


Abstract
Phage Φ29 encodes a DNA-dependent DNA polymerase belonging to the eukaryotic-type (family B) subgroup of DNA polymerases that use a protein as the primer for initiation of DNA synthesis. In one of the most important motifs present in the 3′→5′ exonucleolytic domain of proofreading DNA polymerases, the ExoII motif, Φ29 DNA polymerase contains three amino acid residues, Y59, H61 and F69, which are highly conserved among most proofreading DNA polymerases. These residues have recently been shown to be involved in proper stabilization of the primer terminus at the 3′→5′ exonuclease active site. Here we investigate by means of site-directed mutagenesis the role of these three residues in reactions that are specific for DNA polymerases utilizing a protein-primed DNA replication mechanism. Mutations introduced at residues Y59, H61 and F69 severely affected the protein-primed replication capacity of Φ29 DNA polymerase. For four of the mutants, namely Y59L, H61L, H61R and F69S, interaction with the terminal protein was affected, leading to few initiation and transition products. These findings, together with the specific conservation of Y59, H61 and F69 among DNA polymerases belonging to the protein-primed subgroup, strongly suggest a functional role of these amino acid residues in the DNA polymerase–terminal protein interaction.
INTRODUCTION
Significant advances in understanding the structural biology of DNA polymerases have been made using two different approaches. First, the increasing number of known DNA polymerase structures has expanded to include eukaryotic-like (family B) members like the DNA polymerases from bacteriophage RB69 (1), Thermococcus gorgonarius (2) and the hyperthermophilic archea Thermococcus sp. 9°N-7 (3) and Pyrococcus kodakaraensis KOD1 (4), offering new insights into their replication ability. The most recent members of the crystallographic database from the DNA pol I group (family A) are the DNA polymerase structures of Thermus aquaticus (5), bacteriophage T7 (6) and Bacillus stearothermophilus (7). However, in cases where structural information is missing, a well-established combination of biochemical and genetic approaches has enlarged our knowledge of several DNA polymerase properties, including substrate binding, processivity, fidelity and nucleotidyl transfer (reviewed in 8–11). Φ29 DNA polymerase, a member of the eukaryotic-type family, has been well characterized by means of mutagenesis and a wide variety of biochemical studies (reviewed in 12).
Replication of linear genomes, like that of bacteriophage Φ29 (19 285 bp), cannot be driven by RNA priming because once the primer is removed no DNA polymerase could fill the resulting gap. Thus bacteriophage Φ29 DNA polymerase makes use of a terminal protein (TP) as primer (13,14). Φ29 DNA replication starts by formation of a heterodimer between Φ29 DNA polymerase and a free TP molecule, which is able to recognize and interact with the replication origin at both ends of the TP–DNA template. Then, during initiation, Φ29 DNA polymerase catalyzes, at both ends, the addition of dAMP to the OH group of TP Ser232 using as director the second dTMP of the template strand (15–17). Recovery of the terminal dAMP information is established using a so called ‘sliding-back’ mechanism, based on the 3′-terminal repetition of two dTMPs, allowing the TP–dAMP molecule to be placed opposite the first dTMP (17). A similar mechanism is found for phage PRD1 genome replication, where a ‘stepwise sliding back’ mechanism takes place (18), whereas a ‘jumping back’ step is found for adenovirus initiation of replication (19). For Φ29 DNA replication it has been shown that after sliding back, the same DNA polymerase molecule catalyzes template-directed synthesis of a short elongation product (9 nt) while still maintaining heterodimerization with TP, referred to as the transition step (20). Incorporation of the tenth nucleotide leads to dissociation of the DNA polymerase–TP complex and DNA-primed elongation coupled to strand displacement commences. Elongation proceeds processively and is coupled to strand displacement from both ends without the need for either processivity factors or helicase-like proteins. Once the two replication forks meet, the two partially replicated parental strands separate and full-length DNA synthesis is completed (21).
Φ29 DNA polymerase has served for more than a decade as a model to study DNA polymerase–TP interactions. Thus, the identification of several mutant polymerases defective in the interaction with TP was possible. These mutants also showed impaired behavior in either polymerization or the exonuclease function. In the C-terminal polymerization domain, residues T434 and R438 of motif Tx2GR (22), residues Y226 and G229 of the YxGG/A motif (23) and a residue of the conserved TPR-1 region, D332, were identified as involved in TP binding (24). On the other hand, expression of the C-terminal domain led to a dramatically reduced interaction of TP and DNA polymerase (25), indicating involvement of the N-terminal domain in such an interaction. Thus, in the N-terminal exonuclease domain, residues S122 of the (S/T)Lx2h motif conserved in proofreading DNA polymerases (26) and F65 in the ExoII motif Nx2-3(F/Y)Dx2Ah (26) were shown to be involved in TP interaction (27). Summing up, these results suggest a complex interaction between the DNA polymerase and TP, involving different regions of the polymerase.
In this work we focus on residues Y59, H61 and F69 of Φ29 DNA polymerase, belonging to a well-characterized region in proofreading DNA polymerases, the ExoII motif. Based on our results we propose a function for these three residues in the interaction with TP.
MATERIALS AND METHODS
Nucleotides and proteins
Unlabeled nucleotides were purchased from Pharmacia P-L Biochemicals. [α-32P]dATP (3000 Ci/mmol) was obtained from Amersham International. Φ29 TP was purified as described (28). The wild-type Φ29 DNA polymerase was purified from Escherichia coli NF2690 cells harboring plasmid pJLw2, as described (29). Φ29 DNA polymerase site-directed mutants Y59F/L, H61L/R and F69Y/S were purified as described (30). Φ29 single-stranded (ss)DNA-binding protein (SSB) and Φ29 double-stranded DNA-binding protein (p6), obtained from Φ29-infected Bacillus subtilis cells, were purified as described (31,32).
DNA templates and substrates
Φ29 DNA was obtained by proteinase K treatment of phage particles in the presence of SDS (33), phenol extraction and ethanol precipitation. M13mp18 ssDNA was hybridized to the universal primer in the presence of 0.2 M NaCl and 60 mM Tris–HCl, pH 7.5, and the resulting molecule was used as a primer/template to analyze processive DNA polymerization coupled to strand displacement by Φ29 DNA polymerase. TP-containing Φ29 DNA (Φ29 TP–DNA) was obtained as described (34).
Replication assay (protein-primed initiation plus elongation) using Φ29 TP–DNA as template
The incubation mixture contained, in 25 µl, 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 20 mM ammonium sulfate, 1 mM dithiothreitol, 4% glycerol, 0.1 mg/ml BSA, 20 µM each dCTP, dGTP, dTTP and [α-32P]dATP (1 µCi), 0.5 µg Φ29 TP–DNA, 125 ng purified TP and 5 ng wild-type or mutant Φ29 DNA polymerase. After incubation for 10 min at 30°C, the reaction was stopped by adding 10 mM EDTA, 0.1% SDS and the samples were filtered through Sephadex G-50 spin columns. Relative activity was calculated from the Cerenkov radiation corresponding to the excluded volume. For size analysis, the labeled DNA was denatured by treatment with 0.7 M NaOH and subjected to electrophoresis in alkaline 0.7% agarose gels, as described (35). After electrophoresis, the position of unit length Φ29 DNA (19 285 bases) was detected by ethidium bromide staining, and then the gels were dried and autoradiographed. For analysis of the transition products, 100 ng DNA polymerase were used, providing the concentration of dNTP indicated in each case. The samples were analyzed in a 12% SDS–PAGE gel (360 × 280 × 0.5 mm), to obtain enough resolution to distinguish TP bound to the first elongation products.
Φ29 TP–DNA amplification assay
The incubation mixture contained, in 25 µl, 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 20 mM ammonium sulfate, 1 mM dithiothreitol, 4% glycerol, 0.1 mg/ml BSA, 80 µM each dCTP, dGTP, dTTP and [α-32P]dATP (2 µCi), 5 ng Φ29 TP–DNA, 5 ng wild-type or mutant Φ29 DNA polymerase, 5 ng TP, 10 µg Φ29 SSB and 10 µg Φ29 p6. After incubation for 90 min at 30°C, the samples were processed and the amplified DNA analyzed by electrophoresis in alkaline agarose gels, as described (36). After electrophoresis, the position of unit length Φ29 DNA was detected by ethidium bromide staining.
Replication of primed M13 DNA
The incubation mixture contained, in 25 µl, 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 1 mM dithithreitol, 4% glycerol, 0.1 mg/ml BSA, 80 µM each dCTP, dGTP, dTTP and [α-32P]dATP (2.5 µCi), 0.25 µg primed M13mp8 ssDNA and 25 ng wild-type or mutant Φ29 DNA polymerase. After incubation for the indicated times at 30°C, the samples were processed and the synthesized DNA was quantitated and analyzed as described above for the Φ29 TP–DNA replication assay. After electrophoresis, unit length M13mp8 ssDNA was detected by ethidium bromide staining and then the gels were dried and autoradiographed.
TP–dAMP formation (protein-primed initiation assay)
The incubation mixture contained, in 25 µl, 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 20 mM ammonium sulfate, 1 mM dithiothreitol, 4% glycerol, 0.1 mg/ml BSA, 0.2 µM dATP [α-32P]dATP (2.5 µCi), 0.5 µg Φ29 TP–DNA, 125 ng purified TP and 25 ng wild-type or mutant Φ29 DNA polymerase and was incubated for 4 min at 30°C. For the template-independent initiation assay, Φ29 TP–DNA was omitted, 80 ng wild-type or mutant Φ29 DNA polymerase was added, 1 mM MnCl2 was used instead of MgCl2 and the incubation was for 120 min at 30°C. The reactions were stopped by adding 10 mM EDTA and 0.1% SDS, filtered through Sephadex G-50 spin columns and further analyzed by SDS–PAGE as described (34). Quantitation was done by densitometric analysis of the labeled band corresponding to the TP–dAMP complex, detected by autoradiography.
Interference assay for TP binding
Reactions were carried out as described for the template-independent initiation assay, but in the absence of template and using a limiting amount of TP and different proportions of a mixture of wild-type and mutant DNA polymerases. Φ29 DNA polymerase mutant D249E (catalytically inactive but displaying a normal interaction with TP) was used as a positive control for the interference assay, as previously described (37). The amounts of proteins used were as follows: 10 ng TP, 25 ng wild-type DNA polymerase and either 5, 10, 25, 50 or 100 ng each mutant derivative (D249E, Y59F/L, H61L/R and F69Y/S). In all cases incubation was for 2 h at 30°C. After incubation, reactions were stopped and analyzed as indicated for the protein-primed initiation assay.
Analysis of the interaction between TP and DNA polymerase mutants
The incubation mixture contained, in 150 µl, 50 mM Tris–HCl, pH 7.5, 1 mM dithithreitol, 20 mM ammonium sulfate, 3 µg TP and 6 µg wild-type or mutant DNA polymerase. After incubation for 30 min at 4°C, samples were loaded on top of continuous 15–30% glycerol gradients (4 ml) in the presence of 50 mM Tris–HCl, pH 7.5, 20 mM ammonium sulfate, 0.4 M NaCl, 1 mM EDTA and 7 mM 2-mercaptoethanol, and centrifugated at 4°C for 24 h at 58 000 r.p.m. in a Beckman TST 60.4 rotor. Gradients were fractionated and subjected to 12% SDS–PAGE. Gels were stained after electrophoresis with Coomassie Blue.
RESULTS
Conservation of residues in the ExoII motif among protein-primed DNA polymerases
The multiple sequence alignment shown in Figure 1 depicts the highly conserved ExoII motif, present in all DNA polymerases which have proofreading activity. Here we focus on the subgroup of eukaryotic-type DNA polymerases (family B) using a protein-priming mechanism. In this motif, originally described by Bernad et al. (38), resides one of the four catalytic aspartate residues involved in metal binding and catalysis of 3′→5′ exonuclease activity of proofreading DNA polymerases. Moreover, an asparagine, located five residues N-terminally, and a F/Y directly preceding the catalytic aspartate are strongly conserved residues shown to be ssDNA ligands involved in primer terminus stabilization at the 3′→5′ exonuclease active site (26,39). In addition, three more residues, Y59, H61 and F69 for Φ29 DNA polymerase are highly conserved in this motif (Fig. 1). The tyrosine is present in 14 of 17 DNA polymerases of the protein-primed eukaryotic-type subgroup but in only one of the remaining family B DNA polymerases. The histidine is found in 15 of 17 of the protein-primed subgroup whereas it is absent in all other family B group members. Finally, the phenylalanine is present in 12 of 17 protein-primed DNA polymerases and in only 7 of 30 non-protein-primed family B DNA polymerases. Thus, the high level of conservation of these residues led us to introduce site-directed mutations, and we recently showed their involvement in ssDNA binding during 3′→5′ exonucleolysis (30). Moreover, this conservation could imply a role in specific functions intrinsic to the mechanism used by Φ29 DNA polymerase during replication of the viral genome, such as the DNA polymerase–TP interaction. Six mutant derivatives (Y59F/L, H61L/R and F69Y/S), overexpressed and purified as described elsewhere (30), were analyzed using a variety of in vitro assays corresponding to the different stages of the TP-primed Φ29 DNA replication mechanism.
Figure 1.
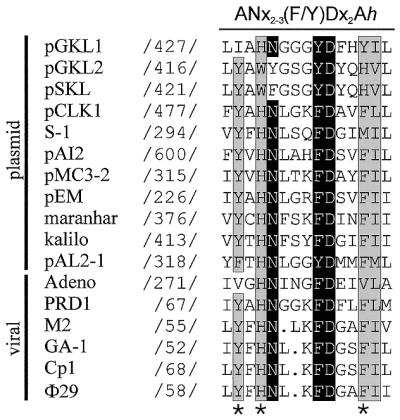
Highly conserved residues among DNA polymerases using a protein as primer. Polymerases included in the alignment correspond to the eukaryotic (family B) proofreading (3′→5′ exonuclease activity) type and are either of plasmid or viral origin. Nomenclature and sequence references are according to de Vega et al. (30). Numbers indicate the position of the first aligned amino acid with respect to the N-terminus of the respective DNA polymerase. Highly conserved residues of the ANx2–3(F/Y)Dx2Ah (ExoII) motif, N, F/Y and D, are white on a black background. Conserved aromatic (A) and aliphatic hydrophobic (h) residues have a gray background. Mutation sites at Φ29 DNA polymerase residues Y59, H61 and F69 are marked with asterisks.
Φ29 DNA polymerase mutants are strongly affected in protein-primed DNA replication
Replication of viral DNA via a protein-primed mechanism involves interaction of DNA polymerase with TP and DNA. During initiation, Φ29 DNA polymerase catalyzes template-directed formation of a covalent TP–dAMP complex at both terminal origins. Subsequent elongation then takes place in a strand displacement manner (21). In our assays we used a minimal Φ29 replication system, making use only of TP–DNA, TP and DNA polymerase. The replication activities of the respective Φ29 DNA polymerase mutants with respect to the wild-type enzyme are given in Table 1. As can be observed, the efficiency displayed by mutants F69Y, F69S, Y59L, H61L and H61R with respect to the wild-type enzyme was reduced 2-, 2-, 3-, 5- and >100-fold, respectively, whereas mutant Y59F displayed essentially wild-type activity. Only exchange of H61 for arginine resulted in products shorter than TP–DNA unit length (19 285 bp, Fig. 2). In Φ29 DNA amplification assays, which more closely resemble the in vivo situation because, in addition to TP and DNA polymerase, we used Φ29 SSB and Φ29 protein p6 in the presence of limiting Φ29 TP–DNA levels, amplification products were only visible when TP–DNA replication was not severely affected. Mutant polymerase Y59F amplified Φ29 TP–DNA at nearly the wild-type level and mutant F69Y displayed 39% of the wild-type activity (Fig. 3). In contrast, mutant polymerases Y59L, H61L and F69S were severely affected in TP–DNA amplification (Fig. 3). For mutant polymerase H61R no signal could be obtained in TP–DNA amplification assays.
Table 1. Enzymatic activities of wild-type and mutant derivatives of F29 DNA polymerasea.
| Parameter assayed | Substrate(s) | F29 DNA polymeraseb |
||||||
|---|---|---|---|---|---|---|---|---|
| wt | T59F | Y59L | H59R | H61L | F69Y | F69S | ||
| F29 TP-DNA replication | F29 TP-DNA, dNTP | 100 | 79 | 34 | <1 | 20 | 58 | 45 |
| F29 TP-DNA amplification | F29 TP-DNA dNTP | 100 | 91 | 3 | <1 | 1 | 39 | 4 |
| Primed M13 DNA replication | Primed M13, dNTP | 100 | 58 | 100 | 9 | 29 | 72 | 87 |
| F29 TP-DNA initiation | F29 TP-DNA, TP, dATP | 100 | 77 | 18 | 25 | 13 | 49 | 22 |
| TP-deoxynucleotidylation | TP, dATP | 100 | 102 | 19 | 10 | 4 | 43 | 7 |
aThe different activity assays were carried out as described in Materials and Methods.
bNumbers indicate the percentage of activity relative to the wild-type enzyme obtained from several experiments.
Figure 2.

Φ29 TP–DNA replication by wild-type and mutant Φ29 DNA polymerases. Autoradiograph of Φ29 TP–DNA replication assay carried out as described in Materials and Methods in the presence of 5 ng wild-type (wt) or mutant Φ29 DNA polymerase. After incubation for 10 min at 30°C, relative activity values were calculated (see Table 1) and the length of the synthesized DNA was analyzed by alkaline agarose gel electrophoresis. The migration position of unit length Φ29 DNA is indicated.
Figure 3.
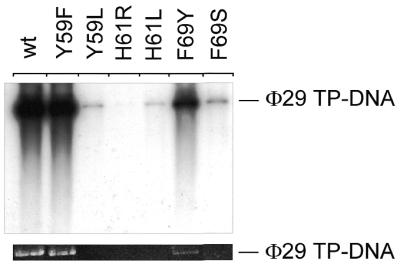
Φ29 DNA polymerase mutants are strongly affected in Φ29 TP–DNA amplification. The assay was carried out as described in Materials and Methods, in the presence of 5 ng wild-type or mutant Φ29 DNA polymerase, 5 ng Φ29 TP and 10 µg each Φ29 DBP and Φ29 SSB. After incubation for 90 min at 30°C, samples were processed and the amplified DNA was analyzed by alkaline agarose gel electrophoresis as described in Materials and Methods. (Top) Autoradiograph of such a gel; (bottom) Φ29 TP–DNA amplification signal detected by ethidium bromide staining. The migration position of unit length Φ29 TP–DNA (19 285 bases) is indicated.
Strand displacement coupled to polymerization capacity of mutant Φ29 DNA polymerases
To separate the specific TP–DNA polymerase interaction at the start of Φ29 TP–DNA replication from the process of nucleotide-primed DNA synthesis, we performed a DNA-primed M13 replication assay (see Materials and Methods). With these assays not only can the polymerization velocity of the mutant DNA polymerases in comparison to the wild-type be assayed, but also their strand displacement ability. In this assay, once the 3′-OH group of the primer was bound, all mutant polymerases could reach M13 unit length during the first round of replication at a similar rate. Replication abilities changed dramatically when DNA polymerases reached the 5′-end of the primer and during the next rounds of replication (a rolling circle type mechanism), which require strand displacement capacity. Comparable to the Φ29 TP–DNA replication assays, mutants Y59F and F69Y were little affected; both replicated M13 DNA several-fold at the same rate and were only slightly affected in the amount of product (Table 1). Mutants Y59L and F69S replicated M13 DNA with high efficiency (100 and 87% of wild-type capacity, respectively), however, substitution of H61 changed the replication ability of Φ29 DNA polymerase dramatically. Due to a poor strand displacement capacity, especially for mutant H61R (not shown), the replication rate was severely decreased (29% for H61L and 9% for H61R; see Table 1).
Poor TP–dAMP production (initiation) of mutants affected in Φ29 TP-DNA replication
Mutant DNA polymerases Y59L, H61L/R and F69S were severely affected in Φ29 TP–DNA replication (see above) but were still able to perform processive DNA-primed replication. However, to start Φ29 DNA replication, Φ29 DNA polymerase has to form a heterodimer with its specific primer molecule, TP. Once the replication origin is recognized by this complex, Φ29 DNA polymerase catalyzes the addition of dAMP to the OH group of TP Ser232, using the second dTMP of the DNA strand as template, a mechanism known as initiation (13,14). In Figure 4 (top) and Table 1 it can be observed that mutants Y59L, H61L/R and F69S catalyzed TP–dAMP formation to a low extent, whereas mutants Y59F and F69Y showed nearly normal initiation capacity. Because the initiation of Φ29 DNA replication is a template-directed event, defects during initiation could be the consequence of a weak affinity for the template DNA. This can be ruled out as Φ29 DNA polymerase is able to catalyze dAMP addition to TP in the absence of template DNA (TP deoxynucleotidylation) (40). The activities of the mutant DNA polymerases under these conditions were similar or even lower than in the presence of template (Fig. 4, bottom, and Table 1), suggesting a specific defect in the Φ29 DNA polymerase–TP interaction. To test this possibility an interference assay was carried out. Hence, functional interaction of mutant DNA polymerases with TP was investigated by competition between wild-type and mutant DNA polymerases for a limited amount of TP in an initiation reaction. As a control we used Φ29 DNA polymerase D249E, catalytically inactive but able to interact normally with TP (37). As shown in Figure 5, the inhibition profile of the control, mutant D249E, followed the theoretical curve, but no competition could be detected for either Y59L, H61L/R or F69S, reflecting a defective interaction with TP. Evidence for a poor interaction of mutant DNA polymerases Y59L, H61L/R and F69S with TP was further obtained by ultracentrifugation, using glycerol gradients. As shown in Figure 6, wild-type Φ29 DNA polymerase (66 kDa) and TP (30 kDa) form a heterodimer of 96 kDa and the two proteins co-sediment in the same fractions. In contrast, the two proteins sedimented separately as monomers when mutant DNA polymerases Y59L, H61R and F69S were used, indicating impairment in the interaction with TP.
Figure 4.

Formation of the TP–dAMP complex catalyzed by wild-type or mutant Φ29 DNA polymerase in the presence or absence of Φ29 TP–DNA. The reactions were carried out as described in Materials and Methods, in the presence of 125 ng TP. The template-dependent reaction (top) was carried out in the presence of 10 mM MgCl2, 0.5 µg TP–DNA and 5 ng wild-type or mutant Φ29 DNA polymerase. Incubation was for 4 min at 30°C. The template-independent reaction (bottom) was performed in the presence of 1 mM MnCl2 and 80 ng mutant or wild-type Φ29 DNA polymerase. Incubation was for 120 min at 30°C. The samples were analyzed by SDS–PAGE and autoradiography. The band corresponds to the TP–dAMP initiation complex. Quantification was by densitometric analysis of the band corresponding to the labeled TP–dAMP complex, detected by autoradiography.
Figure 5.

Mutant Φ29 DNA polymerases Y59L, H61R/L and F69S do not interfere with wild-type polymerase for TP interaction. Reactions were carried out as described for the template-independent initiation assay using a limiting amount of TP and different proportions (5, 10, 25, 50 or 100 ng) of mutant DNA polymerases Y59L, H61R, H61L and F69S, respectively. The amount of TP–dAMP formed under competition conditions is followed relative to the amount formed in the absence of competitor. An inactive polymerase with unmodified TP interaction ability should inhibit formation of the TP–dAMP complex by competition with the wild-type enzyme. None of the indicated mutants showed this behavior. As a control for 100% inhibition, the previously analyzed mutant D249E was used (32), whose inhibition profile resembles the theoretical one. The lines for theoretical and mutant D249E values are least squares fits to a simple exponential decay.
Figure 6.

Interaction between DNA polymerase and TP shown by glycerol gradient centrifugation. Wild-type or mutant DNA polymerase (8 µg) was incubated with TP (4 µg). After incubation for 30 min at 4°C, samples were loaded on top of a continuous 15–30% glycerol gradient (4 ml) and centrifuged as described in Materials and Methods. Gradients were fractionated and subjected to 12% SDS–PAGE. Gels were stained after electrophoresis with Coomassie Blue. Densitometric quantitations in arbitrary units of both Φ29 DNA polymerase (full circles) and Φ29 TP (open circles) are represented.
Multiple abortive products during transition for mutant DNA polymerases
Taking the latter results into account, it was interesting to investigate whether one or all of these four mutant polymerases are affected in transition (the intermediate stage between TP-primed initiation and DNA-primed polymerization), as Φ29 DNA polymerase and TP act as a heterodimer during insertion of the first 9 nt (20). It is known that during transition certain amounts of non-elongated and partially elongated initiation complexes [TP–dAMP and TP–(dNMP)2–11] are produced, as a consequence of the rate limiting transition steps from initiation to elongation mode (20). Such products can only be detected for DNA polymerases lacking 3′→5′ exonuclease activity, because otherwise they are degraded to TP–(dAMP)2. The inability of mutants Y59L, H61L/R and F69S to bind TP effectively was described above and their defective exonuclease activity has been described elsewhere (30). We performed a truncated assay where the nucleotide dCTP was missing during the first part of the reaction, forcing the wild-type DNA polymerase to stop after TP–(dNMP)8 (replication from the left origin of the Φ29 genome) or TP–(dNMP)11 (replication from the right origin) (Fig. 7). Whereas mutant F69S (not shown) and mutants Y59L and H61L reacted essentially like the wild-type polymerase, mutant H61R displayed a very high proportion of partially elongated molecules [TP–(dNMP)2–8]. If these products corresponded to a normal proportion of abortive complexes (only visible due to the affected 3′→5′ exonuclease activity of the mutant), their total number should be comparable to the amount of TP–(dAMP)2 displayed by the wild-type polymerase. However, the amounts of mutant abortive products are higher than the wild-type TP–(dAMP)2 degradation level, suggesting additional problems during this stage for mutant polymerase H61R. In the second part of the assay the missing dCTP was added after 5 min incubation, allowing Φ29 DNA polymerase to fully replicate the template. In contrast to the wild-type and mutant F69S (not shown) DNA polymerases, which were able to elongate all TP–(dNMP)8–11 molecules to full-length TP–DNA, mutants Y59L, H61L and, more drastically, H61R still produced many abortive transition products (Fig. 7).
Figure 7.
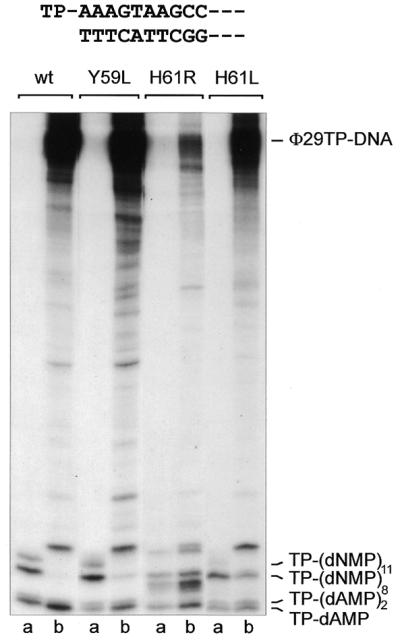
Abortive TP–(dNMP)n products accumulate for DNA polymerase mutants affected in TP binding. The assay was performed as described in Materials and Methods. Samples were analyzed by high resolution SDS–PAGE. The first left-most 10 nt of the Φ29 genome are depicted at the top. Samples contained 20 µM each [α-32P]dATP (1 µCi), dGTP and dTTP. Whereas the first series of samples (a) was stopped after incubation at 30°C for 5 min, dCTP was added to the second series of samples (b) for an additional 5 min incubation at 30°C. The length of different transition products and the position corresponding to full-length Φ29 TP–DNA are indicated.
DISCUSSION
All attempts to improve our understanding of Φ29 DNA polymerase–TP interactions are based on biochemical characterization of mutants of these two proteins and on extrapolation of the resolved three-dimensional structures of closely related DNA polymerases, such as those from RB69 (1), T.gorgonarius (2), Thermococcus sp. 9°N7 (3) and P.kodakaraensis KOD1 (4). The overall structure of Φ29 DNA polymerase is most likely similar to these family B-like DNA polymerases, but residues involved in TP binding lack analogy as none of the known structures consists of a polymerase able to perform protein-primed DNA replication.
Sequence aligments of the ANx2–3(F/Y)Dx2Ah (ExoII) motif, originally described by Bernad et al. (38), show its high conservation among DNA polymerases (see Fig. 1) (30). The importance of the ExoII motif for DNA replication originates from the presence of one of the four catalytic aspartates involved in metal binding and catalysis (41) and of two more conserved residues (N62 and F65 in Φ29 DNA polymerase) which are involved in primer terminus stabilization at the 3′→5′ exonuclease active site in proofreading DNA polymerases (27,39). A striking degree of homology for three more residues, Y59, H61 and F69, among members of the protein-primed subgroup suggested a possible involvement in reactions specific for those DNA polymerases using a protein-primed mechanism. The ends of the Φ29 genome, containing a covalently bound TP molecule, constitute the origins of replication. Initiation of DNA replication is triggered by recognition of these origins by a DNA polymerase–TP complex. The TP in this heterodimer acts as primer for the subsequent replication initiation step. Parental and primer TP are able to interact (42), which may suggest that interaction of the heterodimer with the origin is due to protein–protein interactions between two molecules of TP. Recently a putative coiled coil region in the N-terminus of the TP sequence was described that could be involved in these parental and primer TP interactions which might be important for origin recognition (42). Further elongation does not lead to immediate dissociation of DNA polymerase from the primer TP, but results in a transition stage in which the DNA polymerase synthesizes a 5 nt long DNA molecule while still bound to TP and undergoes some structural change during replication of nucleotides 6–9 (20). Finally, DNA polymerase dissociates from the primer TP with insertion of nucleotide 10 and DNA-primed elongation takes place. Φ29 DNA polymerase is then able to replicate the Φ29 genome in a highly processive way by making use of its strand displacement capacity (21).
Mutants Y59L, H61L/R and F69S showed a strong deficiency in their ability to replicate and amplify Φ29 TP–DNA. M13 DNA replication assays showed that conserved residues Y59, H61 and F69 play no role in DNA-primed replication (although mutants H61L and H61R were affected in strand displacement, as described in Results). Protein-primed initiation in the absence and presence of Φ29 template TP–DNA and interference and glycerol gradient experiments revealed that the interaction between TP and these mutant DNA polymerases was affected. The mechanism of protein priming for DNA replication implies a necessary transition for the DNA polymerase from protein-priming mode at the initiation step to DNA-priming mode during the elongation steps. Furthermore, the switch from protein- to DNA-priming polymerization mode during the first steps of Φ29 DNA replication does not take place immediately after the initiation complex is formed (20). This switch, or transition, most probably implies a DNA polymerase conformational change that starts after insertion of 5 nt and ends when nucleotide 9 is inserted, requiring a high stability of the complex at this moment. With insertion of the tenth nucleotide, DNA polymerase and TP dissociate. Hence, mutants that showed a high reduction in Φ29 TP–DNA replication should not only display a high number of abortive but also accumulation of TP–(dNMP)n products that are not properly elongated. Especially, the poor ability to stably interact with TP of mutant polymerase H61R led to short abortive transition products, which partially contained misinserted nucleotides.
No crystal data are available to date for any TP or DNA polymerase that uses a protein-primed mechanism. Nonetheless, we could show for Φ29 DNA polymerase that the conserved Tx2GR motif is important for DNA polymerase–TP interaction (22). Sequence comparison of Φ29 DNA polymerase with crystal data of RB69 polymerase (1) revealed that Tx2GR is part of the palm subdomain of the polymerization domain in these two DNA polymerases. Other residues leading to a non-functional interaction between TP and Φ29 DNA polymerase are D332 of the recently identified C-terminal TPR-1 motif (24) and Y226 and G229 of the YxGG/A motif (23). Finally, isolated expression of the C-terminal part of Φ29 DNA polymerase resulted in a loss of capacity to interact with TP and, as a consequence, led to a >1000-fold decrease in initiation capacity compared to the complete enzyme (25).
Support for the importance of the Φ29 DNA polymerase N-terminus in functional TP binding was shown by mutation of S122 in the hx2SLx2h motif (26) and our present studies of the ExoII motif and its residues Y59, H61 and F69. Interestingly, these three residues are especially conserved among members of the subgroup of family B DNA polymerases that use a protein-primed DNA replication mechanism. Moreover, their involvement in proper stabilization of the primer terminus at the 3′→5′ exonuclease active site (30) has recently been shown, thus suggesting a double function. First, during initiation of Φ29 DNA replication by protein primer binding, a stage in which exonuclease activity is inactive, and second during transition/elongation in primer terminus stabilization at the exonuclease active site, establishing DNA proofreading. Furthermore, it supports an earlier proposal that during the transition step the contacts of DNA polymerase with TP are replaced by interactions with the nascent DNA, leading to a DNA-primed elongation process (27). Comparison with the structures of the T4 and RB69 polymerases (PDB accession nos 1NOY and 1WAJ, respectively) reveals that Φ29 DNA polymerase residue H61 especially might be directly exposed to the cleft in which the DNA primer or TP molecules are bound. Thus, the roles of TP-binding residues would be transient, changing to DNA binding when elongation proceeds and proper stabilization of the primer strand at the 3′→5′ exonuclease active site is required for fidelity.
The overall interaction between a DNA polymerase utilizing a protein-primed DNA replication mechanism and TP is rather complex. It involves a great many residues of different regions in Φ29 DNA polymerase. Therefore, the mechanism will not require a unique subdomain for interaction, but multiple interactions at different locations in DNA polymerase and TP.
Acknowledgments
ACKNOWLEDGEMENTS
This investigation was aided by research grant 5R01 GM27242-22 from the National Institutes of Health, by grant PB98-0645 from the Dirección General de Investigacíon Científica y Técnica, by grant ERBFMX CT97 0125 from the European Union and by an institutional grant from Fundación Ramón Areces. R.E. was a post-doctoral fellow of the European Union.
REFERENCES
- 1.Wang J., Sattar,A.K., Wang,C.C., Karam,J.D., Konigsberg,W.H. and Steitz,T.A. (1997) Crystal structure of a Pol α family replication DNA polymerase from bacteriophage RB69. Cell, 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- 2.Hopfner K.P., Eichinger,A., Engh,R.A., Laue,F., Ankenbauer,W., Huber,R. and Angerer,B. (1999) Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc. Natl Acad. Sci. USA, 96, 3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez A.C., Park,H.W., Mao,C. and Beese,L.S. (2000) Crystal structure of a Pol α family DNA polymerase from the hyperthermophilic archeon Thermococcus sp. 9 degrees N-7. J. Mol. Biol., 299, 447–462. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto H., Nishioka,M., Fujiwara,S., Takagi,M., Imanaka,T., Inoue,T. and Kai,Y. (2001) Crystal structure of DNA polymerase from hyperthermophilic archeon Pyrococcus kodakaraensis KOD1. J. Mol. Biol., 306, 469–477. [DOI] [PubMed] [Google Scholar]
- 5.Li Y., Korolev,S. and Waksman,G. (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J., 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doublié S., Tabor,S., Long,A.M., Richardson,C.C. and Ellenberger,T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature, 391, 251–258. [DOI] [PubMed] [Google Scholar]
- 7.Kiefer J.R., Mao,C., Hansen,C.J., Basehore,S.L., Hogrefe,H.H., Braman,J.C. and Beese,L.S. (1997) Crystal structure of a thermostable Bacillus DNA polymerase I large fragment at 2.1 Å resolution. Structure, 5, 95–108. [DOI] [PubMed] [Google Scholar]
- 8.Brautigam C.A. and Steitz,T.A. (1998) Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol., 8, 54–63. [DOI] [PubMed] [Google Scholar]
- 9.Doublié S. and Ellenberger,T. (1998) The mechanism of action of T7 DNA polymerase. Curr. Opin. Struct. Biol., 8, 704–712. [DOI] [PubMed] [Google Scholar]
- 10.Kunkel T.A. and Bebenek,R. (2000) DNA replication fidelity. Annu. Rev. Biochem., 69, 497–529. [DOI] [PubMed] [Google Scholar]
- 11.Steitz T.A. (1999) DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem., 274, 17395–17398. [DOI] [PubMed] [Google Scholar]
- 12.Blanco L. and Salas,M. (1996) Relating structure to function in phi29 DNA polymerase. J. Biol. Chem., 271, 8509–8512. [DOI] [PubMed] [Google Scholar]
- 13.Salas M. (1999) Mechanisms of initiation of linear DNA replication in prokaryotes. In Setlow,J.K. (ed.), Genetic Engeneering. Kluwer Academic/Plenum Press, New York, NY, Vol. 21, pp. 159–171. [DOI] [PubMed]
- 14.Salas M., Miller,J.T., Leis,J. and DePamphilis,M.L. (1996) Mechanisms for priming DNA synthesis. In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 131–176.
- 15.Blanco L. and Salas,M. (1984) Characterization and purification of a phage phi29-encoded DNA polymerase required for the initiation of replication. Proc. Natl Acad. Sci. USA, 81, 5325–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hermoso J.M., Méndez,E., Soriano,F. and Salas,M. (1985) Location of the serine residue involved in the linkage between the terminal protein and the DNA of phage phi29. Nucleic Acids Res., 13, 7715–7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Méndez J., Blanco,L., Esteban,J.A., Bernad,A. and Salas,M. (1992) Initiation of phi29 DNA replication occurs at the second 3′ nucleotide of the linear template: a sliding-back mechanism for protein-primed DNA replication. Proc. Natl Acad. Sci. USA, 89, 9579–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caldentey J., Blanco,L., Bamford,D.H. and Salas,M. (1993) In vitro replication of bacteriophage PRD1 DNA. Characterization of the protein-primed initiation site. Nucleic Acids Res., 21, 3725–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King A.J. and Van der Vliet,P.C. (1994) A precursor terminal protein trinucleotide intermediate during initiation of adenovirus DNA-replication—regeneration of molecular ends in vitro by a jumping back mechanism. EMBO J., 13, 5786–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Méndez J., Blanco,L. and Salas,M. (1997) Protein-primed DNA replication: a transition between two modes of priming by a unique DNA polymerase. EMBO J., 16, 2519–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanco L., Bernad,A., Lázaro,J.M., Martín,G., Garmendia,C. and Salas,M. (1989) Highly efficient DNA synthesis by the phage phi29 DNA polymerase. Symmetrical mode of DNA replication. J. Biol. Chem., 264, 8935–8940. [PubMed] [Google Scholar]
- 22.Méndez J., Blanco,L., Lázaro,J.M. and Salas,M. (1994) Primer-terminus stabilization at the Φ29 DNA polymerase active site—mutational analysis of conserved motif Tx(2)GR. J. Biol. Chem., 269, 30030–30038. [PubMed] [Google Scholar]
- 23.Truniger V., Blanco,L. and Salas,M. (1999) Role of the “YxGG/A” motif of phi29 DNA polymerase in protein-primed replication. J. Mol. Biol., 286, 57–69. [DOI] [PubMed] [Google Scholar]
- 24.Dufour E., Méndez,J., Lázaro,J.M., de Vega,M., Blanco,L. and Salas,M. (2000) An aspartic acid residue in TPR-1, a specific region of protein-priming DNA polymerases, is required for the functional interaction with primer terminal protein. J. Mol. Biol., 304, 289–300. [DOI] [PubMed] [Google Scholar]
- 25.Truniger V., Lázaro,J.M., Salas,M. and Blanco,L. (1998) Phi29 DNA polymerase requires the N-terminal domain to bind terminal protein and DNA primer substrates. J. Mol. Biol., 278, 741–755. [DOI] [PubMed] [Google Scholar]
- 26.de Vega M., Lázaro,J.M., Salas,M. and Blanco,L. (1998) Mutational analysis of phi29 DNA polymerase residues acting as ssDNA ligands for 3′-5′ exonucleolysis. J. Mol. Biol., 279, 807–822. [DOI] [PubMed] [Google Scholar]
- 27.de Vega M., Blanco,L. and Salas,M. (1998) Phi29 DNA polymerase residue Ser122, a single-stranded DNA ligand for 3′-5′ exonucleolysis, is required to interact with the terminal protein. J. Biol. Chem., 273, 28966–28977. [DOI] [PubMed] [Google Scholar]
- 28.Zaballos A., Lázaro,J.M., Méndez,E., Mellado,R.P. and Salas,M. (1989) Effects of internal deletions on the priming activity of the phage Φ29 terminal protein. Gene, 83, 187–195. [DOI] [PubMed] [Google Scholar]
- 29.Lázaro J.M., Blanco,L. and Salas,M. (1995) Purification of Φ29 DNA polymerase. Methods Enzymol., 262, 42–49. [DOI] [PubMed] [Google Scholar]
- 30.de Vega M., Lázaro,J.M. and Salas,M. (2000) Phage Φ29 DNA polymerase residues involved in the proper stabilization of the primer-terminus at the 3′-5′ exonuclease active site. J. Mol. Biol., 304, 1–9. [DOI] [PubMed] [Google Scholar]
- 31.Martín G., Lázaro,J.M., Méndez,E. and Salas,M. (1989) Characterization of phage Φ29 protein p5 as a single-stranded DNA binding protein. Function in Φ29 DNA–protein p3 replication. Nucleic Acids Res., 17, 3663–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pastrana R., Lázaro,J.M., Blanco,L., García,J.A., Méndez,E. and Salas,M. (1985) Overproduction and purification of protein p6 of Bacillus subtilis phage Φ29: role in the initiation of DNA replication. Nucleic Acids Res., 13, 3083–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inciarte M.R., Lázaro,J.M., Salas,M. and Viñuela,E. (1976) Physical map of bacteriophage Φ29. Virology, 74, 314–323. [PubMed] [Google Scholar]
- 34.Peñalva M.A. and Salas,M. (1982) Initiation of phage phi29 DNA replication in vitro: formation of a covalent complex between the terminal protein, p3 and 5′-dAMP. Proc. Natl Acad. Sci. USA, 79, 5522–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonnell M.W., Simon,M.N. and Studier,F.W. (1994) Analysis of restriction fragments of T7 DNA and determination of molecular weights by electrophoresis in neutral and alkaline agarose gels. J. Membr. Biol., 110, 119–146. [DOI] [PubMed] [Google Scholar]
- 36.Blanco L., Lázaro,J.M., de Vega,M., Bonnin,A. and Salas,M. (1994) Terminal protein-primed DNA amplification. Proc. Natl Acad. Sci. USA, 91, 12198–12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blasco M.A., Lázaro,J.M., Blanco,L. and Salas,M. (1993) Phi29 DNA polymerase active site. Residue Asp249 of conserved amino acid motif “Dx2SLYP” is critical for synthetic activities. J. Biol. Chem., 268, 24106–24113. [PubMed] [Google Scholar]
- 38.Bernad A., Blanco,L., Lázaro,J.M., Martín,G. and Salas,M. (1989) A conserved 3′-5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell, 59, 219–228. [DOI] [PubMed] [Google Scholar]
- 39.de Vega M., Lázaro,J.M., Salas,M. and Blanco,L. (1996) Primer terminus stabilization at the 3′→5′ exonuclease active site of Φ29 DNA polymerase. Involvement of two amino acid residues highly conserved in proofreading DNA polymerases. EMBO J., 15, 1182–1192. [PMC free article] [PubMed] [Google Scholar]
- 40.Blanco L., Bernad,A., Esteban,J.A. and Salas,M. (1992) DNA-independent deoxynucleotidilation of the Φ29 terminal protein by the Φ29 DNA polymerase. J. Biol. Chem., 267, 1225–1230. [PubMed] [Google Scholar]
- 41.Derbyshire V., Pinsonneault,J.K. and Joyce,C.M. (1995) Structure-function analysis of the 3′-5′ exonucleases of DNA polymerases. Methods Enzymol., 262, 363–385. [DOI] [PubMed] [Google Scholar]
- 42.Serna-Rico A., Illana,B., Salas,M. and Meijer,W.J.J. (2000) The putative coiled coil domain of the phi29 terminal protein is a major determinant involved in recognition of the origin of replication. J. Biol. Chem., 275, 40529–40538. [DOI] [PubMed] [Google Scholar]