

Abstract
Complement system is one of the most important defense mechanisms of the innate immune system. In addition to their roles in immune regulation, complement proteins are also involved in neurodevelopment and adult brain plasticity. Complement dysregulation has been shown in neurodevelopmental disorders including schizophrenia and autism spectrum disorder as well as in mood disorders. A number of clinical as well as genetic studies suggest the role of complement proteins in the cortical thinning and excessive synaptic pruning frequently associated with schizophrenia. The changes in complement proteins are also associated with the pathophysiology of autism spectrum disorder, major depressive disorder and bipolar disorder, but warrant further research. In addition, rodent models suggest a strong case for complement system in anxiety-like behavior. In this article, we review the recent findings on the role of complement system in neuropsychiatric disorders. The possible uses for future complement targeted therapies are also discussed.
Keywords: Complement, Immune system, Depression, Schizophrenia, ASD, Synaptic plasticity, Anxiety
Introduction
Traditionally thought as a liver-secreted set of soluble proteins that travel by blood, the complement system works to ‘complement’ the immune system and protect the body (Bordet, 1909). Studies now show evidence for complement intersecting with normal cell physiology and maintenance, not just T cell effector function. Over 50 blood- and lymph- circulating proteins work to detect and remove pathogens. The classical, lectin, and alternative pathways have been identified for the trigger and process of pathogen/cellular debris removal. The complement system performs 3 main functions: (1) to promote phagocytosis of bacteria, molecular debris, and foreign molecules via neutrophils or monocytes, (2) to induce an inflammatory response using small anaphylatoxin proteins (C3a, C4a, C5a) derived from cleavage of complement proteins, (3) pore formation in lipid membrane of invading pathogens. C1q, C1r, and C1s combine to form C1 complement protein complex. This complex activates the classical pathway by cleavage of C3 into active C3a and C3b (Sarma and Ward, 2011). C1q induces a pro-inflammatory response characterized by increased levels of tumor necrosis factor-alpha and interleukin-6 secreted by human microglial cells (Veerhuis et al., 2005).
Complement system in neuropsychiatric disorders
In neurodevelopment, the complement system mediates proliferation of progenitor cells, neuronal migration, and synaptic pruning (Coulthard et al., 2018; Gorelik, Sapir, Haffner-Krausz, et al., 2017; Gorelik, Sapir, Woodruff, et al., 2017). Complement product C5a helps neural progenitor proliferation through protein kinase C pathway. Inhibition of C5a-C5aR signaling resulted in changes of neocortex morphology and behavior (Coulthard et al., 2017). Conversely, inhibition of C3a-C3aR signaling enhanced progenitor proliferation. C3aR knockout mice showed normal neurogenesis, but memory was still impaired (Coulthard et al., 2018). This might be from compensatory mechanisms since C3aR was deleted from birth. The C1 complex, which initiates the classical pathway, is present in neurons and microglia to tag synapses for proper developmental pruning (Stevens et al., 2007). This allows for redundant synapses to be removed by microglia and is essential for normal brain development/synaptic plasticity (Parkhurst et al., 2013; Paolicelli et al., 2011). Mice that lack C1q or C3 exhibited more synaptic inputs in the lateral geniculate nucleus and overlap of contralateral and ipsilateral tracts (Stevens et al., 2007; Chu et al., 2010). These studies show the importance of the classical pathway of the complement system in early postnatal synaptic refinement.
In zebrafish, C3 and C3aR regulate cell migration in the developing neural crest. Knockdown of C3aR in zebrafish caused neural crest cells to move towards abnormal locations. Neural crest cells in zebrafish are known to release C3, and C3aR works to attract these cells to their correct location (Carmona-Fontaine et al., 2011). Without these complement proteins, animals exhibited changes in cortical layering from deficient cell migration (Coulthard et al., 2018; Gorelik, Sapir, Haffner-Krausz, et al., 2017; Gorelik, Sapir, Woodruff, et al., 2017). Another way the complement system is activated in by mannose-associated serine protease 1 and 2 (MASP-1/MASP-2) in the lectin pathway. Knockout and knockdown for MASP-1 and MASP-2, and C3 showed neuronal migration impairment, but interestingly, were reversed by overexpression of C3a or administration of C3aR agonist (Coulthard et al., 2018; Gorelik, Sapir, Haffner-Krausz, et al., 2017; Gorelik, Sapir, Woodruff, et al., 2017). The importance of the complement system in neurodevelopment underscores its link to neuropsychiatric conditions. Accordingly, a number of studies conducted in rodents and humans indicate that abnormal complement system leads to altered anatomy/physiology in the brain.
Complement system and schizophrenia
The Diagnostic and Statistical Manual of Mental Disorders (DSM-5) defines schizophrenia (SZ) as a mental disorder characterized by delusions, hallucinations, or disorganized speech and can include ‘grossly disorganized/catatonic behavior’ and ‘diminished emotional expression’. Complement dysregulation has been linked to risk of SZ. Genetic variations of complement component 4 (C4) have been linked to complement-mediated synaptic pruning and thinning of cortex (Sekar et al., 2016). An increased expression of C4A, a constituent of C4, was found in the post-mortem brain samples of SZ subjects when compared to healthy controls (Sekar et al., 2016). Using phosphorus magnetic resonance spectroscopy in two SZ cohorts, a direct link was found between C4A gene repeats and neuropil (dendrites, axons, synapses, glial cell processes, and microvasculature) contraction. Young adult-onset early-course SZ patients with high C4A gene copy numbers had increased neuropil contraction in the inferior frontal and parietal cortical areas, while adolescent-onset SZ patients had increased neuropil contraction in the dorsolateral prefrontal cortex and thalamus (Prasad et al., 2018). Cortical thinning has been observed in the past as a hallmark of SZ (McCarley et al., 1999; Honea et al., 2005). In a study with 90 SZ patients and 75 healthy controls, peripheral mRNA for C5aR1, CR1, CR3a, CD55, C59, C3, C3b, and C4 were elevated in patient samples when compared to controls. In a subset of patients with high peripheral cytokine levels had a significant increase in peripheral C4A expression. In this same study, a high inflammation index score predicted a decrease in temporal lobe cortical thickness (Ji et al., 2022). Increased C5 and SERPING1 (encodes C1 inhibitor) mRNA levels were seen in adult Swedish twins with a total of 22 individuals diagnosed with SZ and 13 with bipolar disorder. This was associated with a decrease in superior frontal cortical thickness (Allswede et al., 2018).
The Schizophrenia Working Group of the Psychiatric Genomics Consortium performed a genome wide association study with 36,989 SZ patients and 113,075 controls. A single-nucleotide polymorphism was identified for SZ-risk in chromosome 8 in the CUB and Sushi multiple domains 1 (CSMD1) (Ripke et al., 2014). The CSMD1 protein has been shown to inhibit the complement system by promoting C3b/C4b degradation and preventing MAC formation (Escudero-Esparza et al., 2013), interestingly, only in the classical and lectin pathways (Kraus et al., 2006). Serum and plasma of SZ patients showed higher concentrations of complement proteins compared to controls (Mayilyan et al., 2008; Arakelyan et al., 2011; Sória et al., 2012). Other studies looking into serum C3 and C4 concentrations reported mixed results in SZ patients (Maes et al., 1997; Cazzullo et al., 1998; Li et al., 2016), indicating a need for standardization of complement protein collection and detection. Variations and copy number of the C4A gene are the most discussed findings for the role of complement system in SZ. In contrast, the roles of other complement components in cortical thinning/aberrant synapse pruning often reported in SZ need further investigation.
Complement system and autism spectrum disorder
The DSM-5 defines autism spectrum disorder as persistent deficits in social communication and interaction, restricted repetitive patterns of behaviors, interests, or activities, and symptoms presenting in early development. Our earlier findings showed that knockdown of C3 in the PFC of mice results in social interaction deficits and repetitive behavior (Fagan and Crider et al., 2017). In postmortem PFC samples from autism spectrum disorder (ASD) subjects, mRNA levels of C2, C5, and MASP-1 were significantly elevated, while levels of C1q, C3, and C4 were significantly lowered (Fagan and Crider et al., 2017). Reduced C4 levels were also observed in astrocytes derived from ASD patients, suggesting that C4 might be needed for proper synaptic pruning and lack of C4 may lead to greater brain connectivity seen in ASD (Mansur et al., 2021). Accordingly, increased dendritic spine density has been reported in the cortex of ASD patients (Hutsler and Zhang, 2010). In ASD patients, plasma levels of C1q and C3 proteins were elevated (Corbett et al., 2007; Momeni et al., 2012). Also, in the periphery, complement factor I, which degrades C3b, was increased in ASD patients (Momeni et al., 2012). Overall, increased complement activity can be seen peripherally in ASD subjects, but more studies are needed to determine the relationship between peripheral and CNS complement in ASD. Current literature in this area shows inconclusive results, especially when comparing peripheral and CNS complement.
Complement system and mood disorders
The DSM-5 defines major depressive disorder as a mood disorder expressing frequent and persistent depressed mood, decreased interest or pleasure in most activities, change in appetite, reduced cognition and physical activity, fatigue, feelings of worthlessness or excessive guilt, and recurring thoughts of death. Bipolar disorder is subdivided into Bipolar I, seen by manic episodes lasting at least a week, including excessive self-esteem, decreased need for sleep, increased desire to talk, racing thoughts, distractibility, and restlessness. Bipolar II is characterized by depressive episodes, lasting two weeks or more, alternating with hypomanic episodes. Serum levels of C1q have been observed to be significantly higher in major depressive disorder (MDD) patients (Yao and Li, 2020). In contrast, a lack of C1q in mice led to learned helplessness behavior (Madeshiya et al., 2022). Plasma levels of complement factor H (CFH), a regulator of the alternative pathway, were increased in drug-naïve MDD patients compared to healthy controls (Tang et al., 2021), while in another study CFH plasma levels were decreased (Chen et al., 2016). In postmortem PFC samples of depressed subjects, C3 mRNA levels were elevated, while inhibition of C3a signaling in mice protected them from stress-induced depressive-like behavior (Crider et al., 2018; Tripathi et al., 2021). CSF C5 levels were higher in MDD subjects compared to healthy controls (Ishii et al., 2018).
Serum levels of C3a and C5a were found increased in patients with bipolar disorder (BD), as measured by ELISA (Reginia et al., 2018). These complement components are essential for inducing proinflammatory signaling. In BD patients, higher serum concentrations of C1q, C3(b), C4, factor B and factor H were observed (Yu et al., 2021). In another study, chronic BD patients (n = 22) had a significantly higher peripheral monocyte mRNA expression of C1q, C4, and factor B, while having lower levels of serum C4, factor B, and C5b-9 when compared to first-time BD patients (n = 24) (Akcan et al., 2018). Although the above studies indicate a potential role of complement proteins in mood disorders, additional studies are warranted to establish a strong connection between complement system and mood disorders.
Complement system and anxiety disorders
The DSM-5 defines anxiety disorders as disorders that share an “excessive fear and anxiety related behavioral disturbances”. These fear and anxiety phenotypes include excessive reactions to current threats (fear) and to future threats (anxiety) and may include avoidance behaviors to reduce fear/anxiety. In rodent studies, mice lacking C3 or C3ar1 were tested for anxious-like behavior in the elevated plus maze (EPM), a validated test for anxiety in rodents. This test uses the innate rodent tendency to explore new areas and fear of exposed areas (Pellow et al., 1985; Waif and Frye, 2007; Westacott et al., 2022). Increased anxious behavior was exhibited by mice lacking C3ar1 (C3ar1 KO) who spent less time in the open arms per entry when compared to control (WT) mice, and mice lacking C3 (C3 KO). These findings were confirmed by the elevated zero maze, a variation of the EPM. Additionally, in the open field test, only C3ar1 KO mice spent less time in the center area, indicating increased anxious behavior (Westacott et al., 2022). In a separate cohort, plasma corticosterone levels were assayed 30 min after performing EPM. Basal corticosterone was not different between groups, but once exposed to EPM, plasma corticosterone increased 6–15 fold in all groups, with the greatest increase in C3ar1 KO mice. To differentiate between loss of C3 and C3ar1, Westacott and colleagues also investigated learned/conditioned fear. A neutral cue was associated and predicted an adverse outcome in the fear-potentiated startle paradigm (Davis, 2006; Campeau et al., 1997). Learned fear was measured by behavioral response to a noise in the presence of a cue (conditioned stimulus) previously administered with a mild foot shock (unconditioned stimulus). Fear learning was enhanced in C3 KO mice when compared to WT and C3ar1 KO mice as indicated by increased reactivity to startle stimulus (Westacott et al., 2022). Together, the above findings suggest that a lack of C3ar1 promotes anxiety-like behavior in mice. It is important to note that the mouse models used in the above studies were global knock out mice and therefore, functional compensation during development cannot be ruled out.
Complement system and trauma
Trauma activates the complement system as a first line of defense. Trauma patients may die from their primary injury or even from post-injury complement-mediated inflammation and cell damage from the terminal complement complex C5b-9 (Kohl, 2006; Burk et al., 2012). In a cohort of thirty-three patients with severe injuries (Injury Severity Score (ISS) > 25), trauma patients had a higher concentration of sC5b-9 at admission (Li et al., 2019). A positive correlation was found between sC5b-9 levels and systemic inflammatory response syndrome, a whole-body immune response with major complications. Complement components C4d, C3d, and C5b-9 were found on red blood cells (RBCs) in multiple types of traumas. There was a significant correlation found in patients with an ISS score over 9 and deposition of the C4d and C5b-9 on RBC surfaces for at least 72 h (Satyam et al., 2020). Other traumas, such as traumatic brain injury, spinal cord injury, and burn injury have been linked to dysregulation of the complement system. In rodents, a controlled brain contusion study in rats found significant C3 expression and T-cell infiltration at the injury site (Bellander et al., 2010; Chakraborty et al., 2018). Together, these studies suggest a potential role of complement system in trauma-induced CNS complications.
Complement system and therapeutics
Various components of the complement system have been targeted therapeutically including C5, C5aR, C1q, and C3 in both clinical and preclinical trials. Eculizumab, an antibody which inhibits C5 and terminal pathway initiation, is already on the market to treat atypical hemolytic uremic syndrome and paroxysmal nocturnal hemoglobinuria, both defined by chronic lysis of red blood cells (Wang et al., 1995; Ricklin and Lambris, 2007). A soluble form of complement receptor 1 (sCR1) was developed for helping patients to recover after coronary artery bypass graft surgery (TP10; Avant Immunotherapeutics), capable of inhibiting the classical and alternative pathways. Compstatin is a commercially available peptide that stops C3 from being cleaved into active C3a and C3b. A co-crystal structure of compstatin and C3c suggests inhibitory activity is related to disruption of convertase formation (Janssen et al., 2007). C5aR has been targeted to develop a specific antagonist because C5 signaling is associated with many complement-related disorders. PMX-53 is a cyclic peptidomimetic that acts as a C5aR antagonist and has completed phase 2 clinical trials (Kohl, 2006; Ricklin and Lambris, 2007). Recombinant human mannose-binding lectin has been used to boost the lectin pathway in patients with multiple myeloma undergoing chemotherapy and transplantations (Petersen et al., 2006). Although there are no clinical data on the use of complement therapy in neuropsychiatric disorders, the results from the above clinical trials would be helpful in planning complement-target based therapeutic approach in neuropsychiatric disorders.
Conclusions and perspectives
Synaptic plasticity plays a key role in memory and learning, and the loss of synapses lead to disruption of neuronal circuits, which is the underlying cause of many psychiatric disorders (Wang et al., 2018; Van Spronsen and Hoogenraad, 2010). The relationship between synaptic density and psychiatric symptoms has been demonstrated in a number of preclinical models and clinical studies. For example, chronic stress resulted in reduced synaptic density in rodents (Duman and Aghajanian, 2012). Imaging and postmortem studies showed deficits in brain regional volume (Price and Drevets, 2010; Bora et al., 2012; Koolschijn et al., 2009) and synaptic density (Feyissa et al., 2009; Kang et al., 2012; Duric et al., 2013) in depressed subjects. Similarly, lower levels of dendritic spines (Garey et al., 1998; Glantz and Lewis, 2000) and decreased expression of synaptic markers (Davidsson et al., 1999; Matosin et al., 2016; Halim et al., 2003; Eastwood et al., 2000; Funk et al., 2017; Osimo et al., 2019) were consistently reported in schizophrenia. As discussed above, complement proteins pay a major role in the regulation of synaptic plasticity. In addition to their primary roles to protect against infections, complement proteins maintain homeostasis in the brain by tagging damaged synapses and maintaining synaptic plasticity in development and throughout life. Although both schizophrenia and ASD are considered neurodevelopmental disorders resulting from atypical neural development, the complement system may function in different ways during neurodevelopment in these conditions. An increase in complement activation could lead to accelerated removal of functional synapses resulting in schizophrenia. On the other hand, a deficient complement system during neurodevelopment could result in weak synaptic pruning which may leads to increased dendritic spine density in ASD. Also, it is important to consider other mechanisms including the environmental factors influencing the development of these psychiatric conditions.
Does the activation of peripheral complement system affect the brain plasticity? The intact blood-brain barrier (BBB) restricts the access of complement proteins from the periphery. The local production of complement proteins is important for the immune regulation and homeostasis in the healthy brain. Accordingly, a number of studies have reported the expression of complement proteins in neuronal as well as non-neuronal cells in the brain. However, under chronic inflammatory conditions, a leaky BBB could permit the access of peripheral complement proteins to the brain (Fig. 1). Also, cytokines released in the periphery as a result of complement activation can influence the brain function via neural and/or humoral routes (Romanovsky et al., 2005; Hopkins, 2007). Therefore, blocking complement activation in the periphery may attenuate the increased inflammatory signaling and thereby regulate disease processes in psychiatric conditions. However, the complexity of the complement system as well as their importance in innate immune defense suggest that anti-complement therapies should be considered based on the complement status of the subjects, and their safety and potential side-effects should be rigorously and carefully monitored.
Fig. 1.
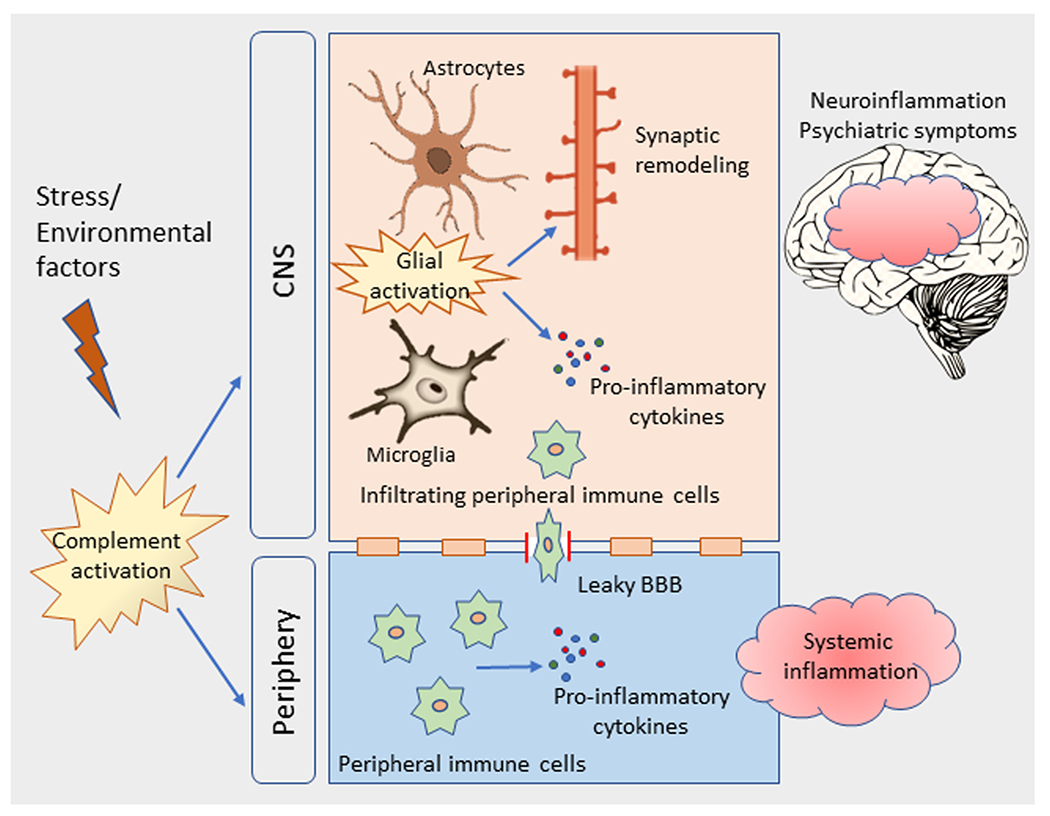
Schematic diagram showing the role of complement activation in chronic stress-mediated neuroinflammation and psychiatric symptoms. Stress or other environmental factors trigger complement activation in the central nervous system (CNS) and in the periphery. In the CNS, activated glial cells release pro-inflammatory cytokines and trigger excessive synaptic pruning. Activated peripheral immune cells release pro-inflammatory cytokines causing systemic inflammation. Also, peripheral immune cells infiltrate into the brain through leaky blood brain barrier. Both peripheral and CNS complement activation can lead to inflammation in the brain, and mediate psychiatric symptoms.
Acknowledgments
The authors acknowledge the funding support from US National Institute of Health/National Institute of Mental Health (NIMH) grants (MH120876 and MH121959), and the Merit Review Award (BX004758) from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development to AP. The contents do not represent the views of the Department of Veterans Affairs or the United States Government. AP acknowledges the funding support from Louis A Faillace Endowed Chair in Psychiatry.
Abbreviations:
- ASD
autism spectrum disorder
Footnotes
Conflict of interest
AP received research funding from Acadia Pharmaceuticals.
References
- Akcan U, Karabulut S, Ismail Küçükali C, Çakir S, Tüzün E, 2018. Bipolar disorder patients display reduced serum complement levels and elevated peripheral blood complement expression levels. Acta Neuropsychiatr. 30 (2), 70–78. 10.1017/NEU.2017.10. [DOI] [PubMed] [Google Scholar]
- Allswede DM, Zheutlin AB, Chung Y, Anderson K, Hultman CM, Ingvar M, Cannon TD, 2018. Complement gene expression correlates with superior frontal cortical thickness in humans. Neuropsychopharmacology 43 (3). 10.1038/npp.2017.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakelyan A, Zakharyan R, Khoyetsyan A, Poghosyan D, Aroutiounian R, Mrazek F, Petrek M, Boyajyan A, 2011. Functional characterization of the complement receptor type 1 and its circulating ligands in patients with schizophrenia. BMC Clin. Pathol 11 10.1186/1472-6890-11-10 (10–10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellander BM, Lidman O, Ohlsson M, Meijer B, Piehl F, Svensson M, 2010. Genetic regulation of microglia activation, complement expression, and neurodegeneration in a rat model of traumatic brain injury. Exp. Brain Res 205 (1), 103–114. 10.1007/S00221-010-2342-Z. [DOI] [PubMed] [Google Scholar]
- Bora E, Fornito A, Pantelis C, Yücel M, 2012. Gray matter abnormalities in Major Depressive Disorder: a meta-analysis of voxel based morphometry studies. J. Affect. Disord 138 (1–2) 10.1016/j.jad.2011.03.049. [DOI] [PubMed] [Google Scholar]
- Bordet J, 1909. The complement-deviation reaction of Bordet-Gengou. Journ. Med. Bord [Google Scholar]
- Burk AM, Martin M, Flierl MA, Rittirsch D, Helm M, Lampl L, Bruckner U, Stahl GL, Blom AM, Perl M, Gebhard F, Huber-Lang M, 2012. Early complementopathy after multiple injuries in humans. Shock 37 (4), 348–354. 10.1097/SHK.0B013E3182471795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau S, Falls WA, Cullinan WE, Helmreich DL, Davis M, Watson SJ, 1997. Elicitation and reduction of fear: behavioural and neuroendocrine indices and brain induction of the immediate-early gene c-fos. Neuroscience 78 (4). 10.1016/S0306-4522(96)00632-X. [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Theveneau E, Tzekou A, Tada M, Woods M, Page KM, Parsons M, Lambris JD, Mayor R, 2011. Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev. Cell 21 (6), 1026–1037. 10.1016/J.DEVCEL.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzullo CL, Saresella M, Roda K, Calvo MG, Bertrando P, Doria S, Clerici M, Salvaggio A, Ferrante P, 1998. Increased levels of CD8+ and CD4+45RA+ lymphocytes in schizophrenic patients. Schizophr. Res 31 (1), 49–55. 10.1016/S0920-9964(97)00153-9. [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Karasu E, Huber-Lang M, 2018. Complement after trauma: suturing innate and adaptive immunity. Front. Immunol 9 (SEP) 10.3389/fimmu.2018.02050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Deng-Feng Z, Zhi-Guo W, Dai-Hui P, Jun C, Jianliang N, Wenxin T, Lin X, Yong-Gang Y, Yi-Ru F, 2016. Complement factor H and susceptibility to major depressive disorder in Han Chinese. Br. J. Psychiatry 208 (5), 446–452. 10.1192/BJP.BP.115.163790. [DOI] [PubMed] [Google Scholar]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA, 2010. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. USA 107 (17), 7975–7980. 10.1073/PNAS.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett BA, Kantor AB, Schulman H, Walker WL, Lit L, Ashwood P, Rocke DM, Sharp FR, 2007. A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol. Psychiatry 12, 292–306. 10.1038/sj.mp.4001943. [DOI] [PubMed] [Google Scholar]
- Coulthard LG, Hawksworth OA, Conroy J, Lee JD, Woodruff TM, 2018. Complement C3a receptor modulates embryonic neural progenitor cell proliferation and cognitive performance. Mol. Immunol 101, 176–181. 10.1016/J.MOLIMM.2018.06.271. [DOI] [PubMed] [Google Scholar]
- Coulthard LG, Hawksworth OA, Li R, Balachandran A, Lee JD, Sepehrband F, Kurniawan N, Jeanes A, Simmons DG, Wolvetang E, Woodruff TM, 2017. Complement C5aR1 signaling promotes polarization and proliferation of embryonic neural progenitor cells through PKCζ. J. Neurosci 37 (22), 5395–5407. 10.1523/JNEUROSCI.0525-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crider A, Feng T, Pandya CD, Davis T, Nair A, Ahmed AO, Baban B, Turecki G, Pillai A, 2018. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain Behav. Immun 70, 246–256. 10.1016/J.BBI.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidsson P, Gottfries J, Bogdanovic N, Ekman R, Karlsson I, Gottfries CG, Blennow K, 1999. The synaptic-vesicle-specific proteins rab3a and synaptophysin are reduced in thalamus and related cortical brain regions in schizophrenic brains. Schizophr. Res 40 (1) 10.1016/S0920-9964(99)00037-7. [DOI] [PubMed] [Google Scholar]
- Davis M, 2006. Neural systems involved in fear and anxiety measured with fear-potentiated startle. Am. Psychol 61 (8) 10.1037/0003-066X.61.8.741. [DOI] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK, 2012. Synaptic dysfunction in depression: potential therapeutic targets. Science 338 (6103). 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duric V, Banasr M, Stockmeier CA, Simen AA, Newton SS, Overholser JC, Jurjus GJ, Dieter L, Duman RS, 2013. Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int. J. Neuropsychopharmacol 16 (1) 10.1017/S1461145712000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastwood SL, Cairns NJ, Harrison PJ, 2000. Synaptophysin gene expression in schizophrenia: investigation of synaptic pathology in the cerebral cortex. Br. J. Psychiatry 176 (MAR.). 10.1192/bjp.176.3.236. [DOI] [PubMed] [Google Scholar]
- Escudero-Esparza A, Kalchishkova N, K urbasic E, Jiang WG, Blom AM, 2013. The novel complement inhibitor human CUB and Sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J. 27 (12), 5083–5093. 10.1096/FJ.13-230706. [DOI] [PubMed] [Google Scholar]
- Fagan K, Crider A, Ahmed AO, Pillai A, 2017. Complement C3 expression is decreased in autism spectrum disorder subjects and contributes to behavioral deficits in rodents. Mol. Neuropsychiatry 3, 19–27. 10.1159/000465523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B, 2009. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 33 (1). 10.1016/j.pnpbp.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk AJ, Mielnik CA, Koene R, Newburn E, Ramsey AJ, Lipska BK, McCullumsmith RE, 2017. Postsynaptic density-95 isoform abnormalities in schizophrenia. Schizophr. Bull 43 (4) 10.1093/schbul/sbw173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TRE, Hirsch SR, 1998. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 65 (4). 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA, 2000. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57 (1), 65–73. 10.1001/ARCHPSYC.57.1.65. [DOI] [PubMed] [Google Scholar]
- Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, Reiner O, 2017. Developmental activities of the complement pathway in migrating neurons. Nat. Commun 8. 10.1038/NCOMMS15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik A, Sapir T, Woodruff TM, Reiner O, 2017. Serping1/C1 inhibitor affects cortical development in a cell autonomous and non-cell autonomous manner. Front. Cell. Neurosci 11 10.3389/FNCEL.2017.00169/BIBTEX (169–169). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim ND, Weickert CS, McClintock BW, Hyde TM, Weinberger DR, Kleinman JE, Lipska BK, 2003. Presynaptic proteins in the prefrontal cortex of patients with schizophrenia and rats with abnormal prefrontal development. Mol. Psychiatry 8 (9). 10.1038/sj.mp.4001319. [DOI] [PubMed] [Google Scholar]
- Honea R, Crow TJ, Passingham D, Mackay CE, 2005. Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am. J. Psychiatry 162 (12). 10.1176/appi.ajp.162.12.2233. [DOI] [PubMed] [Google Scholar]
- Hopkins SJ, 2007. Central nervous system recognition of peripheral inflammation: a neural, hormonal collaboration. Acta Biomed. 78 (Suppl. 1). [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H, 2010. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309. 10.1016/j.brainres.2009.09.120. [DOI] [PubMed] [Google Scholar]
- Ishii T, Hattori K, Miyakawa T, Watanabe K, Hidese S, Sasayama D, Ota M, Teraishi T, Hori H, Yoshida S, Nunomura A, Nakagome K, Kunugi H, 2018. Increased cerebrospinal fluid complement C5 levels in major depressive disorder and schizophrenia. Biochem. Biophys. Res. Commun 497 (2), 683–688. 10.1016/J.BBRC.2018.02.131. [DOI] [PubMed] [Google Scholar]
- Janssen BJC, Halff EF, Lambris JD, Gros P, 2007. Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J. Biol. Chem 282 (40), 29241–29247. 10.1074/JBC.M704587200/ATTACHMENT/F37BC4B9-A38C-4850-AF3F-C4D6E30964A3/MMC1.PDF. [DOI] [PubMed] [Google Scholar]
- Ji E, Boerrigter D, Cai HQ, Lloyd D, Bruggemann J, O’Donnell M, Galletly C, Lloyd A, Liu D, Lenroot R, Weickert TW, Shannon Weickert C, 2022. Peripheral complement is increased in schizophrenia and inversely related to cortical thickness. Brain Behav. Immun 101. 10.1016/j.bbi.2021.11.014. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, Lepack A, Majik MS, Jeong LS, Banasr M, Son H, Duman RS, 2012. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med 18 (9) 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl J, 2006. The role of complement in danger sensing and transmission. Immunol. Res 34 (2), 157–176. 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- Koolschijn PCMP, Van Haren NEM, Lensvelt-Mulders GJLM, Hulshoff Pol HE, Kahn RS, 2009. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum. Brain Mapp 30 (11) 10.1002/hbm.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus DM, Elliott GS, Chute H, Horan T, Pfenninger KH, Sanford SD, Foster S, Scully S, Welcher AA, Holers VM, 2006. CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J. Immunol 176 (7), 4419–4430. 10.4049/JIMMUNOL.176.7.4419. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang Q, Li N, Wang F, Xiang H, Zhang Z, Su Y, Huang Y, Zhang S, Zhao G, Zhou R, Mao L, Lin Z, Cai W, Fang Y, Xie B, Zhao M, Hong W, 2016. Plasma levels of Th17-related cytokines and complement C3 correlated with aggressive behavior in patients with schizophrenia. Psychiatry Res. 246. 10.1016/j.psychres.2016.10.061. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhao Q, Liu B, Dixon A, Cancio L, Dubick M, Lucca JD, 2019. Early complementopathy predicts the outcomes of patients with trauma. Trauma Surg. Acute Care Open 4. 10.1136/tsaco-2018-000217 (217–217). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeshiya AK, Whitehead C, Tripathi A, Pillai A, 2022. C1q deletion exacerbates stress-induced learned helplessness behavior and induces neuroinflammation in mice. Transl. Psychiatry 12 (1), 1–8. 10.1038/s41398-022-01794-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M, Delange J, Ranjan R, Meltzer HY, Desnyder R, Cooremans W, Scharpé S, 1997. Acute phase proteins in schizophrenia, mania and major depression: modulation by psychotropic drugs. Psychiatry Res. 66 (1), 1–11. 10.1016/S0165-1781(96)02915-0. [DOI] [PubMed] [Google Scholar]
- Mansur F, Teles E Silva AL, Gomes AKS, Magdalon J, de Souza JS, Griesi-Oliveira K, Passos-Bueno MR, Sertié AL, 2021. Complement c4 is reduced in ipsc-derived astrocytes of autism spectrum disorder subjects. Int. J. Mol. Sci 22 (14) 10.3390/IJMS22147579/S1 (7579–7579). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matosin N, Fernandez-Enright F, Lum JS, Engel M, Andrews JL, Gassen NC, Wagner KV, Schmidt MV, Newell KA, 2016. Molecular evidence of synaptic pathology in the CA1 region in schizophrenia. Npj Schizophr. 2. 10.1038/npjschz.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayilyan KR, Weinberger DR, Sim RB, 2008. The complement system in schizophrenia. Drug News Perspect. 21 (4) 10.1358/DNP.2008.21.4.1213349 (200–200). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarley RW, Wible CG, Frumin M, Hirayasu Y, Levitt JJ, Fischer IA, Shenton ME, 1999. MRI anatomy of schizophrenia. Biol. Psychiatry 45 (9). 10.1016/S0006-3223(99)00018-9 (1099–1099). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momeni N, Bergquist J, Brudin L, Behnia F, Sivberg B, Joghataei MT, Persson BL, 2012. A novel blood-based biomarker for detection of autism spectrum disorders. Transl. Psychiatry 2 (3). 10.1038/tp.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osimo EF, Beck K, Reis Marques T, Howes OD, 2019. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol. Psychiatry 24 (4). 10.1038/s41380-018-0041-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT, 2011. Synaptic pruning by microglia is necessary for normal brain development. Science 333 (6048), 1456–1458. 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, Hempstead BL, Littman DR, Gan WB, 2013. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 155 (7), 1596–1609. 10.1016/J.CELL.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellow S, Chopin P, File SE, Briley M, 1985. Validation of open: closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods 14 (3), 149–167. 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- Petersen KA, Matthiesen F, Agger T, Kongerslev L, Thiel S, Cornelissen K, Axelsen M, 2006. Phase I safety, tolerability, and pharmacokinetic study of recombinant human mannan-binding lectin. J. Clin. Immunol 26 (5), 465–475. 10.1007/S10875-006-9037-Z. [DOI] [PubMed] [Google Scholar]
- Prasad KM, Chowdari KV, D’Aiuto LA, Iyengar S, Stanley JA, Nimgaonkar VL, 2018. Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients–a pilot study. Transl. Psychiatry 8 (1), 1–12. 10.1038/S41398-018-0181-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Drevets WC, 2010. Neurocircuitry of mood disorders. Neuropsychopharmacology 35 (1). 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginia A, Kucharska-Mazur J, Jabłoński M, Budkowska M, Dołȩgowska B, Sagan L, Misiak B, Ratajczak MZ, Rybakowski JK, Samochowiec J, 2018. Assessment of complement cascade components in patients with bipolar disorder. Front. Psychiatry 9. 10.3389/FPSYT.2018.00614/BIBTEX (614–614). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD, 2007. Complement-targeted therapeutics. Nat. Biotechnol 25 (11), 1265–1275. 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, Lee P, Bullk-Sullivan B, Collier DA, Huang H, Pers TH, Agartz I, Agerbo E, Albus M, Alexander M, Amin F, Bacanu SA, Begemann M, Belliveau SA, O’Donovan MC, et al. , 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511 (7510). 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanovsky AA, Almeida MC, Aronoff DM, Ivanov AI, Konsman JP, Steiner AA, Turek VF, 2005. Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front. Biosci 10 (Suppl. 1) 10.2741/1690. [DOI] [PubMed] [Google Scholar]
- Sarma JV, Ward PA, 2011. The complement system. Cell Tissue Res. 343 (1), 227–235. 10.1007/S00441-010-1034-0/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyam A, Andreo K, Lapchak PH, Dalle Lucca JJ, Davis RB, Tsokos MG, Shapiro NI, Tsokos GC, 2020. Complement deposition on the surface of RBC after trauma serves a biomarker of moderate trauma severity: a prospective study. Shock 53 (1). 10.1097/SHK.0000000000001348 (16–16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar A, Bialas AR, De Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE, Daly MJ, Carroll MC, Stevens B, McCarroll SA, 2016. Schizophrenia risk from complex variation of complement component 4. Nature 530 (7589), 177–183. 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sória L, dos S, Gubert C, de M, Ceresér KM, Gama CS, Kapczinski F, 2012. Increased serum levels of C3 and C4 in patients with schizophrenia compared to eutymic patients with bipolar disorder and healthy. Braz. J. Psychiatry 34 (1), 119–120. 10.1590/S1516-44462012000100022. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AMM, Lambris JD, Smith SJ, John SWM, Barres BA, 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131 (6), 1164–1178. 10.1016/J.CELL.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Tang W, Liu H, Chen L, Zhao K, Zhang Y, Zheng K, Zhu C, Zheng T, Liu J, Wang D, Yu L, Fang X, Zhang C, Su KP, 2021. Inflammatory cytokines, complement factor H and anhedonia in drug-naïve major depressive disorder. Brain Behav. Immun 95, 238–244. 10.1016/J.BBI.2021.03.022. [DOI] [PubMed] [Google Scholar]
- Tripathi A, Whitehead C, Surrao K, Pillai A, Madeshiya A, Li Y, Khodadadi H, Ahmed AO, Turecki G, Baban B, Pillai A, 2021. Type 1 interferon mediates chronic stress-induced neuroinflammation and behavioral deficits via complement component 3-dependent pathway. Mol. Psychiatry 26 (7), 3043–3059. 10.1038/S41380-021-01065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Spronsen M, Hoogenraad CC, 2010. Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep 10 (3) 10.1007/s11910-010-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerhuis R, Boshuizen RS, Morbin M, Mazzoleni G, Hoozemans JJM, Langedijk JPM, Tagliavini F, Langeveld JPM, Eikelenboom P, 2005. Activation of human microglia by fibrillar prion protein-related peptides is enhanced by amyloid-associated factors SAP and C1q. Neurobiol. Dis 19 (1–2), 273–282. 10.1016/J.NBD.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Walf AA, Frye CA, 2007. Estradiol decreases anxiety behavior and enhances inhibitory avoidance and gestational stress produces opposite effects. Stress 10 (3). 10.1080/00958970701220416. [DOI] [PubMed] [Google Scholar]
- Wang X, Christian KM, Song H, Ming G. li, 2018. Synaptic dysfunction in complex psychiatric disorders: from genetics to mechanisms. Genome Med. 10 (1) 10.1186/S13073-018-0518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rollins SA, Madri JA, Matis LA, 1995. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc. Natl. Acad. Sci. USA 92 (19), 8955–8959. 10.1073/PNAS.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westacott LJ, Humby T, Haan N, Brain SA, Bush EL, Toneva M, Baloc AI, Moon AL, Reddaway J, Owen MJ, Hall J, Hughes TR, Paul Morgan B, Gray WP, Wilkinson LS, 2022. Complement C3 and C3aR mediate different aspects of emotional behaviours; relevance to risk for psychiatric disorder. Brain Behav. Immun 99, 70–82. 10.1016/J.BBI.2021.09.005. [DOI] [PubMed] [Google Scholar]
- Yao Q, Li Y, 2020. Increased serum levels of complement C1q in major depressive disorder. J. Psychosom. Res 133 10.1016/J.JPSYCHORES.2020.110105 (110105–110105). [DOI] [PubMed] [Google Scholar]
- Yu H, Ni P, Tian Y, Zhao L, Li M, Li X, Wei W, Wei J, Du X, Wang Q, Guo W, Deng W, Ma X, Coid J, Li T, 2021. Association of Plasma Complement System With Brain Structure Deficits in Bipolar and Major Depressive Disorders. ⟨ 10.21203/RS.3.RS-922528/V1⟩. [DOI] [PubMed] [Google Scholar]