

ABSTRACT
Adeno-associated virus (AAV) vectors are one of the leading platforms for gene delivery for the treatment of human genetic diseases, but the antiviral cellular mechanisms that interfere with optimal transgene expression are incompletely understood. Here, we performed two genome-scale CRISPR screens to identify cellular factors that restrict transgene expression from recombinant AAV vectors. Our screens revealed several components linked to DNA damage response, chromatin remodeling, and transcriptional regulation. Inactivation of the Fanconi anemia gene FANCA; the human silencing hub (HUSH)-associated methyltransferase SETDB1; and the gyrase, Hsp90, histidine kinase, and MutL (GHKL)-type ATPase MORC3 led to increased transgene expression. Moreover, SETDB1 and MORC3 knockout improved transgene levels of several AAV serotypes as well as other viral vectors, such as lentivirus and adenovirus. Finally, we demonstrated that the inhibition of FANCA, SETDB1, or MORC3 also enhanced transgene expression in human primary cells, suggesting that they could be physiologically relevant pathways that restrict AAV transgene levels in therapeutic settings.
IMPORTANCE Recombinant AAV (rAAV) vectors have been successfully developed for the treatment of genetic diseases. The therapeutic strategy often involves the replacement of a defective gene by the expression of a functional copy from the rAAV vector genome. However, cells possess antiviral mechanisms that recognize and silence foreign DNA elements thereby limiting transgene expression and its therapeutic effect. Here, we utilize a functional genomics approach to uncover a comprehensive set of cellular restriction factors that inhibit rAAV-based transgene expression. Genetic inactivation of selected restriction factors increased rAAV transgene expression. Hence, modulation of identified restriction factors has the potential to enhance AAV gene replacement therapies.
KEYWORDS: adeno-associated virus, AAV, viral vector, transgene silencing, HUSH complex, SETDB1, MORC3, Fanconi anemia, DNA damage, SMC5-SMC6, CRISPR screen, restriction factor, virus-host interaction
INTRODUCTION
Adeno-associated virus (AAV) provides a powerful and versatile platform for gene delivery to treat a variety of human genetic diseases. AAV is a small single-stranded DNA virus with a genome size of ~4.7 kb from the Parvoviridae family. Its genome consists of two open reading frames, rep and cap, which are flanked by inverted terminal repeat (ITR) sequences (1, 2). The ITRs are the only cis-acting elements required for genome packaging, which enables the generation of recombinant AAV (rAAV) vectors containing a reporter or therapeutic transgene in place of the endogenous rep and cap genes (3). rAAV vectors have been clinically approved for the treatment of monogenic diseases, such as lipoprotein lipase deficiency, Leber congenital amaurosis, and spinal muscular atrophy, and there are over 150 ongoing clinical studies for additional rAAV-based therapies (4).
Despite the recent clinical successes, rAAV-based gene therapies face several challenges, as follows: (i) large-scale manufacturing of rAAV for high-dose administration (>1014 viral particles per patient) is expensive and difficult; (ii) immunological barriers exist, such as the presence of pre-existing neutralizing antibodies against AAV or the triggering of a robust humoral immune response upon treatment that prevent (repeated) administration; (iii) hepatic and neuronal genotoxicity has been observed in a number of high-dose administrations; and (iv) transgene expression can be lost over time due to the clearance of transduced cells through a cytotoxic T cell response and/or potential silencing of transgene expression (4–6). Increased efficiency in rAAV transgene expression could therefore allow for a reduction in the therapeutic doses needed, which consequently could decrease costs, elicit fewer neutralizing antibodies, reduce the risk of genotoxicity, and lower AAV capsid-directed T lymphocyte-mediated cytotoxicity. However, the precise host cellular pathways that restrict rAAV transgene expression are incompletely understood.
RESULTS
Genome-wide CRISPR screens identify rAAV restriction factors.
To identify cellular restriction factors of rAAV transgene expression that are likely to be conserved across multiple cell types, we performed two parallel genome-wide screens using CRISPR knockout (KO) in A549 human lung adenocarcinoma cells and CRISPR interference (CRISPRi) in K562 myelogenous leukemia cells. We used a nonsaturating multiplicity of infection (MOI) so that the inhibition of a repressor would lead to an increase of expression of a fluorescent reporter transgene. Based on rAAV serotype 2 (rAAV2) MOI titrations, we used 10,000 viral genomes (vg)/cell for the A549 screen and 50,000 vg/cell for the K562 screen (see Fig. S1A in the supplemental material). We performed two rounds of rAAV transduction and fluorescence-activated cell sorter (FACS)-based enrichment of the top 10 to 20% fluorescent CRISPR library cells, followed by an analysis of the guide RNA (gRNA) representation via next-generation sequencing (Fig. 1A). Analysis with MaGeCK, a computational tool that uses robust rank aggregation (RRA) to identify positively or negatively selected genes, resulted in significant enrichment of 20 genes in the A549 screen and of 88 genes in the K562 screens with a cutoff of −log(enrichment score) of >5 (Fig. S1B and C; see Table S1 and S2 in the supplemental material). Nearly all individual gRNAs were enriched for the top 15 genes of both screens, suggesting that the increased transgene expression phenotypes resulting from the genetic perturbations were robust (Fig. S1D). A comparison of the enrichment scores from the A549 and K562 CRISPR screens highlighted that several Fanconi anemia (FA) genes, the histone methyltransferase SETDB1, the GHKL-type ATPase MORC3, the SUMO interacting motifs containing 1 (SIMC1), the DNA damage response gene ATM, and additional genes (SMCHD1, MTF1, DCLRE1A, DAXX, FAM208A, and ZRANB2), were enriched in both data sets (Fig. 1B). STRING analysis on the top 100 hits from both screens identified functional gene clusters and cellular pathways, including the FA pathway, the human silencing hub (HUSH) complex containing the SETDB1 histone methyltransferase, the SMC5-SMC6 complex, the CCR4-NOT complex, and genes linked to DNA damage repair and chromatin regulation (Fig. 1C; see Table S3 in the supplemental material).
FIG 1.

CRISPR screens reveal cellular factors that restrict rAAV transgene expression. (A) Schematic of genome-wide CRISPR screens using CRISPR KO in A549 human lung adenocarcinoma cells and CRISPRi in K562 myelogenous leukemia cells. Cells were transduced with rAAV2 expressing a fluorescent reporter gene (GFP or tdTomato) and the top 10 to 20% fluorescent cells were sorted. After 5 to 7 days, a second round of transduction and sorting was performed before cells were collected for analysis of sgRNA enrichment by next-generation sequencing. (B) MaGeCK gene enrichment scores of the A549 CRISPR KO screen (y axis) versus MaGeCK gene enrichment scores of the K562 CRISPRi screen (x axis). Genes with a −log10 (enrichment score) of >3 in both screens are highlighted. (C) STRING network for the top 100 hits from both the A549 and the K562 CRISPR screens. Genes were clustered based on high-confidence interactions (STRING confidence cutoff, 0.7), and the major gene network is displayed. CRISPR screen enrichment scores for each gene are shown using a color scale. Line thickness indicates the confidence of interaction. (D) Heatmap of gene rankings in A549 and K562 CRISPR screens for selected genes related to DNA damage response and other cellular processes.
The FA pathway is a biochemical network of 19 genes which are responsible for DNA repair of interstrand cross-links and stabilization of stalled replication forks, which can impede DNA replication and transcription (7). Most FA core complex components (FANCA-FANCM), which are responsible for lesion recognition and binding, were robustly enriched in both rAAV screens, while the FA factors mediating lesion resolution and the homologous recombination machinery were not or only partially enriched (Fig. 1D). Another DNA damage repair factor that scored highly in both screens is the serine/threonine protein kinase ATM (Fig. 1B and D). ATM activates checkpoint signaling upon double-strand breaks, apoptosis, and genotoxic stresses, thereby acting as a DNA damage sensor (8). Loss of the FA pathway and ATM is synthetic lethal, suggesting closely linked cellular roles (9). Besides DNA damage response genes, many factors were linked to chromatin and transcriptional regulation (Fig. 1C and D). The histone methyltransferase SETDB1 is recruited by the HUSH complex, composed of FAM208A (also known as TASOR), MPHOSPH8, and PPHLN1, to heterochromatic loci and foreign DNA elements, such as retroviruses and transposons, to mediate transcriptional repression (10, 11). Other HUSH-associated factors (MORC2, ZNF638, and ATF7IP) required for recruitment and full epigenetic silencing activity were also enriched in both screens (12–14). The SMC5-SMC6 complex, highly ranked in the K562 screen, was shown previously to play a role in silencing of extrachromosomal HBV DNA as well as unintegrated HIV-1 DNA (Fig. 1C and D) (15, 16). Finally, MORC3 is a nuclear matrix protein with ATPase activity and has been linked to gene silencing, including of viral pathogens (17–19). Overall, the screens identified numerous candidate restriction factors for rAAV transgene repression, which have canonical roles in DNA damage repair and chromatin regulation.
The Fanconi anemia pathway suppresses transgene expression from rAAV vectors.
The FA pathway has to our knowledge not been functionally associated with the regulation of AAV transgene expression. To validate the impact of the FA pathway on rAAV transgene expression, we transduced three sets of FANCA-expressing and matched FANCA-deficient cells. Dual gRNA/Cas9-mediated deletion of the N terminus of FANCA in UM-SCC-01 cells significantly increased the percentage of rAAV2-tdTomato-positive cells (rAAV2+) as well as their median fluorescence intensity (MFI) relative to wild-type (WT) cells (Fig. 2A and B). This effect was reversed in FANCA transgene-complemented derivatives of the KO cells (Fig. 2A and B). Furthermore, transduction of mouse ear fibroblasts (MEFs) derived from WT or FANCA KO 129S4/SvJaeSor mice showed a similar increase of rAAV transgene expression in the FANCA-deficient background (Fig. 2C and D). Lastly, we obtained FA patient-derived head and neck squamous cell carcinoma cells (“974 cells”), which harbor a FANCA frameshift mutation, as well as corresponding FA patient cells that were lentivirally complemented with an intact FANCA cDNA copy (20). Complementation with FANCA cDNA led to a substantial decrease of rAAV transgene expression relative to the unaltered FA patient-derived 974 cells (Fig. 2E and F). As the FA pathway plays a role in DNA repair, we next tested whether mutations in FANCA affect the stability of rAAV genomes in transduced cells. Quantitative PCR (qPCR) showed that intracellular rAAV genome copy numbers were not significantly different in FANCA KO relative to those of WT or FANCA transcomplemented UM-SCC1 cells (Fig. 2G). Together, these data support that the FA pathway plays a role in the transgene expression from rAAV vectors, independent of rAAV genome stability.
FIG 2.

The Fanconi anemia pathway represses rAAV transgene expression. rAAV2-tdTomato infection rate (A) or MFI (B) of human WT, FANCA KO, and KO UM-SCC1 cells complemented with FANCA transgene (Tg). Cells were infected with rAAV2 (MOI, 10,000) for 48 h, and tdTomato expression was analyzed by flow cytometry. rAAV2-tdTomato infection rate (C) or MFI (D) of mouse ear fibroblasts (MEF) derived from WT or FANCA KO 129S4/SvJaeSor mice. Cells were infected with rAAV2 (MOI, 100,000) for 24 h, and tdTomato expression was analyzed by flow cytometry. rAAV2-tdTomato infection rate (E) or MFI (F) of FA patient-derived head and neck squamous cell carcinoma cells (974 cells), which harbor a FANCA frameshift mutation, as well as of 974 cells that were complemented with FANCA transgene (Tg). Cells were infected with rAAV2 (MOI, 10,000) for 48 h, and tdTomato expression was analyzed by flow cytometry. (G) Quantification of intracellular rAAV genome copies. Transduced cells (MOI, 10,000) were harvested after 48 h, DNA was extracted, and AAV and cellular RPPH1 DNA copy numbers were determined by qPCR. Data are combined from 2 independent experiments with 3 biological replicates each. All FACS data are shown as the mean 6 SD. from biological triplicates. P values were determined by analysis of variance (ANOVA) with post hoc Tukey’s test for A and B and unpaired t test for C to F and are defined as follows: ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P <0.001; and ****, P < 0.0001.
Knockout of MORC3 and SETDB1 enhances rAAV transgene expression.
Next, we sought to validate the impact of the (GHKL)-type ATPase MORC3 and the histone methyltransferase SETDB1 on rAAV, as previous reports have shown antiviral functions for other DNA viruses (10, 14, 18, 19). We generated two clonal A549 KO cells per gene. The MORC3 KO clones harbored frameshift mutations in exon 1, while the SETDB1 KO clones contained a frameshift or large deletion (−21bp) in exon 2 (see Fig. S2A in the supplemental material). The generated KO cell lines did not show any growth defects (Fig. S2B). Infection of KO cells with rAAV2-tdTomato showed a substantial increase in rAAV2+ cells (by 16 to 19% in MORC3 KO and by 22 to 23% in SETDB1 KO cells 3 days postinfection [dpi]) as well as in MFI (by 2.7 to 3.3 times in MORC3 KO and by 5.4 to 6.8 times in SETDB1 KO cells 3 dpi) relative to WT cells (Fig. 3A and B; see Fig. S3 in the supplemental material). rAAV transgene expression remained higher in KO than that in WT cells over 8 days. Signal decreased under all conditions at later time points likely due to the dilution of episomal rAAV DNA during cell proliferation. Lentiviral complementation of MORC3 KO cells with MORC3 cDNA lowered the percentage of rAAV+ cells and MFI to levels observed in WT cells (Fig. 3C and D), supporting that rAAV transgene expression is indeed modulated by MORC3 activity. Similarly, complementation of SETDB1 KO cells with SETDB1 cDNA decreased both the percentage of tdTomato+ cells (albeit to a lesser degree) and MFI (Fig. 3E and F). Next, we tested whether deletion of MORC3 or SETDB1 allowed for achievement of comparable rAAV transduction rates with reduced rAAV MOI. Across a range of 100 to 100,000 vg/cell, MORC3 and SETDB1 KO cells exhibited significantly higher transgene expression levels than WT cells, demonstrating that only ~1/10 of the rAAV amount was needed in KO cells to achieve similar transgene expression levels as in WT cells (Fig. 3G and H).
FIG 3.

Knockout of MORC3 and SETDB1 increases rAAV2 transgene expression. Percentage (A) or MFI (B) of rAAV2-positive cells over time. WT, MORC3 KO, or SETDB1 KO A549 cells were infected with tdTomato-expressing rAAV2 (MOI, 10,000), and cells were measured by flow cytometry at day 1, 3, 6, and 8 postinfection. Percentage (C) or MFI (D) of rAAV2-positive WT, MORC3 KO, or cDNA-complemented MORC3 KO cells. Cells were infected with tdTomato-expressing rAAV2 (MOI, 10,000), and cells were measured by flow cytometry at 24 hours postinfection (hpi). Percentage (E) or MFI (F) of rAAV2-positive WT, SETDB1 KO, or cDNA-complemented SETDB1 KO cells. Cells were infected with tdTomato-expressing rAAV2 (MOI, 10,000), and cells were measured by flow cytometry at 24 hpi. Percentage (G) or MFI (H) of rAAV2-positive WT, MORC3 KO, or SETDB1 KO cells infected with different multiplicities of infection (MOI) and measured by flow cytometry at 24 hpi. (I) Quantification of intracellular rAAV genome copies in WT A549, MORC3 KO (clone 1), and SETDB1 KO (clone 1) cells. Transduced cells (MOI, 10,000) were harvested after 48 h, DNA was extracted, and AAV and cellular RPPH1 DNA copy numbers were determined by qPCR. Data are combined from 2 independent experiments with 3 biological replicates each. FACS data are shown as the mean ± SD from biological triplicates (A to) or from biological duplicates (G and H). P values were determined by ANOVA with post hoc Tukey’s test and are defined as follows: ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
To explore whether the restriction factors act on rAAV genome stability or transcriptional regulation, we measured whether rAAV genome copy numbers differed between WT, MORC3 KO, and SETDB1 KO cells using qPCR. While the rAAV2 genome copy number was slightly lower in MORC3 KO cells at 48 h post-transduction, it was increased by 1.7 times in SETDB1 KO cells (Fig. 3I). Additional quantification of transgene mRNA levels via reverse transcriptase quantitative PCR (RT-qPCR) showed that expression was moderately increased by 1.1 to 1.3 times in MORC3 KO cells and by >2 times in SETDB1 KO cells (see Fig. S4A in the supplemental material). Together, these data indicate that MORC3 likely modulates rAAV transgene expression independently of effects on viral vector genome copies, as MORC3 KO exhibited higher tdTomato levels despite having lower rAAV genome copies. In contrast, deletion of the histone methyltransferase SETDB1 increased rAAV genome copies, which could largely explain the increase in tdTomato fluorescent levels. However, SETDB1 KO led to an even larger increase in transgene mRNA levels, which means that there could be additional restriction on the transcriptional level.
Different rAAV serotypes are used in research and clinical applications, often depending on the target cell type or tissue. We therefore assessed whether MORC3 and SETDB1 also affected transgene expression from different rAAV serotypes. We infected cells with rAAV1, rAAV5, and rAAV6 vectors, which only lowly expressed the green fluorescent protein (GFP) transgene in WT A549 cells. In contrast, GFP expression was increased drastically in MORC3 and SETDB1 KO cells (Fig. 4A and B). Moreover, rAAV vector genomes can be single stranded (ssAAV), which requires second-strand synthesis prior to gene expression (as were all rAAV vectors used above), or they can be constructed with self-complementary genomes (scAAV). scAAV vectors form intramolecular double-stranded DNA (dsDNA) and thus do not require cell-mediated synthesis of the second strand. We observed that the presence of MORC3 and SETDB1 also restricted transgene expression from scAAV genomes, similarly as for ssAAV vectors, which suggests that these cellular restriction factors do not affect processes in the second-strand synthesis (Fig. 4C and D).
FIG 4.

MORC3 and SETDB1 restrict transgene expression from different rAAV serotypes as well as self-complementary rAAV vectors. Percentage (A) or MFI (B) of WT, MORC3 KO, or SETDB1 KO A549 cells infected with rAAV serotypes 1, 5, or 6 (MOI, 50,000) and analyzed by flow cytometry at 3 dpi. Percentage (C) or MFI (D) of WT, MORC3 KO, or SETDB1 KO A549 cells infected with scAAV serotype 2 (MOI, 10,000) and analyzed by flow cytometry at 3 dpi. All data are shown as the mean ± SD from biological triplicates. P values were determined by ANOVA with post hoc Tukey’s test for each rAAV and are defined as follows: ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001.
Transgene expression from lentiviral, adenoviral, and plasmid vectors is inhibited by MORC3 and SETDB1.
In addition to rAAV, other viruses, such as lentivirus and adenovirus, are used for clinical gene delivery applications (21). Interestingly, several recent studies suggested that MORC3 and SETDB1 can suppress the expression of retroviral elements (10, 19, 22). Consistent with previous studies, both MORC3 and SETDB1 KO increased lentivirus transduction rates and the MFI of encoded GFP across multiple lentiviral dilutions (Fig. 5A and B). This effect was overall comparable as that for rAAV; however, in contrast to rAAV, deletion of MORC3 had a stronger effect on lentiviral transgene expression (e.g., ~12 times increase in MFI at 1:3 lentivirus dilution) than the deletion of SETDB1 (~8 times increase in MFI). RT-qPCR of lentiviral mRNAs showed that transgene expression changes were higher than for rAAV2 in MORC3 KO and SETDB1 KO cells, suggesting stronger cellular restriction (Fig. S4B). During infection with GFP-expressing adenovirus 5 (AdV5-GFP), the percentage of GFP+ cells and the MFI of GFP were also increased in MORC3 and SETDB1 KO relative to those of WT cells (Fig. 5C and D). Moreover, transfection of a GFP-expressing plasmid also led to an increased MFI in MORC3 and SETDB1 KO cells relative to A549 WT cells, although the overall percentage of GFP+ cells did not change (Fig. 5E and F). In contrast, expression of a fluorescent protein from transfected mRNA-liposomes was unchanged in MORC3 and SETDB1 KO relative to that of WT cells (Fig. 5G and H). Together, these data suggest that MORC3 and SETDB1 broadly restrict foreign DNA elements, such as viral vectors and plasmid DNA.
FIG 5.

MORC3 and SETDB1 repress transgene expression from lentiviral, adenoviral, and plasmid vectors. Percentage (A) or MFI (B) of WT, MORC3 KO, or SETDB1 KO A549 cells transduced with different dilutions of a GFP-expressing lentivirus. Fluorescent cells were measured by flow cytometry at 3 days post-transduction. Percentage (C) or MFI (D) of WT, MORC3 KO, or SETDB1 KO A549 cells infected with different MOI of a GFP-expressing adenovirus 5 (AdV5-GFP) vector. Cells were analyzed by flow cytometry at 24 hpi. Percentage (E) or MFI (F) of WT, MORC3 KO, or SETDB1 KO A549 cells transfected with a GFP-encoding plasmid and analyzed by flow cytometry at 24 h post-transfection. Percentage (G) or MFI (H) of WT, MORC3 KO, or SETDB1 KO A549 cells transfected with in vitro-transcribed red fluorescent protein (RFP) mRNA and analyzed by flow cytometry at 24 h post-transfection. All data are shown as the mean ± SD from biological triplicates.
Knockdown of MORC3 and SETDB1 in primary human fibroblasts leads to increased transgene expression.
Finally, we explored whether inhibition of MORC3 or SETDB1 activity also affected rAAV transgene expression in untransformed, primary human cells. For this purpose, we performed small interfering RNA (siRNA) knockdown in BJ fibroblasts. Transfection with MORC3 or SETDB1 siRNAs prior to rAAV2-tdTomato transduction increased the percentage of tdTomato+ cells relative to cells transfected with a nontargeting siRNA from 10% to 54% for MORC3 knockdown and to 18% for SETDB1 knockdown (Fig. 6A). RT-qPCR confirmed the successful knockdown of MORC3 and SETDB1 mRNA levels, although residual expression may still restrict rAAV transgene expression to a certain degree (Fig. 6B). Overall, this finding suggests that the rAAV restriction factors identified in the genome-wide CRISPR screens could have a physiologically relevant function in inhibiting rAAV-based therapies in clinical settings. Additional validation of screen hits beyond FANCA, MORC3, and SETDB1 is needed as well as confirmation in additional primary cells and in vivo models.
FIG 6.
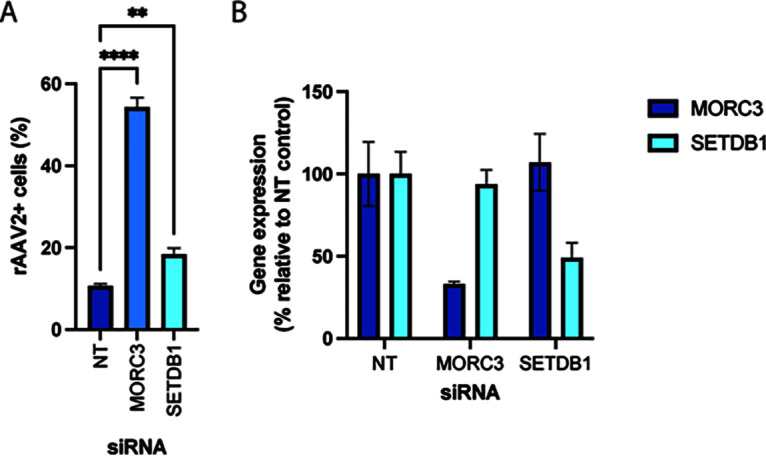
Knockdown of MORC3 and SETDB1 in primary human fibroblasts increases rAAV transgene expression. (A) Percentage of rAAV2-tdTomato-positive BJ fibroblasts that were transfected with a nontarget siRNA (NT) or siRNAs against MORC3 and SETDB1 at 4 days prior to rAAV transduction. Cells were analyzed by flow cytometry at 36 h post-transduction. Data are shown as the mean ± SD from biological triplicates. P values were determined by ANOVA with post hoc Tukey’s test and are defined as follows: ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. (B) Quantification of MORC3 and SETDB1 mRNA levels at 4 days after siRNA transfection. Data are shown as the mean ± SEM from biological triplicates.
DISCUSSION
Our study provides a comprehensive picture of the cellular factors that limit gene expression from AAV vectors in human cells. The majority of the identified host genes have known functions in DNA damage sensing and repair, chromatin remodeling, and transcriptional regulation. The most prominent hit across both CRISPR screens was the FA pathway, which is critical for the resolution of interstrand cross-links or stalled replication forks (7). It is plausible that the ITRs in the AAV genome mimic interstrand cross-links or stalled replication forks and are thus recognized by the FA machinery, which subsequently may impede second-strand synthesis of or transcription from the rAAV genome. Interestingly, another study found that deletion of FANCM led to enhanced AAV template-mediated homologous recombination (23). This finding further supports that the FA machinery engages with AAV genomes to interfere with both transgene expression as well as AAV template-mediated DNA repair. Other DNA damage response genes enriched in our data set, e.g., the ATM kinase and the MRN complex (consisting of MRE11A/RAD50/NBN), have been shown to interact with the ITRs in the AAV genome and to suppress transgene expression (24–26). Interestingly, studies with other DNA viruses suggested proviral effects for the FA pathway, which could be due to distinct differences in their genome structure and life cycle (27–30). Future studies are warranted to mechanistically investigate how the FA pathway interferes with the rAAV vectors and whether FA components bind directly to the AAV genome.
Moreover, we found that knockout of the HUSH-associated histone methyltransferase SETDB1 substantially increased transgene expression from rAAV and other viral vectors, such as lentivirus and adenovirus. However, this effect may be due partially or largely to an increase of intracellular rAAV genome copies. The exact mechanism of how SETDB1 regulates rAAV genome stability and whether it additionally regulates transcriptional activity from vector genomes need to be studied further. In mammalian cells, SETDB1 is recruited by the HUSH complex to regulate heterochromatin and has been described to mediate silencing of human immunodeficiency virus (HIV) and other retroviral sequences by depositing repressive H3K9me3 histone marks (10, 11, 14, 31, 32). Importantly, SETDB1 is able to silence both integrated and unintegrated retroviral DNA (10, 14). rAAV genomes exist typically as unintegrated episomes and are thought to be bound by histones (33). Recently, it was also shown that the HUSH machinery recognizes and silences a broad range of long, intronless transgenes (34). SETDB1 and the HUSH complex may thus be major contributors to rAAV chromatinization and silencing and potentially tagging heterochromatinized episomes for degradation. The crucial role for SETDB1 and the HUSH complex is further underscored by the enrichment of other factors in our screens. ATF7IP interacts with SETDB1 and protects it from proteasomal degradation (13). The DNA-binding protein ZNF638 (also known as NP220) is responsible for the recruitment of the HUSH complex to unintegrated murine retroviral DNA and may also act similarly on rAAV genomes (14). MORC2 was shown to mediate transgene silencing and maintain chromatin compactness upon recruitment by the HUSH complex (12). Finally, the role of the HUSH complex as an rAAV restriction factor was also corroborated by a recent study. Consistent with our data, it showed that knockout of HUSH components resulted in increased rAAV transgene levels and that the deletion of ZNF638 (NP220) decreased repressive H3K9me3 histone marks on AAV genomes (35).
In addition to the HUSH-associated MORC2, MORC3, another Microchidia CW-type zinc finger protein, was the top hit in the A549 screen and was strongly enriched in the K562 screen. MORC proteins are highly conserved, reside in the nucleus, contain a GHKL ATPase domain, and have been linked to heterochromatin condensation and gene silencing (17, 36). Previously, MORC3 was shown to restrict herpesviruses and endogenous retroviruses (ERVs) (18, 19). Mechanistic studies revealed that MORC3 bound to ERV sequences and that knockout of MORC3 resulted in ERV derepression, reduced H3K9me3 levels, and increased chromatin accessibility (19). According to our results, we propose that MORC3 is a critical rAAV restriction factor that silences rAAV genomes through possible chromatin regulation.
Additional screen hits have also been linked to chromatin regulation and gene silencing. The ATRX/DAXX complex is responsible for the deposition of H3.3 histones at heterochromatic regions and has been shown to associate with both SETDB1 and MORC3 (19, 37, 38). Moreover, the CHAF1 complex, together with the histone chaperon ASF1, delivers newly synthesized H3/H4 dimers to replication forks and was shown recently to mediate silencing of unintegrated HIV-1 DNA (39, 40). Overall, these chromatin modifiers with described roles in foreign DNA silencing may therefore also be involved in rAAV repression.
Lastly, the SMC5-SMC6 complex, which is critical for genome stability and DNA repair (41), has emerged as a component of antiviral responses. It has been linked recently to transcriptional restriction of numerous DNA viruses, such as hepatitis B virus, unintegrated retroviral DNA, papillomavirus, and Epstein-Barr virus (15, 16, 42, 43). Our data suggest that SMC5-SMC6 can also potently inhibit rAAV vectors. During DNA repair, this complex is recruited to DNA lesions by FAM178A (also known as SLF2) and SLF1 (44). A recent preprint demonstrated that SIMC1, a top hit in both CRISPR screens and largely with unknown function, forms a distinct complex with FAM178A to localize SMC5/SMC6 to viral replication centers (45). Therefore, it is plausible that rAAV is repressed by recruitment of the SMC5-SMC6 complex via FAM178A/SIMC1.
Overall, the identification of multiple components of specific cellular pathways in our CRISPR screens indicates that they are high-confidence candidates for rAAV restriction factors. Of note, two previous siRNA screens for regulators of rAAV transduction in HeLa cells showed enrichment of a subset of the same restriction factors as in our CRISPR screens, including ATF7IP, CHAF1A, DAXX, NPAT (a positive regulator of ATM), and MORC3 (previously named ZCWCC3) in one study as well as ATF71P, CHAF1A, SETDB1, NPAT, and the transcription factor MTF1 in another study (46, 47). Thus, our work corroborates some previously identified candidate restriction factors and expands the network of cellular factors that interfere with rAAV transgene expression.
MATERIALS AND METHODS
Cell culture.
A549 and K562 were obtained from ATCC and were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Omega Scientific), penicillin-streptomycin (Gibco), nonessential amino acids (Gibco), and l-glutamine (Gibco) at 37°C and 5% CO2. HEK293ft cells were obtained from Thermo Scientific and were cultured as described above. WT, FANCA KO and transcomplemented FANCA KO UM-SCC1 cells, WT and FANCA KO mouse ear fibroblasts, and FANCA-deficient patient-derived 974 cells with and without FANCA complementation were kindly provided by the Fanconi Anemia Research Fund at Oregon Health & Science University and were cultured as described above. Normal human BJ fibroblasts were obtained from ATCC (CRL-2522) and were cultured in knockout Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented with 15% fetal bovine serum, 16.5% Medium 199 (Gibco), penicillin-streptomycin, and l-glutamine at 37°C and 5% CO2.
Viruses.
The following rAAV vectors were obtained from the University of North Carolina (UNC) School of Medicine Gene Therapy Center Vector Core: AAV serotype 2 expressing GFP under a chimeric CMV-chicken β-actin (CBA) promoter (rAAV2-GFP); AAV serotype 2 expressing tdTomato under a CMV immediate enhancer/β-actin (CAG) promoter (rAAV2-tdTomato); AAV serotype 1, 5, or 6 expressing GFP under a CMV promoter (rAAV1/5/6-GFP); and self-complementary AAV serotype 2 expressing GFP under a CMV promoter (scAAV2-GFP). Lentivirus expressing GFP was produced by cotransfection of HEK293ft cells with the following plasmids: plenti-CMV-Puro-DEST (Addgene; 17452; gift from Eric Campeau and Paul Kaufman) where GFP cDNA was cloned into the EcoRV site using NEBuilder HiFi DNA assembly master mix, pCMV-dR8.2 dvpr (Addgene; 8455; gift from Bob Weinberg), pCMV-VSV-G (Addgene; 8454; gift from Bob Weinberg), and pAdVAntage (Promega). Supernatants were collected at 48 h post-transfection, filtered, and stored at −80°C until use. Adenovirus serotype 5, clone Ad5-CMV-hACE2/RSV-eGFP (NR-52390), was kindly provided through BEI Resources.
CRISPR screens.
A549 cells were stably transduced with lentivirus from lentiCas9-BLAST (Addgene; 52962; gift from Feng Zhang) and subsequently selected using blasticidin. Next, a total of 240 million A549-Cas9 cells were transduced with the lentivirus of the human GeCKO v2 library (Addgene; 1000000049; gift from Feng Zhang) (48) at a MOI of 0.4 and selected using puromycin for 7 days. A total of 60 million cells of the A549 CRISPR KO library were collected for genomic DNA (gDNA) extraction to assess the reference gRNA distribution. Another 60 million cells were transduced with rAAV2-GFP at a MOI of 10,000 vg/cell, which resulted in an ~45 to 50% transduction rate. The top 10 to 15% GFP+ cells were sorted 3 dpi and replated. After 7 days, the A549 cells were retransduced with 10,000 vg/cell and the top 10 to 15% GFP+ cells were sorted again. A total of ~8 million cells was collected for gDNA extraction.
K562-dCas9-KRAB cells were a gift from Jonathan Weissman. A total of 240 million cells were transduced with lentivirus from the human genome-wide CRISPRi-v2 (Addgene; 83969; gift from Jonathan Weissman) (49) at a MOI of 0.4 using spin inoculation (1,000 × g at 33°C for 2 h) in 6-well plates. Subsequently, cells were selected using puromycin for 5 days. A total of 100 million K562 CRISPRi library cells were collected for gDNA extraction to assess the initial gRNA distribution. Another 60 million cells were spin infected (1,000 × g at 33°C for 2 h) with AAV2-GFP at a MOI of 50,000 vg/cell. The top 15 to 20% GFP+ cells (out of a total %GFP+ of 60%) were sorted and then continued to be cultured. After 7 days, another round of spin infection with AAV2-tdTomato and FACS of the top 10 to 15% tdTomato+ cells were performed. A total of 16 million cells were collected for gDNA extraction.
For both A549 and K562 cells, gDNA was isolated using the Qiagen DNA blood maxi kit (for reference cells) or multiple QIAamp DNA minikit columns (for sorted cells). CRISPR gRNA encoding DNA sequences were amplified in a two-step nested PCR using the HiFi HotStart ReadyMixPCR kit (Kapa Biosystems). In the first PCR step, 36 to 48 reactions and 12 reaction mixtures containing 6 μg gDNA were set up for the reference and sorted samples, respectively, and amplified for 16 cycles. Reactions for each sample were pooled and mixed. In the second PCR step, 4 reaction mixtures containing 5-μL PCR1 product were amplified for 12 cycles using indexed primers. PCR products were gel purified using the QIAquick gel extraction kit and sequenced on an Illumina NextSeq 500 instrument using custom sequencing primers. Primers sequences are listed in Table S4 in the supplemental material.
For analysis, demultiplexed FASTQ files were aligned to the gRNA reference table and enrichment of each gRNA was calculated by comparing the relative abundance in the selected and unselected cell population. Gene enrichment analysis was performed using MaGeCK (50). For STRING analysis (51), the top 100 enriched genes from each CRISPR screen were used as the input, and gene interactions were computed using a STRING confidence score of 0.4. MaGeCK scores were overlaid using Cytoscape 3.9.0 (52), and only the major gene network is displayed in Fig. 1.
Generation of KO cell lines.
DNA oligonucleotides (Integrated DNA Technologies) containing gRNA sequences complementary to MORC3 or SETDB1 genomic loci were annealed and ligated into pX458 (Addgene; 48138; gift from Feng Zhang). A549 cells were transfected with pX458 constructs using Lipofectamine 3000, and 2 days later, GFP-positive cells were single-cell sorted into 96-well plates containing complete media. After several weeks, gDNA was isolated from obtained cell clones using the QuickExtract solution (Lucigen), the gRNA-targeted sites were PCR amplified, PCR products were Sanger sequenced, and sequences were aligned to reference sequences using Geneious Prime. A list of all used gRNA oligonucleotide and genotyping primer sequences can be found in Table S4.
Lentiviral complementation of KO cells.
For the generation of transcomplemented cell lines, lentivirus was generated as described above using plenti-CMV-Puro-DEST containing MORC3 (OriGene Technologies; RC210530) or SETDB1 (OriGene Technologies; RC226620) cDNA sequences. KO cells were transduced in the presence of Polybrene and subsequently selected using puromycin.
Flow cytometry.
Cells were plated in 96-well plates and infected the next day. At indicated time points, cells were trypsinized and analyzed by flow cytometry using a Cytoflex S flow cytometer (Beckman Coulter). At least 4,000 cells were recorded per sample, and cells were gated based on forward scatter-side scatter (FSC/SSC), FSC-height/FSC-area (FSC-H/FSC-A; singlets), and finally phycoerythrin (PE; tdTomato) or fluorescein isothiocyanate (FITC; GFP) to determine the percentage of fluorescent (infected) cells using FlowJo 10 software. Moreover, the median fluorescence intensity (MFI) of the fluorescent cells was determined using FlowJo 10. In all experiments, uninfected cells served as a negative control for gate setting.
Quantification of intracellular AAV genome copies.
UM-SCC1 or A549 cell lines were plated in 12-well plates and transduced with rAAV2-tdTomato at a MOI of 10,000 vg/cell the next day. At 48 h post-transduction, cells were washed with phosphate-buffered saline (PBS) 3 times, trypsinized, and pelleted by centrifugation. Cellular and rAAV DNA was extracted from cell pellet using the DNeasy blood and tissue kit (Qiagen). A total of 2 μL of purified DNA was used as the input to quantify AAV genome copies via qPCR on a Bio-Rad CFX96 Touch system. AAV genomes copies were determined by amplification with primers targeting the AAV2 ITR region and normalized to the cellular RPPH1 locus copy number using IDT PrimeTime qPCR primers. qPCR primer sequences can be found in Table S4.
RT-qPCR.
For quantification of transgene expression, AAV- or lentivirus-transduced cells were harvested using the Power SYBR green cells-to-CT kit (Invitrogen). After reverse transcription, quantitative PCR was performed on a Bio-Rad CFX96 Touch system and viral transgene levels (GFP or tdTomato) were normalized to cellular 18S levels. For quantification of siRNA knockdown of MORC3 and SETDB1 mRNA levels, cells were harvested 4 days post-siRNA transfection using the Power SYBR green cells-to-CT kit (Invitrogen). IDT PrimeTime qPCR primers for MORC3 and SETDB1 were used, and expression levels were normalized to 18S. QPCR primer sequences can be found in Table S4.
Cell proliferation assay.
WT, MORC3 KO, and SETDB1 KO cells were seeded in 96-well plates at a density of 8,000 cells per well. At 24, 48, and 72 h postplating Cell Titer Glo 2.0 solution (Promega) was added on top of the cell media and incubated for 10 min. After that step, 100 μL was transferred to a white 96-well microplate and luminescence was measured using on an EnVision plate reader (PerkinElmer).
siRNA transfection.
ON-TARGETplus nontargeting control pool, ON-TARGETplus human MORC3 siRNA SMARTpool, and ON-TARGETplus human SETDB1 siRNA SMARTpool were obtained from Horizon Discovery.
A total of 10,000 BJ fibroblasts were plated in 96-well plates to reach ~50% confluence by the next day. Then, cells were transfected with siRNA pools using 0.3 μL RNAiMax reagent (Invitrogen), 30 nM siRNA, and 20 μL Opti-MEM (Gibco) per well of a 96-well plate. At 4 days post-transfection, cells were transduced with rAAV, and rAAV transgene expression was quantified by flow cytometry at 36 h post-transduction. In parallel, transfected but untransduced cells were harvested for analysis of mRNA knockdown.
Data availability.
The raw sequencing data for the A549 CRISPR KO and K562 CRISPRi screens are available at NCBI under the BioProject identifier (ID) PRJNA852611.
ACKNOWLEDGMENTS
We thank Nicole Paulk (UCSF), Jan Carette (Stanford University), and James Zengel (Stanford University) for helpful discussions; the Biohub Genomics Platform for help with sequencing of the CRISPR screen samples; the UNC Gene Therapy Center for providing rAAV vectors; Jonathan Weissman (UCSF) and Feng Zhang (Broad Institute) for providing the CRISPRi and CRISPR KO library plasmids; and Leslie Wakefield and the Fanconi Anemia Research Fund at Oregon Health & Science University for providing FANCA KO and transcomplemented cell lines.
A.M.N. and A.S.P. performed all experiments and analyzed data. A.S.P. designed the study and wrote the manuscript.
We have declared no competing interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Andreas S. Puschnik, Email: andreas.puschnik@czbiohub.org.
Felicia Goodrum, The University of Arizona.
REFERENCES
- 1.Srivastava A, Lusby EW, Berns KI. 1983. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J Virol 45:555–564. 10.1128/JVI.45.2.555-564.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samulski RJ, Muzyczka N. 2014. AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol 1:427–451. 10.1146/annurev-virology-031413-085355. [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin SK, Collis P, Hermonat PL, Muzyczka N. 1988. Adeno-associated virus general transduction vectors: analysis of proviral structures. J Virol 62:1963–1973. 10.1128/JVI.62.6.1963-1973.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D, Tai PWL, Gao G. 2019. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 18:358–378. 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colella P, Ronzitti G, Mingozzi F. 2018. Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther Methods Clin Dev 8:87–104. 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greig JA, Breton C, Martins KM, Zhu Y, He Z, White J, Bell P, Wang L, Wilson JM. 2022. Loss of transgene expression limits liver gene therapy in primates. bioRxiv. 10.1101/2022.03.24.485675.. [DOI]
- 7.Ceccaldi R, Sarangi P, D'Andrea AD. 2016. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17:337–349. 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 8.Maréchal A, Zou L. 2013. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5:a012716. 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai M-Y, Dunn CE, Chen W, Kochupurakkal BS, Nguyen H, Moreau LA, Shapiro GI, Parmar K, Kozono D, D'Andrea AD. 2020. Cooperation of the ATM and Fanconi anemia/BRCA pathways in double-strand break end resection. Cell Rep 30:2402–2415.e5. 10.1016/j.celrep.2020.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Göttgens B, Dougan G, Dawson MA, Lehner PJ. 2015. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348:1481–1485. 10.1126/science.aaa7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, Wysocka J. 2018. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553:228–232. 10.1038/nature25179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tchasovnikarova IA, Timms RT, Douse CH, Roberts RC, Dougan G, Kingston RE, Modis Y, Lehner PJ. 2017. Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat Genet 49:1035–1044. 10.1038/ng.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, Lehner PJ. 2016. ATF7IP-mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep 17:653–659. 10.1016/j.celrep.2016.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y, Wang GZ, Cingöz O, Goff SP. 2018. NP220 mediates silencing of unintegrated retroviral DNA. Nature 564:278–282. 10.1038/s41586-018-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decorsière A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, Livingston CM, Niu C, Fletcher SP, Hantz O, Strubin M. 2016. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531:386–389. 10.1038/nature17170. [DOI] [PubMed] [Google Scholar]
- 16.Dupont L, Bloor S, Williamson JC, Cuesta SM, Shah R, Teixeira-Silva A, Naamati A, Greenwood EJD, Sarafianos SG, Matheson NJ, Lehner PJ. 2021. The SMC5/6 complex compacts and silences unintegrated HIV-1 DNA and is antagonized by Vpr. Cell Host Microbe 29:792–805.e6. 10.1016/j.chom.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP, Hale CJ, Feng W, Michaels SD, Frand AR, Pellegrini M, Dekker J, Kim JK, Jacobsen SE. 2012. MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336:1448–1451. 10.1126/science.1221472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sloan E, Orr A, Everett RD. 2016. MORC3, a component of PML nuclear bodies, has a role in restricting herpes simplex virus 1 and human cytomegalovirus. J Virol 90:8621–8633. 10.1128/JVI.00621-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groh S, Milton AV, Marinelli LK, Sickinger CV, Russo A, Bollig H, de Almeida GP, Schmidt A, Forné I, Imhof A, Schotta G. 2021. Morc3 silences endogenous retroviruses by enabling Daxx-mediated histone H3.3 incorporation. Nat Commun 12:5996. 10.1038/s41467-021-26288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Webster ALH, Sanders MA, Patel K, Dietrich R, Noonan RJ, Lach FP, White RR, Goldfarb A, Hadi K, Edwards MM, Donovan FX, Jung M, Sridhar S, Fedrigo O, Tian H, Rosiene J, Heineman T, Kennedy JA, Bean L, Rosti O, Tryon R, Gonzalez A-M, Rosenberg A, Luo J-D, Carrol T, Velleuer E, Rastatter JC, Wells SI, Surrallés J, Bagby G, MacMillan ML, Wagner JE, Cancio M, Boulad F, Scognamiglio T, Vaughan R, Koren A, Imielinski M, Chandrasekharappa S, Auerbach AD, Singh B, Kutler DI, Campbell PJ, Smogorzewska A. 2021. Fanconi anemia pathway deficiency drives copy number variation in squamous cell carcinomas. bioRxiv. 10.1101/2021.08.14.456365. [DOI]
- 21.Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. 2021. Viral vector platforms within the gene therapy landscape. Sig Transduct Target Ther 6:53. 10.1038/s41392-021-00487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins PL, Kyle KE, Egawa T, Shinkai Y, Oltz EM. 2015. The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc Natl Acad Sci USA 112:8367–8372. 10.1073/pnas.1422187112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Alencastro G, Puzzo F, Pavel-Dinu M, Zhang F, Pillay S, Majzoub K, Tiffany M, Jang H, Sheikali A, Cromer MK, Meetei R, Carette JE, Porteus MH, Pekrun K, Kay MA. 2021. Improved genome editing through inhibition of FANCM and members of the BTR dissolvase complex. Mol Ther 29:1016–1027. 10.1016/j.ymthe.2020.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zentilin L, Marcello A, Giacca M. 2001. Involvement of cellular double-stranded DNA break binding proteins in processing of the recombinant adeno-associated virus genome. J Virol 75:12279–12287. 10.1128/JVI.75.24.12279-12287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz RA, Palacios JA, Cassell GD, Adam S, Giacca M, Weitzman MD. 2007. The Mre11/Rad50/Nbs1 complex limits adeno-associated virus transduction and replication. J Virol 81:12936–12945. 10.1128/JVI.01523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lentz TB, Samulski RJ. 2015. Insight into the mechanism of inhibition of adeno-associated virus by the Mre11/Rad50/Nbs1 complex. J Virol 89:181–194. 10.1128/JVI.01990-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karttunen H, Savas JN, McKinney C, Chen Y-H, Yates JR, Hukkanen V, Huang TT, Mohr I. 2014. Co-opting the Fanconi anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth. Mol Cell 55:111–122. 10.1016/j.molcel.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cherubini G, Naim V, Caruso P, Burla R, Bogliolo M, Cundari E, Benihoud K, Saggio I, Rosselli F. 2011. The FANC pathway is activated by adenovirus infection and promotes viral replication-dependent recombination. Nucleic Acids Res 39:5459–5473. 10.1093/nar/gkr084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spriggs CC, Laimins LA. 2017. FANCD2 binds human papillomavirus genomes and associates with a distinct set of DNA repair proteins to regulate viral replication. mBio 8:e02340-16. 10.1128/mBio.02340-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu S, Phan AT, Mao D, Wang X, Gao G, Goff SP, Zhu Y. 2022. HIV-1 exploits the Fanconi anemia pathway for viral DNA integration. Cell Rep 39:110840. 10.1016/j.celrep.2022.110840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robbez-Masson L, Tie CHC, Conde L, Tunbak H, Husovsky C, Tchasovnikarova IA, Timms RT, Herrero J, Lehner PJ, Rowe HM. 2018. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res 28:836–845. 10.1101/gr.228171.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang GZ, Goff SP. 2017. Transcriptional silencing of Moloney murine leukemia virus in human embryonic carcinoma cells. J Virol 91:e02075-16. 10.1128/JVI.02075-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith-Moore S, Neil SJD, Fraefel C, Linden RM, Bollen M, Rowe HM, Henckaerts E. 2018. Adeno-associated virus Rep proteins antagonize phosphatase PP1 to counteract KAP1 repression of the latent viral genome. Proc Natl Acad Sci USA 115:E3529–E3538. 10.1073/pnas.1721883115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seczynska M, Bloor S, Cuesta SM, Lehner PJ. 2022. Genome surveillance by HUSH-mediated silencing of intronless mobile elements. Nature 601:440–445. 10.1038/s41586-021-04228-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das A, Vijayan M, Walton EM, Stafford VG, Fiflis DN, Asokan A. 2022. Epigenetic silencing of recombinant AAV genomes by NP220 and the HUSH complex. J Virol 96:e02039-21. 10.1128/jvi.02039-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li D-Q, Nair SS, Kumar R. 2013. The MORC family. Epigenetics 8:685–693. 10.4161/epi.24976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xue Y, Gibbons R, Yan Z, Yang D, McDowell TL, Sechi S, Qin J, Zhou S, Higgs D, Wang W. 2003. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA 100:10635–10640. 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoelper D, Huang H, Jain AY, Patel DJ, Lewis PW. 2017. Structural and mechanistic insights into ATRX-dependent and -independent functions of the histone chaperone DAXX. Nat Commun 8:1193. 10.1038/s41467-017-01206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volk A, Crispino JD. 2015. The role of the chromatin assembly complex (CAF-1) and its p60 subunit (CHAF1b) in homeostasis and disease. Biochim Biophys Acta 1849:979–986. 10.1016/j.bbagrm.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geis FK, Sabo Y, Chen X, Li Y, Lu C, Goff SP. 2022. CHAF1A/B mediate silencing of unintegrated HIV-1 DNAs early in infection. Proc Natl Acad Sci USA 119:e2116735119. 10.1073/pnas.2116735119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uhlmann F. 2016. SMC complexes: from DNA to chromosomes. Nat Rev Mol Cell Biol 17:399–412. 10.1038/nrm.2016.30. [DOI] [PubMed] [Google Scholar]
- 42.Yiu SPT, Guo R, Zerbe C, Weekes MP, Gewurz BE. 2022. Epstein-Barr virus BNRF1 destabilizes SMC5/6 cohesin complexes to evade its restriction of replication compartments. Cell Rep 38:110411. 10.1016/j.celrep.2022.110411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bentley P, Tan MJA, McBride AA, White EA, Howley PM. 2018. The SMC5/6 complex interacts with the papillomavirus E2 protein and influences maintenance of viral episomal DNA. J Virol 92:e00356-18. 10.1128/JVI.00356-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Räschle M, Smeenk G, Hansen RK, Temu T, Oka Y, Hein MY, Nagaraj N, Long DT, Walter JC, Hofmann K, Storchova Z, Cox J, Bekker-Jensen S, Mailand N, Mann M. 2015. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 348:1253671. 10.1126/science.1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oravcová M, Nie M, Zilio N, Maeda S, Jami-Alahmadi Y, Lazzerini-Denchi E, Wohlschlegel JA, Ulrich HD, Otomo T, Boddy MN. 2022. The Nse5/6-like SIMC1-SLF2 complex localizes SMC5/6 to viral replication centers. bioRxiv. 10.1101/2022.05.17.492321. [DOI] [PMC free article] [PubMed]
- 46.Hölscher C, Sonntag F, Henrich K, Chen Q, Beneke J, Matula P, Rohr K, Kaderali L, Beil N, Erfle H, Kleinschmidt JA, Müller M. 2015. The SUMOylation pathway restricts gene transduction by adeno-associated viruses. PLoS Pathog 11:e1005281. 10.1371/journal.ppat.1005281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mano M, Ippodrino R, Zentilin L, Zacchigna S, Giacca M. 2015. Genome-wide RNAi screening identifies host restriction factors critical for in vivo AAV transduction. Proc Natl Acad Sci USA 112:11276–11281. 10.1073/pnas.1503607112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, Weissman JS. 2016. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5:e19760. 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS. 2014. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15:554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C. 2021. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49:D605–D612. 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4. Download jvi.01948-22-s0001.pdf, PDF file, 0.7 MB (754.2KB, pdf)
Table S1. Download jvi.01948-22-s0002.xlsx, XLSX file, 1.2 MB (1.2MB, xlsx)
Table S2. Download jvi.01948-22-s0003.xlsx, XLSX file, 1.1 MB (1.1MB, xlsx)
Table S3. Download jvi.01948-22-s0004.xlsx, XLSX file, 0.05 MB (50.8KB, xlsx)
Table S4. Download jvi.01948-22-s0005.xlsx, XLSX file, 0.01 MB (10.2KB, xlsx)
Data Availability Statement
The raw sequencing data for the A549 CRISPR KO and K562 CRISPRi screens are available at NCBI under the BioProject identifier (ID) PRJNA852611.