10) from the isotopic steady state labeling of G3P, Equations 11, 12, and 13.
| (Equation 11) |
| (Equation 12) |
| (Equation 13) |
Given the isotopic steady state labeling of G3P and DHAP, isotopic steady state labeling of NADH and the relative value of f5 to f10 is inferred by minimizing the error between observed and predicted G3P labeling.
Next, by using the isotopic steady state labeling of malate, lactate, and GAP with the inferred labeling of NADH, we can calculate the relative values of f2, f7, and f8 to the total production flux of malate, lactate, and NADH , Equations 14, 15, and 16.
| (Equation 14) |
| (Equation 15) |
| (Equation 16) |
With these relationships, we can now write equations for f1, f2, f5, f7, f8, f9, f10, f11, and f12 in terms of the MDH1, GPD1/GPD1L, and LDH fluxes (f3, f4, andf6) and the relative fluxes , and , Equations 17, 18, 19, 20, 21, 22, 23, and 24, reducing the system to three unknown fluxes and the concentration of NADH.
| (Equation (17) |
| (Equation 18) |
| (Equation 19) |
| (Equation 20) |
| (Equation 21) |
| (Equation 22) |
| (Equation 22) |
| (Equation 23) |
| (Equation 24) |
To solve the system given by Equations 1, 2, 3, and 4 and Equations 17, 18, 19, 20, 21, 22, 23, and 24, an exponential model (Eq. 25) was first numerically fit to the labeling dynamics of GAP.
| (Equation 25) |
Then, by using the isotopic steady state labeling measurements, , and were calculated. f3, f4, f6, and [NADH] were then numerically estimated to minimize the sum of squared errors between the observed labeling dynamics and predicted labeling dynamics of malate, lactate, and G3P. The solution space of the optimization was constrained by the rate of glucose consumption (fglucose consumption) and the rate of lactate excretion (flactate excretion), Equations 26, 27, and 28.
| (Equation 26) |
| (Equation 27) |
| (Equation 28) |
The optimization was carried out by using the Nelder-Mead algorithm within the Python package SciPy (Virtanen et al., 2020). The unknown parameters (f3, f4, f6, [NADH]) were initialized to random values uniformly distributed in the interval (0,1). The measured absolute concentrations were used directly and not optimized. The solution giving the lowest sum of squared errors was kept and the optimal parameters were extracted.
Monte Carlo confidence interval estimation
To estimate confidence intervals for the flux calculations described above, a Monte Carlo procedure based on a previously published methodology was employed (Liu et al., 2018). A new dataset was sampled by fitting a t-distribution to the unlabeled percentage of each metabolite at each time point. With these distributions, three random variates were drawn from each distribution to create a “new” dataset of the same dimensions and statistical characteristics as the original. With this new dataset, the same optimization procedure was used to find and extract the flux estimates. This procedure was repeated 500 times to create a probability distribution for the calculated flux values that accounts for the variability in the measured values. Of the 500 flux sets, only flux sets that had a sum of squared errors less than twice that of the initial optimization using the measured dataset were kept. Solutions with larger errors were assumed to have converged to a local minimum. After removing these solutions, the remaining fluxes were randomly downsampled to 200 values each. For each flux (f3, f4, f6), the 25th and 75th percentiles were taken as the empirical interquartile ranges.
Quantification and statistical analysis
Experimental data are displayed as mean ± SE. Statistics were calculated by using an unpaired, two-tailed Student’s t test or a one-way analysis of variance (ANOVA) followed by Dunnett's test as specified in each figure legend.
Supplementary Material
Highlights.
Fermentation correlates with NADH shuttle flux but not proliferation in cancer cells
The rate of glycolysis outpaces the flux of NADH shuttles in proliferating cells
Saturating NADH shuttle activity drives lactate production during aerobic glycolysis
The fraction of glucose-derived NADH oxidized in mitochondria is cell type-dependent
Acknowledgments
We thank Dr. Cong-Hui Yao for discussion related to experimental design. Financial support was received for this work from NIH awards R35ES028365 (G.J.P.) and R24OD024624 (G.J.P.) as well as the Edward Mallinckrodt Jr. Foundation (G.J.P.) and the Pew Charitable Trusts (G.J.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
G.J.P. is a scientific advisory board member for Cambridge Isotope Laboratories. The Patti laboratory has a collaborative agreement with Thermo Fisher Scientific and Agilent Technologies.
References
- Ahn CS, and Metallo CM (2015). Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan HF, Walter KE, Luengo A, Madreiter-Sokolowski CT, Stryeck S, Lau AN, Al-Zoughbi W, Lewis CA, Thomas CJ, Hoefler G, et al. (2018). Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab 28, 706–720.e706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst P (2020). The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life 72, 2241–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand K, Leibold W, Luppa P, Schoerner C, and Schulz A (1986). Metabolic alterations associated with proliferation of mitogen-activated lymphocytes and of lymphoblastoid cell lines: evaluation of glucose and glutamine metabolism. Immunobiology 173, 23–34. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, and Mak TW (2011). Regulation of cancer cell metabolism. Nat Rev Cancer 11, 85–95. [DOI] [PubMed] [Google Scholar]
- Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, and Satrustegui J (2007). Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem 282, 7098–7106. [DOI] [PubMed] [Google Scholar]
- Contreras L, and Satrústegui J (2009). Calcium signaling in brain mitochondria: interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J Biol Chem 284, 7091–7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, and Miller DM (2009). Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM). Mol Cancer 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellows LK, Boutelle MG, and Fillenz M (1993). Physiological Stimulation Increases Nonoxidative Glucose-Metabolism in the Brain of the Freely Moving Rat. J Neurochem 60, 1258–1263. [DOI] [PubMed] [Google Scholar]
- Fernandez-de-Cossio-Diaz J, and Vazquez A (2017). Limits of aerobic metabolism in cancer cells. Sci Rep 7, 13488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PT, Raichle ME, Mintun MA, and Dence C (1988). Nonoxidative glucose consumption during focal physiologic neural activity. Science 241, 462–464. [DOI] [PubMed] [Google Scholar]
- Gerriets VA, and Rathmell JC (2012). Metabolic pathways in T cell fate and function. Trends Immunol 33, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H, and Kehinde O (1975). An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell 5, 19–27. [DOI] [PubMed] [Google Scholar]
- Guo T, Luna A, Rajapakse VN, Koh CC, Wu Z, Liu W, Sun Y, Gao H, Menden MP, Xu C, et al. (2019). Quantitative Proteome Landscape of the NCI-60 Cancer Cell Lines. iScience 21, 664–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hanse EA, Ruan C, Kachman M, Wang D, Lowman XH, and Kelekar A (2017). Cytosolic malate dehydrogenase activity helps support glycolysis in actively proliferating cells and cancer. Oncogene 36, 3915–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedeskov CJ (1968). Early effects of phytohaemagglutinin on glucose metabolism of normal human lymphocytes. Biochemical Journal 110, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, et al. (2006). Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther 5, 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston K, Pachnis P, Tasdogan A, Faubert B, Zacharias LG, Vu HS, Rodgers-Augustyniak L, Johnson A, Huang F, Ricciardo S, et al. (2021). Isotope tracing reveals glycolysis and oxidative metabolism in childhood tumors of multiple histologies. Med (N Y) 2, 395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TJ, Souza AL, Clish CB, and Puigserver P (2011). A Hypoxia-Induced Positive Feedback Loop Promotes Hypoxia-Inducible Factor 1α Stability through miR-210 Suppression of Glycerol-3-Phosphate Dehydrogenase 1-Like. Molecular and Cellular Biology 31, 2696–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, and Neely JR (1979). Control of maximum rates of glycolysis in rat cardiac muscle. Circ Res 44, 166–175. [DOI] [PubMed] [Google Scholar]
- Kozak LP (1985). Interacting genes control glycerol-3-phosphate dehydrogenase expression in developing cerebellum of the mouse. Genetics 110, 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, and Locasale JW (2016). The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Su X, Quinn WJ 3rd, Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, et al. (2018). Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab 27, 1067–1080 e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, Ferreira R, Naamati A, Ali A, Lewis CA, et al. (2021). Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell 81, 691–707 e696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna A, Elloumi F, Varma S, Wang Y, Rajapakse VN, Aladjem MI, Robert J, Sander C, Pommier Y, and Reinhold WC (2021). CellMiner Cross-Database (CellMinerCDB) version 1.2: Exploration of patient-derived cancer cell line pharmacogenomics. Nucleic Acids Res 49, D1083–D1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, et al. (2012). Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 15, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R (2015). Bringing Warburg to lymphocytes. Nat Rev Immunol 15, 598. [DOI] [PubMed] [Google Scholar]
- O'Donnell JM, Kudej RK, LaNoue KF, Vatner SF, and Lewandowski ED (2004). Limited transfer of cytosolic NADH into mitochondria at high cardiac workload. Am J Physiol Heart Circ Physiol 286, H2237–2242. [DOI] [PubMed] [Google Scholar]
- Oh SJ, Gu DR, Jin SH, Park KH, and Lee SH (2016). Cytosolic malate dehydrogenase regulates RANKL-mediated osteoclastogenesis via AMPK/c-Fos/NFATc1 signaling. Biochem Biophys Res Commun 475, 125–132. [DOI] [PubMed] [Google Scholar]
- Ou X, Ji C, Han X, Zhao X, Li X, Mao Y, Wong LL, Bartlam M, and Rao Z (2006). Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1). J Mol Biol 357, 858–869. [DOI] [PubMed] [Google Scholar]
- Park JO, Rubin SA, Xu YF, Amador-Noguez D, Fan J, Shlomi T, and Rabinowitz JD (2016). Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol 12, 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer T, Schuster S, and Bonhoeffer S (2001). Cooperation and competition in the evolution of ATP-producing pathways. Science 292, 504–507. [DOI] [PubMed] [Google Scholar]
- Phypers B, and Pierce JT (2006). Lactate physiology in health and disease. Continuing Education in Anaesthesia Critical Care & Pain 6, 128–132. [Google Scholar]
- Rabinovich S, Silberman A, Adler L, Agron S, Levin-Zaidman S, Bahat A, Porat Z, Ben-Zeev E, Geva I, Itkin Mv et al. (2020). The mitochondrial carrier Citrin plays a role in regulating cellular energy during carcinogenesis. Oncogene 39, 164–175. [DOI] [PubMed] [Google Scholar]
- Ratner PL, Fisher M, Burkart D, Cook JR, and Kozak LP (1981). The role of mRNA levels and cellular localization in controlling sn-glycerol-3-phosphate dehydrogenase expression in tissues of the mouse. J Biol Chem 256, 3576–3579. [PubMed] [Google Scholar]
- Safer B, Smith CM, and Williamson JR (1971). Control of the transport of reducing equivalents across the mitochondrial membrane in perfused rat heart. J Mol Cell Cardiol 2, 111–124. [DOI] [PubMed] [Google Scholar]
- Spriet LL, Howlett RA, and Heigenhauser GJ (2000). An enzymatic approach to lactate production in human skeletal muscle during exercise. Med Sci Sports Exerc 32, 756–763. [DOI] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh JT, Zhu WL, Newgard CB, Casey PJ, and Wang M (2019). Respiratory Capacity and Reserve Predict Cell Sensitivity to Mitochondria Inhibitors: Mechanism-Based Markers to Identify Metformin-Responsive Cancers. Mol Cancer Ther 18, 693–705. [DOI] [PubMed] [Google Scholar]
- Valvona CJ, Fillmore HL, Nunn PB, and Pilkington GJ (2016). The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol 26, 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Liu J, Zhou Y, and Oltvai ZN (2010). Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Syst Biol 4, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yao W, Li J, He Q, Shao Y, and Huang F (2018). Acetyl-CoA from inflammation-induced fatty acids oxidation promotes hepatic malate-aspartate shuttle activity and glycolysis. Am J Physiol Endocrinol Metab 315, E496–E510. [DOI] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Xu K, Yin N, Peng M, Stamatiades EG, Shyu A, Li P, Zhang X, Do MH, Wang Z, Capistrano KJ, et al. (2021). Glycolysis fuels phosphoinositide 3-kinase signaling to bolster T cell immunity. Science 371, 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zhou L, Shi Q, Zhao Y, Lin H, Zhang M, Zhao S, Yang Y, Ling ZQ, Guan KL, et al. (2015). SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J 34, 1110–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao CH, Fowle-Grider R, Mahieu NG, Liu GY, Chen YJ, Wang R, Singh M, Potter GS, Gross RW, Schaefer Jv et al. (2016). Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem Biol 23, 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao CH, Wang R, Wang Y, Kung CP, Weber JD, and Patti GJ (2019). Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski DC, Jamshidi N, Corbett AJ, Bordbar A, Thomas A, and Palsson BO (2017). Systems biology analysis of drivers underlying hallmarks of cancer cell metabolism. Scientific Reports 7, 41241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Rabinowitz JD, and White E (2016). Mitochondria and Cancer. Mol Cell 61, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original Western Blot images have been deposited in Mendeley and are publicly available as of the date of publication. The link is listed in the key resources table.
All original code has been deposited in Github and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze data reported in this study is available from the lead contact upon reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MDHC (H-6) | Santa Cruz Biotechnology | Cat# sc-166879, RRID:AB_10609257 |
| GPD1 Polyclonal antibody | proteintech | Cat# 27943-1-AP, RRID:AB_2881016 |
| GPD1L Polyclonal antibody | proteintech | Cat# 17263-1-AP, RRID:AB_2112359 |
| β-tubulin antibody (HRP conjugated) | Cell signaling | Cat# 5346 RRID:AB_1950376 |
| Goat anti-rabbit secondary antibodies (HRP conjugated) | LI-COR | Cat# 926-80011 RRID:AB_2721264 |
| Anti-mouse secondary antibody (m-IgGκ BP-HRP) | Santa Cruz Biotechnology | Cat# sc-516102, RRID:AB_2687626 |
| Rabbit anti-Ras (G12V), monoclonal | Cell Signaling | Cat.#: 14412, RRID:AB_2714031 |
| Chemicals, peptides, and recombinant proteins | ||
| Sodium 2-oxobutyrate (AKB) | Sigma | K0875 |
| Sodium pyruvate | Sigma | P5280 |
| Critical commercial assays | ||
| BCA protein assay | Thermo Fisher | Cat.#: 23225 |
| SuperScript III First-Strand Synthesis SuperMix | Invitrogen | Cat#18080400 |
| Maxima SYBR Green/ROX qPCR Master Mix (2X) | Thermo Scientific | K0221 |
| NAD/NADH Quantitation Colorimetric Kit | Biovision | Cat#K337 |
| Deposited Data | ||
| Raw image data | This manuscript | DOI: 10.17632/btc9bbcch8.1 |
| Original code | This manuscript | https://github.com/e-stan/NADH_KFP |
| Experimental models: Cell lines | ||
| 3T3 | American Type Culture Collection | RRID:CVCL_0123 |
| PC-3 | American Type Culture Collection | RRID:CVCL_0035 |
| DITNC1 | American Type Culture Collection | |
| BT549 | American Type Culture Collection | RRID:CVCL_1092 |
| HCT 116-Luc2 | American Type Culture Collection | Cat# CCL-247-LUC2, RRID:CVCL_VU38 |
| Experimental Models: Organisms/Strains | ||
| Mouse: 43-56 days old female nude mice | Charles River Laboratories | 490CRATHHO |
| Oligonucleotides | ||
| Negative Control DsiRNA | IDT | Cat.#: 51-01-14-04 |
| DsiRNA: GPD1L (human) | IDT TriFECTa DsiRNA Kit | hs.Ri.GPD1L.13 |
| DsiRNA: MDH1(human) | IDT TriFECTa DsiRNA Kit | hs.Ri.MDH1.13 |
| DsiRNA: GPD1L(mouse) | IDT TriFECTa DsiRNA Kit | mm.Ri.Gpd1l.13 |
| DsiRNA: MDH1(mouse) | IDT TriFECTa DsiRNA Kit | mm.Ri.Mdh1.13 |
| MDH1 primer 1: Sequence: GGT TAC ACC GAG TTT AAG AGC A | IDT PrimeTime® qPCR Primers | Mm.PT.58.5174962 |
| MDH1 primer 2: Sequence: CCA ATA CGA ACT GCC TGA CA | IDT PrimeTime® qPCR Primers | Mm.PT.58.5174962 |
| GPD1 primer 1: Sequence: TGT CAC CGA AGC CAA GC | IDT PrimeTime® qPCR Primers | Mm.PT.56a.32463908 |
| GPD1 primer 2: Sequence: ACC CAA CTT TCG CAT CAC T | IDT PrimeTime® qPCR Primers | Mm.PT.56a.32463908 |
| GPD1L primer 1: Sequence: TCT TGT GGA TGA ACT GGT GAG | IDT PrimeTime® qPCR Primers | Mm.PT.58.7971254 |
| GPD1L primer 2: Sequence: AAC GTG AAA TAT CTC CCA GGA C | IDT PrimeTime® qPCR Primers | Mm.PT.58.7971254 |
| LDHA primer 1: Sequence: CCC TTG AGT TTG TCT TCC ATG A | IDT PrimeTime® qPCR Primers | Mm.PT.58.29860774 |
| LDHA primer 2: Sequence: CTC CCC AGA ACA AGA TTA CAG T | IDT PrimeTime® qPCR Primers | Mm.PT.58.29860774 |
| B2M primer 1: Sequence: GGG TGG AAC TGT GTT ACG TAG | IDT PrimeTime® qPCR Primers | Mm.PT.39a.22214835 |
| B2M primer 2: Sequence: TGG TCT TTC TGG TGC TTG TC | IDT PrimeTime® qPCR Primers | Mm.PT.39a.22214835 |
| Recombinant DNA | ||
| Mdh1_OMu15660D_pcDNA3.1+/C-(K)-DYK | Genscript | Clone ID:OMu15660D ORF Clones (Accession No.):NM_008618.3 (ORF Sequence),1002 bp Vector:pcDNA3.1+/C-(K)-DYK |
| Gpd1_OMu18825D_pcDNA3.1+/C-(K)-DYK | Genscript | Clone ID:OMu18825D ORF Clones (Accession No.):NM_010271.3 (ORF Sequence),1047 bp Vector:pcDNA3.1+/C-(K)-DYK |
| MDH1_OHu13813D_pcDNA3.1+/ C-(K)-DYK | Genscript | Clone ID:OHu13813D ORF Clones (Accession No.):NM_005917.4 (ORF Sequence),1002 bp Vector:pcDNA3.1+/C-(K)-DYK |
| GPD1_OHu20325D_pcDNA3.1+/C-(K)-DYK | Genscript | Clone ID:OHu20325D ORF Clones (Accession No.):NM_005276.4 (ORF Sequence),1047 bp Vector:pcDNA3.1+/C-(K)-DYK |
| Human MDH1res | Genscript | codon-optimized cDNA of human MDH1 in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| Mouse MDH1res | Genscript | codon-optimized cDNA of mouse MDH1 in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| Human GPD1Lres | Genscript | codon-optimized cDNA of human GPD1L in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| Mouse GPD1Lres | Genscript | codon-optimized cDNA of mouse GPD1L in pcDNA3.1+/C-(K)-DYK (siRNA-resistant) |
| GFP_pcDNA3.1+/C-(K)-DYK | Genscript | Vector name: pcDNA3.1+/C-(K)-DYK |
| pCMV-VSV-G | (Yao et al., 2019) | Addgene plasmid # 8454 |
| pCMV delta R8.2 | (Yao et al., 2019) | Addgene plasmid # 12263 |
| pLVX-HRasV12- hygromycin | (Yao et al., 2019) | |
| Purified lentiviral particles for human MDH1 (NM_005917.3) in pReceiver-Lv105 with CMV promoter, puromycin | GeneCopoeia | LPP-C0441-Lv105-050-S |
| Lentifect™ Purified Lentiviral Particles for Negative Control for pReceiver-Lv105 | GeneCopoeia | LP105-025 |
| Software and algorithms | ||
| Python (v3.7) | NA | www.python.org |
| NumPy (v1.19.5) | NA | www.numpy.org |
| Pandas (v1.0.1) | NA | www.pandas.pydata.org |
| SciPy (v1.4.1) | NA | www.scipy.org |
| Matplotlib (v3.1.3) | NA | www.matplotlib.org |
| Other | ||
| Lipofectamine™ RNAiMAX Transfection Reagent | Thermo Fisher | Cat.#: 13778075 |
| Lipofectamine™ 3000 Transfection Reagent | Thermo Fisher | Cat.#: L3000001 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Fisher | Cat.#: 11668019 |