

SUMMARY
The expansion of introns within mammalian genomes poses a challenge for the production of full-length messenger RNAs (mRNA)s, with increasing evidence that these long AT-rich sequences present obstacles to transcription. Here, we investigate RNA polymerase II (RNAPII) elongation at high resolution in mammalian cells and demonstrate that RNAPII transcribes faster across introns. Moreover, we find that this acceleration requires the association of U1 snRNP (U1) with the elongation complex at 5’ splice sites. The role of U1 to stimulate elongation rate through introns reduces the frequency of both premature termination and transcriptional arrest, thereby dramatically increasing RNA production. We further show that changes in RNAPII elongation rate due to AT-content and U1 binding explain previous reports of pausing or termination at splice junctions and the edge of CpG islands. We propose that U1-mediated acceleration of elongation has evolved to mitigate the risks that long, AT-rich introns pose to transcript completion.
eTOC Blurb:
How does RNAPII transcribe long mammalian genes, avoiding premature termination or arrest within expansive introns? Mimoso and Adelman demonstrate that splicing factor U1 snRNP increases RNAPII elongation rate within AT-rich introns, thereby reducing the likelihood of RNAPII termination or arrest. U1 snRNP is thus critical for synthesis of long genes.
Graphical Abstract
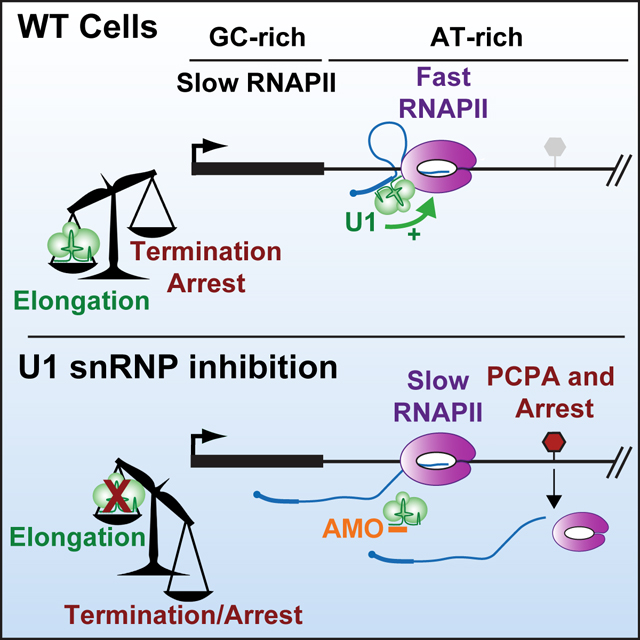
INTRODUCTION
The formation of mature mRNA relies on processive transcription by RNAPII from the transcription start site (TSS) to the transcript end site (TES), a region encompassing 10–100 kb at many mammalian genes. To achieve this level of processivity, the RNAPII elongation complex is stabilized by the ~9 nt hybrid formed between the nascent RNA and DNA template,1 as well as extensive contacts between RNAPII and downstream DNA.2,3 Nonetheless, numerous studies have documented the failure of RNAPII to reach the TES of some mRNAs, resulting in formation of truncated, aberrant transcripts.4–10 Notably, premature termination within introns typically results in retention of intronic sequences within the released RNA. In several diseases, inappropriate translation of these retained introns creates neoantigens that impact cellular function and immune surveillance.11,12
Potential obstacles to productive elongation include sequence elements that can elicit premature termination, stalling, or arrest of RNAPII. Mechanistically, any feature that reduces elongation rate can favor termination, since nucleotide addition and termination are in kinetic competition. Thus, to promote full-length mRNA synthesis, eukaryotic cells encode numerous factors that facilitate elongation.7–9,13 Recent studies have demonstrated that longer genes are more sensitive to perturbation of RNAPII elongation factors, with processivity defects accumulating over gene length.6–9,13,14 Indeed, longer genes, which typically contain several expansive introns, have been reported to exhibit lower expression.15,16 And yet, long genes are enriched in several essential biological processes, such as DNA repair and axon development. Defects in RNAPII elongation could thus impact specific pathways or cell types, in a manner dependent on gene length. In support of this possibility, long genes are more frequently disrupted in neurological disease and cancer progression.17,18
Previous studies of RNAPII elongation proprieties in mammalian cells suggested that RNAPII accelerates across the first ~10kb of the gene body, and that elongation is faster in genes with higher AT content.19,20 However, many questions remain, since these studies employed transcriptional inhibitors to approximate elongation rate which could have resulted in indirect effects on the transcription machinery.19,20 In addition, due to technical limitations, earlier work was biased toward the subset of genes > 100kb, limiting insights into the factors or sequence features that broadly influence elongation rate.
Intriguingly, single-molecule studies using purified RNAPII reported faster elongation rates through GC-rich sequences and suggested that high GC content decreases the likelihood of extended pausing by RNAPII.21 Indeed, AT-rich sequences result in weaker RNA-DNA hybrids that destabilize the elongation complex, and AT-stretches fail to form stable RNA secondary structures that prevent RNAPII backtracking.21–23 Moreover, AT-rich regions inherently contain sequences that resemble polyadenylation signals (PASs, consensus A[A/U]UAAA) typically found at TESs. Recognition of these cryptic PASs by the Cleavage and Polyadenylation (CPA) machinery can elicit cleavage of the nascent RNA and subsequent premature termination of RNAPII.24,25 Thus, data suggesting that RNAPII transcribes AT-rich sequences more rapidly in vivo imply that there are unidentified accessory factors facilitating such elongation in cells, that are not present in purified systems.
The idea that RNAPII transcribes certain sequences, e.g., AT-rich regions, at different rates has direct implications for our understanding of transcription across mammalian introns. Introns, which often comprise of 80–90% of a mammalian gene,26 are largely removed during transcription by the spliceosome27–32, and many groups have reported crosstalk between the splicing and transcription machineries.27 How the spliceosome influences transcription has become an active area of research, and the spliceosome has been implicated in regulating transcription initiation33–36, pause release37, and termination5,38. Furthermore, U1 small nuclear ribonucleoprotein (U1 snRNP, called hereafter U1) has been proposed to stimulate mature mRNA formation in both splicing-dependent and splicing-independent manners.5,23,33,34,39 Although the mechanism of U1 splicing-independent action is unclear, one model suggests that U1 prevents premature termination within introns by blocking recognition of cryptic PASs by the CPA machinery.4,14,24,40–43 The prevention of premature cleavage and polyadenylation (PCPA) by U1 has been reported to influence viral gene expression, neuronal cell activation, and the progression of cancer,5,38,44 raising considerable interest in understanding how this might be achieved.
In this study we investigate RNAPII elongation properties at unprecedented resolution, in the absence of transcription inhibitors, and demonstrate that RNAPII elongates faster across AT-rich introns in mammalian cells. We discover a central role for U1 in this process and demonstrate that without U1 to stimulate RNAPII acceleration in AT-rich regions, RNAPII is susceptible to multiple elongation defects including arrest and premature termination. Overall, our data elucidate how DNA sequence and U1 modulate transcription and reveal new features of RNAPII elongation across long mammalian genes.
RESULTS
RNAPII elongation rate is highly variable across gene bodies
To rigorously define the role of sequence content on RNAPII elongation, we leveraged improved nascent RNA sequencing approaches to measure RNA synthesis and RNAPII density in mouse embryonic stem cells (mESCs). We monitored the rate of RNA synthesis using Transient Transcriptome (TT)-seq, which involves metabolic labeling of RNA during a short pulse of 4sU, followed by selective enrichment of newly synthesized, 4sU-labeled RNA.45 We measured the density of engaged RNAPII with Precision Run-On (PRO)-seq. This strategy allows for single-nucleotide-resolution mapping of active RNAPII complexes through the transcriptional incorporation of a single biotinylated-NTP, which is used to both halt transcription and isolate nascent RNAs.46
Investigation of TT-seq and PRO-seq signals revealed considerable variability across genes. As depicted at the example gene Rbm14, TT-seq signal showed a gradual, but non-uniform, increase within the gene (Figure 1A), in general agreement with reports describing RNAPII acceleration across gene bodies.19 PRO-seq signal decreased sharply downstream of the promoter region, consistent with increased elongation rate as RNAPII escapes from pausing and transitions to productive elongation (Figure 1A). However, PRO-seq signal then increased at locations within the gene body, particularly when RNAPII encountered an internal exon (Figure 1A). Notably, graphing the GC composition across Rbm14 (Figure 1A), revealed that regions with higher GC density, such as promoter regions and exonic sequences, coincided with higher PRO-seq signal.
Figure 1. RNAPII elongation is slower in GC-rich sequences.
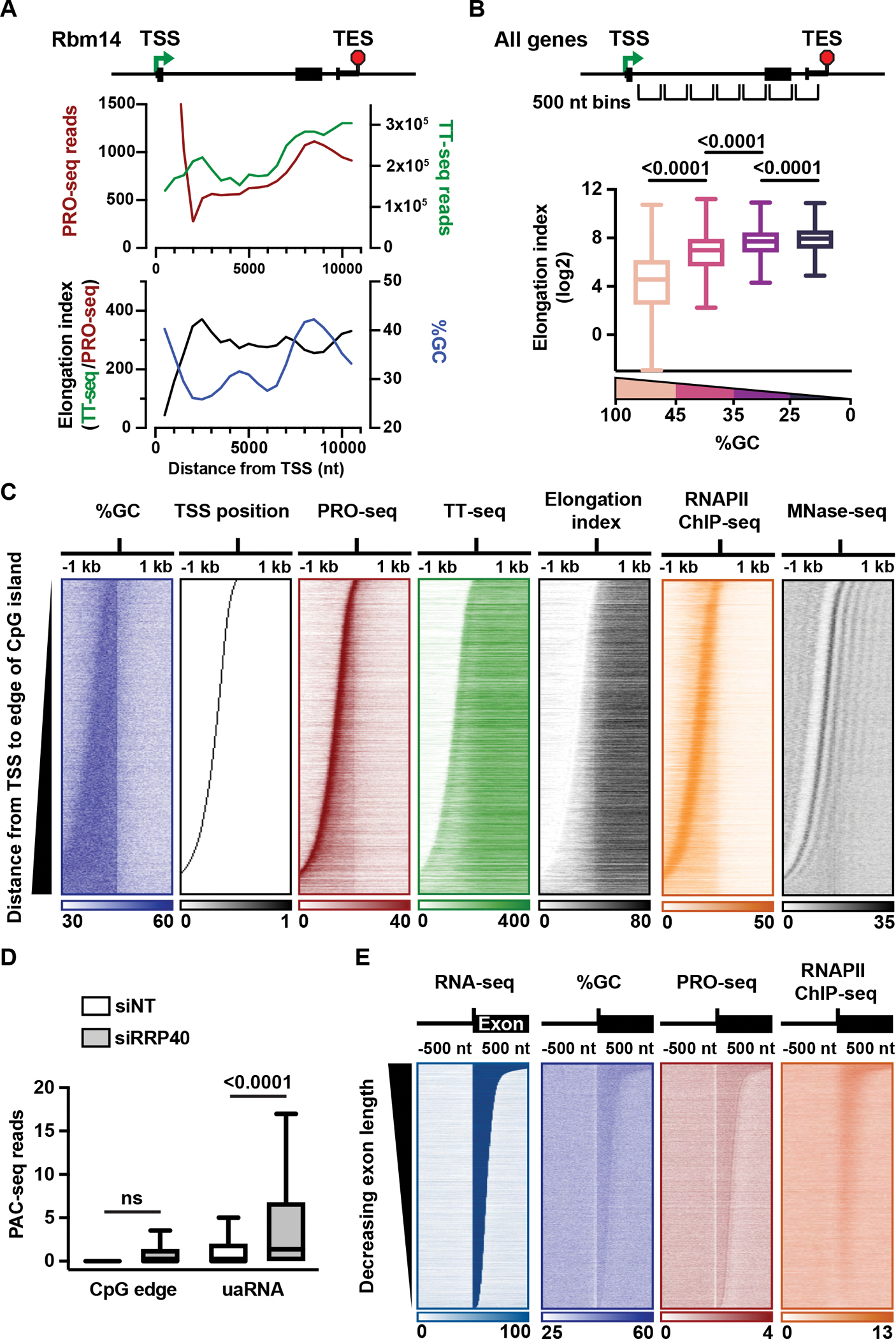
(A) Indicated data are shown in 500 nt bins across an example gene.
(B) Box plot depicts the relationship between elongation index and %GC across active protein coding genes (N = 12,327 intron-containing genes > 1kb). Genes were divided into 500 nt bins starting at the TSS and extending across the gene body, with bins containing the TSS, TES or below read count thresholds removed. Bins were separated into four groups based on %GC (Highest to lowest %GC: N = 2,452; 40,982; 62,740; 3,457). P-values from Mann-Whitney test. (C) Heatmaps of the indicated data are shown for active genes with promoters that overlap a CpG island (N = 9,768). Data is aligned to the downstream edge of the CpG island and summed in 25 nt bins. Genes are ranked by increasing distance from the TSS to the CpG edge.
(D) For genes in (C) with an identified upstream antisense TSS (uaTSS; N = 7,449), PAC-seq reads from control (siNT) and RRP40-depleted (siRRP40) cells were summed in the following regions: CpG edge +/− 500 nt; uaRNA, uaTSS to +1 kb. P-values are from paired t-test.
(E) Heatmaps of indicated data are aligned to the 3’SS of internal introns (N = 28,019) and ranked by length of the downstream exon. Reads are shown in 10 nt bins.
To further investigate the relationship between elongation rate and GC content, we calculated an elongation index across Rbm14, (Figure 1A), by dividing the signal for RNA synthesis (TT-seq) by the signal representing RNAPII density (PRO-seq) (as previously described8,37,47). This elongation index, which allows us to infer relative rates of elongation across genes, reveals that regions with low GC content have higher elongation index, and that a dip in elongation index is observed over the GC-rich internal exon. These data indicate that RNAPII elongation rate can fluctuate both up and down across gene bodies, rather than uniformly increasing, and suggest that the observed fluctuations could be driven by GC content.
Sequence content strongly influences RNAPII elongation behavior
To comprehensively probe the relationship between GC content and RNAPII elongation, we analyzed all 500 nt bins within active protein coding genes (Figures 1B and S1A). This analysis revealed markedly slower elongation indices within regions of high GC content, indicating that the elongation behavior observed at Rbm14 is generalizable. Further, we found slower elongation indices in GC-rich regions independent of RNAPII position within the gene body, and when considering introns and exons separately (Figures S1B, S1C and S1D). These results were recapitulated in human embryonic kidney (HEK293T) cells (Figure S1E).
Given the striking relationship between high GC content and slower transcription elongation, we probed how the high GC content of CpG islands affects RNAPII behavior. Heatmaps of active promoters overlapping a CpG island were aligned at the downstream edge of the CpG island, and rank ordered by distance between the TSS and CpG edge (Figure 1C). PRO-seq signal revealed the anticipated peak of paused RNAPII proximal to the promoter, with continued elevated signal extending to the CpG edge (Figure 1C). Past the CpG edge, a significant drop in PRO-seq signal is observed, in both mESCs and HEK293T cells (Figures 1C, S1F and S1G). To validate our measurement of RNAPII density, we performed RNAPII ChIP-seq. This also shows a significant drop in ChIP-seq signal at the CpG edge (Figure 1C), signifying a bona fide alteration in RNAPII density in this region. The marked reduction in RNAPII signal at the CpG edge has been previously attributed to transcriptional arrest or premature termination.4,48 Importantly, if RNAPII arrests or terminates at the CpG edge, that should yield a concomitant decrease in RNA production. However, we find an increase in TT-seq signal in this region (Figures 1C, S1F and S1G), indicative of continued or even elevated RNA synthesis. Thus, our data argues against the CpG edge serving as a site of pausing or termination. Instead, the elongation index suggests a marked acceleration of RNAPII near the CpG edge (Figures 1C, S1F and S1G). Together, our findings demonstrate that RNAPII accelerates past the CpG edge as RNAPII moves from the GC-rich promoter region into AT-rich sequences downstream.
The CpG edge is not a site of high nucleosome occupancy or premature termination
Previous work has proposed that strongly positioned nucleosomes at the CpG edge present obstacles to RNAPII elongation.4,49,50 To evaluate this possibility, we investigated nucleosome occupancy at the CpG edge. Heatmap analysis of MNase-seq signal revealed a strongly positioned +1 nucleosome located ~100 bp downstream of the TSSs (Figure 1C), supporting a relationship between the position of the +1 nucleosome and the paused RNAPII.51 However, the CpG edge did not correspond to a clear accumulation in MNase-seq signal, implying that the CpG edge is not a site of prominent nucleosome occupancy or positioning. Thus, we find that RNAPII typically encounters the first highly positioned nucleosome well before reaching the CpG edge, arguing against a role for the +1 nucleosome in mediating elongation properties as RNAPII elongates out of the CpG island.
To evaluate levels of CPA-mediated termination near the CpG edge, we isolated RNAs with poly-A tails using Poly-A-Click (PAC)-seq.52 To capture unstable RNAs, these experiments were performed in mESCs depleted of the RNA exosome using siRNA against exosome subunit RRP40 (Figure S2A), and cells treated with a non-targeting siRNA. As anticipated, 95% of genes exhibit PAC-seq signal overlapping the TES under control conditions (Figure S2B), consistent with RNA cleavage and polyadenylation at gene 3’ ends. These experiments also confirmed the occurrence of premature termination and formation of short, unstable polyadenylated RNAs upstream and antisense of mRNA promoters, with PAC-seq reads detectable within these upstream antisense (ua)RNAs primarily after exosome depletion (Figures 1D, S2C, S2D, S2E). However, we observe minimal PAC-seq signal at the CpG edge, even following exosome depletion (Figures 1D and S2F). Together, our data argue against the CpG edge serving as a site of widespread premature termination, and support that the drop in RNAPII density near the CpG edge results from an increase in elongation rate.
RNAPII slows down when transcribing GC-rich exons
Small GC-rich exons within larger AT-rich introns provide an additional opportunity to address the relationship between elongation index and GC content. We thus aligned heatmaps to the 3’ splice sites (SS) of internal exons. RNA-seq signal confirms accurate definition of intron-exon boundaries, and elevated GC content is observed over the length of the exon, as expected (Figure 1E). PRO-seq and RNAPII ChIP-seq signal increase significantly within GC-rich exons as compared to the surrounding AT-rich introns, indicative of slower elongation in exons (Figures 1E and S2G). Importantly, we observed slowed elongation across the length of GC-rich exons, rather than specific pausing of RNAPII at the 5’ or 3’SS, in agreement with recent work.13,32 We conclude that RNAPII elongation rate is highly sensitive to GC content, such that even short internal exons elicit slower elongation.
Inhibition of U1 broadly downregulates intron-containing genes
We next asked whether the splicing machinery might play a role in the increased elongation rates within AT-rich introns. Base pairing of the U1 small nuclear RNA (snRNA) to the 5’SS is the first step in the splicing reaction and occurs shortly after the 5’SS appears in nascent RNA.27,53 92% of genes harbor the 5’SS before, or within 100 nt downstream of the CpG edge (Figure 2A), raising the possibility that U1 could contribute to RNAPII acceleration in this region.
Figure 2. Inhibition of U1 broadly decreases expression of intron-containing genes.
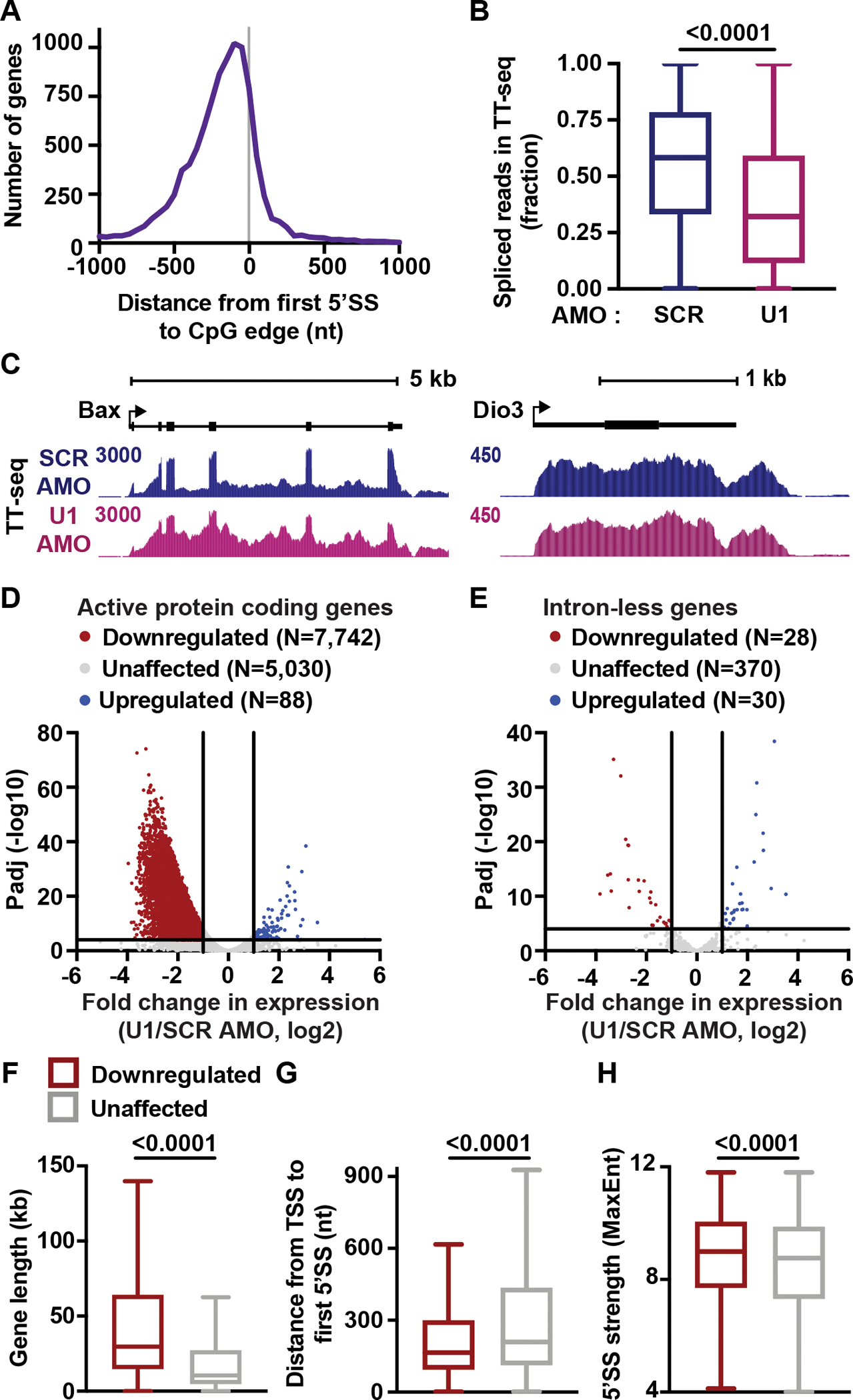
(A) Histogram reporting the distance between 5’SS and CpG edge for genes in Figure 1C. A negative distance indicates that the first 5’SS is upstream of the CpG edge.
(B) Splicing efficiency (SE) for first introns in protein coding genes was calculated as the number of spliced reads divided by the total number of spliced and unspliced reads per intron. The distribution of SE is shown per condition as a box plot. P-values from Wilcoxon test.
(C) TT-seq signal at example genes that have (left) or lack (right) introns.
(D) Volcano plot depicting differentially expressed genes in U1 AMO cells. TT-seq reads were calculated within exons and U1-affected genes were defined by DESeq2 (p <0.0001 and Fold Change >2).
(E) Same as (D) but highlighting intron-less genes.
(F-H) Box plots report the distribution of (F) gene lengths, (G) distances between the TSS and first 5’SS, and (H) the MaxEnt score for first 5’SSs at downregulated and unaffected genes. P-values from Mann-Whitney test.
To functionally deplete U1, we treated mESCs with an antisense morpholino (AMO) complementary to the first 25 nt of the U1 snRNA (U1 AMO). This approach inhibits recognition of the 5’SS by U1, through outcompeting U1 snRNA binding. To determine optimal conditions for U1 inhibition, RNaseH protection assays and Northern blots were performed after electroporating mESCs with U1 AMO or a scrambled (SCR) AMO control (Figure S3A). Treatment of mESCs with 20 μM of U1 AMO for four hours was sufficient to achieve full occupancy of U1 by the complementary AMO (Figure S3B). Notably, these treatments are considerably shorter than those used in past, thereby minimizing indirect effects. To evaluate changes in RNA synthesis after U1 inhibition, we generated TT-seq libraries from SCR and U1 AMO cells and spike-ins were included to enable absolute quantification. Evaluation of intronic reads in TT-seq confirmed that U1 AMO treatment globally reduced splicing efficiency but did not fully abrogate intron removal (Figures 2B and 2C), in agreement with previous work.14
Next, we used TT-seq data to identify protein coding genes whose transcript levels were affected by U1 AMO treatment as compared to control. 7,742 genes, representing ~60% of the active genes interrogated, were significantly downregulated by U1 AMO treatment (Figure 2D). In contrast, only 88 genes were upregulated by U1 AMO. Importantly, investigation of intron-less genes showed that these genes are largely unaffected, with no bias towards downregulation upon U1 inhibition (Figures 2C and 2E). U1 thus plays a positive role specifically at intron-containing genes. We note that our analysis of differentially expressed transcripts following U1 loss employed only reads overlapping exons, to prevent effects of U1 AMO on intron removal from biasing these values. As such, the genes considered unaffected following U1 AMO treatment showed similar exon-level reads but were indeed affected at the level of splicing (Figure S3C).
In agreement with previous work using RNA-seq to define targets of U1,14,41 downregulated genes are significantly longer than unaffected genes (Figure 2F), contain larger first introns, larger total intronic length, and more introns per gene (Figures S3D, S3E and S3F). To further compare downregulated vs. unaffected genes, we evaluated the relative strength and position of the 5’SS. As compared to unaffected genes, downregulated genes have 5’SS motifs that are significantly closer to the TSS and better reflect the consensus sequence (Figures 2G and 2H). The presence of strong, promoter proximal 5’SS motifs at downregulated genes suggests that these genes might have evolved to efficiently recruit U1 during early elongation.
Progressive loss of TT-seq signal across long genes in cells lacking U1
To further explore defects in RNA synthesis following U1 AMO, we generated heatmaps of TT-seq signal across active protein coding genes. Ranking genes by increasing length revealed substantially greater effects of U1 AMO at longer genes (Figure 3A).14 To quantify this effect, we separated genes into quartiles based on length and determined the fold change in RNA synthesis upon U1 AMO treatment for genes in each quartile. TT-seq read density exhibits significant, stepwise decreases upon U1 AMO treatment as gene length increases (Figure 3B). Of note, the longest genes are also the most AT-rich (Figure 3C), and increased AT content at longer genes result from introns representing a larger proportion of gene length (Figure 3D). Investigation of individual genes often revealed a progressive loss of elongating RNAPII across gene bodies, suggesting that long genes can experience a gradual attrition of RNAPII (Figure 3E). These data thus provide opportunities to probe the nature of transcriptional obstacles posed by long stretches of AT-richness in the absence of U1.
Figure 3. U1 AMO causes a progressive loss of TT-seq signal across long genes.
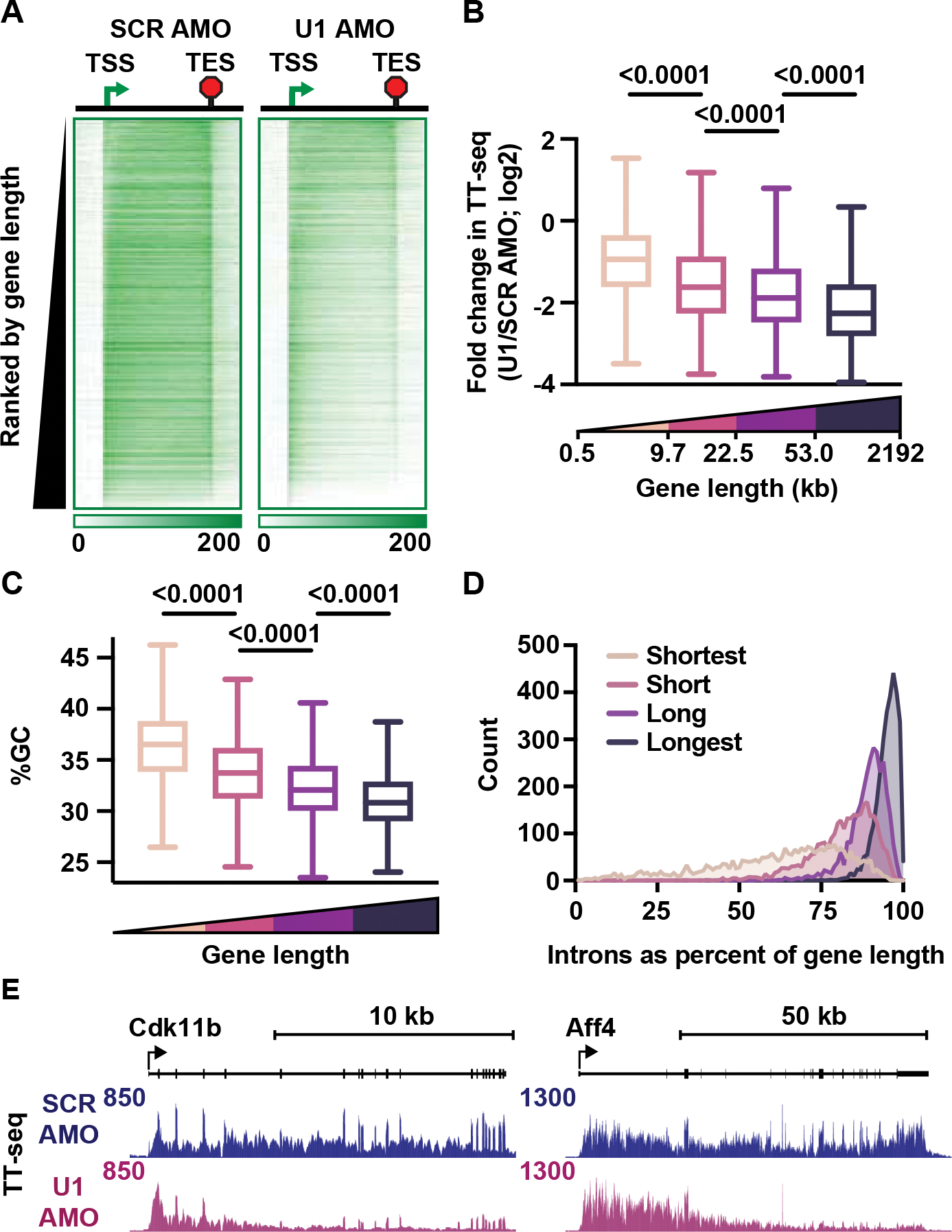
(A) Heatmaps of TT-seq read density at intron-containing genes (N = 12,362), from 2 kb upstream of the TSS to 2 kb downstream of the TES in SCR and U1 AMO conditions. The region between the TSS and TES was scaled by gene length into 100 bins. Genes are ranked by increasing length.
(B-D) Genes in A were divided into quartiles based on gene length (Medians per quartile: 5.28 kb, 14.98 kb, 33.04 kb, 97.45 kb). Box plots depict the (B) fold changes in TT-seq signal (summed between the TSS and TES) between U1 and SCR AMO cells, and (C) average GC content per quartile. P-values from Mann-Whitney test. (D) Total intron length as a percentage of gene length is shown for genes in each quartile as a histogram.
(E) TT-seq signal at example genes.
U1 can stimulate either transcription initiation or elongation, in a gene-specific manner
To interrogate active elongation complexes directly, PRO-seq libraries were generated in SCR or U1 AMO treated cells and spike-ins included to allow for accurate normalization. Notably, most genes downregulated in TT-seq also showed lower PRO-seq signal across gene bodies (Figure S4A), confirming that reduced TT-seq signal after U1 inhibition was driven by impaired transcription rather than altered RNA stability. However, transcriptional defects at downregulated genes were not uniform in U1 AMO cells. Some genes, like Tdqf1 (Figure 4A) showed a complete abrogation of PRO-seq signal across the gene, including initiating or paused RNAPII. In contrast, genes like Spindoc (Figure 4B) exhibited no notable reduction in promoter RNAPII levels, instead showing a progressive loss of PRO-seq signal across the gene body. Our direct, genomic analyses of nascent RNA in U1 AMO cells thus allow us to investigate the gene sets at which U1 stimulates transcription initiation versus elongation.
Figure 4. U1 can stimulate either transcription initiation or elongation.
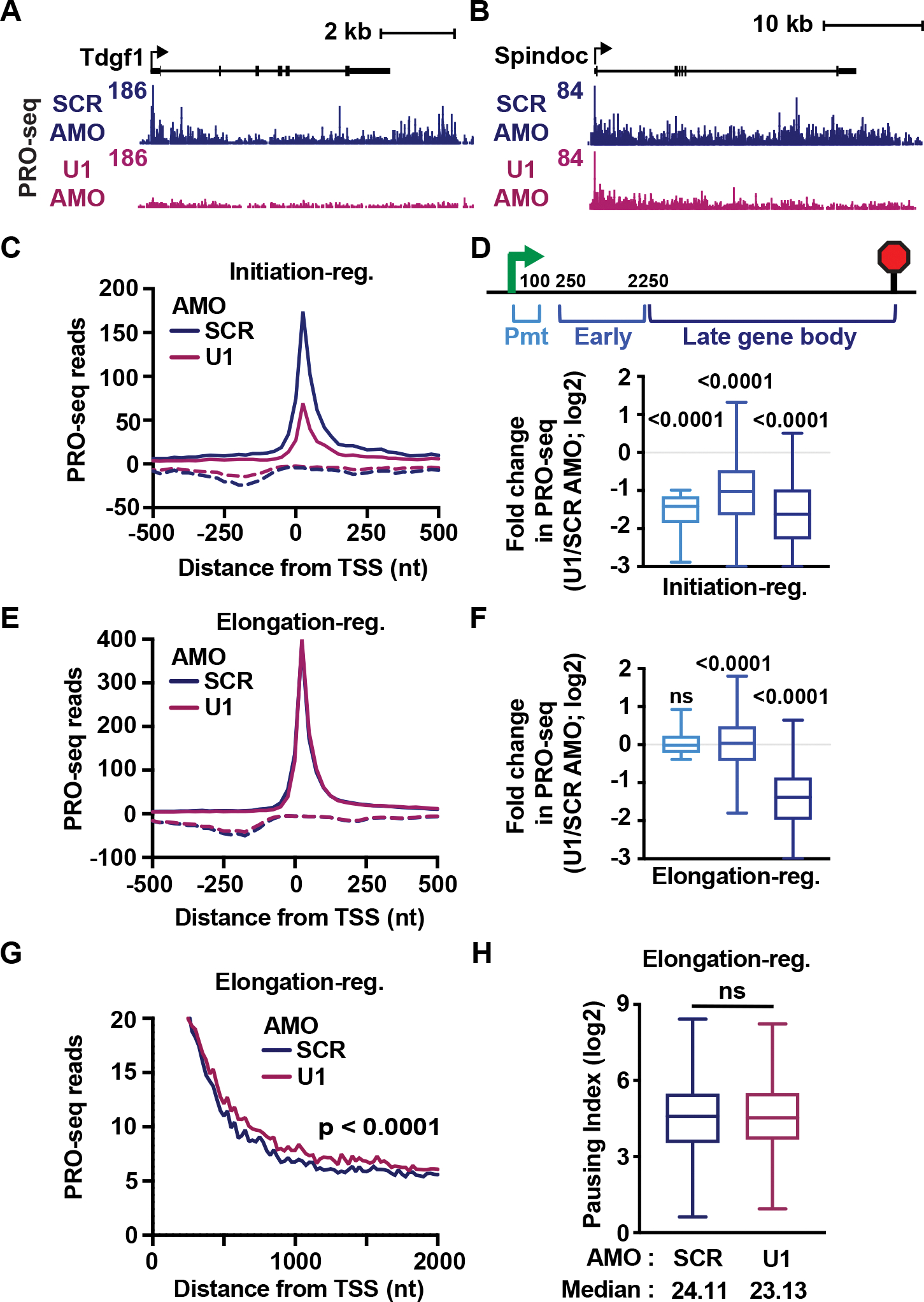
(A-B) PRO-seq signal at an (A) initiation-regulated and (B) elongation-regulated gene.
(C) Average PRO-seq signal per condition at downregulated genes classified as initiation-regulated (N= 1,398). Shown are reads for sense (solid lines) and antisense (dotted lines) strands in 25 nt bins, centered on sense TSSs.
(D) For initiation-regulated genes longer than 2350 nt (N = 1,378), PRO-seq reads were summed in the indicated gene regions. The fold change in PRO-seq signal between conditions is shown as a box plot. P-values from the Wilcoxon test, comparing between SCR and U1 AMO conditions.
(E) Same as (C), but for downregulated genes classified as elongation-regulated (N = 2,696)
(F) Same as (D), but for elongation-regulated genes longer than 2350 nt (N = 2,684).
(G) Same as (E), but zoomed in view of PRO-seq signal. P-value from Wilcoxon test, comparing reads from +500 nt to +2 kb downstream of the TSS.
(H) Pausing index was calculated as the ratio of PRO-seq read density in promoter over early gene body windows. Box plots depict pausing indices at elongation-regulated genes. P-value from Wilcoxon test.
To identify downregulated genes at which U1 AMO suppresses initiation, we selected genes with significant losses in promoter PRO-seq signal in U1 AMO cells (≥2-fold reduction). This revealed 1,398 initiation-regulated genes, which showed lower PRO-seq signal at promoters and across gene bodies (Figures 4C and 4D). We then defined elongation-regulated genes as those downregulated in TT-seq at which U1 inhibition had minimal effect on initiation or establishment of paused RNAPII (<1.3-fold decrease in promoter PRO-seq signal; Figures 4E and 4F promoter window). Interestingly, this set of genes was much larger (N=2,696). For comparison, we also identified a set of unchanged genes, with minimal change (<1.5-fold) in either TT-seq or promoter PRO-seq signal (N=1,004; Figure S4B).
Comparison of these gene groups under control conditions revealed that initiation-regulated genes exhibit lower promoter PRO-seq signals and lower RNA expression than other groups (Figures S4C and S4D). Consistent with low initiation rates, MNase-seq profiles showed narrower, less accessible promoter regions at initiation-regulated genes (Figure S4E) and closer proximity of sense and upstream antisense TSSs (Figure S4F). Previous work has suggested that the splicing machinery can stimulate transcription initiation at both mRNA genes and the cognate uaRNA transcripts,34 supporting models wherein U1 facilitates recruitment of the general transcription machinery.33–35 Consistent with coupling between sense and antisense RNA synthesis,51 we found that loss of U1 selectively reduces PRO-seq signal within uaRNAs adjacent to promoters classified as initiation-regulated (Figures 4C and S4G). Although we did not identify specific sequence features (e.g., core promoter motifs) enriched at initiation-regulated genes, the promoters of initiation-regulated genes are less conserved across mammals than other groups (Figure S4H). This finding is in agreement with reports that splicing stimulates transcription initiation at recently evolved promoters.34 Altogether, we find that initiation-regulated genes have low expression, weak nucleosome depletion and poor sequence conservation. We propose that these genes inherently have slow initiation rates, such that the increased occupancy of the general transcription factors promoted by U1 and/or splicing can stimulate initiation, and thus gene expression.
Inhibition of U1 does not broadly alter pause release
We next focused on elongation-regulated genes, which displayed higher levels of promoter RNAPII and gene expression than initiation-regulated genes under endogenous conditions (Figures S4C and S4D). Interestingly, although U1 AMO treatment elicited significant downregulation of TT-seq and PRO-seq signals across late gene bodies, we observed no reduction in PRO-seq signal within the first 2 kb of these genes (Figures 4F and 4G). In fact, elongation-regulated genes exhibited a modest but statistically significant increase in engaged RNAPII occupancy within this window (Figures 4F, Early window, and 4G). We considered that increased PRO-seq signal within the early gene body might suggest more efficient release of RNAPII from pausing upon U1 AMO treatment, since previous work had suggested that splicing stimulates RNAPII pause release.37 We calculated pausing index, as the ratio of PRO-seq read density at promoters (TSS to +100) over early gene bodies (TSS +250 to +2250). This analysis revealed that U1 AMO treatment caused no significant change in the ratio of paused versus elongating RNAPII at elongation-regulated genes (Figure 4H), arguing against a general requirement for U1 or splicing for the release of paused RNAPII into productive elongation.
RNAPII elongation rate is reduced after U1 inhibition
We next wished to investigate the increased PRO-seq density observed after U1 inhibition within the first several kb of elongation-regulated genes, and across gene bodies at unchanged genes (Figure 5A). This result suggests that RNAPII might elongate more slowly in the absence of U1 association, thereby exhibiting increased residence time within genes. To formally evaluate this possibility, we calculated elongation index across elongation-regulated and unchanged genes. Notably, genes unchanged in TT-seq exhibited a significant increase in PRO-seq read density after U1 inhibition (Figure 5B). This higher density of elongating RNAPII at unchanged genes, in the absence of increased RNA output, yields a broad and significant decrease in elongation index calculated from U1 AMO treated samples (Figure 5B). Similarly, elongation-regulated genes showed significantly slower elongation indices in the absence of U1. To ensure that altered intron retention was not contributing to these findings, these results were confirmed using TT-seq and PRO-seq reads overlapping only introns (Figure S5A).
Figure 5. RNAPII elongation index is reduced in AT-rich regions upon U1 inhibition.
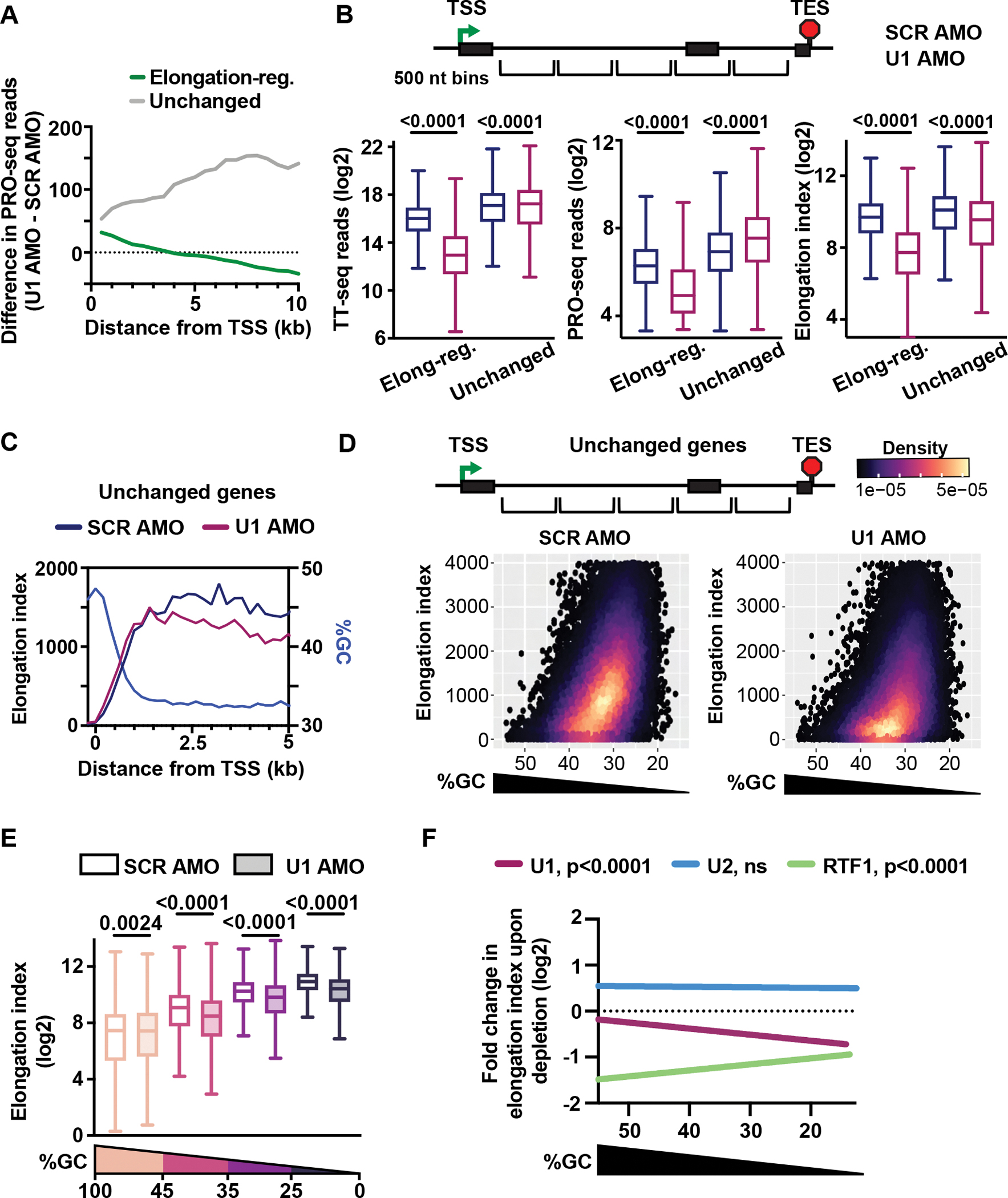
(A) Metagene plot of the difference in PRO-seq reads between SCR and U1 AMO cells for elongation-regulated (N = 2,399) and unchanged (N = 460) genes longer than 10 kb. Reads were summed in 500 nt bins, aligned to TSSs.
(B) Box plots depict TT-seq (left) and PRO-seq (middle) read density, and elongation index (right) for 500 nt bins at elongation-regulated (N= 131,315) and unchanged (N= 22,703) genes. P-values from Wilcoxon test.
(C) Metagene plot of Elongation index and %GC at unchanged genes > 5 kb (N = 692). Reads were summed in 200 nt bins, aligned to TSSs.
(D) Density scatter plot depicting %GC and elongation index at unchanged genes for SCR and U1 AMO cells (500 nt bins).
(E) Bins from (D) were divided into groups by %GC (Highest to lowest %GC: N = 1889, 8881, 9979, 1954). Box plots depict elongation index for indicated conditions. P-values from Wilcoxon test.
(F) Fold change in elongation index was calculated between control cells and those wherein U1, RTF1 or U2 was inhibited, for bins within unchanged genes (defined for each dataset: U1 as in D; RTF1, N = 65,953; U2, N = 32,078). Scatter plots depicting %GC versus fold change in elongation index were generated (see Figure S5C) and the linear trend lines for each dataset are shown here. P-values from F test.
U1 is required for maximal elongation rates in AT-rich regions
Graphing of elongation index across the first 5 kb of unchanged genes revealed a substantial increase downstream of the TSS in SCR AMO cells (Figure 5C), in agreement with work demonstrating an acceleration of transcription as RNAPII transitions into productive elongation.54 However, U1 inhibition prevents RNAPII from achieving peak elongation index (Figure 5C), suggesting that loading of U1 onto the elongation complex at the 5’SS could help RNAPII accelerate within AT-rich intronic sequences. To test this idea, we compared elongation index with respect to GC content in SCR and U1 AMO conditions at unchanged genes (Figure 5D). As anticipated, faster elongation indices were observed within regions with lower GC content under control conditions (Figures 5D and 5E). However, after U1 inhibition, elongation index is reduced, with the greatest effect observed within AT-rich sequences (Figures 5D, 5E and 5F).
Elongation defects after RTF1 and U2 inhibition are distinct from U1 AMO treatment
We next investigated whether faster elongation indices in AT-rich sequences could be driven by other accessory factors. We took advantage of published datasets wherein RTF1, a subunit of the PAF complex, was rapidly depleted using a degron strategy,8 and wherein branchpoint recognition by U2 was inhibited with pladienolide B.37 Notably, these studies utilized TT-seq to evaluate RNA synthesis and mammalian native elongating transcript (mNET)-seq to monitor RNAPII density in human K562 cells. mNET-seq, an analogous method to PRO-seq, involves sequencing of RNAs associated with immunoprecipitated RNAPII.50 Calculation of elongation index using this orthogonal method confirmed slower elongation within regions of higher GC content in K562 cells (Figure S5B). Next, we calculated the fold change in elongation index within unchanged genes for bins separated by GC content. Consistent with prior work, we find that loss of RTF1 globally reduces elongation index (Figure S5C).8 In contrast to U1 AMO, however, we find that the biggest effects occur within regions of higher GC content (Figure 5F). U2 inhibition causes a modest increase in elongation index, in a manner independent of GC content (Figures 5F and S5C). Thus, we conclude that elongation defects after RTF1 depletion and U2 inhibition are different from those observed after U1 inhibition, supporting the idea that each factor affects elongation through a distinct mechanism. Moreover, since slower elongation in AT-rich regions was not observed when blocking splicing through U2 inhibition, our data demonstrates that U1 stimulates elongation in a manner that is independent of splicing.
Without U1, RNAPII is susceptible to premature termination
Having established that inhibition of U1 causes slower elongation across AT-rich regions, we probed the mechanisms underlying decreased expression of elongation-regulated genes. We used a Hidden Markov Model54 to define sites at which the wave of PRO-seq signal representing elongating RNAPII drops off in U1 AMO conditions. Such transition points (TPs) were detected within 48% of elongation-regulated genes (Figure 6A), including those previously reported to undergo PCPA following U1 inhibition in mESCs4 (Figures 6B and S6A). To globally validate the identified TPs, we generated heatmaps of PRO-seq signal aligned to TSSs (Figures 6C and S6B) which confirm a pronounced drop in signal at TPs in U1 AMO cells. We observe TPs at variable distances from the TSS (Figure 6D) with a median distance of 2.8 kb. Since the 5’SS at these genes is located at a median distance of 137 nt downstream from the TSS, U1 should be fully loaded onto RNAPII before it reaches the TP in control cells (Figure 6D).
Figure 6. Without U1, RNAPII is susceptible to premature termination.
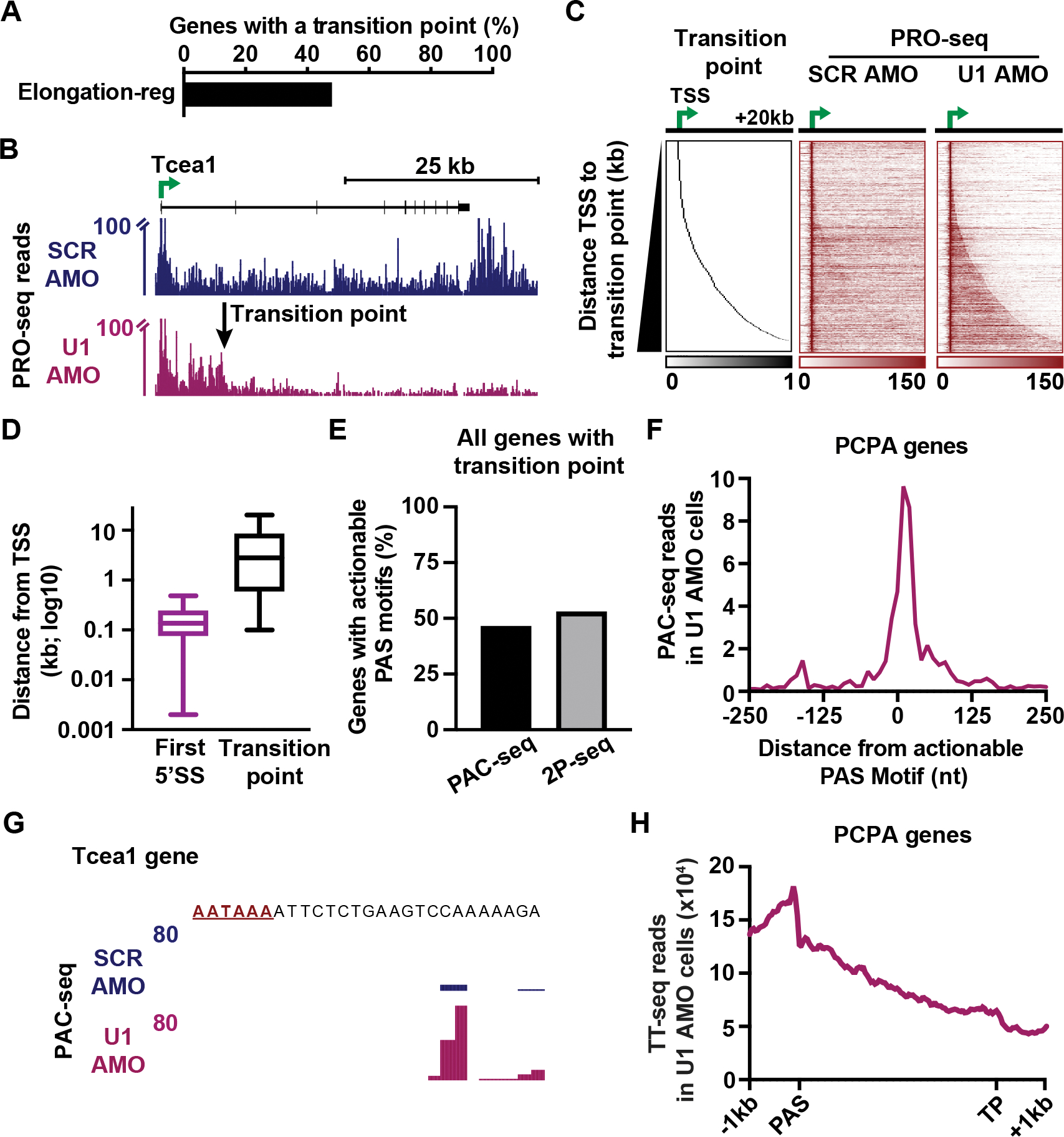
(A) Bar plot depicting the percentage of elongation-regulated genes longer than 10 kb with an identified transition point (N = 1,162 of 2,399 genes).
(B) PRO-seq signal at a gene with a defined TP. Reads are shown in 25 nt bins. Y-axis is truncated to highlight gene body signal.
(C) Heatmaps of PRO-seq at genes with a TP. Data is aligned to TSSs and ranked by distance from the TSS to the TP. Read counts were summed in 250 nt bins.
(D) For genes in C, box plots report the distribution of distances from the TSS to indicated feature.
(E) The percentage of genes with TPs that display an actionable PAS motif in U1 AMO cells based on PAC-seq or 2P-seq.
(F) Metagene plot of PAC-seq signal at actionable PAS motifs in U1 AMO cells. Reads were summed in 10 nt bins.
(G) PAC-seq signal near an actionable PAS motif (red) within the Tcea1 gene.
(H) Metagene plot of TT-seq signal in U1 AMO cells is shown from 1 kb upstream of the actionable PAS to 1 kb downstream of the TP (N = 541 genes with actionable PASs). The region between the PAS and TP was scaled by length into 100 bins.
To identify TPs where the loss of PRO-seq signal following U1 inhibition was a consequence of PCPA, we searched for PAS motifs (using the 10 most common PAS sequences) between the TSS and TP that were associated with PAC-seq reads in U1 AMO cells. Notably, PAC-seq libraries were generated using 4sU-labelled RNA to focus on newly synthesized RNAs. This analysis revealed that 47% of genes with TPs harbor a PAS motif with detectable PAC-seq reads, which we call ‘actionable’ PASs (Figure 6E). This result was confirmed using published data that employed an alternate method (2P-seq) to identify polyadenylated RNAs produced upon U1 inhibition4 (Figure 6E), with strong overlap between datasets (87% of TP genes with actionable PASs by PAC-seq were confirmed by 2P-seq). PAC-seq reads were focused just downstream of the PAS motif (Figure 6F), and PAC-seq signal increased significantly near actionable PASs after U1 AMO treatment compared to control (Figure S6C), as observed at Tcea1 (Figure 6G). Furthermore, TT-seq signal drops near the actionable PAS in U1 AMO cells, consistent with RNA cleavage in this region (Figures 6H). RNA production is reduced beyond the actionable PAS with further decreases at the TP, indicative of transcription termination in this region. The median distance between the actionable PAS and the TP is 3.8 kb (Figure S6D), consistent with distances between PAS motifs and transcription termination sites at gene 3’ ends.55,56 In agreement with earlier work5, TPs associated with PCPA typically occur within introns (94%), and at a median distance of 8.3 kb from the TSS (Figure S6E, PCPA). Altogether, we consider this subset of elongation-regulated genes with actionable PAS motifs to represent genes at which defective U1 and/or 5’ SS recognition leads to PCPA. However, more than half of all TPs fail to show evidence of PCPA (Figure S6F), indicating that premature termination alone cannot explain all the elongation defects observed in U1 AMO cells.
Increased transcriptional arrest is observed in U1 AMO cells
To investigate other elongation defects elicited by U1 inhibition, we evaluated genes with defined TPs, but without PCPA, as observed at Mff and Flrt3 (Figures 7A and S7A). TPs at these genes are predominately located within introns (97%) but were much closer to TSSs than PCPA-associated TPs (Figure S6E, no PCPA; Median distance of 700 nt). We searched for motifs that are enriched around these TPs as compared to mock sites selected at random from elongation-regulated genes without a defined TP. Notably, the distribution of distances between TSSs and mock sites matched those found at TP genes without PCPA to allow for GC content normalization. Interestingly, we found that enriched motifs contain T-stretches and a poly-purine to polypyrimidine pattern (Figures 7B and S7B), both of which resemble previously defined sites of backtracking and transcriptional arrest.57 An arrest motif (RRR(n)YYY(n))57 was found within 150 nt of 91% of TPs without PCPA (Figure 7C), suggesting that TPs at these genes could represent sites of backtracking and arrest of RNAPII. Metagene plots of PRO-seq signal aligned to the arrest site closest to the TP revealed an accumulation of PRO-seq signal upstream and a significant reduction of signal downstream of the arrest motif (Figure 7D and S7C) as observed at Mff and Flrt3. This result is consistent with an increased occurrence of backtracking and arrest at arrest motifs after U1 inhibition. We conclude that the absence of U1 renders the elongation complex prone to transcriptional arrest, consistent with reduced elongation rates resulting in multiple defects in RNA synthesis.
Figure 7. RNAPII undergoes more frequent transcriptional arrest in the absence of U1.
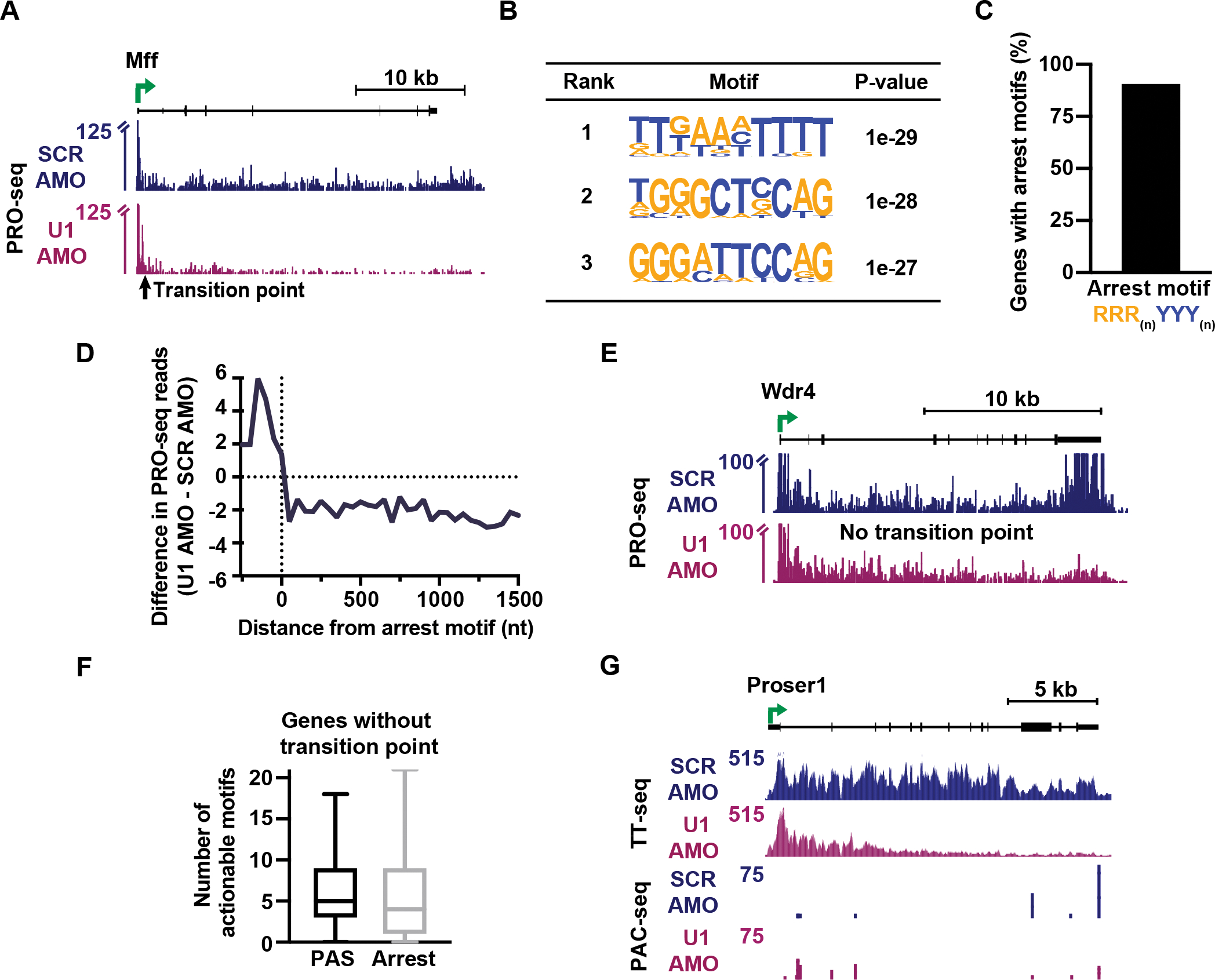
(A) PRO-seq signal at a gene with a TP but no evidence of PCPA. Reads are shown in 25 nt bins. Y-axis is truncated to highlight gene body signal.
(B) Top three enriched motifs within a 100 nt window of the TP at genes without PCPA (N = 621), identified by HOMER.
(C) The percentage of TP genes without PCPA that contain an arrest motif within 150 nt of the TP (N = 565).
(D) Metagene plot of the difference in PRO-seq signal at arrest motifs near TPs at genes without PCPA. To avoid biases from promoter proximal RNAPII signal, only genes with the arrest motif > 400 nt from the TSS are shown (N= 395). Reads were aligned to the final nt of the arrest motif and summed in 50 nt bins.
(E) Same as (A), but for an elongation-regulated gene without a TP.
(F) Box plots showing the number of actionable motifs per elongation-regulated gene without a TP (N = 1,237).
(G) TT-seq and PAC-seq data at an elongation-regulated gene without a TP.
Finally, we focused on the 52% of elongation-regulated genes that do not have a single, defined TP. These genes showed a gradual loss of PRO-seq signal across the gene body, as seen at Wdr4 (Figure 7E). The lack of one dominant TP suggests that RNAPII could be susceptible to arrest and/or termination at multiple sites across these genes. To probe this possibility, we evaluated actionable PAS and arrest motifs usage across these genes and find that genes lacking a TP have a median of 5 actionable PASs (Figure 7F). Proser1 is an example of this behavior, where three actionable PAS motifs are used after U1 AMO treatment and contribute to gene downregulation (Figure 7G). We then defined actionable arrest sites as arrest motifs that elicit ≥ 25% reduction of PRO-seq signal downstream of the motif after U1 AMO treatment. This revealed a median of 4 actionable arrest motifs per elongation-regulated gene without a TP (Figure 7F). Altogether, we conclude that there is not one dominant window where termination or arrest occurs in the absence of U1, but that more than half of affected genes show a gradual attrition of RNAPII during elongation. This continued loss of RNAPII across the gene body agrees with probabilistic termination or arrest throughout the elongation process. In this view, longer genes are more likely to have elongation defects when U1 is perturbed because the extended gene body presents more chances for termination and arrest to occur. To confirm this relationship between elongation index and transcriptional defects, we evaluated PCPA and transcriptional arrest at genes with varying effects on elongation after U1 AMO treatment. These analyses confirm that genes with more pronounced defects in elongation index after U1 inhibition exhibit significantly greater susceptibility to both PCPA and arrest (Figure S7D).
DISCUSSION
In this study, we find that U1 stimulates synthesis of long introns by increasing RNAPII elongation rate. This work answers long standing questions regarding how RNAPII contends with AT-rich introns and how U1 influences gene expression, integrating the two features together to reveal new insights into RNAPII behavior. Based on our data, we propose the following model: To sustain transcription of long genes, RNAPII elongates faster in AT-rich introns compared to GC-rich exons in a manner dependent on U1. Overall, an increase in RNAPII elongation rate over AT-rich introns favors nucleotide addition in kinetic competition over termination or arrest and promotes full length RNA synthesis. Without U1 to stimulate RNAPII elongation rate, the slowed elongation complex is more susceptible to both termination and arrest.
Given our analysis and recent structural work showing a direct interaction between RNAPII and U1,53 we propose that U1 functions as an elongation stimulatory factor. Notably, inhibition of U2 does not elicit similar decreases in elongation index (Figure 5F),37 indicating that the effects observed after U1 inhibition are not an indirect consequence of inhibiting splicing. Instead, our data support a splicing-independent mechanism of transcription stimulation by U1. Future work will be needed to determine how U1 increases elongation rate. For example, U1 could allosterically increase the rate of each NTP addition or prevent backtracking of RNAPII. We envision that interactions of U1 with RNA near the RNA exit channel53 could disfavor re-threading of the RNA into the active site during backtracking. Further, U1-mediated tethering of the 5’SS near RNAPII could create a constrained loop that holds the nascent RNA closer to the active elongation complex, protecting it from binding by the CPA machinery.
We identify a set of genes at which U1 stimulates transcription initiation, in line with earlier work reporting interactions between U1 and the general transcription machinery.33 Our work supports that U1, which associates with RNAPII in early elongation, could help to stabilize the assembly of the pre-initiation complex at these genes, working at both the mRNA and upstream antisense promoters.34 Notably, genes at which U1 increases initiation are those with inherently low expression levels and poor promoter accessibility, suggesting that these promoters are normally weak and rate-limited at the level of transcription initiation. In contrast, a larger group of genes exhibit high levels of promoter-associated RNAPII, indicative of expression regulation during transcription elongation. At these genes U1 AMO has little effect on early phases of transcription and predominantly impacts productive elongation.
We show that, in the presence of U1, elongation index can vary markedly across gene bodies. We observe a slowdown of RNAPII in GC-rich regions using a variety of techniques and biological systems, signifying that this is a global and conserved principle of mammalian transcription. Importantly, RNAPII elongation is slowed over the length of GC-rich exons, rather than pausing specifically at splice sites (Figure 1E). We suspect that the slowing of RNAPII as it enters a GC-rich exon underlies reports of pausing at intron-exon junctions,58 since the change in RNAPII density near 3’SS due to this elongation rate change can be substantial. We propose that the variable %GC and intron content among mammalian genes could underlie the wide range in elongation rates reported for RNAPII in cells,59 and findings that genes with higher intron content exhibit faster elongation rates.19,20 Of note, prior studies have demonstrated that increased elongation rates diminish RNAPII fidelity.60 Intriguingly, our work would suggest that RNAPII fidelity is lower in introns compared to exons. Transcriptional errors might be more readily tolerated in introns since these sequences are excised during splicing whereas exons require high fidelity for translation. Thus, we propose that cells use U1 to stimulate elongation in introns, where the benefit of protecting the polymerase from arrest and termination outweighs the risk of introducing errors into the transcript.
Our work also sheds new light on transcription within CpG islands, and across the CpG edge. Although the edge of the CpG island has been previously proposed to elicit transcriptional arrest or termination, we find instead that the drop in RNAPII density beyond the CpG island results from accelerated elongation. Conceptually, we hypothesize that slow elongation of RNAPII within the CpG island might facilitate the loading of necessary elongation factors before the polymerase enters the first AT-rich intronic sequence.
In summary, we report far-reaching, splicing-independent roles for U1 on the process of transcription. Most critically, the broad occurrence of premature termination and arrest we observe in cells lacking functional U1 would produce truncated transcripts with partial retention of introns. Inappropriate export of such RNAs to the cytoplasm for translation has been shown to generate anomalous or even oncogenic proteins (i.e., neoantigens), which are increasingly implicated in cancer.11,12 Further, given the enrichment of long genes in neuronal function and cell identity61, our demonstration that U1 is required for elongation of long genes gives a fresh perspective on the role of U1 mutations in disease.61–63
Limitations of the Study
Although we minimized the length of U1 AMO treatment compared to previous studies and utilized a SCR AMO control, we cannot rule out secondary effects on gene expression. Additionally, our study took advantage of TT-seq and PRO-seq to infer a relative rate of elongation (as previously described8,37,47). This approach was advantageous to other methods i.e., treating cells with transcription inhibitors that cause cellular stress. However, the calculation of elongation index has limitations. In particular, intron retention caused by U1 AMO would elevate TT-seq signal within introns, and this would increase the elongation index. As such, we are likely under-estimating elongation defects after U1 inhibition using this method. Also, a modest exon bias in TT-seq read distribution (e.g., due to co-transcriptional removal and degradation of introns), could drive an apparent higher elongation index in exons. As such we were unable to confidently use this method to compare elongation indices between exons and introns of similar GC content.
STAR Methods
RESOURCE AVAILABIILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Karen Adelman (Karen_Adelman@hms.harvard.edu)
Materials Availability
Reagents generated in this study are available upon request.
Data and Code Availability
All genomic datasets generated in this study have been deposited to GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
All custom scripts described herein are available on the Adelman Lab GitHub (https://github.com/AdelmanLab). All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
mESCs (CAST/129 hybrid background, female) were cultured on gelatin in KnockOut DMEM (ThermoFisher, 10829018), supplemented with 15% KO serum replacement (ThermoFisher, 10828028), 1X penicillin-streptomycin (MP, TMS-005-C), 1X non-essential amino acids (MP, TMS-001-C), 1% β-ME (MP, ES-007-E), 1X GlutaMAX (ThermoFisher, 35050061), 1000 U/ml LIF (Cell Guidance Systems, GFM200), 1 μM MEK inhibitor (Stemgent, PD0325901), and 3 μM GSK3 inhibitor (Stemgent, CHIR99021). AMO experiments were performed in three clonal mESC lines with a fluorescent splicing reporter at a ncRNA locus, described previously23. mESCs were fed daily and passaged every two days. HEK293Ts were cultured in DMEM (VWR, 45000–312) with 10% FBS (Thermo, 1600044). mESCs and HEK293Ts were cultured at 37°C with 5% CO2. S2 (Drosophila) cells used to generate spike-ins for sequencing experiments were cultured in Shields and Sang M3 medium (Sigma, S3652) supplemented with yeast extract (Sigma, Y-1000), bactopeptone (Difco, 211677) and 10% FBS (Thermo, 1600044). S2 cells were cultured at 27°C. mESC, HEK293T and S2 cells were routinely tested for mycoplasma contamination.
METHOD DETAILS
AMO Delivery
Two million mESCs were electroporated using the Neon 100 μL Kit Transfection System (ThermoFisher Scientific, MPK5000) and the following parameters: Buffer R, Pulse Voltage (v) = 1,200, Pulse Width (ms) = 20 and Pulse Number = 2. mESCs were electroporated with either an antisense morpholino (AMO) complementary to the first 25 nt of the U1 snRNA (U1 AMO: GGTATCTCCCCTGCCAGGTAAGTAT) or a scrambled AMO was used as a control (SCR AMO: CCTCTTACCTCAGTTACAATTTATA).
For all sequencing experiments (n=3 for PRO-seq, n=3 for TT-seq), cells were electroporated with 20 μM of AMO for a total of four hours. For generation of PRO-seq libraries, mESC were electroporated with 20 μM of SCR or U1 AMO, and cells were permeabilized four hours after AMO delivery. For generation of TT-seq libraries, mESC were electroporated with 20 μM of SCR or U1 AMO (n = 3). Three hours and 40 minutes post electroporation, cells were washed twice with PBS and cell media containing 500 μM of 4sU (Sigma-Aldrich, T4509) was added. Cells were returned to the incubator for 20 minutes for a total time of 4-hour exposure of AMO.
RNAseH Protection Assay and Northern Blot
Two million mESCs were electroporated with either SCR or U1 AMO. At harvest, cells were washed twice with PBS, trypsinized, and quenched with DMEM + 10% FBS. Cells were then spun at 1000 RPM for 4 minutes at 4°C. Cells were resuspended in 1 mL cold PBS to remove residual DMEM + 10% FBS. Washed cells were spun at 1000 RPM for 4 minutes at 4°C and resuspended in 41.75 μL of RSB-100 Cell Lysis Buffer (10 mM Tris-Cl pH 7.4, 100 mM NaCl, 2.5 mM MgCl2, 0.5% NP-40, 0.5% Triton X-100). 8.25 μL of the RNAseH Master Mix (Final concentrations, 1X RNaseH Buffer, 3.75 U RNAseH, 5 μM U1 AMO DNA Oligo) was added and the reaction was incubated at 37°C for 25 minutes. 1mL of TRIzol was added and samples were frozen at −80C. RNA was phenol:chloroform extracted and precipitated with isopropanol. U1 snRNA products were visualized by northern blot, as described in Rio et al., 2014.82
Western Blotting
To prepare protein lysates, cells were first washed with PBS, trypsinized, and quenched with DMEM + 10% FBS. Cells were then spun at 1000 RPM for 4 minutes, resuspended in 1 mL cold PBS, and counted. Next, cells were spun at 1000 RPM for 5 minutes at 4°C and resuspended in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with PMSF (1:1000) and 1X Protease Inhibitors (Sigma-Aldrich, 11873580001) at a final cell density of 100,000 cells in 6 μL. Samples were mixed and spun at 10,000 g for 10 minutes at 4°C to pellet insoluble debris. The supernatant was transferred to a new tube, flash frozen, and stored at −80°C. 6 μL of 2X Laemmli buffer supplemented with 5% β-ME was added to 6 μL of lysate. Samples were boiled for 10 minutes at 95°C and loaded onto a 4–20% SDS-PAGE gel (BioRad, 4561096) at room temperature, following manufacturer instructions. Protein was transferred onto a nitrocellulose membrane for 70 minutes at 300 mA at 4°C. The nitrocellulose membrane was blocked in 5% dry milk in 1X TBS-T for 1 hour at room temperature. After which, the membrane was incubated in 5% milk in 1X TBS-T with primary antibody overnight at 4°C. 1:2000 primary antibody dilutions were made to probe for RRP40 (Bethyl Laboratories A303–909A; Rabbit) and Actin (SCBT SC1616; Goat). The next day, blots were washed in 1X TBS-T five times and then incubated in secondary antibody (HRP Goat anti-Rabbit for RRP40, and HRP Donkey anti-Goat for Actin; 1:10,000 dilution). Blots were then washed five times in 1X TBS-T and imaged using SuperSignal West Pico PLUS Chemiluminescent Substrate (ThermoFisher, 34577) on a BioRad ChemiDoc following manufacturer instructions.
RNAi
mESCs were transfected with siRNAs following the RNAiMax (ThermoFisher, 13778075) reverse transfection protocol. First, siRNAs were diluted to 10 μM in H2O. To deplete RRP40, an siRNA pool equally representing four siRNAs from Dharmacon (MQ-064537–01-0010) was generated. For control conditions, mESCs were transfected with Non-targeting Control #2 (D-001210–02-05) from Dharmacon. Next, 9 μL of each 10 μM siRNA stock was diluted in 150 μL of Opti-MEM (ThermoFisher, 31985070). For each reaction, 9 μL of Lipofectamine RNAiMax was diluted in 150 μL Opti-MEM. Next, the diluted siRNAs in Opti-MEM were added to the diluted Lipofectamine in Opti-MEM and mixed. Liposomes were allowed to form for 30 minutes at room temperature. 400,000 cells were added to each reaction. Cells and liposome were mixed and added into a 6 well with media (2 mL final volume). 24 hours post transfection, cells were washed with PBS and feed with fresh media. 48 hours post transfection, cells were harvested for westerns, PAC-seq, and RNA-seq.
PRO-seq library construction and data processing
To generate permeabilized cells for PRO-seq, cells were first washed with room temperature PBS, trypsinized, quenched with cold DMEM + 10% FBS, and immediately placed on ice. All buffers and samples were kept cold on ice and all spins were performed at 1000 RPM and at 4°C unless otherwise noted. Cells were spun down and washed in 10 mL of PBS. Next, cells were spun down and resuspended in 1 mL of Buffer W (10 mM Tris-Cl, pH 8.0, 10 mM KCl, 250 mM sucrose, 5 mM MgCl2, 0.5 mM DTT, 10% glycerol) and passed through a cell strainer (Corning, 352235) to ensure a single cell suspension prior to permeabilization. The 50 mL conical tube was rinsed with an additional 1 mL of Buffer W and the additional 1 mL was passed through the same cell strainer for a final volume of 2 mL Buffer W per sample. The 2 mL single cell suspension was transferred to a 50 mL conical tube. Next, 18 mL of Buffer P (10 mM Tris-Cl, pH 8.0, 10 mM KCl, 250 mM sucrose, 5 mM MgCl2, 0.5 mM DTT, 10% Glycerol, 0.1% Igepal CA-630) was gently added to each sample and cells were nutated for 3 minutes. Cells were then spun for 8 minutes, gently resuspended in 500 μL Buffer F (50 mM Tris-Cl pH 8.3, 40% glycerol, 5 mM MgCl2, 0.5 mM DTT) and transferred to a 1.5mL Lo-Bind Eppendorf tube. The conical tube was rinsed with an additional 500 μL of Buffer F and pooled for a total volume of 1 mL Buffer F per sample. Cells were then spun at 1500 RPM for 4 minutes and resuspended in 200 μL Buffer F. Permeabilized cells were counted, and trypan blue was used to confirm permeabilization efficiency. Cells were resuspended in Buffer F for a final density of 1 million permeabilized cells per 45 μL, flash frozen in liquid nitrogen, and stored at −80°C.
Aliquots of frozen (−80°C) permeabilized cells were thawed on ice and pipetted gently to fully resuspend. For each sample, 1 million permeabilized cells were used for nuclear run-on, and 50,000 permeabilized Drosophila S2 cells was added to each sample for normalization. Nuclear run-on assays and library preparation for mESC libraries were performed as described in Vlaming et al.23 For libraries generated from HEK293Ts, the following modifications were performed: Random hexamer extensions (UMIs) were added to the 3’ end of the 5’ adapter and 5’ end of the 3’ adapter. Adenylated 3’ adapter was prepared using the 5’ DNA adenylation kit (NEB, E2610L) and ligated using T4 RNA ligase 2, truncated KQ (NEB, M0373L; per manufacturer’s instructions with 15% PEG-8000 final) and incubated at 16°C overnight. 180 μL of betaine buffer (1.42 g of betaine brought to 10 mL) was mixed with ligations and incubated 5 minutes at 65°C and 2 minutes on ice prior to addition of streptavidin beads. For all generated libraries, eluted cDNA was amplified 5-cycles (NEBNext Ultra II Q5 master mix (NEB, M0544X) with Illumina TruSeq PCR primers RP-1 and RPI-X) following the manufacturer’s suggested cycling protocol for library construction. A portion of preCR was serially diluted for a test amplification to determine the optimal amplification of final libraries. Pooled libraries were sequenced paired-end using the Illumina NovaSeq platform.
mESC libraries were mapped as follows: using a custom script (trim_and_filter_PE.pl), FASTQ read pairs were trimmed to 41 bp per mate and read pairs with a minimum average base quality score of 20 were retained. Read pairs were further trimmed using cutadapt 1.14 to remove adapter sequences and low-quality 3’ bases (--match-read-wildcards -m 20 -q 10). R1 reads, corresponding to RNA 3’ ends, were then aligned to the spiked in Drosophila genome index (dm6) using Bowtie 1.2.2 (-v 2 -p 6 --best --un), with those reads not mapping to the spike genome serving as input to the primary genome alignment step (mm10, using Bowtie 1.2.2 options -v 2 --best).
For HEK293T libraries, the following mapping pipeline was used: Dual, 6nt Unique Molecular Identifiers (UMIs) were extracted from read pairs using UMI-tools.72 Read pairs were trimmed using cutadapt 1.14 to remove adapter sequences (-O 1 --match-read-wildcards -m 26). The UMI length was trimmed off the end of both reads to prevent read-through into the mate’s UMI, which will happen for shorter fragments. An additional nucleotide was removed from the end of read 1 (R1), using seqtk trimfq (https://github.com/lh3/seqtk), to preserve a single mate orientation during alignment. The paired end reads were then mapped to a combined genome index, including both the spike (dm6) and primary (hg38) genomes, using bowtie2.67 Properly paired reads were retained. These read pairs were then separated based on the genome (i.e., spike-in vs primary) to which they mapped, and both these spike and primary reads were independently deduplicated using UMI-tools.
For all libraries, reads mapping to the reference genome were then sorted, via samtools 1.3.1 (-n), and subsequently converted to bedGraph format using a custom script (bowtie2stdBedGraph.pl) that counts each read once at the exact 3’ end of the nascent RNA. Because R1 in PRO-seq reveals the position of the RNA 3’ end, the “+” and “−“ strands were swapped to generate bedGraphs representing 3’ end positions at single nucleotide resolution.
BedGraphs were normalized using the normalize_bedGraph custom script. For libraries generated from control mESCs and HEK293Ts, no further normalization was performed. For libraries generated under SCR or U1 AMO conditions, we observed a consistent increase in reads mapping to the fly genome (dm6) after U1 inhibition (Average ratio of U1 AMO / SCR AMO = 1.34), indicating a global decrease in RNAPII transcription in U1 AMO cells compared to SCR AMO control. Accordingly, the average ratio of reads mapping to the spike genome between U1 and SCR AMO conditions was used to generate spike normalization factors, as shown below.
| Sample | Total Reads | Reads Mapping to dm6 | Reads Mapping to mm10 | Ratio of the % Spike Return (U1/SCR AMO) | Normalization Factor |
|---|---|---|---|---|---|
| SCR AMO Replicate 1 | 143032721 | 9975634 | 72024239 | 1.392 | 1 |
| U1 AMO Replicate 1 | 152444398 | 16840459 | 81484483 | 1.34 | |
| SCR AMO Replicate 2 | 96631053 | 5157891 | 47270593 | 1.210 | 1 |
| U1 AMO Replicate 2 | 141559066 | 8645133 | 62998303 | 1.34 | |
| SCR AMO Replicate 3 | 160029957 | 10057214 | 75146167 | 1.430 | 1 |
| U1 AMO Replicate 3 | 132869403 | 11170871 | 54672497 | 1.34 |
Combined bedGraphs were generated by summing counts per nucleotide across replicates for each condition (bedgraphs2stdBedgraph). BedGraphs were converted to the bigWig format for visualization.
TT-seq library construction and data processing
After 20-minute 4sU labeling (500 μM), cells were washed with PBS, quickly trypsinized, quenched with cold DMEM + 10% FBS, and immediately placed on ice. All spins were performed at 4°C unless otherwise noted. Cells were spun down for 4 minutes at 1000 RPM, resuspended in 10 mL cold PBS, and counted. Cells were re-spun at 1000 RPM for 4 minutes and resuspended in 2 mL TRIzol. Samples were spiked with 5% 4sU-labeled Drosophila S2 cells (2-hour labeling) resuspended in TRIzol based on cell count.
To generate TT-seq libraries, RNA was phenol:chloroform extracted and ethanol precipitated. Purified RNA was treated with DNaseI (Invitrogen, 18068015) for 30 minutes at room temperature, and ethanol precipitated. RNA integrity was confirmed using the Agilent TapeStation. Next, RNA (50 μg per sample in 50 μL) was chemically fragmented in RNA fragmentation buffer (final concentration: 75 mM Tris Cl, pH 8.3; 112.5 mM KCl; 4.5 mM MgCl2) for 2 minutes at 94°C. To terminate the fragmentation, cold EDTA was added to a final concentration of 50 mM. Samples were incubated on ice for 5 minutes. Fragmentated RNA was then ethanol precipitated, and the fragment size distribution was determined.
Fragmented RNA was biotinylated essentially as described in Duffy et al. 83 with the following modifications: The biotinylation reaction was performed in a total volume of 200 μL and allowed to incubate for 45 minutes in the dark. Excess biotin was removed using chloroform:isoamyl alcohol as per Dölken et al. 84 and MaXtract Heavy gel tubes (Qiagen) were used to separate organic and aqueous phases. Biotinylated RNA was resuspended in 100 μL nuclease-free water and an aliquot of total RNA was taken. In parallel, Dynabeads M-280 Streptavidin (ThermoFisher, 11205D) were rendered RNAse-free in preparation for binding: for each sample, 75 μL of beads were used and treated in batch. Beads were incubated for 10 min in a solution of 100 mM NaOH and 50 mM NaCl, placed on a magnetic stand, and washed twice with 500 μL 100 mM NaCl, twice with 1 X TT-seq wash solution (100 mM Tris-HCl pH 7.5, 10 mM EDTA, 1 M NaCl, 0.05% Tween 20, and 1 μL SuperaseIN RNase Inhibitor (ThermoFisher, AM2694) per 5 mL solution), once in 0.3 X TT-seq wash solution, and finally resuspended in 52 μL 0.3 X TT-seq wash solution per sample, supplemented with 1 μL SuperaseIn RNase Inhibitor.
Biotinylated RNA was heated at 65°C for 5 minutes, placed on ice for 2 minutes, and mixed with 50 μL of prepared beads. Samples were rotated at room temperature in the dark for 30 min. After binding, tubes were placed on a magnetic rack and beads were washed 4 times with 500 μL 1X TT-seq wash solution to remove unbound RNA. To elute, wash solution was removed, beads were resuspended in 50 μL freshly-prepared 0.1 M DTT, and rotated in the dark for 15 min at room temperature. The eluted RNA was recovered, and the elution step was repeated with an additional 50 μL 0.1 M DTT. The combined eluates were purified using the Norgen RNA clean-up and concentration microElute kit (Norgen, 61000) following the manufacturer’s instructions for small RNA enriched samples. Final elution was performed in 14 μL nuclease-free water and the eluate was reapplied to the column for a total of 2 elution steps. 350 ng of enriched RNA was used for library construction with the Illumina TruSeq stranded total RNA kit with RiboZero rRNA depletion, following manufacturer instructions for degraded RNA. Additionally, Superscript III was used for the first strand cDNA synthesis and the reaction was held at 25°C for 10 minutes, 50°C for 15 minutes, 70°C for 15 minutes and held at 4°C. After 5 cycles of PCR, samples were removed from the thermal cycler and a test PCR was performed to determine the optimal number of final cycles. Libraries were pooled and sequenced paired-end 150 bp on the Illumina NovaSeq platform.
Using a custom script (trim_and_filter_PE.pl), FASTQ read pairs were trimmed to 120 bp per mate and read pairs with a minimum average base quality score of 20 retained. Read pairs were further trimmed using cutadapt 1.14 to remove adapter sequences and low-quality 3’ bases (--match-read-wildcards -m 20 -q 10). Reads were first mapped the dm6 version of the Drosophila genome using STAR 2.7.31. Reads not mapping to the spike genome were then used for alignment to mouse (mm10) using parameters --quantMode TranscriptomeSAM GeneCounts --outMultimapperOrder Random --outSAMattrIHstart 0 --outFilterType BySJout -outFilterMismatchNmax 4 --alignSJoverhangMin 8 --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonicalUnannotated --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 --outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0. Duplicates were also removed using STAR. Stranded coverage bedGraph files were generated from deduplicated BAM files using STAR.
BedGraphs were normalized using the normalize_bedGraph custom script. For libraries generated from untreated mESCs and HEK293Ts, no further normalization was performed. For libraries generated under SCR or U1 AMO conditions: We observed a consistent increase in reads mapping to the fly genome (dm6) after U1 inhibition (Average ratio of U1 AMO / SCR AMO = 2.1), indicating a global decrease in RNA synthesis in U1 AMO cells compared to the SCR AMO control. Accordingly, the average ratio of reads mapping to the spike genome between U1 and SCR AMO conditions was used to generate spike normalization factors, as shown below.
| Sample | Total Reads | Reads Mapping to dm6 | Reads Mapping to mm10 | Ratio of the % Spike Return (U1/SCR AMO) | Normalization Factor |
|---|---|---|---|---|---|
| SCR AMO Replicate 1 | 198118400 | 2126083 | 178836866 | 2.336 | 1 |
| U1 AMO Replicate 1 | 224347810 | 5624376 | 199242356 | 2.1 | |
| SCR AMO Replicate 2 | 185260271 | 4435004 | 169923534 | 2.338 | 1 |
| U1 AMO Replicate 2 | 207236497 | 11594050 | 184211926 | 2.1 | |
| SCR AMO Replicate 3 | 209248749 | 6643701 | 188650603 | 1.644 | 1 |
| U1 AMO Replicate 3 | 194832082 | 10169109 | 172859907 | 2.1 |
BedGraph files were converted to the bigWig format, and merged bedGraphs for each experimental condition were generated using bigWigMerge (UCSC tools). Merged bedGraphs were then converted to the bigWig format for visualization.
RNA-seq library construction and data processing
To prepare samples for RNA-seq, cells were first washed with PBS, trypsinized and, quenched with cold DMEM + 10% FBS. Cells were then spun at 1000 RPM for 4 minutes at 4°C, resuspended in 1 mL cold PBS, transferred to a 1.5 mL Eppendorf tube and counted. Next, cells were spun at 1000 RPM for 4 minutes at 4°C and resuspended in TRIzol. Samples were spiked with 5% Drosophila S2 cells (dissolved in TRIzol) based on cell count. Total RNA was phenol:chloroform extracted, treated with DNAseI (Invitrogen, 18068015) for 30 minutes at room temperature, and ethanol precipitated. RNA integrity was confirmed using the Agilent TapeStation.
500 ng total RNA was used for library construction with the Illumina TruSeq stranded total RNA kit with RiboZero rRNA depletion. Manufacturer instructions were followed with the following modifications: Superscript III was used for the first strand cDNA synthesis and the reaction was held at 25°C for 10 minutes, 50°C for 15 minutes, 70°C for 15 minutes and held at 4°C. After 5 cycles of PCR, samples were removed from the thermal cycler and a test PCR was performed to determine the optimal number of final cycles. Libraries were pooled and sequenced paired-end 150 bp on the Illumina NovaSeq platform.
Using a custom script (trim_and_filter_PE.pl), FASTQ read pairs were trimmed to 120bp per mate, and read pairs with a minimum average base quality score of 20 retained. Read pairs were further trimmed using cutadapt 1.14 to remove adapter sequences and low-quality 3’ bases (--match-read-wildcards -m 20 -q 10). Reads were first mapped the dm6 version of the Drosophila genome using STAR 2.7.31. Reads not mapping to the spike genome were then used for alignment to mm10 using parameters --quantMode TranscriptomeSAM GeneCounts --outMultimapperOrder Random --outSAMattrIHstart 0 --outFilterType BySJout --outFilterMismatchNmax 4 --alignSJoverhangMin 8 --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonicalUnannotated --alignIntronMin 20 --alignIntronMax 1000000 -alignMatesGapMax 1000000 --outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0. Duplicates were also removed using STAR. Stranded coverage bedGraph files were generated from deduplicated BAM files using STAR. For siRNA control and RRP40 KD conditions, bedGraphs were depth normalized using normalize_bedGraph (factors shown below).
| Sample | Total Reads | Reads Mapping to mm10 | Normalization Factor |
|---|---|---|---|
| siNT Replicate 1 | 48775342 | 41651403 | 1.05 |
| siNT Replicate 2 | 55675232 | 48248488 | 1.24 |
| siNT Replicate 3 | 59001312 | 48481109 | 1.23 |
| siNT Replicate 4 | 57216035 | 48421672 | 1.24 |
| siRRP40 Replicate 1 | 53509123 | 44922637 | 1.09 |
| siRRP40 Replicate 2 | 63215913 | 53750117 | 1.34 |
| siRRP40 Replicate 3 | 51809399 | 43738287 | 1.09 |
| siRRP40 Replicate 4 | 47527557 | 39992338 | 1.00 |
BedGraph files were converted to the bigWig format, and merged bedGraphs for each experimental condition were generated using bigWigMerge (UCSC tools). Merged bedGraphs were then converted to the bigWig format for visualization.
ChIP-seq library construction and data processing
To prepare a single cell suspension, WT mESCs (n=3) were treated with Accutase and then diluted in PBS (10 mL final volume). mESCs were crosslinked with 1% formaldehyde by agitating at room temperature for 2.5 minutes. The reaction was quenched with glycine (0.125 M final concentration) and cells were agitated on a plate shaker for an additional 5 minutes at room temperature. Cells were then transferred to a conical tube on ice, and the plate was washed with cold PBS. Next, cells were spun at 300 × g for 5 minutes at 4°C, resuspended in cold PBS, and counted. Cells were re-spun at 300 × g for 5 minutes at 4°C and resuspended in Sonication Buffer (20 mM Tris pH 8.0, 2 mM EDTA, 0.5 mM EGTA, 1X protease inhibitors, 0.5% SDS, 0.5 mM PMSF) at a cell density of 1×108 cells per 1 mL. Cells were incubated on ice for 10 minutes, flash frozen in liquid nitrogen and stored at −80°C.
Thawed samples were sonicated for 10 minutes (total “ON” time) using the Qsonica Q800R3 system (70% amplitude, 15 seconds ON/45 seconds off cycles, 150 μL of chromatin per reaction). Sonicated chromatin was then spun at max speed for 10 minutes at 4°C. The supernatant was transferred to a new tube and an aliquot was used to check fragmentation sizes on an agarose gel. Sonicated chromatin from 7.5 million mESCs was used as input for each immunoprecipitation. First, samples were diluted in 1 mL IP Buffer (20 mM Tris pH 8.0, 2 mM EDTA, 0.5% Triton X-100, 150 mM NaCl, 10% glycerol, 5% BSA) and pre-cleared with 100 μL of a 50% slurry of Protein A agarose beads (Millipore, 16–125) equilibrated in IP buffer for 2 hours at 4 °C with rotation. Precleared chromatin was moved to a new tube, and an additional 250 μL of IP buffer was added. 35 μL of an antibody recognizing Rpb3 (Gift from the Wade Lab) was added per reaction and samples were incubated overnight at 4°C with rotation. To isolate chromatin bound by RNAPII, 200 μL of a 50% slurry of Protein A agarose beads equilibrated in IP buffer was added, and samples were incubated at 4°C with rotation for 2 hours. All spins below were performed at 1000 × g for 1 minute at 4°C and all buffers were kept at 4°C. Beads were washed once in Low Salt Buffer (20 mM Tris-HCl pH 8, 2 mM EDTA, 1% v/v Triton X-100, 150 mM NaCl), 3 times in High Salt Buffer (20 mM Tris-HCl pH 8, 2 mM EDTA, 1% v/v Triton X-100, 500 mM NaCl), once in LiCl Salt Buffer (20 mM Tris-HCl pH 8, 2 mM EDTA, 250 mM LiCl plus 1% v/v IGEPAL) and twice in TE. To elute DNA, beads were incubated with 250 μL of elution buffer (1% SDS, 0.1 M NaHCO3) for 15 minutes. After repeating the elution step (500uL final volume), 20 μL of 5 M NaCl was added, and samples were incubated at 65°C overnight to reverse crosslinks. Samples were then treated with Proteinase K (NEB, M027S) for 1 hour. To extract DNA, a phenol-chloroform extraction and ethanol precipitation was performed. ChIP-seq libraries were generated using the NEBNext Ultra II DNA Kit (New England Biolabs), following manufacturer’s instructions. Libraries were sequenced paired-end 150 bp on the Illumina NovaSeq platform.
Using a custom script (trim_and_filter_PE.pl), FASTQ read pairs were trimmed to 50bp per mate, and read pairs with a minimum average base quality score of 20 were retained. Reads were then mapped to the mouse genome (mm10) using bowtie version 1.2.2 (-v2 -k1 --allow-contain -X1000 -p 5 –best). The custom script extract_fragments.pl was used to retain read fragments corresponding to insert sizes of 50–500bp, remove duplicate reads and generate bedGraphs (25nt bins) reporting the read fragment center. The custom script bedgraphs2stdBedGraph was used to merge replicate bedGraphs (n=3).
PAC-seq library construction and data processing
PAC-seq libraries were constructed using the PolyA-ClickSeq Library Kit (ClickSeq Technologies) per manufacturer instructions with the following modifications: After 5 cycles of PCR, samples were removed from the thermal cycler and a test PCR was performed to determine the optimal number of final cycles. For SCR and U1 AMO conditions, libraries were constructed from 250 ng of enriched 4sU-labelled RNA (as described under TT-seq library construction). For untreated mESC, siNT and siRRP40 conditions, 1 μg of total RNA (as described under RNA-seq library construction) was used for library construction. Libraries were pooled and sequenced paired-end 150 bp on the Illumina NovaSeq platform.
PAC-seq libraries were processed as described previously (Custom scripts described herein are available in52), with the following modifications: First, trimmed and filtered reads were mapped to the Drosophila genome (dm6) using HISAT2. Reads not mapping to the spike genome were aligned to the mouse genome (mm10) using HISAT2. For final read processing, a minimum of 5 non-primer/non-template As was required for each unique poly(A) tail and a read count filter per nucleotide position was not applied.
BedGraphs were normalized using the normalize_bedGraph script. For libraries generated under AMO conditions, we observed a consistent increase in reads mapping to dm6 after U1 inhibition (Average ratio of U1 AMO / SCR AMO = 1.41), indicating a global decrease in polyadenylated RNAs in U1 AMO cells compared to the SCR AMO control. Accordingly, the average ratio of reads mapping to the spike genome between U1 and SCR AMO conditions was used to generate spike normalization factors, as shown below.
| Sample | Total Reads | Reads Mapping to dm6 | Ratio of the % Spike Return (U1/SCR AMO) | Final reads mapping to mm10 | Normalization Factor |
|---|---|---|---|---|---|
| SCR AMO Replicate 1 | 40,856,247 | 410674 | 1.195 | 3976345 | 1 |
| U1 AMO Replicate 1 | 58,581,594 | 751044 | 6144267 | 1.41 | |
| SCR AMO Replicate 2 | 40,716,029 | 768947 | 1.722 | 4352088 | 1 |
| U1 AMO Replicate 2 | 39,846,776 | 1165012 | 3548777 | 1.41 | |
| SCR AMO Replicate 3 | 41,256,650 | 781548 | 1.314 | 4892102 | 1 |
| U1 AMO Replicate 3 | 25,393,478 | 547959 | 2402109 | 1.41 |
Libraries generated under siNT and siRRP40 conditions were depth normalized, as shown below.
| Sample | Total Reads | Final reads mapping to mm10 | Normalization Factor |
|---|---|---|---|
| mESC Replicate 1 | 64,478,501 | 11896381 | 1.000 |
| mESC Replicate 2 | 141,325,133 | 26170833 | 2.200 |
| mESC Replicate 3 | 93,423,278 | 17527848 | 1.473 |
| siNT Replicate 1 | 100,373,361 | 19428425 | 1.633 |
| siNT Replicate 2 | 93,197,329 | 18287382 | 1.537 |
| siNT Replicate 3 | 99,967,680 | 18084557 | 1.520 |
| siNT Replicate 4 | 83,042,231 | 15771140 | 1.326 |
| siRRP40 Replicate 1 | 93,662,686 | 17351024 | 1.459 |
| siRRP40 Replicate 2 | 108,324,032 | 19588105 | 1.647 |
| siRRP40 Replicate 3 | 94,176,657 | 17083897 | 1.436 |
| siRRP40 Replicate 4 | 67,623,157 | 12114328 | 1.018 |
Combined bedGraphs were generated by summing counts per nucleotide across replicates for each condition (bedgraphs2stdBedgraph). BedGraphs were converted to the bigWig format for visualization. For Figure S2E, genes containing more than 2 reads in the window TES +/− 500 nt were classified as “containing PAC-seq signal overlapping canonical TESs”.
QUANTIFICATION AND STATISTICAL ANALYSIS
Gene Annotations
RNA-seq and PRO-seq from untreated cells was used to define a data-driven annotation comprising of a single dominant transcription start site (TSS) and transcript end site (TES) per active gene in mESCs, HEK293Ts, and K562 cells. The custom script used to call the dominant gene annotation per cell type is publicly available on the Adelman Lab Github (get_gene_annotations.sh, https://github.com/AdelmanLab/GeneAnnotationScripts). Briefly, HISAT2 v2.2.1 (--known-splicesite-infile) was used to map paired-end RNA-seq reads to the corresponding reference genome (mm10/hg38) and Kallisto was used to quantify reads over individual transcript models to determine transcript expression. TSScall85 was used to generate a list of active TSSs from PRO-seq R2 reads (reflecting the RNA 5’ ends) and determine the dominant TSS per gene. For the dominant TSS and its associated transcript models, the dominant TES is selected based on the average transcript TPM values from Kallisto.
For downstream analysis, a single dominant exon model was determined per gene. First, genes with a single exon annotation were defined as an intronless gene. Genes with 2 or more exon annotations were defined as an intronic gene. For each intronic gene, all transcript models associated with the dominant TSS, and dominant TES cluster were retained. Transcript models where the start of the first intron was upstream of the dominant TSS were removed. For genes containing multiple transcript IDs associated with the dominant TES cluster, the transcript ID used to determine the dominant TES position was favored. If the transcript ID used to define the dominant TES was removed (i.e., more than a 1 nt difference in TES coordinate position), the dominant TES coordinate for the gene model was modified. The list of exon coordinates from the dominant transcript model were retained. The first and last exon coordinates in the GTF were modified to match the position of the dominant TSS and TES. Number of genes investigated per species are listed in the corresponding figure legends.
CpG islands and %G+C content
CpG island coordinates and Gc5base were downloaded from UCSC table browser. Bedtools intersect was used to overlap CpG islands to TSSs. Genes with more than 1 CpG island overlapping the TSS were removed. Number of genes per species with a CpG island overlapping the TSS are listed in the corresponding figure legends.
MNase-seq data processing
FASTQs corresponding to MNase-seq libraries from control mESCs were downloaded from GSE85191. First, FASTQ read pairs were trimmed to 70 nt per mate and read pairs above a minimum quality threshold were retained using the trim_and_filter_PE.pl custom script (-a 1 -b 70 -c 1 -d 70 -m 20 -q sanger). Bowtie version 1.2.2 (-m1 -v2 -X1000 --best -p 5) was used to map trimmed and retained read pairs to the mm10 reference genome. Custom script extract_fragments.pl was used to retain reads corresponding to mono-nucleosome size fragments (read pairs spanning <120bp or >180bp were excluded) and generate bedGraphs reporting the read fragment center (o a -b 1 -min 120 -max 180). The custom script bedgraphs2stdBedGraph was used to merge replicate bedGraphs.
Differential expression analysis
TT-seq reads within exons were summed per gene using featurecounts and DESeq2 was used to generate a list of differentially expressed genes after U1 inhibition. DESeq2 size factors were overwritten to match spike normalization. Significantly affected genes were defined using the following cutoffs: 2-fold change and p < 0.0001.
Classifying transcriptional defects at downregulated genes
To evaluate transcriptional defects at downregulated genes, the following filters were applied: 1) PRO-seq reads were evaluated from the dominant TSS to the dominant TES. Genes that were not at least 10% downregulated in PRO-seq counts in the U1 AMO condition were removed (N=6,229 of 7,714 downregulated protein-coding genes with introns remaining). 2) Promoter PRO-seq signal was evaluated between the TSS to +100nt to minimize overlap with the first 5’SS. Genes with fewer than 15 total promoter read counts between the SCR and U1 AMO conditions were removed (N=6,183 downregulated genes remaining). Downregulated genes with at least a 2-fold decrease in promoter PRO-seq reads were defined as an “U1 initiation-regulated gene” (N=1,398). Downregulated genes with less than a 1.3-fold decrease in Promoter PRO-seq signal were further evaluated for defects in transcription elongation (N=2,696). To evaluate gene body PRO-seq, reads were summed in the TSS+250nt and the TES window and normalized by gene length. P-values for paired comparisons between SCR and U1 AMO conditions were calculated using the Wilcoxon matched-pairs signed rank test. The subset of downregulated genes with less than a 1.3-fold decrease in Promoter PRO-seq signal and significantly lower Gene Body PRO-seq density in the U1 AMO condition were classified as “U1 elongation-regulated genes.”
Splicing efficiency in TT-seq
Splicing efficiency for the first intron in active protein coding genes was calculated using TT-seq fragments that crossed 5’SSs, as the number of spliced reads divided by the total number of spliced and unspliced reads per intron. A custom script to calculate spliced and unspliced reads from TT-seq BAM files is publicly available on the Adelman Lab Github (calculate_fraction_spliced.py; https://doi.org/10.5281/zenodo.7328578).
MaxEnt
UCSC table browser was used to generate a FASTA file of a 9mer sequence corresponding to the 5’SS motif (3 bases in the exon, 6 based in the intron). The FASTA file of 5’SS sequences was run through score5.pl in the Maxentscan suite (https://github.com/Congenica/maxentscan.git) to determine the strength of each 5’SS sequence relative to the consensus 5’SS motif using the Maximum Entropy Model.
Pausing Index
PRO-seq reads were summed (using the make_heatmap script) for elongation-regulated genes in the following windows: Promoter (TSS to +100) and Early Gene Body (TSS +250 to +2250 nt). Pausing Index was calculated as the ratio of Promoter to Early Gene Body PRO-seq read density.
Elongation Index
Active protein-coding genes were divided into 500 nt bins starting at the TSS. 500 nt bins that overlapped the TSS or TES were removed. make_heatmap, as described under “Heatmaps and metagene plots”, was used to calculate PRO-seq and TT-seq read density per 500 nt bin. Elongation index per 500nt bin was calculated by dividing the signal for RNA synthesis (TT-seq read coverage) by the signal representing RNAPII density (PRO-seq 3’ end reads), as described previously.8,37,47 500 nt bins below the following read count thresholds were removed: For Figures 1B, S1A–C and S5B, a minimum of 100 reads counts was required for all relevant datasets. For Figures 5B, 5D–F, and S5A and S5C, a minimum of 10 read counts was required for all relevant datasets. For S1D, a minimum of 2 read counts was required for all relevant datasets. For heatmap and metagene plot representations of elongation index (Figures 1C, S1F–G, 5C), a constant factor of 1 was added to each heatmap to avoid dividing by 0. Scatter density plots reporting the relationship between GC content and elongation index (Figures S1A, 5D and S5C) were generated using the get_density R function (http://slowkow.com/notes/ggplot2-color-by-density) and ggplot2.
Conservation Score
A bigWig file of the mm10 Placental Mammals base wise conversion by PhyloP dataset was downloaded from the UCSC table browser.
Transition Points
A Hidden Markov Model (groHMM)54 was used to identify sites (Transition Point, TP) at which the wave of PRO-seq signal representing elongating RNAPII drops off in U1 AMO conditions (custom script polymeraseWaveBW, https://github.com/dankoc/polymeraseWaves; TSmooth= NA, size = 100, finterWindowSize = 1000). Transition points were called for elongation-regulated genes longer than 10kb (N = 2,399). Transition points identified before the TSS and the first 5’SS were removed from downstream analysis.
Motif analysis
MOODS v1.9.4 (https://github.com/jhkorhonen/MOODS)79 was used to identify PAS (10 most common PAS motifs) and arrest (RRRYYY) motifs between the dominant TSS and TES. Motif matches were restricted to those found within the input strand (+).
An actionable PAS motif was defined as a PAS motif associated with PAC-seq reads (> 0) in U1 AMO-treated cells in the PAS to +100 nt window. To define actionable arrest sites, PRO-seq reads were summed upstream (−100 to −25 nt) and downstream (+25 to +100 nt) of the arrest motif in SCR and U1 AMO cells. Arrest motifs with more than 5 reads in the upstream window under both SCR and U1 AMO conditions were retained. Next, the ratio of PRO-seq signal in the downstream to upstream motif windows was calculated. Actionable arrest motifs after U1 inhibition were defined using the following thresholds: FC ≤ 0.75 after U1 AMO treatment and FC between .75 and 1.25 in SCR AMO conditions.
HOMER81 and MEME80 were used to find sequence motifs around the transition points (TP) of genes with defined TPs, but without evidence of PCPA. The region TP +/− 50nt was used for the list of input sequences. To generate background sequences for HOMER and MEME, mock sites were selected at random from elongation-regulated genes without a defined TP. The location of the mock site was restricted to the gene body to prevent calling sites downstream of the TES at elongation-regulated genes without a defined TP. To enable GC content normalization, the distribution of distances between the TSS and the mock site matched those found at genes without PCPA. Motif searches were restricted to the given strand (RNA sequence).
Heatmaps and metagene plots
Count matrices were generated using the make_heatmap custom script (https://github.com/AdelmanLab/NIH_scripts/tree/main/make_heatmap). Partek Genomics Suite was used to generate visual representations of the indicated heatmaps. Metagene plots were generated by summing reads within bins at each indicated position with respect to the feature of interest (e.g., TSS and CpG edge) and dividing by the number of annotations. For each heatmap, the Bin size and feature used for alignment are indicated in the figure legend. Metagenes were plotted in GraphPad Prism.
Box plots and Statistical Analysis
Box plots were generated in GraphPad Prism and have a line at the median, and whiskers depict 1.5 times the interquartile range. P values were calculated in GraphPad Prism, as indicated in the figure legends.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RRP40 | Bethyl Laboratories | A303-909A |
| Actin | SCBT | SC1616 |
| Rbp3 (for RNAPII ChIP-seq) | Gift Paul Wade Lab, NIEHS, NIH | NA |
| Chemicals, peptides, and recombinant proteins | ||
| LIF | Cell Guidance Systems | Cat # GFM200 |
| PD0325901 | Reprocell | Cat # 04-0006 |
| CHIR99021 | Reprocell | Cat # 04-0004 |
| Biotin-11-NTPs | Perkin Elmer | Cat # NEL54(2/3/4/5)001 |
| 4-thiouridine | Tocris | Cat # 37005 |
| Critical commercial assays | ||
| Illumina TruSeq Stranded Total RNA Library Prep Gold | Illumina | Cat # 20020598 |
| NEB Next Ultra II DNA library kit | NEB | Cat # E7103S |
| PolyA-ClickSeq Library Prep for Illumina Sequencing v1.2 | ClickSeq Technologies | https://clickseqtechnologies.com/ |
| RNA Clean-Up and Concentration Kit | Norgen Biotek Corp | Cat # 61000 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE218135 |
| MNase-seq | Henriques et al., 201864 | GEO: GSE85191 |
| RTF1 dTAG mNETseq and TT-seq (K562) | Žumer et al., 20218 | GEO: GSE159633 |
| PladB 1hr treatment mNETseq and TT-seq (K562) | Caizzi et al., 202137 | GEO: GSE148433 |
| WT mESC PRO-seq and TT-seq | Vlaming et al., 202223 | GEO: GSE178230 |
| 2P-seq (8hr U1 AMO treatment) | Chiu et al., 20184 | GEO: GSE100537 |
| WT mESC RNA-seq | Stein et al., 202247 | GEO: GSE200702 |
| WT K562 RNA-seq and PRO-seq | Blumberg et al., 202165 | GEO: GSE153200 |
| Experimental models: Cell lines | ||
| F121-9 mESCs (CAST/129 background) | Jaenisch/Gribnau labs | 4DNSRMG5APUM |
| mESCs clones with fluorescent splicing reporter system | Vlaming et al., 202223 | NA |
| HEK293T | ATTC | CRL-3216 |
| Oligonucleotides | ||
| SCR AMO (CCTCTTACCTCAGTTACAATTTATA) | Kaida et al., 201040; Gene Tools, LLC | NA |
| U1 AMO (GGTATCTCCCCTGCCAGGTAAGTAT) | Kaida et al., 201040; Gene Tools, LLC | NA |
| siGENOME Non-Targeting siRNA #2 | Dharmacon | D-001210-02-05 |
| siGENOME Mouse Exosc3 siRNA – set of 4 | Dharmacon | MQ-064537-01-0010 |
| Software and algorithms | ||
| bowtie 1.2.2 | Langmead et al., 200966 | N/A |
| Bowtie2 | Langmead and Salzberg., 201267 | |
| STAR 2.7.3a | Dobin et al., 201368 | N/A |
| HISAT2 | Kim et al., 201969 | |
| R | https://www.r-project.org/ | N/A |
| Rstudio | https://www.rstudio.com/ | N/A |
| featureCounts | Liao et al., 201470 | N/A |
| DESeq2 | Love et al., 201471 | N/A |
| Prism | GraphPad | N/A |
| Partek Genomics Suite | https://www.partek.com/ | N/A |
| get_gene_annotations.sh | https://doi-org.ezp-prod1.hul.harvard.edu/10.5281/zenodo.5519927 | N/A |
| make_heatmap | https://doi-org.ezp-prod1.hul.harvard.edu/10.5281/zenodo.5519914 | N/A |
| trim_and_filter_PE.pl | https://doi-org.ezp-prod1.hul.harvard.edu/10.5281/zenodo.5519914 | N/A |
| bowtie2stdBedGraph.pl | https://doi-org.ezp-prod1.hul.harvard.edu/10.5281/zenodo.5519914 | N/A |
| bedgraphs2stdBedGraph | https://doi-org.ezp-prod1.hul.harvard.edu/10.5281/zenodo.5519914 | N/A |
| cutadapt | https://doi-org.ezp-prod1.hul.harvard.edu/10.14806/ej.17.1.200 | N/A |
| UMI-tools | Smith et al., 201772 | N/A |
| Kallisto | Bray et al., 201673 | N/A |
| Calculate_fraction_spliced.sh | https://doi.org/10.5281/zenodo.7328578 | N/A |
| samtools | Li et al., 200974 | N/A |
| bedtools | Quinlan and Hall, 201075 | N/A |
| UCSCtools | Kent et al., 201076 | N/A |
| ImageJ | Schneider et al., 201277 | N/A |
| groHMM | Chae et al., 201578 | N/A |
| polymeraseWaves | Danko et al., 201354 | N/A |
| MOODS v 1.9.4 | Korhonen et al., 200979 | N/A |
| MEME Suite | Bailey et al., 201580 | N/A |
| HOMER | Heinz et al., 201081 | N/A |
HIGHLIGHTS.
AT content of the transcribed region strongly impacts RNAPII elongation rate
U1 snRNP enhances transcription initiation or elongation in a gene-dependent manner
RNAPII acceleration in AT-rich sequences requires U1 snRNP
Without U1 snRNP to stimulate elongation, RNAPII is prone to arrest and termination
Acknowledgements
We thank members of the Adelman Lab, S. Buratowski, S. Eddy, P. Sharp and M. Bao for helpful discussions and comments on the manuscript. We thank H. Suzuki and P. Sharp for providing 2P-seq datasets, the HMS Nascent Transcriptomics Core for helpful discussions and generating PRO-seq libraries in HEK293Ts, B. Martin for sharing code to count spliced reads in TT-seq, and the HMS Biopolymers Facility, Bauer Core Facility at Harvard University and Novogene for sequencing. This work was supported by the National Institutes of Health (NIH R01GM139960 to K.A.). C.A.M was supported by the National Science Foundation Graduate Research Fellowship (DGE1745303) and the Sophia H.Y Chang Fellowship.
Footnotes
Declaration of Interests: K.A. received research funding from Novartis not related to this work, is on the SAB of CAMP4 Therapeutics, and is a member of the Advisory Board of Molecular Cell.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Sidorenkov I, Komissarova N, and Kashlev M (1998). Crucial Role of the RNA:DNA Hybrid in the Processivity of Transcription. Mol. Cell 2, 55–64. 10.1016/S1097-2765(00)80113-6. [DOI] [PubMed] [Google Scholar]
- 2.Hahn S (2004). Structure and mechanism of the RNA Polymerase II transcription machinery. Nat. Struct. Mol. Biol. 11, 394. 10.1038/NSMB763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osman S, and Cramer P (2020). Structural Biology of RNA Polymerase II Transcription: 20 Years On Pol II: RNA polymerase II. 10.1146/annurev-cellbio-042020. [DOI] [PubMed] [Google Scholar]
- 4.Chiu AC, Suzuki HI, Wu X, Mahat DB, Kriz AJ, and Sharp PA (2018). Transcriptional Pause Sites Delineate Stable Nucleosome-Associated Premature Polyadenylation Suppressed by U1 snRNP. Mol. Cell 69. 10.1016/j.molcel.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venters CC, Oh JM, Di C, So BR, and Dreyfuss G (2019). U1 snRNP telescripting: Suppression of premature transcription termination in introns as a new layer of gene regulation. Cold Spring Harb. Perspect. Biol. 11. 10.1101/cshperspect.a032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krajewska M, Dries R, Grassetti AV, Dust S, Gao Y, Huang H, Sharma B, Day DS, Kwiatkowski N, Pomaville M, et al. (2019). CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 10. 10.1038/s41467-019-09703-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narain A, Bhandare P, Adhikari B, Backes S, Eilers M, Dölken L, Schlosser A, Erhard F, Baluapuri A, and Wolf E (2021). Targeted protein degradation reveals a direct role of SPT6 in RNAPII elongation and termination. Mol. Cell 81, 3110–3127.e14. 10.1016/J.MOLCEL.2021.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Žumer K, Maier KC, Farnung L, Jaeger MG, Rus P, Winter G, and Cramer P (2021). Two distinct mechanisms of RNA polymerase II elongation stimulation in vivo. Mol. Cell 81, 3096–3109.e8. 10.1016/J.MOLCEL.2021.05.028. [DOI] [PubMed] [Google Scholar]
- 9.Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, and Peterlin BM (2011). The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 25, 2158–2172. 10.1101/GAD.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubbury SJ, Boutz PL, and Sharp PA (2018). CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 564, 141–145. 10.1038/S41586-018-0758-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong KK, and Van Allen EM (2018). Intron retention is a source of neoepitopes in cancer. Nat. Biotechnol. 2018 3611 36, 1056–1058. 10.1038/nbt.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu SX, De Neef E, Thomas JD, Sabio E, Rousseau B, Gigoux M, Knorr DA, Greenbaum B, Elhanati Y, Hogg SJ, et al. (2021). Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 184, 4032–4047.e31. 10.1016/J.CELL.2021.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheridan RM, Fong N, D’Alessandro A, and Bentley DL (2019). Widespread Backtracking by RNA Pol II Is a Major Effector of Gene Activation, 5′ Pause Release, Termination, and Transcription Elongation Rate. Mol. Cell 73, 107–118.e4. 10.1016/j.molcel.2018.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh JM, Di C, Venters CC, Guo J, Arai C, So BR, Pinto AM, Zhang Z, Wan L, Younis I, et al. (2017). U1 snRNP telescripting regulates a size-function-stratified human genome. Nat. Struct. Mol. Biol. 24, 993–999. 10.1038/nsmb.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, and Kondrashov FA (2002). Selection for short introns in highly expressed genes. Nat. Genet. 2002 314 31, 415–418. 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- 16.Urrutia AO, and Hurst LD (2003). The signature of selection mediated by expression on human genes. Genome Res. 13, 2260–2264. 10.1101/GR.641103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sahakyan AB, and Balasubramanian S (2016). Long genes and genes with multiple splice variants are enriched in pathways linked to cancer and other multigenic diseases. BMC Genomics 17, 1–10. 10.1186/S12864-016-2582-9/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopes I, Altab G, Raina P, and de Magalhães JP (2021). Gene Size Matters: An Analysis of Gene Length in the Human Genome. Front. Genet. 12, 30. 10.3389/FGENE.2021.559998/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jonkers I, Kwak H, and Lis JT (2014). Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 2014. 10.7554/ELIFE.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veloso A, Kirkconnell KS, Magnuson B, Biewen B, Paulsen MT, Wilson TE, and Ljungman M (2014). Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 24, 896. 10.1101/GR.171405.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamft B, Bintu L, Ishibashi T, and Bustamante C (2012). Nascent RNA structure modulates the transcriptional dynamics of RNA polymerases. Proc. Natl. Acad. Sci. U. S. A. 109, 8948–8953. 10.1073/PNAS.1205063109/SUPPL_FILE/PNAS.1205063109_SI.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turowski TW, Petfalski E, Goddard BD, French SL, Helwak A, and Tollervey D (2020). Nascent Transcript Folding Plays a Major Role in Determining RNA Polymerase Elongation Rates. Mol. Cell 79, 488–503.e11. 10.1016/J.MOLCEL.2020.06.002/ATTACHMENT/7BF6A3CF-C202-4F25-A618-7BBBFCFC3CEF/MMC4.ZIP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlaming H, Mimoso CA, Field AR, Martin BJE, and Adelman K (2022). Screening thousands of transcribed coding and non-coding regions reveals sequence determinants of RNA polymerase II elongation potential. Nat. Struct. Mol. Biol. 2022 296 29, 613–620. 10.1038/s41594-022-00785-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almada AE, Wu X, Kriz AJ, Burge CB, and Sharp PA (2013). Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 499, 360–363. 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proudfoot NJ (2016). Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science (80-. ). 352, aad9926–aad9926. 10.1126/science.aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCoy MJ, and Fire AZ (2020). Intron and gene size expansion during nervous system evolution. BMC Genomics 21, 1–9. 10.1186/S12864-020-6760-4/FIGURES/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herzel L, Ottoz DSM, Alpert T, and Neugebauer KM (2017). Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 18, 637–650. 10.1038/nrm.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilkinson ME, Charenton C, and Nagai K (2020). RNA Splicing by the Spliceosome. 10.1146/annurev-biochem-091719-064225 89, 359–388. . [DOI] [PubMed] [Google Scholar]
- 29.Alpert T, Straube K, Carrillo Oesterreich F, and Neugebauer KM (2020). Widespread Transcriptional Readthrough Caused by Nab2 Depletion Leads to Chimeric Transcripts with Retained Introns. Cell Rep. 33. 10.1016/J.CELREP.2020.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrillo Oesterreich F, Herzel L, Straube K, Hujer K, Howard J, and Neugebauer KM (2016). Splicing of Nascent RNA Coincides with Intron Exit from RNA Polymerase II. Cell 165, 372–381. 10.1016/j.cell.2016.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herzel L, Straube K, and Neugebauer KM (2018). Long-read sequencing of nascent RNA reveals coupling among RNA processing events. Genome Res. 28, 1008–1019. 10.1101/GR.232025.117/-/DC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reimer KA, Mimoso CA, Adelman K, and Neugebauer KM (2021). Co-transcriptional splicing regulates 3′ end cleavage during mammalian erythropoiesis. Mol. Cell 0. 10.1016/j.molcel.2020.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damgaard CK, Kahns S, Lykke-Andersen S, Nielsen AL, Jensen TH, and Kjems J (2008). A 5′ Splice Site Enhances the Recruitment of Basal Transcription Initiation Factors In Vivo. Mol. Cell 29, 271–278. 10.1016/j.molcel.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 34.Fiszbein A, Krick KS, Begg BE, Burge Correspondence CB, and Burge CB (2019). Exon-Mediated Activation of Transcription Starts Article Exon-Mediated Activation of Transcription Starts. Cell 179. 10.1016/j.cell.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwek KY, Murphy S, Furger A, Thomas B, O’Gorman W, Kimura H, Proudfoot NJ, and Akoulitchev A (2002). U1 snRNA associates with tfiih and regulates transcriptional initiation. Nat. Struct. Biol. 9, 800–805. 10.1038/nsb862. [DOI] [PubMed] [Google Scholar]
- 36.Kameoka S, Duque P, and Konarska MM (2004). p54nrb associates with the 5′ splice site within large transcription/splicing complexes. EMBO J. 10.1038/sj.emboj.7600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caizzi L, Monteiro-Martins S, Schwalb B, Lysakovskaia K, Schmitzova J, Sawicka A, Chen Y, Lidschreiber M, and Cramer P (2021). Efficient RNA polymerase II pause release requires U2 snRNP function. Mol. Cell 81, 1920–1934.e9. 10.1016/J.MOLCEL.2021.02.016. [DOI] [PubMed] [Google Scholar]
- 38.Kaida D (2016). The reciprocal regulation between splicing and 3′-end processing. Wiley Interdiscip. Rev. RNA 7, 499–511. 10.1002/wrna.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furger A, O’Sullivan JM, Binnie A, Lee BA, and Proudfoot NJ (2002). Promoter proximal splice sites enhance transcription. Genes Dev. 16, 2792–2799. 10.1101/gad.983602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, and Dreyfuss G (2010). U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668. 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, et al. (2012). U1 snRNP Determines mRNA Length and Regulates Isoform Expression. Cell 150, 53–64. 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ntini E, Järvelin AI, Bornholdt J, Chen Y, Boyd M, Jørgensen M, Andersson R, Hoof I, Schein A, Andersen PR, et al. (2013). Polyadenylation site–induced decay of upstream transcripts enforces promoter directionality. Nat. Struct. Mol. Biol. 20, 923–928. 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 43.So BR, Di C, Cai Z, Venters CC, Guo J, Oh JM, Arai C, and Dreyfuss G (2019). A Complex of U1 snRNP with Cleavage and Polyadenylation Factors Controls Telescripting, Regulating mRNA Transcription in Human Cells. Mol. Cell 76, 590–599.e4. 10.1016/j.molcel.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh J, Venters CC, Di C, Pinto AM, Wan L, Younis I, Cai Z, Arai C, So BR, and Dreyfuss G (2020). U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun, 10.1101/730515. 10.1101/730515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gregersen LH, Mitter R, and Svejstrup JQ (2020). Using TTchem-seq for profiling nascent transcription and measuring transcript elongation. Nat. Protoc. 15, 604–627. 10.1038/s41596-019-0262-3. [DOI] [PubMed] [Google Scholar]
- 46.Mahat D, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Publ. Gr 11. 10.1038/nprot.2016.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stein CB, Field AR, Mimoso CA, Zhao C, Huang K-L, Wagner EJ, and Adelman K (2022). Integrator endonuclease drives promoter-proximal termination at all RNA polymerase II-transcribed loci. Mol. Cell 0. 10.1016/J.MOLCEL.2022.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kellner WA, Bell JSK, and Vertino PM (2015). GC skew defines distinct RNA polymerase pause sites in CpG island promoters. Genome Res. 25, 1600–1609. 10.1101/GR.189068.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamieniarz-Gdula K, and Proudfoot NJ (2019). Transcriptional Control by Premature Termination: A Forgotten Mechanism. Trends Genet. 10.1016/j.tig.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, and Proudfoot NJ (2015). Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell 161, 526–540. 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, and Adelman K (2015). Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol. Cell 58, 1101–1112. 10.1016/j.molcel.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Routh A, Ji P, Jaworski E, Xia Z, Li W, and Wagner EJ (2017). Poly(A)-ClickSeq: Click-chemistry for next-generation 3’-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res. 45. 10.1093/nar/gkx286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang S, Aibara S, Vos SM, Agafonov DE, Lührmann R, and Cramer P (2021). Structure of a transcribing RNA polymerase II–U1 snRNP complex. Science (80-. ). 371, 305–309. 10.1126/science.abf1870. [DOI] [PubMed] [Google Scholar]
- 54.Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, and Kraus WL (2013). Signaling Pathways Differentially Affect RNA Polymerase II Initiation, Pausing, and Elongation Rate in Cells. Mol. Cell 50, 212–222. 10.1016/J.MOLCEL.2013.02.015/ATTACHMENT/305B6CE2-8E5E-465D-9714-C816404E613F/MMC1.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Core LJ, Waterfall JJ, and Lis JT (2008). Nascent RNA Sequencing Reveals Widespread Pausing and Divergent Initiation at Human Promoters. Science (80-. ). 322, 1845–1848. 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwalb B, Michel M, Zacher B, Hauf KF, Demel C, Tresch A, Gagneur J, and Cramer P (2016). TT-seq maps the human transient transcriptome. Science (80-. ). 352, 1225–1228. 10.1126/science.aad9841. [DOI] [PubMed] [Google Scholar]
- 57.Hawryluk PJ, Újvári AU, and Luse DS (2004). Characterization of a novel RNA polymerase II arrest site which lacks a weak 3’ RNA±DNA hybrid. 10.1093/nar/gkh505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science (80-. ). 339, 950–953. 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muniz L, Nicolas E, and Trouche D (2021). RNA polymerase II speed: a key player in controlling and adapting transcriptome composition. EMBO J. 40, e105740. 10.15252/EMBJ.2020105740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaplan CD (2010). The architecture of RNA polymerase fidelity. BMC Biol. 8, 1–4. 10.1186/1741-7007-8-85/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suzuki H, Kumar SA, Shuai S, Diaz-Navarro A, Gutierrez-Fernandez A, De Antonellis P, Cavalli FMG, Juraschka K, Farooq H, Shibahara I, et al. (2019). Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nat. 2019 5747780 574, 707–711. 10.1038/s41586-019-1650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen PC, Han X, Shaw TI, Fu Y, Sun H, Niu M, Wang Z, Jiao Y, Teubner BJW, Eddins D, et al. (2022). Alzheimer’s disease-associated U1 snRNP splicing dysfunction causes neuronal hyperexcitability and cognitive impairment. Nat. Aging 2022 210 2, 923–940. 10.1038/s43587-022-00290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shuai S, Suzuki H, Diaz-Navarro A, Nadeu F, Kumar SA, Gutierrez-Fernandez A, Delgado J, Pinyol M, López-Otín C, Puente XS, et al. (2019). The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nat. 2019 5747780 574, 712–716. 10.1038/s41586-019-1651-z. [DOI] [PubMed] [Google Scholar]
- 64.Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, and Adelman K (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 32. 10.1101/gad.309351.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blumberg A, Zhao Y, Huang YF, Dukler N, Rice EJ, Chivu AG, Krumholz K, Danko CG, and Siepel A (2021). Characterizing RNA stability genome-wide through combined analysis of PRO-seq and RNA-seq data. BMC Biol. 19, 1–17. 10.1186/S12915-021-00949-X/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, 1–10. 10.1186/GB-2009-10-3-R25/TABLES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357. 10.1038/NMETH.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/BIOINFORMATICS/BTS635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019 378 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/BIOINFORMATICS/BTT656. [DOI] [PubMed] [Google Scholar]
- 71.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. 10.1186/S13059-014-0550-8/FIGURES/2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. 10.1101/GR.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016 345 34, 525–527. 10.1038/NBT.3519. [DOI] [PubMed] [Google Scholar]
- 74.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/BIOINFORMATICS/BTP352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/BIOINFORMATICS/BTQ033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. 10.1093/BIOINFORMATICS/BTQ351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chae M, Danko CG, and Kraus WL (2015). groHMM: A computational tool for identifying unannotated and cell type-specific transcription units from global run-on sequencing data. BMC Bioinformatics 16, 1–16. 10.1186/S12859-015-0656-3/FIGURES/7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Korhonen JH, Palin K, Taipale J, and Ukkonen E Fast motif matching revisited: high-order PWMs, SNPs and indels. 10.1093/bioinformatics/btw683. [DOI] [PubMed] [Google Scholar]
- 80.Bailey TL, and Elkan C (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36. [PubMed] [Google Scholar]
- 81.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. 10.1016/J.MOLCEL.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rio DC, Donald Rio C, Ares M Jr, Hannon GJ, and Nilsen CSHL Press, T.W. (2014). Northern Blots for Small RNAs and MicroRNAs. Cold Spring Harb. Protoc 2014, pdb.prot080838. 10.1101/PDB.PROT080838. [DOI] [PubMed] [Google Scholar]
- 83.Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, and Simon MD (2015). Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol. Cell 59, 858–866. 10.1016/J.MOLCEL.2015.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. (2008). High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14, 1959–1972. 10.1261/RNA.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, and Adelman K (2010). Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327, 335–338. 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic datasets generated in this study have been deposited to GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
All custom scripts described herein are available on the Adelman Lab GitHub (https://github.com/AdelmanLab). All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.